

Abstract
Purpose:
To examine the theoretical/practical utility of the liver-to-blood partition coefficient (Kpuu) for predicting drug-drug interactions (DDIs), and compare the Kpuu-approach to the extended clearance concept AUCR-approach.
Methods:
The Kpuu relationship was derived from first principles. Theoretical simulations investigated the impact of changes in a single hepatic-disposition process on unbound systemic (AUCB,u) and hepatic exposure (AUCH,u) versus Kpuu. Practical aspects regarding Kpuu utilization were examined by predicting the magnitude of DDI between ketoconazole and midazolam employing published ketoconazole Kpuu values.
Results:
The Kpuu hepatic-disposition relationship is based on the well-stirred model. Simulations emphasize that changes in influx/efflux intrinsic clearances result in Kpuu changes, however AUCH,u remains unchanged. Although incorporation of Kpuu is believed to improve DDI-predictions, utilizing published ketoconazole Kpuu values resulted in prediction errors for a midazolam DDI.
Conclusions:
There is limited benefit in using Kpuu for DDI-predictions as the AUCR-based approach can reasonably predict DDIs without measurement of intracellular drug concentrations, a difficult task hindered by experimental variability. Further, Kpuu changes can mislead as they may not correlate with changes in AUCB,u or AUCH,u. The well-stirred model basis of Kpuu when applied to hepatic-disposition implies that nuances of intracellular drug distribution are not considered by the Kpuu model.
Keywords: Kpuu, Liver-to-Blood Partitioning, Drug-Drug Interactions, Well-Stirred Model
INTRODUCTION:
It is generally accepted that estimation of tissue- or target-specific unbound drug concentration is imperative to accurately assess in vivo pharmacological efficacy, drug-drug interaction (DDI) potential and toxicological effects of therapeutic drugs [1]. However, unbound systemic drug concentrations have historically been used as a surrogate to estimate potential pharmacokinetic or pharmacodyamic drug effects in accordance with the free drug theory [2], largely due to the difficulty in accurately determining intracellular drug concentrations. The free drug theory assumes a rapid equilibrium between unbound drug concentration in the blood and the tissues, i.e. that unbound blood concentration is equal to unbound tissue concentration (CB,u = CH,u), here using the liver as our target organ. However, this assumption may not be valid for substrates of active cellular transport, since one may assume that active uptake would result in increased intracellular unbound drug accumulation, whereas efflux would decrease tissue-specific unbound drug concentration. Therefore, differential concentrations of unbound drug in the blood versus tissue are often anticipated and considered crucial in predictions related to drug disposition.
The unbound liver-to-blood partition coefficient (Kpuu) has been developed to provide estimates of unbound intracellular drug concentrations based on the extended clearance model [3]. The value of Kpuu is governed by active and passive drug passage into and out of the liver, as well as by hepatic elimination (metabolism and biliary excretion) and is based on a single unbound drug driving-force concentration in the liver, as depicted in Figure 1A. As we have recently reviewed [4, 5] and others in the field have recognized [6–12], the assumption that a single liver concentration drives the various processes of basolateral efflux, biliary elimination and metabolism implies that the extended clearance model is a well-stirred model construct. Here we derive Kpuu to demonstrate that Kpuu is also a well-stirred model concept when attempting to predict hepatic elimination and question the relevance of Kpuu determinations for predicting drug-drug interactions.
Figure 1: Schematic representation of liver-to-blood partition coefficient (Kpuu) that relates unbound concentration of drug in the blood (CB,u) to unbound concentration of drug in the liver (CH,u).
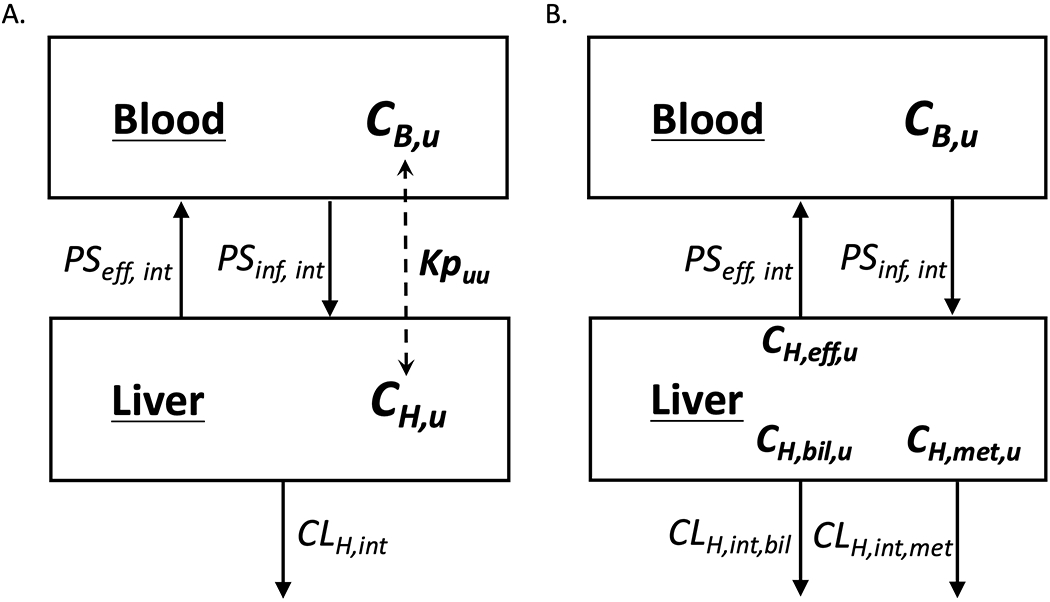
Differential concentrations are determined by the active and passive transporter influx and efflux intrinsic clearances (PSint,inf and PSint,eff) as well as the intrinsic hepatic clearance (CLH,int), which represents the irreversible loss of drug by metabolism and biliary excretion. The average unbound concentration in the liver driving these processes is depicted in (A) and the individual driving force concentrations are depicted in (B).
MATERIALS AND METHODS:
The derivation of Kpuu was conducted based on mass balance principles, with consideration of the amount of drug within an organ with respect to time under steady state conditions due to active plus passive influx and efflux (PSinf,int, PSeff,int), metabolic plus biliary elimination (CLH,int), and fraction unbound of drug in the blood (fu,B) and within the organ (fu,H). The resulting Kpuu relationship was compared with previously derived [4] relationships of systemic and organ exposure (AUCB,u and AUCH,u) following oral dosing, which include considerations of oral bioavailability as indicated by the fraction of oral dose that is absorbed (Fabs) and the fraction that escapes gut elimination (FG).
To illustrate the relevance of Kpuu on predictions of drug-drug interactions, simulations were conducted to explore the impact of up to a 10-fold increase or decrease in an individual disposition process (PSinf,int, PSeff,int, CLH,int or Fabs∙FG) on Kpuu, AUCB,u and AUCH,u using the derived relationships presented here.
Kpuu is commonly employed to estimate intracellular concentrations of perpetrator drugs to predict the inhibitory potential on hepatic disposition processes, such as metabolic or biliary elimination. Here, a metabolic drug interaction between IV midazolam (victim drug) and ketoconazole (perpetrator) [13] was selected to investigate if predictions of changes in AUC were improved by addition of Kpuu to estimate intracellular ketoconazole concentrations. Predicted ratios of midazolam systemic exposure in the ketoconazole versus control phases (AUCR) were calculated using the FDA recommended basic model for reversible inhibition [14]:
| (1) |
where Imax,u is the maximal unbound plasma concentration of the inhibitor drug ketoconazole and Ki is the unbound inhibition constant of ketoconazole on cytochrome P450 (CYP) 3A4-mediated midazolam metabolism. The Ki value was calculated by averaging reported inhibition constants of ketoconazole on CYP3A4-mediated formation of 1-hydroxymidazolam, and was found to be 0.061 μM as summarized by Greenblatt et al. [15]. The value for Imax,u was calculated by multiplying ketoconazole fu,plasma by the observed maximum ketoconazole plasma concentration in the clinical study investigated [13]. The ketoconazole fu,plasma utilized was 0.029 [16] and Imax was estimated to be 3.0 μg/ml (5.6 μM) from visual inspection of published IV plasma concentration time profiles [13]. Measured human ketoconazole Kpuu values from the literature [7, 17, 18], as well as simulated Kpuu values between 0.1 and 10, were utilized to adjust Imax,u (i.e., [Imax,u] ∙ Kpuu) to account for intracellular ketoconazole concentrations in contact with hepatic CYP3A4 in prediction of the magnitude of the ketoconazole-midazolam drug-drug interaction. The ketoconazole Kpuu values identified from the literature and the methodologies employed for Kpuu determination are listed as follows: 0.32 (extended clearance method) [18]; 0.58 (extended clearance method) [7]; 0.72 (homogenization method) [18]; 0.97 (temperature method) [18]; 1.04 (temperature method) [17]; 3.18 (homogenization method) [17]; and 4.67 (log D 7.4 method) [18].
RESULTS AND DISCUSSION:
The Unbound Liver-to-Blood Partition Coefficient (Kpuu) is Only Consistent with the Well-Stirred Model of Hepatic Elimination When Correlated with Hepatic Elimination Parameters
The liver model in Figure 1B depicts the various hepatic processes that govern liver-to-blood drug partitioning, with the reasonable consideration that the driving-force hepatic concentration for basolateral efflux (CH,eff) may not necessarily be equal to the apical concentration driving biliary elimination (CH,bil) nor the average hepatic concentration driving metabolic elimination (CH,met). Solving for the change in total hepatic drug amount (AH) with time (i.e. the mass balance relationship) gives Eq. 2:
| (2) |
where PSint values represent the total of both active and passive basolateral influx (inf) and efflux (eff) into and out of the liver, fu,B is the unbound fraction of drug in the blood, fu,H is the unbound fraction of drug within the liver, CLH,int,met is the intrinsic metabolic clearance, CLH,int,bil is the intrinsic biliary secretion clearance, CB is the total concentration of drug in the blood, and CH is the total drug concentrations in the liver driving basolateral efflux (eff), apical biliary elimination (bil), and metabolism (met).
Although differentiating the various concentrations driving their respective hepatic processes may be a reasonable assumption (as depicted in Figure 1B), there are currently no reliable analytical methods available to differentiate these various intracellular concentrations. Further, the utility of this model is limited when attempting to calculate a single partition coefficient to predict clinically relevant outcomes, especially when multiple processes determine hepatic concentration. But by adopting the well-stirred model of hepatic disposition, it follows that the hepatocyte is a ‘well-stirred’ compartment and a single hepatic concentration (CH) drives all intracellular processes as depicted in Figure 1A and as previously elucidated [5] given by Eq. 3:
| (3) |
where CLH,int is the sum of the intrinsic biliary secretion and intrinsic metabolism clearances.
At steady state, the change in total drug amount over time inside the liver is equal to zero, therefore Eq. 3 can be solved for the ratio of unbound concentration of drug in the liver to that in the blood (CH,u/CB,u), in other words, Kpuu.
| (4) |
Equation 4 is widely utilized throughout the industry to predict relevant unbound liver concentrations at steady-state [3,6,12,18–21].
The Kpuu relationship is a well-stirred model concept when related to liver transport or elimination processes since it is based on the mass balance relationship (presented as Eq. 2) where CH is the single liver concentration that drives biliary elimination, metabolic clearance, and efflux of drug back into the blood. Only in the well-stirred model does a single concentration drive all hepatic processes at steady state; therefore, Kpuu when expressed in terms of elimination parameters is not consistent with alternate hepatic disposition models, such as the parallel tube or axial dispersion models, where the concentrations at the basolateral and apical hepatocyte membranes driving efflux and biliary elimination, respectively, as well as the average concentration driving metabolism, are all assumed to be different concentrations (Eq. 1; Figure 1A). Recognition that Kpuu is based on the well-stirred model has been noted previously by the International Transporter Consortium [1] and as we have recently reviewed [4, 5], it is well-recognized throughout the field that inclusion of transporters in calculations of hepatic elimination are only consistent with the well-stirred model. Therefore, utilization of Kpuu must be accompanied with appreciation that the apparent Kpuu value is a mere estimation of degree of partitioning based on a useful but simplified model of whole-hepatocyte cytosolic drug concentration. This limitation, amongst others, will be discussed in further detail in a subsequent section.
Questioning the Utility of Kpuu for Drug-Drug Interaction or Pharmacogenomic Variance Predictions
Estimation of Kpuu is often utilized in attempts to improve pharmacokinetic or pharmacodynamic predictions, as cytosolic unbound drug concentrations are more relevant than systemic concentrations for predictions of tissue-specific potency or toxicity and drug disposition (such as metabolic or biliary elimination). The International Transporter Consortium has outlined these useful applications of Kpuu in a 2013 review article and the authors conclude that “The intracellular concentration of unbound form of a drug is an important parameter for predicting drug efficacy, toxicity, and DDIs” [1]. We agree that determination of Kpuu is undoubtedly relevant for predicting drug potency or toxicity, as pharmacodynamic effects are driven by unbound intracellular drug concentrations, however the aspects related to the importance of Kpuu in drug-drug interaction prediction should be further clarified.
To examine the utility of Kpuu in drug-drug interactions predictions, we take the integral of the concentrations over all time for the numerator and denominator of the two middle terms of Eq. 4.
| (5) |
where AUCu is the area under the concentration time curve of unbound drug at the respective sites following either an oral or IV dose.
As we have recently demonstrated [4] and many others in the field have previously recognized, following oral dosing the equations describing systemic and hepatic AUCu are given by:
| (6) |
| (7) |
where Fabs is the fraction of the dose absorbed intact and FG is the fraction of the dose that escapes intestinal elimination. As expected, dividing Eq. 7 by Eq. 6 results in Equation 5, the Kpuu relationship. The same Kpuu relationship would be derived utilizing the more complicated equations for AUCH,u and AUCB,u following IV dosing from reference 4.
Examination of Equations 6 and 7 indicates that unbound AUCu in the liver and blood following oral dosing are not a function of protein binding, therefore any changes in protein binding (either hepatic or systemic) will have little clinical relevance on pharmacodynamic outcomes such as efficacy and toxicity [22]. According to these equations, hepatic DDIs will only occur if the perpetrator drug affects Fabs, FG, CLH,int, PSinf,int or PSeff,int. Therefore, knowledge of intracellular unbound concentrations via measurements of fu,B, fu,H or Kpuu will not provide any relevant information regarding predictions of clinically significant changes in systemic or organ drug exposure resulting from DDIs or pharmacogenomics variance. Such changes are simply a multiple of how CLH,int, PSinf,int, PSeff,int, Fabs and FG change for an orally dosed victim drug and knowledge of Kpuu is unnecessary to make that prediction. Therefore, we emphasize that evaluation of AUCR (AUC ratios expressed as AUCinteraction/AUCcontrol) is a more useful approach than evaluating rate determining steps, β or Kpuu in predictions of drug-drug interactions.
Recently, we have critically assessed the pharmacokinetic changes expected for transporter substrates [5] and the impact of changes in CLH,int and FG on the pharmacokinetics of metabolized victim drugs [23] in drug-drug and pharmacogenomic interactions utilizing this extended clearance concept AUCR-based approach. Utilization of Eqs. 6 and 7 provides a clearer understanding of the effect of perpetrator drugs on the magnitude of DDIs than Kpuu-based analysis. For hepatic uptake transporter substrates, a perpetrator drug that markedly inhibits or induces relevant uptake transporters will require a human DDI study to quantitate the effect on systemic concentrations (independent of whether the drug is eliminated by metabolism or not), however, no clinically relevant intrahepatic interaction will be expected. For drugs that are eliminated by metabolism, a perpetrator that significantly inhibits or induces metabolism will require a human DDI study to quantitate the systemic concentration effect, with recognition that both hepatic and intestinal metabolism may be affected. Intrahepatic concentrations will also be affected, therefore DDI studies for metabolic interactions should also measure changes in organ-specific pharmacodynamic outcomes (such as efficacy and toxicity) as changes in both CLH,int and FG will influence intrahepatic concentrations. For drugs that are eliminated into the bile, perpetrator drugs with the potential to inhibit apical biliary efflux transporters within the hepatocyte may result in clinically significant systemic concentration changes requiring a human DDI study, as well as potential intrahepatic concentration changes requiring consideration of potential pharmacodynamic changes. Inhibition of basolateral efflux (PSeff,int) can impact both the systemic and intrahepatic concentrations of all drugs, regardless of their major route of elimination. In summary, changes in intracellular unbound concentrations as a result of drug-drug or pharmacogenomic interactions can reasonably be predicted based on the AUCR-based approach without knowing Kpuu or fu,H.
An excellent example of quantitative drug-drug interaction predictions of OATP1B1 substrates based on the extended clearance AUCR values approach was conducted by Varma et al. [24]. In that study, Kpuu was not used in predictions of DDI, rather, the theoretical changes in Kpuu were derived using Eq. 4 by considering predicted changes in the individual clearance and transport parameters (under the assumption that PSeff,int was only comprised of passive processes) and these changes were compared to observed AUCR values. The analysis categorizes interactions in four groups based on direction of change in AUCR versus Kpuu: (a) increased systemic exposure and increased Kpuu, (b) increased systemic exposure but decreased Kpuu, (c) decreased systemic exposure and decreased Kpuu, and (d) decreased systemic exposure but increased Kpuu, highlighting that changes in Kpuu are not always in the same direction as systemic exposure. For perpetrators that inhibit hepatic active uptake (i.e., active portion of PSinf,int) via OATP1B such as cyclosporine and rifampin (single dose), the expected increase in AUCR of victim OATP substrates is observed based on examination of Eq. 6. Consideration of Eq. 7 would result in understanding that intrahepatic exposure remains unchanged when uptake is inhibited, which is important since efficacy of the statins (prototypical OATP1B substrates) relies on unbound intrahepatic concentrations to drive efficacy. However, if the Kpuu relationship (Eq. 5) had been utilized, inhibition of PSinf,int would predict a decreased unbound liver-to-blood drug concentrations of the victim drug, which may potentially mislead the investigator into thinking that unbound intracellular exposure has decreased, when in reality the reduced Kpuu value is a result of the increase in systemic unbound concentrations and that this change is not relevant for statin’s effects. Varma et al. [24] acknowledge this point by indicating that “these results have potential implications for clinical practice – particularly using statins. Arguably, dose adjustments based on plasma exposure during comedication may avoid systemic adverse events such as myopathy and rhabdomyolysis, but could lead to lack of clinical efficacy due to reduced hepatic concentrations”.
To further illustrate the impact of changes in a single disposition process (such as PSinf,int as discussed by Varma et al. [24]) on observed AUCB,u, AUCH,u and Kpuu values, simulations were conducted to vary a single parameter by 10-fold in each direction (Figure 2). Changes in CLH,int, PSinf,int, PSeff,int, or Fabs∙FG were examined (x-axis) and resulting fold-change in observed AUCB,u, AUCH,u and Kpuu are depicted (y-axis). Changes in PSinf,int (Fig. 2B) or PSeff,int (Fig. 2C) result in no change in unbound liver exposure (blue lines), however, unbound systemic exposure (red lines) and Kpuu (green lines) are observed to change inversely. Changes in Fabs∙FG (Fig. 2D) result in no change in Kpuu due to proportional changes in both systemic and liver unbound exposure. Increases in CLH,int (Fig. 2A) result in decreases in all three parameters to different degrees, with an observed linear impact on unbound liver exposure (blue line). These simulations exemplify why utilization of Kpuu may mislead and why examination of AUC ratios utilizing Eqs. 6 and 7 should preferentially be utilized.
Figure 2: Expected Fold Difference Outcomes for Systemic Unbound Exposure (AUCB,u), Hepatic Unbound Exposure (AUCH,u) and Kpuu Based on Changes in (A) CLint,H, (B) PSinf,int, (C) PSeff,int or (D) Fa·Fg.
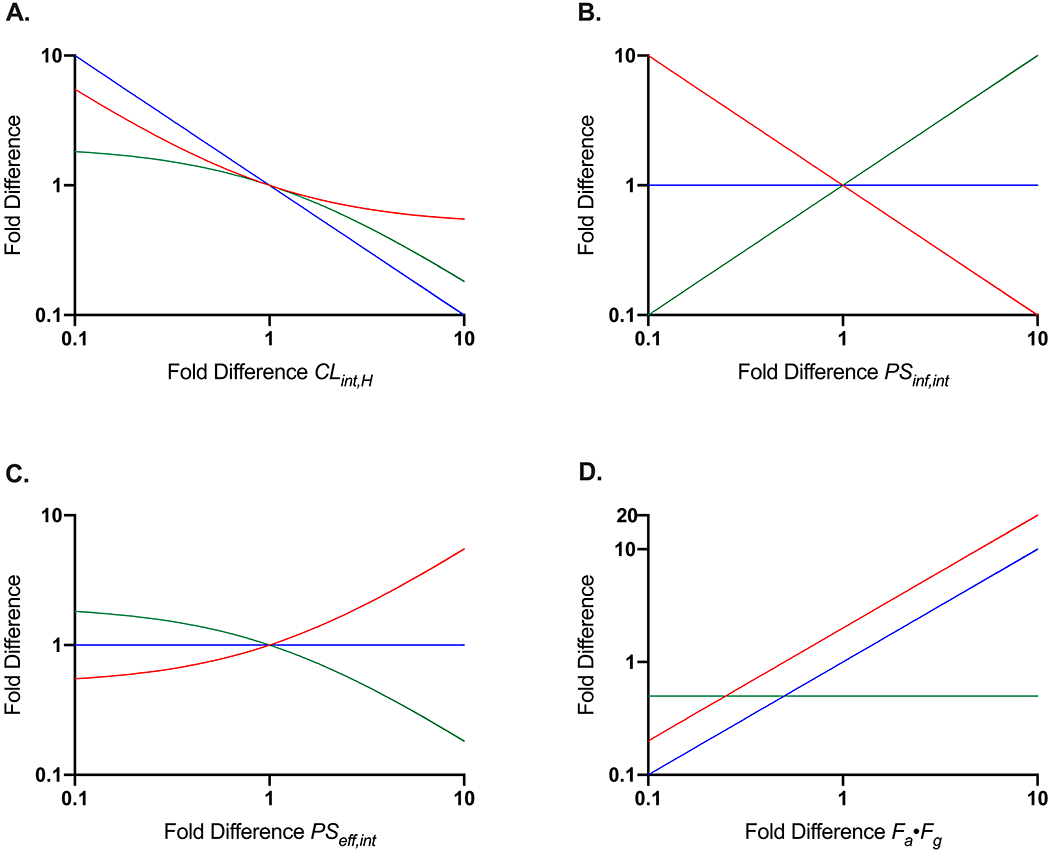
Resulting changes in AUCB,u are indicated by the red lines, changes in AUCH,u are indicated by the blue lines, and changes in Kpuu are indicated by the green lines.
We further emphasize that Eqs. 6 and 7 describe unbound drug exposure, and that multiplying both sides of these equations by fu,B or fu,H, respectively, results in the relationships for total AUC:
| (8) |
| (9) |
Examination of these relationships (Eqs. 8 and 9) further emphasizes that changes in protein binding will definitely impact total drug concentrations, however, these changes will not alter unbound systemic or hepatic concentrations that ultimately drive efficacy and toxicity. The nuanced importance of this concept can best be illustrated when considering drug level monitoring, where a change in protein binding would result in altered total blood concentrations and may ultimately influence a clinician to make a dose adjustment. However, consideration of the unbound exposure relationships (Eqs. 6 and 7) reveal that unbound exposure had not changed and such dose adjustments could lead to lack of efficacy or safety.
The Appropriate Role of Kpuu in Predicting PK/PD and Drug-Drug Interactions
As mentioned above, Kpuu is useful in predicting pharmacodynamic (PD) drug effects driven entirely by unbound intracellular drug concentrations, such as drug efficacy or toxicity associated with a specific organ [21,25–27]. This is particularly relevant for statins, as drug efficacy is a function of intrahepatic concentrations, however systemic or muscle exposure may drive undesirable myopathy side effects. Evaluation of Kpuu may also be useful when estimating free liver concentrations based on readily measurable plasma concentrations in a clinical study. Thus, an in vitro measurement of Kpuu has the potential to allow for estimations of drug exposure within the organ, a concentration that is extremely difficult to measure, which can help inform potential for pharmacological and adverse effects.
Determination of Kpuu may also be helpful in improving predictions of pharmacokinetic drug disposition (i.e. in prediction of hepatic drug elimination). Recently, Riccardi et al. [28] demonstrated improved clearance predictions for transporter and enzyme substrates involving in vitro hepatocyte clearance determinations in the presence of 4% bovine serum albumin, to account for protein-facilitated uptake mechanisms, as recently described by Bowman and Benet [29]. A modified version of Kpuu that accounts for unbound drug partitioning between the liver tissue and the liver plasma was utilized in the mathematical model and was estimated with consideration of measured partitioning between the hepatocytes and the protein-augmented buffer, resulting in improved clearance predictions.
With respect to drug interaction prediction, Kpuu can be utilized in improving predictions of inhibitory potential of an intracellular perpetrator drug, but only in regards to processes driven by intracellular concentrations, i.e. metabolic elimination (CLint,H), biliary elimination (CLint,bil) and basolateral efflux (PSint,eff), since active uptake processes (PSint,inf) into the liver would be driven by systemic perpetrator concentrations (Fig. 1). Predictions of inhibitory potential of perpetrator drugs are routinely performed and according to the FDA Draft Guidance [14] the change in systemic exposure of victim drug as a result of a perpetrator can estimated by Eq. 1, reflecting a basic interaction with a reversible inhibitor of a single hepatic disposition process. More complex models involving time-dependent inhibitors or inhibition of multiple pathways are commonly integrated in physiologically-based pharmacokinetic (PBPK) modeling approaches, and are essentially based on integration of individual specific models for each process into the AUCR relationships presented in Eqs. 6 and 7 to predict overall AUC changes in complex drug-drug interactions [24,30,31]. Determinations of Kpuu are theoretically valuable in accounting for the true unbound concentration of an intracellular inhibitor in order to more accurately predict drug interaction potential, but the uncertainty of Kpuu measurements via the different procedures employed belies the usefulness of this approach as we show below.
Recently, Iwasaki et al. [17] introduced Kpuu into their PBPK models to potentially improve the predictability of twenty-two CYP-mediated DDIs. The authors determined Kpuu by three methods (temperature, homogenization and in vivo rat studies) and incorporated these values into their predictive models; model outcomes of these simulations were compared to that of simulations conducted without consideration of Kpuu (i.e., Kpuu = 1). These authors concluded that the accuracy of DDI predictions improved upon inclusion of Kpuu, however, although root mean square error (RMSE) showed a moderate improvement (5.12 versus 2.31-3.91), there was no change in average fold error (AFE) (1.45 versus 1.36-1.43) nor in percent within 2-fold (86.4% versus 81.8-100%). The purported improvement of DDI predictions based on RMSE values appears to rely entirely on one interaction (itraconazole-triazolam; AUCR = 27.1) for which drug interaction potential was underpredicted. It is telling that the experimental determinations of Kpuu for itraconazole in that report vary greatly between methodologies utilized, with values ranging from 4.16 to 22.6. Aspects related to variability in Kpuu value due to the intricate methodologies required will be discussed in further detail subsequently.
To investigate if predictions of AUCR for a simple metabolic interaction could be improved with implementation of Kpuu to assess relevant inhibitory concentrations, the CYP3A4-mediated interaction between IV midazolam (victim drug) and ketoconazole (perpetrator) [13] was predicted utilizing Eq. 1. Maximum unbound concentration (Imax,u) of ketoconazole was determined to be 0.16 μM and average unbound inhibitory constant (Ki) was 0.061 μM, resulting in a predicted AUCR of 3.6, as compared to the observed ratio of 5.1 as indicated by the solid red line in Figure 3. Published values of ketoconazole Kpuu were utilized to attempt to improve prediction of unbound intracellular ketoconazole concentrations in contact with CYP3A4 and these values were supplemented by simulations of Kpuu ranging from 0.1 to 10, as indicated by the solid black line. To achieve a predicted AUCR of 5.1, a Kpuu value of 1.5 is necessary, however the reported literature Kpuu values ranged almost 15-fold, from 0.32 – 4.67. Thus, depending on the methodology employed, drastically different predictions are achieved. Further, a 4.4-fold difference in Kpuu value for the homogenization method was observed between labs (0.72 [18] vs. 3.18 [17]), highlighting the large degree of inter-lab variability associated with these measurements.
Figure 3. Predicted Magnitude of Metabolic Drug-Drug Interaction Between Ketoconazole (Perpetrator) and Midazolam (Victim) Based on Tsunoda et al. [13].
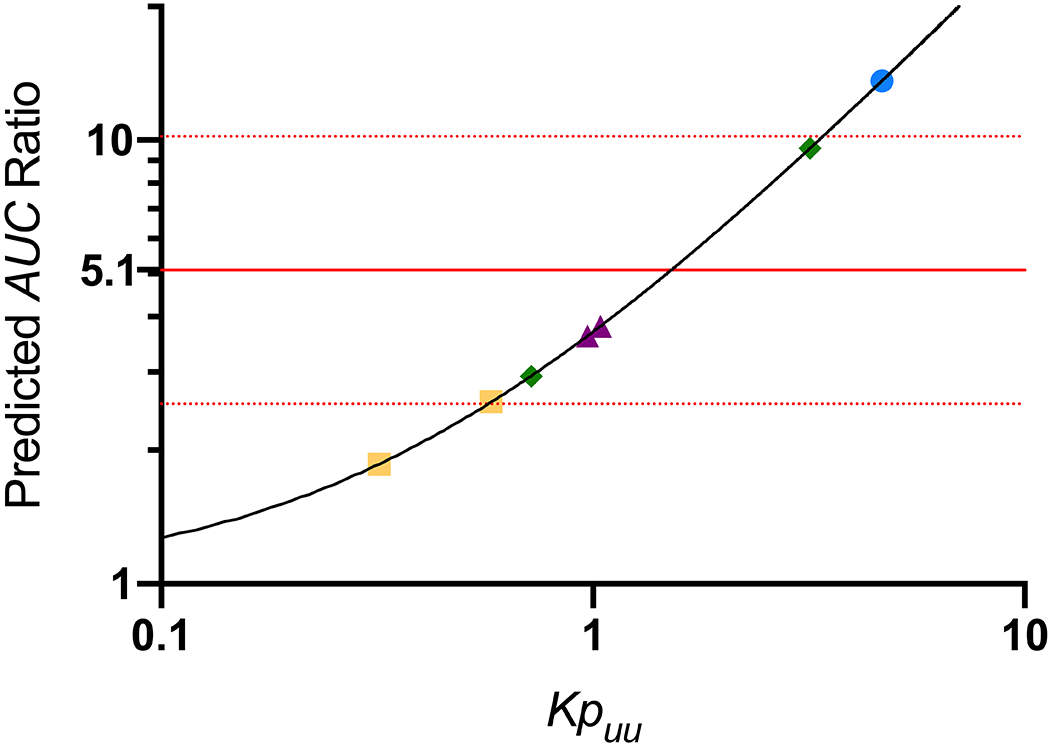
The ratio of AUC for the interaction phase divided by the control phase is plotted on the y-axis. The observed clinical AUC ratio of 5.1 is indicated by the horizontal red line (red dotted lines indicate two-fold differences from this value). Predictive midazolam AUC ratios for simulations incorporating Kpuu values (ranging from 0.1 to 10) that would be observed for reversible CYP3A4 inhibition by ketoconazole are indicated by the black line. Experimental measures of Kpuu from the literature are presented as symbols on this line: purple triangles, temperature method; green diamonds, homogenization method; blue circle, Log D method; yellow squares, extended clearance method.
Limitations in the Utility of Kpuu Values
Although utilization of Kpuu values should theoretically improve the optimization and development of novel therapeutics, significant limitations related to Kpuu methodology result in limited benefits of its implementation, including (1) its basis upon the well-stirred model, which ignores the nuances of intracellular-drug distribution, (2) labor-intensive determination methodologies that are (3) prone to a high degree of variability in outcome with respect to experimental methodology employed and inter-lab variability.
First, the simplification of driving-force concentrations may result in noteworthy limitations in the utility of Kpuu. If subcellular drug localization is not uniform, Kpuu may over- or under-estimate true unbound drug concentrations relevant for the process-of-interest. For instance, discerning any differences between potential inhibition of hepatic metabolism, basolateral transport and apical transport processes poses a challenge when only the average intracellular concentration is known. Further, Kpuu cannot account for the effects of subcellular drug accumulation, which may occur due to pH or electrochemical differences between cytosol and organelles (particularly in lysosomes and mitochondria). If drug accumulates in subcellular compartments, measurement of apparent Kpuu would result in an overestimate of true intracellular cytosolic drug concentration and confound predictions of hepatic disposition of processes associated with unbound cytosolic drug concentrations. The impact of subsequent subcellular drug partitioning on the apparent Kpuu value has been investigated by a number of groups, with particular focus on lysosomal trapping, which is known to affect basic compounds (Table I) [18,32]. For basic drugs, that is diltiazem, erythromycin, imatinib, propranolol, and verapamil, a significant decrease in apparent Kpuu value was observed when lysosomal trapping was inhibited by chloroquine. The largest Kpuu decrease was observed for imatinib (5.3-fold reduction) when lysosomal trapping was inhibited, highlighting that traditional measurements of apparent Kpuu would have significantly overpredicted cytosolic unbound drug concentration by approximately 5-fold. Data summarized in Table I support that this trend is not as apparent for acidic drugs (such as diclofenac, indomethacin and simvastatin acid). The value of Kpuu for verapamil varied greatly between reports (5.7 versus 0.7), highlighting the issues surrounding inter-lab variability. Further in the Riede et al. report [18], erythromycin Kpuu values varied 100-fold between determination methodologies (homogenization method (0.11) versus temperature method (11.5)) within the same lab, demonstrating the need for reliable and consistent methodologies for Kpuu determination. In summary, these studies highlight that subcellular drug accumulation may result in inflated apparent Kpuu values, resulting in an overestimation of true unbound intracellular cytosolic drug concentrations, as well as potential underestimation of subcellular accumulation, which may be relevant for certain pharmacological targets, such as for respiratory indications with targets located in the lung [26]. Until significant advancements in experimental and analytical methodology to detect the nuances of subcellular drug distribution, utilization of Kpuu in pharmacokinetic or pharmacodynamic predictions should be accompanied with recognition of the inherent limitations of the simple but useful single-concentration well-stirred model assumption.
Table I:
Published Kpuu Values with Respect to Subcellular Partitioning
| Drug | Charge Class | Kpuu | Kpuu + Chloroquine | Chloroquine Conc. (μM) | Fold Difference in Kpuu | Method [Reference] |
|---|---|---|---|---|---|---|
| Diclofenac | Acid | 2.6 | 2.2 | 0.5 | 0.85 | Homogenization Methoda; [32] |
| 2.6 | 5 | 1.0 | ||||
| 2.7 | 50 | 1.0 | ||||
| Indomethacin | Acid | 1.3 | 1.4 | 0.5 | 1.1 | Homogenization Methoda; [32] |
| 1.6 | 5 | 1.2 | ||||
| 1.5 | 50 | 1.2 | ||||
| Simvastatin Acid | Acid | 0.70 | 1.3 | 0.5 | 1.9 | Homogenization Methoda; [32] |
| 1.6 | 5 | 2.3 | ||||
| 1.1 | 50 | 1.6 | ||||
| Diltiazem | Basic | 2.9 | 1.9 | 0.5 | 0.66 | Homogenization Methoda; [32] |
| 1.2 | 5 | 0.41 | ||||
| 0.63 | 50 | 0.22 | ||||
| Erythromycin | Basic | 11.5 | 3.36 | 0.5 | 0.29 | Temperature Method; [18] |
| 0.11 | 0.06 | 0.5 | 0.55 | Homogenization Method; [18] | ||
| Imatinib | Basic | 2.71 | 0.56 | 0.5 | 0.21 | Temperature Method; [18] |
| 1.30 | 0.25 | 0.5 | 0.19 | Homogenization Method; [18] | ||
| Propranolol | Basic | 4.7 | 3.3 | 0.5 | 0.70 | Homogenization Methoda; [32] |
| 1.6 | 5 | 0.34 | ||||
| 1.4 | 50 | 0.30 | ||||
| Verapamil | Basic | 5.7 | 5.3 | 0.5 | 0.93 | Homogenization Methoda; [32] |
| 3.7 | 5 | 0.65 | ||||
| 1.8 | 50 | 0.32 | ||||
| Verapamil | Basic | 0.73 | 1.05 | 0.5 | 1.4 | Temperature Method; [18] |
| 0.67 | 0.41 | 0.5 | 0.61 | Homogenization Method; [18] |
Values were digitized from Figure 5 of Mateus et al. [32]
We emphasize that measurement of Kpuu is not a trivial task, either involving (1) determination of total drug partitioning (Kp) that is further corrected by measures or predictions of incubational and hepatic binding (fu,inc and fu,H, respectively) or by (2) measurement of individual hepatic disposition intrinsic clearances for incorporation into Eq. 4, which must be conducted under multiple experimental conditions to isolate each process. Therefore, it is to be expected that experimental outcome may be plagued with variability issues. In a subsequent publication, we will evaluate the reliability of human in vitro Kpuu measurements with respect to inter- and intra-lab variability, experimental methodology, and with consideration of the theoretical Kpuu values expected for transport substrates (versus drugs with no clinical significant transporter involvement for which values are expected to be close to or less than 1). As demonstrated in this manuscript for a limited dataset, our evaluation will report significant variability in measurements for the same drug across different methodologies and by different labs.
CONCLUSIONS:
Although Kpuu may be useful in improving predictions of pharmacodynamics, there is limited benefit of utilization of Kpuu in improving drug-drug interaction predictions. In DDI predictions of victim drug, dependence on extended clearance concept-based AUCR equations (Eqs. 6 and 7) is a more reasonable approach, as changes in Kpuu can potentially mislead an investigator to incorrectly conclude that the Kpuu change has resulted in altered intrahepatic concentrations, an aspect crucial for tissue-specific efficacy or toxicity. Further, utilization of the AUCR equations in DDI prediction can reasonably predict the magnitude of DDIs and does not require any measurement of Kpuu or fu,H, potentially difficult tasks plagued with a high degree of variability between laboratories and between methodologies. The appropriate use of Kpuu is in improving predictions related to pharmacodynamics (i.e. efficacy and toxicity), drug disposition (hepatic clearance or biliary elimination) or in characterizing the inhibitory potential of perpetrator drugs for processes driven by intracellular unbound drug concentrations. Consideration that Kpuu is based on a well-stirred model interpretation of hepatic elimination must be taken into account, as nuances of intracellular drug distribution are not considered by the Kpuu model. Finally, a significant degree of variability in Kpuu values has been suggested in the literature and therefore utilization of this difficult-to-measure theoretical value may result in a large prediction error depending on the particular methodology used. This necessitates the development of reliable and consistent experimental Kpuu determination methodologies to support its role in improving predictive models related to drug disposition.
ACKNOWLEDGEMENTS AND DISCLOUSURES:
This work was supported in part by a Mary Ann Koda-Kimble Seed Award for Innovation. Ms. Sodhi was supported in part by an American Foundation for Pharmaceutical Education Predoctoral Fellowship, NIGMS grant R25 GM56847 and a Louis Zeh Fellowship. Dr. Benet is a member of the UCSF Liver Center supported by NIH grant P30 DK026743. All authors contributed to the writing, derivations, simulations and analysis of this manuscript. The authors declare no conflict of interest.
ABBREVIATIONS:
- AH
amount of drug in the liver
- AUCR
area under the concentration time curves expressed as the ratio of interaction to control
- AUCB,u
area under the concentration time curve of unbound drug in the blood following an oral dose
- AUCH,u
area under the concentration time curve of unbound drug in the liver following an oral dose
- CB
total concentration of drug in the blood
- CB,u
unbound drug concentration in blood
- CH
total concentration of drug in the liver
- CH,bil
total concentration of drug in the liver driving biliary excretion
- CH,eff
total concentration of drug in the liver driving basolateral efflux
- CH,met
total concentration of drug in the liver driving metabolism
- CLH,int
hepatic intrinsic clearance (sum of metabolic intrinsic clearance and intrinsic biliary secretion)
- CLH,int,bil
hepatic intrinsic biliary secretion
- CLH,int,met
hepatic intrinsic metabolic clearance
- CH,u
unbound drug concentration in the liver
- CYP
Cytochrome P450
- DDI
drug-drug interaction
- Fabs
fraction absorbed
- FG
fraction escaping intestinal elimination
- fu,B
fraction of unbound drug in blood
- fu,H
fraction of unbound drug in liver
- fu,inc
fraction of unbound drug in an in vitro incubation
- fu,plasma
fraction of unbound drug in plasma
- Imax,u
maximal unbound plasma concentration of inhibitor drug
- Ki
unbound inhibition constant of inhibitor drug
- Kpuu
unbound liver-to-blood partition coefficient
- PBPK
physiologically-based pharmacokinetic
- PD
pharmacodynamics
- PSeff,int
basolateral efflux (both active and passive) intrinsic clearance
- PSinf,int
basolateral influx (both active and passive) intrinsic clearance
REFERENCES
- 1.Chu X, Korzekwa K, Elsby R, Fenner K, Galetin A, Lai Y, et al. Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther. 2013;94(1):126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trainor GL. The importance of plasma protein binding in drug discovery. Expert Opin Drug Discov. 2007;2(1):51–64. [DOI] [PubMed] [Google Scholar]
- 3.Camenisch G, Riede J, Kunze A, Huwyler J, Poller B, Umehara K. The extended clearance model and its use for the interpretation of hepatobiliary elimination data. ADMET & DMPK. 2015;3(1):1–14. [Google Scholar]
- 4.Benet LZ, Bowman CM, Liu S, Sodhi JK. The extended clearance concept following oral and intravenous dosing; theory and critical analysis. Pharm Res. 2018;35(12):242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benet LZ, Bowman CM, Sodhi JK. How transporters have changed basic pharmacokinetic understanding. AAPS J. 2019;21(6):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barton HA, Lai Y, Goosen TC, Jones HM, El-Kattan AF, Gossed JR, et al. Model-based approaches to predict drug-drug interactions associated with hepatic uptake transporters: preclinical, clinical and beyond. Expert Opin Drug Metab Toxicol. 2013;9(4):459–472. [DOI] [PubMed] [Google Scholar]
- 7.Camenisch G, Umehara K. Predicting human hepatic clearance from in vitro drug metabolism and transport data: a scientific and pharmaceutical perspective for assessing drug-drug interactions. Biopharm Drug Dispos. 2012;33(4):179–194. [DOI] [PubMed] [Google Scholar]
- 8.El-Kattan AF, Varma MVS. Navigating transporter sciences in pharmacokinetics characterization using the extended clearance classification system. Drug Metab Dispos. 2018;46(5):729–739. [DOI] [PubMed] [Google Scholar]
- 9.Patilea-Vrana G, Unadkat JD. Transport vs. metabolism: what determines the pharmacokinetics and pharmacodynamics of drugs? Insights from the extended clearance model. Clin Pharmacol Ther. 2016;100(5):413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sirianni GL, Pang KS. Organ clearance concepts: new perspectives on old principles. J Pharmacokinet Biopharm. 1997;25(4):449–470. [DOI] [PubMed] [Google Scholar]
- 11.Varma MV, Steyn SJ, Allerton C, El-Kattan AF. Predicting clearance mechanism in drug discovery: extended clearance classification system (ECCS). Pharm Res. 2015;32(12):3785–3802. [DOI] [PubMed] [Google Scholar]
- 12.Webborn PJH, Parker AJ, Denton RL, Riley RJ. In vitro-in vivo extrapolation of hepatic clearance involving active uptake: theoretical and experimental aspects. Xenobiotica. 2007;37(10-11):1090–1109. [DOI] [PubMed] [Google Scholar]
- 13.Tsunoda SM, Velez RL, von Moltke LL, Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin Pharmacol Ther. 1999;66(5):461–471. [DOI] [PubMed] [Google Scholar]
- 14.U.S. Food and Drug Administration, Center for Drug Evaluation and Research. In vitro metabolism- and transporter-mediated drug-drug interaction studies: guidance for industry. Silver Spring, MD; 2017. [Google Scholar]
- 15.Greenblatt DJ, Venkatakrishnan K, Harmatz JS, Parent SJ, von Moltke LL. Sources of variability in ketoconazole inhibition of human cytochrome P450 3A in vitro. Xenobiotica. 2010;40(10):713–720. [DOI] [PubMed] [Google Scholar]
- 16.Martínez-Jordá R, Rodriguez-Sasiain JM, Suárez E, Calvo R. Serum binding of ketoconazole in health and disease. Int J Clin Pharmacol Res. 1989;10(5):271–276. [PubMed] [Google Scholar]
- 17.Iwasaki S, Kosugi Y, Zhu AZX, Nakagawa S, Sano N, Funami M, et al. Application of unbound liver-to-plasma concentration ratio to quantitative projection of cytochrome P450-mediated drug-drug interactions using physiologically based pharmacokinetic modelling approach. Xenobiotica. 2019;49(11):1251–1259. [DOI] [PubMed] [Google Scholar]
- 18.Riede J, Camenisch G, Huwyler J, Poller B. Current in vitro methods to determine hepatic Kpuu: a comparison of their usefulness and limitations. J Pharm Sci. 2017;106(9):2805–2814. [DOI] [PubMed] [Google Scholar]
- 19.Li Z, Di L, Maurer TS. Theoretical considerations for direct translation of unbound liver-to-plasma partition coefficient from in vitro to in vivo. AAPS J. 2019;21(3):43. [DOI] [PubMed] [Google Scholar]
- 20.Nordell P, Winiwarter S, Hilgendorf C. Resolving the distribution-metabolism interplay of eight OATP substrates in the standard clearance assay with suspended human cryopreserved hepatocytes. Mol Pharmaceut. 2013;10(12):4443–4451. [DOI] [PubMed] [Google Scholar]
- 21.Riede J, Poller B, Huwyler J, Camenisch G. Assessing the risk of drug-induced cholestasis using unbound intrahepatic concentrations. Drug Metab Dispos. 2017;45(5):523–531. [DOI] [PubMed] [Google Scholar]
- 22.Benet LZ, Hoener B-a. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115–121. [DOI] [PubMed] [Google Scholar]
- 23.Benet LZ, Bowman CM, Koleske ML, Rinaldi CL, Sodhi JK. Understanding drug-drug interaction and pharmacogenomic changes in pharmacokinetics for metabolized drugs. J Pharmacokinet Pharmacodyn. 2019;46(2):155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varma MV, Bi YA, Kimoto E, Lin J. Quantitative prediction of transporter- and enzyme-mediated clinical drug-drug interactions of organic anion-transporting polypeptide 1B1 substrates using a mechanistic net-effect model. J Pharmacol Exp Ther. 2014;351(1):214–223. [DOI] [PubMed] [Google Scholar]
- 25.Loryan I, Sinha V, Mackie C, Van Peer A, Drinkenburg W, Vermeulen A, et al. Mechanistic understanding of brain drug disposition to optimize the selection of potential neurotherapetuics in drug discovery. Pharm Res. 2014;31(8):2203–2219. [DOI] [PubMed] [Google Scholar]
- 26.Ufuk A, Assmus F, Francis L, Plumb J, Damian V, Gertz M, et al. In vitro and in silico tools to assess extent of cellular uptake and lysosomal sequestration of respiratory drugs in human alveolar macrophages. Mol Pharmaceut. 2017;14(4):1033–1046. [DOI] [PubMed] [Google Scholar]
- 27.Tsmandouras N, Dickinson G, Guo Y, Hall S, Rostami-Hodjegan A, Galetin A, et al. Development and application of a mechanistic pharmacokinetic model for simvastatin and its active metabolite simvastatin acid using an integrated population PBPK approach. Pharm Res. 2015;32(6):1864–1883. [DOI] [PubMed] [Google Scholar]
- 28.Riccardi KA, Tess DA, Lin J, Patel R, Ryu S, Atkinson K, et al. , A novel unified approach to predict human hepatic clearance for both enzyme- and transporter-mediated mechanisms using suspended human hepatocytes. Drug Metab Dispos. 2019;47(5):484–492. [DOI] [PubMed] [Google Scholar]
- 29.Bowman CM, Benet LZ. An examination of protein binding and protein-facilitated uptake relating to in vitro-in vivo extrapolation. Eur J Pharm Sci. 2018;123:502–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gertz M, Cartwright CM, Hobbs MJ, Kenworthy KE, Rowland M, Houston JB, et al. Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug-drug interaction potential. Pharm Res. 2013;30(3):761–780. [DOI] [PubMed] [Google Scholar]
- 31.Rowland Yeo K, Walsky RL, Jamei M, Rostami-Hodjegan A, Tucker GT. Prediction of time-dependent CYP3A4 drug-drug interactions by physiologically based pharmacokinetic modelling: impact of inactivation parameters and enzyme turnover. Eur J Pharm Sci. 2011;43(3):160–173. [DOI] [PubMed] [Google Scholar]
- 32.Mateus A, Matsson P, Artursson P. Rapid measurement of intracellular unbound drug concentrations. Mol Pharmaceut. 2013;10(6):2467–2478. [DOI] [PubMed] [Google Scholar]