

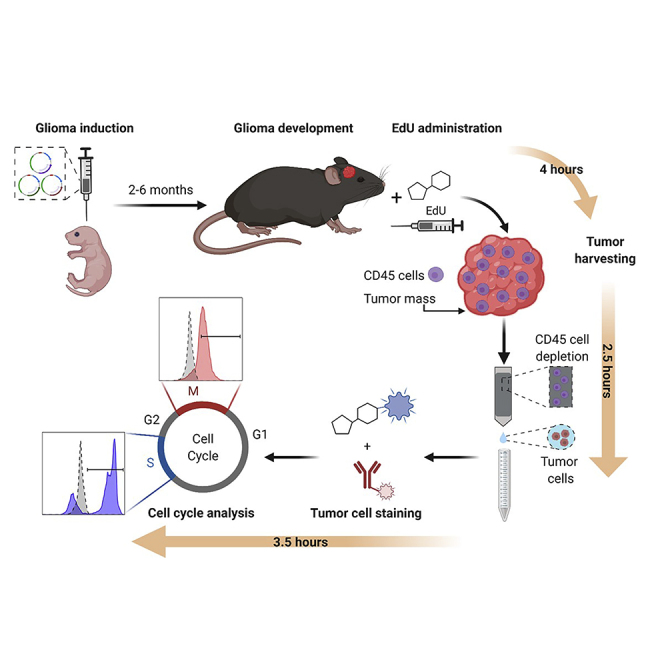
Summary
Malignant gliomas are the most common and aggressive primary brain tumor in adults, and high mitotic rates are associated with their malignancy. Gliomas were modeled in mice using the Sleeping Beauty system to encode genetic lesions recapitulating the human disease. The presented workflow allows the study of the proliferation of glioma cells in vivo, enabling the identification of different phases of the cell cycle, with the advantage that 5-ethynyl-2′-deoxyuridine staining does not involve denaturation steps and samples do not require histological processing.
For complete details on the use and execution of this protocol, please refer to Núñez et al. (2019).
Graphical Abstract
Highlights
-
•
An optimized protocol to analyze in vivo cell cycle progression in glioma models
-
•
Genetically engineered gliomas are modeled in mice using the Sleeping Beauty method
-
•
EdU incorporation and histone 3-serine-10 phosphorylation are measured in tumor cells
-
•
Analysis is done by flow cytometry and samples do not require histological processing
An optimized protocol to analyze in vivo cell cycle progression in genetically engineered gliomas that are modeled in mice using the Sleeping Beauty method. EdU incorporation and histone 3-serine-10 phosphorylation are analyzed by flow cytometry, enabling the assessment of cells undergoing S or M phase, respectively.
BEFORE YOU BEGIN
In vivo profiling of glioma cells’ cell cycle analysis is performed in a genetically engineered glioma model in mice utilizing the Sleeping Beauty (SB) transposon system (Wiesner et al., 2009, Calinescu et al., 2015, Koschmann et al., 2016, Núñez et al., 2019, Garcia-Fabiani et al., 2020, Comba et al., 2020). To achieve this, plasmids encoding the different genetic alterations of interest, which will be stably inserted into the genome of mouse brain cells, need to be constructed. Those sequences encoding for genetic alterations, i.e. the transposons, are flanked by inverted repeats/directed repeats (IR/DR) that will be recognized by the SB transposase. This enzyme will integrate these transposons into the genome in a “cut and paste” manner (Calinescu et al., 2015). One advantage of this method is that the expression of these genes is coupled to the expression of fluorescent proteins, so that when gliomas develop, tumors can be recognized under a stereo-zoom microscope equipped with a fluorescent light (Figure 1A). These plasmids are co-injected along with a plasmid encoding for the SB transposase. This plasmid also contains the coding sequence for the Luciferase enzyme flanked by IR/DR, which will get inserted into the host genome as well (Figure 1A). This enables the study of plasmid uptake and tumor growth by bioluminescence. Refer to (Wiesner et al., 2009, Calinescu et al., 2015) for a detailed cloning protocol of SB-compatible plasmids (i.e. pKT-IRES-Katushka). Depending on the type of genetic alterations introduced, tumors will develop and animals will reach the humane endpoint within 2 to 6 months after the SB procedure (Calinescu et al., 2015, Koschmann et al., 2016, Núñez et al., 2019, Comba et al., 2020) (Table 1). In vivo 5-ethynyl-2′-deoxyuridine (EdU) staining is performed when the tumor is large, i.e., when bioluminescence reaches a signal of 106 photons/s/cm2/sr, but before the tumor burden endpoint.
Figure 1.

Sleeping Beauty Transposase Method to Model Glioma in Mice
(A) Generic plasmid maps used to generate gliomas in mice using the SB transposase method. GFP and Katushka are green and red fluorescent proteins, respectively. miR-30 sequences flank a shRNA encoding sequence designed to silence specific genes. The DNA transposons to be inserted are flanked by inverted repeats/direct repeats (IR/DR), which are recognized by the transposase.
(B) These sequences are then randomly integrated into the host genomic DNA sites, between bases T and A.
(C) Schematic of a 1 day-old mouse pup, showing the coordinates for plasmid injection into the lateral ventricle, at 1.5 mm rostral and 0.8 mm lateral to the lambda and 1.5 mm ventral.
(D) From left to right: bioluminescence scanning of a mouse pup 1 day after SB plasmid injection, when a large tumor has developed (106 photons/s/cm2/sr) and at tumor burden endpoint (107 photons/s/cm2/sr).
Table 1.
Plasmid Combinations to Generate Sleeping Beauty-Based Gliomas in Mouse
| Plasmid Combination | Addgene Cat. No. | Genetic Alteration | Name | Median Survival (DPI) | Reference |
|---|---|---|---|---|---|
| pT2/SB100x-Luc | 20207 | Luciferase expression | NP | 83–95 | (Calinescu et al., 2015, Koschmann et al., 2016, Núñez et al., 2019, Comba et al., 2020) |
| pT/CAGGS-NRASV12 | 20205 | NRASG12V overexpression | |||
| pT2/shP53 | 124261 | p53 knock down | |||
| pT2/SB100x-Luc | 20207 | Luciferase expression | NPA | 69–80 | (Koschmann et al., 2016, Núñez et al., 2019, Comba et al., 2020) |
| pT/CAGGS-NRASV12 | 20205 | NRASG12V overexpression | |||
| pT2/shP53 | 124261 | p53 knock down | |||
| pT2/shAtrx-GFP4 | 124259 | Atrx knock down | |||
| pT2/SB100x-Luc | 20207 | Luciferase expression | NPAI | 163 | (Núñez et al., 2019) |
| pT/CAGGS-NRASV12 | 20205 | NRASG12V overexpression | |||
| pT2/shP53 | 124261 | p53 knock down | |||
| pT2/shAtrx-GFP4 | 124259 | Atrx knock down | |||
| pKT-IDH1(R132H)-Katushka | 124257 | IDH1R132H overexpression | |||
| pT2/SB100x-Luc | 20207 | Luciferase expression | NPAH | 162 | (Garcia-Fabiani et al., 2020) |
| pT/CAGGS-NRASV12 | 20205 | NRASG12V overexpression | |||
| pT2/shP53 | 124261 | p53 knock down | |||
| pT2/shAtrx-GFP4 | 124259 | Atrx knock down | |||
| pKT-H3.3(G34R)-Katushka | - | H3.3G34R overexpression | |||
| pT2/SB100x-Luc | 20207 | Luciferase expression | NPAF | 142 | (Comba et al., 2020) |
| pT/CAGGS-NRASV12 | 20205 | NRASG12V overexpression | |||
| pT2/shP53 | 124261 | p53 knock down | |||
| pT2/shAtrx-GFP4 | 124259 | Atrx knock down | |||
| pT2-shFYN-GFP4 | - | Fyn knock down | |||
| pT2/SB100x-Luc | 20207 | Luciferase expression | NPF | 131 | (Comba et al., 2020) |
| pT/CAGGS-NRASV12 | 20205 | NRASG12V overexpression | |||
| pT2/shP53 | 124261 | p53 knock down | |||
| pT2-shFYN-GFP4 | - | Fyn knock down | |||
| pT2/SB100x-Luc | 20207 | Luciferase expression | NPD | 63–69 | (Wiesner et al., 2009, Calinescu et al., 2015, Comba et al., 2020) |
| pT/CAGGS-NRASV12 | 20205 | NRASG12V overexpression | |||
| pT2-shp53-PDGFβ-GFP4 | - |
p53 knock down PDGFβ overexpression |
|||
| pT2/SB100x-Luc | 20207 | Luciferase expression | NPDF | 108 | (Comba et al., 2020) |
| pT/CAGGS-NRASV12 | 20205 | NRASG12V overexpression | |||
| pT2-shp53-PDGFβ-GFP4 | - |
p53 knock down PDGFβ overexpression |
|||
| pT2-shFYN-GFP4 | - | Fyn knock down | |||
| pT2/SB100x-Luc | 20207 | Luciferase expression | NL | 30 | (Wiesner et al., 2009, Calinescu et al., 2015) |
| pT/CMVSV40-LgT | 20204 | SV40-LgT overexpression | |||
| pT/CAGGS-NRASV12 | 20205 | NRASG12V overexpression |
As described in the text, one-day-old mouse pups are injected with a plasmid combination to introduce different genetic alterations into the sub-ventricular zone brain cells. The specific genetic background generated in these cells will define the aggressiveness of the tumor developed and the median survival of tumor bearing mice, measured as days post injection (DPI).
Breeding Cage Setup
Timing: 20–30 days
-
1.
Set up a breeding cage three to four weeks before the planned date for SB-injections.
-
2.
Once pregnancy is confirmed, place the male mouse in a separate cage and monitor the female daily until pups are born.
-
3.
At post-natal day 1, proceed with the SB protocol.
Plasmid Preparation for SB Procedure
-
4.
Prepare the plasmid DNA mix with the in vivo transfection reagent (in vivo jetPEI), which will be injected into the lateral ventricle of 1 day-old mouse pups.
CRITICAL: The quality of the DNA to be injected is critical. DNA solutions must be sterile, endotoxin free, and highly concentrated for a small volume injection. Up to five plasmids can be injected per DNA/jetPEI mix.
-
a.
DNA mix: add 2.8 μg of the plasmid encoding the SB transposase (pT2/SB100x-Luc) and 5.75 μg of each plasmid encoding for the transposons. Add 10 μL of 10% glucose and sterile water up to 20 μL.
-
b.
In vivo jetPEI mix: the amount of transfection reagent depends on the amount of total DNA used and it can be calculated using the following formula:
| μL of in vivo jetPEI = [(μg DNA x 3) x N/P ratio]/150, |
where N/P is the optimal ratio of nitrogen residues in the polyethyleneimine (PEI) to the phosphate residues in the DNA, 3 the number of nanomoles of anionic phosphate in 1 μg of nucleic acid, and 150 (mM) the concentration of nitrogen residues in the in vivo jetPEI solution. The optimal N/P factor for in vivo transfections is usually 7.
Add 10 μL of 10% glucose and sterile water up to 20 μL.
-
c.
Combine both mixes by pipetting. Vortex the mixture briefly, spin down and incubate it at 25°C for 20 min.
-
5.
Prepare the stereotaxic frame to be used to perform the intraventricular injections
-
a.
Place the 10 μL syringe with a 30G hypodermic needle into the micropump. Make sure that the injection system is working properly by withdrawing and releasing 10 μL of water.
-
b.
Cool down the stereotactic neonatal stage to 2°C–8°C with a slurry of dry ice placed in alcohol.
-
c.
Prepare a bucket with wet ice to induce anesthesia on the pups.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Histone H3 (Ser10) - PE conjugated (Clone 11D8) | Biolegend | Catalog # 650807 |
| Experimental Models: Organisms/Strains | ||
| Mouse (C57BL/6J) | Jackson Lab | # 000664 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM/F12 | Thermo Fisher Scientific - Gibco | Catalog # 11330032 |
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific - Gibco | Catalog # 14190144 |
| BSA | Millipore Sigma | Catalog # A2153-100G |
| N2 Supplement (100 X) | Thermo Fisher Scientific - Gibco | Catalog # 17502048 |
| B27 Supplement (50 X) | Thermo Fisher Scientific - Gibco | Catalog # 17504044 |
| Anti-Anti (100X) | Corning | Catalog # 30-004-CI |
| Normocin | InvivoGen | Catalog # ant-nr-1 |
| MACS BSA Stock Solution | Miltenyi Biotec | Catalog # 130-091-376 |
| autoMACS Rinsing Solution | Miltenyi Biotec | Catalog # 130-091-222 |
| mouse CD45 MicroBeads | Miltenyi Biotec | Catalog # 130-052-301 |
| EGF | PeproTech | Catalog # AF-100-15 |
| FGF | PeproTech | Catalog # 100-18B |
| In vivo jetPEI (includes 10% glucose solution) | Polyplus | Catalog # 201-10G |
| D-Luciferin Potassium Salt | Goldbio | Catalog # LUCK -3G |
| Red Blood Cell (RBC) lysis buffer (10X) | Biolegend | Catalog # 420301 |
| Fetal Bovine Serum (FBS) USDA Pure Premium | Peak Serum | Catalog # PS-FB3 |
| Critical Commercial Assays | ||
| Click-iT™ Plus EdU Pacific Blue™ Flow Cytometry Assay Kit | Thermo Fisher Scientific - Invitrogen | Catalog # C10636 |
| Recombinant DNA | ||
| pT2/SB100x-Luc | Addgene | Catalog # 20207 |
| pT/CAGGS-NRASV12 | Addgene | Catalog # 20205 |
| pT2/shP53 | Addgene | Catalog # 124261 |
| pKT-IDH1(R132H)-Katushka | Addgene | Catalog # 124257 |
| Software and Algorithms | ||
| FlowJo_V10 | Becton Dickinson | N/A |
| Other | ||
| LS Columns | Miltenyi Biotec | Catalog # 130-042-401 |
| QuadroMACS Separator | Miltenyi Biotec | Catalog # 130-090-976 |
| MACS MultiStand | Miltenyi Biotec | Catalog # 130-042-303 |
| Pre-Separation Filters | Miltenyi Biotec | Catalog # 130-041-407 |
| 10 μl syringe | Hamilton | 700 Series |
| 30 G hypodermic needle | Hamilton | Catalog # 7803-07. Specifications: 30GA RN 6PK 1.25" 15° |
| Cell strainers | Alkali Scientific Incorporated | Catalog # MT4070 |
| BD FACSAria™ III Flowcytometer | Becton, Dickinson & Company | N/A |
| Just for Mouse Stereotaxic Instrument | Stoelting | Catalog # 51730 |
| Quintessential Stereotaxic Injector (QSI) | Stoelting | Catalog # 53311 |
| IVIS Spectrum In Vivo Imaging System | Perkin Elmer | Catalog # 124262 |
| Allegra 6R Refrigerated Benchtop Centrifuge | Beckman Coulter | N/A |
| Small Animal Heat Lamp | Morganville Scientific | Catalog # HL0100 |
| Stereomicroscope System SZX7 (equipped with fluorescent lamp) | Olympus | N/A |
| 50 mL Olympus Polystyrene Tubes | Genesee Scientific Corporation | Catalog # 21-108 |
| 15 mL Olympus Polystyrene Tubes | Genesee Scientific Corporation | Catalog # 21-103 |
| CellPro™ Flow Cytometry Tubes without cap, sterile, 12x75mm | Alkali Scientific Incorporated | Catalog # CT6414 |
| Covidien Monoject 1 mL tuberculin Syringes | Fisher Scientific | Catalog # 22-257-154 |
| Friedman Bone Rongeurs | Stoelting | Catalog # 52160P |
| Micro Dissecting Scissors, 4.25 inch, Straight, 24mm, Sharp | Stoelting | Catalog # 52132-22P |
| Brown-Adson Forceps, 4.7in, Side Teeth | Stoelting | Catalog # 52104-31P |
| BD Precision Glide Hypodermic Needles 26G | Fisher Scientific | Catalog # 14-826-15 |
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maria G. Castro, mariacas@med.umich.edu.
Materials Availability
Catalog numbers for the available plasmids used to generate the genetically engineered mouse glioma models using the Sleeping Beauty method are detailed in Table 1. Those plasmids that are not currently available in Addgene will be provided by the Lead Contact upon request with no restrictions.
Data and Code Availability
No datasets or code were generated during this study.
MATERIALS AND EQUIPMENT
Media and Reagent Preparation for the In Vivo Cell Cycle Analysis
-
•
Neurosphere (NS) media
| Reagent | Final Concentration | Amount |
|---|---|---|
| DMEM/F12 | - | 480 mL |
| Anti-Anti 100 X | 1 X (1 IU/mL Penicillin, 100 μg/mL Streptomycin, 0.25 μg/mL Amphotericin B) | 5 mL |
| N2 supplement 100 X | 1 X | 5 mL |
| B27 supplement 50 X | 1 X | 10 mL |
| Normocin | 100 μg/mL | 1 mL |
Prepare 500 mL of NS media in advance and store it at 4 ˚C. At the time of tumor processing, prepare 50 mL of complete media by adding epidermal growth factor and fibroblast growth factor (EGF and FGF) to a final concentration of 20 ng/mL. Use 5 mL for tumor collection and keep the rest on ice for tissue dissociation.
-
•
BSA-EDTA buffer for magnetic sorting of CD45+ cells
| Reagent | Final Concentration | Amount |
|---|---|---|
| MACS 10% BSA Stock Solution | 0.5% BSA | 1 mL |
| autoMACS Rising Solution | 2 mM Ethylenediaminetetraacetic acid (EDTA) | 19 mL |
Prepare the buffer and check that the pH is 7.2. Keep the buffer cold, at 4–8°C during the procedure.
-
•
Luciferin recipe for tumor volume monitoring
| Reagent | Final Concentration | Amount |
|---|---|---|
| Luciferin | 30 mg/mL | 1 grs |
| DPBS | - | 33 mL |
Make 1 mL aliquots and store at -20°C.
-
•
Prepare Red Blood Cell Lysis Buffer 1X (RBCLB) by diluting 1 mL of 10X RBCLB in 9 mL of Milli-Q water. Keep the buffer cold, at 4–8°C during the procedure.
-
•
Prepare reagents necessary for EdU and Click-iT reactions, following the manufacturer’s instructions:
-
○
EdU dilution to be injected: add 10 mL of PBS to Component A from the kit to obtain a 1 mg/mL dilution.
-
○
BSA-PBS buffer: 1% BSA in DPBS, pH 7.1–7.4.
-
○
EdU buffer additive 10X: add 2 mL of deionized water to component G from the kit and mix until the solution is clear.
-
○
Wash reagent 1X: add 50 mL of Component E from the kit to 450 mL of BSA-PBS buffer to prepare 500 mL of saponin-based washing buffer.
-
•
Prepare flow buffer: 2% FBS in DPBS.
STEP-BY-STEP METHOD DETAILS
Injection of SB-Based Plasmids into the Lateral Ventricle of Neonatal Mice to Generate Glioma In Situ
This workflow for inducing brain tumors in neonatal day 1 pups has been adopted from (Wiesner et al., 2009) and is extensively used in our group to develop low or high grade glioma models harboring different genotypes (Calinescu et al., 2015, Koschmann et al., 2016, Núñez et al., 2019, Garcia-Fabiani et al., 2020, Comba et al., 2020). Brain tumors are induced by transposon-mediated integration of genetic alterations delivered in plasmid DNA into the host genome, without the need of viral vectors (Figures 1A and 1B) (Table 1). The tumors arise from the glial cells next to the ependymal layer of the lateral ventricle, where plasmids are injected (Koschmann et al., 2016, Núñez et al., 2019). Gliomas generated with the SB-based method recapitulate salient features of the human disease (Calinescu et al., 2015, Koschmann et al., 2016, Núñez et al., 2019), thus representing a versatile and reliable model to study brain tumor’s biology and the efficacy of therapeutic agents.
-
1.
Load the syringe with 10 μL of the DNA/jetPEI solution.
-
2.
Induce anesthesia by hypothermia by placing the mouse pup wrapped in a gauze on wet ice for 2 min.
-
3.
Place the pup on the previously cooled stereotaxic stage and immobilize the head using the ear bars. Wipe the head with ethanol 70%.
-
4.
Adjust the microdrive screws to lower the needle close to the head and place it in the midline intersection of the parietal and occipital bones (lambda).
-
5.
Adjust the stereotaxic coordinates to 0.8 mm lateral and 1.5 mm rostral to the lambda (Figure 1C).
-
6.
Lower the needle until it touches the skin, record the dorso-ventral coordinates.
-
7.
Lower the needle 1.5 mm to pierce the skin and skull. At this position, the tip of the needle should be inside the lateral ventricle (Figure 1C).
-
8.
Using an automatic injector, inject 0.75 μl of the DNA/jetPEI solution at a rate of 0.5 μl/min.
-
9.
After the DNA/jetPEI solution has been injected, keep the pup in the frame for 1 min.
-
10.
Anesthetize the next pup by repeating step 2.
-
11.
Slowly and gently lift the needle to pull it out from the brain.
-
12.
Place the pup under a heating lamp until normal breathing resumes, monitoring that it recovers its mobility and its rosy color. Apply a gentle lung massage if necessary to aid the breathing recovery.
-
13.
Once the pup is breathing and moving normally, place it back in the cage with the mother and monitor the mother’s response to the pups. Start the injection on the next pup, already anesthetized in step 10.
-
14.
After all the injections are done, monitor the cage for another 30 min to ensure that the mother is nurturing the pups.
-
15.
Place cage back to the animal room and monitor tumor growth.
Tumor Monitoring by Bioluminescence Imaging and EdU Administration
Depending on the genetic alterations introduced into the mouse brain cells, animals will display tumor burden symptoms in two or more months after the SB procedure (Table 1). These symptoms consist of weight loss, ruffled fur, hunched posture, impaired mobility or other abnormalities in movement. Usually, tumor progression can be monitored by bioluminescence two or three weeks before signs of tumor burden become evident. Endpoint tumors commonly display a bioluminescence of 107–108 photons/s/cm2/sr (Figure 1D). EdU injections should be performed before mice show signs of tumor burden; when tumor is easily detectable by bioluminescence but is not yet necrotic or hemorrhagic. For this procedure, EdU is injected when animals display 106 photons/s/cm2/sr IVIS signal (Figure 1D).
-
16.
Measure tumor volume using an IVIS bioluminescence imager.
-
a.
Anesthetize the animals in the anesthesia chamber with oxygen/isoflurane flow (1.5–2.5% isoflurane).
-
b.
Inject 100 μl of Luciferin solution (30 mg/mL) intraperitoneally and start a timer for 5 min.
-
c.
Place the animal back to the chamber and wait for 3–4 min.
-
d.
Start oxygen/isoflurane flow in the bioluminescence chamber and place the animal in the slot equipped with the glass nose cones.
-
e.
Place the animal in the bioluminescence chamber and when the timer goes off acquire a series of 6 images, at 2 min intervals with an automatic exposure time, median binning, and an open aperture of f = 1.
-
f.
Select the area of interest (i.e. the head) with an oval figure and measure the luminescence intensity using the calibrated unit photons/s/cm2/sr.
-
g.
If the animal has a 106 photons/s/cm2/sr signal, proceed to EdU administration.
-
17.
EdU injection
-
a.
Inject 10 mg/kg of the EdU dilution intraperitoneally. For instance, for mice weighting 30 g, inject 300 μl of the EdU dilution.
-
b.
Wait 3 h and proceed with the next steps.
Tumor Dissection and Depletion of CD45 Cells
-
18.
3 h after EdU injection, euthanize the animal with an overdose of isoflurane. Examine animal responsiveness by toe and tail pinch.
-
19.
Decapitate the animal’s head and gently dissect the brain from the skull.
-
20.
Place the brain into a Petri-dish on wet ice. Since the SB plasmids encoding for the oncogenes also encode for fluorescent proteins (Figure 1A), tumor tissue can be visualized and accurately dissected from the brain using a stereo-zoom microscope equipped with a fluorescent light illumination system (Figure 2A).
Figure 2.

Images Depicting Critical Steps of Tumor Dissection and CD45+ Cell Depletion Procedure
(A) Images showing a mouse SB-generated glioma under a stereo-zoom microscope equipped with a fluorescent light illumination system. In this example, the tumor was generated with SB plasmids encoding for genes coupled to GFP (middle picture) and Katushka (right picture) expression as reporter genes. Scale bars, 0.5 cm.
(B) Image showing a 50 mL conical tube, with a cell strainer on the top and a pestle, used to disintegrate the tumor tissue.
(C) Image illustrating MACS CD45-depletion column set up.
-
21.
Dissect the tumor from the brain using scalpels.
-
22.
Place the tumor pieces into a 50 mL conical tube with 5 mL of NS complete media.
-
23.
Using a cell strainer on a 50 mL conical tube and the sterile pestle of a 1 mL syringe, disaggregate the tumor tissue. Rinse the strainer with 10 mL of NS media through the strainer. Continue homogenizing the tissue through the strainer. Rinse two more times with 10 mL of NS media. Keep the single cell suspension on ice (Figure 2B).
-
24.
Centrifuge the cells at 300 xg for 5 min at 25°C.
-
25.
Remove supernatant and add 1 mL of 1X cold RBCLB. Incubate 1 min on ice. To stop cell lysis, add 9 mL of DPBS.
-
26.
Centrifuge the cells at 300 xg for 5 min at 25°C.
-
27.
Discard supernatant and resuspend the pellet in 90 μl of cold BSA-EDTA buffer.
-
28.
Count the cells using trypan blue and a hemocytometer.
-
29.
Add 10 μl of CD45 MicroBeads per 107 total cells. Mix well by pipetting.
-
30.
Incubate beads-cell suspension for 15 min at 4°C.
-
31.
Add 1 mL of BSA-EDTA buffer to wash the cells and centrifuge at 300 xg for 10 min at 4°C.
-
32.
Discard the supernatant and resuspend up to 108 cells in 500 μl of cold BSA-EDTA buffer.
-
33.
Place the LS column on a suitable MACS separator.
-
34.
Precondition the LS column by rinsing it with 3 mL of BSA-EDTA buffer and letting all the solution to go through it.
-
35.
Place a 15 mL collection tube below the column and add the pre-separation filter on top of it.
Note: Pre-separation filters on the top of the columns are important to prevent cell clumping and column clogging.
-
36.
Apply the cell suspension onto the filter and collect the CD45- cells that pass through the column (Figure 2C).
-
37.
Once the suspension stops dripping, wash the column once with 3 mL of cold BSA-EDTA buffer, collecting the eluate in the same tube (first eluate).
-
38.
To further deplete CD45+ cells from this suspension, pass this first eluate through a second pre-conditioned column placed in the magnetic field, with a pre-separator filter on the top. Collect the eluted cells.
-
39.
When the entire cell suspension has gone through the column, apply 3 mL of cold BSA-EDTA buffer to rinse it, collecting the eluted cells (second eluate). These cells are mostly tumor cells, depleted of immune cells.
-
40.
Pellet the CD45-depleted tumor cells by centrifuging the second eluate at 300 xg for 10 min at 4°C.
Note: This method allows the separation of tumor cells from immune cells with a substantial amount of viable cells. The number of CD45+ cells in the tumor enriched cell fraction is negligible (Garcia-Fabiani et al., 2020).
Flow Staining: Click-iT Reaction and Phospho-Ser10-Histone 3 Intracellular Staining
EdU is a thymidine analog that is incorporated into DNA during active DNA synthesis. The detection of EdU is based on a click reaction between a picolyl azide, which is coupled to a fluorescent dye, and an alkyne (EdU nucleoside). In this protocol, we used a Pacific Blue-coupled azide. Moreover, the phosphorylation of the serine residue 10 in the histone 3 tail (p-Ser10-H3) is a critical event in mitosis. Histone-3-Serine-10 phosphorylation is important for chromosome condensation, and starts at late G2 phase, peaks at metaphase and decreases from late anaphase to early telophase (Kim et al., 2017). To study cell cycle characteristics, we performed EdU and p-Ser10-H3 intracellular staining on tumor cells.
Note: It is highly recommended to use single stained compensation controls and single stained samples as controls, i.e., “EdU only” and “p-Ser10-H3 only” samples. Also, keep unstained cells for compensation set up.
-
41.
Add 3 mL of 1% BSA-PBS buffer to wash the cells and centrifuge at 300 xg for 5 min at 4°C. Remove supernatant.
-
42.
For 106 cells, add 100 μL of Click-iT fixative (Component D) and mix well by pipetting.
-
43.
Incubate the samples in fixative for 15 min at 25°C, protected from light.
-
44.
Add 3 mL of 1% BSA-PBS buffer to wash the cells and centrifuge at 300 xg for 5 min at 4°C. Remove supernatant.
-
45.
Resuspend the pellets in 100 μL of 1X Click-iT permeabilization and wash reagent. Mix well.
-
46.
Incubate the samples 15 min at 25°C.
-
47.
Add 1 μL of p-Ser10-H3-PE antibody resuspended in 100 μL 1X Click-iT permeabilization and wash reagent to each sample.
-
48.
Incubate the samples for 30 min at 25°C, protected from light.
-
49.
Add 3 mL of 1% BSA-PBS buffer to wash the cells and centrifuge at 300 xg for 5 min at 4°C. Remove supernatant.
-
50.
Add 100 μL of 1X Click-iT permeabilization and wash reagent. Mix well.
-
51.
Prepare Click-iT EdU buffer additive 1X: dilute Click-iT EdU buffer additive 10X 1:10 in deionized water. For each reaction 50 μL of Click-iT EdU buffer additive 1X are necessary.
-
52.
For each reaction, prepare the following cocktail:
| Component | Volume for 1 Reaction (μL) |
|---|---|
| PBS, DPBS or TBS | 438 |
| Copper protectant (Component F) | 10 |
| Fluorescent dye picolyl azide | 2.5 |
| Click-iT EdU buffer additive 1X | 50 |
| TOTAL | 500.5 |
Note: Use the Click-iT cocktail within 15 min of preparation.
-
53.
Add 500 μL of the Click-iT cocktail to each sample with the fixed cells and mix well. At this point, the total volume is 600 μL.
-
54.
Incubate the reaction for 30 min at 25°C, protected from light.
-
55.
Add 3 mL of 1X Click-iT permeabilization and wash reagent to wash the cells and centrifuge at 300 xg for 5 min at 4°C. Remove supernatant.
-
56.
Resuspend cells in flow buffer and analyze.
Pause Point: Once the cells are fixed, data acquisition by flow cytometry can be performed the next day. Store stained cells at 4°C protected from light.
Data Acquisition
Use the single stained samples and the unstained sample to perform laser compensation. Once the laser set up is done, proceed to run the samples.
The gating strategy is to perform doublet discrimination first by gating in a SSC-W vs SSC-H plot and then FSC-W vs FSC-H plot (Figure 3A), before EdU and p-Ser10-H3 positive cell identification.
Figure 3.

Flow Cytometry Gating Strategy Used to Identify EdU+ and p-Ser10-H3+ Glioma Cells
(A) Total cells are gated to exclude cellular debris (top left plot). Then, doublet discrimination gating is performed to filter out cellular aggregates (top middle and right plots). Lower panel: Left histogram: glioma cells positive for EdU (black=unstained control, blue=EdU (Pacific Blue staining)). Right histogram: glioma cells positive for p-Ser10-H3 expression (black=unstained control, red=p-Ser10-H3 (PE staining)).
(B) Representative results for p-Ser10-H3 and EdU staining of tumor cells isolated from SB gliomas. Tumors were generated by the introduction of the following genetic alterations: NRASG12V overexpression, p53 and Atrx silencing for NPA; or NRASG12V overexpression, p53 and Atrx silencing, and IDH1R132H overexpression for NPAI (Table 1). Results are shown as the % of positive cells (left table) or the median fluorescent intensity (MFI) (right table) ± SD, for each staining. NPAI tumors are less aggressive than NPA tumors and cells show different cell cycle characteristics.
To identify EdU positive cells, plot single cells as a histogram to analyze the distribution of the dye (i.e., Pacific Blue) (Figure 3A, lower left panel). To identify p-Ser10-H3 positive cells, cells were plotted on a separate histogram and PE staining was measured (Figure 3A, lower right panel).
-
57.
Run the single stained controls and set the gates.
-
58.
Run samples, collecting at least 30,000 events.
-
59.
Analyze data using FlowJo software.
EXPECTED OUTCOMES
In general, proliferating cells going through S phase of the cell cycle incorporate EdU while DNA is being synthetized. After the Click-iT reaction, cells in the S phase will be positive for the fluorochrome bound to the azide and the cell population of interest can be easily detected by flow cytometry. After EdU incorporation and Click-iT reaction, two peaks in the stain’s histogram can be identified: one peak with low or no staining, i.e. cells not undergoing S phase; and a second peak with positive staining levels, i.e. cells undergoing S phase (Figure 3A, lower left panel).
To identify cells going through mitosis, p-Ser10-H3 intracellular staining was performed. The analysis can be done in the same way as the EdU analysis. Cells going through mitosis will be stained with anti-p-Ser10-H3 antibody (Prigent and Dimitrov, 2003, Li et al., 2005, Núñez et al., 2019). These cells can be easily detected as a single positive peak in the histogram, whereas cells that are unstained for the marker constitute the negative population of cells, i.e. not undergoing mitosis (Figure 3A, lower right panel).
The genetic background of the glioma induced by the SB method will define the aggressiveness of the tumor and will have an impact on the cell cycle characteristics and on the proportion of tumor cells undergoing S and M phases (Figure 3B).
LIMITATIONS
-
•
Tumor bearing mice displaying an IVIS signal of 108–109 photons/s/cm2/sr will be negative for EdU and p-Ser10-H3 staining. This IVIS signal usually corresponds to maximum tumor burden. At this stage glioma cells are not proliferative and tumor tissue starts to become necrotic. Cells should only be harvested during an exponential growth stage to accurately characterize cell cycle profiles.
-
•
Animals bearing tumors that display IVIS signal of 104–105 photons/s/cm2/sr are not suitable for EdU and p-Ser10-H3 staining. At this IVIS signal the tumor is still very small. In this case, not enough tumor cells can be acquired from a single animal for flow cytometry analysis.
-
•
In order to draw accurate conclusions about the tumor cell cycle profile, tumor cells could be stained with additional cell surface markers such as CD133 or CD44 (Brown et al., 2017). In the case of SB-derived gliomas, tumor cells could also be identified by staining for the fluorescent reporter proteins associated to the expression of the oncogenes introduced.
-
•
CD45+ cell depletion method reliably depletes immune cells from the tumor homogenate (Garcia-Fabiani et al., 2020). In case these reagents cannot be acquired, immune cells could be discriminated from the glioma cells during flow cytometry by staining the tumor cell suspension with an anti-CD45 antibody coupled to a fluorochrome. Right after tumor dissociation, wash the samples with flow buffer and stain 1x106 cells in 200 μL of flow buffer with 1 μL of anti-CD45 antibody fluorochrome-conjugated. Incubate for 30 min on ice, protected from light. Wash twice with flow buffer and continue with cell fixation, p-Ser10-H3 intracellular staining and Click-iT reaction. EdU and p-Ser10-H3 positive cells should then be identified within the CD45-negative population.
-
•
We were not able to perform this method on orthotropic implantable brain tumor model. We believe that this could be related to different properties of SB-based tumors versus implanted tumors regarding the penetration of the EdU compound.
TROUBLESHOOTING
Problem
Low or no EdU labeling.
Potential Solution
The time elapsed between EdU administration and animal euthanasia should be determined empirically and it could vary amongst the type of tumor being harvested and its anatomic location.
Problem
Non-specific/high fluorescent staining of p-Ser10-H3.
Potential Solution
To minimize non-specific p-Ser10-H3 staining, blocking CD16/CD32 antibody could be utilized. This reagent contains immunoglobulins that will bind to the Fc-receptor, thereby blocking the non-specific binding of p-Ser10H3 to other receptors on the tumor cell. Also, adding and additional washing step after the incubation of the sample with the antibody might be added to further eliminate unspecific binding.
Problem
No p-Ser10-H3 staining.
Potential Solution
The concentration of p-Ser10-H3 antibody should be determined empirically. Antibody titration could be performed in fixed and permeabilized cells.
Intracellular proteins may be lost if excessive cell permeabilization occurred before sufficient cross-linking is attained. This could be the case if methanol is used as a fixative instead of formaldehyde.
Problem
Cellular clumping during flow cytometry
Potential Solution
Vortex the sample gently at low speed before running the sample.
Clumps can also be removed by filtering the sample through a 70 μm nylon mesh before running it in the flow cytometer.
Problem
High side scatter signal
Potential Solution
This could be due to lysed or broken cells. Ensure that samples are fresh and prepared according to the workflow detailed above. Do not vortex cells at a high speed before data acquisition.
Acknowledgments
This work was supported by NIH/NINDS grants, R37-NS094804, R01-NS105556, and 1R21NS107894 to M.G.C.; NIH/NINDS grants R01-NS076991 and R01-NS096756 to P.R.L.; the Department of Neurosurgery, the Rogel Cancer Center, Program in Cancer Hematopoiesis and Immunology (CHI), the ChadTough Foundation, Pediatric Brain Tumor Foundation, and Leah’s Happy Hearts to M.G.C. and P.R.L.; RNA Biomedicine grant F046166, Forbes Foundation grant, University of Michigan Medical School, Rogel Cancer Center Scholar, and University of Michigan Medical School to M.G.C.; and American Brain Tumor Association Basic Research Fellowship “in Memory of Bruce and Brian Jackson” to M.B.G.F.
Author Contributions
M.G.C. and P.R.L. conceived the research question and provided funding; M.B.G.F. and P.K. performed the experiments; M.B.G.F. prepared the figures; M.B.G.F. and P.K. wrote the manuscript under the supervision of M.G.C. and P.R.L. All authors read and edited the final version of this manuscript.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Maria B. Garcia-Fabiani, Email: mgarciaf@umich.edu.
Maria G. Castro, Email: mariacas@med.umich.edu.
References
- Brown D.V., Filiz G., Daniel P.M., Hollande F., Dworkin S., Amiridis S., Kountouri N., Ng W., Morokoff A.P., Mantamadiotis T. Expression of cd133 and cd44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS One. 2017;12:e0172791. doi: 10.1371/journal.pone.0172791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calinescu A.A., Nunez F.J., Koschmann C., Kolb B.L., Lowenstein P.R., Castro M.G. Transposon mediated integration of plasmid DNA into the subventricular zone of neonatal mice to generate novel models of glioblastoma. J. Vis. Exp. 2015:52443. doi: 10.3791/52443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comba A., Dunn P.J., Argento A.E., Kadiyala P., Ventosa M., Patel P., Zamler D.B., Núñez F.J., Zhao L., Castro M.G. Fyn tyrosine kinase, a downstream target of receptor tyrosine kinases, modulates antiglioma immune responses. Neuro Oncol. 2020:noaa006. doi: 10.1093/neuonc/noaa006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Fabiani M.B., Comba A., Kadiyala P., Haase S., Núñez F.J., Altshuler D., Lowenstein P.R., Castro M.G. Isolation and characterization of immune cells from the tumor microenvironment of genetically engineered pediatric high-grade glioma models using the sleeping beauty transposon system. Methods Enzymol. 2020;632:369–388. doi: 10.1016/bs.mie.2019.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-Y., Jeong H.S., Chung T., Kim M., Lee J.H., Jung W.H., Koo J.S. The value of phosphohistone h3 as a proliferation marker for evaluating invasive breast cancers: A comparative study with ki67. Oncotarget. 2017;8:65064. doi: 10.18632/oncotarget.17775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschmann C., Calinescu A.A., Nunez F.J., Mackay A., Fazal-Salom J., Thomas D., Mendez F., Kamran N., Dzaman M., Mulpuri L., Krasinkiewicz J., Doherty R., Lemons R., Brosnan-Cashman J.A., Li Y., Roh S., Zhao L., Appelman H., Ferguson D., Gorbunova V., Meeker A., Jones C., Lowenstein P.R., Castro M.G. Atrx loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 2016;8:328ra328. doi: 10.1126/scitranslmed.aac8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.W., Qin Y., Chen J.T., Hao Z., Liu R.M., Huang X.T. Dynamic distribution of ser-10 phosphorylated histone h3 in cytoplasm of mcf-7 and cho cells during mitosis. Cell Res. 2005;15:120–126. doi: 10.1038/sj.cr.7290276. [DOI] [PubMed] [Google Scholar]
- Núñez F.J., Mendez F.M., Kadiyala P., Alghamri M.S., Savelieff M.G., Garcia-Fabiani M.B., Haase S., Koschmann C., Calinescu A.A., Kamran N. Idh1-r132h acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019;11:eaaq1427. doi: 10.1126/scitranslmed.aaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigent C., Dimitrov S. Phosphorylation of serine 10 in histone h3, what for? J. Cell Sci. 2003;116:3677–3685. doi: 10.1242/jcs.00735. [DOI] [PubMed] [Google Scholar]
- Wiesner S.M., Decker S.A., Larson J.D., Ericson K., Forster C., Gallardo J.L., Long C., Demorest Z.L., Zamora E.A., Low W.C., Santacruz K., Largaespada D.A., Ohlfest J.R. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res. 2009;69:431–439. doi: 10.1158/0008-5472.CAN-08-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets or code were generated during this study.