

Summary:
In vitro growth of alveolar soft part sarcoma (ASPS) and establishment of an ASPS cell line, ASPS-1, are described in this study. Using a recently developed xenograft model of ASPS derived from a lymph node metastasis, organoid nests consisting of 15 to 25 ASPS cells were isolated from ASPS xenograft tumors by capture on 70 μm filters and plated in vitro. After attachment to the substratum, these nests deposited small aggregates of ASPS cells. These cells grew slowly and were expanded over a period of 3 years and have maintained characteristics consistent with those of both the original ASPS tumor from the patient and the xenograft tumor including (1) presence of the alveolar soft part locus-transcription factor E3 type 1 fusion transcript and nuclear expression of the alveolar soft part locus-transcription factor E3 type 1 fusion protein; (2) maintenance of the t(X;17)(p11;q25) translocation characteristic of ASPS; and (3) expression of upregulated ASPS transcripts involved in angiogenesis (ANGPTL2, HIF-1-α, MDK, c-MET, VEGF, and TIMP-2), cell proliferation (PRL, PCSK1), metastasis (ADAM9), as well as the transcription factor BHLHB3 and the muscle-specific transcripts TRIM63 and ITGβ1BP3. This ASPS cell line forms colonies in soft agar and retains the ability to produce highly vascularized ASPS tumors in NOD.SCID/NCr mice. Immunohistochemistry of selected ASPS markers on these tumors indicated similarity to those of the original patient tumor as well as to the xenografted ASPS tumor. We anticipate that this ASPS cell line will accelerate investigations into the biology of ASPS including identification of new therapeutic approaches for treatment of this slow growing soft tissue sarcoma.
Keywords: sarcoma, ASPS, alveolar soft part sarcoma cell line (ASPS-1)
Alveolar soft part sarcoma (ASPS), first described in 1952,1 is an exceedingly rare soft tissue sarcoma accounting for <1% of all soft tissue sarcomas2,3 and primarily affects young children and adolescents. ASPS is refractory to both existing chemotherapy and radiation, and is, therefore, a candidate neoplasm for the development of novel therapies which focus on biological targets inherent to ASPS. ASPS characteristically exhibits a nonreciprocal chromosomal translocation, der(17)t(X;17)(p11;q25),4 which fuses the N-terminal region of the alveolar soft part locus (ASPL) gene, located at 17q25, to the C-terminal region of the transcription factor E3 (TFE3), located at Xp11.5 This translocation results in the expression of 2 distinct fusion transcripts, ASPL-TFE3 types 1 and 2, and their respective chimeric proteins which are thought to function as transcription factors. In an effort to gain insight into the biology of ASPS and to identify drug targets for this chemoresistant sarcoma we have sought to develop, over the past 5 years, in vitro and in vivo models of the disease. Propagation of ASPS from ASPS tumors obtained from patients has proved exceedingly difficult owing to both the slow growth rate of the tumor and its challenging histological makeup which includes areas of necrotic cells. Recently we have described a xenograft model of ASPS, established in immunocompromised mice,6 which maintains characteristics consistent with the original ASPS tumor including tumor histology, expression of the ASPL-TFE3 type 1 fusion transcript and ASPL-TFE3 type 1 fusion protein, maintenance of the t(X;17)(p11;q25) translocation characteristic of ASPS, stable expression of many ASPS gene transcripts, and the development and maintenance of a functional vascular network. As an extension of these efforts we report here the establishment and characterization of ASPS-1, a cell line of this rare soft tissue sarcoma.7 This ASPS cell line provides a novel tool which will facilitate investigation into the biology of the disease and aid in the identification and development of ASPS therapeutics.
MATERIALS AND METHODS
ASPS Tumor Procurement and in Vivo Growth of ASPS
A lymph node metastasis was obtained at the time of surgery from a 39-year-old female patient with a previously confirmed diagnosis of ASPS. Informed consent was obtained under NCI clinical research protocol 05-C-N138 and acquisition of tumor was assisted by the Alliance Against Alveolar Soft Part Sarcoma. The research protocol followed in this study is in compliance with the Helsinki Declaration for conduct of research using human patients. This lymph node metastasis has been successfully grown in immunocompromised, sex-matched NOD.SCID mice and developed into a xenograft model of ASPS.6
Establishment of ASPS-1: Isolation of ASPS Nests and in Vitro Culture of ASPS Cells
ASPS xenograft tumors were harvested 7 months after subcutaneous implantation into female NOD.SCID mice. ASPS cells were prepared by mincing small tumor fragments in Dulbecco modified Eagle’s medium Nutrient Mixture F-12 (DMEM: F-12) (1:1 vol/vol; Mediatech, Herndon, VA) containing 10% fetal bovine serum (Hyclone Laboratories, Logan, UT), 100 units/mL penicillin, 100 μg/mL streptomycin, 2.5 μg/mL fungizone, and 100 ng/mL DNase (Sigma-Aldrich, St Louis, MO). This method used for cell preparation from these ASPS xenograft tumors results in a mixture of both nests of ASPS cells consisting of 15 to 25 cells as well as individual tumor and stromal cells. The mixture was transferred to a 15 mL conical centrifuge tube and undissociated tumor fragments were removed by settling at unit gravity for 1 minute. The supernatant, containing both individual ASPS cells and nests of ASPS cells, was overlayed onto 70 μm filters (Becton Dickenson, Bedford, MA). The filters were gently rinsed in 2 mL DMEM: F12 (1:1 vol/vol) containing 10% fetal bovine serum. Under these conditions nests of ASPS cells are captured on the filters whereas single ASPS cells and stromal cells pass through the filters in the eluant. ASPS nests were eluted from the filters with DMEM: F12 (1:1 vol/vol) containing 10% fetal bovine serum and transferred to 6-well culture dishes. The cultures derived from wells showing selective growth of tumor cells were routinely examined over the next 3 years at every fifth passage for the presence of: (1) the t(X;17)(p11;q25) translocation characteristic of ASPS, (2) the type 1 ASPL-TFE3 fusion transcript, (3) signature ASPS genes, and (4) positive nuclear staining with antibodies to the type 1 ASPL-TFE3 fusion protein.
The methodologies used for these determinations have been extensively described.5,6,8 Although trypsin was not used in isolating ASPS nests from the xenograft tumor, established ASPS-1 cell cultures are routinely passaged using 0.05% trypsin and 0.1% ethylenediaminetetraacetic acid in calcium and magnesium-free phosphate-buffered saline (PBS).
In Vitro Growth Kinetics and Colony Formation of ASPS-1
ASPS-1 cells were seeded in 24-well plates at a concentration of 1×104 cells/well. At appropriate intervals cells were fixed in situ by the addition of 0.5 mL of 50% trichloroacetic acid per well and cell mass quantitated using the sulforhodamine B (SRB) protein assay.9 Briefly, plates were placed at 41C for 30 minutes, rinsed with water, and air dried. trichloroacetic acid-fixed cells were stained for 30 minutes with 0.4% SRB (Sigma, St Louis, MO) dissolved in 1% acetic acid. At the end of the staining period, SRB was removed and cultures were quickly rinsed 4 times with 1% acetic acid to remove unbound dye. After rinsing, the plates were air dried and protein-bound dye was solubilized with 10 mM unbuffered Tris base (pH 10.5) for 5 minutes on a shaker. Absorbance was read on a Bio-Tec Elx800 platereader (Bio-tek Instruments, Inc, Winooski, VT) at 564 nm. Curves of absorbance versus protein concentration were linear to 1.5 to 2.0 OD units.
For evaluation of colony forming potential, ASPS-1 cells were harvested, resuspended in ice-cold medium, and filtered through 40 μm filters to remove multicellular aggregates. Two volumes of cell suspension were diluted with 1 volume of 2% agarose, both in medium, and 0.5 mL of the resulting cell suspension overlayed onto a solid support matrix of 0.5 mL of medium containing 0.7% agarose in a 12-well plate. After equilibration at 41C for 10 minutes to solidfy the medium containing ASPS-1 cells, 1 mL of 371C medium, containing 0.7% agarose, was added over the cells and allowed to solidify. Finally, medium was overlayed onto the multicomponent assay system.
Microarray Data Acquisition and Analysis
Total RNA from patient, xenograft, and ASPS-1 cells were isolated and reversed transcribed as previously described.6,8 Microarray data was collected at Expression Analysis, Inc (Durham, NC) using the GeneChip Human U133 plus 2.0 Array (Affymetrix, Santa Clara, CA), according to standardized operating procedures. The following cDNA samples were analyzed in duplicate: ASPS patient tumor, a xenograft of the same material and 5 passage stages of the ASPS-1 cell line (p5, p10, p15, p20, and p25). Affymetrix raw data files (CEL) are available upon request. The Genesifter suite (VizX labs, Seattle, WA) was used for analysis of microarray data as previously described.6,8 Pairwise analysis was performed comparing universal RNA arrays with ASPS arrays. The following criteria were applied to filter the differentially expressed transcript list, a fold change of greater than 3, and a Wilcoxon rank sum test where P <0.05 with Benjamini-Hochberg estimation of false discovery rate.
Immunocytochemistry of Selected ASPS-1 Proteins
ASPS-1 cells were grown in 6-well plates. Growth medium was removed and cell monolayers were rinsed in PBS and fixed in 4% paraformaldehyde in PBS for 10 minutes at room temperature. Cells were permeabilized with −20°C methanol for 5 minutes and then blocked with 1% bovine serum albumin (BSA) in PBS for 30 minutes. The cells were incubated in primary antibody in 1% BSA in PBS for 60 minutes. Wells were rinsed in PBS and incubated in a fluorescein isothiocyanate-conjugated secondary antibody for 60 minutes. Wells were rinsed again in PBS and examined using a Leica DM IRB microscope equipped with BioQuant 98 software (BioQuant Life Science, Nashville, TN) with excitation at 488 nm. Antibody only controls (primary and fluorescein isothiocyanate secondary) were included in each experiment. The following antibodies were used: HIF-1-α, ITGβ1BP3, ASPL-TFE3 type 1, prolactin, TRIM 63, Midkine, c-MET, VEGF, and PCSK1. Antibody sources and dilutions are listed in Supplementary Table 1, Supplemental Digital Content 1, http://links.lww.com/JPHO/A2.
In Vivo Tumorigenicity and Immunohistochemistry of Selected Markers in Tumors Produced by ASPS-1 Cells
For evaluation of tumorigenicity in immunocompromised mice, ASPS-1 cells were grown in vitro, harvested, and suspended in medium containing 1% BSA and 100 ng VEGF/mL. Two volumes of medium containing cells were diluted with 1 volume of high protein Matrigel containing VEGF (final VEGF concentration 100 ng/mL), mixed and placed on ice to prevent solidification. The resulting suspension of ASPS-1 cells was injected subcutaneously into female NOD.SCID mice. ASPS-1 tumors were harvested 3 months after implantation and processed for diagnostic histology and immunohistochemistry as previously described.6,8 The following antibodies were used: HIF-1-α, ITGβ1BP3, CD34, ASPL-TFE3 type 1, ASPL-TFE3 type 2, Prolactin, TRIM63, Midkine, c-MET, VEGF, and PCSK1. Antibody sources, dilutions, methods used for antigen retrieval, and positive controls are listed in Supplementary Table 2, Supplemental Digital Content 2, http://links.lww.com/JPHO/A3.
Short Tandem Repeat Genotyping of ASPS-1 and the Original Patient Tumor
To verify that the ASPS-1 tumor cell line was derived from the original patient tumor, short tandem repeat genotyping was performed. The results were consistent with origin from the patient tumor and did not match the genotype of any known cell line based on a search of major short tandem repeat databases (American Type Culture Collection, Sanger Institute, Japanese Collection of Research Bioresources). Genotyping results are shown in Supplementary Table 3, Supplemental Digital Content 3, http://links.lww.com/JPHO/A4.
RESULTS
Establishment and Characterization of ASPS-1
Isolation of ASPS Nests and in Vitro Culture of ASPS Tumor Cells
The characteristic architecture of ASPS tumors consisting of organoid nests of ASPS cells surrounded by a delicate endothelium,1 prompted initial attempts to disaggregate ASPS tumors avoiding techniques which use trypsin and collagenases. This approach proved to be successful in isolating intact ASPS nests from ASPS xenograft tumors resulting in a mixture of ASPS nests as well as both individual ASPS and stromal cells. To avoid culturing rapidly proliferating stromal cells with the much slower growing ASPS tumor cells, nests of ASPS tumor cells were isolated on filters, washed free of contaminating stromal cells, and plated in vitro. As can be seen in Figure 1 nests of ASPS cells attached to the substratum and, over a period of 7 to 14 days deposited ASPS cells. Over the ensuing 6 to 8 months the ASPS cells slowly proliferated resulting in a monolayer which subsequently exhibited the formation of small 3-dimensional nests of cells.
FIGURE 1.

Establishment of ASPS-1: isolation of ASPS nests and in vitro culture of ASPS cells. ASPS xenograft tumors were harvested 7 months after subcutaneous implantation into female NOD.SCID mice as described in Materials and Methods. Nests of ASPS cells were collected on 70 μm filters. The patterns of growth observed on days 4, 7, 14, and 8 months after plating of ASPS nests are illustrated. ASPS indicates alveolar soft part sarcoma.
Establishment of ASPS-1 was accomplished over a period of several years. The first phase, propagation of cells from ASPS nests and early passage, was extremely slow. Cultures containing nests of ASPS-1 cells were subcultured, using trypsin disaggregation, first into 6-well plates (passage 1), observed for 3 months and were then subcultured at intervals of 7 months (passage 2) and 6 months (passage 3). This initial period of very slow growth of ASPS-1 cells, encompassing nearly 1.5 years, resulted in the ASPS-1 cell line currently used for research. As the growth rate increases with higher passage number we routinely use cells between passages 14 and 25. Cultures are subcultured at a split ratio of 1:3 and exhibit a doubling time of approximately 3 days (Figure, Supplemental Digital Content 4, http://links.lww.com/JPHO/A5).
ASPS-1 Cells Express the ASPL-TFE3 Type 1 Fusion Transcript
Two ASPL-TFE3 fusion transcripts have been described.5 Both the lymph node metastasis derived from the original patient8 as well as the ASPS xenograft tumor model developed from it6 expressed the ASPL-TFE3 type 1 fusion transcript. The ASPL-TFE3 type 1 fusion transcript has been continuously expressed for 25 passages in vitro encompassing a period of 3 years (Fig. 2).
FIGURE 2.

The ASPL-TFE3 type 1 fusion transcript is expressed in ASPS-1 cells. ASPL-TFE3 fusion transcripts were detected using a forward primer corresponding to nt 548 to 569 of ASPL (AAAGAAGTCCAAGTCGGGCCA) and a reverse primer corresponding to nt 972 to 993 in exon 4 of TFE3 (CGTTTGATGTTGGGCAGCTCA). Expected fragment sizes were 195 bp for ASPL-TFE3 type 1 and 300 bp for ASPL-TFE3 type 2.5 ASPS indicates alveolar soft part sarcoma; ASPL-TFE3, alveolar soft part locus-transcription factor E3.
ASPS-1 Cells Exhibit the t(X;17)(q11;p25) Translocation Characteristic of ASPS
Analysis of metaphase chromosomes of ASPS-1 tumor cells (passage 14) revealed a near triploid modal chromosome number and the presence of numerous chromosomal rearrangements including the characteristic t(X;17)(q11;p25) translocation reported for this tumor.4,5 The spectral karyotype was converted in SKYGRAM format from an analyzed metaphase that was positive for the translocation (Fig. 3B). Fluorescence in-situ hybridization analysis of metaphase cells using whole chromosome paints for X and 17 further confirmed the presence of the translocation (Fig. 3B).
FIGURE 3.

ASPS-1 cells exhibit a near triploid karyotype and the der(17)t(X;17)(p11;q25) chromosomal translocation characteristic of ASPS. Panel A, ASPS-1 SKY. Numerous chromosomal aberrations are present in this near triploid karyotype including the characteristic t(X;17)(q11;p25) translocation. Chromosome number=63. Panel B, FISH analysis of ASPS-1. Three copies of the t(X;17)(q11;p25) translocation, 1 normal X chromosome, and 2 deleted X chromosomes are present. X chromosome (green) and chromosome 17 (orange). A detailed description of the methods used for both SKY and FISH has been published.6 SKY and FISH were performed on passage 14 ASPS-1 cells. ASPS indicates alveolar soft part sarcoma; FISH, fluorescence in-situ hybridization; SKY, spectral karyotype.
Gene Expression Profile of ASPS-1 Cells
A comprehensive gene expression analysis on 7 ASPS patients8 identified several classes of genes which were differentially expressed relative to universal RNA, a pool of nontumor RNA. These 58 genes were subdivided into groups according to function (angiogenesis, growth factors, developmental, tissue restricted, drug resistance, steroid biosynthesis, metastasis, apoptosis, transcription factors, unknowns) resulting in a gene profile of ASPS. As part of our efforts to characterize ASPS-1 we have compared this subset of ASPS genes from the patient tumor, the xenograft from which it was derived, and ASPS-1. A heat map is presented in Figure 4 which illustrates their expression in the patient tumor, the xenograft tumor harvested 7 months after implantation into NOD.SCID mice,6 and passages 5 to 25 of ASPS-1. The majority of ASPS signature genes identified in patient tumors has been maintained in both the xenograft tumor and ASPS-1. However, several transcripts were not identified in the xenograft model including STAB1, ABCB5, CYP17A1, CH13L2, NTSR2, and POSTN. It is apparent that a critical subset of angiogenesis genes of this highly vascular sarcoma (ANGPTL2, HIF-1-α, MDK, c-MET, TIMP-2 and VEGF) were consistently expressed in the patient tumor, the xenograft, and ASPS-1. With the exception of MYF6 and NEB there is consistent expression of tissue restricted genes in the xenograft and ASPS-1. ITGβ1BP3, an antimyogenic differentiation gene,10,11 is consistently expressed in the xenograft tumor and ASPS-1.
FIGURE 4.

Comparative ASPS gene expression in patient, xenograft, and ASPS-1 Cells. cDNAs derived from ASPS primary tumor, xenografted tumor, and successive passages of the ASPS-1 cell line were hybridized to duplicate U133 plus 2.0 microarrays and independently subjected to pairwise analysis versus array data from universal RNA as described in Materials and Methods. For the 7 transcript lists generated, a subset of differentially expressed mRNAs previously implicated in ASPS pathogenesis was extracted and a heatmap produced showing fold change across the set. ASPS indicates alveolar soft part sarcoma; ASPL-TFE3, alveolar soft part locus-transcription factor E3.
Expression of Selected Markers by ASPS-1 Cells in Vitro
ASPS-1 cells exhibit strong nuclear staining for the ASPL-TFE3 type 1 fusion protein (Fig. 5) using an antibody directed against a 25-mer peptide spanning the ASPL-TFE3 type 1 fusion.12 A detailed gene expression profile of fresh primary and metastatic ASPS tumors indicated that genes involved in angiogenesis (HIF-1-α, MDK, c-MET, TIMP-2, and VEGF), metastasis (ADAM9), cell proliferation (PRL, PCSK1), and several tissue restricted genes (ITGβ1BP3 and TRIM63) were highly expressed in the disease.8 In this study we have examined ASPS-1 cells for expression of genes associated with angiogenesis (HIF-1-α, c-Met, VEGF, and MDK), cell proliferation (PRL, PCSK1), metastasis (ADAM9), and differentiation (ITGβ1BP3). As can be seen in Figure 5, ASPS-1 cells, grown in vitro, express these marker proteins. Nuclear and cytoplasmic staining was observed for the angiogenesis markers (HIF-1-α, c-MET, MDK, and VEGF); it is noteworthy that while the cytoplasmic staining observed for HIF-1-α, c-MET, and MDK was very diffuse, the staining of ASPS-1 for VEGF appeared also in granules. Staining for the metastasis marker ADAM9 was primarily nuclear whereas that of PRL was primarily cytoplasmic. Nuclear and diffuse cytoplasmic staining of ASPS-1 cells for ITGβ1BP3 and TRIM63 was apparent.
FIGURE 5.

ASPS-1 cells express markers involved in angiogenesis, metastasis, cell proliferation, and skeletal muscle. Cells were grown in coverglass chambers processed as described in Materials and Methods. Illustrated ASPS-1 proteins are ASPL-TFE3 type 1 fusion protein; ADAM9 (metastasis); PRL and PCSK1 (cell proliferation); ITGB1BP3 and TRIM63 (tissue restricted); and HIF-1-α, MDK, c-MET and VEGF (angiogenesis). ASPS indicates alveolar soft part sarcoma; ASPL-TFE3, alveolar soft part locus-transcription factor E3.
In Vivo Tumorigenicity and Immunohistochemistry of ASPS-1 Generated Tumors
ASPS-1 cells produce tumors in approximately 90% of NOD.SCID/NCr mice. Tumors were apparent 2–3 months after subcutaneous injection of ASPS-1 cells. These tumors exhibit morphology comparable to both the original patient tumor and the xenograft tumor from which ASPS-1 was derived, stain positively with periodic acid-Schiff/Diastase, and exhibit a complex vascular network (Fig. 6). Immunohistochemistry of selected ASPS marker proteins indicated strong nuclear staining for the ASPL-TFE3 type 1 fusion protein.12 Staining with antibody to the ASPL-TFE3 type 2 fusion protein was negative. ASPS tumors generated from ASPS-1 cells stained positively for PRL, PCSK1, HIF-1-α, MDK, c-MET, VEGF, and CD34. Staining for the tissue-restricted markers ITGβ1BP3 and TRIM63 were also positive (Fig. 6).
FIGURE 6.
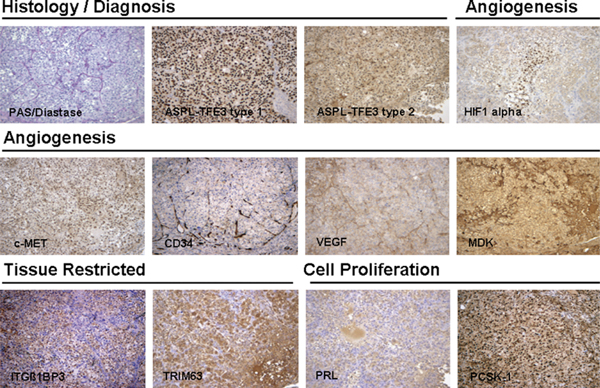
In vivo tumorigenicity and immunohistochemistry of selected markers in ASPS tumors produced by ASPS-1. ASPS-1 cells were injected subcutaneously into female NOD.SCID mice and tumors harvested 3 months later. Tumors were fixed in 10% neutral-buffered formalin and examined for both diagnostic (periodic acid-Schiff/Diastase, nuclear reactivity to ASPL-TFE3 type 1, and ASPL-TFE3 type 2 antibodies) and immunohistochemical markers (PRL, ITGB1BP3, TRIM63, HIF-1-α, MDK, c-MET, and VEGF) as described previously.6,8,12 ASPS indicates alveolar soft part sarcoma; ASPL-TFE3, alveolar soft part locus-transcription factor E3.
DISCUSSION
Since the first description of ASPS more than 50 years ago,1 little progress has been made in the development of new therapeutic strategies for the treatment of this sarcoma. This can be attributed, in part, to the lack of models to study the disease. The development of in vitro and in vivo models has been hindered primarily by the difficulty in acquiring tumors for study of this very rare malignancy.2,3 Additional impediments to ASPS model development include the slow growing nature of ASPS and the histological makeup of the tumor which includes many highly necrotic areas. In the face of these challenges it is hardly surprising that our understanding of ASPS pathobiology remains rudimentary.
Over the past 5 years an effort in our laboratory, centered on obtaining fresh ASPS tumors for research, has resulted in a detailed analysis of the gene expression profile of primary and metastatic ASPS8 and the development of a xenograft model of the disease.6 In this study, we have used this ASPS xenograft model to establish an ASPS cell line, ASPS-1, and characterized it with emphasis on comparing it to both the patient tumor and the xenograft tumor from which it was derived. Using a primarily mechanical dissociation technique, nests of 15 to 25 ASPS cells were isolated from xenograft tumors, captured on filters, and plated in vitro. The cells were expanded, over a period of 3 years, and characterized with respect to the ASPL-TFE3 fusion transcript, presence of the t(X;17)(p11;q25) translocation characteristic of ASPS,4,5 and expression of several signature genes and their respective proteins. Using this cell line, additional studies were undertaken to determine whether ASPS-1, grown in vitro, was capable of producing tumors in immunocompromised mice and whether these tumors retain the histological and molecular characteristics of ASPS.
Generation of this cell line model for the study of ASPS will have a direct impact on several therapeutic rationales currently being explored for the treatment of this disease. Most notably, there is continued interest in the use of antiangiogenics as ASPS therapeutics. Recent studies from our group6,8 and the Lazar et al laboratory13 have delineated upregulation of ASPS genes associated with the angiogenic phenotype. These include HIF-1-α, angiopoietin, midkine, c-MET, and VEGF. HIF-1-α, the master regulator of responses to hypoxia, is known to control several upregulated angiogenesis-related ASPS genes including angiopoietin 2,14 VEGF,15 c-Met,15 and midkine16 and prompted an in vivo therapeutic study in the recently described xenograft model of ASPS6 using topotecan, a topoisomerase 1 drug with antiangiogenic properties17,18 and a potent inhibitor of HIF-1-α,19–21 with the VEGF inhibitor bevacizumab.22 Results from this initial study showed a striking growth delay of 70% and a net log cell kill of 0.7.
A similarly tractable therapeutic approach involves direct targeting of the ASPL-TFE3 fusion. This tumor-specific hybrid transcription factor has been shown to bind to the c-MET promotor resulting in c-Met phosphorylation and down stream activation of the PI3K/Akt signaling pathway.23 Immunohistochemical evidence 24 confirmed the presence of c-Met, phosphorylated c-Met, phosphorylated MAPK/extracellular signal-regulated kinase, and phosphorylated serine/threonine specific protein kinase in ASPS. These observations suggest that downregulation of the ASPL-TFE3 fusion gene and its expressed protein may be a viable strategy in the development of new therapeutics. Preliminary data25 from our laboratory suggest that using siRNA directed against the breakpoint of ASPL-TFE3 results in downregulation of the ASPL-TFE3 type 1 fusion protein and retards the growth of ASPS-1.
Although the tissue of origin of ASPS is somewhat controversial, a myogenic origin is supported by our gene expression profiling of ASPS patients8 and resulted in identification of a novel ASPS drug target, ITGβ1BP3, which was markedly upregulated. This protein, found predominantly in muscle, is a negative regulator of terminal myogenesis10,11 and catalyzes the phosphorylation of nicotinamide riboside and nicotinic acid riboside to nicotinamide mononucleotide and nicotinic acid mononucleotide, respectively. That this antidifferentiation protein is consistently over expressed in ASPS patient tumors,8 the ASPS xenograft tumor,6 and ASPS-1 (Fig. 4) suggests that strategies designed to reduce it may result in terminal differentiation of ASPS tumor cells.
An additional ASPS therapeutic target, PCSK1, was identified8 as an upregulated transcript in ASPS patient tumors. PCSK1 is a member of the subtilisin-like proprotein convertase family responsible for cleaving precursor proteins posttranslationally, at pairs of basic amino acids, into biologically active products. An extensive literature (for reviews see references 26–28) outlines the biological specificity of the proprotein convertases. A detailed description of proprotein convertase expression in cancer and convertase substrates,28 which include matrix metalloproteinases, integrins, and growth factors (PDGFA, PDGFB, VEGF, TGF-B, IGF-1, IGF-2, and FGF-23), has resulted in attempts to identify convertase inhibitors.29–31
A paradoxical finding in our work on ASPS has been the consistent upregulation of 1 of the 4 TIMP family members, tissue inhibitor of metalloproteinase-2 (TIMP-2). Upregulation of TIMP-2 occurs in ASPS tumors from patients,8 ASPS xenografts,6 and in ASPS-1 (Fig. 4, current study). The context in which TIMP-2 functions in highly vascular ASPS tumors is unknown. This soluble extracellular molecule is believed to be the only TIMP family member to both inhibit and activate MMPs.32 Seminal work by the Stetler-Stevenson laboratory33 demonstrated that TIMP-2 inhibition of human microvascular endothelial cell proliferation in vitro and in vivo is independent of its MMP inhibitory activity; the antiangiogenic effects of TIMP-2 were due to its binding to the β1 subunit of the TIMP-2 receptor α3β1 on endothelial cells resulting in suppression of FGFR-1 and KDR (VEGF-R2) phosphorylation after exposure to FGF-2 and VEGF-A. Depressed phosphorylation is mediated by SRC homology 2 proteintyrosine phosphatases. The binding of TIMP-2 to the β1 subunit of the α3β1 receptor on endothelial cells raises the possibility of a similar interaction with the upregulated ASPS antidifferentiation protein, ITGβ1BP3.
Finally, considerable value will be accrued from using the ASPS-1 cell line and xenograft to study the efficacy of a broad range of approved and investigational chemotherapeutics and combination therapies. As a novel paradigm for slow growing sarcomas, it will be interesting to see whether results obtained in this model recapitulate clinical observations for this and other sarcomas. In conclusion, development of a xenograft-compatible ASPS cell line will facilitate in vitro investigations into the biology of ASPS, extending to identification and validation of drug targets, and preclinical in vivo evaluation of novel approaches for treatment of this unique malignancy.
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, the National Institutes of Health under contract N01-CO-12400, the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute, and The Alliance Against Alveolar Soft Part Sarcoma (TAAASPS). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US Government. National Cancer Institute (NCI)-Frederick is accredited by Association for Assessment and Accreditation of Laboratory Animal Care International (AALAC).
International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the “Guide for Care and Use of Laboratory Animals” (National Research Council, 1996; National Academy Press; Washington, DC).
Footnotes
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Website, www.jpho-online.com.
REFERENCES
- 1.Christopherson WM, Foote FW Jr, Stewart FW. Alveolar soft-part sarcomas: structurally characteristic tumors of uncertain histogenesis. Cancer. 1952;5:100–111. [DOI] [PubMed] [Google Scholar]
- 2.Folpe AL, Deyrup AT. Alveolar soft part sarcoma: a review and update. J Clin Pathol. 2006;59:1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soft Tissue sarcomas-SEER pediatric monograph. http://www.seer.cancer.gov.
- 4.Heimann P, Devalck C, Debusscher C, et al. Alveolar soft-part sarcoma: further evidence by FISH for the involvement of chromosome 17q25. Genes Chromosome Cancer. 1998;23:194–197. [PubMed] [Google Scholar]
- 5.Ladanyi M, Lui MY, Antonescu CR, et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20:48–57. [DOI] [PubMed] [Google Scholar]
- 6.Vistica DT, Hollingshead M, Borgel SD, et al. Therapeutic vulnerability of an in vivo model of alveolar soft part sarcoma (ASPS) to antiangiogenic therapy. J Pediatr Hematol Oncol. 2009;31:561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kenney S, Vistica DT, Stockwin L, et al. Establishment and characterization of ASPS-1, a novel cell line derived from metastatic alveolar soft part sarcoma. Proc Am Assoc Cancer Res. 2009;50:978. [Google Scholar]
- 8.Stockwin LH, Vistica DT, Kenney S, et al. Gene expression profiling of alveolar soft part sarcoma (ASPS). BMC Cancer. 2009;9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anti-cancer drug screening. J Natl Cancer Inst. 1990;82:1107–1112. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Mayne R, Wu C. A novel muscle specific beta 1 integrin binding protein (MIBP) that modulates myogenic differentiation. J Cell Biol. 1999;147:1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Rao H, Burkin D, et al. The muscle integrin binding protein (MIBP) intereacts with alpha7 beta1 integrin and regulates cell adhesion and laminin matrix deposition. Dev Biol. 2003;261:209–219. [DOI] [PubMed] [Google Scholar]
- 12.Vistica DT, Krosky PM, Kenney S, et al. Immunohistochemical discrimination between the ASPL-TFE3 fusion proteins of alveolar soft part sarcoma. J Pediatr Hematol Oncol. 2008;30: 46–52. [DOI] [PubMed] [Google Scholar]
- 13.Lazar AJF, Das P, Tuvin D, et al. Angiogenesis-promoting gene patterns in alveolar soft part sarcoma. Clin Cancer Res. 2007;13:7314–7321. [DOI] [PubMed] [Google Scholar]
- 14.Giaccai AJ, Rankin EB. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15:678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rocha S. Gene regulation under low oxygen: holding your breath for transcription. Trends Biochem Sci. 2007;32:389–397. [DOI] [PubMed] [Google Scholar]
- 16.Reynolds PR, Mucenski ML, LeCras TD, et al. Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J Biol Chem. 2004;279:37124–37132. [DOI] [PubMed] [Google Scholar]
- 17.O’Leary JJ, Shapiro RL, Ren CJ, et al. Antiangiogenic effects of camptothecin analogues 9-amino-20(S)-camptothecin, topotecan, and CPT-11 studied in the mouse cornea model. Clin Cancer Res. 1999;5:181–187. [PubMed] [Google Scholar]
- 18.Clements MK, Jones CB, Cumming M, et al. Antiangiogenic potential of camptothecin and topotecan. Cancer Chemother Pharmacol. 1999;44:411–416. [DOI] [PubMed] [Google Scholar]
- 19.Rapisarda A, Uranchimeg B, Scudiero DA, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62:4316–4324. [PubMed] [Google Scholar]
- 20.Rapisarda A, Uranchimeg B, Sordet O, et al. Topoisomerase 1 mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res. 2004;64:1475–1482. [DOI] [PubMed] [Google Scholar]
- 21.Rapisarda A, Zalek J, Hollingshead M, et al. Schedule-dependent inhibition of hypoxia-inducible factor-1 alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res. 2004;64:6845–6848. [DOI] [PubMed] [Google Scholar]
- 22.Ferrara N, Hillan KJ, Novatny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333:328–335. [DOI] [PubMed] [Google Scholar]
- 23.Tsuda M, Davis IJ, Argani P, et al. TFE3 fusions activate MET signaling by transcriptional up-regulation, defining another class of tumors as candidates for therapeutic MET inhibition. Cancer Res. 2007;67:919–929. [DOI] [PubMed] [Google Scholar]
- 24.Lazar AJ, Lahat G, Myers SE, et al. Validation of potential therapeutic targets in alveolar soft part sarcoma: an immunohistochemical study utilizing tissue microarray. Histopathology. 2009;55:750–755. [DOI] [PubMed] [Google Scholar]
- 25.Vistica DT, Kenney S, Burkett S, et al. Therapeutic targeting of the ASPL-TFE3 gene fusion in ASPS-1, a novel cell line derived from metastatic alveolar soft part sarcoma (ASPS). Proc Am Assoc Cancer Res. 2010;51:828. [Google Scholar]
- 26.Seidah NG, Chretien M. Pro-protein convertases of subtilisin/kexin family. Methods Enzymol. 1994;244:171–188. [DOI] [PubMed] [Google Scholar]
- 27.Rouille Y, Duguay SJ, Lund K, et al. Proteolytic processing mechanisms in the biosynthesis of neuroendocrine peptides: the subtilisin-like proprotein convertases. Front Neuroendocrinol. 1995;16:322–361. [DOI] [PubMed] [Google Scholar]
- 28.Khatib AM, Siegfried G, Chretien M, et al. Proprotein convertases in tumor progression and malignancy. Am J Pathol. 2002;160:1921–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basak A. Inhibitors of proprotein convertases. J Mol Med. 2005;83:844–855. [DOI] [PubMed] [Google Scholar]
- 30.Bontemps Y, Scamuffa N, Calvo F, et al. Potential opportunity in the development of new therapeutic agents based on endogenous and exogenous inhibitors of the proprotein convertases. Med Res Rev. 2007;27:631–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lahlil R, Calvo F, Khatib AM. The potential anti-tumorigenic and anti-metastatic side of the proprotein convertase inhibitors. Recent Pat Anticancer Drug Discov. 2009;4:83–91. [DOI] [PubMed] [Google Scholar]
- 32.Stetler-Stevenson WG. The tumor microenvironment: regulation by MMP-independent effects of tissue inhibitor of metalloproteinases-2. Cancer Metastasis Rev. 2008;27:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seo DW, Li H, Guedez L, et al. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell. 2003; 114:171–180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.