

Abstract
Peptide-tuned self-assembly of macromolecular agents (>500 Da) such as therapeutic peptides offers a strategy to improve the properties and biofunctions of degradable nanomaterials, but the tough requirement of macromolecular therapeutics delivery and a lack of understanding of peptide-based self-assembly design present high barriers for their applications. Herein, we developed a new strategy for nanoengineering macromolecular drugs by an elaborate peptide, termed PSP (VVVVVHHRGDC), capable of directly conjugating with cargo to be a PSP-cargo monomer as building block tending to self-assemble into a well-defined nanoshell with tumor-triggered shape and charge switch. As a proof of concept, conjugation PSP to a D-peptide activator of tumor suppressor p53 termed DPMI (1492.5 Da) generated hollow spheres ~80 nm in diameter named PSP-DPMI that disintegrated only in the acidic microenvironment of tumor tissues, followed by integrin-mediated cellular uptake of PSP-DPMI monomers. Importantly, PSP-based self-assembly successfully endowed the DPMI with long circulation time and high cancer-cell-specific intracellular accumulation. PSP-DPMI nanoshells potently inhibited tumor growth in vitro and in vivo by the p53 restoration, while maintaining a highly favorable in vivo safety profile. Out of conventional encapsulation and conjugation, our study showcases a clinically viable novel method to nanoengineer macromolecular agents such as peptide for anticancer therapy and provides a hazard-free alternative strategy for the theranostics delivery.
Graphical Abstract
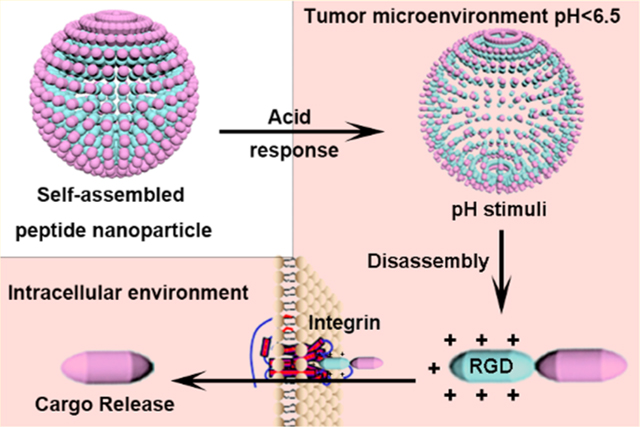
INTRODUCTION
Macromolecular therapeutics, such as peptides, proteins, and nucleic acids, comprising many small organic molecules (<500 Da), possess many attributes including high specificity as well as optimum activity in physiological conditions.1 Although macromolecular therapeutics have been regarded as a potent class of new drugs for a lot of different indications based on their specificity and relatively low side effects, they are challenging for noninvasive delivery as they are incapable to permeate cell membrane, poorly absorbed in the diseased area, and rapidly metabolized in the body owing to their charged structure and large steric hindrance.1,2 To overcome it, benefitted by the enhanced permeation and retention (EPR) effect, an increasing number of delivery platforms for cancer therapy with nanoscale were developed to improve the tumor specificity of the therapeutics.3 However, these nanocarriers have common problems in complex components and incompatible organic syntheses.4,5 Crucially, during the chemical synthesis, organic solvents and toxic reagents most likely incorporated into the carrier-cargo system, thereby posing harmful side effects.6 Moreover, delivery systems assembled from amphiphilic polymers, such as lipidosome (comprising phosphatidylcholine and PEG or analogous combination of a hydrophobic material and a hydrophilic one) and self-assembled polymer, inescapably were perplexed by the relatively low drug encapsulation efficiency due to the hydrophobic interaction between macromolecules and carriers.2,7 Therefore, there is a critical need to develop alternative strategies for more effective and safe delivery of macro-molecular agents.
With favorable pharmacological advantages, including excellent biocompatibility, high permeability, low immunogenicity, and rapid clearance, peptides are attractive alternatives to synthetic polymers as a favorable building block of the self-assembled nanostructure.8,9 More importantly, peptides can self-assemble into a variety of well-defined nanostructures, many of which are favorable for the biomedical applications such as drug delivery.10 Despite there existing a great success of peptide-based nanoparticles in cancer therapy, these particles were still hindered by the poor tumor specificity and short circulation time, leading to poor therapeutic efficacy and inevitable side effects.11,12 A promising approach to overcome these obstacles is to endow these nanoparticles with tumor microenvironment (TME) response,13–15 which is normally able to mediate the specific formation or breakage of certain nanostructure.16 During this process, some physicochemical properties such as size, rigidity, or charge can be precisely manipulated, resulting in the active or passive tumor specificity.17 Among them, size and surface charge mainly dictate the suitability and efficiency of self-assembled peptide-based nanoparticles as a delivery vehicle.18–22 Large nanoparticles typically have favorable pharmacokinetics and high degrees of vascular extravasation at the expense of tumor penetration in the tumor interstitial space.23 In contrast, small nanoparticles exhibit better tumor penetration ability but have a shortened circulation half-life,24,25 thereby causing a decrease in overall tumor accumulation.26 Although negative charge can prolong circulation time and minimize clearance of nano-particles, it can also increase the charge repulsion to isocharged cell membrane, thereby blocking the cellular internalization.20 On the other hand, strong positive charge enables nano-particles to efficiently internalize into tumor cells.27,28 Thus, an ideal delivery system should be relatively large and negatively charged in its initial state to achieve a longer circulation half-life and selective extravasation, but switchable, upon TME response at the tumor site, to small and positively charged particles to facilitate tumor penetration.
Self-assembled peptide-based nanoparticles with TME-triggered size and charge switch, consequently, will supply a viable method to deliver anticancer macromolecular therapeutics. However, when cargos are integrated into the peptide-based self-assembled nanoparticles, the final structure and response of the nanosystems are difficult to predict because the physicochemical characteristics of cargo and carrier may interfere with each other, thereby resulting in the failure of peptide self-assembly and inopportune disassembly.29 Thus, it is necessary to develop novel strategies for minimizing the geometrical indeterminacy and keeping the response characteristics of peptide-based self-assembled nanoparticles after cargo loading. In general, most of the TME-triggered self-assembly drug delivery systems release cargo extracellularly,30 which are suitable for small-molecular therapeutics because of their intrinsic cytomembrane penetrability. However, they fail to deliver the impenetrable macromolecule agents. In addition, most of current studies mainly focus on tumor-site targeting via TME response, but they are always unable to distinguish tumor cells from the matrix cells, resulting in the missing of targeted drugs. Thus, tumor microenvironment-triggered shape and charge switch of bionanomaterials as a promising drug delivery system will be expected to specifically target cancer cells through fusing tumor responsiveness into accelerated cellular internalization of cancer cells.
Herein, to overcome the above technical obstacles for the design of peptide-based nanodelivery system and endow macromolecular therapeutics with long circulation time, strong tumor targeting, and cytomembrane penetrability, we developed a pH-responsive and self-assembling peptide (PSP) of amphiphilic nature for macromolecular therapeutic modification, nanoengineering the drugs based on intermolecular electrostatic forces and hydrophobic interactions (Figure 1). By this strategy, the problem of macromolecule encapsulation will be well solved through directly conjugating cargos with the PSP module as building block of the self-assembled nanoshells, thereby greatly improving the efficiency of drug loading. PSP consists of an N-terminal hydrophobic moiety (VVVVV), a pH-responsive segment (HH), and a C- terminal hydrophilic sequence (RGDC), which will endow the PSP module with the ability to self-assemble into nanoshells, thereby targeting tumor sites through the EPR effect and TME response,31,32 and disassemble in the acidic tumor microenvironment to be taken up by tumor cells through RGD-integrin-mediated endocytosis. As a proof of concept, a typical macromolecular therapeutic without tumor enrichment and cytomembrane penetrability, DPMI (a dodecameric D-peptide can activate p53, a tumor suppressor, sequence: DTDADWDY-DADNDFDEDADLDLDR),33,34 was N-terminally conjugated to the C-terminal Cys residue of PSP via a bifunctional linker. Under physiological conditions (pH 7.4), PSP-DPMI kept the self-assembly into nanoshells as the PSP module and disintegrated at pH 6.5 where His will be protonated. After internalization through RGD-integrin-mediated endocytosis, the degradable accessory, PSP peptide, is expected to be rapidly degraded by endosomal/lysosomal peptidase,35,36 thereby releasing the DPMI to activate the p53 signaling (Figure 1). Our design likely affords a “smart” strategy to efficiently deliver macromolecular therapeutics intracellularly for clinical use.
Figure 1.
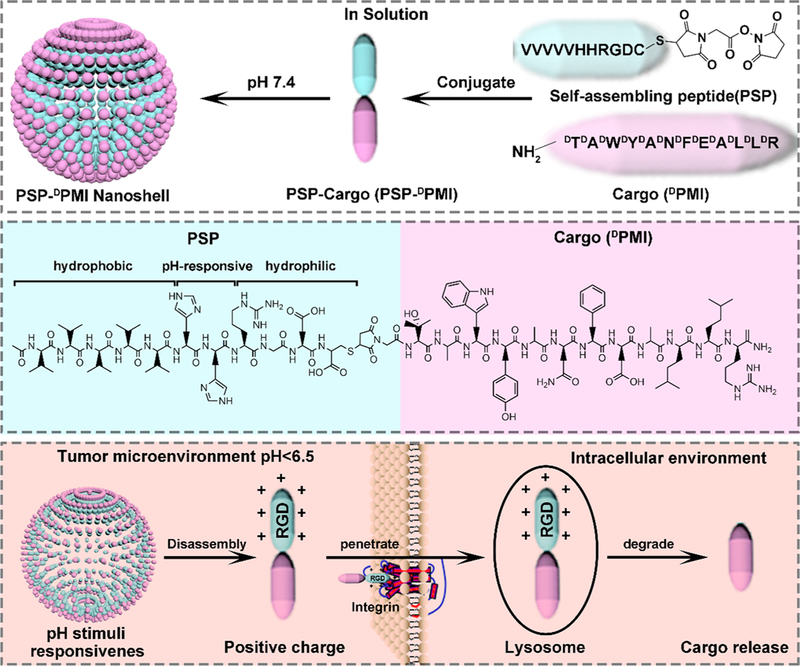
Schematic illustration of self-assembly synthetic procedure and the properties of pH-responsive charge reversal and size change of PSP-DPMI spherical shells.
RESULTS AND DISCUSSION
Design and Preparation of PSP.
The unique opportunities afforded by protein and peptide self-assembly serve as important actors to perform the biological functions, ranging from unicellular prokaryotes to unicellular and multicellular eukaryotes.37 Although natural assemblies can be repurposed to perform new biological functions,38 this strategy is limited to the structure of existing proteins and peptides, which may not be suited to a given application, such as anticancer therapy and drug delivery. To overcome this limitation, the methods for designing novel self-assembled peptide- and protein-based nanobiomaterials are of considerable interest37 and have made good advances in targeted drug delivery and nanocarrier engineering.40 However, until now, very few attempts have been made to develop a self-assembled peptide, which simultaneously possesses a dual role of TME-triggered size and charge switch and highly specific and effective tumor cell internalization. Toward this end, 10 derivatives of the peptide-amphiphile with a hydrophilic integrin ligand were prepared as shown in Figure S1A. Each molecule consisted of three parts (Figure S1B): the first one is a five/six-L-valine chain with hydrophobic side chains, providing the hydrophobic and hydrogen bonding driving force for self-assembly; the second one is the cationic region formed by electropositive amino acids, determining the property of pH-responsive charge reversal and size change; and the last one is the hydrophilic headgroup region of the molecule (the C-terminal end, whose sequence is RGDC), consisting of a RGD ligand for cancer cell adhesion and a Cys for cargo conjugated site.
To test the effect of different cationic regions on self-assembly, all molecules (nos. 1–10) with the peptide sequence VVVVV(V) (XX/XXXX)RGDC were dissolved in PBS buffer (pH 6.0, 0.05 mg/mL). Through adjusting the pH to 7.4 and measuring the hydrate particle size of all molecules by dynamic light scattering (DLS, Figure S1A), we found that only two molecules (no. 6 and no. 9) formed nanoparticles with the diameter more than 50 nm, whereas others either formed precipitate (nos. 1–3) or remained monomolecular (no. 4, no. 5, no. 7, no. 8, and no. 10). Given that nanoparticles with the size from 60 to100 nm are usually favorable for EPR-induced tumor targeting,31 the molecule no. 6 was the best one as PSP tail (Figure S1A, B). Through further analyzing the charge characteristics based on different pH by PROTEIN CALCULATOR41 and pH titration, we found that the molecule no. 6 was neutral at normal pH 7.4 and was quickly converted to the positive charge in acid environment (pH < 7) (Figure S1C,D). This was also supported by the surface charge measured by ZetaCAD at different pH values in water that the surface charge of molecule no. 6 increased from –1.6 mV at pH 7.4 to 16 mV at pH 6.0 (Figure 2A). The pH-responsive charge reversal will endow this molecule with great potential to be an ideal PSP tail. Importantly, the sequence of molecule no. 6 was easy to synthesize by solid phase peptide synthesis (SPSS) with high yield and purity (Figure 2B,C), further promoting its application potential. Moreover, considering that the β-branched hydrophobic amino acid, isoleucine, and the γ-branched one, leucine, tend to form intermolecular hydrogen bonding as valine, we synthesized molecule nos. 11 and 12 substituting valine to isoleucine or leucine, respectively. As shown in Figure S2, although molecule nos. 11 and 12 inherited the pH-responsive charge reversal of molecules no. 6, their sizes were too large to be an appropriate nanomedicine.
Figure 2.
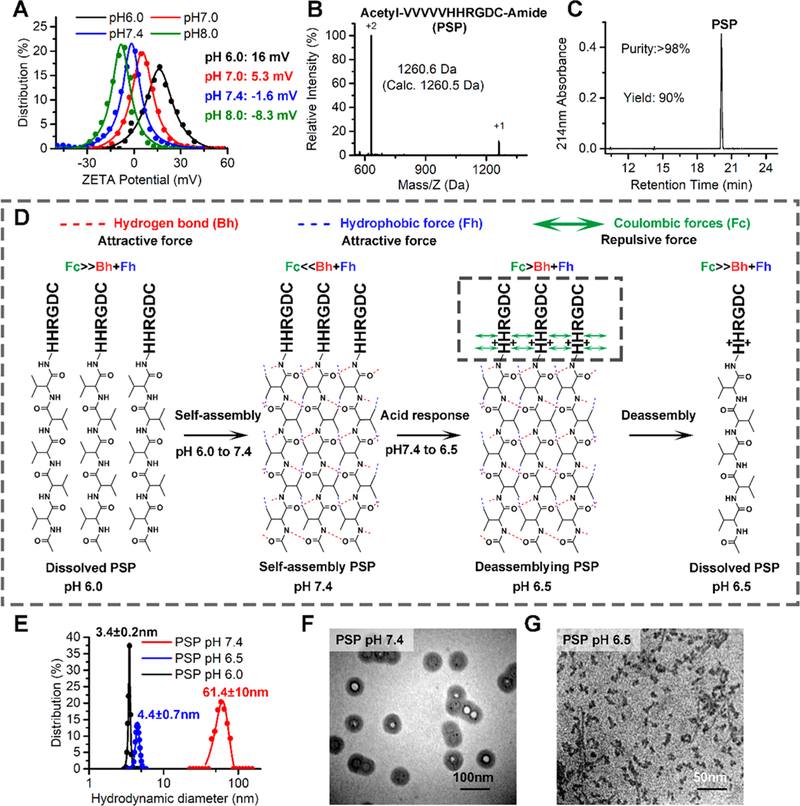
Design and preparation of PSP. (A) The surface charge measured by ZetaCAD at pH 6.0, 7.0, 7.4, and 8.0 of PSP (0.05 mg/mL) in PBS buffer at 37 °C. (B) Molecular weight of PSP characterized by ESI-MASS. (C) Characterization of the synthesized PSP by HPLC. (D) Schematic illustration of the process of self-assembly and pH response of PSP. (E) Hydrodynamic distributions of PSP at pH 6.0, 6.5, and 7.4, which were measured by DLS. (F, G) TEM image of PSP at pH 7.4 and pH 6.5.
During the process of peptide self-assembly, the formations of shape-specific architectures are mainly derived by the combined action of hydrogen bonding and hydrophobic interaction.10,42 Thus, in the present study, the process of self-assembly and pH response should obey the physical principles as follows (Figure 2D). First, the PSP tail (molecule no. 6) benefitted from intermolecular electrostatic repulsion to form relatively monodisperse without aggregation at pH 6.0, which will help dissolve some hydrophobic cargo into buffer. When the pH was adjusted to 7.4, the decreased surface charge resulted in the disappearance of intermolecular electrostatic repulsion. Thus, like surfactants, the specific structure of the PSP tail was able to self-assemble into double-decker shells with rigid hydrophobic endothecium and hydrophilic outer sphere. Meanwhile, intermolecular H-bonding and hydrophobic interactions between the five L-valine backbones resulted in the β-sheets formation (Figure S1E), thereby interlocking the adjacent molecule in place.43,44 Finally, when encountering the acidic tumor microenvironment (pH < 6.5), charge reversal increased the intermolecular electrostatic repulsion over the resultant force of H-bonding and hydrophobic interaction to promote the disassembly of shells. Taken together, these pH-sensitive properties will endow the PSP shells with ultrasensitive size and charge-switchable responsiveness to tumor acidic pH, resulting in deep tumor penetration and increased cellular uptakes.45 To evaluate its pH sensitivity, the PSP shells were incubated in PBS buffers with different pH values at 6.0, 6.5, and 7.4, respectively, and the changes in their size were measured by DLS. As shown in Figure 2E, the particle size presented a sharp transformation from ~60 nm (pH 7.4) to ~5 nm (pH 6.5), suggesting that the PSP tail possess the superdynamic transition responded to the pH. The TEM image further presented some details of the morphology features and the pH-triggered size change of the PSP. As shown in the Figure 2F, well-defined spheroidal morphology was observed at pH 7.4, and confines between the hollow core and the peptide shell could be found in all particles, although some particles showed a white core, while others exhibited dark because the low contrast of the peptide-based materials made it difficult to adjust the focal length best matching all the particles in the scene. Adjusting the pH below 6.5, the hollow shell structure of the PSP deformed and resolved into small amorphous pieces (Figure 2G). Moreover, benefitted by the synthesis of PSP by L-amino acids, PSP could be degraded after the pH response, which will endow the PSP with more biosecurity.
Integration of DPMI into the Building Block of a Self-Assembled Nanoshell.
The central challenge in designing peptide-based self-assembling nanobiomaterials still remains, which is to directly encode the functional-dependent information necessary into the peptide or protein building blocks.39 In physiological conditions, macromolecular agents, like peptides, always possess high specificity and potent activity and are, thereby, rich in all necessary information for biofunction.1 Thus, direct integration of macromolecular therapeutics into the building block will be a plausible strategy for designing self-assembling nanobiomaterials, particularly anticancer nanomedicine. DPMI is a p53 activator consisting of D-amino acids, which can resist enzyme degradation. Thus, using DPMI as an example to conceptually prove the function of the PSP will avoid disturbance by poor stability of the peptides. The strategy for integrating DPMI into the self-assembling building block (PSP module) is outlined in Figure 3A. Briefly, amino-acetyl PSP tail and carboxyamide DPMI were synthesized by SPSS using Fmoc chemistry. The difference is that, for PSP synthesis, coupling reactions were performed using L-enantiomeric Fmoc-amino acids, but for PMI, they were performed by D-enantiomorph (Figure 3B). A bifunctional linker, N-succinimidyl maleimidoacetate, was also introduced to the sulfydryl of PSP for cargo (DPMI) conjugation (Figure 3C). After that, PSP-DPMI was obtained by a mild reaction between PSP-NHS and the amino of DPMI (Figure 3D).
Figure 3.
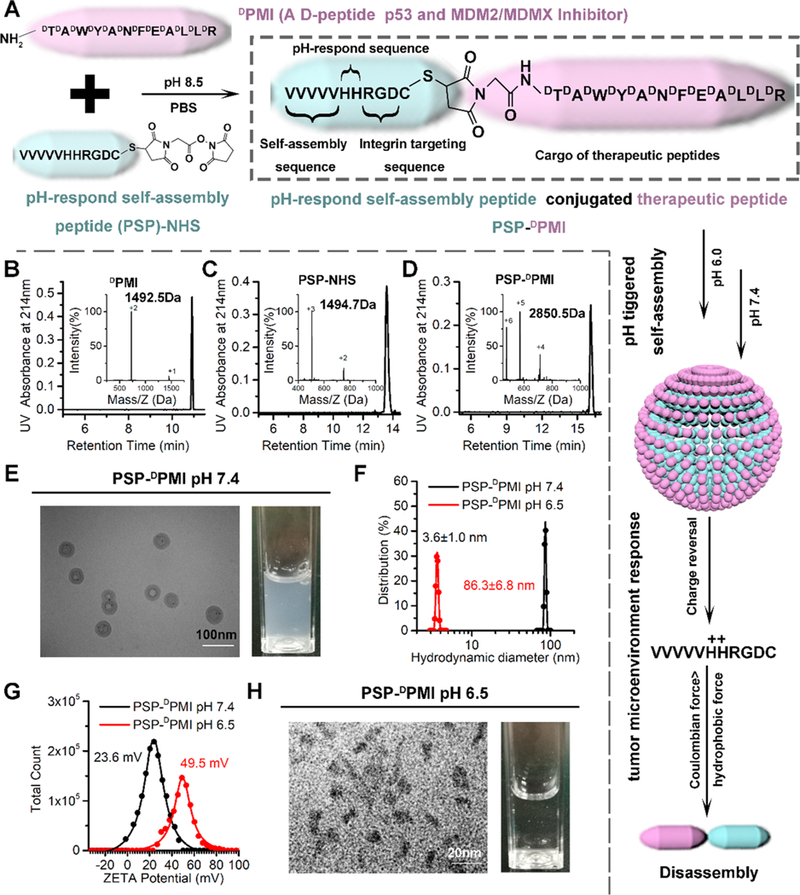
Preparation and characterization of PSP-DPMI spherical shell. (A) The strategy for the preparation of PSP-DPMI. Characterization of synthesized DPMI (B), PSP-NHS (C), and PSP-DPMI (D) by HPLC and ESI-MASS. (E) Transmission electron micrograph (TEM) of PSP-DPMI at pH 7.4 and the light picture of PSP-DPMI (0.2 mg/mL) solution. (F) Hydrodynamic distributions of PSP-DPMI (at pH 7.4 and pH 6.5), which were measured by DLS. (G) The surface charge of PSP-DPMI (at pH 7.4 and pH 6.5), which were measured in PBS buffer by DLS at 37 °C. (H) Transmission electron micrograph (TEM) of PSP-DPMI at pH 6.5 and the light picture of PSP-DPMI (0.2 mg/mL) solution.
To verify whether PSP-DPMI inherits the pH-responsive self-assembly of PSP after cargo conjugation, we dissolved PSP-DPMI in PBS buffer at pH 6.0 of the concentration from 0.05 mg/mL to 1.6 mg/mL and then adjusted the pH to 7.4 to trigger the self-assembly, following by size characterization using DLS. As shown in Figure S3, the highest concentration of PSP-DPMI to self-assemble into appropriate nanoparticles was 0.2 mg/mL, which is high enough to carry out follow-up in vitro and in vivo experiments. Moreover, like PSP, PSP-DPMI maintained hollow shell morphology (Figure 3E) and obtained a relatively narrow hydrate size distribution of 86.3 nm (Figure 3F), but larger than the PSP (~60 nm, Figure 2E). To further test the response of PSP-DPMI to pH, we incubated them in a tumor-acidity mimic PBS buffer at pH 6.5 over a predetermined time and observed its electrical and morphological evolution by using ZetaCAD, TEM, and DLS. After a 30 min incubation at pH 6.5, the surface charge increased to 49.5 (Figure 3G), indicating increased intermolecular electrostatic repulsion. Meanwhile, hollow-shell morphology was partially deformed, and small jellied particles appeared in the solution (Figure 3H). In addition, we observed a sharp peak at 3.6 nm by using DLS (Figure 3F), with size comparable to single molecule of PSP-DPMI. These results indicate that PSP-DPMI completely inherits the pH-responsive self-assembly of PSP and keeps ultrasensitive size and charge-switchable responsiveness to tumor acidic pH. To supply more evidence for the inheritance of the responsiveness after peptide conjugation, we linked the L-isomeride ((L)DPMI) of the DPMI to the PSP. As expected, PSP-(L)DPMI showed the similar size and ZETA potential as PSP-(L)dPMI, both at pH 6.5 and at pH 7.4 (Figure S4). Taken together, profiting from the ingenious design of PSP, whose hydrophobic core region for self-assembly is at the N-terminal and keeps it away from the cargo linker at the C-terminal, PSP-DPMI inherits the pH-responsive self-assembly after cargo conjugation. Notably, after self-assembly, the peptide cargos were located at the outside of the nanoshell, which maximally limited the effects of cargo on the self-assembled structure. To further verify the stability, PSP-DPMI was solved in the DMEM medium containing 20% human serum. As shown in Figure S5A, PSP-DPMI could generally maintain its stability after a 24-h incubation and kept its pH response (Figure S5B).
PSP-Based Self-Assembly Endows the DPMI with Cytomembrane Penetrability, Long Circulation Time, and Strong Tumor Targeting.
The size of PSP-DPMI spherical shell (~90 nm) is favorable for the EPR effect operation and tended to extravasate across the vasculature at tumor sites.31,32,46 Moreover, the large size can minimize the rapid clearance of the nanocomplex resulting in prolonged blood circulation time.47,48 Evidently, nanomedicines with smaller size and positive charge exhibit enhanced performance on cellular internalization.23,27 In addition, in the present study, the RGD sequence was incorporated into PSP-DPMI to target integrin α5β1, which is highly expressed in many cancer types, such as colon, liver, and lung cancers,10 further enhancing their accumulation at the tumor sites and cellular uptake (Figure 4A). Meanwhile, the response of PSP-DPMI to tumor extracellular pH turning into smaller size and positive charge further help its cellular internalization. The intracellular PSP will subsequently be degraded, and the DPMI can target to MDM2, thereby reactivating p53 signaling (Figure 4A).
Figure 4.
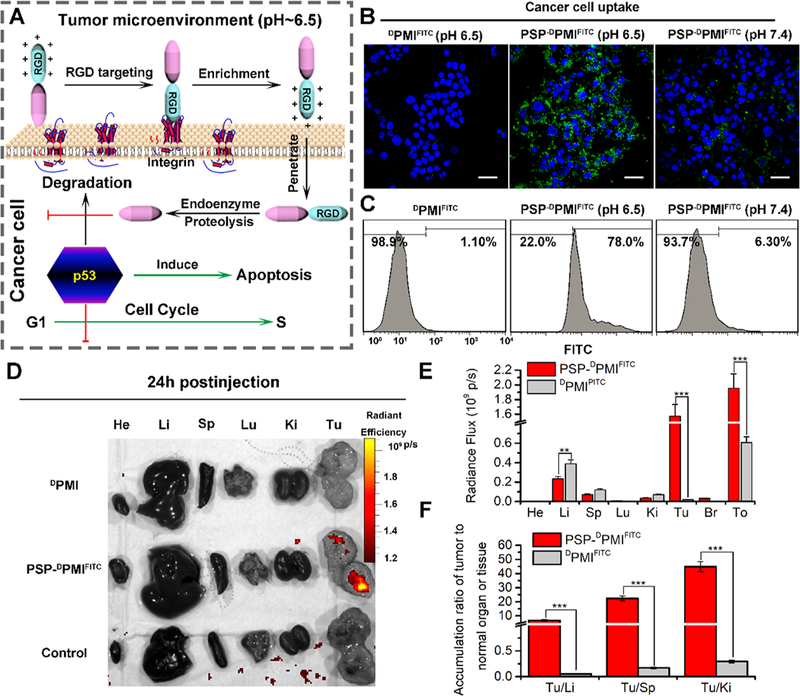
PSP-based self-assembly endows the DPMI with cytomembrane penetrability, long circulation time, and tumor targeting. (A) Schematic depiction for targeting function of RGD motif and the response of PSP-DPMI to tumor extracellular pH. (B) CLSM images of HCT116 cells after 6 h of incubation with DPMIFITC or PSP-DPMIFITC (20 μg/mL) at pH 7.4 or pH 6.5 (scale bar: 60 μm). All photos were taken at the same exciting light and detector gain. (C) Flow cytometry analysis was performed to measure cell uptake of DPMIFITC or PSP-DPMIFITC responded to acidic pH. (D) Ex vivo fluorescent images of tumors and major organs from PMI - and PSP- PMI -treated mice at 12 h postinjection. He, heart; Li, liver; Sp, spleen; Lu, lung; Ki, kidneys; Tu, tumor; Br, brain; To, total. Thresholds were appropriately established: 5.0 × 108. (E) Quantitative analysis of the Ex vivo fluorescence intensity in tumors and major organs. Every tissue was exposed at the same exciting light and normalized by the PBS control group. All data are shown by mean ± s.d. (n = 3/group). (F) Tumor-to-background (normal organ or tissue) ratios for DPMIFITC and PSP-DPMIFITC at 24 h postinjection. PSP-DPMIFITC showed improved tumor selectivity over the surrounding normal organs or tissues compared to DPMIFITC (n = 3). p values were calculated by t-test (**, p < 0.01; ***, p < 0.001).
To test cellular uptake of DPMI, fluorescein isothiocyanate (FITC) was first conjugated to the ε-amino of K9 (A9 mutate to K9) in DPMI A9K (DPMIFITC) as described our previous study.49 FITC-labeled PSP-DPMI (PSP-DPMIFITC) was then synthesized strictly according to the protocol of PSP-DPMI. Next, HCT116 p53+/+ cells were incubated with 20 μg/mL DPMIfitc or PSP-dPMIfitc at pH 7.4 and pH 6.5, respectively. As shown in Figure 4B, PSP-DPMIFITC-treated cells presented low levels of cellular uptakes (<10%) at pH 7.4, whereas the percentage of FITC-positive cells at pH 6.5 was dramatically increased and reached more than 75% after a 6-h treatment. In addition, DPMIFITC-treated cancer cells (Figure 4B,C) and PSP-dPMIfitc normal cells (Figure S6) did not show any cellular internalization at pH 6.5, further indicating that the PSP tail endows the DPMI with an specifically enhanced ability to internalize cancer cells.
To verify the applicability of PSP-DPMI in vivo, mice bearing HCT116 p53+/+ xenografted tumors were modeled to explore the tumor specificity and body circulation of PSP-DPMI by intraperitoneal injection. Two weeks after infection with 1 × 106 HCT116 p53+/+ cells, mice were divided into three biodistribution-test groups, randomly (DPMIFITC, PSP-DPMIfitc and PBS control; n = 3/group). The quantity and distribution of DPMIFITC or PSP-DPMIFITC in tumor and normal tissues were then determined by tracking the FITC fluorescent signals (Figure 4D and Figure S7). Facilitated by the property of pH-responsive charge reversal and size change, 24 h after injection, PSP-DPMIFITC expectedly showed 3.25-fold overall fluorescent signals than DPMIFITC (Figure 4E), suggesting that the pH-sensitive spherical shells prolong the blood circulation time. This was supported by the pharmacokinetic study, in which the half-life of the PSP-DPMI in blood was 7.3 h, while DPMI was not found after 2 h (Figure S8). More importantly, due to the EPR effect, tumor-microenvironment sensitivity, and RGD targeting, PSP-DPMIfitc was primarily accumulated in tumor rather than normal tissues (Figure 4F). In detail, strong fluorescent signals were observed in the tumor after 24 h of injection, whereas very low (kidney and liver) or hardly any (brain, lung, spleen, and heart) fluorescent signals were observed in normal tissues (Figure 4D,E). Furthermore, all tumor-to-background (normal organ or tissue) ratios for PSP-DPMIFITC were significantly higher than those for DPMIFITC (Figure 4F), indicating that the PSP tail improves tumor selectivity over the surrounding normal organs or tissues of DPMIFITC. Moreover, PSP-DPMI was also demonstrated to selectively target tumor after intravenous administration (Figure S9), by which PSP-DPMI efficiently concentrated into tumor sites 6 h postinjection.
PSP-Based Nanoshells Awaken the Function of DPMI and Inhibit Cancer Cell Growth via Reactivating the p53 Signaling.
It is generally known that many human cancers are attributable to the malfunction of tumor suppressor p53.50–52 Therefore, DPMI is expected to reactivate p53 in the present study, thereby inducing cell cycle arrest and apoptosis.34 To do this, we first treated the breast cancer cell line MCF7, melanoma cell line A375, colon cancer cell line HCT116 p53+/+ (carried wild-type p53), and the isogeneic HCT116 p53–/– (p53 deletion) with serial concentrations from 9.8 nM to 20.0 μM of PSP-DPMI, PSP, DPMI, and Nutlin3 (a small molecule antagonist of MDM253 as a positive control). It also should be noted that integrin (Figure S10) and MDM2/MDMX were highly expressed in all these cell lines, and the first three cell lines harbor wild-type p53. Next, we evaluated the effect of the above treatments on cell viability using the MTT assay. As shown in Figure 5A–D, PSP-DPMI significantly inhibited the growth of these three p53-positive cells in a dose-dependent manner, and its inhibitory effect on cell viability was even better than Nutlin3. Expectedly, the PSP tail did not exhibit any inhibitory effect, even at 20.0 μM, the highest concentration used in the test. In addition, it was consistent with our previous study that DPMI failed to show any cytotoxicity against these three cell lines (even at 20 μM) (Figure 5A–D), which was attributed to its inability to traverse cell membrane.34 In a sharp contrast to the findings in HCT116 p53+/+ cells, HCT116 p53–/– cells were highly resistant to treatment of PSP-DPMI and Nutlin3 (Figure 5D), indicating that antitumor activity of PSP-DPMI is p53 dependent.
Figure 5.
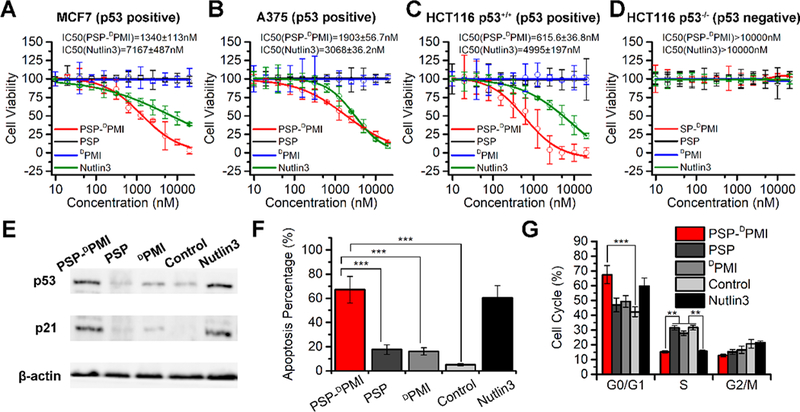
In vitro anticancer efficacy of PSP-DPMI via reactivating the p53 signaling. (A–D) Dose–response curves of the indicated four cell lines with different treatments for 72 h, which were measured by MTT assay. (E) Western blot analysis of p53 and p21 in HCT116 p53+/+ cells. Cells were treated with PSP-DPMI (200 nM) and Nutlin3 (5 μM) for 24 h, and β-actin was used as loading control. (F) HCT116 p53+/+ cells were treated with 200 nM PSP-DPMI or 5 μM Nutlin3 for 48 h, and the FACS analysis was performed to measure cell apoptosis. (G) Cell cycle distributions of HCT116 cells with different treatments for 24 h. Data were shown as mean ± s.d. p values were calculated by t-test or ANOVA test (**, p < 0.01; ***, p < 0.001).
Next, to further determine the effect of PSP-DPMI on the activity of p53 signaling, we explored the expression of p53 in these three cell lines (HCT116 p53+/+, MCF7, and A375) and its downstream target p2154,55 in HCT116 p53+/+ cells with different treatments using Western blot analysis. As shown in Figure 5E and Figure S11, compared to PSP, DPMI, and PBS control, PSP-DPMI (200 nM) and Nutlin3 (5 μM) significantly increased the expression of p53 or p21. Correspondingly, PSP-DPMI significantly induced cell apoptosis compared to PSP, DPMI, and PBS control, even a little better than 5 μM Nutlin3 (Figures 5F and S12). Similarly, 200 nM PSP-DPMI and 5 μM Nutlin3 led to an arrest in G0/G1 phase in HCT116 p53+/+ cells compared to other treatments (Figures 5G and S13). Taken together, our data demonstrate that PSP-DPMI inhibits cancer cell growth via reactivating the p53 signaling, even better than commercial MDM2 inhibitor Nutlin3.
Antitumor Activity of PSP-DPMI in a Colon Cancer Xenograft Model.
To test therapeutic efficacy of PSP-DPMI, mice bearing xenografted HCT116 p53+/+ tumors were divided into five groups (n = 6/group) and received a 13-day treatment regimen by injecting PSP-DPMI, PSP, DPMI, doxorubicin (DOX), Nutlin3, and PBS control every other day (dosage: 3 mg/kg for PSP, DPMI, PSP-DPMI, and Nutlin3; 1.5 mg/mg for DOX), respectively. DOX, a first-line chemotherapy, was used as a positive control. As shown in Figure 6B, free DPMI peptide and PSP did not display any therapeutic effect compared to PBS control. In a sharp contrast, PSP-DPMI suppressed tumor growth significantly compared to PBS control, similar to DOX treatment (Figure 6B). On day 13, we isolated the tumors and studied pathomorphological changes. As expected, we observed histological morphological integrity of tumor tissues in the control, DPMI-, and PSP-treated mice, whereas misshapen tumors and cells were found in the DOX- and PSP-DPMI-treated tumors (Figure 6C). The above conclusions were also supported by statistical data on tumor weight (Figure 6D). Similar to the first-line chemotherapy drug DOX, the average tumor weight of PSP-DPMI-treated mice was significantly less than that of control mice and Nutlin3-treated mice (Figure 6C,D). Next, we performed the TUNEL assay to evaluate the apoptosis in xenograft tumors. The results showed that the amounts of apoptotic cells in xenograft tumors were significantly increased upon PSP-DPMI, Nutlin3, and DOX treatment compared to the control, whereas PSP and DPMI almost did not affect cell apoptosis (Figure 6E). Taken together, our data provide strong evidence that the PSP tail can enhance tumor targeting and anticancer efficacy of DPMI.
Figure 6.
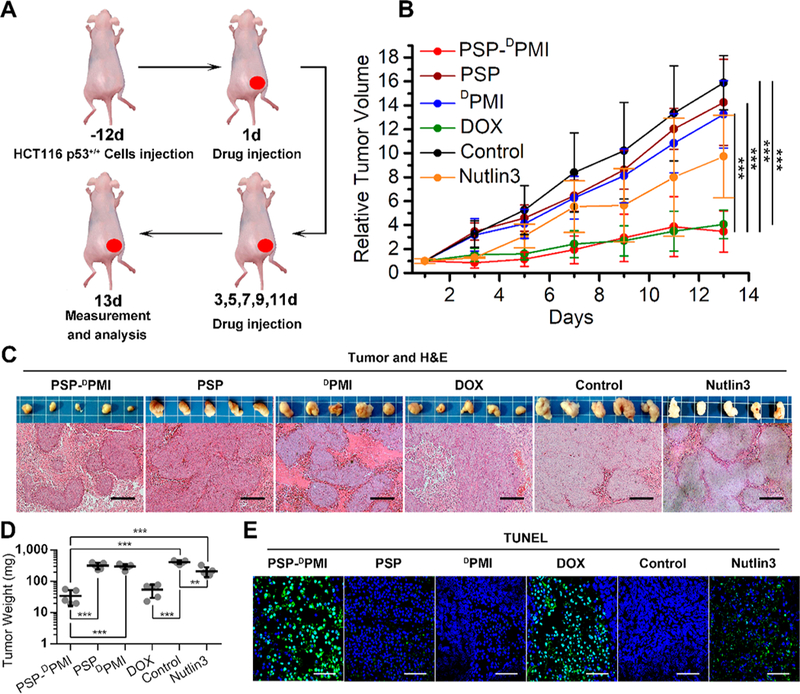
In vivo antitumor activity of PSP-DPMI. (A) Schematic depiction for tumor targeting and therapeutic efficacy of PSP-DPMI in a colon cancer xenograft model by intraperitoneal injection. (B) Tumor growth curves in nude mice subcutaneously inoculated with 1 × 10 HCT116 p53+/+ cells into the right flank. The indicated treatments were administered on days 1, 3, 5, 7, 9, 11, and 13. A statistical analysis was performed using a nonparametric Kruskal–Wallis test. Data are presented as mean ± s.e. (n = 5). (C) Photographs (upper panels) and H&E staining (lower panels) of xenograft tumors from mice with the indicated treatments. Scale bar: 50 μm. (D) Tumor weight of each mouse with the indicated treatments. Data are presented as mean ± s.e. (n = 5). (E) The TUNEL (×200) staining of representative xenograft tumors from mice with the indicated treatments. Scale bar: 60 μm. p values were calculated by t-test or ANOVA test (***, p < 0.001).
Considering that DPMI can reactivate the p53 signaling through blocking interaction between p53 and MDM2/ MDMX (Figure 7A), we performed immunohistochemistry staining in three serial sections from each tumor to assess the levels of p53, p21, and Ki67 proteins (Figure 7B). Expectedly, the levels of p53 and its downstream target p21 were significantly increased in the PSP-DPMI-treated tumors relative to control, PMI- and DPMI-treated tumors (Figure 7C, D). Similarly, the percentage of Ki-67 (a marker of proliferation) positive cells was dramatically decreased in the PSP-DPMI-treated tumors relative to control tumors, whereas PMI- and DPMI treatment almost did not affect the percentage of Ki-67 positive cells relative to control (Figure 7E). Altogether, these findings suggest that PSP-DPMI exhibits potent tumor suppression both in vitro and in vivo via the reactivation of p53 signaling.
Figure 7.
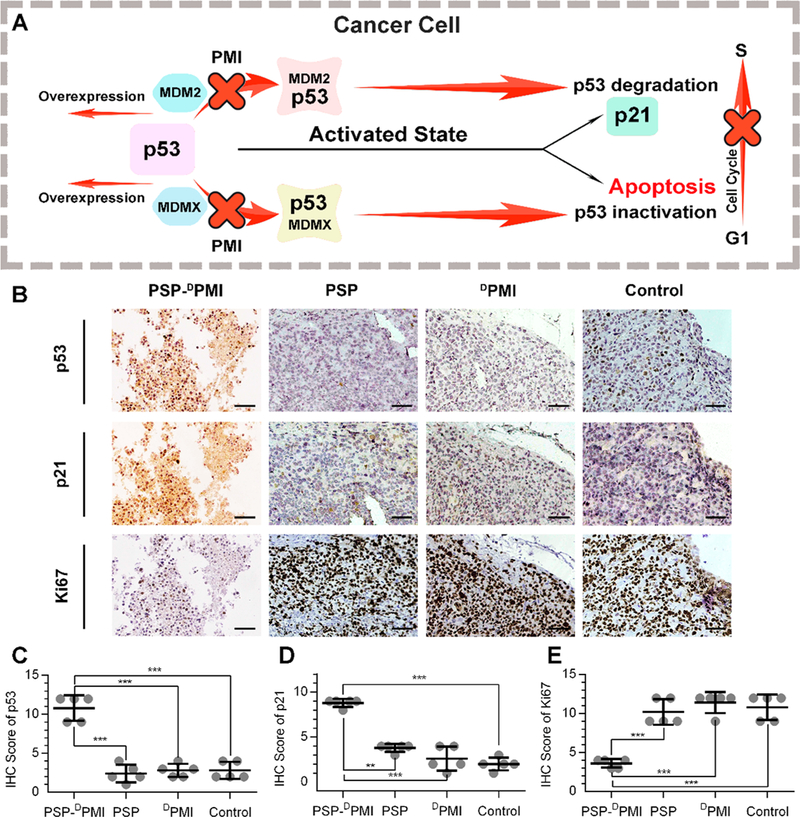
Molecular mechanism underlying tumor-inhibitory effect of PSP-DPMI. (A) Schematic diagram for antitumor activity of PSP-DPMI via reactivating p53 signaling. (B) Representative immunohistochemistry (IHC) staining for p53, p21, and Ki67 in xenograft tumors from mice with the indicated treatments. Three consecutive tissue sections of each tumor were stained, and the same areas were shown in the image. IHC score for p53 (C), p21(D), and ki67 (E) was awarded by two independent observers according to the percentage and intensity of the stained cells. Scale bar: 50 μm. p values were calculated by t-test (**, p < 0.01; ***, p < 0.001).
In Vivo Safety Evaluation of PSP-DPMI.
Without exception, systemic toxicity from off-target drugs is a big challenge for the development of cancer drugs, for which serious side effects and strict dose constraints will result in the failure in the stages of development and clinic trial.56 In addition, due to the intrinsic pharmacological obstacles of macromolecular therapeutics, they have to been packed inside various vehicles to deliver into tumor cells. Unfortunately, the added benefit of delivering cargo comes with harmful side effects from the limited biocompatibility of vehicles.4–6 To minimize these off-targeted and vehicle-associated side effects, a combined strategy was designed in the PSP-DPMI spherical shells. In brief, with the help of the EPR effect, tumor-microenvironment sensitivity and RGD targeting, PSP-DPMI can specifically accumulate at the tumor sites and be preferentially retained at least 24 h after injection. These properties dramatically decrease the probability of off-target effects, thereby eliminating side effects and extending dose range (Figure 8A). In addition, PSP was synthesized by nature amino acid, its monomer can be degraded rapidly after the TME-triggered delivery, and additional cytotoxic vehicles were not introduced into this system, further improving the safety of PSP-based nanoshells.
Figure 8.
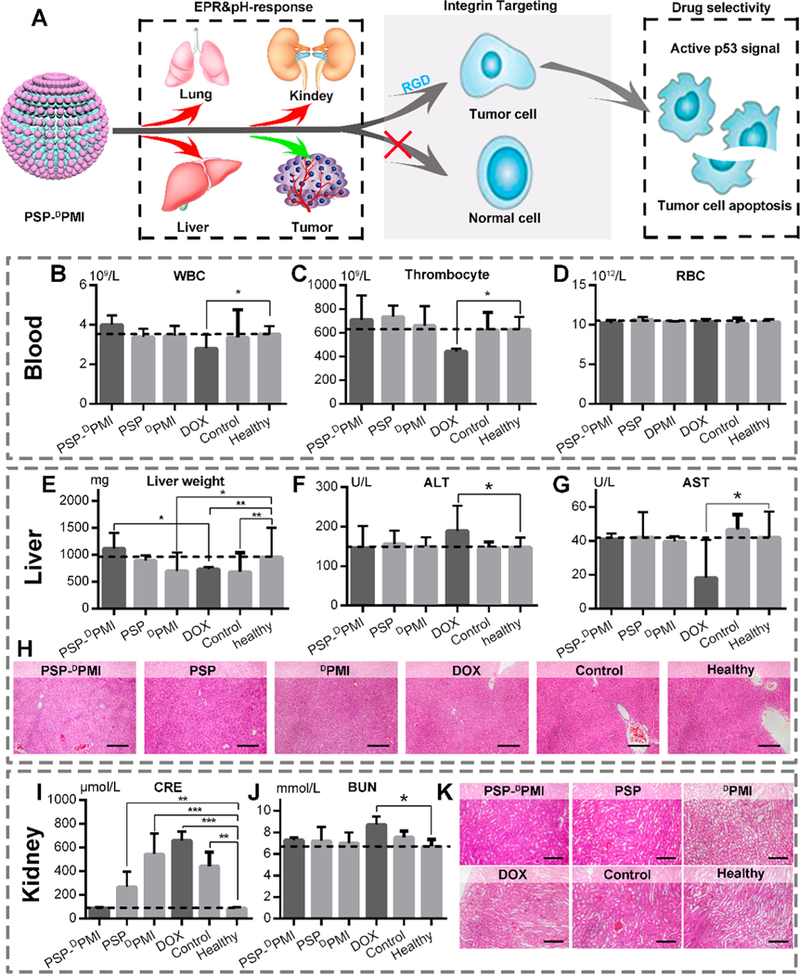
Safety evaluation of PSP-DPMI in vivo. (A) Diagrammatic view of tumor specificity of PSP-DPMI. (B–D) The count of white blood cells (WBCs), thrombocyte, and red blood cells (RBCs) in mice with the indicated treatments. (E) Liver weight of mice with the indicated treatments. (F, G) The activities of two serum enzymes related to liver function in mice with the indicated treatments. ALT, alanine transaminase; AST, aspartate aminotransferase. (H) The representative H&E staining of liver sections in mice with the indicated treatments (scale bar: 50 μm). (I, J) The measurement of renal function indicators in mice with the indicated treatments. CRE, serum creatinine; BUN, blood urea nitrogen. (K) The representative H&E staining of kidney sections in mice with the indicated treatments (scale bar: 50 μm). Data were presented as mean ± s.e. p values were calculated by t-test (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
To verify biosafety of PSP-DPMI in vivo, comprehensive drug toxicity testing and disease diagnosis were carried out during and after anticancer treatments with DOX as a control. Accumulating evidence has demonstrated that DOX as a first-line chemotherapy drug has some undesirable off-targeted effects;57,58 thus, it is appropriate to be a positive control in the biosafety studies. As expected, our data showed that body weight of PSP-DPMI-, PSP-, and DPMI-treated mice did not show a significant difference from control mice after a 13-day administration, whereas DOX-treated mice presented significant decrease of body weight (Figure S14). In addition, DOX-treated mice showed a certain level of leukopenia and thrombopenia, whereas other mice remained as healthy as the control or healthy mice (Figure 8B–D). Given that liver and kidney are the main organs of drug metabolism, we roundly evaluated the drug toxicity to these two organs. First, we determined the effect of drug treatments on liver weight and function. As shown in Figure 8E, control and DPMI- and DOX-treated mice showed a decreased liver weight compared to healthy mice, whereas liver weight was no significant difference between PSP-DPMI-treated and healthy mice, further supporting excellent biosafety of PSP-DPMI. In addition, aberrant levels of two important liver enzymes alanine transaminase (ALT) and aspartate aminotransferase (AST) were found in the DOX-treated mice compared to control or healthy mice. Conversely, PSP-DPMI almost did not affect the levels of these two enzymes (Figure 8F,G). This was also supported by histopathological observations of liver sections (Figure 8H). Next, the kidney function was examined by serum creatinine (CRE, Figure 8I), blood urea nitrogen (BUN, Figure 8J), and the histological changes (H&E staining, Figure 8K). Similarly, PSP-DPMI had no harmful effects on the kidney, whereas the DOX-treated mice showed the symptoms of glomerular lesions (Figure 8I–K). Moreover, DOX-treated mice showed a serious spleen failure, whereas the health of the spleen was maintained in the PSP-DPMI-treated mice (Figure S15). Notably, all treatments used in the present study did not cause some other common drug toxicities, such as myocardial damage (Figure S16), acute sepsis (Figure S17), and allergic lung resistance (Figure S18). Collectively, PSP-DPMI is sufficiently safe and has a great potential for clinical application.
It is the fact that although the ground-breaking discovery of p53 was 30 years ago, p53 agonist have only recently entered clinical trials.59,60 Nutlins, a kind of ramification from imidazoline, have been identified as a potential molecule to reactivate p53 and used in various phases of clinical trials for several cancers.61,62 However, nutlins have also been shown to have some toxicity to B lymphocytes, peripheral blood mononuclear cells, and bone marrow hematopoietic progenitors.57 More unfortunately, thrombocytopenia and neutropenia have been seen in many patients in a clinical trial of RG7112 (a member of the nutlin family) in liposarcoma, and all patients experienced at least one adverse event, such as nausea, vomiting, asthenia, diarrhea, and thrombocytopenia.63 Evidently, on-target activation of PUMA and NOXA by p53 may be a major cause of thrombocytopaenia and neutropenia through inducing the apoptosis of the relevant cell populations.59 Besides, nutlins also have been demonstrated to have some off-target biological effects, such as induction of DNA damage or cell differentiation in a p53-independent manner. In short, the side effects of nutlins severely limit the process of clinical trials and fetter the development of p53 activators. Fortunately, PSP-DPMI fabricated in the present study will offer a new strategy to overcome these side effects and bring to light p53 anticancer peptide therapeutics.
CONCLUSIONS
In the present study, we construct a well-defined self-assembled nanospherical shell with ultrasensitive size and charge-switchable responsiveness to tumor acidic pH by a PSP tail, thereby realizing the nanoengineering of anticancer peptides and endowing them with the pharmacological necessities including cytomembrane penetrability, long circulation time and strong tumor targeting. As an initial proof of concept, a p53-activating D-enantiomeric peptide termed DPMI is modified by a PSP tail and self-assembled into stable hollow spheres (PSP-DPMI). Taking full advantage of the EPR effect, tumor-microenvironment sensitivity, and RGD targeting, PSP-DPMI can specifically accumulate at the tumor sites with long residence time. Via systematic in vitro and in vivo experiments, we demonstrate that PSP-DPMI has potent antitumor activity and excellent biosafety. More importantly, except for peptides, other macromolecule therapeutic drugs such as small proteins and nucleotide drugs also can be integrated into these PSP derived building blocks and self-assemble to form anticancer nanospherical shells. In summary, this general strategy will simultaneously overcome the technical obstacles for self-assembled nanoparticles to be macromolecular drug carriers and the pharmaceutical obstacles for macromolecular therapeutics to be used in clinical settings through the peptide-tuned self-assembly of macromolecular drugs into a TME triggered shape- and charge-switchable nanoshell.
EXPERIMENTAL SECTION
Fabrication of PSP-DPMI Spherical Shell.
N-Succinimidyl maleimidoacetate was the first to be conjugated with the sulfydryl at as prepared PSP by chemical synthesis.64 In detail, 5 mg of N-succinimidyl maleimidoacetate and 5 mg of PSP were dissolved in 10 mL of PBS-acetonitrile solution (50% volume of PBS, pH 7.4, and 50% volume acetonitrile). After 30 min of stirring, the PSP-NHS was collected by HPLC and lyophilization. PSP-DPMI was prepared by a mild reaction between PSP-NHS (0.1 mM) and the amino of DPMI (0.15 mM) in PBS buffer (pH 8.5). After 30 min of magnetic stirring, the purified PSP-DPMI was obtained after liquid chromatography purification and lyophilization. In the spherical shell, PSP-DPMI was dissolved at a high concentration (0.2 mg/mL) in a PBS solution at pH 5.0 and then adjusted the pH to 7.4 to trigger the self-assembly. The PSP self-assembly was performed using the same protocol as described for PSP-DPMI.
Hydrodynamic Size and Zeta Potential Measurement.
The Malvern Zetasizer Nano ZS system were used to measure hydrodynamic size distribution and zeta potential of the peptide nanostructure. Peptide was solved in PBS buffer at the concentration from 0.05 mg/mL to 0.2 mg/mL, then 1 mL of this solution was transferred into a 1 cm cell, and the hydrodynamic size distribution was measured by dynamic light scattering (DLS) at 20 or 37 °C. For zeta potential measurement, the peptides (0.10 mg/mL, 1 mL) were incubated with PBS at different pH values at 37 °C for 30 min and measured by dynamic light scattering (DLS). Data was analyzed by Dispersion Technology Software V5.03, and the results of each sample were conducted from three or four independent measurements.
Cellular Uptake of PSP-DPMI.
DPMI and PSP-DPMI were first labeled by FITC and redispersed in McCoy’s 5A medium (50 μg/ mL). Meanwhile, HCT116 cells were cultured in the McCoy’s 5A medium with 10% FBS for 24 h. Next, the culture medium was replaced by the medium containing FITC-labeled DPMI and PSP-DPMI (DPMIFITC and PSP-DPMIFITC) at pH 7.4 or pH 6.5, and cells were further cultured at 37 °C for 6 h. After incubation, the cells were washed by PBS three times to remove the excess peptide. Cellular uptake of DPMI and PSP-DPMI was detected by confocal laser scanning microscopy (CLSM, FV1200, Olympus) and flow cytometer (BD Biosciences, NJ). For cell imaging, Hoechst33342 (Molecular probes) was used as the control. To prevent cells from falling, cells were fixed with paraformaldehyde before observation. As for CLSM imaging, the excitation was provided by the continuous wave laser at 405 nm for Hoechst33342 (3.15 mW) and 543 nm for FITC (0.7 mW), respectively. Of note, 2 mM HEPES-HCL was used to adjust the pH of the cell culture medium to 6.5.
Establishment of Colon Cancer Xenograft Model.
All experimental procedures involving animals were conducted in accordance with Institution Guidelines and were approved by the Laboratory Animal Center of Xi’an Jiaotong University as our previous protocol.65
In Vivo Biodistribution Analysis.
FITC was first conjugated to the amino group at the N-terminal of DPMI to construct DPMIFITC and PSP-PMIFITC as our precious protocol.66 Next, tumor-bearing mice were divided into two groups (n = 3/group) randomly and injected with 200 μL of DPMIFITC (1 mg/mL) or PSP-DPMIFITC (1 mg/mL), respectively. The distribution image of the FITC-labled peptide in the tumor-bearing mice was taken by an IVIS Spectrum in vivo imaging system (Xenogen, U.S.A., irradiation wavelength: λex = 500 nm, detected wavelength: λem = 520 nm) as in our precious reports.65 To emphasize the peptide-accumulated areas, we set a threshold (Figure 4D: 5 × 108; Figure S7: 1 × 109) and represent intense fluorescence above it. All fluorescence imaging in this work was carried out with the same device.
Evaluation of in Vivo Antitumor Activity.
PSP, DPMI, PSP-DPMI, and Nutlin-3 were administered at 3 mg/kg. Considering that 1.5 mg/kg is the maximum safe dose of DOX for BALB/c nude mice, DOX was administered at a dose of 1.5 mg/kg. Animal treatment and evaluation were the same as in our previous work.65 Additionally, blood routine examination, detection, and analysis on indexes of liver and kidney function and some biochemical enzyme indexes were carried out at the Clinical Laboratory, the First Affiliated Hospital of Xi’an Jiaotong University, according to standard clinical laboratory procedures.
Statistical Analysis.
Statistical analyses were carried out by two-sided Student’s t-test for the comparation between two groups. ANOVA test was used for the comparison of more than two groups. p < 0.05 stands for the significance. Event—time plots were made using the Kaplan—Meier technique. Data were expressed as mean ± s.d. or s.e.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No. 81572627 to P. H.), the Clinical Research Award of the First Affiliated Hospital of Xi’an Jiaotong University (No. XJTU1AF-CRF-2017–003 to P.H. and W.H.), and National Institutes of Health Grants (No. CA167296 and CA219150 to W.L.). The authors gratefully acknowledge financial support from China Scholarship Council (to W.H. and J.Y.) and also thank Prof. Bert Vogelstein (Ludwig Center at John Hopkins, U.S.A.) for kindly providing human colon cancer cell line HCT116 p53+/+ carrying wild-type p53 and the isogenic HCT116 p53–/– cell line with both p53 alleles deleted.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemma-ter.8b02572.
General information protocols of the necessary cell and animal experiments and 18 supplementary figures (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Azad N; Rojanasakul Y Macromolecular Drug Delivery In Biopharmaceutical Drug Design and Development; Wu-Pong S, Rojanasakul Y, Eds.; Springer: New York, 2008; pp 293–323. [Google Scholar]
- (2).Ye J; Shin MC; Liang Q; He H; Yang VC 15 Years of ATTEMPTS: A Macromolecular Drug Delivery System Based on the CPP-Mediated Intracellular Drug Delivery and Antibody Targeting. J. Controlled Release 2015, 205, 58–69. [DOI] [PubMed] [Google Scholar]
- (3).Bechet D; Couleaud P; Frochot C; Viriot M-L; Guillemin F; Barberi-Heyob M Nanoparticles as Vehicles for Delivery of Photodynamic Therapy Agents. Trends Biotechnol 2008, 26, 612–621. [DOI] [PubMed] [Google Scholar]
- (4).Ruan R; Chen M; Zou L; Wei P; Liu J; Ding W; Wen L Recent Advances in Peptides for Enhancing Transdermal Macro-molecular Drug Delivery. Ther. Delivery 2016, 7, 89–100. [DOI] [PubMed] [Google Scholar]
- (5).Kakkar A; Traverso G; Farokhzad OC; Weissleder R; Langer R Evolution of Macromolecular Complexity in Drug Delivery Systems. Nat. Rev. Chem. 2017, 1, 0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gaspar R; Duncan R Polymeric Carriers: Preclinical Safety and the Regulatory Implications for Design and Development of Polymer Therapeutics. Adv. Drug Delivery Rev. 2009, 61, 1220–1231. [DOI] [PubMed] [Google Scholar]
- (7).Pathak Y Recent Developments in Nanoparticulate Drug Delivery Systems In Drug Delivery Nanoparticles Formulation and Characterization; Pathak Y, Thassu D, Eds.; CRC Press: Boca Raton, 2016; pp 19–33. [Google Scholar]
- (8).Zou Q; Liu K; Abbas M; Yan X Peptide-Modulated Self-Assembly of Chromophores toward Biomimetic Light-Harvesting Nanoarchitectonics. Adv. Mater. 2016, 28, 1031–1043. [DOI] [PubMed] [Google Scholar]
- (9).Liu K; Xing R; Zou Q; Ma G; Mohwald H; Yan X Simple Peptide-Tuned Self-Assembly of Photosensitizers towards Anticancer Photodynamic Therapy. Angew. Chem. Int. Ed. 2016, 55, 3036–3039. [DOI] [PubMed] [Google Scholar]
- (10).Habibi N; Kamaly N; Memic A; Shafiee H Self-Assembled Peptide-Based Nanostructures: Smart Nanomaterials toward Targeted Drug Delivery. Nano Today 2016, 11, 41–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Marcucci F; Stassi G; De Maria R Epithelial-Mesenchymal Transition: a New Target in Anticancer Drug Discovery. Nat. Rev. Drug Discovery 2016, 15, 311–325. [DOI] [PubMed] [Google Scholar]
- (12).Han K; Zhang J; Zhang W; Wang S; Xu L; Zhang C; Zhang X; Han H Tumor-Triggered Geometrical Shape Switch of Chimeric Peptide for Enhanced in vivo Tumor Internalization and Photodynamic Therapy. ACS Nano 2017, 11, 3178–3188. [DOI] [PubMed] [Google Scholar]
- (13).Paris JL; Cabanas MV; Manzano M; Vallet-Regi M Polymer-Grafted Mesoporous Silica Nanoparticles as Ultrasound-Responsive Drug Carriers. ACS Nano 2015, 9, 11023–11033. [DOI] [PubMed] [Google Scholar]
- (14).Zhou N; Cao X; Du X; Wang H; Wang M; Liu S; Nguyen K; Schmidt-Rohr K; Xu Q; Liang G; Xu B Hyper–Crosslinkers Lead to Temperature-and pH-Responsive Polymeric Nanogels with Unusual Volume Change. Angew. Chem., Int. Ed. 2017, 56, 2623–2627. [DOI] [PubMed] [Google Scholar]
- (15).Wang H; Zhao S; Agarwal P; Dumbleton J; Yu J; Lu X; He X Multi-Layered Polymeric Nanoparticles for pH-Responsive and Sequenced Release of Theranostic Agents. Chem. Commun. 2015, 51, 7733–7736. [DOI] [PubMed] [Google Scholar]
- (16).Kanapathipillai M; Brock A; Ingber DE Nanoparticle Targeting of Anti-cancer Drugs that Alter Intracellular Signaling or Influence the Tumor Microenvironment. Adv. Drug Delivery Rev. 2014, 79, 107–118. [DOI] [PubMed] [Google Scholar]
- (17).Ge Z; Liu S Functional Block Copolymer Assemblies Responsive to Tumor and Intracellular Microenvironments for Site-Specific Drug Delivery and Enhanced Imaging Performance. Chem. Soc. Rev. 2013, 42, 7289–7325. [DOI] [PubMed] [Google Scholar]
- (18).Mitragotri S; Lahann J Physical Approaches to Biomaterial Design. Nat. Mater. 2009, 8, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tang L; Yang X; Yin Q; Cai K; Wang H; Chaudhury I; Yao C; Zhou Q; Kwon M; Hartman JA; et al. Investigating the Optimal Size of Anticancer Nanomedicine. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 15344–15349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Blanco E; Shen H; Ferrari M Principl es of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol. 2015, 33, 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yan J; He W; Li N; Yu M; Du Y; Lei B; Ma PX Simultaneously Targeted Imaging Cytoplasm and Nucleus in Living Cell by Biomolecules Capped Ultra-Small GdOF Nanocrystals. Biomaterials 2015, 59, 21–29. [DOI] [PubMed] [Google Scholar]
- (22).Zhang J; Yan J; Yang Q; Yan Y; Li S; Wang L; Li C; Lei B; Yang G; He W Arginine-Modified Dual Emission Photo-luminescent Nanocrystals for Bioimaging at Subcellular Resolution. J. Biomater. Appl 2017, 32, 533–542. [DOI] [PubMed] [Google Scholar]
- (23).Chauhan VP; Jain RK Strategies for Advancing Cancer Nanomedicine. Nat. Mater. 2013, 12, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Popovic Z; Liu W; Chauhan VP; Lee J; Wong C; Greytak AB; Insin N; Nocera DG; Fukumura D; Jain RK; Bawendi MG A Nanoparticle Size Series for in vivo Fluorescence Imaging. Angew. Chem. 2010, 122, 8831–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chauhan VP; Stylianopoulos T; Martin JD; Popovic Z; Chen O; Kamoun WS; Bawendi MG; Fukumura D; Jain RK Normalization of Tumour Blood Vessels Improves the Delivery of Nanomedicines in a Size-dependent Manner. Nat. Nanotechnol 2012, 7, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Choi HS; Liu W; Liu F; Nasr K; Misra P; Bawendi MG; Frangioni JV Design Considerations for Tumour-Targeted Nanoparticles. Nat. Nanotechnol. 2010, 5, 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Albanese A; Tang PS; Chan WC The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [DOI] [PubMed] [Google Scholar]
- (28).Davis ME; Shin DM; Chen Z Nanoparticle Therapeutics: an Emerging Treatment Modality for Cancer. Nat. Rev. Drug Discovery 2008, 7, 771–782. [DOI] [PubMed] [Google Scholar]
- (29).De La Rica R; Matsui H Applications of Peptide and Protein-Based Materials in Bionanotechnology. Chem. Soc. Rev. 2010, 39, 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Quader S; Kataoka K Nanomaterial-Enabled Cancer Therapy. Mol. Ther. 2017, 25, 1501–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Maeda H; Nakamura H; Fang J The EPR Effect for Macromolecular Drug Delivery to Solid Tumors: Improvement of Tumor Uptake, Lowering of Systemic Toxicity, and Distinct Tumor Imaging in vivo. Adv. Drug Delivery Rev. 2013, 65, 71–79. [DOI] [PubMed] [Google Scholar]
- (32).Torchilin V Tumor Delivery of Macromolecular Drugs Based on the EPR Effect. Adv. Drug Delivery Rev. 2011, 63, 131–135. [DOI] [PubMed] [Google Scholar]
- (33).Liu M; Pazgier M; Li C; Yuan W; Li C; Lu W A Left-Handed Solution to Peptide Inhibition of the p53-MDM2 Interaction. Angew. Chem. Int. Ed. 2010, 49, 3649–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liu M; Li C; Pazgier M; Li C; Mao Y; Lv Y; Gu B; Wei G; Yuan W; Zhan C; et al. D-Peptide Inhibitors of the p53-MDM2 Interaction for Targeted Molecular Therapy of Malignant Neoplasms. Proc. Natl. Acad. Sci U. S. A. 2010, 107, 14321–14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).McGregor DP Discovering and Improving Novel Peptide Therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [DOI] [PubMed] [Google Scholar]
- (36).Werle M; Bernkop-Schnurch A Strategies to Improve Plasma Half Life Time of Peptide and Protein Drugs. Amino Acids 2006, 30, 351–367. [DOI] [PubMed] [Google Scholar]
- (37).Abbas M; Zou Q; Li S; Yan X Self-Assembled Peptide-and Protein-Based Nanomaterials for Antitumor Photodynamic and Photothermal Therapy. Adv. Mater. 2017, 29, 1605021. [DOI] [PubMed] [Google Scholar]
- (38).Howorka S Rationally Engineering Natural Protein Assemblies in Nanobiotechnology. Curr. Opin. Biotechnol 2011, 22, 485–491. [DOI] [PubMed] [Google Scholar]
- (39).King NP; Bale JB; Sheffler W; McNamara DE; Gonen S; Gonen T; Yeates TO; Baker D Accurate Design of Co-Assembling Multi-Component Protein Nanomaterials. Nature 2014, 510, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Eskandari S; Guerin T; Toth I; Stephenson RJ Recent Advances in Self-Assembled Peptides: Implications for Targeted Drug Delivery and Vaccine Engineering. Adv. Drug Delivery Rev. 2017, 110, 169–187. [DOI] [PubMed] [Google Scholar]
- (41).Putnam C Protein Calculator v3.4. http://protcalc.sourceforge.net (accessed Feb. 2, 2018).
- (42).Zhao F; Ma ML; Xu B Molecular Hydrogels of Therapeutic Agents. Chem. Soc. Rev. 2009, 38, 883–891. [DOI] [PubMed] [Google Scholar]
- (43).Lifson S; Sander C Antiparallel and Parallel β-Strands Differ in Amino Acid Residue Preferences. Nature 1979, 282, 109–111. [DOI] [PubMed] [Google Scholar]
- (44).Wibowo SH; Sulistio A; Wong EH; Blencowe A; Qiao GG Functional and Well-Defined β-Sheet-Assembled Porous Spherical Shells by Surface-Guided Peptide Formation. Adv. Funct. Mater. 2015, 25, 3147–3156. [Google Scholar]
- (45).Li H-J; Du J-Z; Du X-J; Xu C-F; Sun C-Y; Wang HX; Cao Z-T; Yang X-Z; Zhu Y-H; Nie S; Wang J Stimuli-Responsive Clustered Nanoparticles for Improved Tumor Penetration and Therapeutic Efficacy. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Maeda H The Enhanced Permeability and Retention (EPR) Effect in Tumor Vasculature: the Key Role of Tumor-Selective Macromolecular Drug Targeting. Adv. Enzyme Regul 2001, 41, 189–207. [DOI] [PubMed] [Google Scholar]
- (47).Chambers E; Mitragotri S Prolonged Circulation of Large Polymeric Nanoparticles by Non-Covalent Adsorption on Erythrocytes. J. Controlled Release 2004, 100, 111–119. [DOI] [PubMed] [Google Scholar]
- (48).Zhang L; Gu F; Chan J; Wang A; Langer R; Farokhzad O Nanoparticles in Medicine: Therapeutic Applications and Developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [DOI] [PubMed] [Google Scholar]
- (49).Zhan C; Zhao L; Wei X; Wu X; Chen X; Yuan W; Lu W-Y; Pazgier M; Lu W An Ultrahigh Affinity d-Peptide Antagonist Of MDM2. J. Med. Chem. 2012, 55, 6237–6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wade M; Li Y-C; Wahl GM MDM2, MDMX and p53 in Oncogenesis and Cancer Therapy. Nat. Rev. Cancer 2013, 13, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kubbutat MH; Jones SN; Vousden KH Regulation of p53 Stability by Mdm2. Nature 1997, 387, 299–303. [DOI] [PubMed] [Google Scholar]
- (52).Zhang Q; Zeng SX; Lu H Targeting p53-MDM2-MDMX Loop for Cancer Therapy In Mutant p53 and MDM2 in Cancer; Deb SP, Deb S, Eds.; Springer: New York, 2014; pp 281–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Vassilev LT; Vu BT; Graves B; Carvajal D; Podlaski F; Filipovic Z; Kong N; Kammlott U; Lukacs C; Klein C; et al. In vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [DOI] [PubMed] [Google Scholar]
- (54).Xiong Y; Hannon GJ; Zhang H; Casso D; Kobayashi R; Beach D p21 is a Universal Inhibitor of Cyclin Kinases. Nature 1993, 366, 701–704. [DOI] [PubMed] [Google Scholar]
- (55).Bunz F; Dutriaux A; Lengauer C; Waldman T; Zhou S; Brown J; Sedivy J; Kinzler K; Vogelstein B Requirement for p53 and p21 to Sustain G2 Arrest after DNA Damage. Science 1998, 282, 1497–1501. [DOI] [PubMed] [Google Scholar]
- (56).Khanna I Drug Discovery in Pharmaceutical Industry: Productivity Challenges and Trends. Drug Discovery Today 2012, 17, 1088–1102. [DOI] [PubMed] [Google Scholar]
- (57).Coates A; Abraham S; Kaye SB; Sowerbutts T; Frewin C; Fox R; Tattersall M On the Receiving End—Patient Perception of the Side-Effects of Cancer Chemotherapy. Eur. J. Cancer Clin. Oncol. 1983, 19, 203–208. [DOI] [PubMed] [Google Scholar]
- (58).de Boer-Dennert M; De Wit R; Schmitz P; Djontono J; v Beurden V; Stoter G; Verweij J Patient Perceptions of the Side-Effects of Chemotherapy: the Influence of 5HT3 Antagonists. Br. J. Cancer 1997, 76, 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Khoo KH; Verma CS; Lane DP Drugging the p53 Pathway: Understanding the Route to Clinical Efficacy. Nat. Rev. Drug Discovery 2014, 13, 217–236. [DOI] [PubMed] [Google Scholar]
- (60).Bykov VJ; Eriksson SE; Bianchi J; Wiman KG Targeting Mutant p53 for Efficient Cancer Therapy. Nat. Rev. Cancer 2018, 18, 89–102. [DOI] [PubMed] [Google Scholar]
- (61).Burgess A; Chia KM; Haupt S; Thomas D; Haupt Y; Lim E Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, DOI: 10.3389/fonc.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhao Y; Aguilar A; Bernard D; Wang S Small-Molecule Inhibitors of the MDM2-p53 Protein-Protein Interaction (MDM2 Inhibitors) in Clinical Trials for Cancer Treatment: Miniperspective. J. Med. Chem. 2015, 58, 1038–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ray-Coquard I; Blay J-Y; Italiano A; Le Cesne A; Penel N; Zhi J; Heil F; Rueger R; Graves B; Ding M; et al. Effect of the MDM2 Antagonist RG7112 on the P53 Pathway in Patients with MDM2-Amplified, Well-Differentiated or Dedifferentiated Liposarcoma: an Exploratory Proof-of-Mechanism Study. Lancet Oncol. 2012, 13, 1133–1140. [DOI] [PubMed] [Google Scholar]
- (64).Fields GB; Fields CG Solvation Effects in Solid-Phase Peptide Synthesis. J. Am. Chem. Soc. 1991, 113, 4202–4207. [Google Scholar]
- (65).Yan J; He W; Yan S; Niu F; Liu T; Ma B; Shao Y; Yan Y; Yang G; Lu W; et al. Self-Assembled Peptide-Lanthanide Nanoclusters for Safe Tumor Therapy: Overcoming and Utilizing Biological Barriers to Peptide Drug Delivery. ACS Nano 2018, 12, 2017–2026. [DOI] [PubMed] [Google Scholar]
- (66).Niu F; Yan J; Ma B; Li S; Shao Y; He P; Zhang W; He W; Ma PX; Lu W Lanthanide-doped nanoparticles conjugated with an anti-CD33 antibody and a p53-activating peptide for acute myeloid leukemia therapy. Biomaterials 2018, 167, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.