

Abstract
There is currently a lack of precise predictive biomarkers for patient selection in clinical trials of inhibitors targeting replication stress (RS) response proteins ATR and CHK1. The objective of this study was to identify novel predictive biomarkers for the response to these agents in treating non-small cell lung cancer (NSCLC). A genome-wide loss-of-function screen revealed that tumor suppressor PPP2R2A, a B regulatory subunit of protein phosphatase 2 (PP2A), determines sensitivity to CHK1 inhibition. A synthetic lethal interaction between PPP2R2A deficiency and ATR or CHK1 inhibition was observed in NSCLC in vitro and in vivo and was independent of p53 status. ATR and CHK1 inhibition resulted in significantly increased levels of RS and altered replication dynamics, particularly in PPP2R2A-deficient NSCLC cells. Mechanistically, PPP2R2A negatively regulated translation of oncogene c-Myc protein. c-Myc activity was required for PPP2R2A deficiency-induced alterations of replication initiation/RS and sensitivity to ATR/CHK1 inhibitors. We conclude that PPP2R2A deficiency elevates RS by upregulating c-Myc activity, rendering cells reliant on the ATR/CHK1 axis for survival. Our studies show a novel synthetic lethal interaction and identify PPP2R2A as a potential new predictive biomarker for patient stratification in the clinical use of ATR and CHK1 inhibitors.
Keywords: ATR and CHK1 inhibitor, PPP2R2A, replication stress, lung cancer, c-Myc
Introduction
Non-small cell lung cancer (NSCLC) accounts for approximately 85% of lung cancer cases. It is the most common cancer and has the highest mortality. NSCLC is often driven by oncogenes that are either not amenable to direct therapeutic intervention or when targeted therapies or immunotherapy are available, only a small fraction of patients respond to them. Additionally, treatment resistance invariably develops (1), thus highlighting the urgent need for the development of novel treatment strategies.
Phosphatase 2 (PP2A) is responsible for the majority of Serine/Threonine phosphatase activity in human cells (2). The PP2A holoenzyme consists of a catalytic subunit C, a scaffold subunit A, and one of at least 18 associated variable regulatory proteins (B subunits) classified into 4 families (B, B′, B′′, B′′′), each consisting of several isoforms and splice variants. It is estimated that there are between 75 and 100 unique PP2A holoenzymes (3). The substrate specificity of this phosphatase is determined by the specific PP2A complexes. Although functions of the core enzyme, PP2A AC, have been extensively studied, the roles of the regulatory subunits, particularly PPP2R2A (also called B55 α), have not been widely reported. Although limited functional studies of PPP2R2A have been performed, this gene appears to act as a tumor suppressor and is frequently deleted or under-expressed in a wide range of human cancers. Loss of heterozygosity (LOH) of the region coding for PPP2R2A leads to reduced PPP2R2A expression in 60% of prostate cancers, 46% of ovarian cancers, and 43% of lung adenocarcinomas, a major subtype of NSCLC (4). Given that reduced PPP2R2A expression is associated with poor outcomes (5–7), treatments for PPP2R2A-deficient cancers represent an unmet clinical need.
Proteins required for the DNA damage response (DDR) are promising targets for cancer therapy. Currently, inhibitors targeting proteins that are important for the response to replication stress (RS), a branch of DDR that challenges the normal process of DNA replication, are under extensive investigation. RS can be induced by exogenous RS-inducing agents and deregulation of oncogenes, such as c-Myc, Ras, and cyclin E (8). Ataxia-telangiectasia-mutated-and-Rad3-related kinase (ATR) and its major downstream effector checkpoint kinase 1 (CHK1) are key components of the RS response. ATR/CHK1 signaling prevents the entry of cells harboring damaged or incompletely replicated DNA into mitosis when the cells are exposed to DNA damaging agents, such as radiation therapy or chemotherapeutic drugs (9), especially in cells with a deficiency in the cell cycle G1 phase checkpoint. This situation is frequently observed in tumors (e.g. loss of TP53). In addition, ATR/CHK1 suppresses RS caused by oncogene activation to a less toxic level. Thus, cancer cells with high levels of oncogene-induced RS depend heavily on ATR or CHK1 for survival. These unique properties of ATR and CHK1 make them ideal therapeutic targets (9).
Inhibitors targeting ATR and CHK1 are currently being tested either paired with radiotherapy or with a variety of genotoxic chemotherapies, or as single agents in clinical studies. The majority of current clinical trials of ATR and CHK1 inhibitors have not taken molecular features of the cancers into consideration, which could significantly reduce the efficacy of these agents. To date, only limited efficacy has been noted in clinical trials when these agents are combined with standard cancer therapy (10–12). p53 was thought to be a predictive biomarker of the response to CHK1 inhibitor in combination with radiotherapy or chemotherapy based upon preclinical studies; however, p53 status was not associated with the treatment response to CHK1 inhibitors in a clinical trial (10). Interestingly, recent studies suggest that ATR and CHK1 inhibition could be used as monotherapies (13–15). Cancer cells with high levels of oncogene-induced RS are more sensitive to ATR and CHK1 inhibitors without exogenous DNA damage agents in combination (14,15). For example, presence of an extra CHK1 allele limits oncogene (RAS/E1A)-induced RS/DNA damage and promotes transformation (16). Suppression of ATR function in oncogenic KRAS-expressing cells synergistically increases genomic instability and cell death induced by DNA damage (17). Moreover, ATR/CHK1 inhibitors can specifically target Myc (c-Myc)-driven cancer cells (18–21). CHK1 inhibitors may be used as a single agent to target lymphoma cells and small-cell lung cancers (SCLC) expressing c-Myc (18,22,23). Excitingly, a recent phase II clinical trial suggests that 33% (8 out of 24) of BRCA wild-type recurrent high-grade serous ovarian cancer patients had partial responses to Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor. Additionally, this response is associated with high expression of oncogene cyclin E, providing first-in-class proof-of-concept that ATR and CHK1 inhibitors can be used singularly and target cancer cells with oncogene expression (13). Nevertheless, it remains unknown whether tumor suppressor genes affect the sensitivity to ATR and CHK1 inhibitors as a monotherapies.
To determine the factors affecting the sensitivity to ATR and CHK1 inhibitors, we performed a high-throughput screen using a pooled shRNA library in the NSCLC cell line H1299. Here, we show that PPP2R2A is a top hit in our screen. Using in vitro MTT and clonogenic assays and an in vivo xenograft model of NSCLC, we validated the screen result by showing that ATR and CHK1 inhibitors are synthetic lethal to NSCLC cells with reduced PPP2R2A expression. ATR or CHK1 inhibition leads to increased RS, increased replication initiations, and decreased replication fork speed, particularly in the NSCLC cells with PPP2R2A deficiency. At the molecular level, PPP2R2A negatively regulates the expression of c-Myc protein independently of its degradation. PPP2R2A deficiency-induced c-Myc expression depends upon the activity of protein translation initiation. Lastly, PPP2R2A deficiency-induced RS and enhanced sensitivity to ATR and CHK1 inhibitors were abrogated when c-Myc was inactivated. We conclude that PPP2R2A deficiency leads to elevated RS by upregulating c-Myc activity, causing the cells to rely heavily on the ATR/CHK1 axis for survival. Thus, PPP2R2A-deficient NSCLC cells can be specifically targeted by ATR or CHK1 inhibition, and PPP2R2A represents a new potential predictive biomarker to identify patients who may respond to ATR or CHK1 inhibitors.
Materials and Methods
Genome-wide lentiviral shRNA screening
The human GIPZ whole genome library (RHS6083, GE Dharmacon) and H1299 cells were used for the genome-wide lentiviral shRNA screening. The screening procedure was conducted according to the manufacturer’s instructions. One µM of LY2603618 (APExBIO) was used for CHK1 inhibition. Genomic DNA extraction, PCR amplification of shRNA, and PCR product purification were performed according to the RHS6083 protocols. Purified amplicons from different samples were pooled and analyzed with a Bioanalyzer (Agilent) and qPCR, and then sequenced on an Illumina HiSeq 2500 platform on high output mode. TrimGalore was used for trimming and quality evaluation of the sequences from the shRNA samples. Sequences that passed the quality filters were aligned to the shRNA library sequences provided by Dharmacon (GE). The R package DESeq was used to determine whether there was differential expression. An initial significance cutoff was applied to the DESeq output, and only shRNAs that had an adjusted P-value < 0.05 false discovery rate (FDR) were considered.
Cell lines, plasmids, and inhibitors
H1299, HEK-293T, and A549 cells were cultured in DMEM medium (Hyclone); SK-MES-1 cells were cultured in MEM medium (Hyclone); and H838 and H1437 cells were cultured in 1640 medium (Hyclone) supplemented with 10% bovine growth serum (BGS; Hyclone) in a humidified atmosphere containing 5% CO2 at 37°C. Cells which had been passaged ten times or less were used for experiments. All cells were purchased from ATCC in 2016 and were authenticated via STR profiling by the MCIC Genomics core at Ohio State Univeristy in 2020. All cell lines were determined to be Mycoplasma-free using the LookOut® Mycoplasma PCR Detection Kit (MP0035, Sigma) in 2020.
All shRNAs were purchased from Sigma-Aldrich. pMIG FLAG-PPP2R2A was obtained from Addgene (plasmid 13804), and pBabe puro-PPP2R2A was generated by PCR amplification of full-length PPP2R2A from pMIG FLAG-PPP2R2A and its subsequent subcloning into pBabe puro (Addgene). pBABEpuro-c-Myc and pBABEpuro-c-MycS62A plasmids were gifts from Dr. Peter J. Hurlin (Oregon Health and Science University). The CHK1 inhibitor LY2603618 was purchased from APExBIO Technology (A8638), and the ATR inhibitor AZD6738 (S7693), PP2A inhibitor LB-100 (S7357), c-Myc inhibitor 10058-F4 (S7153), and mTOR inhibitor INK128 (S2811) were obtained from Selleckchem.
MTT
Real-time quantitative reverse transcription-PCR
Real-time quantitative reverse transcription-PCR (qRT-PCR) was conducted as described in our previous publications (15,24). Primers are listed in the supplementary materials.
Immunofluorescence assays
Immunofluorescence assays were performed as described previously (24,25). The antibodies used for immunofluorescence were γ-H2AX (JBW301, 1:500, Millipore) and p-RPA32 (S4/S8) (A300-245A, 1:500, Bethyl).
Comet assay
The Neutral Comet Assay was performed using the Comet Assay kit from Trevigen, following the manufacturer’s instructions. Comets were analyzed using TriTek CometScore software ver.2.0.0.38.
Immunoblotting
Immunoblotting was conducted as previously described (15,25). The primary antibodies used for western blot were c-Myc (SC-40, 1:500, Santa Cruz Technology); phospho-c-Myc (#13748, c-Myc-pSer62, 1:500; Cell Signaling Technology); phospho-c-Myc (Y011034, c-Myc-pThr58, 1:1000; Applied Biological Materials Inc.); RPA2 (Clone NA18, 1:100, Calbiochem/EMD Millipore); anti-β-actin (Clone AC-74, 1:50000, Sigma-Aldrich); anti-CHK1 (G-4, 1:200, Santa Cruz Technology); phospho-CHK1 antibody (#133D3, CHK1-pSer345, 1:500, Cell Signaling Technology); CDC45 (G-12 sc55569, 1:200, Santa Cruz Technology); γ-H2Ax (ser139, clone JBC301, 1:500, Millipore); rabbit polyclonal antibody phosphor RPA32 Ser4/Ser8 (BL647, 1: 1000, Bethyl; Histone H3 (#9715, 1:1000, Cell Signaling Technology); PPP2R5A (ab89621,1:1000, Abcam); eIF4E (A2162, 1:1000, Abclonal); Phospho-eIF4E (#9741, eIF4E-Ser209, 1:1000, Cell Signaling Technology); 4E-BP1 (#9452, 1:1000, Cell Signaling Technology); Phospho-4E-BP1 (#2855, 4E-BP1-Thr36/47, 1:1000, Cell Signaling Technology); Phospho-p70S6K antibody (#9205, p70S6K-Thr389, 1:1000, Cell Signaling Technology); p70S6K (#9207, 1:1000, Cell Signaling Technology); Cdc45 (H-300 clone, SC20685, 1:50, Santa Cruz Technology); and phospho-histone H3 (Ser10); (#9706, 1:100, Cell Signaling Technology).
DNA fiber assays
DNA fiber assays were performed as previously described (24,25). The antibodies used for the DNA fiber assay were BrdU antibody (#347580, 1:200, BD Biosciences) and CldU antibody (ab6326, 1:400, Abcam).
Cycloheximide assay
Cycloheximide assay was performed as previously described (3).
Xenograft studies
Both male and female athymic nude mice (Strain code: 553, NCI Frederick), 4–5 weeks of age, were used for this study. The mice were bred at Ohio State University. Xenografts were established by subcutaneous injections of A549 cells (4 × 106 cells) into both flanks of the mice. Tumor diameters were measured with digital calipers, and the tumor volume in mm3 was calculated using the following formula: Volume = (width)2 × length/2. Once tumor volume reached 100–150 mm3, the mice were treated with vehicle control or a CHK1 inhibitor (25 mg/kg of LY2603618) via intraperitoneal injection twice a day for 3 days, followed by 4 days of rest. An ATR inhibitor (50 mg/kg; AZD6738) was administered for 20 consecutive days via oral gavage as described previously (26). All mice were maintained under barrier conditions, and the experiments were conducted using protocols and conditions approved by the Institutional Animal Care and Use Committee of The Ohio State University.
Results
Genome-wide shRNA screen identifies PPP2R2A as a CHK1 inhibitor-sensitizing gene.
To identify novel determinants that sensitize cells to CHK1 inhibitors, we conducted a high-throughput screen using the Decode Pooled Lentiviral shRNA library in the p53-deficient NSCLC cell line H1299. Cells were infected with the GIPZ human shRNA library encompassing 95,700 lentiviral shRNAs that target 18,205 unique human protein-coding genes. The screening workflow is outlined in Fig. 1A. LY2603618 is the first highly selective and potent CHK1 inhibitor (CHKi). The effects of the shRNAs were quantified as standardized Z scores and are presented in a heatmap, with negative scores indicating sensitization and positive scores indicating resistance (Fig. 1B). The Z value distribution is displayed in Fig. 1C. Notably, PPP2R2A (B55 α), PPP2R1A (PP2A Aα), and PPP2R2D (B55 δ), which encode three subunits of PP2A, were identified as new hits (Fig. 1C). We selected PPP2R2A as a major focus of this study because this gene was the most frequent hit, had a relatively strong Z score, and reduced expression as a result of PPP2R2A LOH is seen at a high frequency in lung cancer (4). In addition, analysis of publicly available lung cancer patient gene expression data suggests that low PPP2R2A expression is associated with poor prognosis in these patients (Fig. S1A–D). Analysis of The Cancer Genome Atlas (TCGA) data also suggests that PPP2R2A homozygous deletion is not associated with amplification/in-frame driver mutations in EGFR, ALK, or ROS1 (Fig. S1E), which are the most common oncogene drivers that have been targeted in NSCLC (27), suggesting that the available therapies targeting these oncogene drivers may not be suitable for treating low-expressing PPP2R2A lung cancer.
Fig. 1. Genome-wide shRNA screening identifies PPP2R2A as a CHK1 inhibitor sensitivity gene.
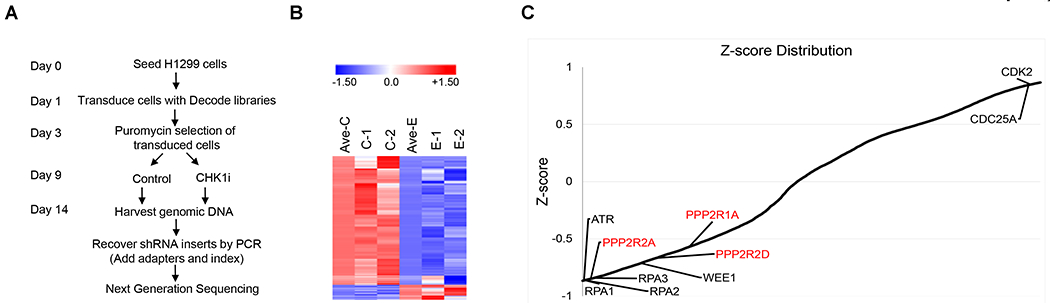
(A) Screening workflow. gDNA was isolated from the reference and experimental populations of transduced cells. CHK1i, CHK1 inhibitor (LY2603618). (B) The heatmap depicts the Z scores for two replica screens (C1/C2 for replicas of the control group and E1/E2 for replicas of the treatment group). (C) Plot of average Z scores. Several well-established ATR/CHK1 inhibitor sensitivity or resistance genes are indicated in black. Newly identified hits in our screen are indicated in red.
Synthetic lethal interaction between PPP2R2A deficiency and ATR/CHK1 inhibition in vitro and in vivo.
To validate our results, we directly assessed the relationship between PPP2R2A deficiency and CHK1 and ATR inhibitor sensitivity using MTT assays. H1299 (p53 null) cells and A549 (p53 wild type) cells depleted of PPP2R2A by shRNA exhibited significant suppression of proliferation following LY2603618 (CHK1i) and AZD6738 (ATRi) treatment. In contrast, cells treated with CHK1 or ATR inhibitor alone or untreated PPP2R2A knockdown cells continued to proliferate, although at a slower rate than the control cells (shcon) (Fig. 2A–D). To examine the potential synergistic interaction between PPP2R2A inhibition and CHK1 inhibition, we performed a modified MTT assay based upon CompuSyn, in which only inhibitors rather than shRNA knockdown can be used. Because a PPP2R2A inhibitor is not available, we used the PP2A inhibitor LB-100, which is currently being tested in clinical trials as an anti-tumor drug. Combination index analysis suggested that LB-100 and LY2603618 act synergistically, since their combination indices were lower than 0.9 at all doses tested (Fig. S2A, B). Similar results were found for AZD6738 in both H1299 (Fig. S2C) and A549 cells (Fig. S2D). In addition, the synthetic lethality between PPP2R2A deficiency and ATR/CHK1 inhibition was further confirmed by a colony formation assay in H1299 cells (Fig. S2 E–F). Thus, ATR/CHK1 axis inhibition acts synergistically with the loss of PPP2R2A expression and PP2A inhibition, regardless of p53 status.
Fig. 2. Synthetic lethality between PPP2R2A deficiency and CHK1 or ATR inhibition.
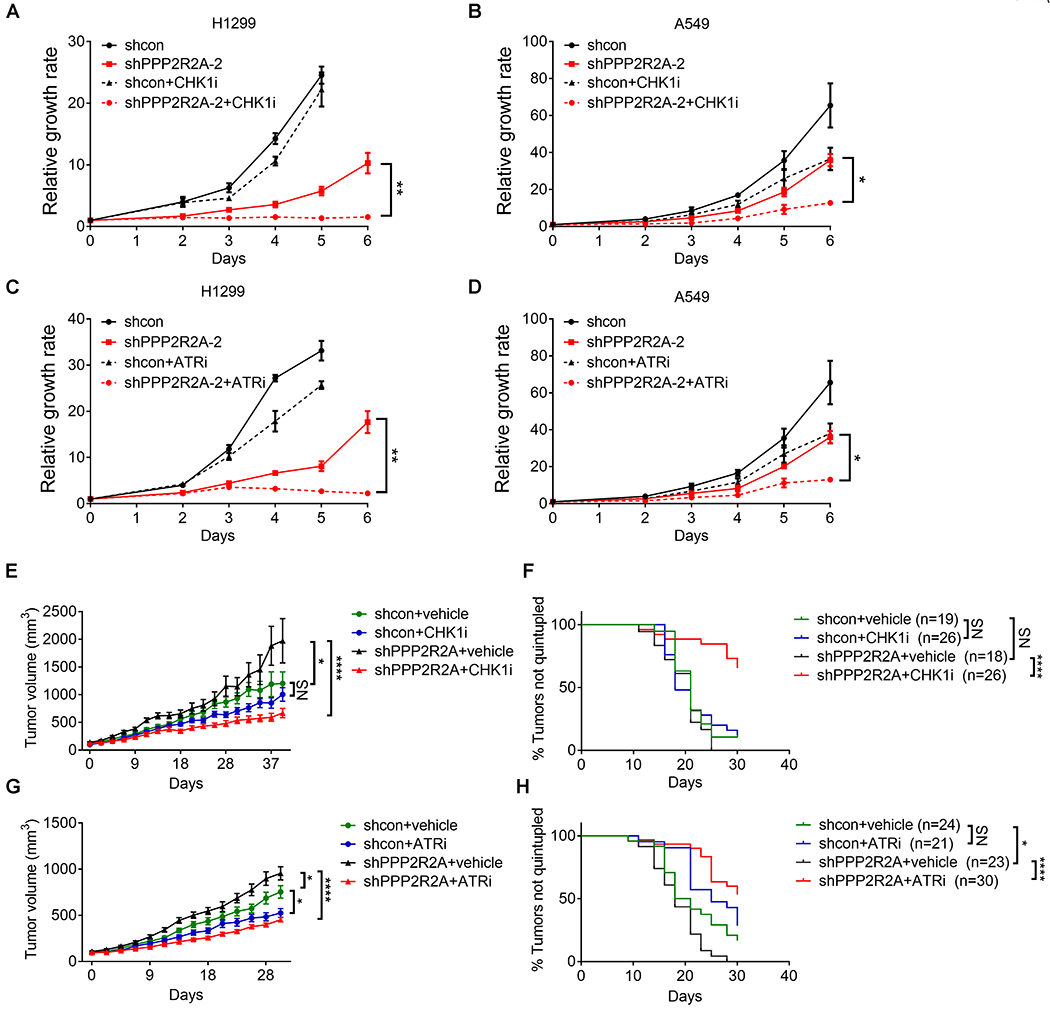
(A-D) CHK1 inhibition by LY2603618 (1 μM) and ATR inhibition by AZD6738 (1 μM) inhibited the proliferation of H1299 cells (A, C) and A549 cells (B, D) depleted of PPP2R2A as detected by MTT assay. Data are the mean ± SEM of three independent experiments. The cell number of the control group (shcon) of H1299 cells at day 6 (A, C) are not included because the cell growth had plateaued. *P < 0.05, **P < 0.01, Student’s t test (right panel). (E–H) CHK1-ATR inhibition led to tumor volume reduction and prolonged survival in PPP2R2A-defective xenografts. The mice were treated with either vehicle or intraperitoneal CHK1 inhibitor LY2603618 (25 mg/kg) via intraperitoneal injection twice a day for 3 days followed by 4 days of rest for three cycles (E, F). For ATR inhibition, vehicle or AZD6738 (50 mg/kg) was administered for 20 consecutive days via oral gavage (G, H). Tumor volumes are mean ± SEM (E, G). Statistical significance was determined by one-way ANOVA followed by Bonferroni’s post hoc analysis for multiple comparisons. *P < 0.05, ***P < 0.001, and ****P < 0.0001. Kaplan–Meier analysis was used for overall survival (F, H). n = number of tumors. Significance was determined by the Mantel–Cox test. *P < 0.05 and ****P < 0.0001.
To further verify our results, we determined the sensitivity of CHK1 and ATR inhibitors in cell lines with high and low PPP2R2A expression. H1437 and SK-MES-1 express lower levels of PPP2R2A due to LOH of the PPP2R2A-coding region, whereas PPP2R2A expression remains intact in A549 and H838 cells (4). SK-MES-1 and H1437 cells were more sensitive compared to A549 and H838 cells (Fig. S3A–C). Correspondingly, expression of exogenous PPP2R2A (Fig. S3D) led to increased resistance to CHK1 and ATR inhibitors in H1437 and SK-MES-1 cells (Fig. S3E–H). Thus, low expression of PPP2R2A as a result of LOH is associated with sensitivity to CHK1 and ATR inhibition.
Next, we further validated our results using an in vivo assay. CHK1 inhibition significantly inhibited tumor growth (Fig. 2E) and increased overall survival in mice bearing tumors derived from A549 cells with stable PPP2R2A knockdown, whereas CHK1 inhibition was not effective in control cells with intact PPP2R2A expression (Fig. 2F). Similar results were also seen with the ATR inhibitor (Fig. 2G, H). Thus, ATR and CHK1 inhibition specifically target cancer cells with PPP2R2A deficiency. Of note, PPP2R2A knockdown xenografts grow faster compared to those derived from control cells, further supporting its role as a tumor suppressor (Fig. 2E, G). However, PPP2R2A knockdown leads to slow growth of cancer cells in vitro (Fig. 2A–B), which supports the hypothesis that PPP2R2A deficiency leads to a RS response, resulting in decreased cell growth in vitro. Multiple synergistic pathways are induced during tumorigenesis, which can overcome this barrier, and PPP2R2A appears to have tumor-suppressive functions in vivo. For instance, oncogene-induced RS triggers a RS response and senescence/apoptosis (28), but cancer cells overcome these suppressive responses using compensatory mechanisms. In addition, the tumor microenvironment provided by our in vivo assay may also contribute to the differences in the results between the in vitro and in vivo assays.
PPP2R2A deficiency leads to increased replication initiation, which is further enhanced by ATR/CHK1 inhibition.
We hypothesized that PPP2R2A deficiency may activate oncogene activities, which subsequently cause RS by dysregulating replication initiation, rendering the cells more sensitive to ATR and CHK1 inhibition. To test this hypothesis, we first determined the status of replication initiation using a DNA fiber assay according to the protocol indicated in Fig. 3A. The percentage of new origin firing was increased in the PPP2R2A-depleted cells compared to the control cells (Fig. S4A). PPP2R2A deficiency promotes the enrichment of CDC45 in chromatin, which is a replication initiation rate limiting factor and is associated with oncogene-induced replication initiation (29,30) (Fig. S4B). CDC45-mediated over-firing could decrease the elongation rate and induce fork asymmetries (29). Correspondingly, a reduced fork speed was observed in cells with PPP2R2A deficiency (Fig. S4C). Therefore, PPP2R2A deficiency can lead to upregulated initiation of replication in the absence of exogenous DNA damage agents. Given the synthetic lethality between PPP2R2A defects and ATR/CHK1 inhibition (Fig. 2), we next measured CHK1 and ATR inhibition-induced replication initiation in cells with or without PPP2R2A knockdown. The percentages of newly fired origins increased significantly in the cells treated with either CHK1 inhibitor (Fig. 3B) or ATR inhibitor (Fig. 3C), which is consistent with previous reports that ATR/CHK1 signaling suppress replication initiation (31,32). CHK1 or ATR inhibition leads to a much higher level of replication initiations in PPP2R2A-depleted cells compared to control cells with intact PPP2R2A expression (Fig. 3B, C). Furthermore, ATR/CHK1 inhibition caused a remarkable increase in non-extractable CDC45 in PPP2R2A-deficient cells, whereas much less CDC45 chromatin recruitment was observed in the control cells (Fig. 3D). Accordingly, the elongation rates decreased dramatically in the cells treated with CHK1 (Fig. 3E) and ATR inhibitors (Fig. 3F), particularly in the PPP2R2A-deficient cells. Representative DNA fiber staining is shown in Fig. S5. Cumulatively, our data suggest that PPP2R2A deficiency leads to dysregulated replication dynamics by increasing replication initiations and decreasing replication fork speed. ATR/CHK1 inhibition leads to a further increase in replication initiations and decreased replication fork speed. These results are consistent with a synergistic interaction between PPP2R2A deficiency and CHK1 or ATR inhibition in suppressing cancer cell proliferation and tumor growth (Fig. 2).
Fig. 3. ATR/CHK1 inhibition leads to dysregulated replication fork dynamics, particularly in cells with PPP2R2A deficiency.
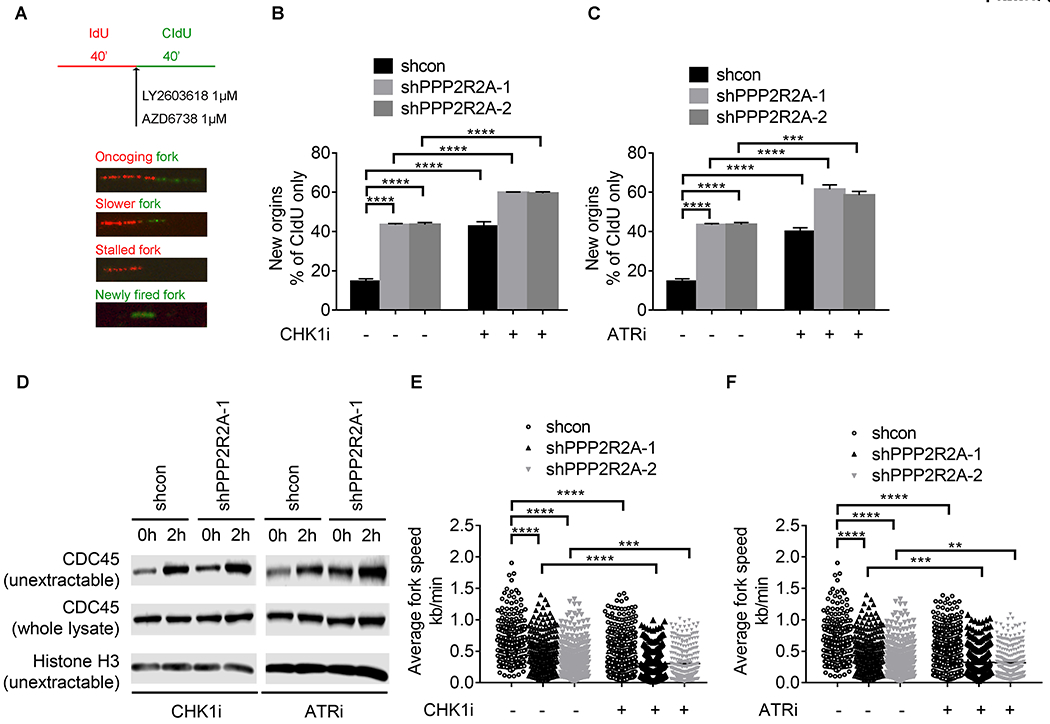
(A) Schematic representation of the DNA fiber assay in the presence of CHK1 and ATR inhibitors in H1299 cells. CHK1 inhibition by LY2603618 (B) and ATR inhibition by AZD6738 (C) significantly increases the rate of replication initiations. (D) ATR/CHK1 inhibition leads to increased chromatin loading of CDC45 in PPP2R2A-defective cells. (E, F), ATR/CHK1 inhibition significantly decreases the average replication fork speeds, particularly in cells depleted of PPP2R2A. Data in B, C, E, and F are the mean ± SEM of three independent experiments. Statistical significance was determined by one-way ANOVA followed by Bonferroni’s post hoc analysis for multiple comparisons. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
ATR/CHK1 inhibition increases RS, particularly in cells with reduced PPP2R2A expression.
Deregulated replication initiations cause RS. To determine the consequences of the increased replication initiation rate due to PPP2R2A deficiency (Fig. 3), we first evaluated PPP2R2A deficiency-induced RS by analyzing the phosphorylation of RPA2 (pRPA2) and histone protein H2AX (γ-H2AX), markers for RS and/or DNA double strand breaks (DSBs) by western blot. Prior to CHK1 inhibition, PPP2R2A depletion alone increased pRPA2, γ-H2AX, and p-Ser 345 CHK1 expression compared to control cells (Fig. S6A), indicating that PPP2R2A depletion caused spontaneous RS and activated ATR/CHK1 signaling in the absence of exogenous DNA damage. In addition, increased formation of pRPA2/γ-H2AX and 53BP1 foci (Fig. S6B, C) and increased tail moment as seen by immunostaining and comet assay under neutral conditions (Fig. S6D, E) further confirmed the presence of PPP2R2A knockdown-induced RS. Although we could not exclude the possibility that the increased expression and foci of pRPA2 and γ-H2AX in PPP2R2A knockdown cells is due to reduced phosphatase activity of PPP2R2A, the result from the neutral comet assay, which can directly detect DSBs, suggests that PPP2R2A deficiency leads to increased DSBs. Therefore, PPP2R2A deficiency leads to spontaneous RS/DSBs, which is consistent with the increased replication initiation observed in the PPP2R2A knockdown cells (Fig. S4A). Next, we examined whether ATR/CHK1 inhibition induced RS in the presence and absence of PPP2R2A deficiency. Consistent with previous reports (31,33) that CHK1 inhibition alone results in RS, CHK1 inhibition increased p-RPA2 and γ-H2AX protein expression (Fig. 4A). The inhibition was monitored via detection of p-CHK1-Ser345 expression (Fig. 4A). Greater increases in CHK1 inhibition-induced p-RPA2 and γ-H2AX protein (Fig. 4A) and CHK1-ATR inhibition-induced p-RPA2 and γ-H2AX foci (Fig. 4B, C, S7B) were observed in PPP2R2A knockdown cells. A similar result was observed using a second PPP2R2A shRNA (Fig. S7C). A comet assay under neutral conditions was conducted to detect DSBs (Fig. 4D, S7A). The amount of DSBs in the cells treated with CHK1i was dramatically higher in the PPP2R2A-deficient cells compared to control cells (Fig. 4D and Fig. S7A). Staining of pan-γ-H2AX, a marker for unrepaired DNA damage/apoptosis, was also increased after ATR/CHK1 inhibitor treatment, and PPP2R2A knockdown led to a greater increase in pan-γ-H2AX staining after ATR/CHK1 inhibition (Fig. S8A, B, S9A, B). Taken together, these results suggest that PPP2R2A deficiency leads to increased RS in the absence of exogenous DNA damage agents, triggering ATR/CHK1 activation. ATR/CHK1 inhibition leads to a greater increase in the level of RS in PPP2R2A-deficient NSCLC cells. This result aligns with the greater toxicity of CHK1 and ATR inhibitors and the dramatic upregulation of ATR/CHK1 inhibition-induced replication initiation, especially in the PPP2R2A-deficient cells (Fig. 2, 3).
Fig. 4. ATR/CHK1 inhibition leads to increased replication stress, particularly in cells with reduced PPP2R2A expression.
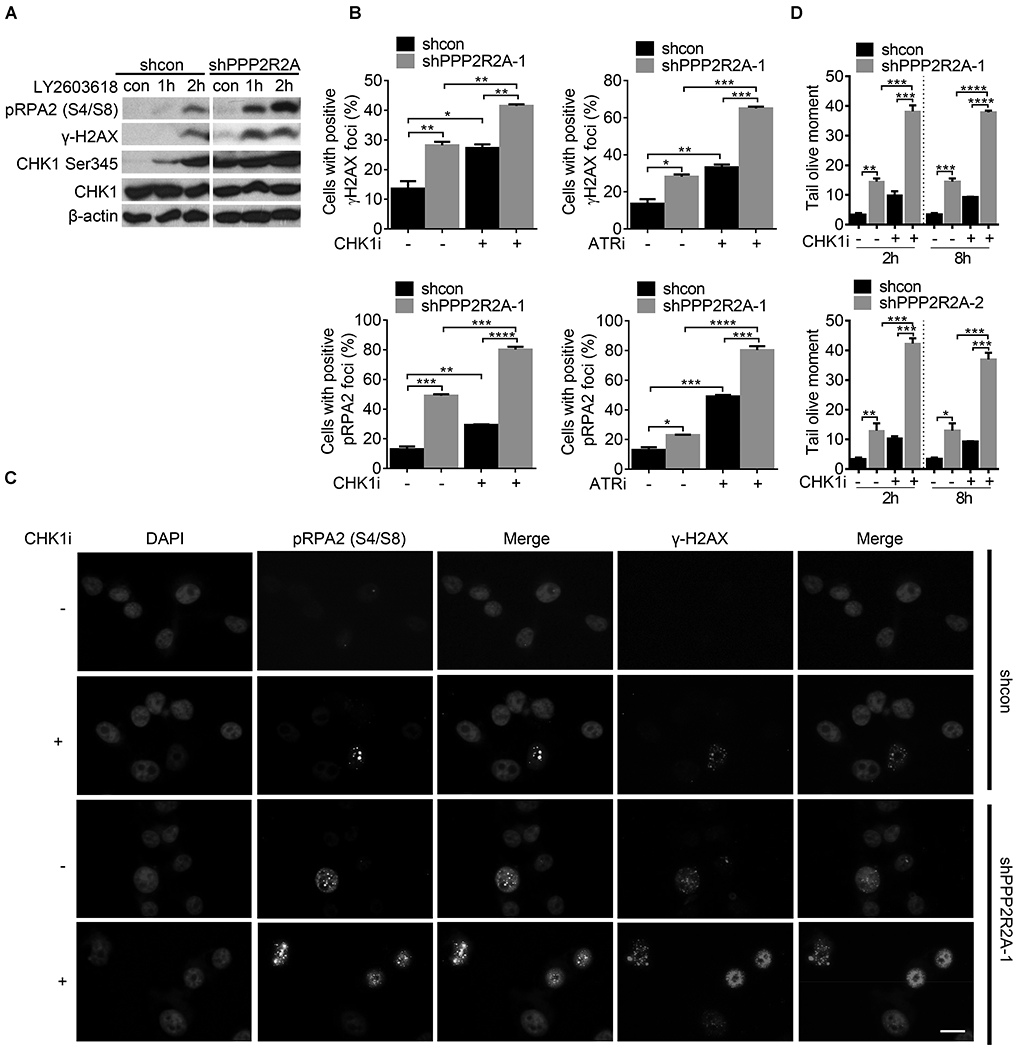
(A) p-RPA2, p-CHK1, and γ-H2AX protein expression increased after CHK1 inhibition. H1299 cell lysate was collected 1 and 2 h after CHK1 inhibition (LY2603618, 1 μM). (B) The percentages of cells with positive γ-H2AX and p-RPA2 foci (≥5) in H1299 cells with or without PPP2R2A knockdown using immunofluorescence assay. Cells were collected and fixed after treatment with the CHK1 inhibitor LY2603618 (1 μM) or the ATR inhibitor AZD6738 (1 μM) for 2 h. (C) Representative imaging of γ-H2AX and p-RPA2 foci. Scale bars=100 μm. (D) Quantification of olive tail moment. H1299 cells were treated with LY2603618 (1 μM) for 2 h or 8 h. All data are presented as the mean ± SEM. ***P < 0.001, by one-way ANOVA followed by Bonferroni’s post hoc test. All data are representative of three independent experiments.
PPP2R2A deficiency leads to increased expression of c-Myc protein.
Next, we attempted to address the question of how PPP2R2A deficiency leads to spontaneous RS. Although not all oncogenes induce RS, c-Myc is a well-known oncogene which causes RS (30,34,35) and is reported to be a putative substrate of PP2A-B56α (PPP2R5A) (3). PPP2R2A knockdown via two independent shRNAs led to increased c-Myc protein expression in two NSCLC cell lines (Fig. 5A). In support of the results that PPP2R2A reduction led to increased c-Myc expression and replication stress/DNA damage (Fig. 5A, S6A), reconstitution of PPP2R2A expression in H1437 cells with low expression of PPP2R2A led to decreased expression of c-Myc and γ-H2AX protein (Fig. 5B,C). The expression of p-RPA2 and γ-H2AX were used as controls to ensure that RS was generated in the experimental conditions. The increase in c-Myc expression was not related to changes in the c-Myc transcript (Fig. 5D). Thus, the upregulation of c-Myc protein in PPP2R2A-deficient cells occurs independently of c-Myc transcription. B56α is the only B regulatory subunit of PP2A that has been reported to negatively regulate c-Myc phosphorylation (S62) (3). B56α depletion led to increased c-Myc protein expression (Fig. 5E), which is consistent with a previous report (3). However, PPP2R2A knockdown had no effect on B56α expression and led to increased c-Myc expression in the B56α-depleted cells (Fig. 5E). Thus, these results suggest that PPP2R2A deficiency negatively regulates c-Myc protein expression independently of c-Myc transcription and B56α.
Fig. 5. PPP2R2A depletion-induced replication stress is associated with increased c-Myc expression.
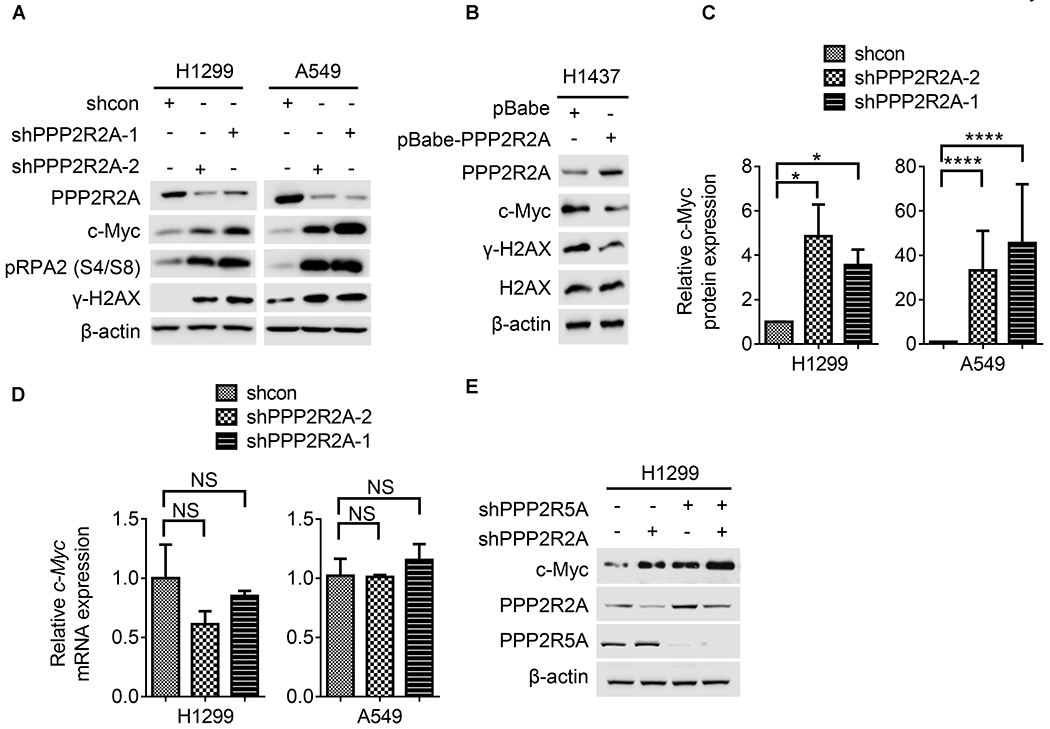
(A) PPP2R2A downregulation by two different shRNAs leads to increased replication stress and c-Myc in H1299 and A549 cells. (B) Stable overexpression of PPP2R2A in H1437 cells decreased the expression of c-Myc and γ-H2AX. (C) Statistical analysis of the protein expression levels of c-Myc relative to β-actin in H1299 and A549 cells with or without PPP2R2A knockdown. Data are presented as the mean ± SEM (n=3). (D) Statistical analysis of mRNA levels of c-Myc in H1299 and A549 cells with or without PPP2R2A knockdown. Data were normalized by the amount of GAPDH mRNA, data are the mean ± SEM (n=3). The statistical significance of C and D was determined by one-way ANOVA followed by Bonferroni’s post hoc analysis for multiple comparisons. *P < 0.05 and ****P < 0.0001. (E) PPP2R2A knockdown leads to upregulation of c-Myc in the PPP2R5A-depleted cells by shRNA.
PPP2R2A knockdown-induced c-Myc expression depends upon increased protein translation initiation.
c-Myc protein stability is controlled by a series of sequential phosphorylation events on two highly conserved residues, T58 and S62, which exert opposing effects on c-Myc protein stability—S62 phosphorylation stabilizes c-Myc and T58 phosphorylation destabilizes c-Myc. S62 phosphorylation stabilizes c-Myc by inhibiting ubiquitination-dependent degradation (36). T58 phosphorylation requires prior S62 phosphorylation (37). This dually phosphorylated form of c-Myc associates with the phosphorylation-directed prolyl isomerase Pin1, which can catalyze a cis-to-trans conformational change in the phospho-S62-P63 peptidyl bond of c-Myc. This form of c-Myc is then a target for PP2A, which dephosphorylates S62, resulting in an unstable, T58-single phosphorylated form of c-Myc that is then a substrate for proteasome-mediated ubiquitination and degradation (38). We speculated that PPP2R2A may dephosphorylate S62 and lead to the alteration of protein stability. Increased p-c-Myc-S62 was present in the PPP2R2A knockdown cells (Fig. 6A). The increased p-c-Myc-S62 as a consequence of PP2A’s inability to dephosphorylate this residue can also result in increased c-Myc phosphorylation at T58 due to the hierarchical nature of c-Myc phosphorylation (3). We indeed found that PPP2R2A deficiency led to increased p-c-Myc-T58 (Fig. 6A). Surprisingly, PPP2R2A knockdown using different shRNAs did not alter the stability of c-Myc protein in either H1299 (Fig. 6B, C) or A549 cells (Fig. 6D). Given that the degradation of c-Myc is inhibited in mitotic cells (39), we determined the rate of c-Myc protein degradation in synchronized cells. H1299 and A549 cells with or without PPP2R2A knockdown were first synchronized in the late G1 phase using the well-established double thymidine block (40) (Fig. S10A). Nearly 80% of the cells were synchronized in the G1/S phase (Fig. S10B). Similar to the results from the unsynchronized cells (Fig. 6B–D), the degradation rate of c-Myc protein was almost identical in the cells with or without PPP2R2A knockdown (Fig. S10C, D). Given that HEK-293T, a kidney cancer cell line, was used in most of the studies which showed that PP2A regulates c-Myc stability (3,41), we next examined c-Myc stability using this cell line. Interestingly, a prolonged half-life of c-Myc protein was observed in HEK-293T cells depleted of PPP2R2A compared to control cells (Fig. S10E), indicating that PPP2R2A regulates c-Myc protein stability in a cell context-dependent manner. Thus, we concluded that PPP2R2A deficiency leads to increased c-Myc protein expression without affecting its stability in NSCLC cells.
Fig. 6. PPP2R2A knockdown-induced c-Myc expression depends on the translational initiation activity but is independent of the regulation of c-Myc degradation.
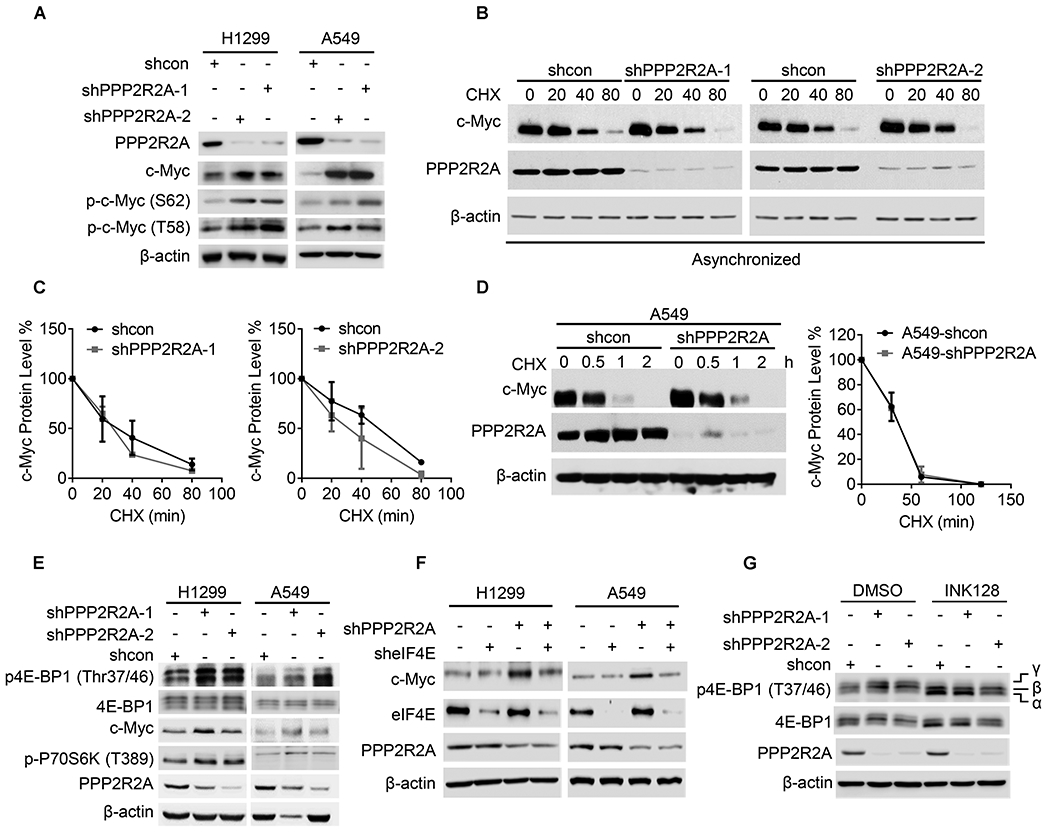
(A) PPP2R2A deficiency leads to increased p-c-Myc-S62 and p-c-Myc-T58 protein expression, as detected by western blot. (B, C) H1299 cells with or without PPP2R2A knockdown with two independent shRNAs were treated with cycloheximide (100μg/ml) at the indicated time points (B). c-Myc protein expression was quantified in three independent experiments (C). (D) The c-Myc protein degradation rate was analyzed in A549 cells with or without PPP2R2A knockdown (left panel). c-Myc protein expression was quantified in three independent experiments (right panel). (E) PPP2R2A deficiency leads to increased p-4E-BP1 and p-p70S6K expression. (F) eIF4E knockdown abrogates PPP2R2A deficiency-induced c-Myc expression. (G) mTOR inhibitor abrogated PPP2R2A deficiency-induced p-4E-BP1 expression. A549 cells with or without PPP2R2A knockdown were treated with mTOR inhibitor (INK128, 100nM) for 6h by using two shRNAs. The α-β-γ isoforms represent the phosphorylation status of 4E-BP1 with α being hypophosphorylated and γ being hyperphosphorylated.
Phosphorylation of 4E-BP1 is important for the activation of translation initiation factor eIF4E. Both elF4E and p70S6K are targets of mTOR (mammalian target of rapamycin) and are important for protein translation initiation. Dephosphorylation of 4E-BP1 stimulates 4E-BP1 binding to eIF4E, thus blocking catabolite gene activator protein-dependent translation initiation. 4E-BP1 is inactivated by phosphorylation via the mTOR pathway, thus facilitating protein synthesis. eIF4E exerts gene-specific effects, and its expression causes a selective increase in the translation of poorly translated mRNAs characterized by lengthy, G-C rich, highly structured 5-UTRs which often encode proteins whose levels are tightly maintained, including c-Myc (42,43). 4E-BP1 can be dephosphorylated by PP2A-C in vitro (44). In addition, 4E-BP1 and p70S6K are potential substrates of PPP2R2A (45), and PPP2R2A-containing PP2A holoenzymes are believed to act upon this substrate to negatively regulate translation (46). Therefore, we hypothesized that PPP2R2A deficiency leads to increased p-4E-BP1 and p-p70S6K, and increased c-Myc expression due to their lack of dephosphorylation triggering subsequent translation. Indeed, much larger amounts of highly phosphorylated, and thus more slowly migrating 4E-BP1 were observed in the PPP2R2A knockdown cells compared to the control cells (Fig. 6E). PPP2R2A knockdown also increased p70S6K phosphorylation (Fig. 6E). In support of our hypothesis that translation activity is required for PPP2R2A deficiency-induced c-Myc protein expression, inhibition of translation activity by eIF4E knockdown abrogated PPP2R2A deficiency-induced c-Myc expression (Fig. 6F) and mTOR inhibition by INK128 abolished PPP2R2A deficiency-induced p-4E-BP1 expression (Fig. 6G). Taken together, these results suggest that PPP2R2A depletion increases c-Myc expression via upregulation of translation activity by increasing the phosphorylation of translation initiation factors without affecting c-Myc protein stability.
Of note, although inhibition of translation initiation by eIF4E knockdown abrogated PPP2R2A deficiency-induced c-Myc expression, eIF4E knockdown alone did not cause a decrease in c-Myc protein expression (Fig. 6F). We postulate that there may be two reasons for this. First, eIF4E expression is low and its activity is normally inhibited by 4E-BP1. Therefore further knockdown of eIF4E may not cause any significant decrease in c-Myc translation, although overexpressing eIF4E can result in an increase in c-Myc protein expression (47). Second, c-Myc translation initiation can occur either via an eIF4E-mediated cap-dependent mechanism or via an eIF4E-independent internal ribosome entry segment (IRES) pathway (48,49). Deficiency in cap-dependent translation as a result of reduced eIF4E expression may trigger the other pathway to compensate. This regulation may be cell context-dependent, since eIF4E knockdown did not result in increased c-Myc expression in H1299 cells (Fig. 6F). Regardless of these different mechanisms, inhibition of translation initiation by eIF4E knockdown abrogated PPP2R2A deficiency-induced c-Myc expression.
c-Myc is required for PPP2R2A deficiency-induced RS and sensitivity to ATR/CHK1 inhibition.
Given that PPP2R2A deficiency leads to increased expression of c-Myc, we hypothesized that PPP2R2A deficiency-induced replication initiation, RS, and the sensitivity to ATR and CHK1 inhibition is dependent upon c-Myc. To test this hypothesis, we first determined how c-Myc inhibition by the pharmaceutical inhibitor 10058-F4 affects PPP2R2A knockdown-induced replication initiation. As we anticipated, c-Myc inhibition abrogated the PPP2R2A deficiency-induced replication initiation, which was demonstrated by the DNA fiber assay (Fig. 7A). To verify this result, we further evaluated if c-Myc inhibition affects PPP2R2A deficiency-induced γ-H2AX and p-RPA2 foci. Upon PPP2R2A knockdown, the proportions of cells with positive pRPA2 and γ-H2AX foci were significantly decreased after c-Myc inhibition (Fig. 7B, S11). Thus, c-Myc activity is required for PPP2R2A deficiency-induced RS.
Fig. 7. c-Myc inhibition abrogates the PPP2R2A knockdown-induced replication stress and the sensitivity to ATR/CHK1 inhibition.
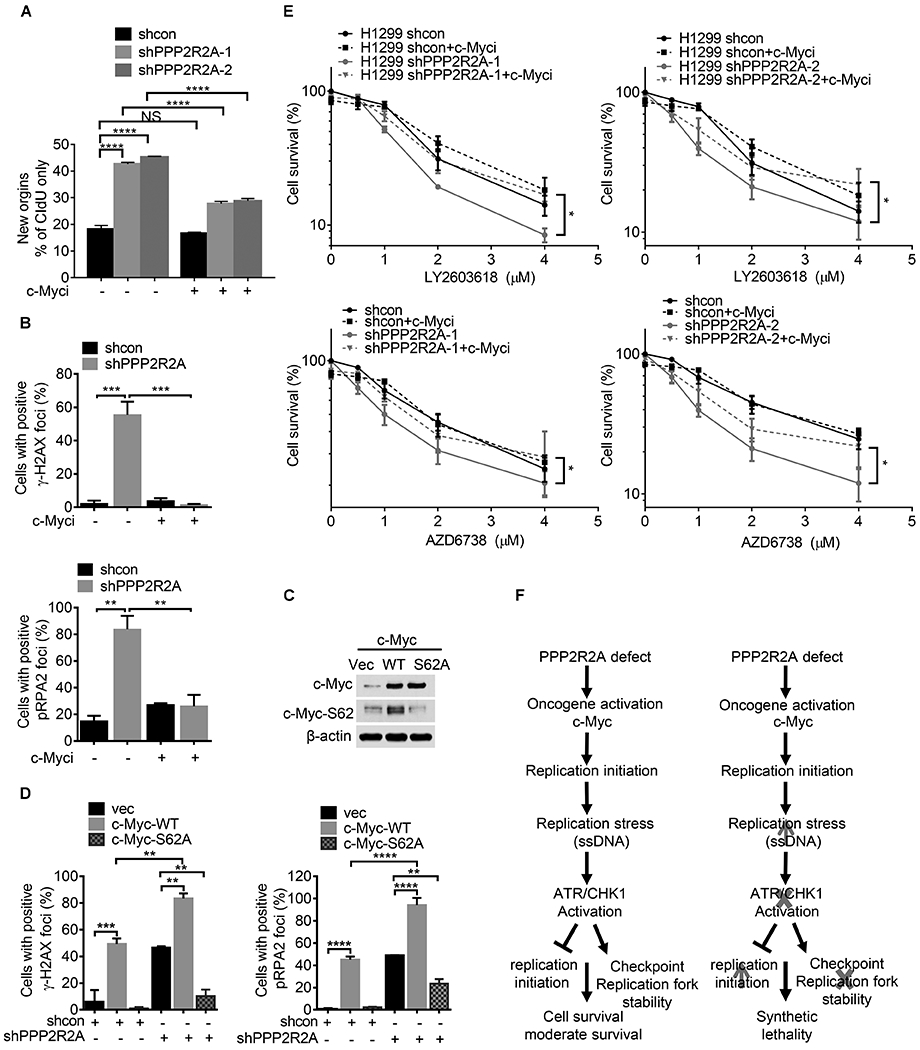
(A) c-Myc inhibition by a pharmaceutical inhibitor abolishes PPP2R2A deficiency-induced replication initiations. H1299 cells were pulse-labeled with IdU for 40 min and released into CIdU for 40 min. 20 μM of c-Myc inhibitor 10058-F4 or DMSO as control was present during the pulse-labeling process. Statistical significance was determined by one-way ANOVA followed by Bonferroni’s post hoc analysis for multiple comparisons. ****P < 0.0001. (B) c-Myc inhibition abrogate PPP2R2A deficiency-induced γ-H2AX (upper panel) and p-RPA2 (lower panel) foci in H1299 cells. The cell containing ≥5 foci is considered as positive. Cells were collected and fixed after 10058-F4 (20 μM) treatment for 24 h after PPP2R2A knockdown. Data represent the mean ± SEM (n=3). ***P < 0.001, by one-way ANOVA followed by Bonferroni’s post hoc analysis. (C) Stable overexpression of phospho-defective c-Myc-S62A decreases c-Myc-S62 phosphorylation in H1299 cells. (D) c-Myc-S62A mutant expression abrogate PPP2R2A knockdown-induced γ-H2AX (left) and p-RPA2 (right) foci in H1299 cells. Data represent the mean ± SEM. **P < 0.01 and ****P < 0.0001, by one-way ANOVA followed by Bonferroni’s post hoc analysis. (E) c-Myc inhibition abrogates the PPP2R2A deficiency-induced sensitivity to ATR or CHK1 inhibitors. LY2603618 (1 μM) or AZD6738 (1 μM) were added to H1299 cells at the indicated doses with or without 10058-F4 (20 μM) treatment for 48 h. Data represent the mean ± SEM (n=3). Statistical significance was determined by two-way ANOVA followed by Bonferroni’s post hoc analysis for multiple comparisons. *P < 0.05. (F) The working model on the synthetic lethality between PPP2R2A deficiency and ATR/CHK1 axis inhibition.
Phosphorylation of c-Myc-S62 is important for both c-Myc stability and c-Myc oncogenic activity (50–52).To exclude any non-specific effects of the c-Myc inhibitor, we next determined the impact of c-Myc-S62A mutant expression on replication initiation and RS induced by PPP2R2A deficiency. Cells expressing c-Myc were generated by transfection of H1299 cells with c-Myc-WT and c-MycS62A constructs. Consistent with previous publications, the c-Myc-S62A mutant leads to increased c-Myc expression despite the fact that S62A is important for c-Myc stabilization (Fig. 7C). Mutation of c-Myc at S62 prevents the phosphorylation of T58, which is important for c-Myc degradation (53). We found that stable expression of c-Myc-WT resulted in an increased proportion of cells with positive γ-H2AX and p-RPA2 foci, which is consist with previous results (34) (Fig. 7D). In contrast, mutant c-Myc-S62A expression fails to increase the percentage of cells with positive γ-H2AX and p-RPA2 foci (Fig. 7D). By using the generated cell lines with stable expression of c-Myc-WT and c-Myc-S62A (Fig. 7C), we found that c-Myc-S62A expression significantly reduced the fraction of the cell population with positive γ-H2AX and p-RPA2 foci triggered by PPP2R2A deficiency. The representative immunostaining is presented in Fig. S12. Last, we determined if c-Myc is important for PPP2R2A deficiency-induced sensitivity to ATR and CHK1 inhibitors. Indeed, c-Myc inhibition abrogated the PPP2R2A deficiency-induced sensitivity to CHK1 inhibitor and ATR inhibitor in H1299 (Fig. 7E). Taken together, these results suggest that c-Myc activity is required for PPP2R2A deficiency-induced replication initiation and subsequent spontaneous RS and enhanced sensitivity to ATR and CHK1 inhibition. These results support our hypothesis that PPP2R2A deficiency leads to increased RS via increased c-Myc activity that renders the cells more sensitive to ATR/CHK1 inhibition (Fig. 7F). Densitometric quantitation of western blot results in this study are shown in Fig. S13A–P, S14A–N, S15A–J.
Discussion
Here, we identified novel synthetic lethal interactions between PPP2R2A deficiency and CHK1 or ATR inhibition. In addition, we also provide new insights into the mechanisms through which PPP2R2A deficiency leads to spontaneous RS/DNA damage as a result of upregulated replication initiation, in the absence of extrinsic DNA damaging agents by upregulating c-Myc activity. Lastly, we suggest PPP2R2A as a novel predictive biomarker to guide the development of ATR and CHK1 inhibitor trials.
A novel synthetic lethal interaction between PPP2R2A deficiency and ATR/CHK1 axis inhibition.
Our genome-wide screen discovered PPP2R2A, a B regulatory subunit of PP2A, as a new candidate gene that is associated with ATR/CHK1 inhibitor sensitivity (Fig. 1, 2). Although ATR/CHK1 inhibition causes minimal or no genotoxic damage or apoptosis in lung adenocarcinoma cells (NSCLC) in which the PPP2R2A status is not known (54), our results strongly suggest that NSCLC cancer cells are sensitive to ATR and CHK1 inhibitors when PPP2R2A is defective. ATR/CHK1 inhibition causes greater tumor growth suppression in PPP2R2A low-expressing cells compared to cells with intact PPP2R2A (Fig. 2, Fig. S2–3). The synergetic interaction was observed in both p53 wild type and mutant cells. Of note, there is a possibility that the therapeutic effectiveness of CHK1/ATR inhibitors in treating PPP2R2A-deficient cancer is partially due to more rapid tumor growth (Fig. 2E, G). However, the rapid tumor growth rate is not the sole reason since the cells deficient in PPP2R2A are still sensitive to ATR/CHK1 inhibition in vitro, although PPP2R2A deficiency leads to a slow growth rate (Fig. 2A–D). This result suggests that other intrinsic mechanisms, such as increased replication stress, may contribute to the sensitivity of PPP2R2A-deficient cells to ATR/CHK1 inhibition. In addition, we noticed that ATR/CHK1 inhibition causes slow growth without complete shrinkage (Fig. 2E,G). Therefore, to maximize the outcome of ATR/CHK1 inhibitor treatment, combination therapy with standard care, immunotherapy, or targeted therapy may be needed. In summary, our study provides strong evidence supporting the hypothesis that ATR and CHK1 inhibitors can specifically target PPP2R2A-deficient NSCLC.
PPP2R2A deficiency-induced spontaneous RS depends upon c-Myc activity.
Different B subunits have partially redundant functions, but each trimer of PP2A has been suggested to have specific physiological activities resulting from both cell type and tissue-specific expression of the regulatory subunits and their cognate substrates (55). PP2A is important for cell recovery from RS induced by ribonucleotide reductase inhibitor hydroxyurea via regulation of DNA repair (56), and PPP2R2A is required for dephosphorylation of DDR protein ATM following exposure to ionizing radiation (4). However, it is not clear how PP2A/PPP2R2A deficiency induces endogenous RS in the absence of extrinsic RS and DNA damaging agents. Several B subunit-containing PP2A complexes have been implicated in tumor suppression. Although MAPK, c-Myc, Wnt, and PI3K signaling have been implicated as key PP2A-associated substrates in regulating tumorigenesis (2,55), the role of B subunits, PPP2R2A in particular, in regulating oncogene signaling has not been widely studied. We found that PPP2R2A deficiency leads to endogenous RS by upregulation of c-Myc (Fig. 4–6), a well-known oncogene which can cause RS (30,34,35). c-Myc activity is required for PPP2R2A deficiency-induced RS and the enhanced sensitivity to ATR and CHK1 inhibitors (Fig. 7). Therefore, our results suggest that defects in PPP2R2A can lead to activation of oncogene c-Myc and subsequent RS, providing the molecular basis for the sensitivity to ATR and CHK1 inhibition in PPP2R2A-deficient NSCLC.
Our study established the role of c-Myc in PPP2R2A deficiency-induced spontaneous RS. However, we cannot exclude the possibility that PPP2R2A deficiency-induced RS can be caused by other oncogene pathways as well. In addition, mechanisms other than oncogene pathways may also be important for PPP2R2A deficiency-induced RS. For instance, the role of PP2R2A in the dephosphorylation of DDR proteins involved in cell cycle checkpoints and DNA repair likely contributes to RS. However, dephosphorylation of DDR proteins by PP2A/PPP2R2A is subsequent to the generation of RS/DNA damage. It is possible that in the absence of exogenous DNA damage agents, PPP2R2A deficiency lead to an increase in spontaneous RS/DNA damage via dephosphorylation of oncogenic proteins, such as c-Myc, which subsequently activate DDR signaling. PPP2R2A deficiency further abrogates the recovery from RS via impaired deposphorylation of DDR proteins (p-ATM, γ-H2AX and p-RPA2) (4). Lastly, PPP2R2A deficiency leads to impaired homologous recombination repair, which can contribute to RS as well (4). Thus, PPP2R2A deficiency leads to increased RS through multiple mechanisms, which involve both the generation of RS and the recovery from RS.
PPP2R2A deficiency-induced c-Myc expression depends upon increased translation activity.
c-Myc is a putative downstream substrate of B subunit B56α. B56 α-containing PP2A holoenzyme negatively regulates c-Myc stability by dephosphorylating pSer62 on c-Myc. A recent study demonstrated that Eya3 can alter the regulation of c-Myc by PP2A. Eya3 expression increases c-Myc stability by enabling PP2A-PPP2R2A to dephosphorylate pT58 on c-Myc. Eya3 expression leads to decreased p-T58-c-Myc but increased p-S62-c-Myc levels (41). Therefore, although PP2A is considered to be tumor suppressor, it may stimulate tumor progression when Eya3 binds to it (41). Indeed, in pancreatic cancer, PPP2R2A seems to act as an oncogene (57). Thus, the functions of PPP2R2A may be cell context-based. We initially hypothesized that PPP2R2A negatively controls c-Myc stability via dephosphorylation of c-Myc, a common mechanism that contributes to increased c-Myc stability. Surprisingly, PPP2R2A has no effect on c-Myc protein stability in NSCLC cancer cells (Fig. 6, S10). PPP2R2A reduction leads to an increase in total c-Myc protein expression and also elevated pT58 and pS62 levels (Fig. 6). The increased phosphorylation of c-Myc in cells with PPP2R2A deficiency could be a passive event as a result of the increased total c-Myc expression.
Our study suggests that PPP2R2A deficiency leads to increased p-4E-BP1 and p-p70S6K, which are two putative substrates of PPP2R2A (45) and are important for protein translation initiation and ribosome biogenesis, respectively. 4E-BP1 can be dephosphorylated by PP2A-C in vitro (44). Dephosphorylation of 4E-BP1 promotes 4E-BP1 binding to eIF4E, thus blocking catabolite gene activator protein-dependent translation initiation (46). Therefore, PPP2R2A deficiency likely leads to increased p-4E-BP1 by dephosphorylation and triggers a subsequent increase in translation activity, leading to the increase in c-Myc protein expression. In support of this hypothesis, PPP2R2A deficiency leads to increased phosphorylation of 4E-BP1. In addition, PPP2R2A deficiency-induced c-Myc expression depends upon eIF4E and mTOR activity (Fig. 6). The finding that PPP2R2A deficiency leads to c-Myc expression at the translational level is of great interest, as it suggests that the increased p-4E-BP1 as a result of PPP2R2A deficiency is at least partially due to the suppression of PPP2R2A-mediated dephosphorylation. With our discoveries arise several intriguing questions. First, further mechanistic studies are needed to clarify whether 4E-BP1 and p70S6K are direct downstream substrates of the PPP2R2A-containing PP2A complex. Over 100 binding proteins of B55α (PPP2R2A) and B55β have been found in Xenopus egg extracts, including eIF4A, eIF4G, and eIF4E (58). Although it is not clear if PPP2R2A directly dephosphorylates eIF4E, PP2A negatively regulates eIF4F assembly and cap-dependent translation through inhibition of Mnk and eIF4E phosphorylation (59). In addition, c-Myc is susceptible to translation regulation by the Mnk-eIF4E pathway. Thus, PPP2R2A deficiency induces an increase in protein translational activity at multiple levels by dephosphorylation of a variety of factors that are required for protein translation. A comprehensive understanding of the coordinated interplay among PPP2R2A, c-Myc, 4E-BP1, eIF4E, and other proteins required for protein translation is needed.
Dysregulation of replication initiation contributes to PPP2R2A deficiency-induced RS and the synergistic interaction between PPP2R2A deficiency and ATR/CHK1 axis inhibition.
Oncogenic activation is a major source of endogenous RS. Oncogenes induce RS via aberrant replication initiation as a result of origin firing increase, replication–transcription collisions, reactive oxygen species, and defective nucleotide metabolism (60). Although it is not fully understood how increased replication initiation by oncogenes leads to increased RS, it may be related to CDK2-mediated deregulation of replication initiation/firing and subsequent deletion of the dNTP pool and ssDNA/DSB formation (61), and an increased number of active replication fork-induced collisions or interference between replication and transcription machineries. c-Myc induces RS by deregulation of replication initiation (35). Our study supports the hypothesis that PPP2R2A deficiency causes RS at least partially via regulation of c-Myc.
Cancer cells with high levels of RS are sensitive to ATR/CHK1 inhibition because ATR/CHK1 signaling is critical to prevent RS from reaching toxic levels. This occurs via multiple mechanisms, including inhibition of replication initiation and activation of cell cycle checkpoints and DNA repair pathways (9). Our study suggests that ATR/CHK1 inhibition causes a significant increase in replication initiation, particularly in PPP2R2A-deficient cells, which is consistent with the increase in RS seen in PPP2R2A-deficient cells (Fig. 4). These results suggest that ATR and CHK1 inhibition-induced replication initiation contributes to increased RS, especially in PPP2R2A-deficient NSCLC. Thus, we propose that deficiency of PPP2R2A leads to increased oncogene-induced RS via upregulation of c-Myc activity, perhaps due to increased replication initiation, activating ATR and CHK1 for survival (Fig. 7F). It is worth noting that our study could not distinguish between early/middle and late replication initiation in the cells with PPP2R2A deficiency or ATR/CHK1 inhibition. It has been demonstrated that c-Myc overexpression alters the spatiotemporal program of replication initiation in early-replicating origins in the Xenopus system (30), and work from the Bakkenist group suggests that ATR/CHK1 suppresses all replication initiations that occur in early, middle, and late stages in unchallenged cells (32). As shown in the model proposed in Fig. 7F, the dysregulation of replication initiation contributes to PPP2R2A deficiency-induced RS and the observed synergistic interaction between PPP2R2A deficiency and ATR/CHK1 axis inhibition. Although we did not study the effect of interruption of the cell cycle checkpoint and DNA repair by ATR/CHK1 inhibition, these mechanisms may also be important for the synthetic lethality between PPP2R2A deficiency and ATR/CHK1 inhibition.
PPP2R2A deficiency as a biomarker of ATR and CHK1 inhibitor sensitivity.
Extensive preclinical data support the clinical application of ATR and CHK1 inhibitors for targeted cancer therapy with or without combination with radiotherapy and chemotherapy. However, compelling results from clinical trials are lacking for most of the combinations (10–12,62–64). A recent study has demonstrated that CHK1 inhibitor targets cisplatin-resistant high grade-serous ovarian cancer, which most likely is associated with oncogene cyclin E overexpression (13). Although the PPP2R2A status is unknown in this study (13), PPP2R2A expression is frequently downregulated in ovarian cancer due to LOH (4,65). It will be very interesting to study whether PPP2R2A deficiency is associated with the response to CHK1 inhibitor in this setting. We found that ATR and CHK1 inhibitors are advantageous in PPP2R2A-defective NSCLC, and PPP2R2A deficiency can be used as a new biomarker to predict the efficacy of ATR and CHK1 inhibition. Considering that PPP2R2A deficiency occurs at a high frequency in a wide range of human cancer, that PP2A activity is also often disrupted in human cancers by high expression of endogenous PP2A inhibitor and other non-genomic mechanisms (55), and that low PPP2R2A expression is associated with poor prognosis, this novel approach of targeting a subset of PPP2R2A-deficient NSCLC by ATR and CHK1 inhibitor could have a significant impact on improving patient survival using a precision medicine-based approach.
Supplementary Material
Statement of Significance.
This study reveals new approaches to specifically target PPP2R2A-deficient lung cancer cells and provides a novel biomarker which will significantly improve treatment outcome with ATR and CHK1 inhibitors.
Acknowledgments
The work described herein was supported by grants (R21CA226317, R21CA245590 and R01CA240374) and the Lung Cancer Discovery Award to J. Zhang from the National Cancer Institute and American Lung Association, respectively, and Pilot Awards from the Case Comprehensive Cancer Center and VeloSano Bike to Cure and NCI Comprehensive Cancer Center Support Grant P30 CA016058 to J. Zhang. This research was also supported by The Ohio State University Comprehensive Cancer Center Basic Science Award (CCC Fund 500734) to Z. Qiu.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Neel DS, Bivona TG. Resistance is futile: overcoming resistance to targeted therapies in lung adenocarcinoma. npj Precision Oncology 2017;1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochimica et biophysica acta 2009;1795:1–15 [DOI] [PubMed] [Google Scholar]
- 3.Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Molecular and cellular biology 2006;26:2832–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalev P, Simicek M, Vazquez I, Munck S, Chen L, Soin T, et al. Loss of PPP2R2A inhibits homologous recombination DNA repair and predicts tumor sensitivity to PARP inhibition. Cancer research 2012;72:6414–24 [DOI] [PubMed] [Google Scholar]
- 5.Ruvolo PP, Qui YH, Coombes KR, Zhang N, Ruvolo VR, Borthakur G, et al. Low expression of PP2A regulatory subunit B55alpha is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia 2011;25:1711–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beca F, Pereira M, Cameselle-Teijeiro JF, Martins D, Schmitt F. Altered PPP2R2A and Cyclin D1 expression defines a subgroup of aggressive luminal-like breast cancer. BMC cancer 2015;15:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Z, Kurimchak A, Nikonova AS, Feiser F, Wasserman JS, Fowle H, et al. PPP2R2A prostate cancer haploinsufficiency is associated with worse prognosis and a high vulnerability to B55alpha/PP2A reconstitution that triggers centrosome destabilization. Oncogenesis 2019;8:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaillard H, Garcia-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer 2015;15:276–89 [DOI] [PubMed] [Google Scholar]
- 9.Qiu Z, Oleinick NL, Zhang J. ATR/CHK1 inhibitors and cancer therapy. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology 2018;126:450–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scagliotti G, Kang JH, Smith D, Rosenberg R, Park K, Kim SW, et al. Phase II evaluation of LY2603618, a first-generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non-small cell lung cancer. Investigational new drugs 2016;34:625–35 [DOI] [PubMed] [Google Scholar]
- 11.Wehler T, Thomas M, Schumann C, Bosch-Barrera J, Vinolas Segarra N, Dickgreber NJ, et al. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer 2017;108:212–6 [DOI] [PubMed] [Google Scholar]
- 12.Seto T, Esaki T, Hirai F, Arita S, Nosaki K, Makiyama A, et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer chemotherapy and pharmacology 2013;72:619–27 [DOI] [PubMed] [Google Scholar]
- 13.Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. The Lancet Oncology 2018;19:207–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Lai J, Du Z, Gao J, Yang S, Gorityala S, et al. Targeting radioresistant breast cancer cells by single agent CHK1 inhibitor via enhancing replication stress. Oncotarget 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang X, Pan Y, Qiu Z, Du Z, Zhang Y, Fa P, et al. RNF126 as a Biomarker of a Poor Prognosis in Invasive Breast Cancer and CHEK1 Inhibitor Efficacy in Breast Cancer Cells. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24:1629–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Contreras AJ, Gutierrez-Martinez P, Specks J, Rodrigo-Perez S, Fernandez-Capetillo O. An extra allele of Chk1 limits oncogene-induced replicative stress and promotes transformation. J Exp Med 2012;209:455–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilad O, Nabet BY, Ragland RL, Schoppy DW, Smith KD, Durham AC, et al. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer research 2010;70:9693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoglund A, Nilsson LM, Muralidharan SV, Hasvold LA, Merta P, Rudelius M, et al. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clinical cancer research : an official journal of the American Association for Cancer Research 2011;17:7067–79 [DOI] [PubMed] [Google Scholar]
- 19.Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nature structural & molecular biology 2011;18:1331–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proceedings of the National Academy of Sciences of the United States of America 2011;108:3336–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 2012;31:1661–72 [DOI] [PubMed] [Google Scholar]
- 22.Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nature structural & molecular biology 2011;18:1331–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer research 2017;77:3870–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Lai J, Du Z, Gao J, Yang S, Gorityala S, et al. Targeting radioresistant breast cancer cells by single agent CHK1 inhibitor via enhancing replication stress. Oncotarget 2016;7:34688–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Deng O, Feng Z, Du Z, Xiong X, Lai J, et al. RNF126 promotes homologous recombination via regulation of E2F1-mediated BRCA1 expression. Oncogene 2016;35:1363–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, et al. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Molecular cancer therapeutics 2017;16:566–77 [DOI] [PubMed] [Google Scholar]
- 27.Tsao AS, Scagliotti GV, Bunn PA Jr., Carbone DP, Warren GW, Bai C, et al. Scientific Advances in Lung Cancer 2015. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2016;11:613–38 [DOI] [PubMed] [Google Scholar]
- 28.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006;444:638–42 [DOI] [PubMed] [Google Scholar]
- 29.Kohler C, Koalick D, Fabricius A, Parplys AC, Borgmann K, Pospiech H, et al. Cdc45 is limiting for replication initiation in humans. Cell Cycle 2016;15:974–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srinivasan SV, Dominguez-Sola D, Wang LC, Hyrien O, Gautier J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep 2013;3:1629–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Molecular and cellular biology 2005;25:3553–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moiseeva TN, Yin Y, Calderon MJ, Qian C, Schamus-Haynes S, Sugitani N, et al. An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proceedings of the National Academy of Sciences of the United States of America 2019;116:13374–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai Y, Chen S, Pei XY, Almenara JA, Kramer LB, Venditti CA, et al. Interruption of the Ras/MEK/ERK signaling cascade enhances Chk1 inhibitor-induced DNA damage in vitro and in vivo in human multiple myeloma cells. Blood 2008;112:2439–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cottini F, Hideshima T, Suzuki R, Tai YT, Bianchini G, Richardson PG, et al. Synthetic Lethal Approaches Exploiting DNA Damage in Aggressive Myeloma. Cancer discovery 2015;5:972–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, et al. Non-transcriptional control of DNA replication by c-Myc. Nature 2007;448:445–51 [DOI] [PubMed] [Google Scholar]
- 36.Junttila MR, Westermarck J. Mechanisms of MYC stabilization in human malignancies. Cell Cycle 2008;7:592–6 [DOI] [PubMed] [Google Scholar]
- 37.Lutterbach B, Hann SR. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Molecular and cellular biology 1994;14:5510–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nature cell biology 2004;6:308–18 [DOI] [PubMed] [Google Scholar]
- 39.Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Molecular and cellular biology 2000;20:2423–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen G, Deng X. Cell Synchronization by Double Thymidine Block. Bio Protoc 2018;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L, Zhou H, Li X, Vartuli RL, Rowse M, Xing Y, et al. Eya3 partners with PP2A to induce c-Myc stabilization and tumor progression. Nature communications 2018;9:1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin CJ, Malina A, Pelletier J. c-Myc and eIF4F constitute a feedforward loop that regulates cell growth: implications for anticancer therapy. Cancer research 2009;69:7491–4 [DOI] [PubMed] [Google Scholar]
- 43.De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004;23:3189–99 [DOI] [PubMed] [Google Scholar]
- 44.Moreno CS, Lane WS, Pallas DC. A mammalian homolog of yeast MOB1 is both a member and a putative substrate of striatin family-protein phosphatase 2A complexes. The Journal of biological chemistry 2001;276:24253–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li S, Brignole C, Marcellus R, Thirlwell S, Binda O, McQuoid MJ, et al. The adenovirus E4orf4 protein induces G2/M arrest and cell death by blocking protein phosphatase 2A activity regulated by the B55 subunit. J Virol 2009;83:8340–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peterson RT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proceedings of the National Academy of Sciences of the United States of America 1999;96:4438–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Benedetti A, Rhoads RE. Overexpression of eukaryotic protein synthesis initiation factor 4E in HeLa cells results in aberrant growth and morphology. Proceedings of the National Academy of Sciences of the United States of America 1990;87:8212–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stoneley M, Chappell SA, Jopling CL, Dickens M, MacFarlane M, Willis AE. c-Myc protein synthesis is initiated from the internal ribosome entry segment during apoptosis. Molecular and cellular biology 2000;20:1162–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nanbru C, Lafon I, Audigier S, Gensac MC, Vagner S, Huez G, et al. Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. The Journal of biological chemistry 1997;272:32061–6 [DOI] [PubMed] [Google Scholar]
- 50.Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene 1994;9:59–70 [PubMed] [Google Scholar]
- 51.Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M, et al. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer research 2011;71:925–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benassi B, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M, et al. c-Myc phosphorylation is required for cellular response to oxidative stress. Molecular cell 2006;21:509–19 [DOI] [PubMed] [Google Scholar]
- 53.Fan-Minogue H, Cao Z, Paulmurugan R, Chan CT, Massoud TF, Felsher DW, et al. Noninvasive molecular imaging of c-Myc activation in living mice. Proceedings of the National Academy of Sciences of the United States of America 2010;107:15892–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doerr F, George J, Schmitt A, Beleggia F, Rehkamper T, Hermann S, et al. Targeting a non-oncogene addiction to the ATR/CHK1 axis for the treatment of small cell lung cancer. Scientific reports 2017;7:15511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kauko O, Westermarck J. Non-genomic mechanisms of protein phosphatase 2A (PP2A) regulation in cancer. Int J Biochem Cell Biol 2018;96:157–64 [DOI] [PubMed] [Google Scholar]
- 56.A genomics-based classification of human lung tumors. Science translational medicine 2013;5:209ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hein AL, Seshacharyulu P, Rachagani S, Sheinin YM, Ouellette MM, Ponnusamy MP, et al. PR55alpha Subunit of Protein Phosphatase 2A Supports the Tumorigenic and Metastatic Potential of Pancreatic Cancer Cells by Sustaining Hyperactive Oncogenic Signaling. Cancer research 2016;76:2243–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang F, Zhu S, Fisher LA, Wang W, Oakley GG, Li C, et al. Protein interactomes of protein phosphatase 2A B55 regulatory subunits reveal B55-mediated regulation of replication protein A under replication stress. Scientific reports 2018;8:2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y, Yue P, Deng X, Ueda T, Fukunaga R, Khuri FR, et al. Protein phosphatase 2A negatively regulates eukaryotic initiation factor 4E phosphorylation and eIF4F assembly through direct dephosphorylation of Mnk and eIF4E. Neoplasia 2010;12:848–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kotsantis P, Petermann E, Boulton SJ. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer discovery 2018;8:537–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013;155:1088–103 [DOI] [PubMed] [Google Scholar]
- 62.Webster JA, Tibes R, Morris L, Blackford AL, Litzow M, Patnaik M, et al. Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk Res 2017;61:108–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Daud AI, Ashworth MT, Strosberg J, Goldman JW, Mendelson D, Springett G, et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33:1060–6 [DOI] [PubMed] [Google Scholar]
- 64.Infante JR, Hollebecque A, Postel-Vinay S, Bauer TM, Blackwood EM, Evangelista M, et al. Phase I Study of GDC-0425, a Checkpoint Kinase 1 Inhibitor, in Combination with Gemcitabine in Patients with Refractory Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23:2423–32 [DOI] [PubMed] [Google Scholar]
- 65.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463:899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.