

SUMMARY
Epigenetic gene regulation shapes neuronal fate in the embryonic nervous system. Post-embryonically, epigenetic signaling within neurons has been associated with impaired learning, autism, ataxia and schizophrenia. Epigenetic factors are also enriched in glial cells. However, little is known about epigenetic signaling in glia and nothing is known about the intersection of glial epigenetic signaling and presynaptic homeostatic plasticity. During a screen for genes involved in presynaptic homeostatic synaptic plasticity, we identified an essential role for the histone acetyltransferase and deubiquitinase SAGA complex in peripheral glia. We present evidence that the SAGA complex is necessary for homeostatic plasticity, demonstrating involvement of four new genes in homeostatic plasticity. This is also evidence that glia participate in presynaptic homeostatic plasticity, invoking previously unexplored intercellular, homeostatic signaling at a tripartite synapse. We show, mechanistically, SAGA signaling regulates the composition of and signaling from the extracellular matrix during homeostatic plasticity.
Keywords: Glia, Peripheral glia, SAGA, Histone acetyltransferase, Histone deubiquitinase, Epigenetic regulation, Multiplexin, Endostatin, Extracellular matrix, Intercellular signaling, Synaptic homeostasis, Homeostatic plasticity, Neuromuscular junction, Epilepsy, Schizophrenia, Autism
INTRODUCTION
The homeostatic modulation of presynaptic neurotransmitter release, termed presynaptic homeostatic potentiation (PHP), is a fundamental form of neural regulation. It is highly conserved from Drosophila to human (Davis, 2013; Davis and Muller, 2015). PHP can be initiated by the loss or inhibition of postsynaptic glutamate receptors and is expressed as a compensatory enhancement of presynaptic vesicle release (Frank et al., 2006; Petersen et al., 1997). Large-scale forward genetic screens have identified numerous genes that function within the pre- and postsynaptic elements that are essential for the initiation and expression of presynaptic homeostatic plasticity. Yet, to date, there has been no evidence that glia participate in the mechanisms of PHP, nor has there been any evidence for the participation of epigenetic gene regulation.
Epigenetic regulation of gene expression has a profound impact on neuronal cell fate specification and neural circuit development in the embryonic nervous system (Baizabal et al., 2018; Yao et al., 2016). The importance of epigenetic control of gene expression is evident based on the numerous neurodevelopmental, neuropsychiatric and neurodegenerative disorders that have been associated with loss of epigenetic controls including Rett Syndrome, Fragile X, Autism, Schizophrenia, Spinocerebellar Ataxia (SCA), and Amyotrophic Lateral Sclerosis (ALS) (Amir et al., 1999; Coffee et al., 1999; De Rubeis et al., 2014; Jimenez-Pacheco et al., 2017; Lindblad et al., 1996; Sun et al., 2016; Tsankova et al., 2007). It has become clear that epigenetic gene regulation is dynamic and sensitive to changes in neuronal function in both the developing and mature nervous systems. Epigenetic factors that function within neurons have been demonstrated to influence both Hebbian and non-Hebbian neural plasticity (Guzman-Karlsson et al., 2014). Specifically, levels of histone acetylation and DNA methylation within neurons have been associated with, and are required for, normal long-term potentiation (LTP) and memory formation (Graff and Tsai, 2013; Guzman-Karlsson et al., 2014). Multiple neuronal epigenetic factors have also been linked to non-Hebbian postsynaptic quantal scaling (Benevento et al., 2016; Blackman et al., 2012; Meadows et al., 2015; Yu et al., 2015).
The bulk of existing information underscores the importance of epigenetic signaling within neurons. Less is known about epigenetic gene regulation in glia. This is particularly true with respect to Hebbian and homeostatic plasticity, but also applies to neurological and psychiatric disease. This gap in knowledge exists despite the fact that glia are key players that control many aspects of neural development including synaptogenesis, synapse refinement, circuit maturation and quantal scaling (Beattie et al., 2002; Christopherson et al., 2005; Eroglu et al., 2009; Singh et al., 2016; Stellwagen and Malenka, 2006; Stogsdill et al., 2017) and are increasingly linked to neurodegnerative pathology (Hong et al., 2016; Stevens et al., 2007). The evidence in favor of an essential function for epigenetic signaling in glia exists primarily in the context of nerve injury (Arthur-Farraj et al., 2017; Koreman et al., 2018; Staszewski and Prinz, 2014). As yet, there is no direct evidence demonstrating that epigenetic signaling or glia is necessary for either the induction or expression of PHP.
Here we demonstrate that multiple genes encoding members of the Spt-Ada-Gcn5 Acetyltransferase and Deubiquitinase (SAGA) complex are specifically required in peripheral glia for presynaptic homeostatic plasticity. The SAGA complex is a prominent histone acetyltransferase protein complex that is conserved from yeast to human (Helmlinger and Tora, 2017; Lee and Workman, 2007). Remarkably, SAGA-dependent signaling is not constrained to the synapse, but appears to propagate throughout the extracellular space surrounding the synapse, axon, soma and dendrites. This represents a shift in the composition of the extracellular environment of the entire neuron during homeostatic plasticity. We propose that glial-dependent, epigenetic specification of the extracellular neuronal environment may be a general mechanism that is necessary for the life-long maintenance of homeostatic plasticity, existing for months in Drosophila (Mahoney et al., 2014), years in mice (Plomp et al., 1992) and decades in human (Cull-Candy et al., 1980).
RESULTS
The SAGA Complex Components Ada2b and Sgf11 are Necessary for PHP.
The SAGA complex in Drosophila is composed of approximately twenty proteins, encompassing histone acetyltransferase and deubiquitinase catalytic activities (Weake and Workman, 2012). An ongoing effort to screen for genes involved in homeostatic plasticity identified a potential function for ada2b during PHP and this was subsequently expanded to a test of four genes, all components of the SAGA complex: two acetyltransferases ada2b (ada2b1, (Qi et al., 2004)) and gcn5 (gcn5E333st/gcn5Q186st, (Carre et al., 2005)) and two deubiquitinases sgf11 (sgf11e01308, (Weake et al., 2008)) and non-stop ((Weake et al., 2008), Figure 1A). First, we demonstrated that loss of these individual genes impairs the rapid induction of synaptic homeostasis. Bath application of 15μM Philanthotoxin (PhTX) induced a ~50% reduction of the average miniature excitatory postsynaptic potential amplitude (mEPSP amplitude) at the neuromuscular junction (NMJ; Figure 1B–1D). In wild-type, an increase in presynaptic release (quantal content; Figure 1C and 1F) was observed that offset the change in mEPSP amplitude and restored postsynaptic excitation to baseline values (Figure 1E). By contrast, there is no change in quantal content in the ada2b, gcn5 or sgf11 mutants (Figure 1C and 1F). As a consequence, average excitatory postsynaptic potential (EPSP) amplitude at mutant synapses is significantly smaller in the presence of PhTX compared to the absence of PhTX (Figure 1E). Thus, three independent genes, all members of the SAGA complex, encompassing two different enzymatic activities, are essential for the rapid induction of PHP.
Figure 1. SAGA Complex Gene Mutations Block PHP.
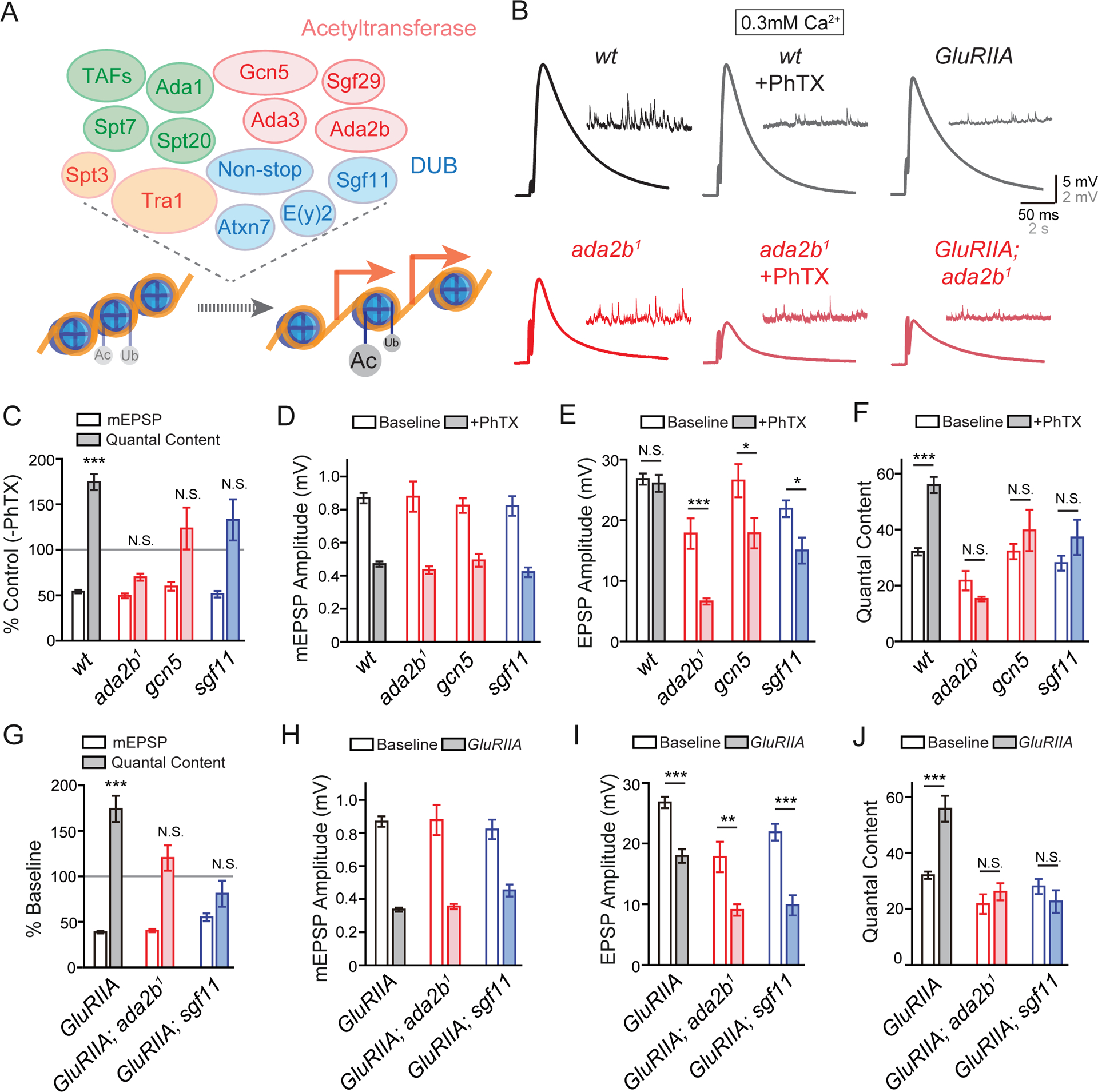
(A) Diagram of the Acetyltransferase (red) and Deubiquitinase (DUB, blue) modules in the SAGA complex in Drosophila (upper panel). Other components include: structural (green) and Transcription Factor (TF) Binding and Transcription factor Binding Protein (TBP) Binding modules (orange). Acetylation and deubiquitination of histone H3 leads to relaxed chromatin structure and active gene transcription (red arrows, lower panel).
(B) Representative EPSP and mEPSP traces in wild-type (wt, black) and the ada2b1 mutant (ada2b1, red) in the absence (−PhTX) and the presence of philanthotoxin (+PhTX). At far right the GluRIIA mutant and GluRIIA;ada2b1 double mutant are shown.
(C) mEPSP amplitudes (open bars) and presynaptic release (quantal content, filled bars). Data for each genotype is presented as the percent change in PhTX compared to the same genotype recorded in the absence of PhTX. Genotypes: wild-type (wt), ada2b (ada2b1), gcn5 (gcn5E333st/gcn5Q186st), sgf11 (sgf11e01308). Mean ± SEM; ***p < 0.001, N.S. not significant; Student’s t-test for pair-wise comparisons within a genotype +/− PhTX.
(D) – (F) Non-normalized values for analysis of the mutants in (C). Average mEPSP amplitude (D), EPSP amplitude (E) and presynaptic release (quantal content, F) in the absence (open) and presence (filled bars) of PhTX. Genotypes as in (C). Mean ± SEM; *p < 0.05, ***p < 0.001, N.S. not significant; Student’s t-test for pair-wise comparisons within a genotype +/− PhTX.
(G) – (J) Data presented for indicated genotypes as in (C) – (F). The following genotypes are presented: GluRIIA (gray bars), GluRIIA;ada2b1 double mutants (red bars), GluRIIA;sgf11e01308 mutants (blue bars). Mean ± SEM; **p < 0.01, ***p < 0.001, N.S. not significant; Student’s t-test for pair-wise comparisons.
Next, we assessed whether the SAGA complex is required for the long-term maintenance of PHP, induced by genetic deletion of the postsynaptic glutamate receptor subunit GluRIIA (Petersen et al., 1997) (Figure 1G and 1I). We examined two different GluRIIA and SAGA double mutant combinations: GluRIIA;ada2b1 and GluRIIA;sgf11e01308. In both double mutants, the mEPSP amplitudes are significantly decreased (Figure 1G and 1H) and the homeostatic regulation of presynaptic release is completely blocked (Figure 1G and 1J). As a consequence, EPSP amplitudes are significantly decreased in the double mutants compared to the same genotype alone at baseline. Based on these data, we propose that the SAGA complex is necessary for both the rapid induction and maintenance of presynaptic homeostasis.
Ada2b is Necessary in Peripheral Glia for PHP.
In order to examine the specific cell type in which the Drosophila SAGA complex functions during presynaptic homeostasis we tested SAGA activity in motoneurons, muscle and in peripheral glia using tissue-specific, RNAi-mediated, gene knockdown (Figure 2). Specifically, we expressed UAS-ada2b RNAi in motoneurons (OK371-Gal4), muscle (MHC-Gal4), perineurial glia (Egr-Gal4, (Keller et al., 2011); NP6293-Gal4, (Awasaki et al., 2008; Stork et al., 2012)) and astrocyte-like glia (Alrm-Gal4, (Doherty et al., 2009; Stork et al., 2012)). We demonstrated that PhTX-induced synaptic homeostasis was completely normal when ada2b-RNAi was expressed in either mononeurons or muscle (Figure 2C and S1). However, when ada2b-RNAi was expressed in perineurial glia, we found a complete block of PHP (Figure 2C, 2D and S1). We also demonstrated that perineurial glia, identified by expression of Egr-Gal4 and NP6293-Gal4, extended to the NMJ (Figure 2B). Although these glia do not extend along the full length of the presynaptic nerve terminal, they are localized in close proximity to presynaptic boutons at the NMJ (Figure 2B). This is consistent with prior ultrastructural data examining glial morphology at the site of nerve-muscle contact in third-instar larvae and other prior reports (Brink et al., 2012; Keller et al., 2011; Kerr et al., 2014).
Figure 2. Loss of SAGA Complex Gene Expression within Glia Impairs PHP.
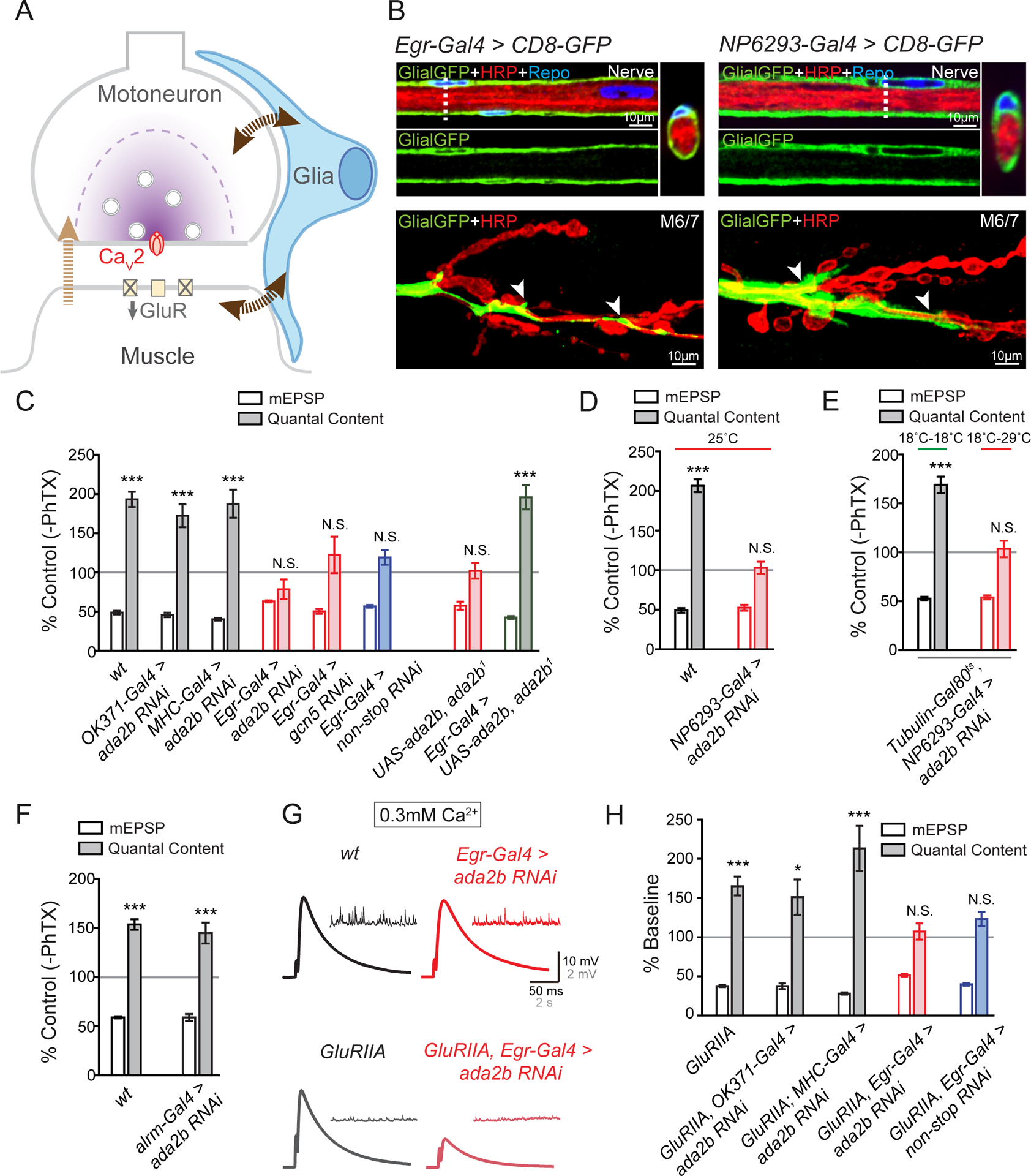
(A) Schematic of the Drosophila NMJ. Inhibition of postsynaptic glutamate receptor sensitivity leads to a calcium-channel dependent increase of presynaptic release in motoneurons. Glia (blue) are present at the NMJ. In addition to a retrograde signal from muscle to motoneurons (light brown arrow), signaling to and from glia (dark brown arrows) could also contribute to presynaptic homeostatic plasticity.
(B) Representative confocal sections of peripheral glia surrounding the axon bundle in the peripheral nerve. Glial-specific expression of UAS-CD8-GFP (Egr-Gal4>CD8-GFP, Glial-GFP, green, left panel; NP6293-Gal4>CD8GFP, Glial-GFP, green, right panel), neuronal membrane (HRP, red), and glial nuclei (immunolabeling for Repo, blue). Dotted vertical line indicated site of optical cross section, shown at upper panel on the right, rotated 90°. Bottom, the distribution of glia membrane at the NMJ of muscles 6 and 7 (M6/7). White arrows indicate the localization of glia at the NMJ.
(C) mEPSP amplitudes (open bars) and presynaptic release (quantal content, filled bars). Data for each genotype is presented as the percent change in PhTX compared to the same genotype recorded in the absence of PhTX. Genotypes as follows: wild-type (wt, gray bars), motoneuron-specific knockdown of ada2b (OK371-Gal4>ada2b RNAi, gray bars), muscle-specific knockdown of ada2b (MHC-Gal4>ada2b RNAi, gray bars), glial-specific knockdown of ada2b (Egr-Gal4>ada2b RNAi, red bars), glial-specific knockdown of gcn5 (Egr-Gal4>gcn5 RNAi, red bars), glial-specific knockdown of non-stop (Egr-Gal4>non-stop RNAi, blue bars), heterozygous UAS-ada2b in the homozygous ada2b1 mutant background without a Gal4 driver (UAS-ada2b,ada2b1), and glial-specific expression of UAS-ada2b in ada2b1mutant (Egr-Gal4>UAS-ada2b, ada2b1). Mean ± SEM; ***p < 0.001, N.S. not significant; Student’s t-test.
(D) mEPSP amplitudes (open bars) and presynaptic release (quantal content, filled bars). Data for each genotype is presented as the percent change in PhTX compared to the same genotype recorded in the absence of PhTX. Genotypes as follows: wild-type (wt, gray bars), perineurial glial-specific knockdown of ada2b (NP6293-Gal4>ada2b RNAi, red bars). All animals are raised at 25°C. Mean ± SEM; ***p < 0.001, N.S. not significant; Student’s t-test. See also Figures S1 and S2.
(E) mEPSP amplitudes (open bars) and presynaptic release (quantal content, filled bars). Data is presented as the percent change in PhTX compared to the same genotype recorded in the absence of PhTX. Perineurial glial-specific knockdown of ada2b (Tubulin-Gal80ts, NP6293-Gal4>ada2b RNAi) at permissive (18°C–29°C, red line) and non-permissive temperatures (18°C–18°C, green line). Mean ± SEM; ***p < 0.001, N.S. not significant; Student’s t-test.
(F) mEPSP amplitudes (open bars) and presynaptic release (quantal content, filled bars) in astrocyte-specific knockdown of ada2b (Alrm-Gal4>ada2b RNAi). Data is presented as the percent change in PhTX compared to the same genotype recorded in the absence of PhTX. Mean ± SEM; ***p < 0.001, N.S. not significant; Student’s t-test.
(G) Representative traces for EPSP and spontaneous mEPSP for wild-type (wt, black), GluRIIA mutant (GluRIIA, gray), glial-specific knockdown of ada2b (Egr-Gal4>ada2b RNAi, red) and glial-specific knockdown of ada2b in the GluRIIA mutant background (GluRIIA,Egr-Gal4>ada2b RNAi, light red).
(H) Quantification as in (C). Data for each genotype is expressed as percent change compared to the same genotype recorded in the absence of the GluRIIA mutation. The following genotypes are presented: GluRIIA (gray bars), GluRIIA,OK371-Gal4>ada2b RNAi (gray bars), GluRIIA;MHC-Gal4>ada2b RNAi (gray bars), GluRIIA,Egr-Gal4>ada2b RNAi (red bars), and GluRIIA,Egr-Gal4>non-stop RNAi (blue bars). Mean ± SEM; *p < 0.001, ***p < 0.001, N.S. not significant; Student’s t-test. See also Figure S1 and S2.
We have performed a series of control experiments to confirm a role for ada2b within perineurial glia during post-embryonic development. First, we confirmed the identity and specificity of the perineurial glial Gal4 drivers (Figure S2). We also characterized the identity of Egr positive cells in the peripheral glia. The percentage of total peripheral glia that are labeled by Egr-Gal4 is similar to NP6293-Gal4, a confirmed perineurial Gal4 driver (Awasaki et al., 2008; Stork et al., 2012) (Figure S2B and S2C). Moreover, when we used both Gal4 drivers to express UAS-GFP with a nucleus localization sequence (UAS-GFP.nls), the ratio of GFP positive nuclei to all glial nuclei is unchanged, indicating that Egr-Gal4 and NP6293-Gal4 label largely overlapping, if not the same population of peripheral glia (Figure S2B). Both Egr-Gal4 and NP6293-Gal4 block PHP when used to express UAS-ada2b RNAi, arguing for peripheral glial-specific activity that is likely localized to perineurial glial cells, as defined by these Gal4 lines.
Next, we addressed the possibility that ada2b functions during early embryonic development to affect synapse function. To address this issue, we used a temperature sensitive Tubulin-Gal80ts driver, combined with NP6293-Gal4, to induce the expression of UAS-ada2b RNAi only during post-embryonic larval development. We demonstrate that presynaptic homeostatic plasticity is still impaired at the NMJ when ada2b is disrupted in perineurial glia following the completion of embryonic development (Figure 2E; temperature shift 18–29°C initiated 30 hours after egg laying).
Next, we performed tissue-specific genetic rescue experiments. We expressed UAS-ada2b specifically in glia in the ada2b1 mutant and fully restored PHP in the ada2b1 mutant background (Figure 2C). Note that PhTX-induced synaptic homeostasis remained completely blocked in ada2b1 mutants harboring only the UAS-ada2b transgene without a source of Gal4 present (Figure 2C), emphasizing that the rescue of PHP is due to Gal4-mediated overexpression of UAS-ada2b in glia.
Finally, we asked whether the SAGA complex also functions in other glial cell types within the central nervous system during PHP. In particular, astrocyte-like glial cells populate the Drosophila CNS (Stork et al., 2012) and astrocytes are known to participate in other forms of homeostatic plasticity in mammalian cultured neurons (Stellwagen and Malenka, 2006). We used the previously characterized astrocyte-like glial Gal4 driver Alrm-Gal4 (Doherty et al., 2009; Stork et al., 2012) to drive the expression of UAS-ada2b RNAi and observed normal homeostatic plasticity. These data suggest that ada2b is not necessary in astrocyte-like glia, within the CNS for synaptic homeostasis at the NMJ (Figure 2F).
Additional SAGA Components, Non-Stop and Gcn5, are Necessary for PHP.
We have extended our analysis of the SAGA complex to include two additional genes: non-stop and gcn5. The non-stop gene encodes a deubiquitinase enzyme and gcn5 has acetyltransferase activity (Figure 1). When we used Egr-Gal4 to drive the expression of either UAS-gcn5 RNAi or UAS-non-stop RNAi, we found that the rapid induction of homeostatic plasticity was blocked (Figure 2C). These data are consistent with a general requirement for the activity of SAGA complex for the normal expression of PHP.
Finally, we tested the sustained expression of PHP. When either UAS-ada2b RNAi was driven in motoneurons or muscles in the GluRIIA mutant background, we observed normal PHP (Figure 2H). However, when UAS-ada2b RNAi was driven in glia (GluRIIA, Egr-Gal4>ada2b RNAi), the sustained expression of homeostasis was blocked (Figure 2G and 2H). We observed similar impairment of sustained homeostasis when non-stop was disrupted specifically in glia in the GluRIIA mutant background (Figure 2H). Two conclusions can be made at this point. First, our data demonstrate the essential participation of peripheral glia in both the rapid induction and sustained expression of PHP. Second, we demonstrate that multiple components of the SAGA complex are essential for presynaptic homeostatic plasticity, acting within peripheral glia.
Normal Glial and Synapse Morphology.
We examined the morphology of the NMJ and surrounding glia. Loss of ada2b expression in glia (Egr-Gal4>ada2b RNAi) had no effect on the shape or distribution of glia within the peripheral nerve or at the NMJ of muscles 6/7 (Figure 3A–3C). Total active zone number, estimated by quantification of Brp puncta number (Marie et al., 2004) is either unaltered following loss of ada2b expression in glia (Egr-Gal4>ada2b RNAi; abdominal segment 3), or slightly increased (abdominal segment 2) compared to wild-type (Figure 3D). There is a small increase of the total postsynaptic area (region of Dlg immunostaining) at the NMJ following expression of ada2b RNAi in glia, but only in abdominal segment 3 (Figure 3E). Finally, there is a slight increase of total bouton number in segment 2 but no change of Brp density in the ada2b RNAi mutant (Figure 3F and 3G). In conclusion, although small differences in NMJ anatomy are observed, there are no consistent changes that could account for the block of PHP following glial-specific depletion of ada2b expression. Finally, we performed a further control to ensure that Egr-Gal4 line does not itself interfere with PHP. Eiger (Egr) is the Drosophila homologue of mammalian TNF-α, important for homeostatic scaling in mice (Stellwagen and Malenka, 2006). We examined PhTX-induced presynaptic homeostasis in an egr null mutation (egrΔ25; (Keller et al., 2011)) and demonstrated that PHP was completely normal (Figure S3). This experiment also effectively rules out Eiger as a potential candidate for glial-neuronal signaling downstream of the SAGA complex in peripheral glia for PHP.
Figure 3. Glial-specific Impairment of ada2b Does Not Affect Glia or Synapse Morphology.
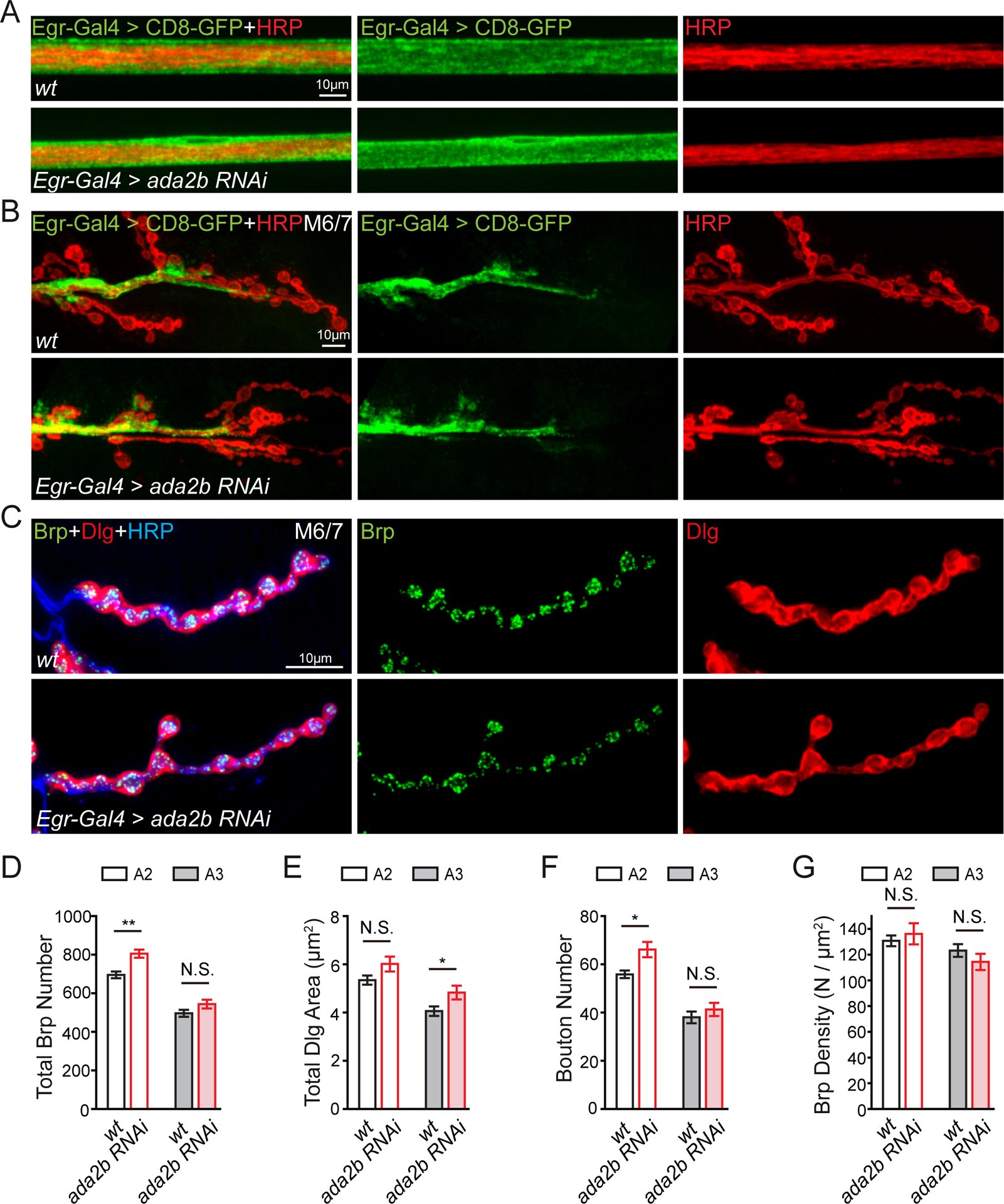
(A) Representative confocal images of peripheral glia surrounding a peripheral nerve. Glia = UAS-CD8-GFP (green) and neuronal membrane = HRP (red). Glial-specific knockdown of ada2b (Egr-Gal4>ada2b RNAi) compared to wild-type (wt).
(B) Representative confocal images of peripheral glia at the NMJ in wild-type (wt) and ada2b mutants (Egr-Gal4>ada2b RNAi), labeling as in (A).
(C) NMJ as in (B) immunolabeled with anti-Bruchpilot (Brp, green, presynaptic), anti-discs large (Dlg, red, postsynaptic) and the neuronal membrane (anti-HRP, blue).
(D) – (G) The total number presynaptic Brp puncta (D), total area of Dlg (E), total number of synaptic boutons (F), and Brp density (Brp number/Dlg area, G) at muscle 6/7 in abdominal segment 2 (A2, open bars) and segment 3 (A3, filled bars). Genotypes and sample size: Egr-Gal4>ada2b RNAi, N=7 NMJ for A2, N=7 NMJ for A3, n=4 animals, red bars); wild-type (wt, N=7 NMJ for A2, N=7 NMJ for A3, n=4 animals, black bars). Mean ± SEM; *p < 0.05, **p < 0.01, N.S. not significant; Student’s t-test. See also Figure S3.
Modulation of SAGA-dependent H3K9 and H3K14 Acetylation during PHP.
The next question we address is whether SAGA complex activity is altered, in peripheral glia, following muscle-specific disruption of postsynaptic glutamate receptors. Lysines at position 9 and 14 in histone H3 (H3K9 and H3K14) are direct targets of SAGA complex-dependent acetylation (Carre et al., 2005; Qi et al., 2004; Weake et al., 2008). We acquired antibodies specific for acetylation of H3K9 (H3K9Ac) and H3K14 (H3K14Ac) and assessed the abundance of these epigenetic markers in peripheral glia (Figure S4). The nuclei of peripheral glia are distributed throughout the peripheral nerves that extend from the CNS to the musculature. Both anti-H3K9Ac and anti-H3K14Ac immunostaining label the nuclei of peripheral glia (Figure S4). If this labeling is due to SAGA complex activity, then it should be diminished in the ada2b mutant. We demonstrated that H3K9Ac and H3K14Ac staining intensity is significantly reduced in the ada2b1 mutant background compared to wild-type (Figure 4A–4F). Finally, we demonstrated that ada2b-dependent H3K9Ac and H3K14Ac staining occurs in Egr-Gal4 positive peripheral glia, the same peripheral glia that are necessary for PHP. Thus, ada2b is essential for H3K9Ac and H3K14Ac acetylation in Egr positive perineurial glia.
Figure 4. Glial H3K9Ac and H3K14Ac are Enhanced in Egr Positive Cells in the GluRIIA Mutant.
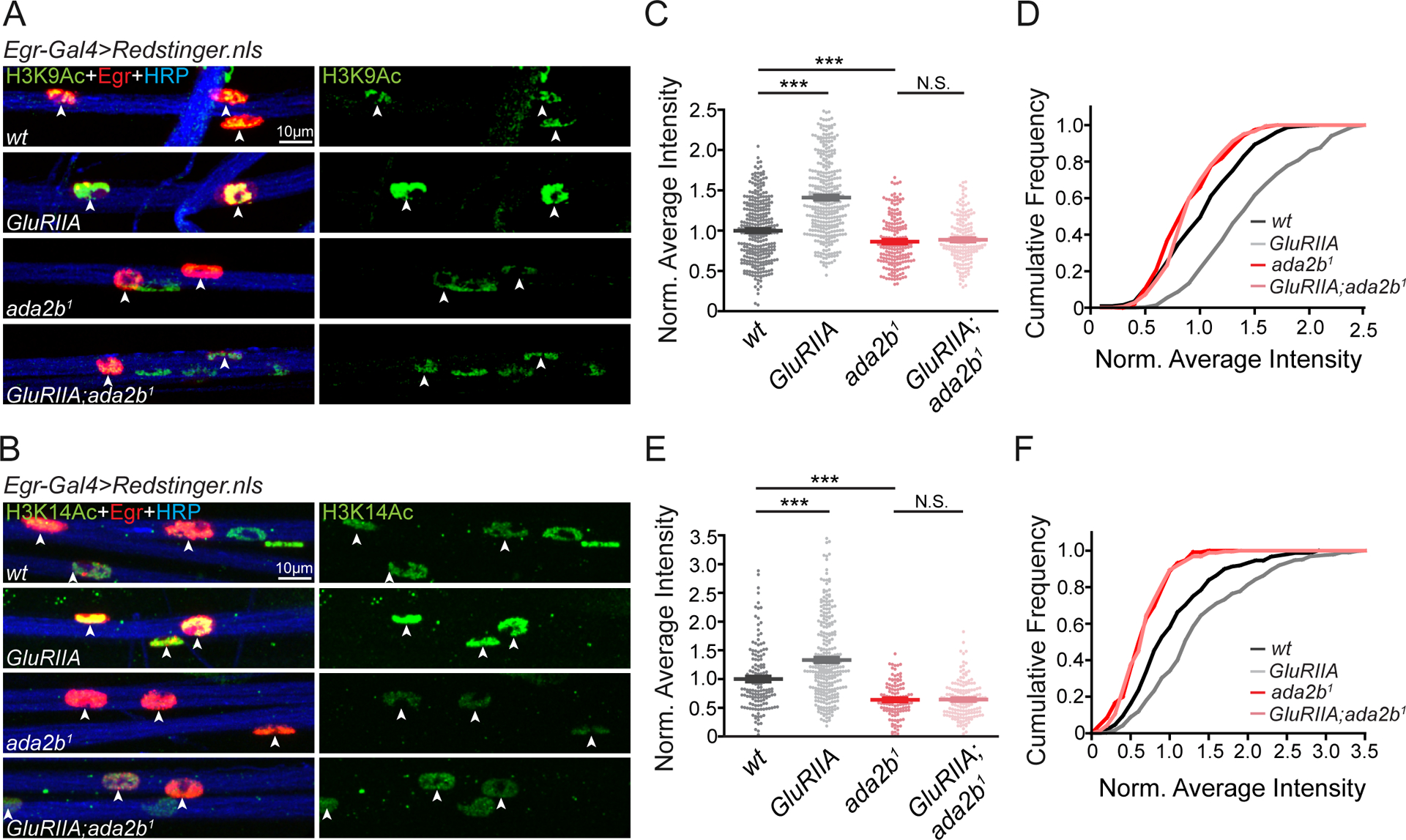
(A) – (B) Representative confocal images of acetylated H3K9 (H3K9Ac, A) and acetylated H3K14 (H3K14Ac, B) in glial nuclei on peripheral nerves. Acetylated H3K9 or H3K14 (H3K9/14Ac, green), Egr positive glial nuclei (Egr-Gal4>Redstinger.nls, red) and neuronal membrane (HRP, blue) are shown for wild-type (wt), GluRIIA mutants, ada2b1 mutants, and GluRIIA;ada2b1 double mutants.
(C) – (D) Quantification of average H3K9Ac fluorescence intensity (C) and cumulative distribution of average H3K9Ac intensity (D) within Egr positive glial nuclei. Average intensities for wild-type (wt, N=309 nuclei, n=6 animals, black), GluRIIA mutants (GluRIIA, N=349 nuclei, n=6 animals, gray), ada2b1 mutants (ada2b1, N=188 nuclei, n=4 animals, red) and GluRIIA;ada2b1 double mutants (GluRIIA;ada2b1, N=179 nuclei, n=4 animals, light red) are normalized to wild-type values. Mean ± SEM; ***p < 0.001; N.S. not significant; One-way ANOVA, Bonferroni’s multiple comparison test.
(E) – (F) Quantification of average H3K14Ac fluorescence intensity (E) and cumulative distribution of average H3K14Ac intensity (F) within Egr positive glial nuclei. Average intensities for wild-type (wt, N=148 nuclei, n=4 animals, black), GluRIIA mutants (GluRIIA, N=284 nuclei, n=4 animals, gray), ada2b1 mutants (ada2b1, N=115 nuclei, n=4 animals, red) and GluRIIA;ada2b1 double mutants (GluRIIA;ada2b1, N=160 nuclei, n=4 animals, light red) are normalized to wild-type values. Mean ± SEM; ***p < 0.001; N.S. not significant; One-way ANOVA, Bonferroni’s multiple comparison test. See also Figure S4.
Next, we asked whether H3K9Ac and H3K14Ac staining is altered in the background of the GluRIIA mutant. It is important to emphasize that the GluRIIA subunit of the AMPA/Kainate glutamate receptor is only expressed in muscle (Petersen et al., 1997). We demonstrated that both H3K9Ac and H3K14Ac staining intensity in peripheral glia was significantly increased in the GluRIIA mutant compared to wild-type (Figure 4A–4F). This argues for previously unexplored, PHP-associated signaling from muscle to peripheral glia, inducing a change in histone acetylation. In order to associate the change in glial histone acetylation to the mechanisms of PHP, we demonstrated that enhanced H3K9Ac and H3K14Ac staining intensity was abolished in the GluRIIA;ada2b1 double mutant, co-incident with the ada2b-dependent block of PHP. Since a muscle-specific perturbation (GluRIIA deletion mutation) induces an Ada2b-dependent, glial-specific epigenetic change, our data argue for the existence of a signaling system that, directly or indirectly, connects muscle to peripheral glia during PHP. We acknowledge the possibility that other epigenetic codes, such as histone methylation and DNA methylation patterns may also be altered in the GluRIIA mutant background.
Independent Evidence that Peripheral Glia Participate in PHP.
To provide further evidence of a required function of peripheral glia during PHP, we sought other ways to impair glial function, independent of the SAGA complex mutations. The Sar1-GTPase is necessary for secretory trafficking from the endoplasmic reticulum (ER) to the Golgi apparatus (Ye et al., 2007). Depleting Sar1 should generally impair the secretion of signaling molecules in peripheral glia. We expressed UAS-sar1 RNAi in perineurial glia using Egr-Gal4 as well as NP6293-Gal4 (Awasaki et al., 2008; Stork et al., 2012) and assayed PHP (Figure S5). First, we demonstrated that perineurial glia were normally distributed and were present at the NMJ when Sar1 was knocked down in perineurial glia, although there were changes in glial morphology at the NMJ (Figure S5F). Next, we demonstrated impaired basal synaptic transmission (Figure S5B and S5E), highlighting a role for Sar1-mediated secretory trafficking in glia during the development or maintenance of presynaptic release at baseline. Finally, we demonstrated a complete block of PHP following application of PhTX to the NMJ, suggesting that secretory trafficking in peripheral glia was necessary for PHP (Figure S5B–S5E). As a control, we probed synapse morphology at the NMJ when expressing UAS-sar1 RNAi in glia (Figure S5F–S5I). Although there was a slight increase of bouton numbers at the NMJ of muscle 6/7 (abdominal segment 2), the total number of presynaptic active zones remains normal (Figure S5F–S5I). There are many caveats to this experiment, given the non-specific disruption of secretory trafficking. None-the-less, the data demonstrate a requirement for peripheral glia in neuromuscular PHP.
Multiplexin is Secreted by Perineurial Glia and is Required for PHP.
There is reason to hypothesize that Multiplexin, a component of the extracellular matrix that is necessary for PHP (Wang et al., 2014) could be derived from glia. Multiplexin is the homologue of mammalian Collagen XVIII and can be cleaved to produce a conserved, matrix-derived signaling molecule, referred to as a matrikine, named Endostatin (O’Reilly et al., 1997; Wang et al., 2014). Although Endostatin is essential for PHP, the tissue that secretes and deposits Multiplexin at the NMJ remains unknown. More specifically, we reported that when transgenic Endostatin was supplied by either the neuron or muscle cell, it was sufficient, in either case, to support PHP in a multiplexin mutant (Wang et al., 2014). But, in this experiment, an artificial signal peptide drives constitutive secretion of Endostatin from either nerve or muscle. The endogenous source of Multiplexin was never determined, a question that can only be addressed by tissue-specific depletion of Multiplexin. Furthermore, it remains unknown whether, or how, Multiplexin secretion is altered during PHP.
Recently, a transcriptome profiling study identified Drosophila multiplexin as a downstream target gene of the SAGA complex in the Drosophila visual system (Ma et al., 2016). Thus, we hypothesized that SAGA complex-dependent expression and secretion of Multiplexin within glia could be a primary mechanism involved in PHP. To test this hypothesis, we used two independent methodologies. First, we examined the endogenous expression pattern of Multiplexin using a Multiplexin protein-trap line harboring a Mimic-PT.GFSTF.0 inserted within the multiplexin locus (Nagarkar-Jaiswal et al., 2015). The Mimic-PT.GFSTF.0 cassette is inserted in an intron close to the 3’ end of the gene, and the GFP-tagged protein reports the endogenous expression of the Multiplexin protein. Multiplexin-tagged-GFP is observed in cells localized on the surface of the ventral nerve cord (data not shown) and in cells surrounding all peripheral nerves (Figure 5A). Multiplexin-GFP was also observed at the NMJ, consistent with the anatomical position of peripheral glia in axons and at the NMJ (Figure 5A and 5B). We then probed the co-localization of Multiplexin-GFP with perineurial and subperineurial glia (Figure S6). We visualized peripheral glia by expressing UAS-tdTomato using the Egr-Gal4 driver and simultaneously visualized Multiplexin-GFP. Multiplexin-GFP shows precise co-localization with tdTomato expressing glia within the peripheral nerve and at the NMJ (Figure S6A and S6C). In contrast, when we drive the expression of UAS-tdTomato using subperineurial glial Gal4 driver Moody-Gal4 (Schwabe et al., 2005; Stork et al., 2012), Multiplexin-GFP is concentrated in cells localized outside the subperineurial glial layer on the peripheral nerves, suggesting that Multiplexin is strongly expressed in perineurial glia in the periphery (Figure S6B).
Figure 5. Ada2b-dependent Expression of Multiplexin in Perineurial Glia.

(A) – (B) Representative confocal images for Multiplexin expression on peripheral nerves (upper panels) and at the NMJ of muscle 6/7 (lower panels). Endogenous Multipelxin is labeled by the dmpMi protein trap (DMP, green). Peripheral nerves and the presynaptic membrane at the NMJ are immunolabeled with HRP (red). Multiplexin-GFP is greatly diminished when UAS-GFP RNAi is expressed in the dmpMi protein trap line using pan glial-specific Gal4 drivers (A, Repo-Gal4>GFP RNAi, dmpMi), or perineurial glial-specific Gal4 drivers (B, Egr-Gal4>GFP RNAi, dmpMi; NP6293-Gal4>GFP RNAi, dmpMi) compared to the control (dmpMi).
(C) Representative confocal images for extracellular Multiplexin (Surface DMP, green). Endogenous Multiplexin is secreted into the ECM and is present throughout the NMJ in proximity to active zones (white arrows, inset; Brp, red) in wild-type animals (wt).
(D) – (E) Representative confocal images (D) and quantification (E) of average fluorescence intensity of Multiplexin-GFP (dmpMi , GFP signal indicated by white arrows) at the NMJ of wild-type (wt, black, N=8 NMJ, n=3 animals) and ada2b1 mutants (ada2b1, red, N=11 NMJ, n=3 animals). Normalized average intensity is shown. Mean ± SEM; *p < 0.05; Student’s t-test. See also Figure S5 and S6.
As a second approach, we tested whether perineurial glia, identified by Egr-Gal4 or NP6293-Gal4, are the sole source of Multiplexin. To do so, we selectively depleted the endogenously-tagged Multiplexin-GFP protein in glia by expressing UAS-GFP RNAi. The UAS-GFP RNAi was expressed with a pan-glial-specific Gal4 driver (Repo-Gal4 (Sepp et al., 2001; Stork et al., 2012)), as well as the two perineurial glia-specific Gal4 drivers (Egr-Gal4 and NP6293-Gal4). We found that Multiplexin-GFP was completely abolished, regardless of which Gal4 driver was used (Figure 5A and 5B). Thus, Multiplexin protein that resides in peripheral nerve as well as at the NMJ is solely derived from the perineurial glia identified by Egr-Gal4 or NP6293-Gal4.
Next, to complement the above analyses, we used non-cell-permeabilizing conditions to immuno-label Multiplexin, thereby testing the abundance of the secreted form of Multiplexin protein (Figure 5C). We previously demonstrated the specificity of our non-cell permeabilizing protocol (Wang et al., 2014). The Multiplexin antibody that we used recognizes the C-terminal Endostatin domain of Multiplexin (Harpaz et al., 2013). Antibody specificity was verified by the lack of signal in a multiplexin null mutant and a C-terminal deletion mutant (data not shown). In our experiment, the NMJ was co-labeled for surface Endostatin (without permeabilization) and the active zone component Brp (after fixation and permeabilization, Figure 5C). This double-labeling protocol was also previously verified (Wang et al., 2014). We found that extracellular Multiplexin formed punctuate structures that were distributed throughout the presynaptic nerve terminal and these structures were often present in close proximity to presynaptic active zones (Figure 5C). Thus, endogenous Multiplexin protein is secreted and is present at or in close proximity to neurotransmitter release sites.
Finally, we tested whether the abundance of Multiplexin protein, at the NMJ, was dependent upon SAGA complex function. We estimated total Multiplexin protein by staining for Multiplexin-GFP in the GFP protein trap (Mimic-PT.GFSTF.0; Figure 5D) using permeabilizing conditions. We found that synaptic Multiplexin-GFP is reduced by ~50% in the ada2b1mutant compared to wild-type (Figure 5D and 5E), demonstrating that Multiplexin protein levels were strongly influenced, but not completely dependent upon, the activity of ada2b. Thus, Ada2b is essential for PHP and controls the abundance of Multiplexin, a required PHP protein.
Perineurial Glia-derived Multiplexin is Required for PHP.
We then determined whether glia are the relevant source of Multiplexin that is necessary for the rapid induction and sustained expression of PHP. We knocked down multiplexin, using UAS-multiplexin RNAi, and did so in either motoneurons (OK371-Gal4) or glia (using each of the following drivers individually: Egr-Gal4, NP6293-Gal4 or Repo-Gal4). We observed a complete block of synaptic homeostasis when we expressed UAS-multiplexin RNAi in glia using either pan-glial (Repo-Gal4) or when using either of the perineurial-specific glial (NP6293-Gal4 and Egr-Gal4) drivers (Figure 6A, 6B and S7). Note that when multiplexin expression was knocked down in motoneurons, PHP was normal (Figure 6B and S7), a finding that effectively controls for the use of the RNAi line.
Figure 6. Glial-derived Multiplexin is Required for PHP.
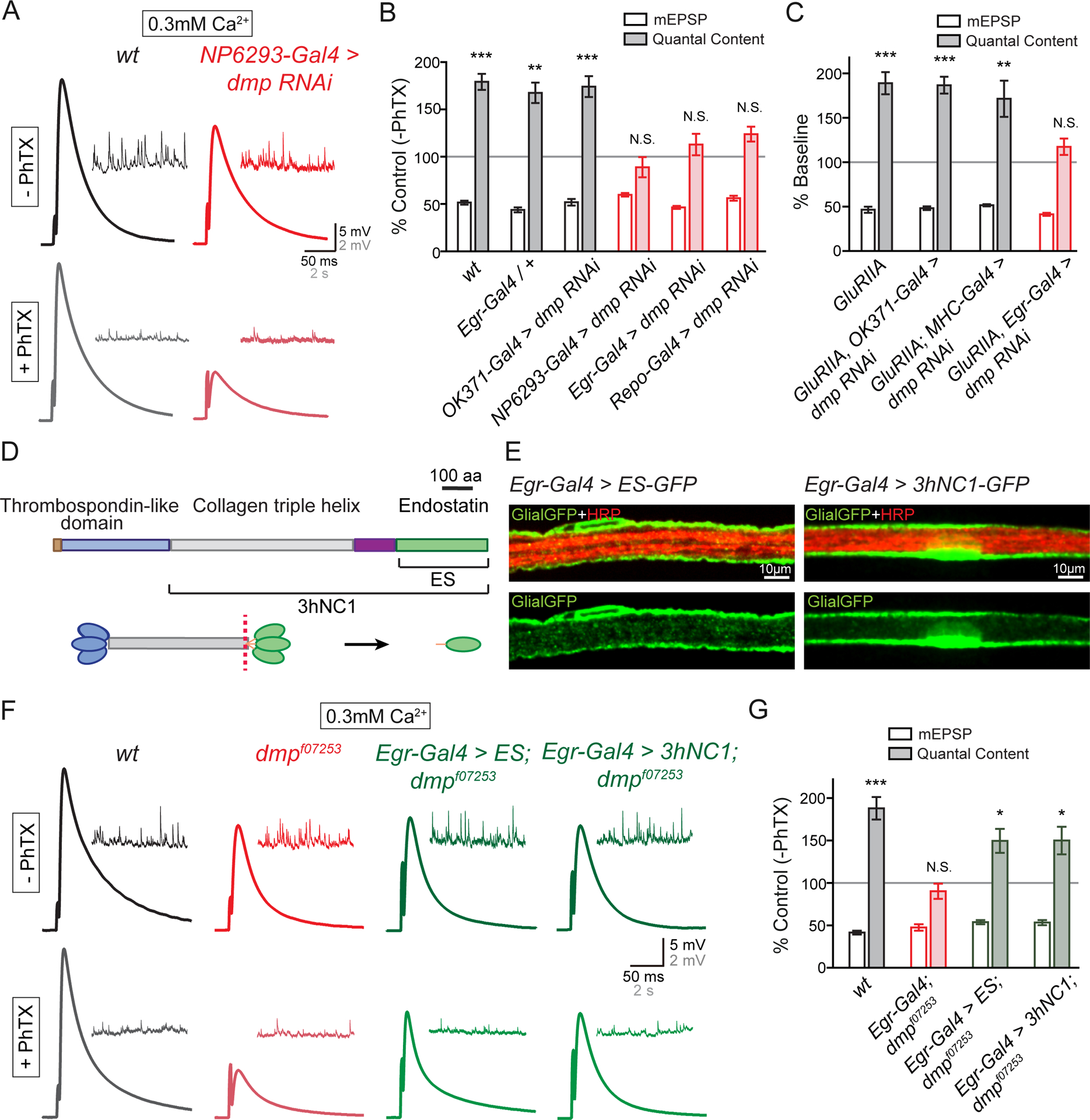
(A) Representative EPSP and mEPSP traces in wild-type (wt, black) and at the NMJ when UAS-dmp RNAi is driven by the perineurial glial-specific Gal4 driver NP6293-Gal4 (red) in the absence (−PhTX) and the presence of PhTX (+PhTX).
(B) Percent change in mEPSP (open bars) and quantal content (filled bars) in the presence of PhTX, as in Figure 1C. Genotypes: wild-type (wt, gray bars), synapses bearing heterozygous Egr-Gal4 (Egr-Gal4/+, gray bars), UAS-dmp RNAi driven by motoneuron-specific Gal4 (OK371-Gal4>dmp RNAi, gray bars), UAS-dmp RNAi driven by perineurial glial-specific Gal4 (NP6293-Gal4>dmp RNAi, red), UAS-dmp RNAi driven by Egr-Gal4 (Egr-Gal4>dmp RNAi, red), UAS-dmp RNAi driven by pan-glial Gal4 (Repo-Gal4>dmp RNAi, red bars). Mean ± SEM; **p < 0.01, ***p < 0.001, N.S. not significant; Student’s t-test.
(C) Quantification of mEPSP (open) and quantal content (filled). Each genotype is presented as percent change due to the presence of the GluRIIA mutant. Gentoypes: GluRIIA (GluRIIA, gray bars), UAS-dmp RNAi driven by motoneuron-specific Gal4 in GluRIIA mutants (GluRIIA, OK371-Gal4>dmp RNAi, gray bars), UAS-dmp RNAi driven by muscle-specific Gal4 in GluRIIA (GluRIIA; MHC-Gal4>dmp RNAi, gray bars), UAS-dmp RNAi driven by glial-specific Gal4 in GluRIIA (GluRIIA, Egr-Gal4>dmp RNAi, red). Mean ± SEM; **p < 0.01, ***p < 0.001, N.S. not significant; Student’s t-test.
(D) Diagram of the Multiplexin protein, the source of Endostatin. Thrombospondin-like domain (blue), Collagen triple helix (gray) and Endostatin domain (ES, green) of Multiplexin (upper panel). Uncleaved middle-length isoform of Multiplexin (3hNC1) and cleaved form (Endostatin, abbreviated ES) are indicated. C-terminal domain of Multiplexin can be cleaved proteolytically (red dotted line, lower panel) at hinge region to release monomers of Endostatin (ES, green oval).
(E) Representative confocal images of perineurial glia expressing UAS-ES-GFP (Egr-Gal4>ES-GFP, Glial-GFP, green, left panels) and UAS-3hNC1-GFP (Egr-Gal4>3hNC1-GFP, Glial-GFP, green, right panels) driven by Egr-Gal4 on the peripheral nerve. Neuronal membrane is indicated by anti-HRP labeling (HRP, red).
(F) Representative EPSP and mEPSP traces in wild-type (wt, black), multiplexin mutants (dmpf07253, red), UAS-ES driven by Egr-Gal4 in dmpf07253 mutants (Egr-Gal4>ES;dmpf07253, green), and UAS-3hNC1 driven by Egr-Gal4 in dmpf07253 mutants (Egr-Gal4>3hNC1;dmpf07253, green) in the absence (−PhTX) and presence of philanthotoxin (+PhTX).
(G) Quantification of mEPSP amplitude (open bars) and quantal content (filled bars), normalized as in (A). Genotypes: wild-type (wt, gray bars), dmpf07253 mutants bearing heterozygous Egr-Gal4 (Egr-Gal4/+;dmpf07253, red bars), UAS-ES driven by Egr-Gal4 in dmpf07253 mutant (Egr-Gal4>UAS-ES;dmpf07253, green bars), and UAS-3hNC1 driven by Egr-Gal4 in dmpf07253 mutant (Egr-Gal4>UAS-3hNC1;dmpf07253, green bars). Mean ± SEM; *p < 0.05, ***p < 0.001, N.S. not significant; Student’s t-test. See also Figure S7.
An identical data set was collected using the GluRIIA mutant to induce PHP. We observed impaired homeostasis only when multiplexin was specifically depleted in glia (Figure 6C and S7). Finally, we complemented our tissue-specific RNAi experiments with tissue-specific genetic rescue. We expressed the full-length Multiplexin (UAS-3hNC1, Figure 6D) or the C-terminal Endostatin domain of Multiplexin (UAS-ES, Figure 6D; (Meyer and Moussian, 2009)) in the multiplexin null mutant (dmpf07253) using a peripheral glial-specific Gal4 driver (Egr-Gal4). We observe a strong GFP signal when we drive the expression of UAS-3hNC1-GFP or UAS-ES-GFP using Egr-Gal4, demonstrating robust expression of these transgenes (Figure 6E). Glial-specific expression of UAS-3hNC1 (Egr-Gal4>3hNC1;dmpf07253) or UAS-Endostatin (Egr-Gal4>ES;dmpf07253) rescues PHP in the multiplexin null mutant, whereas the heterozygous Egr-Gal4 driver alone (Egr-Gal4/+;dmpf07253) does not (Figure 6F and 6G). Based on these experiments, we conclude that peripheral glia, and perineurial glia more specifically, are the relevant source of Multiplexin protein, necessary for the both the rapid induction and sustained expression of PHP.
SAGA Mediates the Up-regulation of Multiplexin in the GluRIIA Mutant.
As a final set of experiments, we asked whether Multiplexin transcript and protein were modulated during the long-term expression of PHP, caused by the GluRIIA mutation, and whether this modulation was sensitive to SAGA mutations. First, we demonstrated a ~50% increase of Multiplexin protein expression at the NMJ of the GluRIIA mutant compared to wild-type (Figure 7A, 7B and 7E). To our surprise, we also observed a wide-spread increase in Multiplexin (Figure 7C–7E) that correlates with the wide-spread changes in Ada2b-dependent histone acetylation within peripheral glia in the GluRIIA mutant background (Figure 4A–4F). Next, we extended this observation by demonstrating a ~100% increase of multiplexin mRNA level in the GluRIIA mutant larval brain compared to wild-type (Figure 7F). As predicted, the increase in multiplexin mRNA is restored to baseline levels in the GluRIIA;ada2bEP double mutant, indicating that the elevated multiplexin gene expression is Ada2b-dependent. Since the ada2b allele used in this experiment (ada2bEP) also blocks PHP in the GluRIIA mutant background (Figure S8), it argues that Ada2b-dependent modulation of multiplexin expression and protein are linked to the expression of PHP.
Figure 7. SAGA-dependent Enhancement of Peripheral Multiplexin Expression during Homeostatic Plasticity.
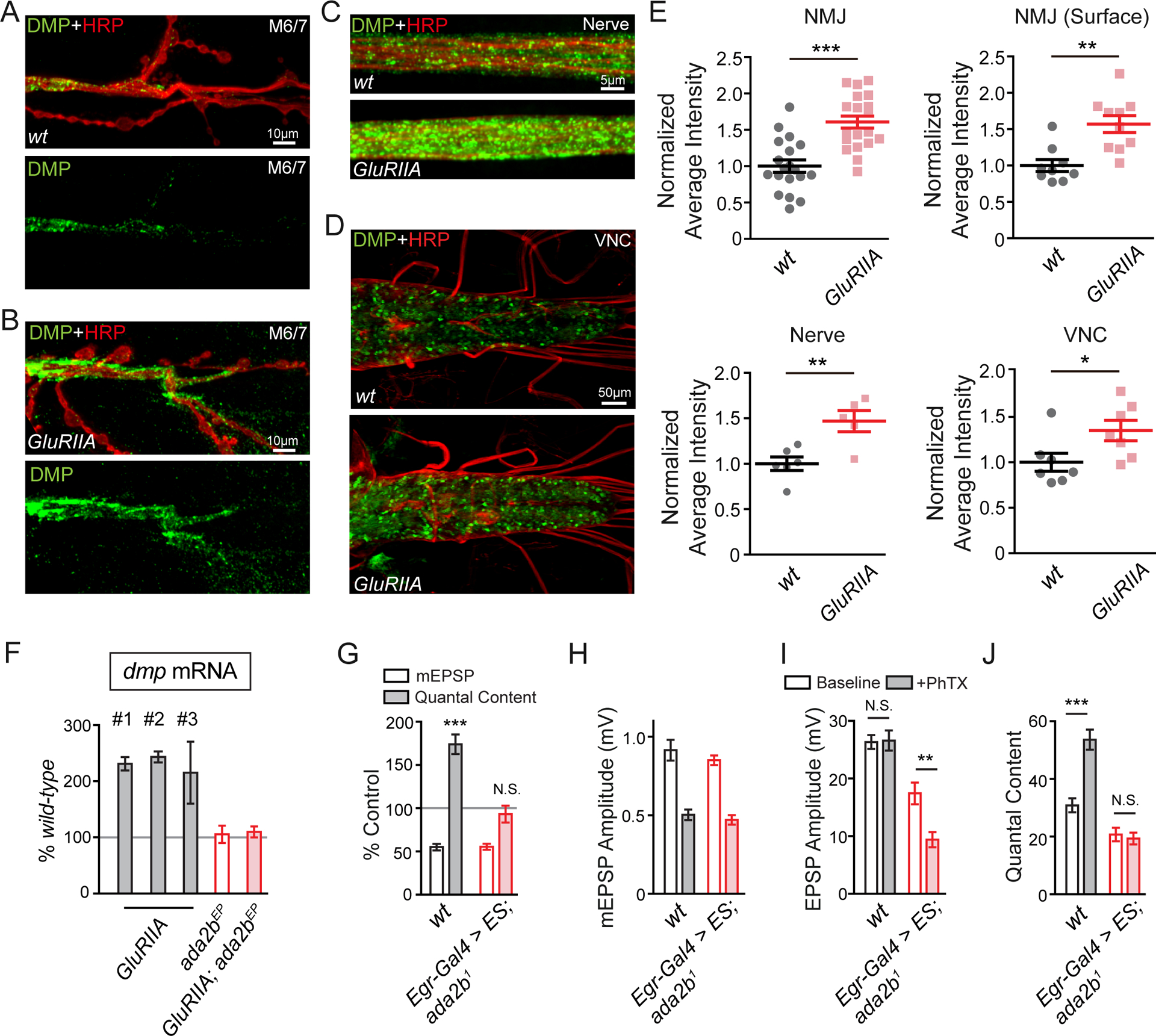
(A) – (D) Representative confocal images of Multiplexin protein at the NMJ on muscle 6/7 (A and B), the peripheral nerve (C, nerve) and the ventral verve cord (D, VNC) in wild-type and GluRIIA mutants. Total endogenous Multiplexin is labeled by dmpMi protein trap (DMP, green) and the neuronal membrane is indicated by anti-HRP immunolabeling (HRP, red).
(E) Quantification of average fluorescence intensity of total endogenous Multiplexin at the NMJ (NMJ, wt N=19 synapses, n=7 animals; GluRIIA N=20 synapses, n=7 animals), on the peripheral nerve (nerve, wt n=6 animals; GluRIIA n=5 animals) and in the VNC (VNC, wt n=7 animals; GluRIIA n=7 animals) in the wild-type control and GluRIIA mutant animals. The average fluorescence intensity of extracellular endogenous Multiplexin is quantified at the NMJ (NMJ surface, wt N=9 synapses, n=3 animals; GluRIIA N=10 synapses, n=3 animals) in wild-type and GluRIIA mutants. Mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001; Student’s t-test.
(F) Quantification of dmp mRNA level by Q-PCR. The dmp transcript levels in GluRIIA (three independent measures, #1 – #3), ada2bEP mutant (ada2bEP, red empty bar), GluRIIA;ada2bEP double mutants (GluRIIA;ada2bEP, red filled bar) are normalized to wild-type values.
(G) mEPSP amplitudes (open bars) and quantal content (filled bars) recorded in PhTX and normalized to each genotype in the absence of PhTX. Genotypes: wild-type (wt, gray bars) and UAS-ES driven by Egr-Gal4 in ada2b1 mutant (Egr-Gal4>ES,ada2b1, red bars). Mean ± SEM; ***p < 0.001, N.S. not significant; Student’s t-test.
(H) – (J) Non-normalized values in the absence (open bars) and presence (filled bars) of PhTX. Average mEPSP amplitude (C), EPSP amplitude (D), and quantal content (E) are presented for genotypes as in (G). Mean ± SEM; **p < 0.01***p < 0.001, N.S. not significant; Student’s t-test. See also Figure S8.
Finally, we address whether Multiplexin is the only glial-derived signal that is important for PHP. In other words, is SAGA-dependent modulation of Multiplexin sufficient to explain the entire glia and SAGA-dependent control of PHP. We hypothesize that Multiplexin is, actually, only one component of a more complex SAGA and glial-specific signaling event. Indeed, glial-specific expression of a secreted form of Endostatin that is sufficient to rescue the multiplexin mutant (Figure 6F and 6G), fails to restore PHP in the ada2b1 mutant (Figure 7G–7J).
DISCUSSION
Based on our accumulated data, we propose the existence of a previously unexplored homeostatic signaling system, originating in muscle following perturbation of muscle-specific glutamate receptors. This muscle-derived signal, of unknown identity, activates SAGA-dependent epigenetic changes that occur throughout glial cells in the peripheral nerves, both within the nerves themselves and in close proximity to the neuromuscular active zones. The glial epigenetic signaling response is essential for PHP, as demonstrated by the required function of four new PHP genes, all components of the SAGA complex. We provide evidence that one element of the SAGA-dependent epigenetic signaling is enhanced deposition of the extracellular matrix protein Multiplexin, a required factor for both short and long-term PHP. We hypothesize that this represents a stable change of extracellular matrix composition that sustains a homeostatic change in motor function (Figure 8).
Figure 8. Model for Glia Signaling in PHP.
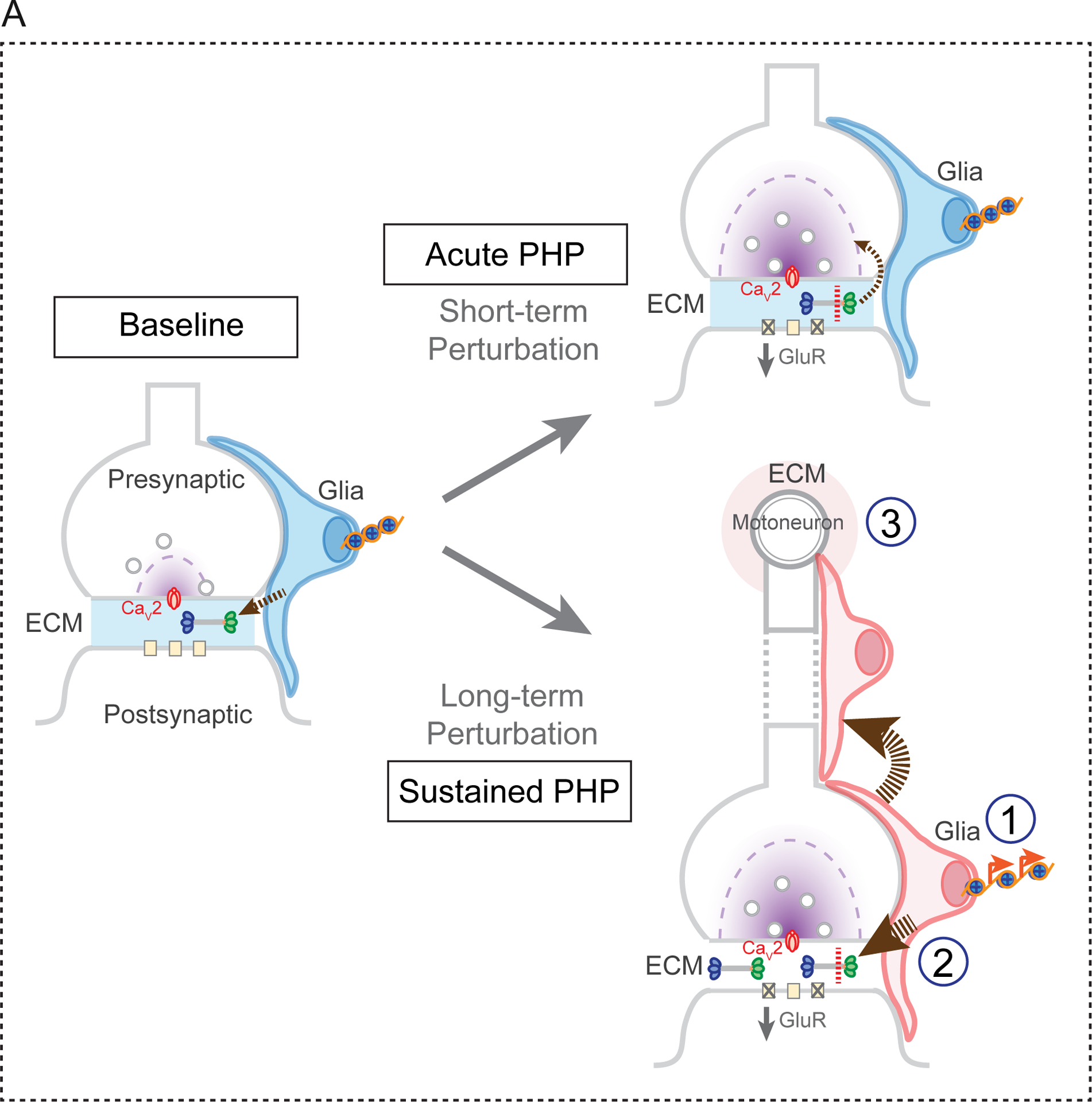
The SAGA complex is required for both the rapid induction and sustained expression of PHP. At baseline, glial secrete Multiplexin, which is deposited within the extracellular matrix (ECM) at the NMJ (arrow, left panel). Application of PhTX (short-term perturbation) results in the proteolytic cleavage of Multiplexin and the release of Endostatin, which is essential for the induction of PHP (acute PHP, upper right panel). A sustained perturbation (long-term perturbation), such as the GluRIIA mutation, induces epigenetic signaling in peripheral glia, a process that is necessary for the sustained expression of PHP (lower right panel). Within peripheral glia, members of the SAGA complex are required for secretion of elevated levels of Multiplexin and other, as yet unidentified, factors that function either locally (1), along the peripheral nerve (2) or within the ventral nerve cord where the motoneuron cell bodies reside (3).
Role of the Extracellular Matrix
We demonstrate enhanced deposition of Multiplexin in the GluRIIA mutant background that is Ada2b-dependent and correlates with increased histone acetylation throughout peripheral glia. Remarkably, elevated Multiplexin is not restricted to the NMJ, or even to the peripheral nerves. This is evidence of a systemic response, affecting the entirety of the peripheral nerve and ventral nerve cord, in response to the muscle-specific deletion of a non-essential glutamate receptor subunit, GluRIIA. Many possibilities exist for this systemic response. It certainly implies the existence of a homeostatic signaling from muscle to glia. But, it also argues that this signal is either broadly released and diffusible, or is a signal that can be propagated through glial-glial junctions (Figure 8), originating at the NMJ and extending throughout the peripheral nerves. Finally, another possibility is that signaling from muscle to nerve (Orr et al., 2017) could induce secondary signaling from the motoneurons to surrounding glia, with glia mediating feedback signaling including the deposition of increased levels of Multiplexin (Figure 8).
Why is there a systemic response? There exists precedent for dynamic epigenetic signaling in the nervous system, including the molecular mechanisms that control circadian rhythm (Aguilar-Arnal and Sassone-Corsi, 2015; Etchegaray et al., 2003; Koike et al., 2012). One possibility is that the spread of the Multiplexin signal throughout the periphery, extending to the CNS, might be preparatory for additional perturbations. For example, if a perturbation was to be environmental, immunological or injury related, as opposed to muscle-specific elimination of a glutamate receptor, then it might be advantageous to prepare surrounding tissue for the eventual spread of such a persistent perturbation. This is speculative, but is consistent with theoretical models of homeostasis, adaptation, resilience and allostasis in other systems (Karatsoreos and McEwen, 2011). We also note the formal possibility that a systemic response could be initiated centrally and propagate to the periphery, something that could be addressed in the future.
There is yet another possibility. The systemic remodeling of the ECM could influence the induction of Hebbian-type plasticity within the CNS following disruption of transmission at the NMJ. In the mammalian central nervous system, glia and ECM structures surrounding neurons control synaptic transmission, neuron excitability and neuronal states (Poskanzer and Yuste, 2016; Sorg et al., 2016; Volterra and Meldolesi, 2005; Wang and Fawcett, 2012). Previous studies demonstrated that the ECM remodeling can be induced by synaptic signals and ECM signaling influences the expression of Hebbian synaptic plasticity (Dityatev and Schachner, 2003; Tonnesen et al., 2018). These are testable possibilities for future studies.
Epigenetic Signaling, Homeostatic Plasticity and Etiology of Disease.
There is an emerging literature regarding epigenetic regulation of gene expression during Hebbian plasticity (Graff and Tsai, 2013; Levenson and Sweatt, 2005), although a glial-specific function is lacking. By contrast, very little is known about the function and capacity of epigenetic codes during homeostatic plasticity. And, virtually nothing is known about how the glial epigenome maintains the stability and function of the nervous system. Mutations in epigenetic factors or abnormal epigenetic modifications have been associated with numerous neurodevelopmental, neuropsychiatric and neurodegenerative disorders (Chen et al., 2003; Chen et al., 2012; De Rubeis et al., 2014). In most instances, including autism spectrum disorder, the focus remains on direct effects within neurons and at specific time points during neurodevelopment (Willsey et al., 2013). The importance of these data is emphasized by reports documenting remarkable benefit from HDAC inhibitors in clinical settings (Fischer et al., 2007; Steffan et al., 2001), but the locus of action (neuron vs. glia) remains to be defined. Importantly, there are data for Rett Syndrome indicating a loss of epigenetic control within glia (Lioy et al., 2011). Our results not only emphasize the need for further exploration of glial-dependent epigenetic signaling in neurological and psychiatric disease, but set the stage for determining whether glial-derived epigenetic signaling may be a cellular and molecular link that connects homeostatic plasticity to these devastating disorders.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Graeme Davis (Graeme.Davis@ucsf.edu).
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila stocks were raised at room temperature on standard molasses food. Drosophila alleles used for RNAi knockdown experiments were raised at 25°C. For motoneuron-specific Gal4 expression we used OK371-Gal4 on the second chromosome. For muscle specific expression, we used MHC-Gal4. For glial specific expression, we used pan, perineurial, and subperineurial glial Gal4 drivers as indicated in figure legends. Unless otherwise noted, the w1118 strain was used as a wild-type (wt) control. For temperature-dependent expression experiments, eggs were laid on apple plates and were maintained at 18°C. First-instar larvae with the right genotypes in the experimental group were selected and transferred to 29°C. Larvae in the control group were maintained at 18°C.
METHOD DETAILS
Electrophysiology
Sharp-electrode recordings were made from muscle 6 at abdominal segments 2 and 3 in third-instar larvae using an Axoclamp 2B or Axoclamp 900A amplifier (Molecular Devices). HL3 saline was used (in mM): 70 NaCl, 5 KCl, 10 MgCl2, 10 NaHCO3, 115 Sucrose, 5 Trehalose, 5 HEPES, and 0.3 CaCl2 (unless specified otherwise). EPSP and mEPSP traces were analyzed in Igor Pro (WaveMetrics) with previously published routines and MiniAnalysis (Synaptosoft) (Gavino et al., 2015). For the rapid induction of synaptic homeostasis, larvae were incubated in 15μM Philanthotoxin-433 in an un-stretched, partially dissected preparation (PhTX, Santa Cruz Biotechnology) for 10min (Frank et al., 2006). For each NMJ, the average amplitudes of evoked EPSP are based on the mean peak amplitudes in response to 20–30 individual stimuli. Spontaneous mEPSPs were recorded continuously 60–90s. Quantal content was estimated for each NMJ as the ratio of EPSP amplitude/mEPSP amplitude. The mean value across all NMJ for a given genotype is reported. All statistics analyses were performed in GraphPad Prism.
Immunocytochemistry
Standard immunocytochemistry was performed as previously described (Wang et al., 2014). Briefly for total immunostaining: dissected third instar larvae were fixed with 4% PFA in PBS for 20min and incubated with primary antibody diluted in PBST (PBS with 0.5% Triton-100) overnight at 4°C after six brief washes with PBST. Larvae were then incubated with secondary antibody diluted in PBST for 1.5hr at room temperature and mounted in Vectorshield without DAPI (otherwise specified in Figure Legends or Results, Vector Laboratories) after six brief washes with PBST. For surface Multiplexin immunolabeling, dissected third instar larvae were incubated with primary antibody for 20min at room temperature and fixed with ice-cold ethanol. For surface GFP immunolabeling, dissected third instar larvae were incubated with mouse anti-GFP antibody for 20min at room temperature and fixed with 4% PFA in PBS.
Image Acquisition
Confocal imaging for peripheral nerves and synapses was performed on a Yokagawa CSU22 spinning disk confocal with a 60x/1.4 plan Apochromat objective. Z-stacks of peripheral nerves or NMJ on muscle 6/7 were acquired and maximum projections were used for analysis. Confocol images for the ventral nerve cords were acquired using an Andor Zyla sCMOS camera mounted to a Nikon Ti Microscope with an Andor Borealis CSU-W1 spinning disc confocal with a Nikon Plan Apo 20x/0.75. Deconvolution imaging for synapse morphology was performed using a Plan Apo objective 60x/1.4 (Carl Zeiss) on an Axiovert 200 inverted microscope (Carl Zeiss) equipped with a cooled CCD camera (CoolSNAP HQ; Roper Scientific). Image acquisitions were performed in SlideBook software (Intelligent Imaging Innovation).
Quantitative RT-PCR
Quantitative RT-PCR was performed as previously described (Bergquist et al., 2010; Muller et al., 2012; Younger et al., 2013), with slightly modified procedures. Primer probes specific for real-time PCR detection of Multiplexin (Dmp, Dm01847117_g1) and Ribosomal protein L32 (RpL32, Dm02151827_g1) were designed and developed by Applied Biosystems. The brains were removed from 5 third-instar larvae (3 replicates/genotype) and RNA isolation was performed immediately (RNeasy Plus Micro kit, Qiagen). Potential DNA contamination was removed (TURBO DNA-free, Ambion) and single-strand cDNA libraries were prepared with SuperScript III First-Strand synthesis system (Invitrogen).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Quantification of data are presented as mean ± standard error of the mean (SEM) with the precise N numbers indicated in the figure legends or the supplemental table. Statistical analysis was performed using Prism (5.01, GraphPad) and using paired two-tailed Student’s t test or one-way ANOVA as indicated in the figure legends.
Quantitative Image Analysis
Maximum projections of deconvolved or confocal images were used for analyses. Quantification of Brp and bouton number (Figure 3) was performed as previously described (Frank et al., 2009) with Watershed Segmentation plugin in Fiji software (NIH). For H3K9Ac and H3K14Ac average fluorescence intensity analyses (Figure 4), individual Egr positive nuclei on proximal peripheral nerves were traced and analyzed for each genotype in Fiji (NIH). Mean of the average fluorescence intensity of each nucleus quantified were reported. For Multiplexin-GFP average fluorescence intensity analysis (Figure 7), GFP signal is amplified by mouse anti-GFP antibody and the average fluorescence intensity is measured in ROIs generated by tracing the GFP expressing cells/segments at the NMJ (muscle 6/7) and along peripheral nerves that innervate muscle 6/7. GFP intensity in ventral nerve cord is quantified as the average fluorescence intensity of the VNC (optical lobes not included). All analyses were performed using Fiji software (NIH) and statistics analyses were performed in GraphPad Prism.
DATA AND CODE AVAILABILITY
This study did not generate data sets for public repositories or new data code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-Bruchpilot (1:100) | Developmental Studies Hybridoma Bank | Nc82 |
| rabbit anti-Discs large (1:1000) | (Frank et al., 2006; Hauswirth et al., 2018) | |
| rabbit anti-GFP (1:1000) | Invitrogen | G10362 |
| mouse anti-GFP (1:1000) | Invitrogen clone 3E6 | A-11120 |
| mouse anti-Repo (1:10) | Developmental Studies Hybridoma Bank | 8D12 |
| rabbit anti-H3 acetyl K9 (1:500) | abcam | ChIP grade ab10812 |
| rabbit anti-H3 acetyl K14 (1:200) | abcam | ChIP grade ab52946 |
| Alexa conjugated secondary antibodies (488, cy3, cy5) (1:300) | Jackson Immuno-research laboratories | |
| Alexa Fluor 647 conjugated goat anti-HRP (1:100) | Jackson Immuno-research laboratories | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Philanthotoxin-433 | Santa Cruz Biotechnology | 276684-27-6 |
| Critical Commercial Assays | ||
| RNeasy Plus Micro kit | Qiagen | 74134 |
| TURBO DNA-free | Ambion | AM1907 |
| SuperScript III First-Strand synthesis system | Invitrogen | 18080051 |
| TaqMan® Fast Universal PCR Master Mix | Applied Biosystem | 4352042 |
| Experimental Models: Organisms/Strains | ||
| dmpf07253 | Bloomington | BL19062 CG42543 |
| dmpMi | Bloomington | BL60567 |
| dmp RNAi | Bloomington | BL28299 |
| ada2bEP | Bloomington | BL31778 CG9638 |
| ada2b RNAi | Bloomington | BL31347 |
| ada2b1 | (Qi et al., 2004) | Jerry Workman (Stowers Institute, Kansas City, Missouri) |
| gcn5E333st | Bloomington | BL9333 CG4107 |
| gcn5Q186st | Bloomington | BL9334 |
| gcn5 RNAi | VDRC | v108943 |
| sfg11e01308 | Bloomington | BL17941 CG13379 |
| non-stop RNAi | Bloomington | BL28725 CG4166 |
| GFP RNAi | Bloomington | BL9331 |
| UAS-tdTomato | Bloomington | BL36328 |
| UAS-Redstinger.nls | Bloomington | BL8546 |
| UAS-GFP.nls | Bloomington | BL4776 |
| Tubulin-Gal80ts | Yuh-Nung Jan (University of California, San Francisco) | |
| sar1 RNAi | VDRC | v108458 CG7073 |
| UAS-ada2b-3HA | FlyORF | F000122 |
| UAS-3hNC1 | (Meyer and Moussian, 2009) | Bernard Moussian (Max-Planck-Institute for Developmental Biology, Tubingen, Germany) |
| UAS-ES | (Meyer and Moussian, 2009) | Bernard Moussian (Max-Planck-Institute for Developmental Biology, Tubingen, Germany) |
| UAS-3hNC1-GFP | (Wang et al., 2014) | |
| UAS-ES-GFP | (Wang et al., 2014) | |
| NP6293-Gal4 | (Stork et al., 2012) | Marc Freeman (Vollum Institute, Portland, Oregon) |
| Moody-Gal4 | (Stork et al., 2012) | Marc Freeman (Vollum Institute, Portland, Oregon) |
| Alrm-Gal4 | (Stork et al., 2012) | Marc Freeman (Vollum Institute, Portland, Oregon) |
| Repo-Gal4 | (Keller et al., 2011) | |
| Egr-Gal4 | (Keller et al., 2011) | |
| EgrΔ25 | (Keller et al., 2011) | |
| Oligonucleotides | ||
| Dmp_qPCR primer | Applied Biosystems | Dm01847117_g1 |
| RpL32_qPCR primer | Applied Biosystems | Dm02151827_g1 |
| Software and Algorithms | ||
| GraphPad Prism (5.01) | GraphPad | |
| MiniAnalysis (6.0.3) | Synaptosoft | |
| Igor Pro (6.37) | WaveMetrics | |
| Fiji | NIH | |
| SlideBook | Intelligent Imaging Innovation | |
ACKNOWLEDGMENTS
Work in the laboratory of G.W.D. was supported by NINDS grant R35-NS097212. T.W. was supported by NIH NRSA postdoc fellowship NIH NINDS 1F32NS081884-03.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aguilar-Arnal L, and Sassone-Corsi P (2015). Chromatin landscape and circadian dynamics: Spatial and temporal organization of clock transcription. Proc Natl Acad Sci U S A 112, 6863–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, and Zoghbi HY (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23, 185–188. [DOI] [PubMed] [Google Scholar]
- Arthur-Farraj PJ, Morgan CC, Adamowicz M, Gomez-Sanchez JA, Fazal SV, Beucher A, Razzaghi B, Mirsky R, Jessen KR, and Aitman TJ (2017). Changes in the Coding and Non-coding Transcriptome and DNA Methylome that Define the Schwann Cell Repair Phenotype after Nerve Injury. Cell Rep 20, 2719–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasaki T, Lai SL, Ito K, and Lee T (2008). Organization and postembryonic development of glial cells in the adult central brain of Drosophila. J Neurosci 28, 13742–13753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baizabal JM, Mistry M, Garcia MT, Gomez N, Olukoya O, Tran D, Johnson MB, Walsh CA, and Harwell CC (2018). The Epigenetic State of PRDM16-Regulated Enhancers in Radial Glia Controls Cortical Neuron Position. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, and Malenka RC (2002). Control of synaptic strength by glial TNFalpha. Science 295, 2282–2285. [DOI] [PubMed] [Google Scholar]
- Benevento M, Iacono G, Selten M, Ba W, Oudakker A, Frega M, Keller J, Mancini R, Lewerissa E, Kleefstra T, et al. (2016). Histone Methylation by the Kleefstra Syndrome Protein EHMT1 Mediates Homeostatic Synaptic Scaling. Neuron 91, 341–355. [DOI] [PubMed] [Google Scholar]
- Bergquist S, Dickman DK, and Davis GW (2010). A hierarchy of cell intrinsic and target-derived homeostatic signaling. Neuron 66, 220–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman MP, Djukic B, Nelson SB, and Turrigiano GG (2012). A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J Neurosci 32, 13529–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brink DL, Gilbert M, Xie X, Petley-Ragan L, and Auld VJ (2012). Glial processes at the Drosophila larval neuromuscular junction match synaptic growth. PLoS One 7, e37876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carre C, Szymczak D, Pidoux J, and Antoniewski C (2005). The histone H3 acetylase dGcn5 is a key player in Drosophila melanogaster metamorphosis. Mol Cell Biol 25, 8228–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, and Greenberg ME (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889. [DOI] [PubMed] [Google Scholar]
- Chen YC, Gatchel JR, Lewis RW, Mao CA, Grant PA, Zoghbi HY, and Dent SY (2012). Gcn5 loss-of-function accelerates cerebellar and retinal degeneration in a SCA7 mouse model. Hum Mol Genet 21, 394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, and Barres BA (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. [DOI] [PubMed] [Google Scholar]
- Coffee B, Zhang F, Warren ST, and Reines D (1999). Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet 22, 98–101. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Miledi R, Trautmann A, and Uchitel OD (1980). On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human end-plates. J Physiol 299, 621–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, and Muller M (2015). Homeostatic control of presynaptic neurotransmitter release. Annu Rev Physiol 77, 251–270. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev A, and Schachner M (2003). Extracellular matrix molecules and synaptic plasticity. Nat Rev Neurosci 4, 456–468. [DOI] [PubMed] [Google Scholar]
- Doherty J, Logan MA, Tasdemir OE, and Freeman MR (2009). Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci 29, 4768–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, et al. (2009). Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray JP, Lee C, Wade PA, and Reppert SM (2003). Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 421, 177–182. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, and Tsai LH (2007). Recovery of learning and memory is associated with chromatin remodelling. Nature 447, 178–182. [DOI] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, and Davis GW (2006). Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 52, 663–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, and Davis GW (2009). A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron 61, 556–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavino MA, Ford KJ, Archila S, and Davis GW (2015). Homeostatic synaptic depression is achieved through a regulated decrease in presynaptic calcium channel abundance. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, and Tsai LH (2013). Histone acetylation: molecular mnemonics on the chromatin. Nat Rev Neurosci 14, 97–111. [DOI] [PubMed] [Google Scholar]
- Guzman-Karlsson MC, Meadows JP, Gavin CF, Hablitz JJ, and Sweatt JD (2014). Transcriptional and epigenetic regulation of Hebbian and non-Hebbian plasticity. Neuropharmacology 80, 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpaz N, Ordan E, Ocorr K, Bodmer R, and Volk T (2013). Multiplexin promotes heart but not aorta morphogenesis by polarized enhancement of slit/robo activity at the heart lumen. PLoS Genet 9, e1003597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth AG, Ford KJ, Wang T, Fetter RD, Tong A, and Davis GW (2018). A postsynaptic PI3K-cII dependent signaling controller for presynaptic homeostatic plasticity. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, and Tora L (2017). Sharing the SAGA. Trends Biochem Sci 42, 850–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Pacheco A, Franco JM, Lopez S, Gomez-Zumaquero JM, Magdalena Leal-Lasarte M, Caballero-Hernandez DE, Cejudo-Guillen M, and Pozo D (2017). Epigenetic Mechanisms of Gene Regulation in Amyotrophic Lateral Sclerosis. Adv Exp Med Biol 978, 255–275. [DOI] [PubMed] [Google Scholar]
- Karatsoreos IN, and McEwen BS (2011). Psychobiological allostasis: resistance, resilience and vulnerability. Trends Cogn Sci 15, 576–584. [DOI] [PubMed] [Google Scholar]
- Keller LC, Cheng L, Locke CJ, Muller M, Fetter RD, and Davis GW (2011). Glial-derived prodegenerative signaling in the Drosophila neuromuscular system. Neuron 72, 760–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr KS, Fuentes-Medel Y, Brewer C, Barria R, Ashley J, Abruzzi KC, Sheehan A, Tasdemir-Yilmaz OE, Freeman MR, and Budnik V (2014). Glial wingless/Wnt regulates glutamate receptor clustering and synaptic physiology at the Drosophila neuromuscular junction. J Neurosci 34, 2910–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, and Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koreman E, Sun X, and Lu QR (2018). Chromatin remodeling and epigenetic regulation of oligodendrocyte myelination and myelin repair. Mol Cell Neurosci 87, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, and Workman JL (2007). Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol 8, 284–295. [DOI] [PubMed] [Google Scholar]
- Levenson JM, and Sweatt JD (2005). Epigenetic mechanisms in memory formation. Nat Rev Neurosci 6, 108–118. [DOI] [PubMed] [Google Scholar]
- Lindblad K, Savontaus ML, Stevanin G, Holmberg M, Digre K, Zander C, Ehrsson H, David G, Benomar A, Nikoskelainen E, et al. (1996). An expanded CAG repeat sequence in spinocerebellar ataxia type 7. Genome Res 6, 965–971. [DOI] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, et al. (2011). A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Brennan KJ, D’Aloia MR, Pascuzzi PE, and Weake VM (2016). Transcriptome Profiling Identifies Multiplexin as a Target of SAGA Deubiquitinase Activity in Glia Required for Precise Axon Guidance During Drosophila Visual Development. G3 (Bethesda) 6, 2435–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney RE, Rawson JM, and Eaton BA (2014). An age-dependent change in the set point of synaptic homeostasis. J Neurosci 34, 2111–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie B, Sweeney ST, Poskanzer KE, Roos J, Kelly RB, and Davis GW (2004). Dap160/intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron 43, 207–219. [DOI] [PubMed] [Google Scholar]
- Meadows JP, Guzman-Karlsson MC, Phillips S, Holleman C, Posey JL, Day JJ, Hablitz JJ, and Sweatt JD (2015). DNA methylation regulates neuronal glutamatergic synaptic scaling. Sci Signal 8, ra61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F, and Moussian B (2009). Drosophila multiplexin (Dmp) modulates motor axon pathfinding accuracy. Dev Growth Differ 51, 483–498. [DOI] [PubMed] [Google Scholar]
- Muller M, Liu KS, Sigrist SJ, and Davis GW (2012). RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J Neurosci 32, 16574–16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarkar-Jaiswal S, DeLuca SZ, Lee PT, Lin WW, Pan H, Zuo Z, Lv J, Spradling AC, and Bellen HJ (2015). A genetic toolkit for tagging intronic MiMIC containing genes. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, and Folkman J (1997). Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88, 277–285. [DOI] [PubMed] [Google Scholar]
- Orr BO, Fetter RD, and Davis GW (2017). Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 550, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, and DiAntonio A (1997). Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19, 1237–1248. [DOI] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, and Molenaar PC (1992). Adaptation of quantal content to decreased postsynaptic sensitivity at single end-plates in alpha-bungarotoxin-treated rats. J Physiol 458, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poskanzer KE, and Yuste R (2016). Astrocytes regulate cortical state switching in vivo. Proc Natl Acad Sci U S A 113, E2675–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi D, Larsson J, and Mannervik M (2004). Drosophila Ada2b is required for viability and normal histone H3 acetylation. Mol Cell Biol 24, 8080–8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe T, Bainton RJ, Fetter RD, Heberlein U, and Gaul U (2005). GPCR signaling is required for blood-brain barrier formation in drosophila. Cell 123, 133–144. [DOI] [PubMed] [Google Scholar]
- Sepp KJ, Schulte J, and Auld VJ (2001). Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev Biol 238, 47–63. [DOI] [PubMed] [Google Scholar]
- Singh SK, Stogsdill JA, Pulimood NS, Dingsdale H, Kim YH, Pilaz LJ, Kim IH, Manhaes AC, Rodrigues WS Jr., Pamukcu A, et al. (2016). Astrocytes Assemble Thalamocortical Synapses by Bridging NRX1alpha and NL1 via Hevin. Cell 164, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg BA, Berretta S, Blacktop JM, Fawcett JW, Kitagawa H, Kwok JC, and Miquel M (2016). Casting a Wide Net: Role of Perineuronal Nets in Neural Plasticity. J Neurosci 36, 11459–11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staszewski O, and Prinz M (2014). Glial epigenetics in neuroinflammation and neurodegeneration. Cell Tissue Res 356, 609–616. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, et al. (2001). Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413, 739–743. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, and Malenka RC (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Stogsdill JA, Ramirez J, Liu D, Kim YH, Baldwin KT, Enustun E, Ejikeme T, Ji RR, and Eroglu C (2017). Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 551, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork T, Bernardos R, and Freeman MR (2012). Analysis of glial cell development and function in Drosophila. Cold Spring Harb Protoc 2012, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Poschmann J, Cruz-Herrera Del Rosario R, Parikshak NN, Hajan HS, Kumar V, Ramasamy R, Belgard TG, Elanggovan B, Wong CCY, et al. (2016). Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell 167, 1385–1397 e1311. [DOI] [PubMed] [Google Scholar]
- Tonnesen J, Inavalli V, and Nagerl UV (2018). Super-Resolution Imaging of the Extracellular Space in Living Brain Tissue. Cell 172, 1108–1121 e1115. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, and Nestler EJ (2007). Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 8, 355–367. [DOI] [PubMed] [Google Scholar]
- Volterra A, and Meldolesi J (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6, 626–640. [DOI] [PubMed] [Google Scholar]
- Wang D, and Fawcett J (2012). The perineuronal net and the control of CNS plasticity. Cell Tissue Res 349, 147–160. [DOI] [PubMed] [Google Scholar]
- Wang T, Hauswirth AG, Tong A, Dickman DK, and Davis GW (2014). Endostatin is a trans-synaptic signal for homeostatic synaptic plasticity. Neuron 83, 616–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weake VM, Lee KK, Guelman S, Lin CH, Seidel C, Abmayr SM, and Workman JL (2008). SAGA-mediated H2B deubiquitination controls the development of neuronal connectivity in the Drosophila visual system. EMBO J 27, 394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weake VM, and Workman JL (2012). SAGA function in tissue-specific gene expression. Trends Cell Biol 22, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Christian KM, He C, Jin P, Ming GL, and Song H (2016). Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci 17, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye B, Zhang Y, Song W, Younger SH, Jan LY, and Jan YN (2007). Growing dendrites and axons differ in their reliance on the secretory pathway. Cell 130, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger MA, Muller M, Tong A, Pym EC, and Davis GW (2013). A Presynaptic ENaC Channel Drives Homeostatic Plasticity. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Su Y, Shin J, Zhong C, Guo JU, Weng YL, Gao F, Geschwind DH, Coppola G, Ming GL, et al. (2015). Tet3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat Neurosci 18, 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate data sets for public repositories or new data code.