

Abstract
Clinical translation of therapeutic peptides, particularly those targeting intracellular protein–protein interactions (PPIs), has been hampered by their inefficacious cellular internalization in diseased tissue. Therapeutic peptides engineered into nanostructures with stable spatial architectures and smart disease targeting ability may provide a viable strategy to overcome the pharmaceutical obstacles of peptides. This study describes a strategy to assemble therapeutic peptides into a stable peptide–Au nanohybrid, followed by further self-assembling into higher-order nanoclusters with responsiveness to tumor microenvironment. As a proof of concept, an anticancer peptide termed β-catenin/Bcl9 inhibitors is copolymerized with gold ion and assembled into a cluster of nanohybrids (pCluster). Through a battery of in vitro and in vivo tests, it is demonstrated that pClusters potently inhibit tumor growth and metastasis in several animal models through the impairment of the Wnt/β-catenin pathway, while maintaining a highly favorable biosafety profile. In addition, it is also found that pClusters synergize with the PD1/PD-L1 checkpoint blockade immunotherapy. This new strategy of peptide delivery will likely have a broad impact on the development of peptide-derived therapeutic nanomedicine and reinvigorate efforts to discover peptide drugs that target intracellular PPIs in a great variety of human diseases, including cancer.
Keywords: cancer targeted therapy, immunotherapy, peptide-Au nanohybrids, peptide-derived nanocluster, tumor microenvironment-responsiveness
1. Introduction
Intracellular protein–protein interactions (PPIs) is a crucial and yet inadequately exploited class of therapeutic targets for their essential roles in most of cellular processes and disease initiation and progression.[1] Small molecule drugs have been demonstrated to be highly successful in targeting enzymes, receptors, and ion channels for clinical benefits;[2] however, they generally failed to target PPIs due to their own physicochemical limitations.[3] Peptides, on the other hand, can potently and specifically inhibit PPIs, aided by their large interacting surface of diverse topological structure.[4] In spite of their prominent advantages over small-molecule inhibitors in targeting PPIs, peptide inhibitors suffer from two major pharmacological deficiencies—poor proteolytic stability and low membrane permeability, severely limiting their clinical use.[5]
To overcome these pharmacological hurdles of peptides and improve their therapeutic efficacy, two general strategies have emerged: 1) chemically modifying peptide backbone and/or side chains to enhance proteolytic resistance,[6] and 2) covalently or noncovalently loading peptide cargos to drug delivery vehicles for promoting cellular uptake.[5] The first strategy often entails the use of sidechain-stapled peptides, backbone-cyclized (circular) peptides, D-peptides, peptoids, beta-peptides, or peptide-grafted miniature proteins.[6,7] The second strategy is largely manifested by the development of various peptide drug carriers, including macromolecule micelles, liposomes, and nanomedicine.[5,8] While some success has been achieved in developing peptide therapeutics by using these two strategies,[4,5] significant challenges remain with regard to efficient and targeted delivery of therapeutic peptides into cells of diseased tissues such as solid tumors.
Nanoparticles are particularly attractive as anticancer peptide drug carriers because of their efficient cellular internalization and “enhanced permeability and retention (EPR)” properties.[9] Among various nanoparticles, gold nanoparticle (AuNP)-based nanocarriers have superior merits such as physicochemical stability, biocompatibility, and universality.[9c,10] Thus, AuNP-based therapies have been widely used in clinical trials, and some of them have been approved for clinical use.[11] However, current chemistries for peptide conjugation and encapsulation by nanoparticles often lead to product aggregation and precipitation in aqueous solution, thereby resulting in low drug loading efficiency and failure of delivery.[5,12] Another technical challenge is to ensure efficient spatiotemporal release of peptide cargos at the disease sites.[4,13] Overall, these technical obstacles, if not overcome, will potentially negate therapeutic efficacy and clinical safety of peptide-derived nanomedicine.[13]
B-cell CLL/lymphoma 9 (Bcl9), a transcriptional cofactor overexpressed in human cancers, activates the Wnt/β-catenin signaling via interacting with β-catenin to facilitate its nuclear translocation, thereby promoting tumor progression.[14] Despite the critical role of Wnt/β-catenin pathway in tumorigenesis, it remains a challenge to therapeutically intervene this pathway for clinical benefits because it is also required for the proliferation and differentiation of stem cells.[15] Thus, compounds or modalities that target the Wnt/β-catenin pathway with high specificity for cancer cells will be of great clinical significance.
Toward these ends, we nanoengineered therapeutic peptides into a stable peptide–Au nanohybrid in which the polymeric Au–peptide precursor [Au(I)-S-peptide]n was reduced and assembled into a auric-sphere particle with narrow distribution at low nanoscales. Notably, the peptides themselves were utilized as one of the building blocks of the drug delivery system, thereby greatly improving the loading efficiency. Moreover, to endow delivery system with high targeting ability to tumor, Au–peptide nanohybrid (nanoparticle) was assembled into a tumor microenvironment (TME)-responsive nanocluster with switchable charge and size. In this proof-of-concept study, we copolymerized HAuCl4 and a peptide inhibitor of the β-catenin–Bcl9 interaction to form Au–peptide nanohybrid that self-assembled into higher-order nanoclusters, termed pCluster. The otherwise highly stable pCluster was pH responsive and disintegrated into nanoparticles only in the acidic TME, thereby leading to efficient and tumor-specific cellular uptake of peptide cargos.pCluster potently inhibited not only the growth of various cancer cell lines in vitro via blocking Wnt pathway, but also suppressed tumor growth and metastasis, and synergized with PD1/PD-L1 checkpoint blockade immunotherapy in animal models. Importantly, it maintained a highly favorable biosafety profile. This viable strategy reported here will enable us to develop a class of peptide-derived nanomedicines for cancer therapy, and likely reinvigorate peptide drug discovery efforts for a great variety of diseases including cancer.
2. Results
2.1. Design of Therapeutic Peptides Targeting the Wnt/β-catenin Signaling
β-catenin, the primary signal transducer in the Wnt pathway, is important for embryonic development, tumorigenesis, and tumor immunology.[16] The dysregulation of the Wnt signaling is considered as a major driving force in the progression of many diseases including malignancies.[17] During tumorigenesis, β-catenin interacts with a cofactor Bcl9, shuttling it into nucleus to activate its downstream targets.[18] Thus, the development of β-catenin/Bcl9 inhibitors (BBIs) is attractive for cancer therapy. As the complete and detailed structure of β-catenin/Bcl9 complex provided in Figure S1A (Supporting Information), three hydrophobic and two charged residues in a short peptide termed Bcl9 can interact with a hydrophobic groove in β-catenin.[19] This helix consequently has great potential to be used as a BBI to block the activity of the Wnt signaling (Figure S1A, Supporting Information). To prove this, we synthesized the BBI (351–357Bcl9), as shown in Figure S1B (Supporting Information), and found that it effectively binds to β-catenin with an affinity value of ≈1 × 10−6 M (Figure S1C,D, Supporting Information). In addition, using fluorescence polarization-based competitive binding assay, we demonstrated that the BBI dramatically competed with Bcl9 to bind β-catenin (Figure S1E, Supporting Information). Linking to a cell-penetrating peptide (RRRRRRRR, named R8), BBI-R8 exhibited strong ability to inhibit the proliferation of a hyperactivated-Wnt-signaling cell line Hep3B (Figure S1F, Supporting Information). However, the BBI alone almost did not affect cell proliferation (Figure S1F, Supporting Information), which is presumably attributed to the membrane impenetrability and biologically instability of peptides.[5,20] Collectively, these results indicate that BBI is capable of suppressing the proliferation of cancer cells through blocking the interaction between Bcl9 and β-catenin. Therefore, the BBI peptide may be an ideal example to synthesize cytostatic nanoclusters for cancer therapy.
2.2. Preparation of TME-Responsive pCluster
pClusters were prepared by a three step “one pot” reaction, which proceeds in good yield under mild conditions. In details, 1) the flavescent Au3+ was reduced by the thiol in BBI-SH to form a polymeric structure of [BBI-S-Au(I)]n, termed p(BBI-SH-Au1+); and then, 2) p(BBI-S-Au1+) was reduced to a polymeric Au–peptide nanoparticle (pParticle) as described previously;[21] finally, 3) a cationic polymer poly-L-lysine (PLL) was added to trigger the self-assembly of pParticle into the massive pCluster (Figures 1 and 2A).
Figure 1.
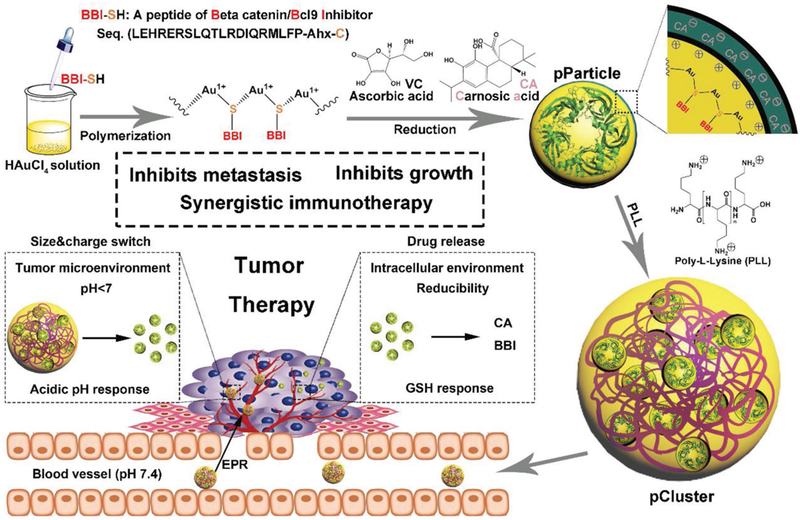
Schematic depiction for synthesis and function of pCluster.
Figure 2.
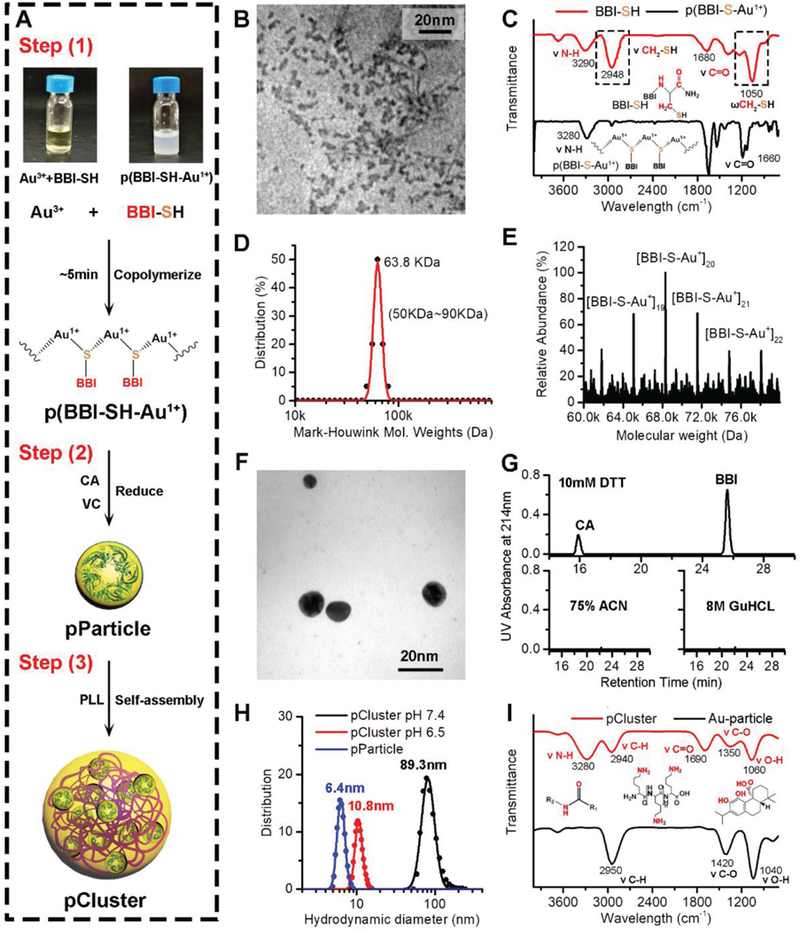
Preparation and characterization of pCluster. A) Schematic depiction for the three-step synthesis of pCluster. B) TEM image of p(BBI-SH-Au+), presenting an irregular amorphous form. C) FTIR spectra of free peptide (BBI-SH) and Au–BBI polymeric structure (p(BBI-S-Au1+)). D) Molecular weight distribution of the [BBI-S-Au+]n polymers measured by the Mark–Houwink–Sakurada method, which uses empirical constants to calculate the molecular weight from the diffusion coefficient determined from the autocorrelation function of the scattered light (DLS). E) Molecular weight measured by ESI mass of the p(BBI-SH-Au+) species. F) TEM image of pParticle. G) HPLC analysis of the pParticle after a 4 h incubation with 10 × 10−3 M dithiothreitol (pH 6.5), 75% acetonitrile (25% water), or 8 M guanidine hydrochloride. pParticle was separated by separation, and the supernatant was detected by HPLC. H) Hydrodynamic distributions of pParticle, pCluster at pH 6.5, and pCluster at pH 7.4 measured by dynamic light scattering, showing that pParticle has a narrow size distribution, and pCluster has a size-switchable property responded to the acidic pH. I) Fourier transform infrared spectra of pCluster and Au-particle, demonstrating the correct chemical structures of pCluster.
After step (1), structural characterization was attempted by performing transmission electron microscopy (TEM) image, in which the p(BBI-S-Au1+) presented an irregular amorphous form under 5 nm (Figure 2B). The formation of polymeric structure was further confirmed by the fourier transform infrared spectra (FTIR), and all absorption peaks of free thiol in BBI-SH spectroscopy disappeared in the p(BBI-S-Au1+) spectroscopy (Figure 2C), indicating that Au+1 ions were bridged by the thiolate sulfur atom of BBI-SH with a coordination number of 2. Meanwhile, molecular weight of the p(BBI-S-Au1+) was systematically investigated by the Mark–Houwink–Sakurada method[22] (Figure 2D) and electrospray ionization mass spectrometry (ESI-MS) (Figure 2E), and several species with masses from 50 K ([BBI-S-Au+]18) to 80 K ([BBI-S-Au+]23) were obtained through the above methods.
During step (2), the solution containing carnosic acid (CA) and ascorbic acid (VC) decomposed the polymeric structure to form the ≈6 nm Au–peptide nanoparticle with a narrow size distribution, termed pParticles (Figure 2F and Figure S2A, Supporting Information). To confirm that the BBI-SH was covalently coalesced into the nanoparticle rather than adsorbed on the surface, pParticles were solved into 75% acetonitrile (ACN) or 8 M GuHCL, respectively, both of which had strong solubility of peptides and could elute the peptides from the solid particles. As shown in Figure 1G, no peptide was found in the eluant of 75% ACN or 8 M GuHCl, while dithiothreitol (DTT), a strong reductant, triggered the release of BBI from pParticle. These data indicate that peptides are covalently bonded to nanoparticles rather than adsorbed on the surface. To further verify it, the TEM image of the pParticles solved in the DTT solution (10 × 10−3 M in phosphate buffered saline (PBS) pH 6.5) were taken (Figure S3A, Supporting Information), in which the decomposed pParticles showed out-of-shaped low-density shadow like the TEM image of free BBI-Sh and CA (Figure S3B, Supporting Information), other than the granuliform Au particle. Taken together, these results demonstrate that pParticle is a uniform spheroidal auric–peptide nanohybrid with narrow size distribution. To prove that the bioactivity of BBI can be maintained after the polymerization, a BBI-free homologous nanohybrid, termed CtrlpParticle, was prepared to compare its antitumor activity with pParticle. As shown in Figure S2B (Supporting Information), pParticle showed stronger in vitro antitumor activity than CtrlpParticle, further supporting antitumor bioactivity of BBI.
To endow nanoparticles with high targeting ability to tumor, pParticles were assembled into a size-switchable and pH-sensitive nanocluster (pCluster) at step (3). During the synthesis of pCluster, a cationic polymer PLL (200 × 10−6 M, pH 7.4) was added dropwise to the solution of pParticle. Self-assembled pClusters with a spherical shape were formed within few minutes due to the interaction between the protonated amine group in PLL and the carboxyl group of CA and BBI. To obtain the most appropriate size and pH sensibility, we used different ratios of CA:BBI to synthesize pCluster, and found that the ratio of CA:BBI = 2:1 was the most viable (Table S1, Supporting Information). Based on this ratio, we measured the size of pCluster by dynamic light scattering (DLS), and determined that its diameter was around 89.3 nm (Figure 2H), which is a feasible size for EPR effect. This was also supported by the TEM observation (Figure S4A, Supporting Information). For comparison, we also prepared the BBI- and CA-free nanoparticle, termed Au-particle. As shown in Figure 1I, FTIR spectroscopy proved successful synthesis of pCluster as evidenced by characteristic absorbance given by BBI, CA, and PLL. In ultraviolet–visible spectra (Figure S4B, Supporting Information), compared to the pParticle, a red shift of characteristic peak in pCluster curve from 540 to 555 nm could be observed, indicating the larger size of pCluster. Taken together, these results strongly support that peptide, CA, and PLL are successfully conjugated to pParticles.
In general, nanoparticles with the size from 60 to 100 nm always possess drawn-out blood circulation and tend to extravasate across the faulty tumor vasculature but enrich at the vicinity of blood vessels rather than distribution in the dense tumor matrix.[23] Although pony-sized nanoparticles (<15 nm) can deeply penetrate into the tumor and extensively distribute in the tumor matrix, they have to suffer from the stochastic diffusion into noncancerous tissue.[23] Thus, to obtain tumor-specific extravasation, pCluster is supposed to maintain the large size in the circulation, while will transform to small size in the tumor acid microenvironment. To prove it, pClusters were incubated in the tumor-acidity mimic buffer (10 × 10−3 M PBS at pH 6.5) for 4 h, following the measurement of surface charge, hydrated particle size, and TEM morphology. Expectedly, the ZETA potential of pClusters reversed from −4.7 mV at pH 7.4 to 30.6 mV at pH 6.5 (Figure S4C, Supporting Information), which was probably ascribed to the protonation of CA and BBI-SH (Figure S4D, Supporting Information). Aided by this charge reversal, pCluster disassembled into small nanoparticles with hydrated diameter of 10.8 nm (Figure 1H), which was in line with the TEM image (Figure S4A, Supporting Information).
The pH-responded pClusters were also sensitive to the elevated intracellular redox milieu, which would endow them with an intracellularly glutathione (GSH)-triggered drug release. To verify it, the high-performance liquid chromatography (HPLC) was used to monitor drug release behaviors after incubation with three kinds of conditions: 1) 10 × 10−3 M PBS at pH 7.4, to mimic normal physiological environment; 2) 10 × 10−3 M PBS at pH 6.5, to mimic acidic TME; and 3) 10 × 10−3 M PBS at pH 6.5 containing 10 × 10−3 M GSH, to mimic intracellular reductive environment at tumor sites. After a 4 h incubation at pH 7.4, pCluster maintained its integrity and stability with <10% CA and <1% BBI-SH release (Figure S4E, Supporting Information). Adjusting pH to 6.5 resulted in the partial CA release (<30% after 4 h), whereas no obvious BBI-SH release was observed under this condition (Figure S4E, Supporting Information). In a sharp contrast, when pClusters were incubated in intracellular redox environment mimic solution, nearly all of the BBI and CA were totally released within an additional 4 h (Figure S4E, Supporting Information).
2.3. Tumor-Specific Accumulation and TME-Triggered Cellular Uptake of pClusters
Small nanoparticles with positive charge can electrostatically attract to the negatively charged cell membrane, thereby triggering cellular internalization.[24] By contrast, large negatively charged particles always suffer from the steric hindrance and charge repulsion, resulting in the failure of cellular internalization.[25] Given the size-convertible and charge-reversible behaviors of pClusters in response to the acidity (Figure 2H and Figure S4D, Supporting Information), the TME switches the pCluster from cellular rejection to cellular internalization (Figure 3A). To prove it, pClusters were labelled with Texas Red, and their cellular internalization was evaluated in Hep3B and HCT116 cells. At pH 7.4, pClusters showed very low intracellular fluorescence in both Hep3B and HCT116 cells (Figure 3B), while cellular internalization was dramatically increased in these cells when pClusters were incubated at pH 6.5 (Figure 3B). These results were proved again by quantitative flow cytometry (Figure 3C). To determine cellular internalization pathway, HCT116 cells were first coincubated with fluorescein isothiocyanate (FITC)-labeled pClusters at 37 or 4 °C (pH 6.5) for 4 h. The results showed that 4 °C incubation completely suppressed cellular internalization of pClusters (Figure S5A,B, Supporting Information), suggesting their cellular uptake is mediated by a adenosine triphosphate (ATP)-dependent pathway. Next, two specific inhibitors of micropinocytosis, Amiloride (targeting to cytoskeleton) and cytochalasin D (targeting to actin), were used to pretreat HCT116 cells. As expected, both of them abolished cellular internalization of pClusters (Figure S5C,D, Supporting Information), further demonstrating that pH-responded pCluster can effectively enter cancer cells as a result of micropinocytosis. Collectively, these findings indicate that pH-responded pCluster internalizes into cells via ATP-dependent micropinocytosis pathway.
Figure 3.
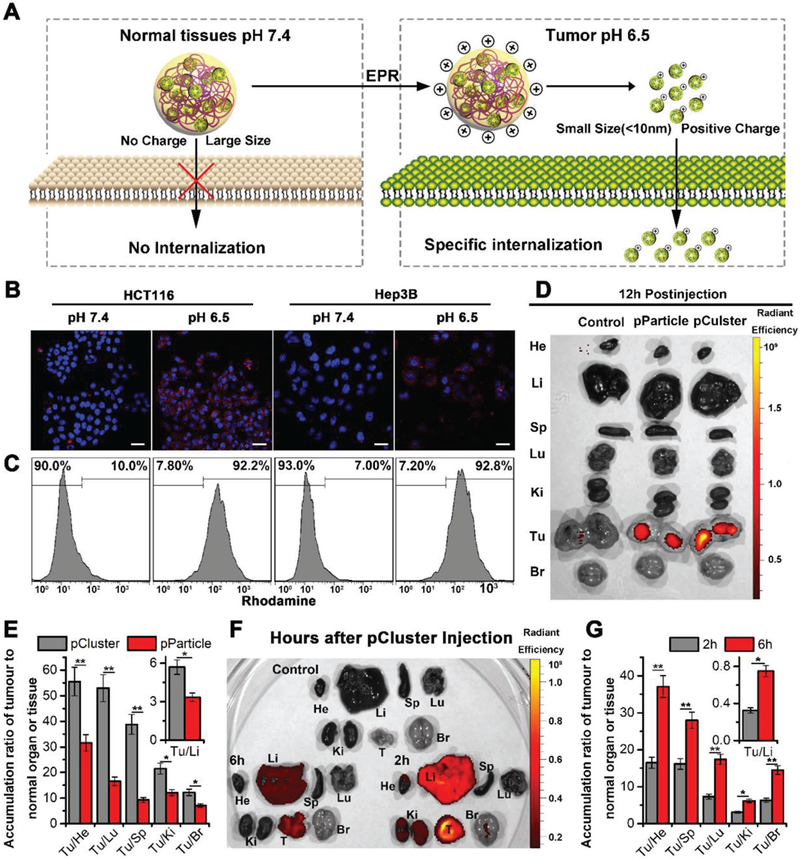
High tumor selectivity of pCluster. A) Schematic depiction for tumor targeting of pCluster. B) Confocal laser scanning microscope (CLSM) images of HCT116 and Hep3B cells after a 6 h incubation with pCluster (20 μg mL−1) at pH 7.4 or pH 6.5, all images were taken under the same exciting light and detector gain (scale bar: 60 μm). C) Cellular uptake of pCluster responded to acidic pH by flow cytometry analysis. D) Ex vivo fluorescent images of tumors and major organs from pParticle- and pCluster-treated mice at 12 h postinjection. He, heart; Li, liver; Sp, spleen; Lu, lung; Ki, kidneys; Tu, tumor; Br, Brain. E) Tumor-to-background (normal organ or tissue) ratios for pParticle and pCluster at 12 h postinjection. pCluster showed much better tumor selectivity than pParticle (n = 3, mean ± s.d.). F) Ex vivo fluorescent images of tumors and major organs from pCluster treated mice at 2 and 6 h postinjection. G) Tumor-to-background (normal organ or tissue) ratios for pCluster at 2 and 6 h postinjection. The latter showed significantly better tumor selectivity than the former (n = 3, mean ± s.d.). p values were calculated by t-test (*p < 0.05; **p < 0.01).
Besides, small size of pH-responded pClusters is supposed to be in favor of the penetration and distribution in the tumor site. To verify this, tumor masses were isolated and incubated with FITC-labeled pCluster at pH 7.4 or 6.5 for 4 h. After fully washing, tumor masses were observed under fluorescence microscope. The results showed that fluorescence could only be detected in the internal area of tumor mass at pH 6.5, while almost did not observe fluorescence signal in intratumoral region at pH 7.4 (Figure S6, Supporting Information), indicating that small nanoparticles are favorable for tumor penetration and distribution.
More importantly, combined TME response with inherent EPR effect, pClusters could selectively enrich in the tumor sites. Via EPR effect, nanoparticles <150 nm can extravasate from the defective “leaky” tumor vasculature and accumulate in cancer cells.[25] In addition, ineffective lymphatic drainage is always accompanied by the increased tumor volume, resulting in the subsequently promoting nanoparticles retention.[26] Thus, to assess biodistributions of pClusters and pParticles in healthy organs and tumor, near-infrared fluorophore Texas Red was conjugated to pClusters and pParticles. As shown in Figure 3D, compared to pParticles, pClusters exhibited a higher accumulation at the tumor site 12 h after injection relative to other organs. Through further analyzing this fluorescence distribution, we found that all ratios of tumor to noncancerous organ for pClusters were statistically significantly higher than that for pParticles (Figure 3E), indicating that pH-sensitive properties improved tumor specificity of pClusters. Biodistributions of pClusters were also examined at 2 and 6 h postinjection (Figure 3F). As shown in Figure 2G, all ratios of tumor to noncancerous organ at 6 h postinjection were significantly higher than the ratios at 2 h postinjection, further supporting pClusters with high targeting ability to tumor. Notably, we almost did not observe the accumulation of pClusters in liver and kidney at 12 postinjection (Figure 3D), demonstrating pClusters with high metabolizability rate.
2.4. pCluster Potently Inhibits Tumor Growth In Vitro and In Vivo
Bcl9 is highly expressed in human cancer cells, and facilitates β-catenin entry into the nucleus, promoting tumor progression by activating Wnt pathway.[27] By our design, BBI can efficiently inhibit this process via blocking the interaction between Bcl9 and β-catenin (Figure S7A, Supporting Information). In addition to being a reductant for pCluster synthesis, CA is also an effective small molecule inhibitor targeting the Wnt signaling through inducing cytoplasmic β-catenin degradation (Figure S7A, Supporting Information).[28] Thus, pCluster is a multiple stimuli-responsive synthetic cytostatic with two different functions to block the Wnt/β-catenin pathway in cancer cells (Figure 4A). First, the thiazolyl blue (MTT) assay was performed to assess the effect of pClusters on the proliferation of three Wnt-activated cancer cell lines: Hep3B (human hepatoma cell line), HCT116 (human colorectal cell line), and F16B10 (murine melanoma cell line) cells. As shown in Figure 4B and Figure S7B–D (Supporting Information), pCluster, pParticle, and CA inhibited the proliferation of these cells in a dose-dependent manner, while Au-Cluster as a control almost did not affect cell proliferation. Notably, pCluster was more potent than CA and pParticle in these three Wnt-activated cancer cell lines (Figure S7B–D, Supporting Information), while it did not show any advantage of inhibitory proliferation in a Wnt-latent lung adenocarcinoma cell line A549 compared to pParticle (Figure S7E, Supporting Information). There is evidence demonstrating that blockade of β-catenin–Bcl9 interaction by CA or peptide inhibitors can result in the instability of β-catenin and subsequent degradation of β-catenin.[20,28] Expectedly, our data showed that β-catenin was significantly decreased upon treatment of pCluster, pParticle, and 10 × 10−6 M CA relative to the control in both Hep3B and HCT116 cells (Figure 4C and Figure S8, Supporting Information), indicating the successful disruption of β-catenin–BCL9 interaction. In line with β-catenin, Cyclin D, a downstream target of the Wnt/β-catenin signaling pathway,[29] was significantly down-regulated by these β-catenin/Bcl9 inhibitors, further proving the bioactivity of these β-catenin antagonists. In addition, given that Cyclin D acts as a cell cycle regulatory factor, we expectedly found that pParticle, CA, and pCluster significantly induced G0/G1 phase arrest in Hep3B and HCT116 cells but not A549 cells compared to Au-Cluster, particularly pCluster (Figure 4D and Figure S9, Supporting Information).
Figure 4.
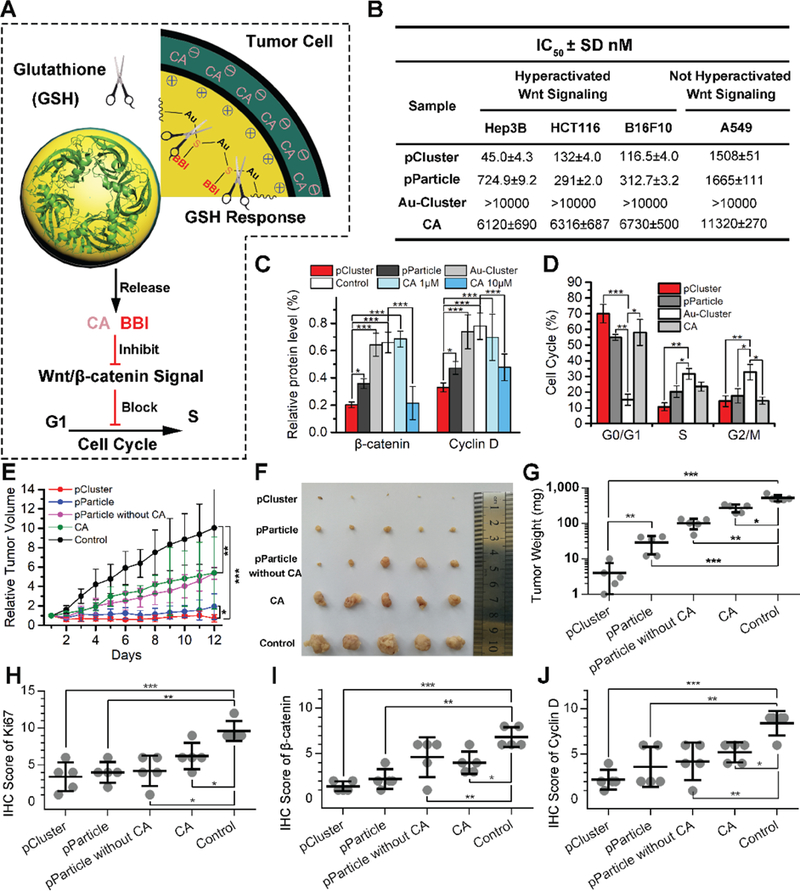
Antitumor activity of pCluster. A) Schematic depiction of GSH-responded drug release and biological functions of pCluster. B) Effects of pCluster, pParticle, Au-Cluster, and carnosic acid (CA) on the growth of Hep3B, HCT116, B16F10, and A549 cells measured by MTS assay (mean ± s.d., n = 4). C) Western blot analysis was performed to monitor the level of Cyclin D and β-catenin in Hep3B cells with the indicated treatments. All results were performed in triplicate and normalized by the loading control, β-actin. D) Cell cycle distributions were analyzed by flow cytometry in Hep3B cells with the indicated treatments for 24 h (n = 3, mean ± s.d.). E) Tumor growth curve of volume according time during the administration (mean ± s.d., n = 5). Statistically significant differences were judged by Kruskal–Wallis test. F) Photographs and G) weight of tumors collected from mice after 12-day administration. Immunohistochemical (IHC) scores of intratumoral protein levels of H) Ki67, I) β-catenin, and J) Cyclin D. p values were calculated by t-test (*p < 0.05; **p < 0.01; ***p < 0.001).
To test in vivo antitumor activity of pCluster, nude mice bearing Hep3B xenograft tumors were used to investigate therapeutic efficacy of our nanoclusters. As shown in Figure 3E, tumor volume in the PBS-treated (control) group increased rapidly during the 12-day treatment, while CA and pParticle without CA moderately inhibited tumor growth. By a sharp contrast, pParticle with CA displayed much higher tumor-inhibitory effect than CA alone or pParticle without CA, indicating that combination of CA and BBI induces synthetic cytostatic effect on tumor growth. Importantly, pCluster exhibited better anti-tumor activity than pParticle (Figure 4E), further demonstrating the superiority of TME-responsive behaviors. At the 12th day, xenograft tumors were isolated and weighed (Figure 4F,G). Statistical data on tumor weight further supported the above conclusions. Next, we performed the immunohistochemical (IHC) assay to measure the levels of Ki67 (a well-known proliferative marker) and evaluate the proliferative ability of tumor. As shown in Figure 4H and Figure S10 (Supporting Information), it can be found that the percentage of Ki67-positive cells in the pCluster-treated tumors was much lower than that in the other tumors. In addition, we expectedly found a significant decrease in β-catenin levels in the pCluster-treated tumors compared to the other tumors by IHC assay (Figure 4I). Meanwhile, we also found that pCluster treatment obviously decreased nuclear β-catenin and increased membranous β-catenin, especially at cell junctions (Figure S10, Supporting Information). Accordingly, there was a significant decrease in Cyclin D levels in the pCluster-treated tumors relative to the other tumors (Figure 4J). Collectively, as an efficient inhibitor of the Wnt signaling, pCluster extremely has the potential to be an applicable nano-therapeutic for anticancer therapy.
2.5. pCluster Maintains a Highly Favorable Biosafety Profile
In the last 30 years, although there have been many groundbreaking discoveries of the conserved Wnt signaling pathway,[30] none of Wnt-targeted therapeutic has yet been approved for clinical application.[15] The major cause is severe side effects from these Wnt inhibitors because blockade of this pathway can also impair physiological functions of normal cells, such as tissue homeostasis and repair.[15] Thus, it will require an accurate act, by which the Wnt signaling can be precisely abrogated at the tumor sites, while did not interfere its physiological roles in normal cells. To this end, a double-targeting strategy was adopted in our design to guarantee tumor enrichment other than normal organs (Figure 5A). As mentioned above, aided by the EPR effect and TME response, pClusters can specifically accumulate in the tumor site, thereby achieving a switchable cellular uptake in response to TME. Thus, this two-level-targeting strategy will greatly reduce the risk of side effects from Wnt inhibitors.
Figure 5.
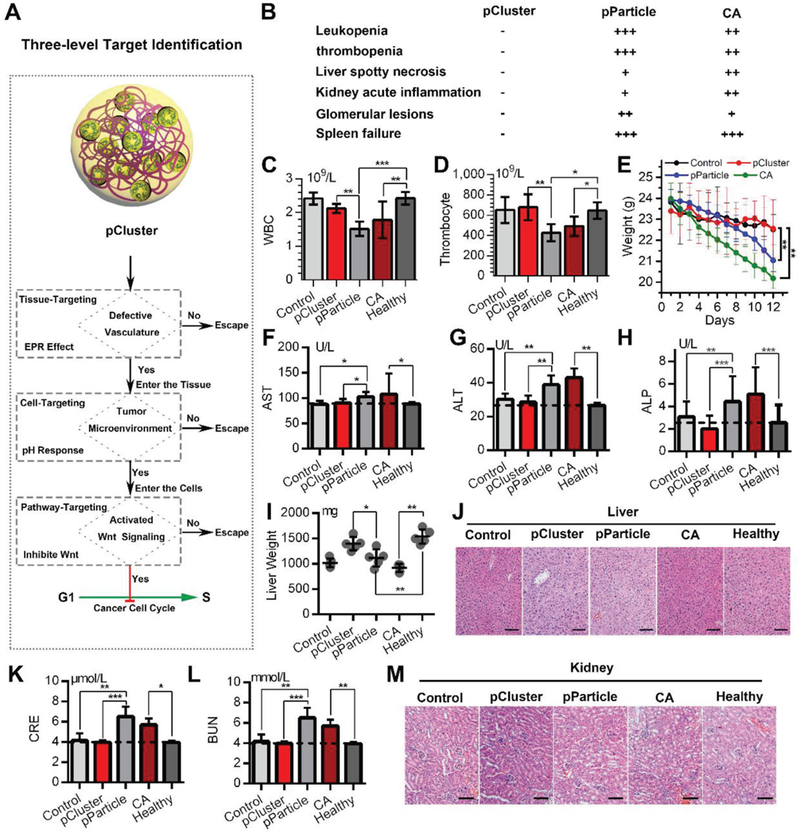
Biosafety evaluation of pCluster. A) Schematic diagram for the safety of cancer therapy benefitted from three-level target identification of pCluster. B) Disease diagnosis in mice with the indicated treatments. C,D) The count of white blood cells (WBCs) and thrombocyte in mice with the indicated treatments. E) The time course of animal weight in each group with the indicated treatments. F–H) The activities of three liver enzymes related to liver function in mice with the indicated treatments. AST: aspartate aminotransferase; ALT: alanine transaminase; ALP: alkaline phosphatase. I) The changes in liver weight of mice with the indicated treatments. J) The representative images of H&E-stained liver sections in mice with the indicated treatments (scale bar: 50 μm). K,L) The measurement of renal function indicators in mice with the indicated treatments. CRE: serum creatinine; BUN: blood urea nitrogen. M) The representative images of H&E-stained kidney sections in mice with the indicated treatments (scale bar: 50 μm). Data were presented as mean ± s.e. p values were calculated by t-test (*p < 0.05; **p < 0.01; ***p < 0.001).
To validate the biosafety of pCluster, we first determined the effect of pCluster on the viability of the peripheral blood mononuclear cells (PBMCs). Expectedly, we did not observe cytotoxic effect of pCluster on PBMCs at pH 6.5 and pH 7.4 (Figure S11, Supporting Information). To further determine the biosafety of pCluster, a series of biosafety assessments were performed during and after pCluster treatment. During administration, pParticle- and CA-treated mice suffered a series of side effects including severe hemocytosis and organic damage, while pCluster-treated mice remained in perfect health (Figure 5B). In details, pParticle- and CA-mice showed low levels of white blood cells and platelets (Figure 5C,D), and rapid loss of body weight relative to control or healthy mice (Figure 5E). In addition, CA and pParticle treatment resulted in changes of liver function and pathology (Figure 5F–J), the symptoms of acute kidney inflammation and glomerular lesions (Figure 5K–M), and spleen failure (Figure S12A,B, Supporting Information). By a sharp contrast, none of these hemocytosis and organic damage was observed in all of the pCluster-treated mice (Figure 5B–M), indicating the favorable biological reliability of pCluster. Notably, all pCluster-, pParticle-, and CA-treated mice did not encounter common side effects of chemotherapeutics, including heart failure (Figure S12C,D, Supporting Information), acute sepsis (Figure S13A,B, Supporting Information), or allergic lung resistance (Figure S13C,D, Supporting Information). Altogether, our data demonstrate that pCluster is avirulent enough for clinical use.
2.6. pCluster Effectively Suppresses Tumor Metastasis
Tumor metastasis is a major determinant of poor patient survival, accounting for 90% of cancer-related death.[31] The Wnt/β-catenin signaling pathway is always hyperactivated during tumor metastasis.[32] Upon activated Wnt signaling, β-catenin can be shuttled by Bcl9 into nucleus to promote tumor cell proliferation and metastasis.[33] With our design, pCluster can effectively block the activity of Wnt/β-catenin pathway, thereby suppressing tumor metastasis (Figure 6A). Cell migration and invasion are two key properties associated with tumor metastasis. Therefore, we first evaluated the effect of pCluster on in vitro migration and invasion abilities of HCT116 and F16B10 cells. Upon a 24 h incubation with 100 × 10−9 M pClusters, the number of migrating/invading cells were significantly less than the control (Figure 6B,C) in both of these two cell lines.
Figure 6.
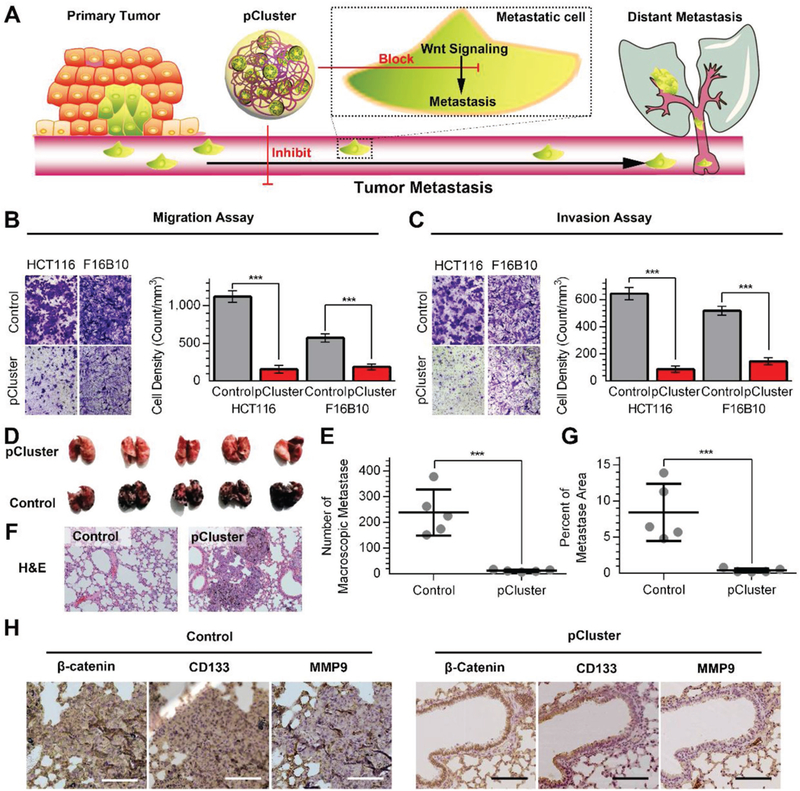
Inhibitory effect of pCluster on tumor metastasis. A) Schematic diagram of pCluster suppressing tumor distant metastasis. B,C) The representative pictures of migrated/invaded cells (left panels). Histograms, corresponding to left panels, show means ± s.d. of cell numbers from three independent assays (right panels). D) The representative photographs of lungs taken 15 days after injection of B16F10 cells. Macroscopic analysis of the lungs confirmed that pCluster decreased the lung colonization by B16F10 cells. E) The number of superficial macroscopic metastases in the lungs (n = 5). F) The representative photographs of the H&E-stained tissue sections of pulmonary metastases. G) The percentage of pulmonary metastatic area analyzed from (F) (n = 5). H) The representative IHC staining of β-catenin, CD133, and MMP9 for three serial sections from pulmonary metastasis tumors (scale bar: 100 μm). p values were calculated by t-test (***p < 0.001).
To determine antimetastasis activity of pCluster in vivo, B16F10 cells (5 × 104 per mouse) were injected into the caudal vein of C57 mice to construct lung metastasis model. From the 4th day after cell injection, mice were consecutively administered pClusters for 14 days at a dose of 1 mg per kg per two days. After administration, lung metastasis was significantly inhibited upon pCluster by either intravenous injection or intraperitoneal injection compared to the control (Figure S14, Supporting Information). To further determine the effect of pCluster on the metastatic tumors at both tissue and protein levels, 5 × 105 B16F10 cells were injected into the caudal vein of each mice to cause more severe pulmonary metastasis. As shown in Figure 6D,E, we indeed found more metastatic nodules in the lung of control mice, and demonstrated that lung metastasis of B16F10 cells was dramatically inhibited by pClusters. This was also supported by the data from the H&E-stained tissue sections (Figure 6F) and metastatic area analysis (Figure 6G). To elucidate the mechanism, the levels of β-catenin and its downstream metastasis-related proteins such as CD133 and matrix metallopeptidase 9 (MMP9) were measured by the IHC assay. In general, β-catenin positively feedbacks to CD133 in tumorigenesis, thereby inhibiting cell differentiation and subsequently showing the properties of cancer stem cells.[34] In addition, the Wnt/β-catenin cascade can also regulate the expression of MMP9, which is closely associated with tumor metastasis.[35] As expected, the expression of β-catenin, CD133, and MMP9 in metastatic lesions was decreased upon pCluster treatment in Figure 6H. Collectively, our data demonstrate that pClusters strongly inhibit tumor metastasis in vitro and in vivo through blocking Wnt/β-catenin signaling.
2.7. pCluster Synergizes the PD-L1 Checkpoint Blockade Immunotherapy
PD-1/PD-L1 signaling pathway induces the cytotoxic T lymphocytes (CTL) exhaustion, as a result of tumor immune evasion.[36]In recent years, PD-1/PD-L1 checkpoint-blockade strategies have been approved to enhance antitumor immunities by inhibiting CTL exhaustion;[37] however, only a fraction of patients respond to this treatment. There is evidence showing that treatment outcome is merely achieved in cancerous persons with a high number of infiltrated CTL cells within the TME.[38] Unfortunately, intratumoral active Wnt/β-catenin cascade can block T-cell infiltration, thereby resulting in the resistance to PD-1/PD-L1-derived immunotherapy.[39] Encouraged by potent inhibition of Wnt/β-catenin signaling by pCluster, we hypothesize that pCluster treatment may eliminate the resistance and has a synergistic effect with PD-L1 checkpoint blockade (Figure 7A, left panel).
Figure 7.
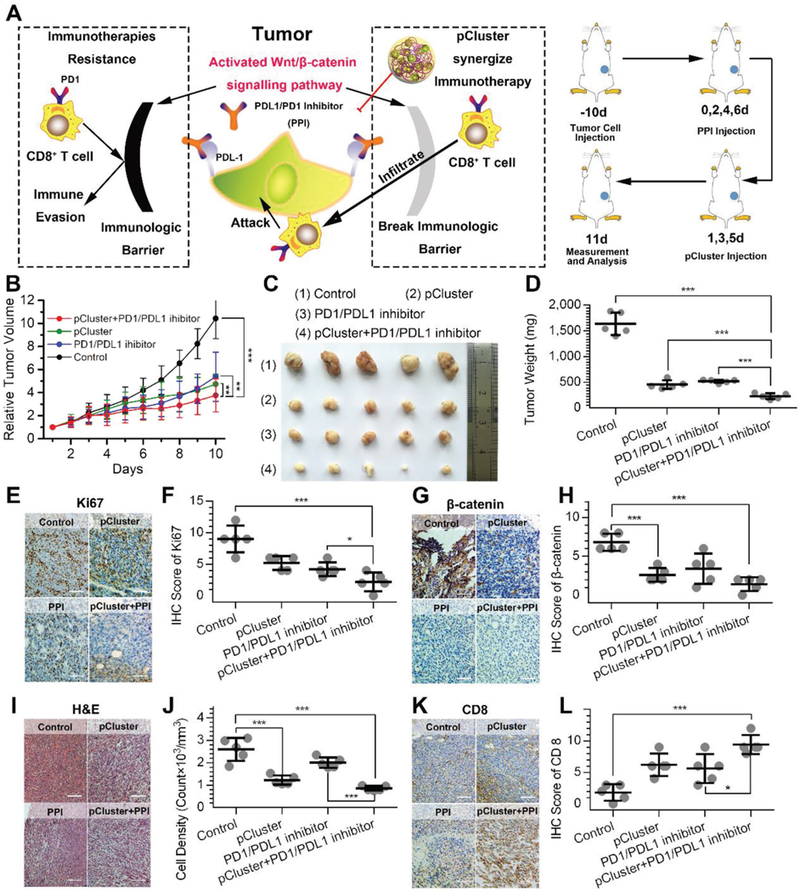
Synergistic effect of pCluster on the PD-L1 checkpoint blockade immunotherapy. A) The proposed mechanism of pCluster synergizing anti-PD-L1 immunotherapy. B) Tumor growth curves in mice with the indicated treatments. A statistical analysis was performed using a nonparametric Kruskal–Wallis test. Data were presented as mean ± s.e. (n = 5). C) Photographs and D) weight of xenograft tumors collected from mice with the indicated treatments. E) Representative IHC staining and F) IHC score for Ki67 in tumor tissues from mice with the indicated treatments (scale bar: 50 μm). G) Representative IHC staining and H) IHC score for β-catenin in tumor tissues from mice with the indicated treatments (scale bar: 50 μm). I) Representative H&E staining and J) tumor cell density of tumor tissues from mice with the indicated treatments (scale bar: 50 μm). K) Representative IHC staining and L) IHC score for CD8 positive T cells in tumor tissues from mice with the indicated treatments (scale bar: 50 μm). p values were calculated by t-test (*p < 0.05; **p < 0.01; ***p < 0.001).
To prove this, a MC38 homografting colon cancer model was established at the flank of normal C57 mice (Figure 7A, right panel). After the tumors reaching 50–80 mm3, all mice were randomly divided into four groups (five mice per group): 1) PBS (Control); 2) pCluster treatment; 3) PD1/PDL1 inhibitor treatment; 4) pCluster plus PD1/PDL1 inhibitor (PPI) treatment. As expected, combined treatment of pCluster and PPI synergistically inhibited tumor growth compared to PPI treatment alone (Figure 7B–D). This was also supported by the results of Ki67-staining in the tumors with the indicated treatments (Figure 7E and F). In addition, we also found that the levels of β-catenin were significantly decreased upon pCluster treatment alone and combined treatment relative to the control (Figure 7G,H), indicating successful blockade of Wnt/β-catenin signaling. Importantly, the inhibition of Wnt/β-catenin pathway caused a significant decrease in tumor cell density (Figure 7I,J), thereby eliminating the barrier of CTL infiltration. Next, we attempted to confirm the elicitation of CTL (CD8+ T-cell) infiltration by pCluster treatment. As shown in Figure 7K,L, the CTL was remarkably enriched in the tumors with combined therapy (group 4) compared to those with PPI monotherapy (group 3). Taken together, our data demonstrate that pCluster can reverse the immunosuppression caused by activated Wnt/β-catenin signaling to synergize the PD-1/PD-L1 checkpoint-blockade therapies, ultimately improving the efficacy of anticancer therapy.
3. Discussion
Human diseases including malignancies are generally caused by aberrant PPIs, either through the dysfunction of essential interactions or the structural variation of protein complex.[40] The PPI modulation can correct aberrant function of the protein complex, thereby paving the way to develop clinical diagnostics and therapeutics.[6] Compared to the interference of gene transcription by siRNA, PPI modulation can keep the normal function of target protein other than destroy the whole protein, thereby greatly reducing possible side effects from the deficiency of target protein. Considering that β-catenin is required for many normal physiological processes in especial of stem cell proliferation and differentiation,[30] thus disturbing β-catenin expression at transcriptional levels will result in the maladjustment of these β-catenin-dependent physiological processes. However, the interaction between β-catenin and Bcl9 specifically occurred in cancer cell,[41] thus modulating β-catenin–Bcl9 interaction other than disturbing β-catenin transcription would be a class of safe anticancer therapy. However, it is extremely difficult to target β-catenin–Bcl9 interaction because of their high, flat, and featureless surface area. While small molecule inhibitors have been proven successful in targeting enzymes, receptors, and ion channels for clinical applications, they are not applicable to modulate PPIs in general because of their limited size.
Peptides, aided by the large interacting surfaces of diverse topological structure, serve as ideal candidates to modulate PPIs.[4] Moreover, it is viable and convenient to design a peptide inhibitor derived from the linear sequence. However, three pharmacological obstacles still hinder their clinical application, including: 1) short half-life of blood circulation, 2) horrible cellular internalization, and 3) limited tumor specificity.[4,5] To overcome these bottlenecks, we successfully developed a clinically practicable strategy through assembling therapeutic peptide and gold ion into a TME-responsive polymeric Au–peptide nanocluster. Synergized with EPR effects, TME responsiveness can endow delivery system with high targeting ability to tumor. During the circulation and at the healthy organ, the large size and negative charge of pCluster can impede its tissue infiltration and cellular uptake. By contrast, responding to acidic TME, pCluster can disassemble into small positively charged nanoparticles, which is favorable of neoplastic retention and cellular internalization. In this proof-of-concept study, we specifically delivered these nanoclusters into the tumor cells, and demonstrated that they potently inhibited tumor growth and metastasis, and synergized the efficacy of PD-L1 inhibitor in several animal models. Compared to exciting strategies such as sidechain-stapling, backbone-cyclizing, D-enantiomerization, and liposome encapsulation, the present strategy can easily and rapidly turn a peptide-derived PPI inhibitor into a tumor-specific therapeutic agent instead of complicated design and priori assess. Considering that peptide is easy to modify, this strategy directly endows the therapeutic peptides whose pKI is at neutral, with TME-responsiveness, which allows the peptides to selectively reach and penetrate the tumor sites with very low toxicity. More importantly, compared to conventional method to link peptides to the surface of nanoparticles, pParticles can take full advantage of the whole volume to load the peptide cargo, thereby fundamentally solving the problem of low cargo loading and the aggregation resulting from the peptide conjugation.
Given that pathologic role of the β-catenin in a broad range of human cancers and preventing antitumor immunity,[20,39] it has become a potential pharmacologic target. However, β-catenin participates in many important homeostatic functions,[42] thus it will be a pressing challenge to simultaneously achieve high anticancer activity and selectivity. Although some therapeutics have been demonstrated to successfully inhibit tumor growth through blocking the Wnt pathway; however, severe side effects such as bone marrow hypoplasia and anemia are unfortunately observed in experimental animals. Profited from the switch of cytomembrane penetration in response to acidic TME (Figure 3B,C), pCluster can penetrate cytomembrane entering cellular inner only at the tumor sites but not in healthy organs such as liver and kidney. Thus, although there exists the accumulation of pClusters in liver, the switch of cytomembrane penetration can keep the healthy cells safe, as supported by our data that side effects were not observed in mice treated with pClusters. Thus, this proof-of-concept study highlights clinical translation potential of this peptide-derived anticancer strategy, thereby achieving a therapeutic window to combat Wnt-driven cancers.
4. Conclusions
In summary, the data presented here provide compelling evidence that this novel TME-responsive peptide-derived clustered nanohybrid (pCluster) is advantageous in delivering therapeutic peptides into tumor sites and overcoming important obstacles to the application of anticancer peptides. Through this strategy, nanoparticles formed by copolymerized Au–peptide can self-assemble into pH-responsive higher-order pCluster, and subsequently disintegrate into Au–peptide nanoparticles only in the tumor sites, thereby achieving efficient tumor-specific cellular uptake of peptide cargo. Our data demonstrate that pClusters potently inhibit tumor growth and metastasis, and synergize the PD-L1 checkpoint blockade immunotherapy through impairing Wnt/β-catenin cascade. With superior biosafety, pCluster has shown great potential in clinical translation as Wnt/β-catenin signaling inhibitor for anticancer peptide therapeutics. More importantly, this viable general strategy will likely have a broad impact on the development of peptide-based nanomedicine for cancer therapy.
5. Experimental Section
Fabrication of pParticle and pCluster:
The tetra chloroauric acid aqueous solution of HAuCl4 XH2O (0.01 M, 1 mL) was mixed with the aqueous solution of BBI-SH (250 × 10−6 M, 8.5 mL). By using 6 M NaOH, the pH of the mixture solution was adjusted between 6.0 and 7.0. A total volume of 500 μL solution containing freshly prepared 0.1 M VC and 0.1 M CA was added with stirring. The pParticles were then prepared after 1 h reaction at 25 °C. Next, the excess reactants were eliminated by dialysis tubing (cutoff, 10 KDa). To synthesize pCluster, 500 μL cationic polymer PLL solution (Mw: 20 kDa, 2 × 10−3 M, pH 7.4) was added dropwise to the above solution of pParticle. The collection of nano-gold cluster fraction was concentrated by centrifugal filtration (molecular weight cut off (MWCO) 50 kDa, Millipore Amico Ultra). After that, pParticles and pClusters were freeze-dried for subsequent experiments.
Quantification of Drug Loading and GSH-Responded Drug Release:
First, the PBS buffer (pH 7.4) was prepared, containing GuHCL (6 M) and DTT (1 M). The prepared buffer can break the bond between the BBI-SH and gold atoms. For quantification of drug loading, pClusters or pParticles were dissolved in the above prepared PBS buffer (pH 7.4), and the amounts of released peptides or CA were then quantified using HPLC. As for the test of GSH-responded drug release, pClusters or pParticles were first added into PBS buffer (pH 7.4) containing 10 × 10−3 M glutathione. After 1 h incubation at room temperature, pClusters or pParticles were removed by 10 000 g centrifuge. The supernatants were authenticated by ESI mass and quantified by HPLC.
In Vivo Biodistribution Analysis:
HCT116 cells (4 × 106 cells per site) were subcutaneously injected into the hip of four- to five-weeks-old male athymic nude mice. Four weeks after inoculation, 200 μL Texas Red-labelled pClusters or pParticles (1 mg mL−1) were injected into the mice via intraperitoneal injection. We took the ex vivo images of mice using the IVIS Spectrum In Vivo Imaging System with an excitation wavelength of 620 nm. According to the experimental plan, the mice were killed humanely at predetermined times, and different organs and tumor were then obtained from each mouse. The fluorescence intensities of all organs and tumor were analyzed by the IVIS Spectrum In Vivo Imaging System.
Xenograft Tumor Model:
According to Institution Guidelines, we designed all animal experiments and obtained the approvement from the Laboratory Animal Center of Xi’an Jiaotong University. The xenograft tumor model was similarly established as mentioned above.[8] When the average tumor volume reached ≈50 mm3, the mice bearing xenograft tumors were then randomly divided into different groups (five mice per group), and the treatment was begun. pCluster, pParticle, pParticle without CA, and CA were administered at 1 mg kg−1 on d 2, 4, 6, and 8, with PBS as a negative control. Tumor volume was calculated, and H&E or immunohistochemical staining was performed as described previously.[43] In addition, all exploration on drug toxicity was followed by the standard clinical laboratory procedures as our previous reports.[44]
Tail Vein Metastasis Model:
We collected and resuspended B16F10 cells in 1640 medium without fetal bovine serum (FBS). Different number of cells were then injected into the vein tail of C57BL/6 mice. The detailed dosages were as follows: 5 × 104 cells per 0.2 mL for comparing therapy efficacy of different route of administration, and 5 × 105 cells per 0.2 mL for assessing inhibitory effect of pCluster on tumor metastasis. 10 mice were randomly and equally divided into two groups: the PSB-treated (Control) groups and pCluster-treated groups. pCluster injection was started 3th day after cell injection. The pCluster-treated mice were intraperitoneally administrated with 1 mg kg−1 pCluster every other two days for consecutive 1 week (4 times injection). The mice in control group were administrated with an equal volume of PBS. The mice were euthanized on the 10th day to compare therapy efficacy between i.v and i.p. administration of pCluster, while the mice were euthanized on the 14th day to evaluate the effect of pCluster on tumor metastasis. The lungs were separated and photographed, and then the numbers of metastatic nodules on the surface of lungs were counted. Following this, paraffin sections of lung tissue were prepared for H&E staining and IHC staining of β-catenin, MMP9, and CD133.
Synergistic Effect of pCluster on PD-L1 Therapy in Animal Model:
First, 5 × 105 MC38 cells were subcutaneously injected into the flank of C57BL/6 mice. After about two weeks, tumor volume of each mice reached ≈100 mm3. Mice were divided into four groups randomly (five mice per group): PBS control; pCluster; PPI; PPI + pCluster. The PPIs (2.5 mg kg−1) were intraperitoneally injected into animals every other day (days 0, 2, 4, and 6) for a total of four injections as previously described,[45] by which this dose of 2.5 mg kg−1 can inhibit tumor growth at a certain degree rather than being too high to mask antitumor effect of pCluster. The dosage of pCluster was consistent with the pCluster dosage (1 mg kg−1) used in the nude mouse model (Figure 4) and the metastasis model (Figure 6), and pClusters (1 mg kg−1) were intraperitoneally injected into animals at days 1, 3, and 5. Tumor sizes were monitored every day. All mice were euthanized at day 11, and the tumors were resected, photographed, and weighted. All tissue samples were then prepared for H&E staining and IHC staining of β-catenin, Ki67, and CD8 as described previously.[8]
Supplementary Material
Acknowledgements
W.H., S.W., and J.Y. contributed equally to this work. This work was supported by NIHR01CA219150 (to W.L.), the Clinical Research Award of the First Affiliated Hospital of Xi’an Jiaotong University (Grant No. XJTU1AF-CRF-2017-003 to P.H. and W.H.), and the National Natural Science Foundation of China (Grant No. 81572627 to P.H.).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Wangxiao He, Key Laboratory for Tumor Precision Medicine of Shaanxi, Province and Department of Endocrinology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, P. R. China; Center for Translational Medicine, Key Laboratory of Biomedical Information, Engineering of Ministry of Education, School of Life Science and Technology, and Frontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an 710049, P. R. China; Institute of Human Virology and Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD 21201, USA.
Simeng Wang, Key Laboratory for Tumor Precision Medicine of Shaanxi, Province and Department of Endocrinology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, P. R. China.
Jin Yan, Department of Biologic and Materials Sciences, Department of Biomedical Engineering, Macromolecular, Science and Engineering Center, Department of Materials Science and Engineering, University of Michigan, Ann Arbor, MI 48109, USA.
Yiping Qu, Key Laboratory for Tumor Precision Medicine of Shaanxi, Province and Department of Endocrinology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, P. R. China.
Liang Jin, Department of Infectious Diseases, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, P. R. China.
Fang Sui, Key Laboratory for Tumor Precision Medicine of Shaanxi, Province and Department of Endocrinology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, P. R. China.
Yujun Li, Key Laboratory for Tumor Precision Medicine of Shaanxi, Province and Department of Endocrinology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, P. R. China.
Weiming You, Department of Oncology, BenQ Medical Center, Nanjing Medical University, Nanjing 210029, P. R. China.
Guang Yang, Department of Oncology, BenQ Medical Center, Nanjing Medical University, Nanjing 210029, P. R. China.
Qi Yang, Key Laboratory for Tumor Precision Medicine of Shaanxi, Province and Department of Endocrinology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, P. R. China.
Meiju Ji, Center for Translational Medicine, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, P. R. China.
Yongping Shao, Center for Translational Medicine, Key Laboratory of Biomedical Information, Engineering of Ministry of Education, School of Life Science and Technology, and Frontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an 710049, P. R. China.
Peter X. Ma, Department of Biologic and Materials Sciences, Department of Biomedical Engineering, Macromolecular, Science and Engineering Center, Department of Materials Science and Engineering, University of Michigan, Ann Arbor, MI 48109, USA
Wuyuan Lu, Institute of Human Virology and Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD 21201, USA.
Peng Hou, Key Laboratory for Tumor Precision Medicine of Shaanxi, Province and Department of Endocrinology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, P. R. China.
References
- [1].Mullard A, Nat. Rev. Drug Discovery 2012, 11, 173. [DOI] [PubMed] [Google Scholar]
- [2].Moffat JG, Vincent F, Lee JA, Eder J, Prunotto M, Nat. Rev Drug Discovery 2017, 16, 531. [DOI] [PubMed] [Google Scholar]
- [3].Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW, Nat. Rev. Cancer 2014, 14, 248. [DOI] [PubMed] [Google Scholar]
- [4].Fosgerau K, Hoffmann T, Drug Discovery Today 2015, 20, 122. [DOI] [PubMed] [Google Scholar]
- [5].Acar H, Ting JM, Srivastava S, LaBelle JL, Tirrell MV, Chem. Soc. Rev. 2017, 46, 6553. [DOI] [PubMed] [Google Scholar]
- [6].Nevola L, Giralt E, Chem. Commun. 2015, 51, 3302. [DOI] [PubMed] [Google Scholar]
- [7].Sewald N, Jakubke H-D, Peptides: Chemistry and Biology, John Wiley & Sons, Hoboken, USA: 2015. [Google Scholar]
- [8] a).Yan J, He W, Yan S, Niu F, Liu T, Ma B, Shao Y, Yan Y, Yang G, Lu W, ACS Nano 2018, 12, 2017; [DOI] [PubMed] [Google Scholar]; b) Yan J, He W, Li N, Yu M, Du Y, Lei B, Ma PX, Biomaterials 2015, 59, 21; [DOI] [PubMed] [Google Scholar]; c) Niu F, Yan J, Ma B, Li S, Shao Y, He P, Zhang W, He W, Ma PX, Lu W, Biomaterials 2018, 167, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9] a).Maeda H, Adv. Enzyme Regul. 2001, 41, 189; [DOI] [PubMed] [Google Scholar]; b) Meng H, Xue M, Xia T, Ji Z, Tarn DY, Zink JI, Nel AE, ACS Nano 2011, 5, 4131; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li Z, Zhu Z, Liu W, Zhou Y, Han B, Gao Y, Tang Z, J. Am. Chem. Soc. 2012, 134, 3322. [DOI] [PubMed] [Google Scholar]
- [10] a).Li Z, Cheng E, Huang W, Zhang T, Yang Z, Liu D, Tang Z, J. Am. Chem. Soc. 2011, 133, 15284; [DOI] [PubMed] [Google Scholar]; b) He W, Huang H, Yan J, Zhu J, J. Appl. Phys. 2013, 114, 204701; [Google Scholar]; c) Paciotti GF, Kingston DG, Tamarkin L, Drug Dev. Res. 2006, 67, 47. [Google Scholar]
- [11] a).Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD, Small 2005, 1, 325; [DOI] [PubMed] [Google Scholar]; b) Wang AZ, Langer R, Farokhzad OC, Annu. Rev Med. 2012, 63, 185. [DOI] [PubMed] [Google Scholar]
- [12].Spicer CD, Jumeaux C, Gupta B, Stevens MM, Chem. Soc. Rev 2018, 47, 3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13] a).Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R, Nat. Nanotechnol. 2007, 2, 751; [DOI] [PubMed] [Google Scholar]; b) Blanco E, Shen H, Ferrari M, Nat. Biotechnol. 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Züllig S, Basler K, Cell 2002, 109, 47. [DOI] [PubMed] [Google Scholar]
- [15].Kahn M, Nat. Rev. Drug Discovery 2014, 13, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Clevers H, Cell 2006, 127, 469. [DOI] [PubMed] [Google Scholar]
- 17.[] Logan CY, Nusse R, Annu. Rev. Cell Dev. Biol. 2004, 20, 781. [DOI] [PubMed] [Google Scholar]
- [18] a).Townsley FM, Cliffe A, Bienz M, Nat. Cell Biol. 2004, 6, 626; [DOI] [PubMed] [Google Scholar]; b) Krieghoff E, Behrens J, Mayr B, J. Cell Sci. 2006, 119, 1453. [DOI] [PubMed] [Google Scholar]
- [19].Sampietro J, Dahlberg CL, Cho US, Hinds TR, Kimelman D, Xu W, Mol. Cell 2006, 24, 293. [DOI] [PubMed] [Google Scholar]
- [20].Takada K, Zhu D, Bird GH, Sukhdeo K, Zhao J-J, Mani M, Lemieux M, Carrasco DE, Ryan J, Horst D, Sci. Transl. Med. 2012, 4, 148ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21] a).Schaaff TG, Knight G, Shafigullin MN, Borkman RF, Whetten RL, J. Phys. Chem. B 1998, 102, 10643; [Google Scholar]; b) Link S, Beeby A, FitzGerald S, El-Sayed MA, Schaaff TG, Whetten RL, J. Phys. Chem. B 2002, 106, 3410. [Google Scholar]
- [22].Kasaai MR, Carbohydr. Polym. 2007, 68, 477. [Google Scholar]
- [23] a).Wang J, Mao W, Lock LL, Tang J, Sui M, Sun W, Cui H, Xu D, Shen Y, ACS Nano 2015, 9, 7195; [DOI] [PubMed] [Google Scholar]; b) Li H-J, Du J-Z, Liu J, Du X-J, Shen S, Zhu Y-H, Wang X, Ye X, Nie S, Wang J, ACS Nano 2016, 10, 6753. [DOI] [PubMed] [Google Scholar]
- [24] a).Mintzer MA, Simanek EE, Chem. Rev 2009, 109, 259; [DOI] [PubMed] [Google Scholar]; b) Zhou Q, Hou Y, Zhang L, Wang J, Qiao Y, Guo S, Fan L, Yang T, Zhu L, Wu H, Theranostics 2017, 7, 1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Acharya S, Sahoo SK, Adv. Drug Delivery Rev. 2011, 63, 170. [DOI] [PubMed] [Google Scholar]
- [26].Jain RK, Stylianopoulos T, Nat. Rev. Clin. Oncol. 2010, 7, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27] a).Fodde R, Brabletz T, Curr. Opin. Cell Biol. 2007, 19, 150; [DOI] [PubMed] [Google Scholar]; b) Holland JD, Klaus A, Garratt AN, Birchmeier W, Curr. Opin. Cell Biol. 2013, 25, 254. [DOI] [PubMed] [Google Scholar]
- [28].de La Roche M, Rutherford TJ, Gupta D, Veprintsev DB, Saxty B, Freund SM, Bienz M, Nat. Commun. 2012, 3, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hoggard LR, Zhang Y, Zhang M, Panic V, Wisniewski JA, Ji H, J. Am. Chem. Soc. 2015, 137, 12249. [DOI] [PubMed] [Google Scholar]
- [30].Anastas JN, Moon RT, Nat. Rev Cancer 2013, 13, 11. [DOI] [PubMed] [Google Scholar]
- [31].Christofori G, Nature 2006, 441, 444. [DOI] [PubMed] [Google Scholar]
- [32] a).Beavon I, Eur. J. Cancer 2000, 36, 1607; [DOI] [PubMed] [Google Scholar]; b) Li X, Xu Y, Chen Y, Chen S, Jia X, Sun T, Liu Y, Li X, Xiang R, Li N, Cancer Lett. 2013, 336, 379. [DOI] [PubMed] [Google Scholar]
- [33].Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T, Nat. Rev Cancer 2005, 5, 744. [DOI] [PubMed] [Google Scholar]
- [34] a).Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, Gingras A-C, Mazitschek R, Neel BG, Stagljar I, Cell Rep. 2012, 2, 951; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R, Nature 2007, 445, 111. [DOI] [PubMed] [Google Scholar]
- [35].Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Nat. Cell Biol. 2000, 2, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pardoll DM, Nat. Rev. Cancer 2012, 12, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37] a).Brahmer JR, Tykodi SS, Chow LQ, Hwu W-J, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh, Odunsi K, N. Engl. J. Med. 2012, 366, 2455; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sunshine J, Taube JM, Curr. Opin. Pharmacol. 2015, 23, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38] a).Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF, Sci. Transl. Med. 2013, 5, 200ra116; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, Wamwea A, Bigelow E, Lutz E, Liu L, J. Immunother. 2015, 38, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Spranger S, Bao R, Gajewski TF, Nature 2015, 523, 231. [DOI] [PubMed] [Google Scholar]
- [40].Ryan DP, Matthews JM, Curr. Opin. Struct. Biol. 2005, 15, 441. [DOI] [PubMed] [Google Scholar]
- [41].Zhao J-J, Lin J, Zhu D, Wang X, Brooks D, Chen M, Chu Z-B, Takada K, Ciccarelli B, Tao J, Cancer Res. 2014, 74, OF1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Barker N, Clevers H, Nat. Rev. Drug Discovery 2006, 5, 997. [DOI] [PubMed] [Google Scholar]
- [43].Wu L, Zhang H, Jiang Y, Gallo RC, Cheng H, Proc. Natl. Acad. Sci. USA 2018, 115, E4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44] a).He W, Yan J, Sui F, Wang S, Su X, Qu Y, Yang Q, Guo H, Ji M, Lu W, Shao Y, Hou P, ACS Nano 2018, 12, 11664; [DOI] [PubMed] [Google Scholar]; b) He W, Yan J, Jiang W, Li S, Qu Y, Niu F, Yan Y, Sui F, Wang S, Zhou Y, Jin L, Li Y, Ji M, Ma PX, Liu M, Lu W, Hou P, Chem. Mater. 2018, 30, 7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chang HN, Liu BY, Qi YK, Zhou Y, Chen YP, Pan KM, Li WW, Zhou XM, Ma WW, Fu CY, Angew. Chem., Int. Ed. 2015, 54, 11760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.