

SUMMARY
Mechanisms resulting in abdominal pain include altered neuro-immune interactions in the gastrointestinal tract, but the signaling processes that link immune activation with visceral hypersensitivity are unresolved. We hypothesized that enteric glia link the neural and immune systems of the gut and that communication between enteric glia and immune cells modulates the development of visceral hypersensitivity. To this end, we manipulated a major mechanism of glial intercellular communication that requires connexin-43 and assessed the effects on acute and chronic inflammation, visceral hypersensitivity, and immune responses. Deleting connexin-43 in glia protected against the development of visceral hypersensitivity following chronic colitis. Mechanistically, the protective effects of glial manipulation were mediated by disrupting the glial-mediated activation of macrophages through the macrophage colony-stimulating factor. Collectively, our data identified enteric glia as a critical link between gastrointestinal neural and immune systems that could be harnessed by therapies to ameliorate abdominal pain.
Graphical Abstract
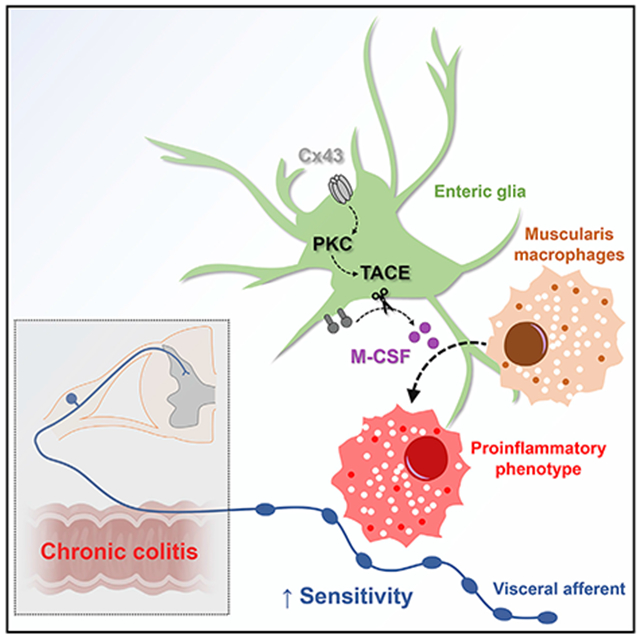
In Brief
Grubišić et al. report that enteric glia regulate macrophage activation and visceral sensitivity following intestinal inflammation through mechanisms that require glial connexin-43 (Cx43) and macrophage colony-stimulating factor (M-CSF) production. Proinflammatory signals induce glial Cx43-dependent M-CSF production through protein kinase C (PKC) and tumor necrosis factor (TNF)-alpha converting enzyme (TACE).
INTRODUCTION
Abdominal pain is the leading gastrointestinal (GI) symptom in the United States and is the dominant symptom of bowel disorders such as irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD) (Chang et al., 2018; Lee et al., 2017a; Mearin et al., 2016; Peery et al., 2015; Rodríguez-Fandiño et al., 2017). Nociceptor sensitivity is the most important factor that gates the transmission of noxious information from the intestine to the brain, and processes that alter nociceptor sensitivity in the periphery play a fundamental role in the generation of abdominal pain and the transition from acute to chronic pain (Brierley and Linden, 2014). Therefore, neuronal plasticity involving the sensitization of sensory nerve fibers in the intestine, known as visceral hypersensitivity, has emerged as a widely accepted mechanism underpinning abdominal pain (Simrén et al., 2018).
Non-neuronal cells such as immune and glial cells play active roles in the pathogenesis and resolution of chronic somatic and visceral pain (Ji et al., 2016). These cells contribute to neuroinflammation and data from human genomics and preclinical animal studies suggest that neuroinflammation is a key process that contributes to visceral hypersensitivity in the intestine (Videlock et al., 2018). However, the mechanisms that link neuroinflammation with immune responses and visceral hypersensitivity are unresolved. Sensory neurons densely innervate the enteric nervous system where they intermingle with enteric glia. Enteric glia are a unique type of peripheral neuroglia that modulate neuron activity in the intestine and bridge neuro-immune interactions (Chow and Gulbransen, 2017; Grubišić and Gulbransen, 2017a). Importantly, communication between nociceptors and enteric glia drives neuroinflammation in the mouse intestine (Delvalle et al., 2018), and activated glial cells contribute to visceral hypersensitivity in mouse models of IBS (Xu et al., 2018). These observations suggest that enteric glia could play a significant role in processes that modify visceral perception (Morales-Soto and Gulbransen, 2019).
Given that immune cells including macrophages, neutrophils, mast cells, and lymphocytes also modulate the activity of sensory neurons (Chavan et al., 2017), we hypothesized that interactions between enteric glia and immune cells contribute to nociceptor sensitization during inflammation. We addressed this question by ablating glial intercellular communication mediated by connexin-43 (Cx43) and studied the effects on visceral sensitivity and immune responses following acute and chronic intestinal inflammation. Our data show that visceral hypersensitivity after chronic inflammation is reduced in animals lacking enteric glial Cx43. This effect is not mediated by modifying the overt inflammatory response but, instead, involves an impaired ability of enteric glia to activate muscularis macrophages via the production of macrophage colony-stimulating factor (M-CSF). In vitro experiments with mouse and human enteric glia show that enteric glia produce M-CSF in response to pro-inflammatory stimuli and that this mechanism requires functional glial Cx43 hemichannels for downstream activation of protein kinase C (PKC) and tumor necrosis factor (TNF)-alpha converting enzyme (TACE), an enzyme that cleaves membrane-bound M-CSF. Together, these data highlight a mechanism whereby enteric glia influence visceral sensitivity through interactions with muscularis macrophages. Targeting the mechanisms involved could benefit the development of therapies to address visceral pain.
RESULTS
Deleting Connexin-43 in Enteric Glia Does Not Affect the Severity of Colitis
In prior work, we identified Cx43 as a major molecular mediator of intercellular communication used by enteric glia (Figure 1A; McClain et al., 2014) and found that deleting Cx43 in enteric glia reduces neuroinflammation in the context of colitis (Brown et al., 2016). Based on these data, we hypothesized that glial mediators released in a Cx43-dependent manner are important to recruit and/or activate immune cells that subsequently drive neuroplastic changes in nociceptors. To this end, we deleted glial Cx43 in Sox10CreERT2;Cx43f/f mice and assessed the effects on intestinal inflammation driven by acute and chronic dextran sodium sulfate (DSS, 2%) models of colitis (Figures 1B and 1C). DSS disrupts epithelial barrier function and drives inflammation through innate immune mechanisms (Kiesler et al., 2015). Despite the prominent role of glial Cx43 signaling in enteric reflexes that coordinate gut motility and secretomotor responses (Grubišić and Gulbransen, 2017a, 2017b; McClain et al., 2014), deleting glial Cx43 did not alter the overall severity of colitis as reflected by changes in body weight and macroscopic damage (p > 0.1635 and 0.2146, respectively, two-way ANOVA for genotype; Figures 1C and 1D). Likewise, pathological damage to the mucosa and submucosal layers was not altered in mutant mice (4.8 ± 0.5 damage score [control] versus 5.3 ± 0.4 damage score [Sox10CreERT2;Cx43f/f] and 7.2 ± 0.3 damage score [control] versus 7.7 ± 0.2 damage score [Sox10CreERT2;Cx43f/f] in acute and chronic DSS treatments, respectively; p = 0.1699, two-way ANOVA for genotype [Figures 1E and 1F]), and we did not observe overt damage to the muscularis and myenteric plexus (Figure 1E). Indeed, all groups had comparable cellular composition in the myenteric plexus (Figure 1G), and the density of myenteric neurons and glial cells were similar in all experimental groups (2,152 ± 204 neurons and 2,401 ± 130 glia per mm2 for acute treatment; Figure 1H). Therefore, the potential inflammatory driving force for neuroplastic changes in visceral sensory neurons is similar in Sox10CreERT2;Cx43f/f mice and control littermates. This was important because it allowed us to study changes in visceral hypersensitivity driven by cell signaling events rather than a gross modification of local or systemic inflammation.
Figure 1. Connexin-43 Deletion in Enteric Glia Does Not Affect the Overall Severity of Acute or Chronic Colitis.
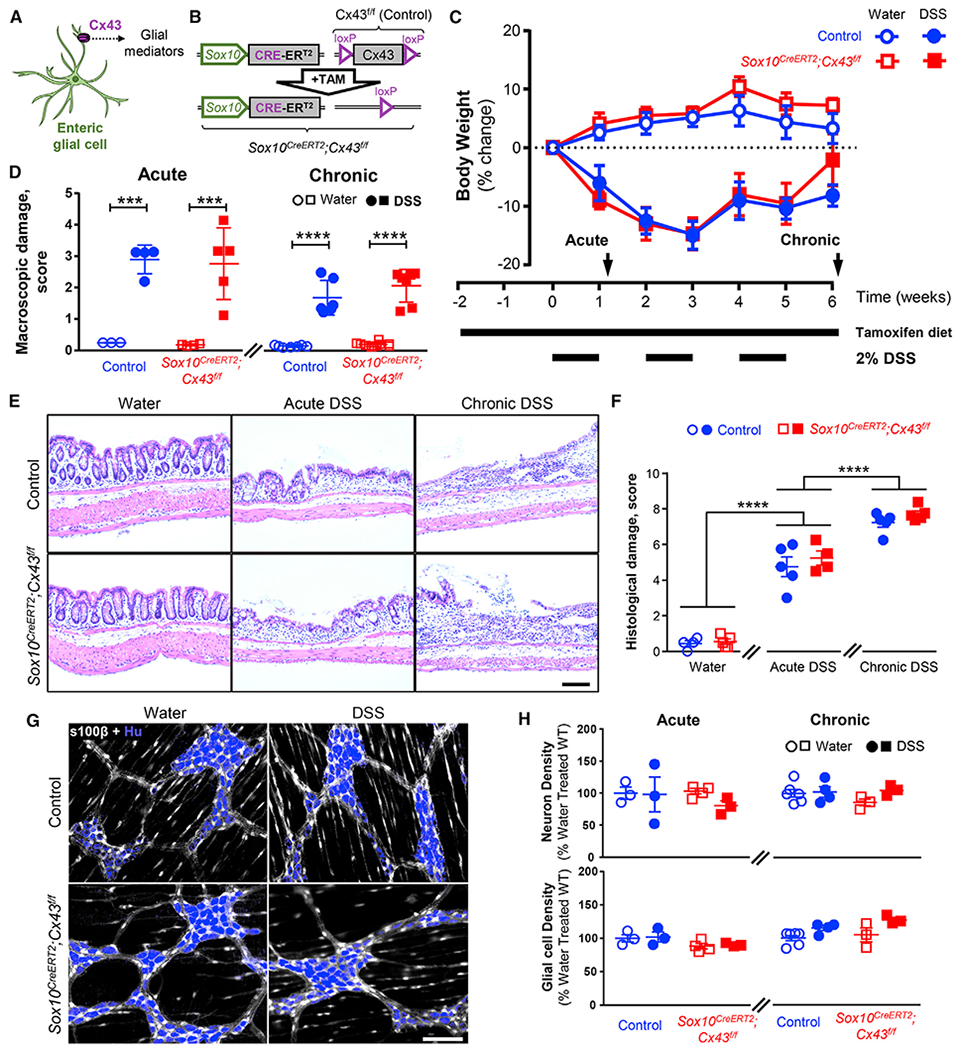
(A) Schematic depicting that Cx43 hemichannels regulate the release of glial mediators.
(B) Schematic depicting the tamoxifen-induced deletion of the Cx43 encoding gene in Sox10CreERT2;Cx43f/f mice.
(C) Quantification of body weight as a measure of colitis severity in control and Sox10CreERT2;Cx43f/f mice. Tamoxifen citrate was administered via diet beginning 2 weeks before the induction of inflammation and throughout the experiment (long horizontal line). Colonic inflammation was induced by 2% dextran sodium sulfate (DSS) in drinking water for 1 week (acute) or intermittently (1 week on/1 off) for 3 rounds (chronic) (short horizontal lines). Experiments were performed at two time points, 3 days after the first course of DSS and a week after the final DSS treatment, to assess the effects of acute and chronic inflammation (arrows). Data are shown as mean ± SEM, n = 5–8 mice.
(D) Macroscopic tissue damage scores at the acute (left) and chronic (right) time points were comparable between the Sox10CreERT2;Cx43f/f and their control littermates. ***p < 0.001; ****p < 0.0001; 2-way ANOVA. Data are shown as mean ± SEM, n = 5–8 mice.
(E) Representative images of hematoxylin-and-eosin-stained colon cross-sections of control (top) and Sox10CreERT2;Cx43f/f mice (bottom) treated with water (left), acute DSS (middle), and chronic DSS (right). Scale bar, 100 μm.
(F) Blinded evaluation of histological staining. See Table S2 for details about histological disease activity scoring. ****p < 0.0001; 2-way ANOVA. Data are shown as mean ± SEM, n = 3–10 mice.
(G and H) The density of myenteric neurons and glial cells is comparable between Sox10CreERT2;Cx43f/f and control mice treated with water or mild acute and chronic DSS models. (G) Representative images of immunolabeling for myenteric neurons (HuC/D, blue) and glia (s100β, gray). Scale bar, 100 μm. (H) Neuronal (top) and glial (bottom) density (cells per ganglionic area) in acute (left) or chronic (right) DSS mice. Hu labeling was used to assess neuron numbers, and s100β was used to identify glial cells and determine the ganglionic area. Data are shown as mean ± SEM, n = 3–6 mice.
Glial Cx43 Deletion Protects against the Development of Visceral Hypersensitivity following Chronic DSS Colitis
We assessed visceral sensitivity by recording visceromotor responses (VMRs) to colorectal distensions (CRDs) (Figures 2A and 2B; Larauche et al., 2010) in healthy and DSS-treated mice (Boué et al., 2014; Lapointe et al., 2015; Reiss et al., 2017; Scanzi et al., 2016). Acute inflammation caused hyperalgesia and increased the magnitude of VMRs to noxious pressures in all animals (Figures 2C and 2D). Neither baseline visceral sensitivity nor the development of acute hyperalgesia required glial Cx43 signaling because neither was affected in Sox10CreERT2;Cx43f/f mice (p = 0.1127 and 0.6614, two-way ANOVA for genotype, Figures 2C and 2D). In contrast, visceral pain following chronic DSS required glial Cx43 signaling, and heightened VMRs were absent in mice lacking glial Cx43 (p = 0.025, two-way ANOVA for genotype, Figures 2C and 2D). These changes are not due to differences in inflammation-induced fibrosis because compliance measurements were comparable (p > 0.4447, two-way ANOVA for genotype) in all groups (Figure 2E). Thus, glial intercellular communication through Cx43 plays an important role in the transition to chronic visceral hypersensitivity following inflammation. CRD primarily tests the sensitivity of mechanosensitive spinal afferents that innervate the seromuscular layer of the mouse colon (Brierley et al., 2018; Feng et al., 2012). Given that the mild colitis driven by our relatively low-dose DSS model was not sufficient to induce significant neurodegeneration in the myenteric plexus (Figures 1G and 1H), we speculated that the observed effects of glial Cx43 signaling could be mediated through neuro-immune signaling rather than direct effects of glia on neuroplasticity.
Figure 2. Cx43 Deletion in Enteric Glia Protects against the Development of Visceral Hypersensitivity following Chronic DSS Colitis.
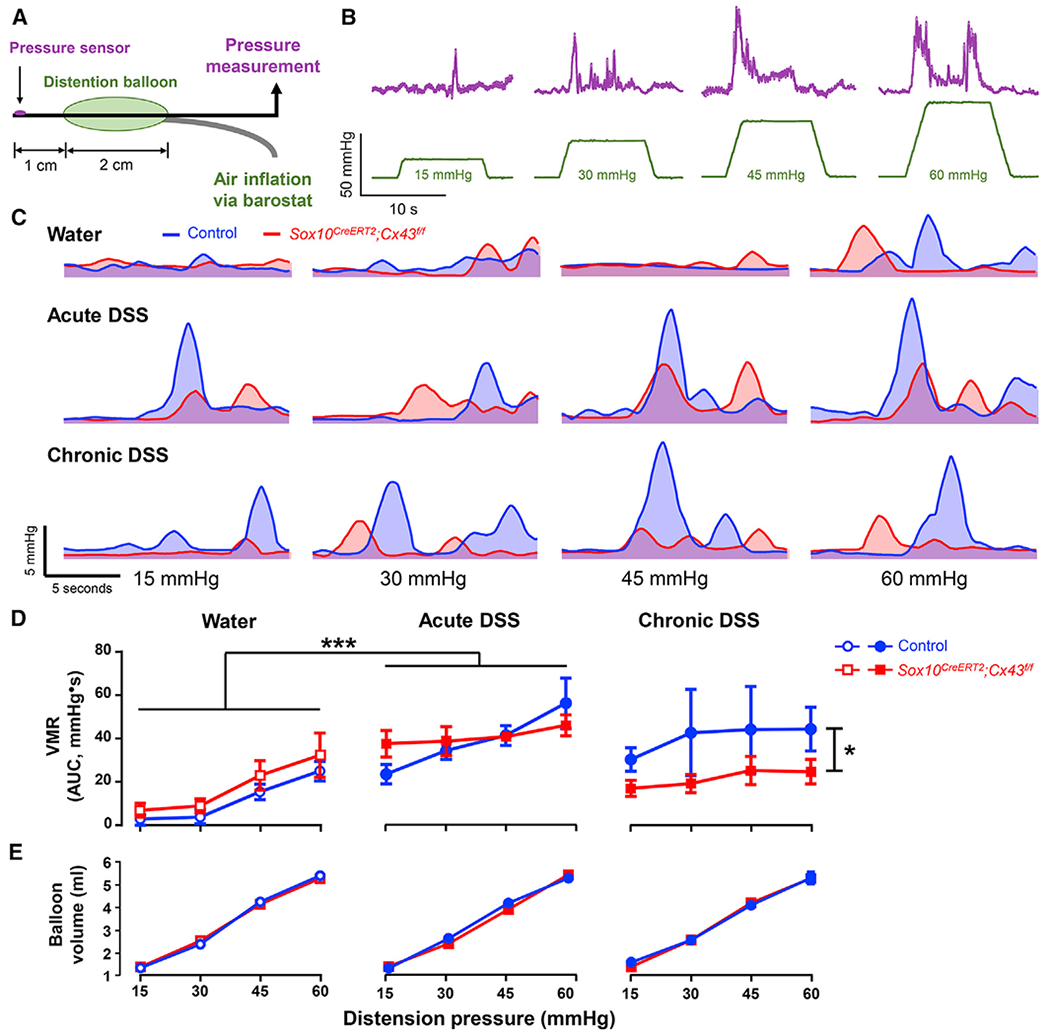
(A) Model of the pressure probe and distension balloon used to record visceromotor responses (VMRs) to colorectal distensions in mice.
(B) Representative pressure recordings (top, magenta) in response to distention of the colorectum (bottom, green).
(C) Smoothed representative traces of VMRs in control (blue) and Sox10CreERT2;Cx43f/f mice (red) treated with water (top row), acute DSS (middle row), or chronic DSS (bottom row). The area under the curve (AUC) was used to quantify the VMRs in (D).
(D and E) VMR (D) and compliance (E) measurements in Sox10CreERT2;Cx43f/f mice (red) and their control littermates (blue) after treatments with water (left), acute DSS (middle), and chronic DSS (left). *p = 0.025; ****p < 0.0001; 2-way ANOVA. Data are shown as mean ± SEM, n = 5–8 mice. (D) Water-treated Sox10CreERT2;Cx43f/f animals and their control littermates had comparable basal VMRs and exhibited similar increases following acute DSS. Mice lacking glial Cx43 do not exhibit heightened VMRs following chronic DSS colitis. (E) Compliance measurements were equal in all groups.
Glial Cx43 Regulates M-CSF Expression during Intestinal Inflammation
We began to assess the potential effects of glial Cx43 signaling on immune responses by assessing serum levels of inflammatory mediators. Serum granulocyte-colony stimulating factor (G-CSF) was significantly increased following acute and chronic inflammation but was not modulated by deleting glial Cx43 (Figure S1). Overall, deleting glial Cx43 had little effect on systemic cytokines/chemokines. Therefore, we focused our efforts on assessing local levels of cytokines and chemokines in the colon (Figure 3A). Several of the 31 cytokines and chemokines assessed were significantly modified by acute and/or chronic DSS colitis. However, only M-CSF was regulated by both inflammation and glial Cx43 signaling as indicated by a failure to increase colonic M-CSF levels during DSS colitis in mice lacking glial Cx43 (Figure 3B). Deleting glial Cx43 also diminished macrophage inflammatory protein (MIP)-1a, eotaxin, and interleukin-2 (IL-2) in both healthy and DSS-treated animals, and several other inflammatory mediators were specifically affected by DSS treatment (increased levels of G-CSF, IL-17, IP-10, MIG, and RANTES; decreased levels of IL-2) or by deleting glial Cx43 (IL-7, IL-9). Based on these data, M-CSF emerged as the most likely candidate underlying the effects of glial Cx43 signaling on visceral hypersensitivity.
Figure 3. Glial Cx43 Regulates M-CSF Expression during Intestinal Inflammation.
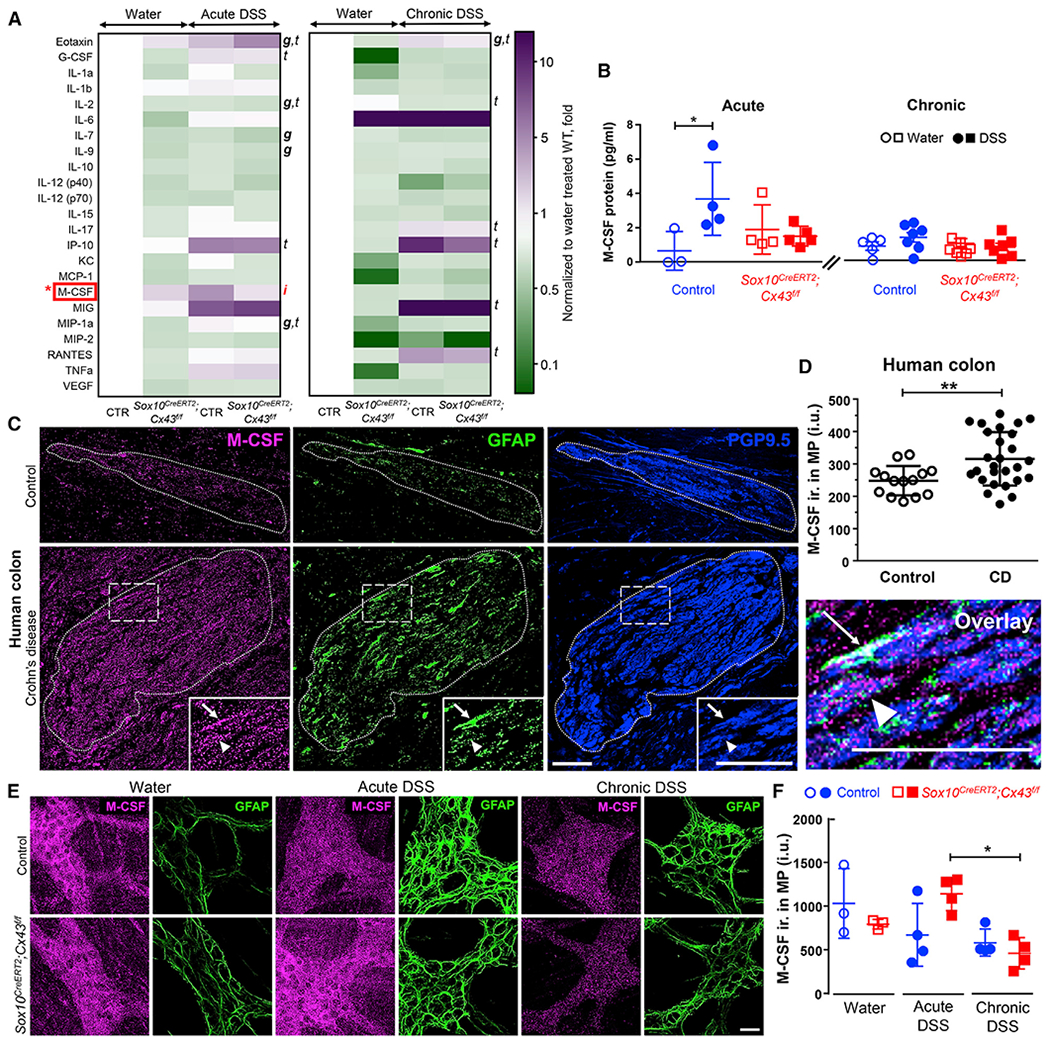
(A and B) Quantification of local cytokine and chemokine production within the mouse colon by a 31-Plex Mouse Cytokine/Chemokine Array. (A) Heatmap showing average relative protein expression after acute (left) or chronic (right) inflammation. (B) Summary data from multiplex arrays showing quantification of M-CSF. Glial Cx43 signaling regulates M-CSF expression during acute colitis. g, genotype; t, treatment; i, interaction; 2-way ANOVA (p < 0.05). *p < 0.05; 2-way ANOVA. Data are shown as mean ± SD, n = 3–7 mice. Granulocyte-colony stimulating factor (G-CSF) was also significantly increased in the sera of DSS-treated animals but was not regulated by glial Cx43 (Figure S1). Transcriptomics data show that mouse enteric glia express M-CSF (Figure S2).
(C) Representative images showing immunolabeling for M-CSF in human myenteric ganglia in colon samples from individuals with Crohn disease and control colon samples from patients that underwent resections for bowel trauma, volvulus, or intestinal bleeding. Dotted lines demarcate the borders of myenteric ganglia, which was defined by PGP9.5 labeling (neurons, blue). Arrows and arrowheads point to glia (GFAP immunolabeling, green) and neurons, respectively. Note that GFAP labeling is increased in Crohn disease samples indicating reactive gliosis. Scale bar, 50 μm.
(D) Quantification of M-CSF labeling in myenteric ganglia from controls without abdominal pain and individuals with Crohn (CD) causing low to high levels of abdominal pain. **p = 0.0016; unpaired Student’s t test with Welch’s correction. Data are shown as mean ± SD, n = 14 and 27 ganglia from 4 and 5 subjects (2 males and 2 or 3 females in each group).
(E and F) Immunolabeling for M-CSF in Sox10CreERT2;Cx43f/f mice and control littermates treated with water, acute DSS, or chronic DSS. Representative images(E) and quantification (F) of M-CSF immunoreactivity (ir., magenta) within the MP, expressed in intensity units (IUs). GFAP (glia, green) was used to identify myentericganglia. Scale bar, 25 μm. *p = 0.0144; 2-way ANOVA, Tukey’s post hoc test. Data are shown as mean ± SD, n = 3–4 mice. Controls for the specificity of the M-CSF antibody were performed in both mouse and human tissues (Figure S3).
M-CSF is a chemokine that drives macrophage recruitment and/or differentiation. Thus, we hypothesized that enteric glia contribute to macrophage activation by producing M-CSF. Glial transcriptome data support this hypothesis by showing that enteric glia express M-CSF and other inflammatory mediators important for macrophage recruitment and activation (Figure S2; Delvalle et al., 2018). To understand how M-CSF expression is regulated during inflammation, we performed immunolabeling experiments with specimens from healthy and inflamed mice, and humans with or without Crohn disease (Figure 3C). M-CSF labeling in the myenteric plexus was localized to both neurons and glial cells. M-CSF immunoreactivity in myenteric ganglia from individuals with Crohn disease (316 ± 16 intensity units [IUs]) was significantly higher (p = 0.0016, unpaired Student’s t test with Welch’s correction) in comparison to healthy controls (248 ± 12 IUs, Figure 3D). In agreement with these changes in human IBD samples, M-CSF immunoreactivity in mouse myenteric ganglia increased during colitis (Figure 3E). Importantly, mice lacking glial Cx43 exhibited lower M-CSF expression (p = 0.0144, two-way ANOVA with Tukey post hoc test) during the transition from acute to chronic DSS colitis (1,138 ± 95 IUs [acute] versus 461 ± 89 IUs [chronic], Figure 3F). Together, these results support the concept that glial Cx43 signaling regulates M-CSF production in the enteric nervous system during chronic intestinal inflammation.
Glial Connexin-43 Signaling Regulates Muscularis Macrophage Activation in the Mouse Colon
Given that glial Cx43 regulates M-CSF, and M-CSF regulates macrophage migration and activity, we tested the hypothesis that glial Cx43 signaling regulates muscularis macrophage phenotype. Immunolabeling data show that enteric glia form the cellular interface between the enteric nervous system and muscularis macrophages (Figure 4A; Videos S1 and S2). This tight anatomical relationship suggests a high potential for cellular crosstalk. To understand how intercellular glial-macrophage communication might affect the activation of glia and macrophages during inflammation, we assessed the expression of glial fibrillary acidic protein (GFAP) and CD68, two markers that reflect reactive gliosis (von Boyen et al., 2004) and pro-inflammatory macrophages (Cipriani et al., 2016), respectively (Figure 4B). GFAP expression was increased to a similar extent in Sox10CreERT2;Cx43f/f mice and their control littermates during acute DSS colitis. Likewise, GFAP expression normalized during chronic inflammation regardless of genotype. CD68 expression surrounding the myenteric plexus increased in control mice during the chronic phase of inflammation (Figure 4C). However, mice lacking glial Cx43 failed to show an increase in CD68 during both acute and chronic phases of colitis (Figure 4C). These findings indicate that glial Cx43 is not required for changes in GFAP associated with reactive gliosis but is required for the activation and/or recruitment of proinflammatory macrophages. Comparable levels of markers of tissue-resident macrophages (F4/80+; p = 0.3861, unpaired t test with Welch’s correction) and newly recruited macrophages (CCR2+; p = 0.1905, Mann Whitney U test) were present in Sox10CreERT2;Cx43f/f and control mice (Figures 4D and 4E), suggesting that changes in CD68 reflect the activation of resident macrophages rather than an increase in macrophage numbers by recruitment. CD68 labels multiple populations of phagocytic cells such as dendritic cells, neutrophils, basophils, mast cells, activated T cells, and a proportion of mature B cells (Naeim, 2008). Therefore, we assessed the colocalization of F4/80 with CCR2 and CD68 (Figure S4) to differentiate macrophage CD68 expression from expression in other potential cell types. We observed less than 5% of overlap between F4/80 and CCR2 in all mice supporting the ability of these labels to identify distinct subsets of macrophages. Half of all CD68 labeling (52% ± 5%) colocalized with F4/80, a marker of tissue-resident macrophages. Deleting glial Cx43 reduced colocalization between CD68 and F4/80 to 29% ± 5% (p = 0.0124, unpaired t test with Welch’s correction) but did not significantly affect CD68 labeling that was not colocalized to F4/80 (p = 0.1549, unpaired t test with Welch’s correction). Together, these results support the conclusion that enteric glia regulate muscularis macrophage activation during colitis through Cx43-dependent signaling.
Figure 4. Enteric Glia Regulate Muscularis Macrophage Activation in the Mouse Colon through Cx43-Dependent Signaling.
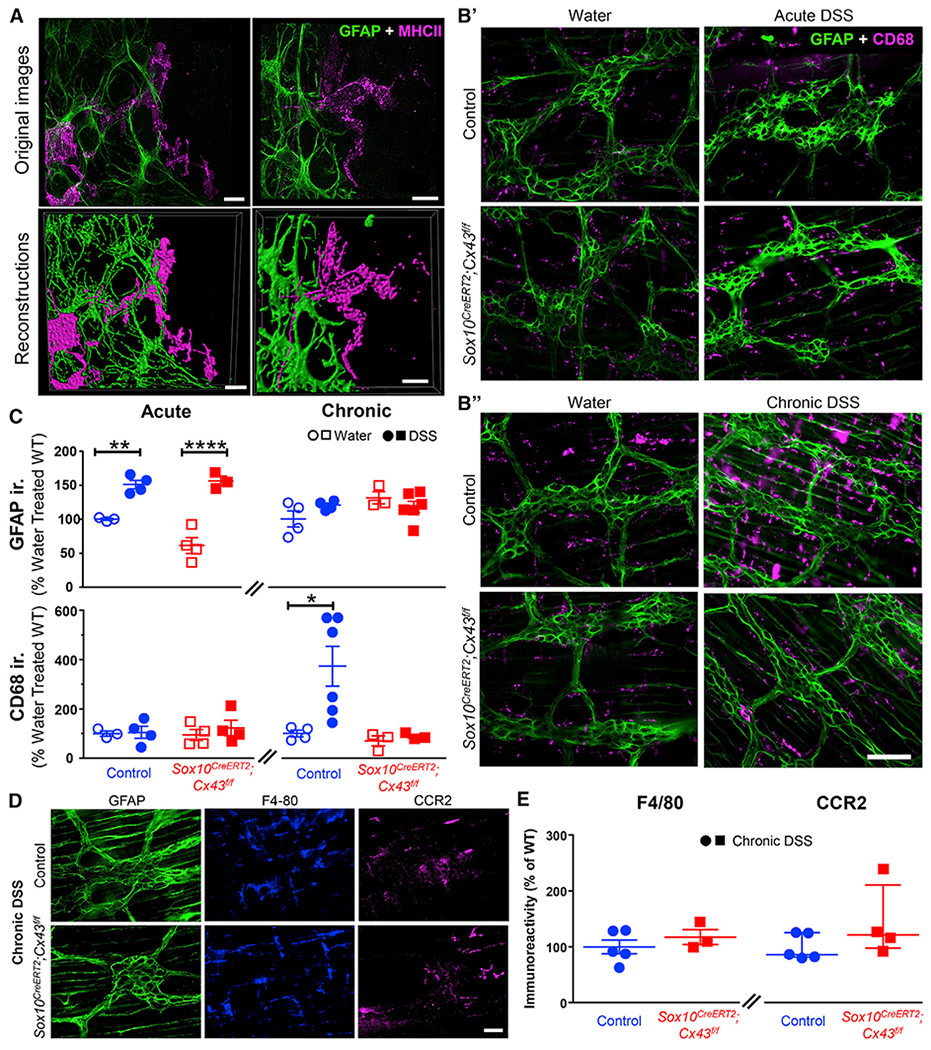
(A) Representative images of immunolabeling for enteric glia (labeled with glial fibrillary acidic protein [GFAP, green]) and muscularis macrophages (labeled with major histocompatibility complex II [MHC-II, magenta]) in the myenteric plexus of the mouse colon. Original images and reconstructions are in the top and bottom rows, respectively. Scale bars, 10 μm.
(B) Representative images showing glial (GFAP, green) and macrophage (CD68, magenta) activation in the myenteric plexus of the mouse colon following acute (B′) or chronic (B″) DSS colitis. Scale bars, 100 μm.
(C) Quantification of GFAP (top) and CD68 (bottom) immunolabeling. Immunoreactivity is normalized to water-treated control littermates. *p < 0.05; 2-way ANOVA. Data are shown as mean ± SEM, n = 3–6 mice.
(D and E) Effects of deleting glial Cx43 on the abundance of resident and newly recruited macrophages. (D) Representative images showing labeling for glia (GFAP), tissue-resident macrophages (F4/80), and newly recruited macrophages (CCR2) in the myenteric plexus of control (top) and Sox10CreERT2;Cx43f/f mice (bottom) following chronic DSS colitis. Scale bar, 50 μm. (E) Quantification of immunolabeling for F4/80 (left) and CCR2 (right) in Sox10CreERT2;Cx43f/f and controls. Welch’s t test (p = 0.386) and Mann–Whitney U test (p = 0.191). Data are shown as mean ± SEM (left) and median ± interquartile range (right), n = 3–5 mice. F4/80 and CCR2 label distinct populations of macrophages, while F4/80 and CD68 co-label muscularis macrophages (Figure S4).
Enteric Glia Are the Primary Source of M-CSF in the Myenteric Plexus and Regulate Muscularis Macrophage Activation through Cx43-Dependent M-CSF Release
Our data to this point show that glial Cx43 signaling regulates M-CSF production and muscularis macrophage activation during colitis. To link these mechanisms, we conducted in vitro experiments where we assessed the relative contribution of glia to M-CSF production in the myenteric plexus, the necessity of M-CSF in muscularis macrophage activation, and the dependence on glial Cx43. Enteric neurons (Muller et al., 2014) and glia have the potential to produce M-CSF, but the relative contributions of the two cell types are not known. To address this issue, we began by using flow cytometry to assess the relative proportions of neurons, glia, and nonneuronal cells that express M-CSF at the level of the myenteric plexus. Myenteric plexus whole mounts were isolated from the colons of healthy Sox10CreERT2;Cx43f/f and control mice, and cell suspensions were prepared following stimulation with the proinflammatory cytokine IL-1β (Figures 5A–5E). M-CSF labeling was performed on non-permeabilized cells to assess the expression of cell-surface, membrane-bound M-CSF. Only 10% ± 1% of the cells expressing the membrane-bound form of M-CSF were identified as neurons based on colabeling with the pan-neuronal marker Hu (Figure 5D). In contrast, 35% ± 3% were positively identified as glia based on colabeling with the glial marker GFAP (Figure 5D). Deleting glial Cx43 or treating cells with IL-1β did not affect the proportions of cells expressing M-CSF (9% ± 1% to 11% ± 1% and 36% ± 1% to 38% ± 2%, respectively). Hence, enteric glia are the major source of biologically active M-CSF in the mouse colon myenteric plexus while neurons play a comparatively minor role under our experimental conditions.
Figure 5. Enteric Glia Are the Primary Source of M-CSF in the Myenteric Plexus and Regulate Muscularis Macrophage Activation through Cx43-Dependent M-CSF Release.
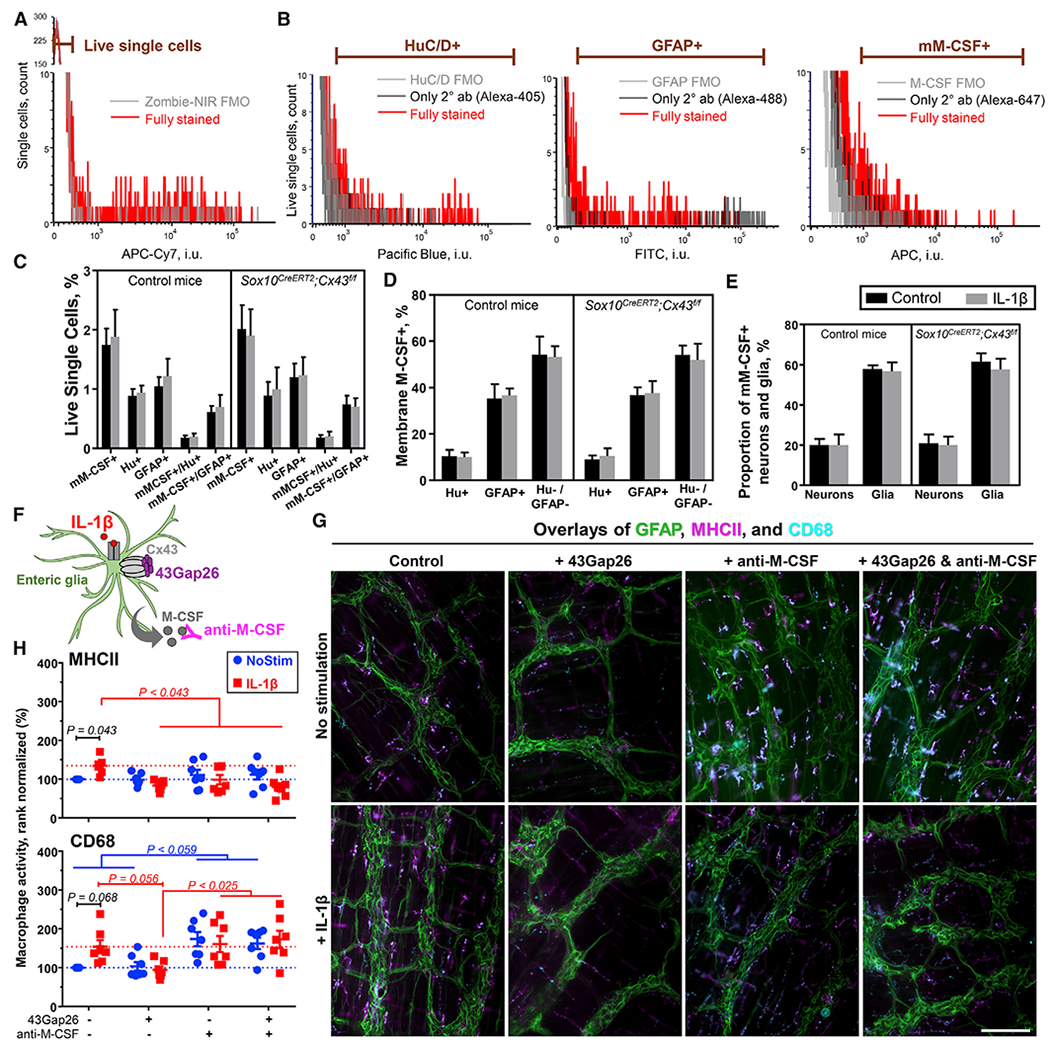
(A–D) Flow cytometry on cells dissociated from the muscular layer of colons harvested from healthy Sox10CreERT2;Cx43f/f mice and their respective controls. Half of each preparation was stimulated overnight with IL-1β (10 ng/mL). (A) The gating strategy for live cells was set using fluorescence minus one (FMO) control. This gate was applied to single-cell events (Figure S5A). (B) Gating strategies for neuronal marker HuC/D (Hu+), glial marker GFAP (GFAP+), and membrane M-CSF (mM-CSF+) were adjusted according to their respective FMO and secondary antibody (2° ab)-only controls. Figures S5B–S5D has dot plots of a fully stained sample and the controls. (C) Proportions of single live cells that are mM-CSF+, Hu+, or GFAP+ or co-express mM-CSF with HuC/D or GFAP. (D) Proportions of mM-CSF+ cells that co-express HuC/D (≈10%), GFAP (≈40%) or no neuron nor glial markers (≈50%).
(E) Quantification of the proportions of enteric neurons (Hu+) and enteric glia (GFAP+) that express mM-CSF (20 and 60%, respectively). Deleting glial Cx43 or treating cells with IL-1β did not affect the proportions of cells expressing M-CSF. Data are shown as mean ± SD, n = 3 control and 5 Sox10CreERT2;Cx43f/f mice.
(F–H) Effects of IL-1β (10 ng/mL), 43Gap26 (100 μM), and anti-M-CSF blocking antibodies (5 μg/mL) on macrophage reactivity in isolated samples of myenteric plexus. (F) Schematic showing drugs site of action: IL-1β binds to the IL-1 receptor, 43Gap26 blocks Cx43 hemichannels, and anti-M-CSF antibody neutralizes M-CSF. (G) Representative images of immunolabeling for glial cells (GFAP, green) and markers of macrophage activation (MHCII, magenta; CD68, cyan) in samples of colon myenteric plexus that were stimulated in vitro with IL-1β (+ IL-1β) in the presence or absence (control) of 43Gap26 (+ 43Gap26), rat anti-M-CSF blocking antibodies (+ anti-M-CSF), or both (+ 43Gap26 & anti-M-CSF). Scale bar, 100 μm. (H) Quantification of MHCII (top) and CD68 (bottom) labeling, normalized to the untreated controls. Dotted lines show average responses from unstimulated (blue) and IL-1β-stimulated controls (red). We analyzed the data with 2-way ANOVA. Data are shown as mean ± SEM, n = 7 mice; each data point is an average of 4 fields of view (FOV, 593 × 444 μm).
Given that glia are the predominant source of M-CSF in the myenteric plexus (Figure 5D) and glial Cx43 signaling is required for macrophage activation (Figure 4) and M-CSF production (Figure 3) during colitis, we tested whether glial Cx43-dependent M-CSF production is required for muscularis macrophage activation in whole-mount preparations of myenteric plexus. Myenteric glia were stimulated with the proinflammatory cytokine IL-1β to drive proinflammatory responses and subsets of preparations were co-incubated with 43Gap26, a Cx43 mimetic peptide that blocks Cx43 hemichannels, anti-M-CSF blocking antibodies, or both 43Gap26 and anti-M-CSF (Figure 5F). Macrophage activation was monitored by assessing MHCII and CD68 expression, which are markers of antigen presentation and phagocytosis, respectively (Figure 5G). 43Gap26 and anti-M-CSF antibodies did not affect basal MHCII expression (p = 0.8023, two-way ANOVA with Tukey post hoc test; Figure 5H, top). IL-1β stimulation increased MHCII expression by 35% ± 8% (p = 0.0434, two-way ANOVA with Sidak post hoc test), and the increase in MHCII required both Cx43 and M-CSF as it was fully blocked by 43Gap26 and anti-M-CSF antibodies, with the combination having no additional effect (p = 0.0428, two-way ANOVA with Tukey post hoc test) (Figure 5H, top). CD68 expression was more variable and many comparisons did not reach statistical significance (Figure 5H, bottom). However, the effects of IL-1β stimulation and 43Gap26 tended to affect CD68 expression similarly as MHCII expression (p < 0.0678, two-way ANOVA). Anti-M-CSF antibodies used alone or in combination with 43Gap26 tended to increase baseline CD68 expression (p < 0.0594, two-way ANOVA) to a similar extent as IL-1β (54%–74%), implying that the antibodies induced a Cx43-independent increase in phagocytic activity. Taken together, our results show that Cx43 hemichannels and M-CSF are required for muscularis macrophage activation, and the lack of additive effects suggests that both Cx43 hemichannels and M-CSF are parts of the same glial signaling pathway.
Glial M-CSF Production Is Regulated by Cx43, PKC, and TACE
To provide additional support for the concept that enteric glia produce M-CSF through Cx43-dependent mechanisms, we conducted in vitro experiments with primary cultures of mouse and human enteric glia where we measured glial M-CSF production and enzymatic activity in the M-CSF molecular production pathway. Cx43 hemichannels have a pore size of approximately 2 nm and can pass molecules that are less than 1 kDa (Harris, 2001). M-CSF is significantly larger, ranging between 44 and 86 kDa (Cosman et al., 1988), so it is unlikely that M-CSF is directly released through Cx43 hemichannels. Basal M-CSF release is regulated at the post-transcriptional level by processes at the cell surface where the protein is cleaved by TACE (Horiuchi et al., 2007). Thus, it is more likely that Cx43 hemichannels indirectly regulate the proteolytic cleavage of M-CSF because glial Cx43 hemichannels mediate intracellular calcium levels in enteric glia (McClain et al., 2014) and PKC mediates TACE activity (Horiuchi and Toyama, 2008). To directly test this hypothesis, we isolated enteric glia from mouse colon myenteric plexus and assessed PKC and TACE activation driven by IL-1β in the presence or absence of 43Gap26 (Figures 6A–6D). IL-1β stimulation increased PKC activity in cell lysates to 255% ± 99% (mean ± SD) of untreated controls and 43Gap26 abolished this effect (p = 0.0008 and p = 0.6256, respectively, ANOVA followed by Dunnett’s multiple comparisons test; Figure 6C). TACE activity was assessed in live-cell suspensions to specifically assess the activity of TACE at the cell membrane where it mediates cleavage the of the membrane-bound M-CSF. Membrane-associated TACE activity was increased to 175% ± 103% (mean ± SD) of untreated controls when cells were treated with IL-1β alone and this effect was blocked by co-incubated with 43Gap26 (p = 0.0342 and p = 0.5649, respectively, ANOVA followed by Dunnett’s multiple comparisons test; Figure 6D). Together, these results show that Cx43 hemichannels are required for IL-1β to activate key elements of M-CSF production such as PKC and TACE in enteric glia.
Figure 6. Glial M-CSF Production Is Regulated by Cx43, PKC, and TACE.
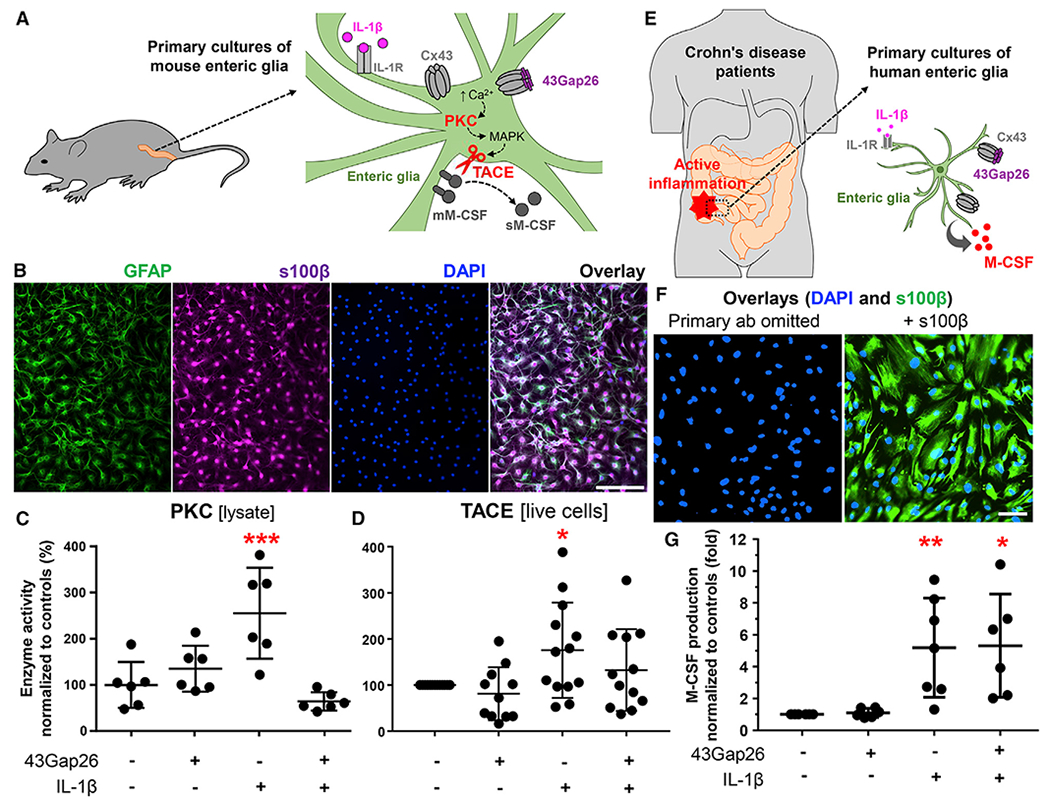
Data from in vitro experiments with a primary mouse (A–D) and human (E–G) enteric glia.
(A) Primary cultures of mouse enteric glia were derived from the colon myenteric plexus. Schematic showing proposed mechanisms underlying IL-1β-induced M-CSF release. IL-1R, IL-1 receptor; Cx43, Cx43 hemichannel; 43Gap26, Cx43 mimetic peptide that binds to extracellular loops of Cx43 and blocks Cx43 hemichannels; ↑Ca2+, increase in cytosolic calcium; PKC, protein kinase C; MAPK, a mitogen-activated protein kinase; TACE, tumor necrosis factor α-converting enzyme; mM-CSF and sM-CSF, membrane-bound and soluble M-CSF.
(B) Representative images of mouse enteric glia cultures labeled with markers of enteric glia (GFAP, green, and s100β, magenta) and counterstained with the nuclear marker DAPI (blue). Scale bar, 100 μm.
(C and D) Quantification of PKC and TACE activity in primary cultures of mouse enteric glia stimulated overnight with IL-1β (+ IL-1β, 1 ng/mL) in the presence or absence of 43Gap26 ±43Gap26, 100 μM). (C) PKC activity in cell lysates was normalized to total protein concentration in the cell lysate and to mean of untreated controls. (D) TACE activity in live-cell suspensions was normalized to cell density and to untreated controls that originated from the same colon. *p = 0.0342; ***p = 0.0008; ANOVA followed by Dunnett’s multiple comparisons test. Data are shown as mean ± SD, n = 6 and 11–13 mice, respectively.
(E) Human enteric glial cells were cultured from segments of the intestine harvested from individuals undergoing resections for Crohn disease. Cultured gliawere incubated with IL-1β to stimulate the production of proinflammatory cytokines, and a subset of cultures was co-incubated with 43Gap26.
(F) Representative images of human enteric glia derived from the distal colon. Images show labeling for s100β (glia, green) and DAPI (nuclei, blue) in samples where the primary anti-s100β antibody was omitted (left) or fully stained (right). Scale bar, 100 μm.
(G) Quantification of M-CSF in supernatants from human enteric glial cultures after IL-1β stimulation (+ IL-1β, 1 ng/mL) and co-incubation with 43Gap26 (+ 43Gap26, 100 μM). Raw M-CSF concentrations were normalized to cell density and to untreated controls that originated from the same culture. *p = 0.0103; **p = 0.0097; ANOVA followed by Dunnett’s multiple comparisons test. Data are shown as mean ± SD, n = 3–4 patients (duplicate cultures derived from involved or noninvolved regions). Besides, proinflammatory signals change the expression of connexin genes in mouse and human enteric glia (Figure S6).
To directly test whether enteric glia release M-CSF in a Cx43-dependent manner in humans, we cultured human enteric glial cells purified from resected samples from individuals with Crohn disease (Figures 6E–6G). As in our mouse samples, we used IL-1β to stimulate proinflammatory reactions in human glia (Figure 6E). IL-1β stimulation induced a 5-fold increase in glial M-CSF production (p = 0.0097, ANOVA followed by Dunnett’s multiple comparisons test; Figure 6G). Blocking Cx43 channels with 43Gap26 did not have a significant effect on IL-1β-stimulated M-CSF release (Figure 6G). These data show that human enteric glia produce M-CSF in response to pro-inflammatory stimuli. However, glial M-CSF production becomes insensitive to Cx43 hemichannel blockade in individuals with chronic intestinal disease. To understand why this occurs, we assessed how intestinal inflammation affects glia connexin expression in mice and humans (Figure S6). Our data show that proinflammatory stimuli significantly alter glial connexin gene profiles. Importantly, changes in Cx26 and Cx30 could reduce the efficacy of 43Gap26 following inflammation.
DISCUSSION
Bidirectional communication between astrocytes and microglia is a well-defined mechanism in the central nervous system that determines the functional fate of both cell types (Jha et al., 2019). For example, astrocyte-derived M-CSF regulates microglial maturation and differentiation in the developing human brain (Liu et al., 1994), and inhibiting astrocyte-to-microglia signaling mediated by M-CSF prevents central sensitization and mechanical hypersensitivity in chronic post-ischemic pain models (Tang et al., 2018). Our data suggest that analogous mechanisms are functional in the intestine, that these mechanisms are important for dictating glial and macrophage phenotype, and that they influence the function of neurons innervating the intestine. Muscularis macrophages and enteric glia are often compared to microglia and astrocytes based on their similar functions and gene expression profiles. Muscularis macrophages express microglia-specific genes and transcription factors that are not expressed by other subsets of tissue macrophages (Verheijden et al., 2015) and enteric glia express traditional astrocytic markers and exhibit similar functional relationships with neurons (Gulbransen and Sharkey, 2012). However, intestinal macrophages and glia are unique cell types with distinct developmental origins and transcriptional profiles (Grubišić and Gulbransen, 2017a; Rao et al., 2015; Verheijden et al., 2015).
Our data show that enteric glia regulate muscularis macrophage activation through Cx43-dependent M-CSF release. Proinflammatory signals stimulate glial M-CSF release through Cx43 hemichannel signaling, cytosolic PKC activity, and cell membrane TACE, an enzyme that cleaves M-CSF from the cell surface (Figure 7). Murine astrocytes also produce M-CSF upon stimulation with proinflammatory stimuli such as IL-1β, TNF-alpha, and lipopolysaccharides that regulate the expression of the M-CSF gene at the posttranscriptional level (Frei et al., 1992). In enteric glia, Cx43 hemichannels likely couple to PKC through intracellular Ca2+ signaling since Cx43 hemichannels regulate glial Ca2+ responses (McClain et al., 2014) and enteric glia express PKC isoforms such as PKCα and PKCβI that require Ca2+ for activation (Furness et al., 2006; Poole et al., 2003). The observation that 43Gap26 and anti-M-CSF-neutralizing antibodies are equally as effective, but not additive, in blocking the effects of IL-1β on macrophage activation supports the conclusion that glial Cx43-dependent M-CSF release regulates macrophage activation. Surprisingly, anti-M-CSF-neutralizing antibodies blocked the increase in antigen presentation driven by IL-1β but stimulated phagocytosis in a Cx43-independent manner. Cells attempting to clear immune complexes (Mantovani et al., 1972) formed by soluble M-CSF and the blocking antibody may explain this observation, but identifying the exact mechanism will require future work. Cx43 hemichannels interact with diverse cellular components such as cytoskeletal proteins, scaffolding proteins, protein kinases, and phosphatases involved in Cx43 channel-independent functions (Olk et al., 2009) that could also play a role in posttranslational processing and intracellular M-CSF trafficking. Soluble M-CSF is generally released by proteolytic cleavage from the cell surface and other forms of M-CSF, such as membrane and extracellular matrix-bound, have more complex posttranslational processing (Pixley and Stanley, 2004). These multiple forms of M-CSF have diverse biological effects on macrophage phenotypes in addition to their distinct mechanisms of posttranslational processing, intracellular trafficking, and release (Liao et al., 2016).
Figure 7. Proposed Mechanisms Whereby Enteric Glia Regulate Visceral Pain through M-CSF Signaling with Macrophages.
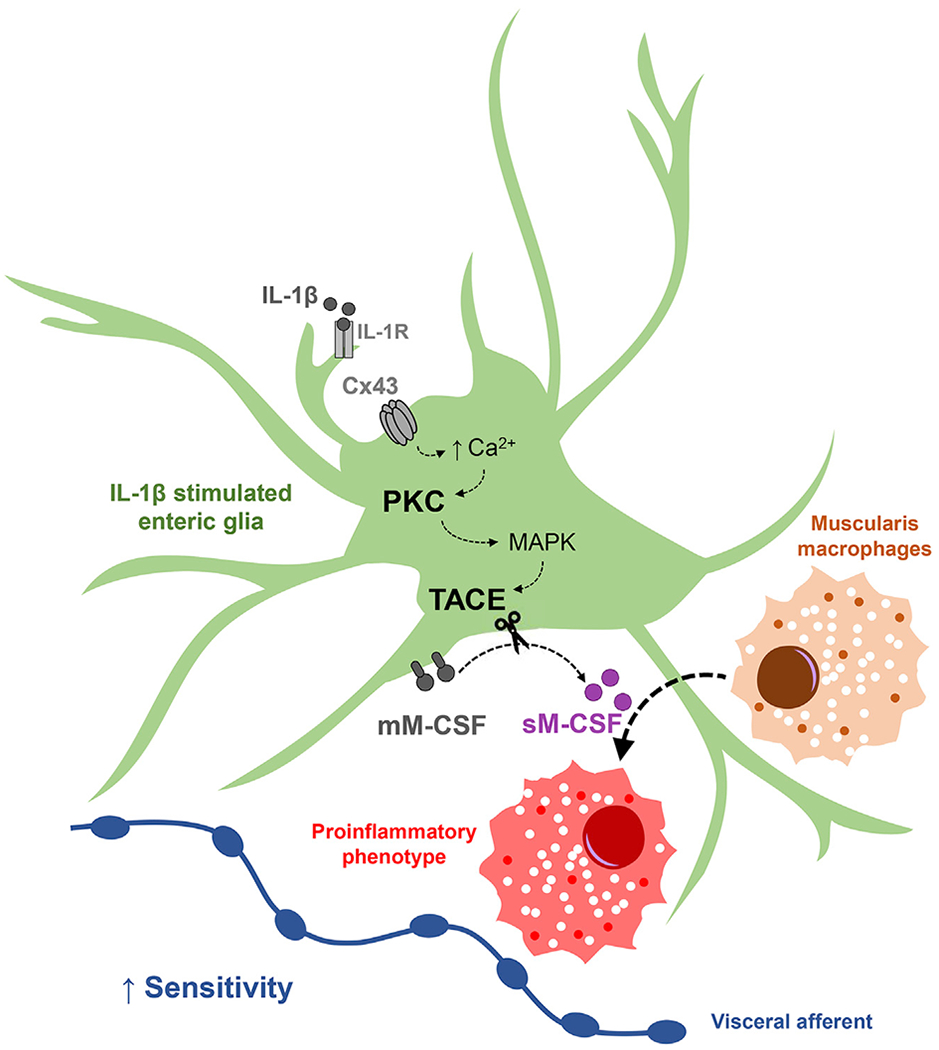
Schematic illustrating signaling mechanisms between enteric glia and macrophages identified in this study. Proinflammatory stimuli such as IL-1β induce glial reactivity and signaling through connexin-43 (Cx43) hemichannels. Cx43 hemichannels mediate calcium (Ca2+) responses in enteric glia (McClain et al., 2014). Glial activity increases signaling through protein kinase C (PKC) and activates tumor necrosis factor α converting enzyme (TACE), possibly via mitogen-activated protein kinase (MAPK) (Horiuchi and Toyama, 2008). Glial Cx43-dependent TACE activation results in proteolytic cleavage of cell membrane M-CSF (mM-CSF), increased release of soluble M-CSF (sM-CSF), and, in turn, macrophage activation. M-CSF produced by neurons and interstitial cells of Cajal could also contribute to macrophage activation. Data shown in this study provide evidence that interactions between enteric glia and macrophages contribute to the development of persistent hypersensitivity of visceral afferents in the intestines.
Human and murine enteric glia both produce M-CSF, but the specific mechanisms involved in its secretion may differ depending on species, experimental condition, and/or disease state. For example, the production of M-CSF by human enteric glia in culture was not affected by the Cx43 mimetic peptide 43Gap26, while the production of M-CSF by enteric glia in mice was dependent upon Cx43 expression and function. One possible explanation for this difference is that deleting the Cx43 gene in mice affects both channel-dependent and -independent functions of Cx43, while 43Gap26 only affects channel-dependent functions. This explanation seems unlikely because 43Gap26 also blocked macrophage activation in isolated preparations of mouse intestine and PKC and TACE activation in cultured murine enteric glia. More likely possibilities include differences in connexin channel expression and function that occur during chronic disease and/or cell-culture conditions. Intestinal inflammation alters the expression profile of connexin channels expressed by glia in mice (Delvalle et al., 2018) and humans (Figure S6), and other connexin isoforms would not be sensitive to blockade by 43Gap26. Inflammation can also induce the formation of heteromeric hemichannels that would render Cx43 insensitive to 43Gap26 (Koval et al., 2014). The loss of sensitivity to Cx43 modulation in glia from diseased individuals is an important observation because this could direct the timing and design of treatments targeting enteric glia. Understanding the mechanisms involved more fully will undoubtedly be an area of focus in future studies.
Our results show that glia-macrophage communication contributes to visceral nociceptor sensitization during chronic colitis (Figure S7). Glial Cx43 regulates colonic M-CSF production during acute intestinal inflammation and muscularis macrophage activation and visceral hypersensitivity following chronic colitis. These acute and chronic changes are linked by unique M-CSF signaling processes and their effects on macrophage functions. Unlike most immune mediators, M-CSF is constantly released during steady-state conditions (Seitz et al., 1994). M-CSF is important for macrophage homeostasis and regulates survival, proliferation, migration, and activation (Pixley and Stanley, 2004). These processes depend on the context and concentration of M-CSF. For instance, low concentrations of M-CSF support macrophage survival but do not induce significant proliferation (Tushinski et al., 1982). Macrophages also degrade M-CSF in a signaling feedback loop (Bartocci et al., 1987; Tushinski et al., 1982), and higher numbers of macrophages require more M-CSF for basic survival. Acute DSS colitis causes an increase in M-CSF expression (Zwicker et al., 2015), and the number of macrophages is increased with a delay of several days after the treatment (Hall et al., 2011). The activity of macrophages, however, is only significantly increased after chronic inflammation (Bento et al., 2012). Thus, glial M-CSF production during acute colitis could regulate macrophage activity in chronic inflammation and consequently transition to chronic visceral sensitivity. These data, however, do not rule out the possibility that enteric glia could influence visceral sensitivity through multiple other mechanisms during other circumstances. For example, enteric glia could modulate nociceptors by releasing neuromodulators such as ATP and IL-1β (Binshtok et al., 2008; Brown et al., 2016; Cook and McCleskey, 2002; Murakami et al., 2009). Glial factors may also influence the expression and/or function of nociceptive channels such as transient receptor potential ankyrin 1, vanilloid 1 and 4 (TRPA1, TRPV1, TRPV4), purinergic P2X3 receptors, tetrodotoxin-insensitive sodium channels Nav1.8 and Nav1.9, and T-type calcium channels Cav3.2 (Beyak et al., 2004; Brierley et al., 2008, 2009; Brierley and Linden, 2014; Cenac et al., 2010; De Schepper et al., 2008; Hockley et al., 2014; Lapointe et al., 2015; Scanzi et al., 2016; Wynn et al., 2004). Additionally, growth factors such as nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and glia-derived neurotrophic factor (GDNF) are secreted by glia in response to proinflammatory cytokines (Delvalle et al., 2018; von Boyen et al., 2006) and could modify nociceptor sensitivity (Dubin et al., 2012; Prato et al., 2017; Wangzhou et al., 2020).
Macrophage maturation is defective in IBD and IBS (Baillie et al., 2017; Rodríguez-Fandiño et al., 2017), and strategies that modulate this process could have broad therapeutic relevance. M-CSF influences several essential functions of macrophages including survival, proliferation, chemotaxis, and activation (Pixley and Stanley, 2004). Multiple cell types in the intestinal muscularis contribute to M-CSF production including neurons (Muller et al., 2014), Interstitial cells of Cajal (Breland et al., 2019; Lee et al., 2017b; Avetisyan et al., 2018), and, as shown here, glia. Interstitial cells of Cajal-derived M-CSF provides an important migratory signal for muscularis macrophages in the developing gut (Avetisyan et al., 2018), and neuronal M-CSF regulates certain immune responses (Muller et al., 2014). Our data show that most M-CSF produced in the muscularis is derived from cells outside the enteric nervous system, potentially Interstitial cells of Cajal. However, the M-CSF which is produced within the enteric nervous system is mainly derived from glia. This conclusion is based on the flow cytometry data showing that glia express the biologically active, cell-surface form of M-CSF to a far greater extent than neurons. Thus, manipulating glial M-CSF may have therapeutic relevance. One approach to perturb glial M-CSF would be to reduce glial Cx43 expression through the use of antisense oligodeoxynucleotides (Becker et al., 2016). However, careful cellular targeting would be necessary because Interstitial cells of Cajal and smooth muscle cells utilize gap junctions composed of Cx43 for cell-cell coupling that is necessary for motor function (Cousins et al., 2003; Döring et al., 2007). Targeting M-CSF itself may also provide therapeutic benefit, and, in support, neutralizing antibodies and inhibitors of M-CSF and M-CSF receptors are under consideration as potential therapies in cancer-associated pain (Cannarile et al., 2017). Of course, this approach reduces all M-CSF regardless of cellular origin, and it is still unclear how M-CSF derived from neurons and Interstitial cells of Cajal affects visceral hypersensitivity.
Glial cells modulate pain sensations at multiple points along the gut-brain axis, and some of the mechanisms we describe here for enteric glia are shared among these other populations of glia. For example, spinal microglia are activated by M-CSF and G-CSF and enhance painful stimuli (Basso et al., 2017; Guan et al., 2016). It is unlikely that these mechanisms contribute to reduced visceral hypersensitivity in our study since serum levels of these chemokines were not significantly affected by either inflammation or deletion of glial Cx43. Cx43 does play an important role in mechanisms whereby astrocytes and satellite glia modulate pain sensations in the central nervous system and dorsal root ganglia (Ji et al., 2013). We avoided modulating Cx43 in these cell types by using the Sox10 promoter to specifically target peripheral glia. Changes to Cx43 expression in satellite glial cells could contribute to the analgesic effects we observed here (Britsch et al., 2001; Retamal et al., 2017), but this scenario is unlikely since Sox10CreERT2;Cx43f/f animals exhibit VMRs comparable to control littermates at baseline conditions and during acute inflammation.
Our study specifically focused on interactions between enteric glia and macrophages, but we do not rule out the possibility that interactions between enteric glia and other classes of immune cells play important roles in inflammatory responses, neuroinflammation, or the modification of visceral sensitivity. For example, mast cells regulate visceral hypersensitivity in IBS (Boeckxstaens, 2018; Lee and Lee, 2016), and enteric glia express receptors for mediators released by activated mast cells such as histamine (Kimball and Mulholland, 1996) and proteases (Garrido et al., 2002). Also, enteric glia express immune mediators that regulate many types of immune cells (Delvalle et al., 2018) including mast cells, antigen-presenting cells, and lymphocytes. Glia-lymphocyte interactions could play a particularly important role in the induction and maintenance of chronic pain because Th1/Th17 cells promote intestinal inflammation and colonic tissue damage but have simultaneous opioid-mediated analgesic activity, thereby reducing abdominal pain (Boué et al., 2014).
In conclusion, our results highlight mechanisms of communication between enteric glia and muscularis macrophages that contribute to the development of visceral pain (Figure 7). These findings provide an insight into the cellular mechanisms that control neuroimmune interactions in the intestine that could ultimately benefit the treatment of visceral hypersensitivity in functional GI disorders.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian Gulbransen (gulbrans@msu.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The cytokine/chemokine multiplex data generated during this study are available in Table S3. The data for Figures S2 and S6A were sourced from a published (Delvalle et al., 2018) and publicly available dataset “Next generation sequencing of distal colon glial cells with DNBS-induced inflammation and neurokinin-2 receptor antagonism utilizing RiboTag mice” (Database: GSE114780).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committees (IACUC) at Michigan State University (MSU), USA, and by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee at Monash University, Australia. Mice of both sexes were used at 8-12 weeks of age and data were pooled together because there were no significant sex differences in prior work on enteric glial Cx43 (Brown et al., 2016; Grubišić and Gulbransen, 2017b; McClain et al., 2014). Mice were maintained in a temperature-controlled environment (Innovive, San Diego, CA; Innocage system with ALPHA-dri® bedding) on a 12-hour light: dark cycle with access to acidified water and food ad libitum.
Transgenic mice were generated on the C57BL/6 genetic background. Sox10CreERT2 transgenic mice were a gift from Dr. Vassilis Pachnis (The Francis Crick Institute, London, England) (Laranjeira et al., 2011). The Sox10 line is well characterized and selectively targets enteric glia (Delvalle et al., 2018; Grubišić and Gulbransen, 2017b; McClain and Gulbransen, 2017). Transgenic mice with an inducible and conditional deletion of Cx43 in neural crest cells marked by SRY-box-containing gene 10 (Sox10)-expressing glia (Sox10::CreERT2+/−/Cx43f/f; hereafter referred to as Sox10CrsERT2;Cx43f/f) were generated in-house by crossing Sox10CreERT2+/− mice with Cx43f/f mice (B6.129S7-Gja1tm1Dlg/J; Jackson Laboratory; RRID: IMSR_JAX:008039). While Cx43 is widely expressed in the gut, our prior data show that this targeting strategy selectively deletes glial Cx43 and does not affect Cx43 in other cell types (Grubišić and Gulbransen, 2017b). Littermates not expressing Cre were used as background controls (hereafter referred to as control). Cre recombinase activity was started 2 weeks before experiments and induced throughout the study by feeding animals tamoxifen citrate in the chow (400 mg/kg). All mice (including controls) were fed with a tamoxifen diet for the same duration to control for the potential effects of tamoxifen. Genotyping was performed by Transnetyx, Inc (Cordova, TN).
Human Tissue
Experimental protocols involving human tissues were approved by the Institutional Review Boards at Michigan State University (IRB# LEGACY02-780S), University of California, Los Angeles (IRB# 11-002504), and the College of Medicine at the Ohio State University (IRB# 2012H0231 and 2017H0441).
Fresh samples of human jejunum were collected from individuals undergoing Roux-en-Y gastric bypass surgery for weight loss as previously described (Fried et al., 2017). Human jejunum samples were fixed overnight in Zamboni’s fixative at 4°C, micro-dissected for whole-mount preparations of longitudinal muscle-myenteric plexus (LMMP) and processed by whole-mount immunohistochemistry.
Specimens of the human colon were collected from individuals undergoing resections for Crohn’s disease with low to high visceral pain status or from patients undergoing resections for bowel trauma, volvulus, intestinal bleeding or diverticulitis without the pain status that served as controls. Samples were formalin-fixed, paraffin-embedded, and cut in serial 5 mm thick sections for immunohistochemical studies.
Human enteric glial cells were isolated from samples of ileum collected from three individuals (two males and one female) with Crohn’s disease (two regions were collected from each patient: an inflammation involved region and noninvolved region) and from an inflammation involved region in ileum from a female patient with Crohn’s disease.
METHOD DETAILS
Dextran Sodium Sulfate (DSS) Induced Colitis
Acute colitis was induced by adding 2% DSS (colitis grade, M.W. 36-50 kDa; MP Biomedical, Solon, OH) to drinking water (2% w/v) for 7 days. Chronic DSS colitis was induced by intermittent exposure to 2% DSS (1 week on/1 off) for 3 intervals. Mice were euthanized 3 days and 7 days after final DSS treatment for acute and chronic time-points, respectively. Macroscopic damage was assessed as described previously (Storr et al., 2009).
Slide and whole-mount immunohistochemistry (IHC)
Paraffin-embedded slides were first deparaffinized by two 10 min incubations with 100% Xylene and one 5 min incubation with Xylene-ethanol (1:1) solution. Slides were gradually hydrated by 5 min incubations in 100% (twice), 95%, 75%, and 50% ethanol, rinsed in distilled water, and boiled (95-100°C) for 20 min in an antigen retrieval buffer (pH 6.0) composed of 10 mM citrate buffer, 0.05% Tween 20 and 2 mM EDTA. Immunolabeling on slides was performed as described below.
Whole-mount preparations of myenteric plexus were prepared by microdissection from mouse distal colon or fresh human jejunum fixed overnight in Zamboni’s fixative or 4% paraformaldehyde at 4°C. Immunolabeling was conducted as described elsewhere (Gulbransen et al., 2012) and is summarized below.
Antibody details are supplied in Table S1. Briefly, slides and whole mounts were rinsed three times (10 min each) in phosphate-buffered saline (PBS) or PBS containing 0.1% Triton X-100 (PBST) followed by a 30 or 60 min incubation in blocking solution [containing 4% normal goat serum (or 5% normal horse serum), 0.1 or 0.4% Triton X-100 and 1% bovine serum albumin]. Primary antibodies were applied overnight at room temperature (RT) orfor 48 hours at 4°C before being rinsed three times with PBS. Secondary antibodies were applied for 1 or 2 hours at RT. Whole mounts were rinsed with 0.1 M phosphate buffer and mounted on slides with bicarbonate-buffered glycerol (consisting of a 1:3 mixture of 142.8 mM sodium bicarbonate and 56.6 mM carbonate to glycerol) or ProLong Diamond anti-fade mounting media (ThermoFisher).
The immunolabeling protocol was modified in a subset of experiments to account for primary antibodies that were raised in the same host (rat anti-CD68 and either rat anti-MHClI or rat anti-F4/80). In this case, a 6-step sequential protocol was used as described elsewhere (https://www.jacksonimmuno.com/technical/products/protocols/double-labeling-same-species-primary/example-c). Briefly, after blocking with normal goat serum, samples were 1) incubated with one of the rat primary antibodies overnight at RT; 2) stained with the secondary conjugated goat anti-rat antibody for 2 h at RT; 3) blocked with rat serum solution (4% rat normal serum, 1% BSA, 0.4% Triton X-100) for 1 h at RT; 4) blocked with donkey anti-rat fab fragments diluted 1:50 in donkey serum solution (4% donkey normal serum, 1% BSA, 0.4% Triton X-100) overnight at RT; 5) incubated the preparations with the other of the rat primary antibodies overnight at RT; 2) stained with the secondary conjugated donkey anti-rat antibody for 2 h at RT. Each step was followed by rinsing three times with PBS.
Image Acquisition and Analysis
Epifluorescence images were acquired through the 20x [(PlanApo, 0.75 numerical aperture (n.a.)] and 40x (PlanFluor, 0.75 n.a.) objectives of an upright epifluorescence microscope (Nikon Eclipse Ni, Melville, NY) with a Retiga 2000R camera (QImaging, Surrey, BC, Canada) controlled by QCapture Pro 7.0 (QImaging). Confocal images were acquired through the 40x (UPLFL, 1.3 n.a.) and 60x (Plan-Apochromat, 1.42 n.a.) oil immersion objectives of an inverted Olympus Fluoview FV1000 microscope (Olympus, Center Valley, PA), or by Leica TCS SP8 laser-scanning confocal microscope using an HC PLAN APO 63x oil immersion objective (1.4 n.a.).
Epifluorescence images were analyzed offline using ImageJ software (National Institutes of Health, Bethesda, MD). Cell counts were performed using the cell counter plug-in of ImageJ software. Enteric neuron and glial cell numbers are presented as ganglionic packing density, calculated by counting the number of HuC/D-immunoreactive neurons or s100β-immunoreactive glia within the defined ganglionic area. Mean gray values of glial fibrillary acidic protein (GFAP), CD68, and macrophage colony-stimulating factor (M-CSF) fluorescence were measured within a defined ganglionic area following background subtraction. Cell counts and ganglionic expression data analysis were performed on a minimum of 10 ganglia per animal and averaged to obtain a value for that animal. N values represent the number of animals in each experiment and data is expressed as percent water control or mean intensity units (IU). Confocal images were processed by MetaMorph 7.0 software (Molecular Devices, LLC, San Jose, CA, United States.). A subset of confocal images was first deconvolved using Huygens Professional software (v17.10, Scientific Volume Imaging, the Netherlands, https://svi.nl/). Deconvolution of the Z stacks was carried out using the following settings: number of iterations = 200, signal to noise ratio (SNR) = 10, quality change threshold = 0.01. Calculated PSFs of each captured channel were used in all deconvolution processing. The deconvolved images were subsequently rendered using Imaris v9.1.2.
Chemicals and Reagents
Unless otherwise stated, chemicals and reagents for this study were purchased from Sigma-Aldrich (St. Louis, MO).
Histology
Sections of the distal colon were fixed overnight in Zamboni’s fixative at 4°C. Tissue was washed with PBS until cleared. Hematoxylin and Eosin (H&E) staining was performed on paraffin-embedded cross-sections of the distal colon by the Investigative Histopathology Laboratory at MSU. A disease activity score was performed in a blinded fashion and modified from previous work (Kim et al., 2012; Wirtz et al., 2017). Criteria for disease activity scoring are identified in Table S2.
Cytokine/ chemokine multiplex array
Mouse colon tissue samples were homogenized on ice using a tissue grinder in a buffer (pH 7.5) containing: Tris/Tris-HCl (25 mM), NaCl (130 mM), KCl (2.7 mM), Tween 20 (5% v/v), and a protease inhibitor cocktail tablet (Sigma S8820). The samples were sonicated with 10 pulses of 1 s intervals and centrifuged at 10,000 x g for 10 minutes at 4°C. Supernatant protein concentrations were measured on a NanoDrop Lite Spectrophotometer (ThermoFisher Scientific) and samples were adjusted to have the same concentration. An aliquot of each sample was further diluted (2-4x) and coded. Mouse 31-Plex Cytokine Array / Chemokine Array was run blindly by Eve Technologies Corp. (Calgary, AB, Canada), and analytes with concentration less than 1 pg/ml were excluded from further analysis. Raw data are available in Table S3.
Visceromotor Responses (VMR) to Colorectal Distention (CRD)
VMR recordings were performed as described and validated elsewhere (Larauche et al., 2010). Briefly, mice were anesthetized using isoflurane inhalation (2.5% and 1.5% in O2 for induction and maintenance, respectively) and a balloon-pressure sensor was inserted into the colon approximately 1 cm from the rectum. The balloon-pressure sensor consisted of a balloon (1 cm width x 2 cm length) secured 1 cm distal from the tip of a pressure transducer catheter (SPR-524 Mikro-Tip catheter; Millar Instruments, Houston, TX, USA). Each balloon was connected to a barostat and the pressure transducer was connected to a preamplifier (model 600; Millar Instruments, Houston, TX, USA). The CRD protocol consisted of a series of three consecutive graded phasic distention periods at a constant pressure of 15, 30, 45, and 60 mm Hg with a 10 s stimulus duration and a 4-minute interstimulus interval. VMR from these trials were recorded and analyzed in a blinded fashion using LabChart 7 software (ADInstruments, Colorado Springs CO).
In situ activation of myenteric glia
Whole-mounts of myenteric plexus (8 preparations per mouse) were prepared from the distal colon on ice-cold DMEM/Nutrient Mixture F-12 (Life Technologies) supplemented with L-glutamine and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES). Media was exchanged after 1 h and antibiotics (penicillin 100 U/mL and streptomycin 100 μg/mL) were added before placing preparation in an incubator (37°C in a 95% O2 / 5% CO2). Subsets of preparations (2 preparations per subset) received 43Gap26 (100 μM), rat anti-M-CSF blocking antibodies (5 μg/ml, Bio-Techne Ltd, MAB4161), both 43Gap26 and antibodies, or vehicle. After a 1 h incubation, IL-1β (10 ng/ml) was added to half of the preparations and all samples were incubated overnight at 37°C. Samples were fixed with 4% paraformaldehyde (1 h, RT) the following day, stained for GFAP, CD68, and MHCII (Novus, NBP2-21789), imaged and analyzed as described above.
Isolation of myenteric plexus cells for flow cytometry and primary cultures
Whole-mount sheets of myenteric plexus from the mouse colon were prepared as previously described (Smith et al., 2013). Briefly, colons were flushed with ice-cold DMEM/Nutrient Mixture F-12 (Life Technologies) supplemented with L-glutamine and HEPES, the remaining mesentery was removed, and colons were cut in half to yield two 3-4 cm segments. The segments were individually placed on plastic rods (≈2 mm in diameter) and cotton swabs wetted with the ice-cold DMEM were used to remove the myenteric plexus and longitudinal muscle layers.
Preparations were rinsed twice with HBSS substituted with HEPES (10 mM) and digested in 2.5 mL of HBSS/HEPES buffer substituted with Liberase TM (0.13 Wunsch units/ml) and DNase I (100 Kuntz units/ml) for 30 min at 37°C with occasional mixing. A gentleMACS Dissociator and gentleMACS C Tubes were used for cell dissociation. The enzyme reaction was stopped by an ice-cold complete medium composed of DMEM/Nutrient Mixture F-12 supplemented with L-glutamine and HEPES, penicillin and streptomycin (100 U/mL and 100 μg/mL), and 10% fetal bovine serum (FBS, Denville Scientific, Inc). Cell suspensions were filtered through a 100 μm cell strainer before culturing or additionally filtered through a 40 μm cell strainer before flow cytometry.
Flow cytometry
Cell suspensions were spun down (10 min at 350 x g and 4°C), resuspended and aliquoted in 1.5 mL centrifuge tubes containing 1 mL of PBS at RT. Cells were resuspended in 100 ul PBS containing Zombie NIR Fixable Viability dye (diluted 1:500) for 15 min at RT, then washed with 1 mL of PBS and spun down at 400 x g for 5 min at RT (following washing/centrifugation steps had the same conditions). Cells were fixed in 4% PFA for 15 min at RT and washed/spun twice before the addition of flow cytometry buffer (FCB, 1% bovine serum albumin in PBS) containing TruStain FcX PLUS (1:100) for 15 min @ RT. Rabbit anti-M-CSF antibodies (1:50, Bioss) were added and incubating overnight at 4°C. Cells were washed twice with PBS and then with PBST (0.1% Triton X-100 in PBS) for 10 min at RT and spun. FCB containing 0.1% Triton X-100 and TruStain FcX PLUS (1:100) was added for 15 min @ RT before adding chicken anti-GFAP (1:100, Aves) and biotin mouse anti-HuC/D (1:50) antibodies and incubating overnight at 4°C. Cells were washed twice with PBST before adding FCB containing 0.1% Triton X-100, donkey anti-rat Alexa 647 (1:200), goat anti-chicken Alexa 488 (1:200), and streptavidin DyLight 405 (1:500) for 1 h at RT. Cells were washed once with PBST and once with FCB before resuspension in 300 μl of FCB and analyzed on a BD LSR II [Becton Dickinson (BD) Biosciences, San Jose, CA] run by BD FACSDiva v8.0.1. Compensation was performed using unstained and single stained (compatible primary and secondary antibody) cells. Each fully stained sample was run in duplicate acquiring 20,000 events. Fluorescence minus one (FMO) controls and cells stained with only one of each of the secondary antibodies were run for proper gating. Post hoc analysis was performed using FCS Express 7 Research Edition (Win64) v7.01.0018. After gating for single live cells (Figure S5; Figure 5A), gatings for M-CSF+, Hu+, or GFAP+ cells were set to include < 1% of pertained FMO or secondary antibody only controls (Figure 5B).
Primary cultures of mouse enteric glia
Enteric glia were isolated and cultured using a modification of protocols described elsewhere (Smith et al., 2013). Enteric glia were isolated as described above and plated in 24-well plates on coverslips coated with poly D-Lysine (100 μg/ml) and laminin (25 μg/ml). Cell suspensions were initially seeded in the complete medium and plates were placed in an incubator (37°C, 95% O2 / 5% CO2). New media composed of DMEM/Nutrient Mixture F-12 supplemented with L-glutamine and HEPES, and substituted with antibiotics (penicillin 100 U/mL and streptomycin 100 μg/mL), GIBCO N-2 Supplement (0.2%), GIBCO G-5 Supplement (0.2%), and mouse NGF-β (0.05%) was exchanged every 2 days. Cells reached 50%–80% confluency with 2 weeks. Experimental drugs including 43Gap26 (100 μM), IL-1β (1 ng/ml), 43Gap26 and IL-1β, or vehicle were added to media the day before collection for protein kinase C (PKC) and tumor necrosis factor α-converting enzyme (TACE) activity assays.
Immunocytochemical identification of glial cells in primary cultures of mouse enteric glia
Cultures of primary mouse enteric glia were seeded onto coated glass coverslips and reached 50%–80% confluency by 2 weeks. Cells were rinsed twice with Hank’s balanced salt solution (HBSS) and fixed with freshly prepared 4% (w/v) paraformaldehyde for 20 min at RT. Coverslips were rinsed three times with PBS and cells were permeabilized with PBST for 10 min at RT. Cells were incubated in blocking solution (4% normal goat serum, 0.1% Triton X-100, and 1% bovine serum albumin) for 30 min at RT. Primary antibodies were applied for 2-3 hours at RT before being rinsed three times with PBS. Secondary antibodies were applied for 1-2 hours at RT. Coverslips were rinsed twice with PBS and once with 0.1 M phosphate buffer and mounted on slides with DAPI Fluoromount-G® (SouthernBiotech, 0100-20).
PKC activity assay
Lysis buffer, made from Tris-buffered saline, Nonidet P 40 Substitute (1%), and SIGMAFAST Protease Inhibitors (used per instructions), was added to each well and plates were placed on ice for 10 min. Cells were scraped and lysate collected in prechilled tubes. Cell lysates were centrifuged at 13,000 x g for 15 min at 4°C and clear supernatants were transferred to new prechilled tubes, snap-frozen on dry ice, and stored at −80°C. Cell lysates were thawed on ice and assessed using a PKC Kinase Activity Assay Kit (used per instructions). An aliquot of each lysate was also assayed with a Bicinchoninic Acid (BCA) Kit for Protein Determination (used per instructions). Sample absorbance (optical density 450 nm or 562 nm, respectively) was recorded using an Infinite M1000 PRO microplate reader (Tecan Group Ltd, Männedorf, Switzerland) run by i-controlTM microplate reader software (Tecan), v1.6.19.2. Raw background-subtracted PKC activity values were normalized to total protein concentration.
TACE activity assay
Accutase was added to each well and plates were incubated at 37°C for 10 minutes to detach cells. Cells were dislodged, placed in tubes, and centrifuged for 5 min at 400 x g at RT. The pellet was resuspended and a SensoLyte ® 520 TACE (α - Secretase) Activity Assay Kit was used to assess TACE activity (per instructions). After adding the TACE substrate solution into each well, the plate was mixed and sample fluorescence intensity was immediately recorded by an Infinite M1000 PRO microplate reader (Tecan) using i-controlTM microplate reader software v1.6.19.2 (Tecan) or Synergy H1 microplate reader (BioTek Instruments, Inc., Winooski, Vermont, USA) using Gen5 software (BioTek Instruments, Inc.). Excitation/emission wavelengths were 490/520 nm. An aliquot of live cell suspension was used for manual cell counting on a hemocytometer to determine cell density. Raw TACE activity values were normalized to the cell density and to untreated controls that originated from the same colon.
Isolation of glial cells from human intestinal surgical specimens, purification and primary cultures of human enteric glia
The procedure was described in detail elsewhere (Liñan-Rico et al., 2016). Briefly, tissue collection was carried out by a surgeon; it was immersed immediately in ice-cold oxygenated Krebs solution and promptly transported to the research facilities within 15 minutes in coordination with the clinical research team and pathology. For isolating myenteric ganglia, tissue was pinned luminal side facing up in aSylgard lined Petri dish under a stereoscopic microscope and the mucosa, submucosa, and most of the circular muscle were dissected away using scissors, and then flipped over to remove longitudinal muscle by dissection. Myenteric plexus tissue was minced (0.1–0.2 cm2 pieces) and dissociated in an enzyme solution (0.125 mg/ml Liberase, 0.5 μg/ml Amphotericin B) prepared in Dulbecco’s modified Eagle’s medium (DMEM)-F12, for 60 minutes at 37°C with agitation. Ganglia were removed from the enzymatic solution by spinning down, and re-suspending in a mixture of DMEM-F12, bovine serum albumin 0.1%, and DNase 50 μg/mL. This solution, containing the ganglia, was transferred to a 100-mm culture dish and isolated single ganglia free of smooth muscle or other tissue components were collected with a micropipette while visualized under a stereoscopic microscope and plated into wells of a 24-well culture plate and kept in DMEM-F12 (1:1) medium containing 10% FBS and a mixture of antibiotics (penicillin 100 U/mL, streptomycin 100 μg/mL, and amphotericin B 0.25 μg/mL) at 37°C in an atmosphere of 5% CO2 and 95% humidity. After cells reach semi-confluence in 3 to 4 weeks, human enteric glia were enriched and purified by eliminating/separating fibroblasts, smooth muscle, and other cells. Glial cell enrichment and purification were achieved by labeling the isolated cells with magnetic microbeads linked to anti-specific antigen, D7-Fib, and passing them through a magnetic bead separation column following the manufacturer instructions (Miltenyi Biotec Inc., San Diego, CA). This purification protocol was performed twice (P1 and P2) to reach a cell enrichment of up to 10,000 fold.
After reaching confluency, the glial cells were cultured for 24 h in 10% FBS media. After 24h the media was exchanged with fresh 0.1% FBS media and collected after 24 h and immediately frozen at −80°C (24 h control supernatants). The glial cultures were then incubated for 1 h in 0% FBS media and this was also collected and immediately frozen down (1 h control supernatants). The glial cells were allowed to recover overnight in 10% FBS media. After the recovery period, the glial cells were incubated with 1 ng/ml of IL-1β in regular cell medium for 6 h and fresh 0.1% FBS media was exchanged and collected for another 24 h (24 h IL-1β stimulated supernatants). This media was exchanged for 0% FBS media and incubated another hour and collected and frozen (1 hr IL-1β stimulated supernatant). The same induction and supernatant collection procedures were performed on subsets of human glia subcultures pre-incubated with a 100 μM of Cx43 mimetic peptide 43Gap26. Cultured cells were counted after the final supernatant collection.
Immunochemical identification of glial cells in primary cultures of human enteric glia
We harvested single ganglia, isolated and cultured glial cells using an approach that yields preparations with > 95% purity (Liñan-Rico et al., 2016). To confirm the identity of glial cells in the cultures, we used immunohistochemistry. Human enteric glia were fixed in 4% paraformaldehyde for 15 minutes at room temperature, rinsed 3 times with cold phosphate-buffered saline (PBS) 0.1 M, and placed at 4°C until further processing. Cells were treated with 0.5% Triton X-100, 10% normal donkey serum in PBS to permeabilize the cells and block nonspecific antibody binding for one hour at room temperature. The primary antibody was diluted in PBS, 0.5% Triton X-100, and 1% normal donkey serum, and were incubated with cells overnight (18–24 h) at 4°C. The next day, preparations were rinsed 3 times in 0.1M PBS and incubated 2 hours at room temperature in secondary antibody diluted in PBS, 0.5% Triton X-100, and 1% normal donkey serum. Polyclonal rabbit anti-S100β antibody (1:200 dil., Abcam, Cambridge, MA), mouse monoclonal anti-α smooth muscle/epithelial actin antibody (1:50–1:500 dil., cat #ab18147; Abcam), rabbit anti-GFAP antibody (1:500 dil., cat #z0334; DAKO, Carpinteria, CA), monoclonal mouse anti-fibroblast / epithelial cell antibody (1:100– 1:500 dil., cat #NB600–777; Novus Biologicals, Littleton, CO) and monoclonal mouse anti-HuC/D antibody (1:50 dil., cat #A21271; ThermoFisher, Cambridge, MA) were used for analysis. Alexa Fluor 488 or 568 donkey anti-mouse or anti-rabbit secondary antibodies were used at a dilution of 1:400 (Cambridge, MA). The omission of primary antibodies was used to test for background staining of the secondary antibodies. Pre-absorption of primary antisera with immunogenic peptides abolished immunoreactivity. Data confirmed previous reports by Turco et al. (2014) and are not shown, except for illustrating that all cells express s100β immunoreactivity.
Quantification of M-CSF from supernatants of cultured human enteric glia
Frozen supernatants were first thawed and purified/concentrated using prewashed Amicon® Ultra-2 mL columns (pore size 10 KDa; UFC201024, MilliporeSigma, Burlington, MA) at the maximum recommended centrifuge speed for 30 min at room temperature. Each concentrate was diluted with Assay Diluent B of the human M-CSF ELISA kit (ELH-MCSF-1, RayBio®, Peachtree Corners, GA) and run in duplicates. Absorbance was measured on the Infinite M1000 PRO microplate reader (wavelength 450 nm; Tecan Group Ltd, Mannedorf, Switzerland) using i-controlTM microplate reader software (Tecan, version 1.6.19.2). Raw concentrations, obtained from the human M-CSF standard dilution curve, were adjusted for the concentration factor and total cell number.
RNA isolation from primary human enteric glia and NanoString nCounter Gene Expression Assay of selected connexins
Cells were lysed in TRIZOL (Life Technologies) and frozen at −80°C. Total RNA isolation was done using the TRIZOL method and after the separation of aqueous and organic phases, an RNA kit (NORGEN Biotek Corp., Ontario, Canada) was used to purify and increase the concentration of the RNA. Gene expression analysis was done using the Nanostring nCounter Analysis System (Nanostring Technologies, Seattle, WA).
Agilent RNA 6000 Nano Chip was used to evaluate the RNA quality. NanoString nCounter technology is based on direct detection of target molecules using color-coded molecular barcodes, providing a digital simultaneous quantification of the number of target molecules. Total RNA (200ng) was hybridized overnight with nCounter Reporter (20 μL) probes in hybridization buffer and excess of nCounter Capture probes (5 μL) at 650C for 16-20 hours. The hybridization mixture containing target/probe complexes was allowed to bind to magnetic beads containing complementary sequences on the capture probe. After each target found a probe pair, excess probes were washed, followed by a sequential binding to sequences on the reporter probe. Biotinylated capture probe-bond samples were immobilized and recovered on a streptavidin-coated cartridge. The abundance of specific target molecules was then quantified using the nCounter digital analyzer. Individual fluorescent barcodes and target molecules present in each sample were recorded with a CCD camera by performing a high-density scan (600 fields of view). Images were processed internally into a digital format and were normalized using the NanoString nSolver Software analysis tool. Counts were normalized for all target RNAs in all samples based on the positive control RNA, to account for differences in hybridization efficiency and posthybridization processing including purification and immobilization. The average was normalized by background counts for each sample obtained from the average of the 8 negative control counts. Subsequently, a normalization of mRNA content was performed based on internal reference housekeeping genes using nSolver software (NanoString Technologies). Nanostring probes for selected connexin and housekeeping genes are in Table S4.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using Prism 7 (GraphPad Software, San Diego, CA) and are shown as mean ± standard error of the mean (SEM) unless stated otherwise such as mean ± standard deviation (SD), mean ± range, and median ± 95% confidence interval. Sample sizes were determined from previous experimental data using a power of 0.8 and a significance level of 0.05. The DSS-treated group was assigned by splitting the same genotype and sex littermates into two cohorts and treating one of them with DSS. The DSS-treated cohort had an extra member if the total of the same genotype and sex littermates was an odd number. Male and female data were pooled and tested for normality using D’Agostino and Pearson or Shapiro-Wilk tests. Outliers were excluded using the ROUT method (Q = 5%). Data were analyzed by two-way analysis of variance (ANOVA) followed by a Tukey, Bonferroni, or Sidak post hoc tests or when applicable a two-tailed Welch’s t test and ANOVA followed by Dunnett’s multiple comparisons test. A P value of less than 0.05 was considered significantly different from the null hypothesis. Statistical details of experiments such as the statistical tests used, the exact value of n, what n represents (e.g., number of animals, number of cells, or number of patients, etc.), the definition of center and dispersion/precision measures (e.g., mean ± SD) can be found in the figure legends, figures, and Results.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AffiniPure Fab Fragment Donkey Anti-Rat IgG (H+L) | Jackson Immuno, West Grove, PA | Cat# 712-007-003; RRID: AB_2340634 |
| biotin mouse anti-HuC/D | Invitrogen, Carlsbad, CA | Cat# A21272; RRID: AB_2535822 |
| chicken anti-GFAP | Abcam, Cambridge, MA | Cat# ab4674; RRID: AB_304558 |
| chicken anti-GFAP | Aves, Tigard, OR | Cat# GFAP87867983; RRID: AB_2313547 |
| donkey anti-chicken Alexa 488 | Jackson Immuno, West Grove, PA | Cat# 703-545-155; RRID: AB_2340375 |
| donkey anti-guinea pig DyLight 405 | Jackson Immuno, West Grove, PA | Cat# 706-475-148; RRID: AB_2340470 |
| donkey anti-rabbit Alexa 594 | Jackson Immuno, West Grove, PA | Cat# 711-585-152; RRID: AB_2340621 |
| donkey anti-rat Alexa 647 | Jackson Immuno, West Grove, PA | Cat# 712-605-150; RRID: AB_2340693 |
| goat anti-chicken Alexa 488 | Invitrogen, Carlsbad, CA | Cat# A-11039; RRID:AB_2534096 |
| goat anti-chicken DyLight 405 | Jackson Immuno, West Grove, PA | Cat# 103-475-155; RRID: AB_2337389 |
| goat anti-rabbit Alexa 488 | Invitrogen, Carlsbad, CA | Cat# A-11034; RRID:AB_2576217 |
| goat anti-rat Alexa 594 | Jackson Immuno, West Grove, PA | Cat# 112-585-003; RRID: AB_2338372 |
| guinea pig anti-PGP9.5 | Neuromics, Edina, MN | Cat# GP14104; RRID: AB_2210625 |
| rabbit anti-CCR2 | Abcam, Cambridge, MA | Cat# ab216863; RRID:AB_2832204 |
| rabbit anti-M-CSF | Bioss, Woburn, MA | Cat# bs-4910R; RRID:AB_2832205 |
| rabbit anti-S100β | Abcam, Cambridge, MA | Cat# ab52642; RRID: AB_882426 |
| rat anti-CD68, clone FA-11 | AbD Serotec, Raleigh, NC | Cat# MCA1957; RRID: AB_322219 |
| rat anti-F4/80 | Abcam, Cambridge, MA | Cat# ab6640; RRID: AB_1140040 |
| rat anti-M-CSF, clone 131614 | Bio-Techne Ltd, Minneapolis, MN | Cat# MAB4161; RRID:AB_2276674 |
| rat anti-MHC class II | BioLegend, San Diego, CA | Cat# 107602; RRID: AB_313317 |
| rat anti-MHC class II | Novus, Centennial, CO | Cat# NBP2-21789; RRID:AB_2828034 |
| streptavidin Alexa 594 | Jackson Immuno, West Grove, PA | Cat# 016-580-084; RRID: AB_2337250 |
| streptavidin DyLight 405 | Jackson Immuno, West Grove, PA | Cat# 016-470-084; RRID: AB_2337248 |
| TruStain FcX PLUS (anti-mouse CD16/32) Antibody | BioLegend, San Diego, CA | Cat# 156604; RRID:AB_2783138 |
| Biological Samples | ||
| Human intestinal tissue | Michigan State University, East Lansing, MI | N/A |
| Human intestinal tissue | The Ohio State University, Columbus, OH | N/A |
| Human intestinal tissue | University of California, Los Angeles, CA | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 43Gap26 | AnaSpec Inc. | AS-62644 |
| ACCUTASE | STEMCELL Technologies Inc, Vancouver, Canada | 07920 |
| Amphotericin B | Life Technologies | 15290018 |
| Anti-fibroblast microbeads, human | Miltenyi Biotec Inc., San Diego, CA | 130-050-601 |
| Bovine serum albumin | Millipore-Sigma | A2153 |
| Corning® Laminin, Mouse, 1 mg | Corning Inc., Corning, NY | 354232 |
| DAPI Fluoromount-G® | SouthernBiotech, Birmingham, AL | 0100-20 |
| Deoxyribonuclease I (DNase I) Type IV | Millipore-Sigma | D5025-15KU |
| Dextran Sodium Sulfate (DSS) | MP Biomedical, Solon, OH | 0216011080 |
| DNase | Millipore-Sigma | D4527 |
| Dulbecco’s modified Eagle’s medium (DMEM)-F12, HEPES, no phenol red | Life Technologies | 11039021 |
| Fetal bovine serum | Life Technologies | 26140-079 |
| Fetal bovine serum | Denville Scientific, Inc., Holliston, MA | FB5001 |
| gentleMACS C Tubes | Miltenyi Biotec Inc. | 130-093-237 |
| gentleMACS Dissociator | Miltenyi Biotec Inc | 130-093-235 |
| GIBCO G-5 Supplement (100X) | Thermo Fisher Scientific | 17503012 |
| GIBCO HBSS (10X), calcium, magnesium, no phenol red | ThermoFisher Scientific Inc., Waltham, MA | 14065056 |
| GIBCO N-2 Supplement (100X) | Thermo Fisher Scientific | 17502048 |
| HEPES | Millipore Sigma | H3375 |
| Isoflurane | Henry Shein, Dublin, OH | 029405 |
| Liberase TH Research Grade | Millipore-Sigma | 5401151001 |
| Liberase TM Research Grade | Millipore-Sigma | LIBTM-RO |
| Mouse NGF-β, 20 μg | Cell Guidance Systems Ltd, St. Louis, MO | GFM11-20 |
| Nonidet P 40 Substitute | Millipore Sigma | 74385 |
| Paraformaldehyde | Millipore Sigma | 158127 |
| Penicillin/ Streptomycin solution 100X | Millipore Sigma | P4333 |
| Picric acid | Millipore Sigma | P6744 |
| Poly-D-Lysine solution, 1.0 mg/mL | MilliporeSigma | A-003-E |
| Protease inhibitor cocktail | Millipore Sigma, Burlington, MA | P8340-5ML |
| Recombinant human IL-1β | InvivoGen | rcyec-hil1b |
| Recombinant human M-CSF | Novus | 216-MC |
| Recombinant mouse IL-1β | R&D Systems | 401-ML-005 |
| SIGMAFAST Protease Inhibitor Tablets | Millipore Sigma | S8820 |
| Tamoxifen citrate in the chow (400 mg/kg) | Envigo | TD.140849 |
| Tris Buffered Saline (TBS), 10X | Fisher BioReagents | BP2471-1 |
| Zombie NIR Fixable Viability Kit | BioLegend, San Diego, CA | 423106 |
| Critical Commercial Assays | ||
| A magnetic bead separation column | Miltenyi Biotec Inc., San Diego, CA | 130-042-201 |
| Amicon Ultra-2 Centrifugal Filter Unit | MilliporeSigma, Burlington, MA | UFC201024 |
| Bicinchoninic Acid (BCA) Kit for Protein Determination | Sigma-Aldrich Company Ltd., St. Louis, MO | BCA1 |
| Mouse 31-Plex Cytokine Array / Chemokine Array | Eve Technologies Corp., Calgary, AB, Canada | SKU: MD31 |
| PKC Kinase Activity Assay Kit | Abcam, Cambridge, MA | ab139437 |
| RayBio® Human M-CSF ELISA Kit | RayBiotech Life, Peachtree Corners, GA | ELH-MCSF-1 |
| SensoLyte ® 520 TACE (α - Secretase) Activity Assay Kit *Fluorimetric* | AnaSpec, Inc., Fremont, CA | AS-72085 |
| Deposited Data | ||
| Next-generation sequencing of distal colon glial cells with DNBS-induced inflammation and neurokinin-2 receptor antagonism utilizing RiboTag mice | Delvalle et al., 2018 | Database: GSE114780 |
| Contributors: Gulbransen B. D., Delvalle N.M., and Dharshika C. | ||
| Cytokine/ chemokine multiplex array raw data | This paper | Table S3 |
| Experimental Models: Organisms/Strains | ||
| B6.129S7-Gja1tm1Dlg/J | Jackson Laboratory, Bar Harbor, ME | RRID: IMSR_JAX:008039 |
| Tg(Sox10-icre/ERT2)93Vpa | Dr. Vassilis Pachnis, The Francis Crick Institute, London, England | MGI ID: 5910373 |
| Oligonucleotides | ||
| hCx26 GGCTGTCTGTTGTATTCATTGTGGTCATAGCACCTAACAACATTGTAGCCTCAATCGAGTGAGACAGACTAGAAGTTCCTAGTGATGGCTTATGATAGCA | This paper | NM_004004.5 |
| hCx30 AGGCACGAAACCACTCGCAAGTTCAGGCGAGGAGAGAAGAGGAATGATTTCAAAGACATAGAGGACATTAAAAAGCAGAAGGTTCGGATAGAGGGGTCGC | This paper | NM_006783.4 |
| hCx31 GGTGGACCTACCTGTTCAGCCTCATCTTCAAGCTCATCATTGAGTTCCTCTTCCTCTACCTGCTGCACACTCTCTGGCATGGCTTCAATATGCCGCGCCT | This paper | NM_001005752.1 |
| hCx43 GCGAACCTACATCATCAGTATCCTCTTCAAGTCTATCTTTGAGGTGGCCTTCTTGCTGATCCAGTGGTACATCTATGGATTCAGCTTGAGTGCTGTTTAC | This paper | NM_000165.3 |
| hCx45 TTGCTGGCAAGGACCGTGTTTGAGGTGGGTTTTCTGATAGGGCAGTATTTTCTGTATGGCTTCCAAGTCCACCCGTTTTATGTGTGCAGCAGACTTCCTT | This paper | NM_001080383.1 |
| hCx47 GGAATGGGGCTCTGGGTTCCTGCCTGTGGCCTGTCTGTCCTCCTCCCTAATTCAGACCCAGCCTCAAGAGGAAAGGGAGTAAAATAAAACTAACTTGTTT | This paper | NM_020435.2 |
| ADA1 GCAGGTGCACAGGGAAGTCATCCCTACACATACTGTCTATGCTCTTAACATTGAAAGGATCATCACGAAACTCTGGCATCCAAATCATGAAGAGCTGCAG | This paper | NM_053053.3 |
| CTNNB1 TCTTGCCCTTTGTCCCGCAAATCATGCACCTTTGCGTGAGCAGGGTGCCATTCCACGACTAGTTCAGTTGCTTGTTCGTGCACATCAGGATACCCAGCGC | This paper | NM_001098210.1 |
| NMNAT1 CCGAGAAGACTGAAGTGGTTCTCCTTGCTTGTGGTTCATTCAATCCCATCACCAACATGCACCTCAGGTTGTTTGAGCTGGCCAAGGACTACATGAATGG | This paper | NM_022787.3 |
| RBP1 TGATCATCCGCACGCTGAGCACTTTTAGGAACTACATCATGGACTTCCAGGTTGGGAAGGAGTTTGAGGAGGATCTGACAGGCATAGATGACCGCAAGTG | This paper | NM_002899.3 |
| Software and Algorithms | ||
| BD FACSDiva v8.0.1 | Becton, Dickinson and Company, Franklin Lakes, NJ | RRID:SCR_001456 |
| FCS Express 7 Research Edition (Win64) v7.01.0018. | De Novo Software, Pasadena, CA | RRID:SCR_016431 |
| Huygens Professional software v17.10 | Scientific Volume Imaging, the Netherlands, | http://svi.nl RRID:SCR_014237 |
| ImageJ and Fiji | National Institutes of Health, Bethesda, MD | RRID:SCR_003070, RRID:SCR_002285 |
| Imaris v9.1.2 | Oxford Instruments, Abingdon, United Kingdom | RRID:SCR_007370 |
| LabChart 7 | ADInstruments, Colorado Springs CO | RRID:SCR_001620 |
| MetaMorph 7.0 | Molecular Devices, LLC, San Jose, CA | RRID:SCR_002368 |
| Prism 7 | GraphPad Software, San Diego, CA | RRID:SCR_002798 |
Highlights.
Enteric glia regulate visceral hypersensitivity during chronic inflammation
Glial mechanisms include connexin-43 and M-CSF production
Glial M-CSF modulates muscularis macrophage activation
Inflammation-induced glial M-CSF production is regulated by Cx43, PKC, and TACE
ACKNOWLEDGMENTS
TThis work was supported by a Crohn’s and Colitis Foundation Research Fellowship Award (577598) to V.G., National Institutes of Health (NIH) R01DK103723, R01DK120862, and Crohn’s and Colitis Foundation Senior Research Award (327058) to B.D.G., NIH R01DK113943 to F.L.C., a grant from Helmsley Charitable Trust (281574.5069091.0010), and salary support from R01 DK108894 to S.C.R. We acknowledge the University of California Los Angeles Translational Pathology Core laboratory for preparation of paraffin-embedded sections for immunohistochemical studies.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108100.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Avetisyan M, Rood JE, Huerta Lopez S, Sengupta R, Wright-Jin E, Dougherty JD, Behrens EM, and Heuckeroth RO (2018). Muscularis macrophage development in the absence of an enteric nervous system. Proc. Natl. Acad. Sci. USA 115, 4696–4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie JK, Arner E, Daub C, De Hoon M, Itoh M, Kawaji H, Lassmann T, Carninci P, Forrest AR, Hayashizaki Y, et al. ; FANTOM Consortium (2017). Analysis of the human monocyte-derived macrophage transcriptome and response to lipopolysaccharide provides new insights into genetic aetiology of inflammatory bowel disease. PLoS Genet. 13, e1006641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartocci A, Mastrogiannis DS, Migliorati G, Stockert RJ, Wolkoff AW, and Stanley ER (1987). Macrophages specifically regulatethe concentration of their own growth factor in the circulation. Proc. Natl. Acad. Sci. USA 84, 6179–6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso L, Lapointe TK, Iftinca M, Marsters C, Hollenberg MD, Kurrasch DM, and Altier C (2017). Granulocyte-colony-stimulating factor (G-CSF) signaling in spinal microglia drives visceral sensitization following colitis. Proc. Natl. Acad. Sci. USA 114, 11235–11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker DL, Phillips AR, Duft BJ, Kim Y, and Green CR (2016). Translating connexin biology into therapeutics. Semin. Cell Dev. Biol 50, 49–58. [DOI] [PubMed] [Google Scholar]
- Bento AF, Leite DF, Marcon R, Claudino RF, Dutra RC, Cola M, Martini AC, and Calixto JB (2012). Evaluation of chemical mediators and cellular response during acute and chronic gut inflammatory response induced by dextran sodium sulfate in mice. Biochem. Pharmacol 84, 1459–1469. [DOI] [PubMed] [Google Scholar]
- Beyak MJ, Ramji N, Krol KM, Kawaja MD, and Vanner SJ (2004). Two TTX-resistant Na+ currents in mouse colonic dorsal root ganglia neurons and their role in colitis-induced hyperexcitability. Am. J. Physiol. Gastrointest. Liver Physiol 287, G845–G855. [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, and Samad TA (2008). Nociceptors are interleukin-1beta sensors. J. Neurosci 28, 14062–14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckxstaens GE (2018). The Emerging Role of Mast Cells in Irritable Bowel Syndrome. Gastroenterol. Hepatol. (N. Y.) 14, 250–252. [PMC free article] [PubMed] [Google Scholar]
- Boué J, Basso L, Cenac N, Blanpied C, Rolli-Derkinderen M, Neunlist M, Vergnolle N, and Dietrich G (2014). Endogenous regulation of visceral pain via production of opioids by colitogenic CD4(+) T cells in mice. Gastroenterology 146, 166–175. [DOI] [PubMed] [Google Scholar]
- Breland A, Ha SE, Jorgensen BG, Jin B, Gardner TA, Sanders KM, and Ro S (2019). Smooth Muscle Transcriptome Browser: offering genome-wide references and expression profiles of transcripts expressed in intestinal SMC, ICC, and PDGFRα+ cells. Sci. Rep 9, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley SM, and Linden DR (2014). Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat. Rev. Gastroenterol. Hepatol 11, 611–627. [DOI] [PubMed] [Google Scholar]
- Brierley SM, Page AJ, Hughes PA, Adam B, Liebregts T, Cooper NJ, Holtmann G, Liedtke W, and Blackshaw LA (2008). Selective role for TRPV4 ion channels in visceral sensory pathways. Gastroenterology 134, 2059–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley SM, Hughes PA, Page AJ, Kwan KY, Martin CM, O’Donnell TA, Cooper NJ, Harrington AM, Adam B, Liebregts T, et al. (2009). The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology 137, 2084–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley SM, Hibberd TJ, and Spencer NJ (2018). Spinal Afferent Innervation of the Colon and Rectum. Front. Cell. Neurosci 12, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, and Wegner M (2001). The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 15, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown IA, McClain JL, Watson RE, Patel BA, and Gulbransen BD (2016). Enteric glia mediate neuron death in colitis through purinergic pathways that require connexin-43 and nitric oxide. Cell. Mol. Gastroenterol. Hepatol 2, 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, and Rüttinger D (2017). Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenac N, Altier C, Motta JP, d’Aldebert E, Galeano S, Zamponi GW, and Vergnolle N (2010). Potentiation of TRPV4 signalling by histamine and serotonin: an important mechanism for visceral hypersensitivity. Gut 59, 481–488. [DOI] [PubMed] [Google Scholar]
- Chang L, Di Lorenzo C, Farrugia G, Hamilton FA, Mawe GM, Pasricha PJ, and Wiley JW (2018). Functional Bowel Disorders: A Roadmap to Guide the Next Generation of Research. Gastroenterology 154, 723–735. [DOI] [PubMed] [Google Scholar]
- Chavan SS, Pavlov VA, and Tracey KJ (2017). Mechanisms and Therapeutic Relevance of Neuro-immune Communication. Immunity 46, 927–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow AK, and Gulbransen BD (2017). Potential roles of enteric glia in bridging neuroimmune communication in the gut. Am. J. Physiol. Gastrointest. Liver Physiol 312, G145–G152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani G, Gibbons SJ, Kashyap PC, and Farrugia G (2016). Intrinsic Gastrointestinal Macrophages: Their Phenotype and Role in Gastrointestinal Motility. Cell. Mol. Gastroenterol. Hepatol 2, 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SP, and McCleskey EW (2002). Cell damage excites nociceptors through release of cytosolic ATP. Pain 95, 41–47. [DOI] [PubMed] [Google Scholar]
- Cosman D, Wignall J, Anderson D, Tushinski J, Gallis B, Urdal D, and Cerretti DP (1988). Human macrophage colony stimulating factor (M-CSF): alternate RNA splicing generates three different proteins that are expressed on the cell surface and secreted. Behring Inst. Mitt 83, 15–26. [PubMed] [Google Scholar]
- Cousins HM, Edwards FR, Hickey H, Hill CE, and Hirst GD (2003). Electrical coupling between the myenteric interstitial cells of Cajal and adjacent muscle layers in the guinea-pig gastric antrum. J. Physiol 550, 829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schepper HU, De Winter BY, Van Nassauw L, Timmermans JP, Herman AG, Pelckmans PA, and De Man JG (2008). TRPV1 receptors on unmyelinated C-fibres mediate colitis-induced sensitization of pelvic afferent nerve fibres in rats. J. Physiol 586, 5247–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvalle NM, Dharshika C, Morales-Soto W, Fried DE, Gaudette L, and Gulbransen BD (2018). Communication Between Enteric Neurons, Glia, and Nociceptors Underlies the Effects of Tachykinins on Neuroinflammation. Cell. Mol. Gastroenterol. Hepatol 6, 321–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döring B, Pfitzer G, Adam B, Liebregts T, Eckardt D, Holtmann G, Hofmann F, Feil S, Feil R, and Willecke K (2007). Ablation of connexin43 in smooth muscle cells of the mouse intestine: functional insights into physiology and morphology. Cell Tissue Res. 327, 333–342. [DOI] [PubMed] [Google Scholar]
- Dubin AE, Schmidt M, Mathur J, Petrus MJ, Xiao B, Coste B, and Patapoutian A (2012). Inflammatory signals enhance piezo2-mediated mechanosensitive currents. Cell Rep. 2, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, La JH, Schwartz ES, and Gebhart GF (2012). Irritable bowel syndrome: methods, mechanisms, and pathophysiology. Neural and neuroimmune mechanisms of visceral hypersensitivity in irritable bowel syndrome. Am. J. Physiol. Gastrointest. Liver Physiol 302, G1085–G1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei K, Nohava K, Malipiero UV, Schwerdel C, and Fontana A (1992). Production of macrophage colony-stimulating factor by astrocytes and brain macrophages. J. Neuroimmunol 40, 189–195. [DOI] [PubMed] [Google Scholar]
- Fried DE, Watson RE, Robson SC, and Gulbransen BD (2017). Ammonia modifies enteric neuromuscular transmission through glial γ-aminobutyric acid signaling. Am. J. Physiol. Gastrointest. Liver Physiol 313, G570–G580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness JB, Hind AJ, Ngui K, Robbins HL, Clerc N, Merrot T, Tjandra JJ, and Poole DP (2006). The distribution of PKC isoforms in enteric neurons, muscle and interstitial cells of the human intestine. Histochem. Cell Biol 126, 537–548. [DOI] [PubMed] [Google Scholar]
- Garrido R, Segura B, Zhang W, and Mulholland M (2002). Presence of functionally active protease-activated receptors 1 and 2 in myenteric glia. J. Neurochem 83, 556–564. [DOI] [PubMed] [Google Scholar]
- Grubišić V, and Gulbransen BD (2017a). Enteric glia: the most alimentary of all glia. J. Physiol 595, 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubišić V, and Gulbransen BD (2017b). Enteric glial activity regulates secretomotor function in the mouse colon but does not acutely affect gut permeability. J. Physiol 595, 3409–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, et al. (2016). Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci 19, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbransen BD, and Sharkey KA (2012). Novel functional roles for enteric glia in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol 9, 625–632. [DOI] [PubMed] [Google Scholar]
- Gulbransen BD, Bashashati M, Hirota SA, Gui X, Roberts JA, MacDonald JA, Muruve DA, McKay DM, Beck PL, Mawe GM, et al. (2012). Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat. Med 18, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall LJ, Faivre E, Quinlan A, Shanahan F, Nally K, and Melgar S (2011). Induction and activation of adaptive immune populations during acute and chronic phases of a murine model of experimental colitis. Dig. Dis. Sci 56, 79–89. [DOI] [PubMed] [Google Scholar]
- Harris AL (2001). Emerging issues of connexin channels: biophysics fills the gap. Q. Rev. Biophys 34, 325–472. [DOI] [PubMed] [Google Scholar]
- Hockley JR, Boundouki G, Cibert-Goton V, McGuire C, Yip PK, Chan C, Tranter M, Wood JN, Nassar MA, Blackshaw LA, et al. (2014). Multiple roles for NaV1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease-derived stimuli. Pain 155, 1962–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, and Toyama Y (2008). Posttranslational regulation of cell-surface colony-stimulating factor-1. Crit. Rev. Immunol 28, 215–227. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Miyamoto T, Takaishi H, Hakozaki A, Kosaki N, Miyauchi Y, Furukawa M, Takito J, Kaneko H, Matsuzaki K, et al. (2007). Cell surface colony-stimulating factor 1 can be cleaved by TNF-alpha converting enzyme orendocytosed in aclathrin-dependent manner. J. Immunol 179,6715–6724. [DOI] [PubMed] [Google Scholar]
- Jha MK, Jo M, Kim JH, and Suk K (2019). Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 25, 227–240. [DOI] [PubMed] [Google Scholar]
- Ji RR, Berta T, and Nedergaard M (2013). Glia and pain: is chronic pain a gliopathy? Pain 154 (Suppl 1), S10–S28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Chamessian A, and Zhang YQ (2016). Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesler P, Fuss IJ, and Strober W (2015). Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol 1, 154–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Shajib MS, Manocha MM, and Khan WI (2012). Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp 60, 3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball BC, and Mulholland MW (1996). Enteric glia exhibit P2U receptors that increase cytosolic calcium by a phospholipase C-dependent mechanism. J. Neurochem 66, 604–612. [DOI] [PubMed] [Google Scholar]
- Koval M, Molina SA, and Burt JM (2014). Mix and match: investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 588, 1193–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe TK, Basso L, Iftinca MC, Flynn R, Chapman K, Dietrich G, Vergnolle N, and Altier C (2015). TRPV1 sensitization mediates postinflammatory visceral pain following acute colitis. Am. J. Physiol. Gastrointest. Liver Physiol 309, G87–G99. [DOI] [PubMed] [Google Scholar]
- Laranjeira C, Sandgren K, Kessaris N, Richardson W, Potocnik A, Vanden Berghe P, and Pachnis V (2011). Glial cells in the mouse enteric nervous system can undergo neurogenesis in response to injury. J. Clin. Invest 121, 3412–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larauche M, Gourcerol G, Million M, Adelson DW, and Taché Y (2010). Repeated psychological stress-induced alterations of visceral sensitivity and colonic motorfunctions in mice: influence of surgery and postoperative single housing on visceromotor responses. Stress 13, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KN, and Lee OY (2016). The Role of Mast Cells in Irritable Bowel Syndrome. Gastroenterol. Res. Pract 2016, 2031480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AD, Spiegel BM, Hays RD, Melmed GY, Bolus R, Khanna D, Khanna PP, and Chang L (2017a). Gastrointestinal symptom severity in irritable bowel syndrome, inflammatory bowel disease and the general population. Neurogastroenterol. Motil 29, Published online May 2017. 10.1111/nmo.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Ha SE, Park C, Park PJ, Fuchs R, Wei L, Jorgensen BG, Redelman D, Ward SM, Sanders KM, and Ro S (2017b). Transcriptome of interstitial cells of Cajal reveals unique and selective gene signatures. PLoS ONE 12, e0176031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Feng W, Wang R, Ma S, Wang L, Yang X, Yang F, Lin Y, Ren Q, and Zheng G (2016). Diverse in vivo effects of soluble and membrane-bound M-CSF on tumor-associated macrophages in lymphoma xenograft model. Oncotarget 7, 1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liñán-Rico A, Turco F, Ochoa-Cortes F, Harzman A, Needleman BJ, Arsenescu R, Abdel-Rasoul M, Fadda P, Grants I, Whitaker E, et al. (2016). Molecular Signaling and Dysfunction of the Human Reactive Enteric Glial Cell Phenotype: Implications for GI Infection, IBD, POI, Neurological, Motility, and GI Disorders. Inflamm. Bowel Dis 22, 1812–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Brosnan CF, Dickson DW, and Lee SC (1994). Macrophage colony-stimulating factor mediates astrocyte-induced microglial ramification in human fetal central nervous system culture. Am. J. Pathol 145, 48–53. [PMC free article] [PubMed] [Google Scholar]
- Mantovani B, Rabinovitch M, and Nussenzweig V (1972). Phagocytosis of immune complexes by macrophages. Different roles of the macrophage receptor sites for complement (C3) and for immunoglobulin (IgG). J. Exp. Med 135, 780–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JL, and Gulbransen BD (2017). The acute inhibition of enteric glial metabolism with fluoroacetate alters calcium signaling, hemichannel function, and the expression of key proteins. J. Neurophysiol 117, 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain J, Grubisic V, Fried D, Gomez-Suarez RA, Leinninger GM, Sevigny J, Parpura V, and Gulbransen BD (2014). Ca2+ responses in enteric glia are mediated by connexin-43 hemichannels and modulate colonic transit in mice. Gastroenterology 146, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mearin F, Lacy BE, Chang L, Chey WD, Lembo AJ, Simren M, and Spiller R (2016). Bowel Disorders. Gastroenterology, S0016-5085(16)00222-5. [DOI] [PubMed] [Google Scholar]
- Morales-Soto W, and Gulbransen BD (2019). Enteric Glia: A New Player in Abdominal Pain. Cell. Mol. Gastroenterol. Hepatol 7, 433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PA, Koscsó B, Rajani GM, Stevanovic K, Berres ML, Hashimoto D, Mortha A, Leboeuf M, Li XM, Mucida D, et al. (2014). Crosstalk between Muscularis Macrophages and Enteric Neurons Regulates Gastrointestinal Motility. Cell 158, 1210. [DOI] [PubMed] [Google Scholar]
- Murakami M, Ohta T, and Ito S (2009). Lipopolysaccharides enhance the action of bradykinin in enteric neurons via secretion of interleukin-1beta from enteric glial cells. J. Neurosci. Res 87, 2095–2104. [DOI] [PubMed] [Google Scholar]
- Naeim F (2008). Principles of Immunophenotyping In Hematopathology, Faramarz Naeim PNR and Grody Wayne W., eds. (Academic Press; ), pp. 27–55. [Google Scholar]
- Olk S, Zoidl G, and Dermietzel R (2009). Connexins, cell motility, and the cytoskeleton. Cell Motil. Cytoskeleton 66, 1000–1016. [DOI] [PubMed] [Google Scholar]
- Peery AF, Crockett SD, Barritt AS, Dellon ES, Eluri S, Gangarosa LM, Jensen ET, Lund JL, Pasricha S, Runge T, et al. (2015). Burden of Gastrointestinal, Liver, and Pancreatic Diseases in the United States. Gastroenterology 149, 1731–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pixley FJ, and Stanley ER (2004). CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 14, 628–638. [DOI] [PubMed] [Google Scholar]
- Poole DP, Hunne B, Robbins HL, and Furness JB (2003). Protein kinase C isoforms in the enteric nervous system. Histochem. Cell Biol 120, 51–61. [DOI] [PubMed] [Google Scholar]
- Prato V, Taberner FJ, Hockley JRF, Callejo G, Arcourt A, Tazir B, Hammer L, Schad P, Heppenstall PA, Smith ES, and Lechner SG (2017). Functional and Molecular Characterization of Mechanoinsensitive “Silent” Nociceptors. Cell Rep 21, 3102–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao M, Nelms BD, Dong L, Salinas-Rios V, Rutlin M, Gershon MD, and Corfas G (2015). Enteric glia express proteolipid protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia 63,2040–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss D, Ceredig RA, Secher T, Boué J, Barreau F, Dietrich G, and Gavériaux-Ruff C (2017). Mu and delta opioid receptor knockout mice show increased colonic sensitivity. Eur. J. Pain 21, 623–634. [DOI] [PubMed] [Google Scholar]
- Retamal MA, Riquelme MA, Stehberg J, and Alcayaga J (2017). Connexin43 Hemichannels in Satellite Glial Cells, Can They Influence Sensory Neuron Activity? Front. Mol. Neurosci 10, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Fandirño OA, Hernández-Ruiz J, López-Vidal Y, Charúa-Guindic L, Escobedo G, and Schmulson MJ (2017). Maturation Phenotype of Peripheral Blood Monocyte/Macrophage After Stimulation with Lipopolysaccharides in Irritable Bowel Syndrome. J. Neurogastroenterol. Motil 23, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanzi J, Accarie A, Muller E, Pereira B, Aissouni Y, Goutte M, Joubert-Zakeyh J, Picard E, Boudieu L, Mallet C, et al. (2016). Colonic overexpression of the T-type calcium channel Cav 3.2 in a mouse model of visceral hypersensitivity and in irritable bowel syndrome patients. Neurogastroenterol. Motil 28, 1632–1640. [DOI] [PubMed] [Google Scholar]
- Seitz M, Loetscher P, Fey MF, and Tobler A (1994). Constitutive mRNA and protein production of macrophage colony-stimulating factor but not of other cytokines by synovial fibroblasts from rheumatoid arthritis and osteoarthritis patients. Br. J. Rheumatol 33, 613–619. [DOI] [PubMed] [Google Scholar]
- Simrén M, Törnblom H, Palsson OS, van Tilburg MAL, Van Oudenhove L, Tack J, and Whitehead WE (2018). Visceral hypersensitivity is associated with GI symptom severity in functional GI disorders: consistent findings from five different patient cohorts. Gut 67, 255–262. [DOI] [PubMed] [Google Scholar]
- Smith TH, Ngwainmbi J, Grider JR, Dewey WL, and Akbarali HI (2013). An in-vitro preparation of isolated enteric neurons and glia from the myenteric plexus of the adult mouse. J. Vis. Exp 78, 50688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storr MA, Keenan CM, Zhang H, Patel KD, Makriyannis A, and Sharkey KA (2009). Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm. Bowel Dis 15, 1678–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Liu L, Xu D, Zhang W, Zhang Y, Zhou J, and Huang W (2018). Interaction between astrocytic colony stimulating factor and its receptor on microglia mediates central sensitization and behavioral hypersensitivity in chronic post ischemic pain model. Brain Behav. Immun 68, 248–260. [DOI] [PubMed] [Google Scholar]
- Turco F, Sarnelli G, Cirillo C, Palumbo I, De Giorgi F, D’Alessandro A, Cammarota M, Giuliano M, and Cuomo R (2014). Enteroglial-derived S100B protein integrates bacteria-induced Toll-like receptor signalling in human enteric glial cells. Gut 63, 105–115. [DOI] [PubMed] [Google Scholar]
- Tushinski RJ, Oliver IT, Guilbert LJ, Tynan PW, Warner JR, and Stanley ER (1982). Survival of mononuclear phagocytes depends on a lineage-specific growth factor that the differentiated cells selectively destroy. Cell 28, 71–81. [DOI] [PubMed] [Google Scholar]
- Verheijden S, De Schepper S, and Boeckxstaens GE (2015). Neuron-macrophage crosstalk in the intestine: a “microglia” perspective. Front. Cell. Neurosci 9, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videlock EJ, Mahurkar-Joshi S, Hoffman JM, Iliopoulos D, Pothoulakis C, Mayer EA, and Chang L (2018). Sigmoid colon mucosal gene expression supports alterations of neuronal signaling in irritable bowel syndromewith constipation. Am. J. Physiol. Gastrointest. Liver Physiol 315, G140–G157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Boyen GB, Steinkamp M, Reinshagen M, Schäfer KH, Adler G, and Kirsch J (2004). Proinflammatory cytokines increase glial fibrillary acidic protein expression in enteric glia. Gut 53, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Boyen GB, Steinkamp M, Reinshagen M, Schäfer KH, Adler G, and Kirsch J (2006). Nerve growth factor secretion in cultured enteric glia cells is modulated by proinflammatory cytokines. J. Neuroendocrinol 18, 820–825. [DOI] [PubMed] [Google Scholar]
- Wangzhou A, Paige C, Neerukonda SV, Dussor G, Ray PR, and Price TJ (2020). A pharmacological interactome platform for discovery of pain mechanisms and targets. bioRxiv, 2020.2004.2014.041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz S, Popp V, Kindermann M, Gerlach K, Weigmann B, Fichtner-Feigl S, and Neurath MF (2017). Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc 12, 1295–1309. [DOI] [PubMed] [Google Scholar]
- Wynn G, Ma B, Ruan HZ, and Burnstock G (2004). Purinergic component of mechanosensory transduction is increased in a rat model of colitis. Am. J. Physiol. Gastrointest. Liver Physiol 287, G647–G657. [DOI] [PubMed] [Google Scholar]
- Xu S, Qin B, Shi A, Zhao J, Guo X, and Dong L (2018). Oxytocin inhibited stress induced visceral hypersensitivity, enteric glial cells activation, and release of proinflammatory cytokines in maternal separated rats. Eur. J. Pharmacol 818, 578–584. [DOI] [PubMed] [Google Scholar]
- Zwicker S, Martinez GL, Bosma M, Gerling M, Clark R, Majster M, Söderman J, Almer S, and Boström EA (2015). Interleukin 34: a new modulator of human and experimental inflammatory bowel disease. Clin. Sci. (Lond.) 129, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cytokine/chemokine multiplex data generated during this study are available in Table S3. The data for Figures S2 and S6A were sourced from a published (Delvalle et al., 2018) and publicly available dataset “Next generation sequencing of distal colon glial cells with DNBS-induced inflammation and neurokinin-2 receptor antagonism utilizing RiboTag mice” (Database: GSE114780).