

Abstract
Mechanisms driving adaptive developmental responses to chronic high-altitude (HA) exposure are incompletely known. We developed a novel rat model mimicking the human condition of cardiopulmonary adaptation to HA starting at conception and spanning the in utero and postnatal timeframe. We assessed lung growth and cardiopulmonary structure and function and performed transcriptome analyses to identify mechanisms facilitating developmental adaptations to chronic hypoxia. To generate the model, breeding pairs of Sprague-Dawley rats were exposed to hypobaric hypoxia (equivalent to 9,000 ft elevation). Mating, pregnancy, and delivery occurred in hypoxic conditions. Six weeks postpartum, structural and functional data were collected in the offspring. RNA-Seq was performed on right ventricle (RV) and lung tissue. Age-matched breeding pairs and offspring under room air (RA) conditions served as controls. Hypoxic rats exhibited significantly lower body weights and higher hematocrit levels, alveolar volumes, pulmonary diffusion capacities, RV mass, and RV systolic pressure, as well as increased pulmonary artery remodeling. RNA-Seq analyses revealed multiple differentially expressed genes in lungs and RVs from hypoxic rats. Although there was considerable similarity between hypoxic lungs and RVs compared with RA controls, several upstream regulators unique to lung or RV were identified. We noted a pattern of immune downregulation and regulation patterns of immune and hormonal mediators similar to the genome from patients with pulmonary arterial hypertension. In summary, we developed a novel murine model of chronic hypoxia exposure that demonstrates functional and structural phenotypes similar to human adaptation. We identified transcriptomic alterations that suggest potential mechanisms for adaptation to chronic HA.
Keywords: high altitude, hypoxia, lung growth, pulmonary hypertension, RNA-Seq
INTRODUCTION
There are more than 140 million people born and raised at high altitude (HA; defined as >8,200 ft above sea level) (48). These individuals typically adapt to chronic hypoxia by increasing oxygen transport to tissues and altering cellular metabolism and somatic growth. The multitude of cardiopulmonary, hematologic, and metabolic adaptations to the decreased alveolar oxygen tension resulting from HA exposure include smaller somatic size, larger lungs, and higher hematocrit levels (3, 18, 44, 51). Although these processes are initially adaptive, if they are exaggerated and/or prolonged, they can result in cardiopulmonary morbidities. For example, some HA residents develop chronic mountain sickness as manifested by excessive erythrocytosis, and ~5% of HA residents develop severe high-altitude pulmonary hypertension (HAPH) with elevated pulmonary artery pressures and right heart failure (5, 51).
Although chronic hypoxia at sea level is an important contributor to advanced lung disease and the development of symptomatic pulmonary hypertension, most HA residents adapt appropriately through modifications in lung growth and develop asymptomatic HAPH (51). However, the mechanisms that underlie these adaptive changes are not completely understood. It is known from studies in HA Tibetans that selective evolutionary genetics play a vital role in the adaptive process as mutations in EPAS1 (endothelial PAS domain-containing protein 1; the gene encoding hypoxia-inducible factor 2) confer protection against aberrant erythropoiesis (6). Interestingly, adaptive responses to chronic hypoxia also occur in subjects without a hereditary link, suggesting a developmental adaptive response to chronic hypoxia. For example, fetuses born at HA exhibit smaller biometric measurements and higher hematocrit levels compared with fetuses born at low altitude (LA) (5, 18, 30). Furthermore, as an adaptation to increase gas exchange, infants and adults at HA exhibit larger lung volumes and greater pulmonary diffusion capacities compared with subjects at LA (39). Although evidence exists that these adaptive processes start in utero, the underlying mechanisms and mediators are poorly understood. In particular, little is known about transcriptome modifications that occur under chronic hypoxia conditions and what role they might play in adaptive lung and pulmonary vascular development.
Several animal studies have evaluated the effects of intermittent or chronic hypoxia on lung development and cardiopulmonary outcomes after birth (10, 17, 24, 27, 57, 75). However, these studies employed hypoxia exposure after delivery, and there are a limited number of studies investigating human cardiopulmonary adaptations to chronic hypoxia and/or HA starting at the time of conception. Additionally, previous rodent studies employed a degree of hypoxia that is more profound than that of humans living at HA. An animal model mimicking the human condition of cardiopulmonary adaptation to HA starting at conception and spanning the entire in utero timeframe would facilitate the study and identification of mechanisms that lead to adaptive phenotypes for chronic hypoxia early in life. Additionally, such a model could be used to better understand mechanisms of maladaptive pulmonary hypertensive phenotypes, such as pulmonary hypertension secondary to chronic lung disease. We therefore developed a novel murine model of simulated moderate HA exposure throughout conception and early life that mimics the human condition related to adaptive lung growth and cardiopulmonary function. We hypothesized that significant transcriptomic alterations occur under chronic hypoxic conditions and that these alterations provide a mechanism for developmental adaptations to chronic hypoxia.
METHODS
Animal care and model.
Studies were performed using male and female Sprague-Dawley rats (Harlan, Indianapolis, IN). Animal protocols were approved by the Indiana University Institutional Animal Care and Use Committee (IACUC; protocol no. 11056-MD/R). Animals received care in compliance with the Guide for the Care and Use of Laboratory Animals. Animals were allowed access to food and water ad libitum throughout the duration of the study.
Age-matched 7-wk-old male or female Sprague-Dawley rats (200–225 g) were acclimated to either room air or hypobaric for 3 wk until they achieved the appropriate breeding age of 10 wk. At 10 wk, each mating pair was placed together in a standard cage for mating in their respective room air or hypoxic environments. A hypobaric hypoxia chamber, depressurized to an equivalent of an inspired oxygen fraction of 15%, was used to simulate a 9,000-ft HA environment. This replicates the altitude of our recent data demonstrating that humans at HA have increased lung volumes very early in life (39).
Males were removed from the cages once the female was visibly pregnant, and females delivered naturally at term. Pups were weaned at 21 days, and physiological experiments and animal euthanization with tissue harvest occurred at 6 wk of age (Fig. 1). The 6-wk time point was chosen so that we could study lung growth and developmental changes before rats reached adulthood but also have large enough animals to feasibly perform the physiological experiments and have adequate tissue for the desired analyses. This time point is comparable to an age of 15–17 yr in humans (28a). Animals exposed to in utero and postnatal room air are denoted RA-RA, and animals exposed to in utero and postnatal hypoxia are denoted H-H. Animal care was provided as described previously (33). All experiments were performed in accordance with recent recommendations (8, 54), including randomization and blinding at the time of measurement and analysis.
Fig. 1.

Timeline of experiments and hypoxia exposure. Breeder pairs were acclimated to their respective environments for 3 wk before mating. Offspring were exposed to their respective in utero and postnatal environments until harvest at 6 wk of age. , fraction of inspired oxygen; H-H, in utero and postnatal hypoxia; RA-RA, in utero and postnatal room air.
Diffusing capacity and alveolar volume.
Diffusion capacity of the lung to carbon monoxide (DLCO) and alveolar volume (VA) were measured in animals anesthetized with intraperitoneal ketamine (95 mg/kg) plus xylazine (10 mg/kg). Animals were subsequently tracheostomized and mechanically ventilated (Flexivent, SCIREQ, Montreal, Canada) on room air. For each animal, the volume required to inflate the lung to 10 cm H2O was determined, and this volume of test gas (0.5% neon; 0.5% carbon monoxide; 21% oxygen; balance nitrogen) was used to manually inflate the lung for a 6-s breath hold and then withdrawn. The concentrations of neon and carbon monoxide in the volume of gas withdrawn from the lung were measured with a gas chromatograph (3000 Micro GC, INFICON, East Syracuse, NY). Measurements were repeated two to four times with at least 4 min between replicates. DLCO and VA were calculated as previously described and expressed under standard temperature and pressure conditions (2, 11, 16, 23). Dead space was presumed negligible due to animal size; therefore, VA was calculated as volume of the test gas multiplied by (concentration of the test gas/concentration of expired neon).
Echocardiographic and hemodynamic assessment.
Rats were anesthetized without supplemental oxygen using 1–2% isoflurane via a nose cone and then placed on a heated platform (22). Two-dimensional short-axis and long-axis images were acquired, and velocity time integral was measured in the right ventricular outflow tract. Stroke volume and cardiac output were calculated based on the velocity time integral as previously described (20, 22). Upon completion of echocardiography, animals were allowed at least 4 h of recovery before undergoing either DLCO testing or hemodynamic assessments. For the H-H group, animals were returned to the hypobaric chamber during recovery.
Right ventricular systolic pressure (RVSP) measurements were obtained in a blinded fashion in subgroups of randomly selected animals. Rats were anesthetized with 1–2% isoflurane and ventilated under 2% isoflurane with 100% supplemental oxygen via a tracheostomy. The right internal jugular vein was cannulated, and a 2 French Millar catheter (Millar Instruments, Houston, TX) was advanced into the right ventricle (RV) as previously described (34). RVSP was obtained with verification of appropriate RV waveform. Animals were then euthanized via exsanguination followed by immediate organ harvest.
Tissue harvest and fixation.
Whole blood was collected before exsanguination, and hematocrit values were obtained via visual measurement of red blood percentage after microcentrifugation. After blood collection, a bilateral pneumothorax was induced, and the lungs were flushed by infusing cold saline through the pulmonary artery until blood was visually cleared from the lungs. A clamp was then placed at the distal right mainstem bronchus. Under 20 cm of pressure, the left lung was inflated via continuous formalin infusion through the existing tracheostomy before being tied off and removed from the animal and submerged in formalin. After 48 h, the lungs were transferred to PBS and lung volumes were measured via water displacement.
After the heart was excised from the carcass, it was partitioned into the RV and left ventricle (LV) plus septum. Tissues were weighed individually for calculation of Fulton index [RV/(LV + septum)].
Pulmonary vascular remodeling and lung morphometry.
After the lung was embedded in paraffin and sectioned, Verhoeff–van Gieson immunohistochemical staining was performed on lung sections as previously described (4a). In a blinded fashion, pulmonary arteries <200 µm in diameter were identified by proximity to terminal bronchioles or alveolar ducts under a ×20 objective as previously described (34). Images were obtained using an Olympus BX41 microscope with Olympus camera. At least 10 vessels per animal were sampled, and the wall fraction [(total area – luminal area)/total area] was calculated using Image J software (22).
To determine whether there were differences in enlargement of air spaces between the two groups, the mean linear intercept (MLI) measurement was utilized to provide an estimate of alveolar size as previously described (13, 58). Briefly, a grid of evenly spaced horizontal lines was overlaid on each image taken at ×20 using light microscopy. For ease of counting, each grid was divided into four quadrants, and the number of times each line intersected an alveolar wall was recorded and averaged. To account for spatial differences in alveolar size, all images were taken at the level of the main stem bronchus. Slides that included large airways or blood vessels were excluded.
RNA-Seq and differential gene expression analysis.
Fresh frozen tissue (~25–50 mg) from male and female rat lung or heart RV was ground with a mortar and pestle kept on dry ice. The pulverized tissue was homogenized in 1 mL TRIzol using the Polytron homogenizer at 20,000 rpm for 1 min on ice. Total RNA was isolated from the TRIzol homogenate after high-speed centrifugation (20 min, 4°C) using Zymo Direct-zol column procedure and included on-column DNase digestion. RNA quality and integrity were determined by UV spectrum ratios and Agilent BioAnalyzer Tapestation. The median lung RNA Integrity Number (RIN) was 9.2 (range: 6.1–10), whereas the RV was slightly lower at 8.1 (range: 5.8–9.6), reflecting relatively poorer quality and yield from RV tissue. Roughly 250 ng of total RNA, based on the Bioanalyzer estimates, was used for next-generation sequencing library preparation.
Libraries were prepared using the KAPA mRNA Hyperprep Dual Index Kit, and both tissues’ libraries were mixed and run on the Illumina HiSeq4000 in one flow cell across five lanes. Paired-end 75 base reads were collected using Illumina’s HiSeq3000/4000 PE cluster/SBS kit. Raw sequence reads were quality controlled using FASTQC (9) and aligned to the rat Rn6.0 genome build using the STAR aligner (12). Median read mapping statistics (31 million raw reads) showed quality control Q30 of 96.3%, uniquely mapped reads (84.2%), of which 55.3% mapped to genes (14 million reads) (37). The Rn6 refGene (UCSC) genome annotation was used.
Differential gene expression analysis was completed for each tissue separately using edgeR with the Robust option (59), and genes were identified using a false discovery rate (FDR) cutoff P value <0.05. The differential expression gene list from each tissue was uploaded into the Ingenuity Pathway Analysis tool (IPA; Qiagen, Build 47547484) to identify significant upstream regulators. Validation of specific gene expression differences was completed using quantitative RT-PCR (qRT-PCR; Applied Biosystems) in both male and female animals. The RNA-Seq data have been deposited at NCBI/GEO (https://www.ncbi.nlm.nih.gov/geo/) under the accession GSE140146.
Statistical analysis.
Analyses were performed to determine whether there was a significant effect of treatment on the outcomes of interest. Analysis of variance (3) was performed to determine whether there was a significant difference in outcomes between RA-RA and H-H treatment groups in an unadjusted model and then run as an A × B factorial model with sex, including an interaction effect. If the interaction effect was nonsignificant, the model was run again with just the two main effects. Analysis of covariance (ANCOVA) models were then performed with pup weight as a covariate to determine whether that addition would attenuate results. All analytic assumptions were verified, and if distributions were skewed, log transformations were performed. For any variable that still did not achieve linearity, generalized estimating equations were used, which enable the model to fit nonlinear data to the appropriate distribution within the exponential family. A P value of less than 0.05 was considered significant. Correlations between gene expression and functional and structural parameters were determined by establishing Pearson’s correlation coefficients. All analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC).
RESULTS
A total of 29 RA-RA animals (2 litters comprising 15 males and 14 females) and 21 H-H animals (2 litters comprising 9 males and 12 females) were born and raised to 6 wk of age. There were zero pup deaths. Across all cardiopulmonary analyses, there were no sex differences or litter differences observed.
In utero and postnatal hypoxia exposure results in lower body weight and higher hematocrit levels.
Body weight and hematocrit levels are sensitive to chronic hypoxia exposure (3, 18, 44, 51). We therefore evaluated whether our degree of simulated moderate high-altitude exposure is sufficient to affect these parameters. We found that H-H animals exhibited 12% lower body weights and 7% higher hematocrit levels compared with RA-RA animals (Fig. 2, A and B).
Fig. 2.

Rats exposed to in utero and postnatal hypoxia (H-H) exhibit lower body weights (A) and higher hematocrit levels (B) compared with animals exposed to in utero and postnatal room air (RA-RA). Error bars represent means ± SE. Analysis by unpaired t test. *P < 0.05, **P < 0.01. The “//” marks at the figure’s origin indicate the y-axis is offset from zero.
In utero and postnatal hypoxia exposure is associated with increased lung growth and greater gas exchange.
Next, we evaluated markers of lung growth and function. H-H animals exhibited larger lungs, as demonstrated by 17% larger alveolar volumes, accompanied by a 44% greater diffusing capacity compared with RA-RA animals (Fig. 3, A and B). Mean linear intercepts (MLIs) of the two groups, on the other hand, did not differ (Fig. 4). We calculated the number of alveoli in the average RA and HA animal by using alveolar volume as measured by gas dilution and MLI as an estimate of alveolar diameter and assuming a spherical shape. We estimated the number of alveoli in the average RA and HA animals to be 43 and 51 million, respectively. Taken together, these data suggest the occurrence of compensatory and adaptive lung growth and improved gas exchange without alveolar enlargement.
Fig. 3.

Rats exposed to in utero and postnatal hypoxia (H-H) demonstrate larger lung volumes and greater gas exchange compared with animals exposed to in utero and postnatal room air (RA-RA). A: lung volume as measured by alveolar volume. B: gas exchange as measured by the diffusing factor for carbon monoxide (DLCO). Error bars represent means ± SE. Analysis by unpaired t test. **P < 0.01, ****P < 0.0001.
Fig. 4.

Rats exposed to in utero and postnatal hypoxia (H-H) are characterized by similar alveolar size compared with animals exposed to in utero and postnatal room air (RA-RA). Representative images of an RA-RA rat (A) with an average number of microns per intercept (MLI) of 58.4 compared with an H-H animal (B) with an average MLI of 58.9. C: MLI data for all animals. Size bar = 50 μm. Error bars represent means ± SE.
In utero and postnatal hypoxia exposure results in increased right ventricular mass and pulmonary vascular remodeling.
Chronic hypoxia exposure is a known inducer of pulmonary hypertension (63); however, previous rodent studies employed a degree of hypoxia that is more profound than that of humans living at HA (7, 42, 46). We therefore evaluated whether a lesser degree of simulated moderate HA exposure is sufficient to induce a pulmonary hypertensive phenotype. Indeed, animals in the H-H group displayed a 22% greater RV mass compared with RA-RA animals, suggestive of pressure increases in the pulmonary vasculature (Fig. 5A). Hemodynamic assessments similarly revealed that animals in the H-H group exhibited a 42% higher RVSP compared with the RA-RA group (Fig. 5B). This was associated with preserved cardiac output, suggesting an adaptive RV remodeling phenotype (Fig. 5, C and D). Analysis of pulmonary vascular remodeling revealed the presence of pulmonary artery wall remodeling in the H-H group compared with the RA-RA group (Fig. 5E). These data suggest the presence of a mild pulmonary hypertensive phenotype with maintained RV adaptation.
Fig. 5.

Animals exposed to in utero and postnatal hypoxia (H-H) exhibit pulmonary hypertension. A: right ventricular (RV) hypertrophy as measured by the Fulton index [weight of RV divided by weight of left ventricle (LV) plus septum: RV/(LV + septum)]. B: pulmonary hypertension assessment via right ventricular systolic pressure (RVSP) measurement. Cardiac output measurements by echocardiography, both adjusted (C) and unadjusted (D) for animal weight. E: pulmonary artery (PA) remodeling as measured by dividing the PA wall area by the entire vessel area after Verhoeff–van Gieson staining of lung. Representative images are shown. Note increase in PA wall thickness (arrows) in H-H group. RA-RA, in utero and postnatal room air. Error bars represent means ± SE. Analysis by unpaired t test. *P < 0.05, **P < 0.01, ***P < 0.001. Size bar = 10 μm. The “//” marks indicate the figure’s y-axis is offset from zero.
RNA-Seq reveals multiple differentially expressed genes in lungs and right ventricles from rats exposed to in utero and postnatal hypoxia versus normoxic controls.
The total RNA processed for next-generation sequencing was from 29 lungs and 25 RVs, divided into roughly equal numbers between 2 litters, RA-RA versus H-H and male versus female. After libraries were created, differential gene expression analysis was completed (59). Using an FDR cutoff P value <0.05, there were 742 and 491 significant differentially expressed genes in lung and RV, respectively. Standard volcano plots for lung and RV are shown in Fig. 6. We then performed separate analyses for each sex in each tissue (Fig. 7, A and B). Interestingly, male lungs revealed a greater number of differentially expressed genes (n = 832) than female lungs (n = 146). Of note, the majority of differentially expressed genes in male lungs were not differentially expressed in female lungs (n = 770; 92.5%), suggesting a sex bias in the hypoxia-induced transcriptome. RV tissue had a much more muted response in terms of number of differentially expressed genes and discrimination based on sex (Venn diagrams in Fig. 7).
Fig. 6.

Differentially expressed genes in rats exposed to in utero and postnatal hypoxia vs. normoxic controls. Volcano plots are shown for rat lung (A) and right ventricle (RV; B). Each dot represents a gene. Red denotes genes with a false discovery rate (FDR) P value < 0.05 (lung n = 742 genes; RV = 491 genes) with blue highlighting genes validated by quantitative RT-PCR (Table 1). x-axis depicts log2[fold change (FC)] ratios, expressed as hypoxia/room air (H-H/RA-RA), while the y-axis is log10(FDR q-value). See Table 1 for gene names.
Fig. 7.

Differentially expressed genes in male vs. female rats exposed to in utero and postnatal hypoxia vs. normoxic controls. Volcano plots are shown for male and female rat lung (A) and male and female right ventricle (RV; B). Red denotes genes with a false discovery rate (FDR) q-value < 0.05. x-axis depicts log2[fold change (FC)] ratios, expressed as hypoxia/room air (H-H/RA-RA), while the y-axis is log10(FDR q-value). Venn diagrams represent the number of differentially expressed genes in each sex as well as the overlap in differentially expressed genes between both sexes.
A number of gene expression changes were validated by qRT-PCR to confirm the reliability of the RNA-Seq data sets (Table 1). Genes were selected by their relevance to predicted pathway changes potentially important in hypoxia or pulmonary hypertension [clock circadian regulator (CLOCK), G protein and MAPK signaling, estrogen metabolism]. Expression patterns of five genes (Per3, Arntl, Rgs4, Tnnt2, and Adora2) matched those predicted from the RNA-Seq data sets in both lung and RV. Several genes tested were validated as differentially regulated in lung (Lyve1, Ntrk2, and Cyp1a1) or RV (Igf2) as anticipated from the RNA-Seq data sets.
Table 1.
Validation of differential expression prediction in RNA-Seq data set by qRT-PCR
| Gene Symbol | Description | Rat Lung HH/RARA |
Rat Lung RV/RARA |
||||||
|---|---|---|---|---|---|---|---|---|---|
| RNA-Seq |
qRT-PCR |
RNA-Seq |
qRT-PCR |
||||||
| Log2(Fold Change) | FDR q-Value | ΔΔCt(HH-RARA) | FDR q-Value | Log2(Fold Change) | FDR q-Value | ΔΔCt(HH-RARA) | FDR q-Value | ||
| Per3 | Period circadian regulator 3 | 1.585 | 5.21E-07 | 1.162 | 3.79E-03 | 1.935 | 2.59E-08 | 1.647 | 1.64E-02 |
| Arntl | Aryl hydrocarbon receptor nuclear translocator-like | −1.096 | 1.31E-04 | −1.985 | 3.79E-03 | −2.452 | 5.79E-09 | −2.534 | 2.09E-03 |
| Rgs4 | Regulator of G-protein signaling 4 | −1.137 | 3.46E-03 | −1.739 | 2.41E-02 | −0.899 | 1.29E-06 | −0.811 | 8.97E-03 |
| Adora2 | Adora 2 | −0.483 | 2.71E-03 | −0.419 | 2.52E-02 | −0.685 | 8.55E-03 | −0.463 | 7.79E-02 |
| Lyve1 | Lymphatic vessel endothelial hyaluronan receptor 1 | −0.967 | 3.98E-11 | −1.240 | 2.09E-03 | ns | ns | ns | ns |
| Ntrk2 | Neurotrophic receptor tyrosine kinase 2 | 2.523 | 2.11E-27 | 2.397 | 1.40E-05 | ns | ns | ns | ns |
| Cyp1a1 | Cytochrome P450, family 1, subfamily a, polypeptide 1 | −2.088 | 2.56E-17 | −2.337 | 5.04E-04 | ns | ns | ns | ns |
| Tnnt2 | Troponin T2, cardiac type | 1.229 | 2.13E-05 | 1.092 | 2.52E-02 | 0.280 | ns | 0.384 | 5.96E-02 |
| Lgf2 | Insulin-like growth factor 2 | 0.427 | 8.30E-03 | 0.432 | ns | −0.980 | 5.33E-04 | −0.535 | 5.96E-02 |
A selection of predicted differentially expressed genes from the RNA-Seq data sets was validated by quantitative RT-PCR (qRT-PCR). Genes were chosen for their relevance to transcriptional changes caused by hypoxia or pulmonary hypertension. Values are expressed as log2[fold change ratios (hypoxia/room air; HH/RA-RA)]. qRT-PCR multiple comparison corrected q-values were computed by the Benjamini–Hochberg method. FDR, false discovery rate; HH, in utero and postnatal hypoxia (n = 7; 4 males, 3 females); ; ns, not significant; RA-RA, in utero and postnatal room air (n = 7; 3 males, 4 females); RV, right ventricle.
Lung Htr4 expression is inversely correlated with DLCO.
To assess for potential relationships between lung and RV mRNA expression and parameters of cardiopulmonary adaptation to HA, we performed correlation analyses between lung mRNA expression and DLCO or hematocrit levels as well as between RV mRNA expression and Fulton index or hematocrit levels. Approximately 20 lung genes were correlated with DLCO (Table 2). Interestingly, we identified the peptidase and angiogenesis regulator high-temperature requirement factor A4 (HtrA4) as the top gene correlated with DLCO (Table 2 and Fig. 8). No lung genes were found to be significantly correlated with hematocrit, and no RV genes were correlated with Fulton index or hematocrit. Subgrouping the correlation analysis by sex did not reveal any significant genes with any of the tested parameters, most likely due to the study being underpowered.
Table 2.
Correlation analysis of rat hypoxia lung RNA expression and diffusing capacity for carbon monoxide
| Gene Symbol | Gene_Description | Lung Expression Versus DLCO Correlation Analysis |
Lung RNA-Seq Analysis |
||
|---|---|---|---|---|---|
| Correlation r | Correlation FDR q-Value | Log2(Fold Change HH/RARA) | FDR q-Value | ||
| Htra4 | HtrA serine peptidase 4 | −0.793 | 0.011 | −1.412 | 6.08E-12 |
| Fam181b | Family with sequence similarity 181, member B | 0.776 | 0.012 | 3.079 | 7.02E-30 |
| Mcpt1l1 | Mast cell protease 1-like 1 | 0.741 | 0.027 | 1.850 | 5.46E-17 |
| Dync1li2 | Dynein, cytoplasmic 1 light intermediate chain 2 | 0.739 | 0.027 | 0.221 | 3.80E-02 |
| Tmem150c | Transmembrane protein 150C | −0.743 | 0.027 | −1.122 | 9.83E-13 |
| Micb | MHC class I polypeptide-related sequence B | 0.684 | 0.045 | 0.382 | 2.93E-02 |
| Scin | Scinderin | −0.694 | 0.045 | −1.366 | 6.40E-08 |
| Gria4 | Glutamate ionotropic receptor AMPA type subunit 4 | 0.687 | 0.045 | 2.401 | 2.16E-07 |
| Sele | Selectin E | −0.685 | 0.045 | −1.516 | 1.05E-06 |
| Cldn10 | Claudin 10 | −0.690 | 0.045 | −0.710 | 5.56E-05 |
| Slc26a10 | Solute carrier family 26, member 10 | 0.686 | 0.045 | 2.960 | 3.82E-25 |
| Lyve1 | Lymphatic vessel endothelial hyaluronan receptor 1 | −0.704 | 0.045 | −0.967 | 3.98E-11 |
| Ntrk2 | Neurotrophic receptor tyrosine kinase 2 | 0.716 | 0.045 | 2.523 | 2.11E-27 |
| Abcd4 | ATP binding cassette subfamily D member 4 | 0.701 | 0.045 | 0.244 | 1.94E-01 |
| Tmc5 | Transmembrane channel-like 5 | −0.684 | 0.045 | −1.236 | 1.50E-05 |
| Ffar3 | Free fatty acid receptor 3 | 0.696 | 0.045 | 3.283 | 3.02E-28 |
| Ms4a8 | Membrane spanning 4-domains A8 | −0.689 | 0.045 | −1.042 | 5.07E-06 |
| Plppr5 | Phospholipid phosphatase related 5 | 0.713 | 0.045 | 2.146 | 5.44E-10 |
| Nell1 | Neural EGFL-like 1 | 0.700 | 0.045 | 0.759 | 4.06E-10 |
| Gadd45g | Growth arrest and DNA-damage-inducible γ | −0.685 | 0.045 | −0.491 | 1.02E-03 |
| Rgs6 | Regulator of G protein signaling 6 | 0.699 | 0.045 | 0.452 | 2.77E-03 |
| Hacd3 | 3-Hydroxyacyl-CoA dehydratase 3 | −0.684 | 0.045 | −0.290 | 8.39E-03 |
| Aoc1 | Amine oxidase, copper containing 1 | −0.689 | 0.045 | −1.383 | 5.58E-05 |
| Prrg4 | Proline rich and Gla domain 4 | −0.704 | 0.045 | −0.589 | 1.44E-02 |
| Ccr6 | C-C motif chemokine receptor 6 | −0.682 | 0.045 | −0.760 | 1.54E-03 |
| Lax1 | Lymphocyte transmembrane adaptor 1 | −0.680 | 0.046 | −0.860 | 3.37E-07 |
Pearson correlation analysis of lung RNA-Seq-derived gene expression and diffusing capacity for carbon monoxide (DLCO) measurements for the animals that had both measurements. Correlation values (r) and multiple comparison corrected q-values are shown for genes with q-values < 0.05, along with their differential gene expression values from the RNA-Seq analysis. FDR, false discovery rate; HH, in utero and postnatal hypoxia; RARA, in utero and postnatal room air.
Fig. 8.
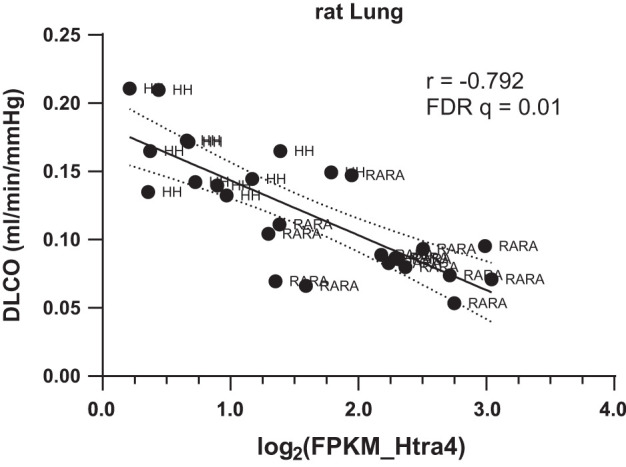
Lung expression of high-temperature requirement factor A4 (Htra4) is inversely correlated with diffusion capacity of the lung to carbon monoxide (DLCO). Relationship between lung expression of Htra4 and DLCO was assessed using Pearson’s correlation analysis. FDR q, false discovery rate q-value; FPKM, fragments per kilobase of exon model per million reads mapped; H-H, in utero and postnatal hypoxia; RA-RA, in utero and postnatal room air.
Common themes of both lung and right ventricle IPA pathway analyses.
Changes in each tissue’s transcriptional program can be compared by examining the similarities of the identified differentially expressed genes (lung vs. RV classifier gene list) and by the upstream regulators (URs) predicted by IPA (lung vs. RV UR output). In these analyses, there are two considerations. First, which genes or predicted URs are in common from both tissues? Second, is there concordance in the direction of change (up/up and down/down vs. up/down and down/up)? Figure 9A shows the relationship of differential gene expression [log2(fold-change)] in the 98 genes found to be in common between lung and RV tissues. Interestingly, there was strong concordance in the fold-change direction found in both tissues (n = 84 genes with concordant expression; χ2 = 1.15E-07).
Fig. 9.

Concordant differential gene expression patterns in hypoxia (H-H) vs. room air (RA-RA) rat lung and right ventricle (RV). A: genes in common that were differentially expressed in H-H vs. RA-RA lung and RV [false discovery rate (FDR) P < 0.05; lung n = 742, RV = 491] fold change (FC) ratios are plotted (n = 98 common genes; n = 84 concordant expression; χ2 = 1.15E-07). FC ratios are expressed as H-H/RA-RA treatment. B: all differentially expressed genes from either lung or RV (FDR P < 0.05; lung n = 742, RV = 491) were inputted into Ingenuity Pathway Analysis (IPA). The predicted upstream regulators (URs) from each tissue were aligned for concordant predictions (n = 155 UR predicted in common). The majority of UR predictions had concordant z-score estimates (up/up or down/down; n = 116; χ2 = 7.40E-06). Dashed lines in A indicate 95% confidence limits of the linear best fit model. Red box in B is bounding UR z-score > |2.0| to visualize the IPA suggested cutoff for calling a pathway activated or inhibited. Note that discordant UR predictions all had a z-score < |2.00|, indicating a weakly predicted transcriptional state for discordant URs.
After the differentially expressed gene list from each tissue was uploaded into IPA, particular focus was placed on potential URs. Using this approach, predictions in several thousand potential URs were performed. We found 155 predicted URs in common between lung and RV tissues. Of these, 116 were concordant in their direction (up/up or down/down; χ2 = 7.40E-06), and the predicted UR z-scores were significantly correlated (correlation coefficient r = +0.59; P < 0.0001; Fig. 9B). These results suggest a strong similarity between the lung and RV global transcriptional patterns in hypoxia.
Table 3 highlights a number of common predicted themes between lung and RV tissue observed. In particular, IPA revealed several common predicted themes in both lung and RV tissue, namely chemical/drug modulators, cytokines, growth factors, receptors, and traditional transcription factors. These mediators have been implicated in hypoxia signaling and/or pulmonary hypertension pathogenesis (15, 19, 21, 31, 36, 55). Specifically, in both H-H lung and RV there was global downregulation of immune response gene expression, including innate immunity [strong inhibition of lipopolysaccharide (LPS)-predicted effect], and macrophage activity (inhibition of IFNG, TNF, IL6, and IL10). β-Estradiol and estrogen receptor 1 (ESR1) were predicted to have an activated signature in both H-H lung and RV. Lastly, Erb-B2 receptor tyrosine kinase 2 (ERBB2), Krüppel-like factor 2 (KLF2), and NF-κ-B inhibitor α (NFKBIA) showed inverse predicted activity profiles in H-H lung and RV. Taken together, these analyses suggest significant similarities in the transcriptional state of the hypoxic lung and hypoxic RV.
Table 3.
Rat H-H/RA-RA lung and right ventricle common top scoring upstream regulators
| Upstream Regulator | Molecule Type | Rat HH/RARA Lung |
Rat RV HH/RARA |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Expr Log Ratio | Adjusted P Value | Activation z-Score | Predicted Activation State | Expr Log Ratio | Adjusted P Value | Activation z-Score | Predicted Activation State | ||
| β-Estradiol | Chemical | 2.27E-26 | 0.572 | 2.85E-13 | 0.770 | ||||
| Dexamethasone | Chemical | 2.54E-17 | 1.114 | 3.45E-16 | 0.034 | ||||
| Lipopolysaccharide | Chemical | 1.72E-23 | −4.662 | Inhibited | 6.66E-20 | −2.132 | Inhibited | ||
| IFNG | Cytokine | 2.99E-12 | −5.049 | Inhibited | 4.29E-12 | −2.702 | Inhibited | ||
| IL10 | Cytokine | 2.17E-09 | −0.894 | 2.71E-07 | −1.056 | ||||
| IL1B | Cytokine | −0.536 | 1.02E-14 | −5.377 | Inhibited | −1.151 | 5.30E-19 | −0.712 | |
| IL6 | Cytokine | −2.614 | 1.91E-16 | −1.441 | 2.71E-07 | −0.593 | |||
| TNF | Cytokine | 7.82E-19 | −6.062 | Inhibited | 3.62E-23 | −2.826 | Inhibited | ||
| TGFB1 | Growth factor | 5.11E-14 | −3.031 | Inhibited | 2.84E-09 | −1.996 | |||
| ESR1 | Receptor | 2.24E-08 | 1.612 | 4.11E-05 | 0.406 | ||||
| ERBB2 | Kinase | 4.57E-09 | 2.520 | Activated | 2.97E-09 | −0.389 | |||
| KLF2 | Transcription regulator | −1.035 | 5.23E-10 | 1.249 | 1.39E-09 | −0.543 | |||
| NFKBIA | Transcription regulator | 2.99E-12 | 1.174 | 4.03E-08 | −1.442 | ||||
Expression (Expr) log ratios indicate genes that were included in the differentially regulated gene lists in the lung or right ventricle (RV) analyses (q-value < 0.05). Activation status (indicated by z-score), as predicted by Ingenuity Pathway Analysis (IPA) tool, indicates strength of activation (positive value) or inhibition (negative value) status. Z-scores > |2.0| are indicated as “Activated” (> +2.0) or “Inhibited” (< −2.0). Most notable is global downregulation of immune response gene expression in both lung and RV tissue. The P value represents the chance that the given differentially expressed gene list fits a predicted pattern expected for a transcriptional regulator, otherwise known as an enrichment estimation. Also displayed is IPA analysis of the pattern of up- and downregulated genes and predicted functional “status” of the upstream regulator. In most cases, the actual predicted gene’s expression is not a component of the uploaded differentially expressed gene list but is a predicted regulator from both of these considerations (enrichment and observed differentially expressed up- or downregulation). ESR1, estrogen receptor α, ERBB2, Erb-B2 receptor tyrosine kinase 2; HH, in utero and postnatal hypoxia; IFNG, interferon γ; IL6, interleukin 6; IL10, interleukin 10; IL1B, interleukin 1β; KLF2, Krüppel-like factor 2; NFKBIA, NF-κ-B inhibitor α; RARA, in utero and postnatal room air; TGFB1, transforming growth factor β1; TNF, tumor necrosis factor α.
Hypoxic pulmonary hypertension transcription factors identified in rat lung and RV.
In addition to common lung and RV upstream regulation themes, we also identified several transcription factors strongly predicted to be activated or inhibited in H-H lung or RV tissue (Fig. 10). In the rat lung, T-box transcription factor 2 (TBX2) and mesenchyme homeobox 2 (MEOX2) were predicted activated, whereas signal transducer and activator of transcription 1 (STAT1) and RELA proto-oncogene, NF-κB subunit (RELA) were predicted inhibited. TBX2 has a potential role for immortalizing cancer (70), MEOX2 regulates limb myogenesis in development (74), and STAT1 and RELA (67) are important in regulating immune response transcriptional programs. In the RV, predicted activated upstream transcription factors identified included mothers against decapentaplegic homolog 7 (SMAD7) and CLOCK. Predicted inhibited transcription factors in the H-H RV were hypoxia-inducible factor 1α (HIF1A) and STAT1, which was also predicted in the H-H lung. Taken together, this demonstrates that both hypoxic lungs and RVs exhibit changes in transcription involved in proliferation, immune responses, circadian rhythm, and hypoxia signaling.
Fig. 10.

Selected upstream transcription factors unique to lung or right ventricle (RV). Predicted activated (A, C) or inhibited (B, D) upstream transcription factors in lungs (A, B) and RVs (C, D) from rats exposed to in utero and postnatal hypoxia vs. normoxic controls. In the lung, T-box transcription factor 2 (TBX2) and mesenchyme homeobox 2 (MEOX2) were predicted activated (A), whereas signal transducer and activator of transcription 1 (STAT1) and proto-oncogene, NF-κB subunit (RELA) were predicted inhibited (B). In the RV, predicted activated upstream transcription factors identified included mothers against decapentaplegic homolog 7 (SMAD7) and clock circadian regulator (CLOCK; C), whereas hypoxia-inducible factor 1α (HIF1A) and STAT1 were predicted inhibited (D). Corrected P values and z-scores for these transcription factors are shown in boxes below each diagram. Hubs identify transcription factors, spokes identify their targets (see legend for further details).
Comparison of the rat hypoxic pulmonary hypertension and human lung pulmonary arterial hypertension transcriptome.
To evaluate the relevance to human pulmonary arterial hypertension, we compared the rat lung and RV H-H/RA-RA upstream regulator predictions with our recently published human lung pulmonary arterial hypertension (29) transcriptome data set (62) (Table 4). Interestingly, the global pattern of immune downregulation, as well as regulation patterns of LPS, ESR1, ERBB2, and NFKBIA, were shared between human and rat, suggesting hypoxia-induced changes in expression patterns that span several species. On the other hand, differences in expression patterns were noted for β-estradiol, dexamethasone, TGFB1 [a strong predicted activator in human lung PAH (35, 71)], and KLF2 (a regulator of proliferative processes in the pulmonary vasculature whose mutation has been linked to heritable PAH) (15), suggesting that these mediators are differently regulated by hypoxia in rat and human.
Table 4.
Commonly activated or inhibited regulators in rat hypoxia lungs and human pulmonary arterial hypertension (67) lungs
| Upstream Regulator | Molecule Type | Rat HH/RARA Lung |
Human PAH/FD Lung |
||
|---|---|---|---|---|---|
| Adjusted P Value | Activation z-Score | Adjusted P Value | Activation z-Score | ||
| β-Estradiol | Chemical | 2.27E-26 | ↑ | 3.18E-16 | Inhibited |
| Dexamethasone | Chemical | 2.54E-17 | ↑ | 1.58E-14 | Inhibited |
| Lipopolysaccharide | Chemical | 1.72E-23 | Inhibited | 5.64E-09 | ↓ |
| IFNG | Cytokine | 2.99E-12 | Inhibited | 4.09E-05 | ↓ |
| IL10 | Cytokine | 2.17E-09 | ↓ | 7.91E-06 | Inhibited |
| IL1B | Cytokine | 1.02E-14 | Inhibited | 1.13E-03 | ↓ |
| IL6 | Cytokine | 1.91E-16 | ↓ | 6.59E-03 | ↓ |
| TNF | Cytokine | 7.82E-19 | Inhibited | 6.00E-11 | ↓ |
| TGFB1 | Growth factor | 5.11E-14 | Inhibited | 1.02E-08 | ↑ |
| ESR1 | Receptor | 2.24E-08 | ↑ | 2.65E-06 | ↑ |
| ERBB2 | Kinase | 4.57E-09 | Activated | 2.08E-03 | ↑ |
| KLF2 | Transcription regulator | 5.23E-10 | ↑ | 1.24E-02 | Inhibited |
| NFKBIA | Transcription regulator | 2.99E-12 | ↑ | 5.76E-03 | ↑ |
Rat lung upstream regulators were compared with predictions from our recently published human PAH transcriptome data set (29). Expression of regulators in hypoxic rat lungs is compared with lungs from rats exposed to in utero and postnatal room air (HH/RARA). Expression of regulators from PAH lungs is compared with expression in failed lung transplant donor (FD) lungs. Adjusted P values indicate degree of pathway enrichment from the differentially expressed gene lists. Z-scores indicate predicted pathway direction with “Inhibited”/”Activated” indicating z-scores > |2.0|, and up or down arrows indicating z-scores < |2.0|. Note a global pattern of immune downregulation that is shared between rat and human, as well as common LPS, ESR1, ERBB2, and NFKBIA regulation patterns. HH, in utero and postnatal hypoxia; RARA, in utero and postnatal room air. See Table 2 for full names of regulators.
DISCUSSION
To our knowledge, this is the first animal model of in utero chronic hypoxia that mimics cardiopulmonary adaptation of humans residing at HA. Our transcriptomic analyses provide novel insight into potential mechanisms for these adaptations. In particular, we describe a phenotype characterized by adaptive mild-to-moderate changes in body weight, hematocrit, alveolar volume, gas exchange, and cardiopulmonary hemodynamics similar to what has been described in humans. Consistent with what we know about human development at HA, animals exposed to chronic hypoxia from conception exhibited lower body weights and higher hematocrit levels compared with their room air counterparts (Fig. 2). Chronically hypoxic animals also had larger lung volumes and higher pulmonary diffusion capacity (Fig. 3), both of which are important physiological mechanisms for adapting to chronic hypoxia that have been described in adults born and raised at HA (25). These animals also displayed a mild and adapted pulmonary hypertensive phenotype (Fig. 5), consistent with the modest pulmonary vascular remodeling seen in humans with chronic hypoxia-induced pulmonary hypertension (4, 63).
Previous animal studies evaluated effects of acute hypoxia (64) or chronic hypoxia (26, 56) during a specific developmental time period, demonstrating that neonatal or perinatal hypoxia causes a restricted pulmonary arterial bed and has long-term implications for pulmonary vasoreactivity later in life. Fewer studies have evaluated the in utero time period alone (7, 42, 46). Our study is unique in that we evaluated cardiopulmonary effects of chronic hypoxia introduced at the earliest time point in life and sustained in the postnatal time period. This study therefore has the potential to determine adaptive and nonpathological responses to chronic hypoxia throughout the entire early lifespan.
Although the effect of sustained hypoxia on lung growth and development early in life was evaluated in previous murine models, these studies differ in the timing and severity of the chronic hypoxia exposure and do not consistently replicate the human condition (43, 55). Postnatal exposure to 12% oxygen from 2–14 days of life resulted in a larger lung volume with larger alveoli in adult mice compared with RA controls (55). Similarly in rats, perinatal exposure to 10% oxygen during the last day of gestation and continued for the pups for 1–2 h after delivery resulted in smaller somatic size and lung volumes, increased MLI, and decreased surface area for gas exchange (43). The murine model with a chronic hypoxia exposure most similar to our study included female rats acclimatized to 13% oxygen for several weeks and maintained in 13% oxygen during pregnancy and delivery, and then evaluated pups raised in the 13% oxygen environment (7, 42). In that study, there were no differences in lung volumes for hypoxic and RA rats at 40 days of age; however, morphometric analysis of the lung parenchyma demonstrated that rats exposed to chronic hypoxia exhibited larger MLI, fewer alveoli, and less parenchymal tissue for gas exchange (7). These morphometric findings contrast with our physiological findings, as well as the physiological and high-resolution computed tomography imaging findings in adult humans born and raised at HA (40). Potential explanations for the differences between the two murine studies with chronic hypoxia exposure include that the previous study: 1) used a greater degree of hypoxia (13% vs. the equivalent of 15% employed in our study), 2) evaluated only 3–5 animals per group with no indication of sex, 3) did not report whether hematocrit was affected, 4) acclimatized only female animals to chronic hypoxia before breeding, and 5) conducted breeding in RA rather than in hypoxia. We believe that our study more closely mimics the condition of human reproduction at HA.
As intended, to replicate the human phenotype, structural and functional changes in our model were modest. MLI, an indirect measurement of alveolar size, did not differ for the two groups, suggesting that the larger lung volumes in the H-H animals are not a function of larger alveoli but rather result from an increased number of alveoli of similar size. This would also increase alveolar surface area for gas exchange, which is consistent with our finding of a higher diffusion capacity in the H-H group. Our H-H animals exhibited evidence of pulmonary hypertension and pulmonary vascular remodeling without a decrease in cardiac output, indicating an adaptive response to chronic hypoxia. This is consistent with the phenotype described in humans residing at HA (3, 18, 44, 51). We do not suggest that this is a model of the more pronounced pulmonary vascular changes with angio-obliterative lesions that are seen in murine models of PAH (1, 66). Although we did not detect any sex differences in structural or physiological parameters analyzed, we did note a sex bias in the lung transcriptome response. Studies with larger animal groups and/or longer time courses may be required to detect potential phenotypical differences between sexes.
Potential mechanisms for the observed cardiopulmonary adaptations are provided by our transcriptomic analyses. We found changes in genes related to circadian rhythm, proliferative signaling, receptor signaling, and cytochrome P450 activity (Fig. 6; Table 1). Of note, the global transcriptional changes observed in the hypoxic lung and RV shared many common features across these different tissues, both in the concordant directionality of fold changes of individual genes as well as predicted upstream regulators (Table 3). For example, we highlighted a large-scale downregulation of immune signature genes in both tissues, which was also observed in human PAH lung tissue (Table 4). Further similarities between hypoxic lung and RV include roles for β-estradiol/ESR1 and TGFB1 pathways, both of which are known to play a role in human hypoxia-induced pulmonary hypertension, PAH, and subsequent RV failure. Interestingly, several upstream regulators identified in rat lungs were also significantly enriched in human PAH lung compared with control tissue (Table 4), suggesting hypoxia-induced expression patterns that span several species. Examples include ESR1 and ERBB2. An interesting dichotomy was observed in male-specific differentially expressed genes of the lung transcriptome not seen in the female lung or in RV tissue of either sex (Fig. 7). Several potential mechanisms could be responsible for this difference even in our long-term adaptation model to HA. First, males may have two clear transcriptional programs between normoxia and HA that are maintained. Second, driven by either evolutionary constraints or lung structure/physiology that is female specific, the female lung may be better adapted to HA (28). Finally, the male response may be more homogenous (thus making it easier to detect differentially expression) due to absence of confounding biological effects (e.g., estrous cycle).
Although our correlation analyses may have been underpowered, we detected a robust negative correlation between lung Htra4 abundance and DLCO (Fig. 8 and Table 2). This is interesting because Htra4’s product Htra4 originates in the placenta, another organ specialized in gas exchange (69). Secreted Htra4 is increased in preeclampsia (a vascular disease characterized by profound endothelial cell dysfunction) and impairs function of cultured human umbilical venous endothelial cells (61, 69). Our data suggest that Htra4 may also negatively affect angiogenesis and gas exchange processes in the chronically hypoxic lung. This will be the focus of future studies in our laboratories.
We also highlighted several strongly predicted upstream transcription factors (Fig. 10). In hypoxic lungs, MEOX2 and TBX2 were predicted to be significantly upregulated, whereas STAT1 and RELA were downregulated. MEOX2 is a homeobox protein involved in regulation of limb myogenesis, the lung overexpression of which reverses hypoxia-induced pulmonary hypertension and inhibits smooth muscle cell proliferation and induces apoptosis in vitro (72). MEOX2 may thus be a modulator of increased muscularization of pulmonary vasculature. TBX2 is a potential driver of lung cancer progression (47). STAT1 and RELA are both involved in immune signaling, with STAT1 being responsive to cytokines and RELA being the most abundant protein partner in the NF-κB complex after translocation to the nucleus. Predicted downregulation of both of these pathways is consistent with the global downregulation of immune-specific genes and pathways. STAT1 has been implicated in suppressing angiogenesis under hypoxia in cancer (73), which may be relevant to vascular endothelial cell changes in pulmonary hypertension. NF-κB complex activity is dependent on oxygen levels in BEAS-2B immortalized bronchial epithelial cells, with decreased activity under hypoxic conditions (29), consistent with our findings.
In the hypoxic RV, the upregulated status of SMAD7 (an inhibitory SMAD) would predict inhibition of TGFβ and bone morphogenetic protein (BMP) pathways, and in fact both had negative z-scores in the IPA analysis. Consistent with this finding, SMAD7 is increased in several rat models of pulmonary hypertension, along with downregulation of BMPR2 protein (45). One unexpected finding was the predicted downregulation of HIF1A. It is possible that this predicted downregulation of HIF1A in hypoxic RV tissue may be secondary to other features of the transcriptome (41). For example, inhibition of HIF1A may be ascribed due to its link to several downregulated genes such as CDKN1A (cyclin inhibitor), EDN1 and ADM (vascular tone), TGFB2 (TGF signaling), and VEGFC (angiogenesis). Another possibility is the interaction between the immune system and HIF1A, where innate immune and inflammation response positively regulate HIF1A in an oxygen-independent manner (49). Because we observed a global downregulation of the immune system in both hypoxic lungs and RVs, this subsequently could lead to a downregulated HIF1A prediction. We corroborated the IPA analysis data by measuring HIF-1α in RV homogenates from RARA and HH animals by Western blot and did not observe an increase in HIF-1α protein (data not shown). Although a lack of HIF-1α upregulation in chronic hypoxia seems counterintuitive, there is evidence that attenuated HIF signaling is associated with favorable adaptations to HA in Tibetans (6, 52). Additionally, experimental data suggest that chronic hypoxia may lead to destabilization of HIF-1α mRNA, mediated at least in part via a concomitant increase in the quantity of natural antisense HIF-1α (aHIF) (38). In fact, a “HIF switch” has been reported, where HIF-1α activity decreases, while HIF-2α and HIF-3α activities increase during chronic hypoxia (60). Such a phenomenon may have been at play in our chronically hypoxic rats and will be studied in the future.
Additionally, CLOCK was predicted to be upregulated in hypoxic RVs, consistent with validated upregulation of Per3 and downregulation of Arntl mRNA (Table 1). The RNA-Seq hypoxic RV data set also revealed upregulation of mRNA of other members of the CLOCK pathway, such as Per1, Per2, and Cry2 (data not shown). Besides daily circadian function, CLOCK is implicated in lung tumorigenesis (50), cell proliferation in cancer (68), pulmonary function in chronic airway diseases (65), and immune regulation (14), features relevant to our chronically hypoxia lung and RV tissues. CLOCK may be a new therapeutic avenue for intervention in hypoxia and/or pulmonary hypertension.
Our study has limitations. Our assessments of somatic growth, diffusing capacity of carbon monoxide, alveolar volume, and pulmonary vascular remodeling were obtained at 6 wk of age and not immediately after birth, preventing us from ascertaining whether hypoxia in utero or sustained hypoxia in the postnatal period was the major determinant of the differences we observed in our study groups. Further studies will evaluate these end points in newborn animals and compare them to findings later in life. Another limitation is that we did not employ a three-dimensional, unbiased assessment to measure the lung volumes, such as stereology. We also used whole lung and RV tissue for our RNA-Seq analyses, which may have diluted potential biological differences and prevents us from determining which specific cell types might play a significant role in the observed adaptations to chronic hypoxia. Lastly, we did not employ a provocative test, such as exercise, to assess for RV dysfunction under additional cardiopulmonary stress, which could provide further insight into adaptive and maladaptive RV phenotypes. This will be the focus of future studies.
In conclusion, we report a novel murine model that replicates the physiological cardiopulmonary adaptation of adults born and raised at HA. The consistency of our animal model with human data generates a strong potential to evaluate molecular mechanisms of adaption to chronic hypoxia that will then assist in identifying novel targets and pathways for the treatment of cardiopulmonary diseases related to chronic hypoxia.
GRANTS
This work was supported in part by the Pulmonary Hypertension Association Barst Fund, Cordelia’s Pediatric Pulmonary Hypertension Research and Mentoring Grant (S.K.), Veterans Affairs Merit Review Award 2 I01 BX002042-05 (T.L.), and NIH National Heart, Lung, and Blood Institute Grants 1R56HL134736-01A1 (T.L.), 1R01HL144727-01A1 (T.L.), and T32 HL091816-08 (M.W.G., R.S.T.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.K., R.S.S., M.W.G., and R.S.T. conceived and designed research; S.K., R.S.S., L.Z., A.J.F., E.A.M., B.H.R., and T.G.C. performed experiments; S.K., R.S.S., L.Z., E.R.S., and J.E.S. analyzed data; S.K., R.S.S., E.R.S., M.I., and R.S.T. interpreted results of experiments; S.K., R.S.S., and L.Z. prepared figures; S.K. and R.S.S. drafted manuscript; S.K., R.S.S., L.Z., T.L., and R.S.T. edited and revised manuscript; S.K., R.S.S., M.W.G., T.L., and R.S.T. approved final version of manuscript.
REFERENCES
- 1.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754, 2010. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 2.Ahlfeld SK, Gao Y, Conway SJ, Tepper RS. Relationship of structural to functional impairment during alveolar-capillary membrane development. Am J Pathol 185: 913–919, 2015. doi: 10.1016/j.ajpath.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aldashev AA, Sarybaev AS, Sydykov AS, Kalmyrzaev BB, Kim EV, Mamanova LB, Maripov R, Kojonazarov BK, Mirrakhimov MM, Wilkins MR, Morrell NW. Characterization of high-altitude pulmonary hypertension in the Kyrgyz: association with angiotensin-converting enzyme genotype. Am J Respir Crit Care Med 166: 1396–1402, 2002. doi: 10.1164/rccm.200204-345OC. [DOI] [PubMed] [Google Scholar]
- 4.Arias-Stella J, Saldana M. The terminal portion of the pulmonary arterial tree in people native to high altitudes. Circulation 28: 915–925, 1963. doi: 10.1161/01.CIR.28.5.915. [DOI] [PubMed] [Google Scholar]
- 4a.Armed Forces Institute of Pathology Laboratory Methods in Histotechnology, edited by Prophet EB, Mills B, Arrington JB, Sobin LH. Washington, D.C.: American Registry of Pathology, 1992. [Google Scholar]
- 5.Ballew C, Haas JD. Hematologic evidence of fetal hypoxia among newborn infants at high altitude in Bolivia. Am J Obstet Gynecol 155: 166–169, 1986. doi: 10.1016/0002-9378(86)90104-3. [DOI] [PubMed] [Google Scholar]
- 6.Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, Li C, Li JC, Liang Y, McCormack M, Montgomery HE, Pan H, Robbins PA, Shianna KV, Tam SC, Tsering N, Veeramah KR, Wang W, Wangdui P, Weale ME, Xu Y, Xu Z, Yang L, Zaman MJ, Zeng C, Zhang L, Zhang X, Zhaxi P, Zheng YT. Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci USA 107: 11459–11464, 2010. doi: 10.1073/pnas.1002443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanco LN, Massaro D, Massaro GD. Alveolar size, number, and surface area: developmentally dependent response to 13% O2. Am J Physiol Lung Cell Mol Physiol 261: L370–L377, 1991. doi: 10.1152/ajplung.1991.261.6.L370. [DOI] [PubMed] [Google Scholar]
- 8.Bonnet S, Provencher S, Guignabert C, Perros F, Boucherat O, Schermuly RT, Hassoun PM, Rabinovitch M, Nicolls MR, Humbert M. Translating research into improved patient care in pulmonary arterial hypertension. Am J Respir Crit Care Med 195: 583–595, 2017. doi: 10.1164/rccm.201607-1515PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breese MR, Liu Y. NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 29: 494–496, 2013. doi: 10.1093/bioinformatics/bts731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campen MJ, Shimoda LA, O’Donnell CP. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J Appl Physiol (1985) 99: 2028–2035, 2005. doi: 10.1152/japplphysiol.00411.2005. [DOI] [PubMed] [Google Scholar]
- 11.Cox AM, Gao Y, Perl AT, Tepper RS, Ahlfeld SK. Cumulative effects of neonatal hyperoxia on murine alveolar structure and function. Pediatr Pulmonol 52: 616–624, 2017. doi: 10.1002/ppul.23654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21, 2013. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunnill MS. Quantitative methods in the study of pulmonary pathology. Thorax 17: 320–328, 1962. doi: 10.1136/thx.17.4.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Early JO, Menon D, Wyse CA, Cervantes-Silva MP, Zaslona Z, Carroll RG, Palsson-McDermott EM, Angiari S, Ryan DG, Corcoran SE, Timmons G, Geiger SS, Fitzpatrick DJ, O’Connell D, Xavier RJ, Hokamp K, O’Neill LAJ, Curtis AM. Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc Natl Acad Sci USA 115: E8460–E8468, 2018. doi: 10.1073/pnas.1800431115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eichstaedt CA, Song J, Viales RR, Pan Z, Benjamin N, Fischer C, Hoeper MM, Ulrich S, Hinderhofer K, Grünig E. First identification of Krüppel-like factor 2 mutation in heritable pulmonary arterial hypertension. Clin Sci (Lond) 131: 689–698, 2017. doi: 10.1042/CS20160930. [DOI] [PubMed] [Google Scholar]
- 16.Fallica J, Das S, Horton M, Mitzner W. Application of carbon monoxide diffusing capacity in the mouse lung. J Appl Physiol (1985) 110: 1455–1459, 2011. doi: 10.1152/japplphysiol.01347.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farahani R, Kanaan A, Gavrialov O, Brunnert S, Douglas RM, Morcillo P, Haddad GG. Differential effects of chronic intermittent and chronic constant hypoxia on postnatal growth and development. Pediatr Pulmonol 43: 20–28, 2008. doi: 10.1002/ppul.20729. [DOI] [PubMed] [Google Scholar]
- 18.Frisancho AR. Developmental adaptation to high altitude hypoxia. Int J Biometeorol 21: 135–146, 1977. doi: 10.1007/BF01553707. [DOI] [PubMed] [Google Scholar]
- 19.Frump AL, Albrecht ME, McClintick JN, Lahm T. Estrogen receptor-dependent attenuation of hypoxia-induced changes in the lung genome of pulmonary hypertension rats. Pulm Circ 7: 232–243, 2017. doi: 10.1177/2045893217702055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frump AL, Goss KN, Vayl A, Albrecht M, Fisher A, Tursunova R, Fierst J, Whitson J, Cucci AR, Brown MB, Lahm T. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol 308: L873–L890, 2015. doi: 10.1152/ajplung.00006.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frump AL, Selej M, Wood JA, Albrecht M, Yakubov B, Petrache I, Lahm T. Hypoxia upregulates estrogen receptor β in pulmonary artery endothelial cells in a HIF-1α-dependent manner. Am J Respir Cell Mol Biol 59: 114–126, 2018. doi: 10.1165/rcmb.2017-0167OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goss KN, Cucci AR, Fisher AJ, Albrecht M, Frump A, Tursunova R, Gao Y, Brown MB, Petrache I, Tepper RS, Ahlfeld SK, Lahm T. Neonatal hyperoxic lung injury favorably alters adult right ventricular remodeling response to chronic hypoxia exposure. Am J Physiol Lung Cell Mol Physiol 308: L797–L806, 2015. doi: 10.1152/ajplung.00276.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham BL, Brusasco V, Burgos F, Cooper BG, Jensen R, Kendrick A, MacIntyre NR, Thompson BR, Wanger J. 2017 ERS/ATS standards for single-breath carbon monoxide uptake in the lung. Eur Respir J 49: 1600016, 2017. doi: 10.1183/13993003.00016-2016. [DOI] [PubMed] [Google Scholar]
- 24.Grover TR, Parker TA, Balasubramaniam V, Markham NE, Abman SH. Pulmonary hypertension impairs alveolarization and reduces lung growth in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 288: L648–L654, 2005. doi: 10.1152/ajplung.00288.2004. [DOI] [PubMed] [Google Scholar]
- 25.Guleria JS, Pande JN, Sethi PK, Roy SB. Pulmonary diffusing capacity at high altitude. J Appl Physiol 31: 536–543, 1971. doi: 10.1152/jappl.1971.31.4.536. [DOI] [PubMed] [Google Scholar]
- 26.Hakim TS, Mortola JP. Pulmonary vascular resistance in adult rats exposed to hypoxia in the neonatal period. Can J Physiol Pharmacol 68: 419–424, 1990. doi: 10.1139/y90-059. [DOI] [PubMed] [Google Scholar]
- 27.Herrera EA, Pulgar VM, Riquelme RA, Sanhueza EM, Reyes RV, Ebensperger G, Parer JT, Valdéz EA, Giussani DA, Blanco CE, Hanson MA, Llanos AJ. High-altitude chronic hypoxia during gestation and after birth modifies cardiovascular responses in newborn sheep. Am J Physiol Regul Integr Comp Physiol 292: R2234–R2240, 2007. doi: 10.1152/ajpregu.00909.2006. [DOI] [PubMed] [Google Scholar]
- 28.Horiuchi M, Kirihara Y, Fukuoka Y, Pontzer H. Sex differences in respiratory and circulatory cost during hypoxic walking: potential impact on oxygen saturation. Sci Rep 9: 9550, 2019. doi: 10.1038/s41598-019-44844-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Jackson Laboratory. Age converter (Online). http://www.age-converter.com/mouse-age-calculator.html. [3 December 2018].
- 29.Jagannathan L, Jose CC, Arita A, Kluz T, Sun H, Zhang X, Yao Y, Kartashov AV, Barski A, Costa M, Cuddapah S. Nuclear factor κB1/RelA mediates inflammation in human lung epithelial cells at atmospheric oxygen levels. J Cell Physiol 231: 1611–1620, 2016. doi: 10.1002/jcp.25262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krampl E, Lees C, Bland JM, Espinoza Dorado J, Moscoso G, Campbell S. Fetal biometry at 4300 m compared to sea level in Peru. Ultrasound Obstet Gynecol 16: 9–18, 2000. doi: 10.1046/j.1469-0705.2000.00156.x. [DOI] [PubMed] [Google Scholar]
- 31.Kumar S, Wei C, Thomas CM, Kim IK, Seqqat R, Kumar R, Baker KM, Jones WK, Gupta S. Cardiac-specific genetic inhibition of nuclear factor-κB prevents right ventricular hypertrophy induced by monocrotaline. Am J Physiol Heart Circ Physiol 302: H1655–H1666, 2012. doi: 10.1152/ajpheart.00756.2011. [DOI] [PubMed] [Google Scholar]
- 33.Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, Justice MJ, Brown MB, Van Demark M, Trulock KM, Dieudonne D, Reddy JG, Presson RG, Petrache I. 17β-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med 185: 965–980, 2012. doi: 10.1164/rccm.201107-1293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lahm T, Frump AL, Albrecht ME, Fisher AJ, Cook TG, Jones TJ, Yakubov B, Whitson J, Fuchs RK, Liu A, Chesler NC, Brown MB. 17β-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 311: L375–L388, 2016. doi: 10.1152/ajplung.00132.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lambers C, Roth M, Zhong J, Campregher C, Binder P, Burian B, Petkov V, Block LH. The interaction of endothelin-1 and TGF-β1 mediates vascular cell remodeling. PLoS One 8: e73399, 2013. doi: 10.1371/journal.pone.0073399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Wei C, Kim IK, Janssen-Heininger Y, Gupta S. Inhibition of nuclear factor-κB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension 63: 1260–1269, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03220. [DOI] [PubMed] [Google Scholar]
- 37.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930, 2014. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 38.Lin Q, Cong X, Yun Z. Differential hypoxic regulation of hypoxia-inducible factors 1α and 2α. Mol Cancer Res 9: 757–765, 2011. doi: 10.1158/1541-7786.MCR-11-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Llapur CJ, Martínez MR, Caram MM, Bonilla F, Cabana C, Yu Z, Tepper RS. Increased lung volume in infants and toddlers at high compared to low altitude. Pediatr Pulmonol 48: 1224–1230, 2013. doi: 10.1002/ppul.22764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Llapur CJ, Martínez MR, Grassino PT, Stok A, Altieri HH, Bonilla F, Caram MM, Krowchuk NM, Kirby M, Coxson HO, Tepper RS. Chronic hypoxia accentuates dysanaptic lung growth. Am J Respir Crit Care Med 194: 327–332, 2016. doi: 10.1164/rccm.201509-1851OC. [DOI] [PubMed] [Google Scholar]
- 41.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 40: 294–309, 2010. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Massaro GD, Olivier J, Dzikowski C, Massaro D. Postnatal development of lung alveoli: suppression by 13% O2 and a critical period. Am J Physiol Lung Cell Mol Physiol 258: L321–L327, 1990. doi: 10.1152/ajplung.1990.258.6.L321. [DOI] [PubMed] [Google Scholar]
- 43.Massaro GD, Olivier J, Massaro D. Short-term perinatal 10% O2 alters postnatal development of lung alveoli. Am J Physiol Lung Cell Mol Physiol 257: L221–L225, 1989. doi: 10.1152/ajplung.1989.257.4.L221. [DOI] [PubMed] [Google Scholar]
- 44.Moore LG, Niermeyer S, Zamudio S. Human adaptation to high altitude: regional and life-cycle perspectives. Am J Phys Anthropol 107, Suppl 27: 25–64, 1998. [DOI] [PubMed] [Google Scholar]
- 45.Murakami K, Mathew R, Huang J, Farahani R, Peng H, Olson SC, Etlinger JD. Smurf1 ubiquitin ligase causes downregulation of BMP receptors and is induced in monocrotaline and hypoxia models of pulmonary arterial hypertension. Exp Biol Med (Maywood) 235: 805–813, 2010. doi: 10.1258/ebm.2010.009383. [DOI] [PubMed] [Google Scholar]
- 46.Murphy JD, Aronovitz MJ, Reid LM. Effects of chronic in utero hypoxia on the pulmonary vasculature of the newborn guinea pig. Pediatr Res 20: 292–295, 1986. doi: 10.1203/00006450-198604000-00003. [DOI] [PubMed] [Google Scholar]
- 47.Nehme E, Rahal Z, Sinjab A, Khalil A, Chami H, Nemer G, Kadara H. Epigenetic suppression of the T-box subfamily 2 (TBX2) in human non-small cell lung cancer. Int J Mol Sci 20: 1159, 2019. doi: 10.3390/ijms20051159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niermeyer S, Andrade Mollinedo P, Huicho L. Child health and living at high altitude. Arch Dis Child 94: 806–811, 2009. doi: 10.1136/adc.2008.141838. [DOI] [PubMed] [Google Scholar]
- 49.Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity 41: 518–528, 2014. doi: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden MG, Jacks T. Circadian rhythm disruption promotes lung tumorigenesis. Cell Metab 24: 324–331, 2016. doi: 10.1016/j.cmet.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Penaloza D, Arias-Stella J. The heart and pulmonary circulation at high altitudes: healthy highlanders and chronic mountain sickness. Circulation 115: 1132–1146, 2007. doi: 10.1161/CIRCULATIONAHA.106.624544. [DOI] [PubMed] [Google Scholar]
- 52.Peng Y, Cui C, He Y, Ouzhuluobu, Zhang H, Yang D, Zhang Q, Bianbazhuoma, Yang L, He Y, Xiang K, Zhang X, Bhandari S, Shi P, Yangla, Dejiquzong, Baimakangzhuo, Duojizhuoma, Pan Y, Cirenyangji, Baimayangji, Gonggalanzi, Bai C, Bianba, Basang, Ciwangsangbu, Xu S, Chen H, Liu S, Wu T, Qi X, Su B. Down-regulation of EPAS1 transcription and genetic adaptation of Tibetans to high-altitude hypoxia. Mol Biol Evol 34: 818–830, 2017. doi: 10.1093/molbev/msw280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Provencher S, Archer SL, Ramirez FD, Hibbert B, Paulin R, Boucherat O, Lacasse Y, Bonnet S. Standards and methodological rigor in pulmonary arterial hypertension preclinical and translational research. Circ Res 122: 1021–1032, 2018. doi: 10.1161/CIRCRESAHA.117.312579. [DOI] [PubMed] [Google Scholar]
- 55.Ramani M, Bradley WE, Dell’Italia LJ, Ambalavanan N. Early exposure to hyperoxia or hypoxia adversely impacts cardiopulmonary development. Am J Respir Cell Mol Biol 52: 594–602, 2015. doi: 10.1165/rcmb.2013-0491OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reeves JT, Leathers JE. Postnatal development of pulmonary and bronchial arterial circulations in the calf and the effects of chronic hypoxia. Anat Rec 157: 641–655, 1967. doi: 10.1002/ar.1091570410. [DOI] [PubMed] [Google Scholar]
- 57.Reinke C, Bevans-Fonti S, Grigoryev DN, Drager LF, Myers AC, Wise RA, Schwartz AR, Mitzner W, Polotsky VY. Chronic intermittent hypoxia induces lung growth in adult mice. Am J Physiol Lung Cell Mol Physiol 300: L266–L273, 2011. doi: 10.1152/ajplung.00239.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robbesom AA, Versteeg EM, Veerkamp JH, van Krieken JH, Bulten HJ, Smits HT, Willems LN, van Herwaarden CL, Dekhuijzen PN, van Kuppevelt TH. Morphological quantification of emphysema in small human lung specimens: comparison of methods and relation with clinical data. Mod Pathol 16: 1–7, 2003. [DOI] [PubMed] [Google Scholar]
- 59.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140, 2010. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Serocki M, Bartoszewska S, Janaszak-Jasiecka A, Ochocka RJ, Collawn JF, Bartoszewski R. miRNAs regulate the HIF switch during hypoxia: a novel therapeutic target. Angiogenesis 21: 183–202, 2018. doi: 10.1007/s10456-018-9600-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singh H, Zhao M, Chen Q, Wang Y, Li Y, Kaitu’u-Lino TJ, Tong S, Nie G. Human HtrA4 expression is restricted to the placenta, is significantly up-regulated in early-onset preeclampsia, and high levels of HtrA4 cause endothelial dysfunction. J Clin Endocrinol Metab 100: E936–E945, 2015. doi: 10.1210/jc.2014-3969. [DOI] [PubMed] [Google Scholar]
- 62.Stearman RS, Bui QM, Speyer G, Handen A, Cornelius AR, Graham BB, Kim S, Mickler EA, Tuder RM, Chan SY, Geraci MW. Systems analysis of the human pulmonary arterial hypertension lung transcriptome. Am J Respir Cell Mol Biol 60: 637–649, 2019. doi: 10.1165/rcmb.2018-0368OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 64.Stenmark KR, Fasules J, Hyde DM, Voelkel NF, Henson J, Tucker A, Wilson H, Reeves JT. Severe pulmonary hypertension and arterial adventitial changes in newborn calves at 4,300 m. J Appl Physiol (1985) 62: 821–830, 1987. doi: 10.1152/jappl.1987.62.2.821. [DOI] [PubMed] [Google Scholar]
- 65.Sundar IK, Yao H, Sellix MT, Rahman I. Circadian clock-coupled lung cellular and molecular functions in chronic airway diseases. Am J Respir Cell Mol Biol 53: 285–290, 2015. doi: 10.1165/rcmb.2014-0476TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, McMahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438, 2001. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 67.Tully JE, Nolin JD, Guala AS, Hoffman SM, Roberson EC, Lahue KG, van der Velden J, Anathy V, Blackwell TS, Janssen-Heininger YM. Cooperation between classical and alternative NF-κB pathways regulates proinflammatory responses in epithelial cells. Am J Respir Cell Mol Biol 47: 497–508, 2012. doi: 10.1165/rcmb.2012-0014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Verlande A, Masri S. Circadian clocks and cancer: timekeeping governs cellular metabolism. Trends Endocrinol Metab 30: 445–458, 2019. doi: 10.1016/j.tem.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, Lim R, Nie G. HtrA4 may play a major role in inhibiting endothelial repair in pregnancy complication preeclampsia. Sci Rep 9: 2728, 2019. doi: 10.1038/s41598-019-39565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wansleben S, Peres J, Hare S, Goding CR, Prince S. T-box transcription factors in cancer biology. Biochim Biophys Acta 1846: 380–391, 2014. doi: 10.1016/j.bbcan.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 71.Woo KV, Ornitz DM, Singh GK. Diagnosis and pathophysiological mechanisms of group 3 hypoxia-induced pulmonary hypertension. Curr Treat Options Cardiovasc Med 21: 16, 2019. doi: 10.1007/s11936-019-0718-3. [DOI] [PubMed] [Google Scholar]
- 72.Xia S, Tai X, Wang Y, An X, Qian G, Dong J, Wang X, Sha B, Wang D, Murthi P, Kalionis B, Wang X, Bai C. Involvement of Gax gene in hypoxia-induced pulmonary hypertension, proliferation, and apoptosis of arterial smooth muscle cells. Am J Respir Cell Mol Biol 44: 66–73, 2011. doi: 10.1165/rcmb.2008-0442OC. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Y, Jin G, Zhang J, Mi R, Zhou Y, Fan W, Cheng S, Song W, Zhang B, Ma M, Liu F. Overexpression of STAT1 suppresses angiogenesis under hypoxia by regulating VEGF-A in human glioma cells. Biomed Pharmacother 104: 566–575, 2018. doi: 10.1016/j.biopha.2018.05.079. [DOI] [PubMed] [Google Scholar]
- 74.Zheng H, Hu Z, Zhai X, Wang Y, Liu J, Wang W, Xue S. Gax regulates human vascular smooth muscle cell phenotypic modulation and vascular remodeling. Am J Transl Res 8: 2912–2925, 2016. [PMC free article] [PubMed] [Google Scholar]
- 75.Zieliński J. Effects of intermittent hypoxia on pulmonary haemodynamics: animal models versus studies in humans. Eur Respir J 25: 173–180, 2005. doi: 10.1183/09031936.04.00037204. [DOI] [PubMed] [Google Scholar]