

ABSTRACT
The inappropriate use of antibiotics in man is driving to insurgence of pathogenic bacteria resistant to multiple drugs (MDR) representing a challenge in critical illness. The interaction of MDR bacteria with host cells can guide molecular perturbations of host transcriptional programmes involving epigenetic-sensitive mechanisms, mainly DNA methylation, histone modifications, and non-coding RNAs leading to pathogen survival. Clinical evidence of epigenetic manipulation from MDR bacteria mainly arises from Mycobacterium tuberculosis as well as Helicobacter pylori, Escherichia coli, Listeria monocytogenes, Pseudomonas aeruginosa, and Legionella pneumophila infection suggesting possible biomarkers of disease. For example, DNA hypermethylation of E-cadherin (CDH1), upstream transcription factor 1/2 (USF1/2), WW domain containing oxidoreductase (WWOX), and mutL homolog 1 (MLH1) genes in gastric mucosa is correlated with malignancy suggesting useful biomarkers of early disease state. Moreover, upregulated circulating miR-361-5p, miR-889, miR-576-3p may be useful biomarkers to discriminate tuberculosis patients. Moreover, Listeria monocytogenes can indirectly induce H3 hyperacetylation leading to inflammation in human endothelial cells whereas Pseudomonas aeruginosa excretes QS 2-AA to directly induce H3 deacetylation leading to bacterial persistence in human monocytes. Remarkably, epigenetic-sensitive drugs may aid to counteract MDR in clinical setting. Trichostatin A, a histone deacetyltransferase inhibitor (HDACi), leads to AMP β-defensin 2 (HBD2) gene up-regulation in human epithelial cells suggesting a useful ‘epi-therapy’ for Escherichia coli-induced intestinal diseases. We update on the most current clinical studies focusing on epigenetic changes involved in bacterial-host interactions and their putative role as biomarkers or drug targets to improve precision medicine and personalized therapy in critical illness and transplantation setting.
KEYWORDS: Bacterial infections, epigenetics, biomarkers, epidrugs, personalized therapy
Introduction
Antimicrobial resistance is posing a threat to the ability to prevent and cure severe bacterial infections in many medical arenas such as organ transplantation, community and healthcare-associated infections, prosthetic implants and overall is compromising the life-saving contribution of antibiotic therapy to modern medicine [1]. The global spread of multi-drug resistant (MDR), extensively drug-resistant (XDR) and even pandrug-resistant bacteria (PDR) is a worldwide hazard, associated with increased morbidity, mortality, and health-care costs [2,3]. Since the initial discovery of the first antibiotics, bacteria have shown the ability to develop sophisticated defence systems to guarantee their survival in host organisms [4]. The indiscriminate and widespread use of antibiotics in man, agriculture, and animal farming, leading to selection of resistant and more virulent bacteria, and the decreased investment in research and discovery of the pharmaceutical companies are the major causes of the current antibiotic resistance crisis [1–4]. Antimicrobial resistance can be intrinsic, acquired, or adaptive, each characterized by different genetic and epigenetic molecular pathways [5–7]. Epigenetic-sensitive mechanisms, including DNA methylation, histone, and non-histone modifications as well as non-coding RNA, such as micro-RNAs (miRNAs) and long non-coding RNAs (lncRNAs), are heritable and acquired modifications able to alter gene expression at different levels without any changes in the primary DNA sequence [8]. Differential genetic and epigenetic mechanisms can contribute to the development of bacterial persistence and resistance [9–11]. Moreover, the host could react to the interaction with pathogens by inducing long-term epigenetic changes leading to improved responses, known as ‘trained immunity’ [12] as well as tolerance or ‘immune paralysis’, a hypo-inflammatory response occurring during and after sepsis [13]. Since epigenetic changes can be easily reverted by small agents, known as ‘epidrugs’, they might provide useful drug targets to prevent and fight MDR infections. Our goal is to update on the most current evidence regarding the role of epigenetic-sensitive mechanisms which guide MDR bacterial-host interactions in man. Since their relevance in critical care, we mainly focus on lipopolysaccharide (LPS) as well as Helicobacter pylori, Escherichia coli, Mycobacterium tuberculosis, Listeria monocytogenes, Pseudomonas aeruginosa, and Legionella pneumophila, suggesting potential molecular indicators contributing to the natural history of critical illness (Table 1). Despite evidence is currently rare, we also emphasize the potential role provided by the epigenome for development of diagnostic, preventive, and therapeutic strategies to fight MDR bacterial infections in transplantation field and discuss future directions.
Table 1.
Some clinical evidence on the role of epigenetic-sensitive mechanisms in bacterial-host interactions.
| Bacterial infection | Interacting host cells | Epigenetic change | Targeted genes and signalling pathways | Putative clinical role | Refs |
|---|---|---|---|---|---|
| DNA methylation | |||||
| H. pilori | Epithelial cells of gastric mucosa | DNA hypermethylation |
|
Diagnostic biomarkers of early gastric carcinogenesis |
[23–27] |
| UPEC | Uroepithelial cells | DNA hypermethylation |
|
Diagnostic biomarkers of urinary tract infections |
[28] |
| M. tuberculosis | Monocyte-derived DCs | DNA hypomethylation in non canonical sites; Gain of histone activation marks (H3K27ac) |
|
Molecular mechanisms of ‘trained immunity’ | [29] |
| Macrophages | DNA hypermethylation |
|
Non-canonical bacterial epigenetic strategies might be innovative drug targets | [30] | |
| Histone modifications | |||||
| L. monocytogenes | Human umbilical venous endothelial cells |
Increased acetylation of H4 and phosphorylation/acetylation of H3 |
|
Biomarkers and drug targets | [38] |
| P. aeruginosa | Monocytes | Global deacetylation of H3K18 |
|
Biomarkers and drug targets | [39] |
| L. pneumophila | Human monocyte/ alveolar epithelial cell lines (A549) |
Promoter genes |
|
Biomarkers and drug targets | [42] |
| MiRNAs | |||||
| M. tuberculosis | Plasma | Differentially expressed circulating miRNAs |
|
Biomarkers of early detection of infection | [43] |
| Plasma | Differentially expressed circulating miRNAs |
|
|
[44] | |
| Plasma | Differentially expressed circulating miRNAs |
|
Biomarkers of active infection | [45] | |
| LPS | Human cultured skin fibroblasts | Differentially expressed cellular miRNAs |
|
Putative useful epi-therapy for wound healing | [46] |
| LncRNAs | |||||
| M. tuberculosis | Peripheral blood mononuclear cells | // |
|
Biomarkers of resistant infection | [47] |
Abbreviations: CCL17: chemokine (C-C motif) ligand 17; CCL18: chemokine (C-C motif) ligand 18; DCs: dendritic cells; H3K14: histone 3 lysine 14; HLA: human leukocyte antigen; IL-1: interleukin 1; IL-6: interleukin 6; IL-16: interleukin 16; IL17: interleukin 17; IL-1β: interleukin-1 β; IRF4: interferon regulatory factor 4; Lnc-RNA: long non-coding RNAs; LPS: lipopolysaccharides; MCP-1: monocyte chemoattractant protein-1; miRNA: microRNAs; ncRNA: non-coding RNA; NFKB1: nuclear factor kappa B subunit 1; NK: natural killer; NFKB: nuclear factor kappa B; SCFA: short chain fatty acids; Sirt2: NAD-dependent deacetylase sirtuin 2; SP1: SP1 transcription factor; TLR5: toll-like receptor 5; TF: transcription factor; UPEC: uropathogenic Escherichia coli; TNF: tumour necrosis factor.
Methodology
We performed electronic searches in PubMed database to identify studies in English published in the last 10 years regarding the possible role of epigenetic-sensitive mechanisms underlying the interactions between MDR bacteria and hosts in man. The search syntax consisted of terms related to DNA methylation, histone acetylation and methylation, miRNAs, and lncRNAs, combined with lipopolysaccharide (LPS) and selected MDR pathogens. The search yielded 16 original articles: 8 on DNA methylation, 3 on histone modifications, 4 on miRNAs and 1 on lnc-RNAs (see Table 1). Moreover, we searched in https://clinicaltrials.gov/and; it resulted that only one clinical trial tested the epidrug vorinostat in evaluating the risk of infections in transplantation.
Bacterial remodelling of the host epigenome
An overview of the epigenetic regulation systems
In this section, we briefly describe the basic mechanisms of action of the major epigenetic-based regulators of MDR bacterial-host interactions.
DNA methylation is a major epigenetic factor influencing gene expression. It refers to the addition of a methyl group on cytosines at 5’ position to form 5-methylcytosine [14,15]. DNA (cytosine-5)-methyltransferases recognize CpG dinucleotides corresponding to the 5’-C-phosphate-G-3’ sequence of nucleotides [14,15]. Moreover, CpG dinucleotides are enriched in ‘CpG islands’ sequences of at least 200 base pairs which are typically undermethylated with low nucleosome occupancy and found at upstream of about 60% of gene promoters to orchestrate the lineage-specific gene expression patterns during developmental stages [15]. This chemical modification is catalysed by three classes of ‘writers’: DNA methyltransferase (DNMT) 1, maintaining the heritable DNA methylation patterns through cell division, and DNMT3a and 3b which establish a de novo DNA methylation patterns during the first stages of embryonic development [14,15]. In the DNMT-catalysed reactions, S-adenosylmethionine (SAM) is the donor of methyl group which is associated to gene repression by two major mechanisms: (1) it obstacles the binding of transcription factors (activators) to their specific sites; (2) it recruits methyl-CpG-binding proteins, such as MECP2 or MBD2 which, in turn, recruit additional proteins able to add silencing modifications to neighbouring histones [16]. In addition, the ten-eleven translocation proteins (TET1, TET2, TET3), a family of iron-dependent oxygenases, act as ‘erasers’ by combining deamination and oxidation of 5-methyl-cytosine to remove 5-methyl-cytosine from DNA [14]. Generally, methylation in the promoter gene is negatively correlated with expression levels, whereas methylation in the gene body is directly associated with active gene expression suggesting a regulation process highly related to the genomic context [17].
The histone acetylation system is regulated by histone acetyltransferases (HATs), bromodomain extraterminal motif (BET) proteins and histone deacetylases (HDACs) [18]. HATs function as ‘writers’ by adding acetyl groups to histones associated with open chromatin conformation providing DNA interaction to RNA polymerase and transcription factors and promotes genes transcription [18]. BET proteins function as ‘readers’, by recognizing acetylated lysine residues in histones, recruiting the positive transcription elongation factor complex (P-TEFb) and thus activating transcription [19]. HDACs serve as ‘erasers’ by deacetylation of histones which are associated with closed chromatin conformation limiting DNA access and therefore gene expression [20]. In mammals, the major class of known HATs are Gcn5, P300, CBP, and PCAF whereas HDACs are classified into four categories including class I (HDAC 1, 2,3, and 8), class II (HDAC 4,5,6,7, 9, and 10), class III (HDACs referred to as sirtuins, Sirt 1–7), and class IV (HDAC 11) [20]. Moreover, histone methylation mainly occurs on the arginine or lysine residues of histones tails and is mediated by arginine methyltransferases (PRMT) and lysine methyltransferases (HMTs), respectively [21]. Among non-coding RNA molecules, miRNAs are endogenous single-stranded non-coding RNAs of 21–22 nucleotides passing from nucleus to cytosol to regulate gene expression at post-transcriptional level [22]. These ribomolecules can promote degradation or translational repression by imperfect base-pairing with the 3’ untranslated region (3’UTR) of multiple target mRNAs leading to downregulation of protein expression [22]. Moreover, lncRNAs are transcripts >200 nucleotides in size which are involved in RNA–protein interactions to carry out their functions by modulating chromatin-modifying complexes and interacting with transcription factors [22].
DNA methylation
In this section, we reported some studies focusing on the role of DNA methylation in MDR bacterial-host interactions in humans. Several evidence reported that Helicobacter pylori, a class I carcinogen in gastric cancer, could induce DNA hypermethylation in epithelial cells of human gastric mucosa [23–27]. These epigenetic changes mainly occur at promoters of some tumour suppressor genes, such as E-cadherin (CDH1), upstream transcription factor 1/2 (USF1/2), WW domain containing oxidoreductase (WWOX), and mutL homolog 1 (MLH1) genes as well as at CpG islands of miRNA genes [23–27]. As downstream effect, Helicobacter pylori could manipulate some pathways involved in the first steps of gastric oncogenic transformation suggesting putative early diagnostic biomarkers [23–27]. However, if Helicobacter pylori uses direct or indirect mechanisms to induces aberrant DNA hypermethylation in gastric epithelial cells remains to be elucidated. Moreover, uropathogenic Escherichia coli (UPEC)- induced DNA hypermethylation, by up-regulation of DNMT1 expression, led to down-regulation of cyclin-dependent kinase inhibitor 2A (CDKN2A) tumour suppressor gene triggering a persistent proliferative state in human uroepithelial cells [28]. These epigenetic signature could act as potential biomarker for recurrent urinary tract infections and could be used to customize treatment of patients with infection-associated uropathies. Moreover, infection of human monocyte-derived dendritic cells (DCs) with a live virulent strain of Mycobacterium tuberculosis was associated with a rapid DNA demethylation, more localized to distal enhancer elements rather than promoter regions [29]. These demethylated sites also correlated with histone activation marks and increased chromatin accessibility leading to an active recruitment of transcription factors, such as nuclear factor kappa B (NFKB) which regulated the transcriptional response to infection in human DCs [29]. Overall, these epigenetic tags may be additional mechanisms underlying the ‘trained immunity’, a sort of memory of first attack in innate immune cells leading to a stronger transcriptional responses upon restimulation and increased resistance to secondary infection [28]. Also, Mycobacterium tuberculosis infection induced a non-CpG methylation in both gene bodies (e.g., introns) and intergenic positions (e.g., repetitive DNA elements) in infected human THP-1-derived macrophages [30]. Taken together, these results suggested that rapid genome-wide DNA methylation changes in non-canonical regions (not CpG islands and promoter) may modulate the host gene expression after Mycobacterium tuberculosis infection suggesting novel molecular routes of bacterial pathogenesis [29,30].
Remarkably, DNA methylation can persist during mitotic and meiotic (transgenerational effect) division leading to inheritance of environmentally acquired risk factors including stress, metabolic abnormalities, nutrient deficiency, and infections responsible for the late onset of diseases in the offspring [31–34]. The transgenerational effect hypothesis overcomes the Mendelian theory of hereditary, for which the genetic factors are the only vectors for the inheritance of disease states providing huge implication for primary prevention [31–34]. Preclinical evidence demonstrated that maternal bacterial infections could induce pathogenic epigenetic-sensitive changes in the foetal tissues affecting their physiological development. In particular, the maternal oral infection with Campylobacter rectus induced hypermethylation of the imprinted insulin like growth factor 2 (IGF2) gene at promoter level leading to a foetal chronic pro-inflammatory state correlated to increased risk for pre-term delivery and growth restriction [35]. Despite early epigenetic alterations may represent the molecular bridge linking maternal infection to pathogenic foetal development, no causal-effect proofs are established and this warrants further research.
Histone acetylation and methylation systems
Bacteria can manipulate host histone acetylation systems by using three routes: (1) direct and indirect effects on epigenetic histone tags, (2) modulation of HAT and HDAC expression, (3) and production of metabolites acting on the acetylation system components [36,37]. Often, targeted genes are involved in inflammatory responses and when HDACs are suppressed, the inflammatory response is increased, but when HDACs are enhanced, the immune response is decreased [36,37]. Bacteria manipulate this to their advantage, either increasing or decreasing (depending on what they require) the immune response in order to be able to survive [36,37]. Indirectly, bacteria can modulate histone tags in host cells through their effects on mitogen-activated protein kinase (MAPK) signalling pathway. Listeria monocytogenes activates MAPK cascade resulting in a simultaneous increased binding of p300/CPB complex (HAT) and reduced binding of HDAC1 in infected human endothelial cells [38]. This switches the host system towards an increased acetylation of H4 and phosphorylation/acetylation of H3 (active markers) at promoter of the interleukin 8 (IL-8) gene leading to overexpression of many immune-related genes as well as recruitment and degranulation of neutrophils [38]. Moreover, Pseudomonas aeruginosa used a direct strategy by excretion of the quorum-sensing (QS) regulated molecule 2-amino acetophenone (2-AA) leading to overexpression of HDAC1 in human monocytes [39]. As downstream effect, a global histone H3K18 hypoacetylation reprogrammed host immune response, including downregulation of tumour necrosis factor (TNF), interleukin-1 β (IL-1β), and monocyte chemoattractant protein-1 (MCP-1) genes increasing bacteria persistence [39]. Some bacteria can modulate the host histone acetylation systems by synthesizing and excreting specific metabolites, named short chain fatty acids (SCFAs), including acetic, propionic, and butyric acids that serve as potent inhibitors of class I/II HDACs [40]. Whereas SCFAs are beneficial products of anaerobic bacterial fermentation in gut microbiota, high concentrations may be dangerous during infection by enhancing expression of regulatory T cells and thus downregulating the degree of the immune response [40]. In contrast to trained immunity, upon LPS insult, macrophages can become tolerant and less responsive to LPS and other stimuli on reinfection leading to the immune paralysis, as clinical phenotype strongly associated with critical illness and sepsis [40]. An epigenomic profiling of histone marks combined with RNA sequencing revealed that sites of histone methylation were more constant whereas both promoters or enhancers were responsive to dynamic acetylation at H3K27 during differentiation of macrophages from monocytes in humans [41].
Although histone acetylation is the most studied, some enzymes that catalyse histone methylation have also been studied. For example, Legionella pneumophila secreted RomA, a SET domain-containing methyltransferase, which caused trimethylation H3K14 and prevented acetylation of this residue leading to downregulation of innate immune response by affecting toll-like receptor 5 (TLR5) and interleukin-6 (IL-6) genes in human monocytes and alveolar epithelial cell lines (A549) [42].
miRNAs and lnc-RNAs
In this section, we summarized some clinical studies emphasizing the role of miRNAs and lncRNAs as putative biomarkers of MDR bacterial infection mainly in tuberculosis (TB) patients. Circulating miR-361-5p, miR-889, and miR-576-3p were differentially expressed in pulmonary TB patients with respect to controls suggesting a useful panel for early detection of infection [43]. Moreover, circulating miR-769-5p, miR-320a, and miR-22-3p were able to distinguish TB patients with respect to controls [44]. In particular, low levels of miR-320a could discriminate ‘responders’ from ‘non responders’ patients suggesting a biomarker for individual drug-resistance [44]. Another case–control study reported that overexpression of exosomal miR-484, miR-425, and miR-96 could have a potential diagnostic power to identify a state of active TB [45]. Furthermore, a decreased expression of miR-211-3p after treatment with increasing LPS dose was correlated to a lower rate of cell proliferation in human cultured skin fibroblasts suggesting a novel molecular network underlying the delay in wound healing after infection [46]. Since overexpression of miR-211-3p reverted LPS effects, this molecule may be a novel therapeutic strategy that should be tested in animal models [46]. For the first time, 712 lncRNAs and 454 mRNAs were identified as differentially expressed in peripheral blood mononuclear cells from MDR-TB with respect to drug-sensitive TB and healthy controls suggesting that virulent MDR-TB strains can escape immune system by affecting specific molecular networks [47].
Characterizing non-coding RNAs-mRNA pairs and their underlying pathobiological mechanisms could reveal novel molecular determinants to understand and fight MDR infection [48]. However, more large studies and rigorous validation approaches are needed.
Promising ‘epidrugs’ for the treatment of MDR bacterial infections
Generally, HDAC inhibitors (HADCi) recognize the zinc-containing domain of HDACs and block substrate binding to suppress their ‘erasing’ activity [49]. Mainly, HDACi can increase the expression of antimicrobial peptides (AMPs), as key effectors of the innate immunity owing to the direct attack against membranes of different types of microorganisms [36]. Endogenously, AMPs are synthesized by epithelial cells which are the first line of defence in the airway and enteric tract when encountering foreign organisms [50]. Trichostatin A (TSA) belongs to class I and class II HDACi and upregulates the AMP β-defensin 2 (HBD2) gene expression by increasing phosphorylation of histone 3 on serine S10 at promoter level in human colonic epithelial cell line, upon challenge with Escherichia coli (Figure 1(a)) [50]. This epigenetic mechanism is advantageous because does not enhance inflammatory cytokines (e.g., interleukin-8) avoiding tissue damage associated with inflammation [50]. Furthermore, HDACi affects several other aspects of the immune response that might be relevant to host responses to MDR bacteria. For example, tubastatin A, as target HDAC6i, promoted intracellular bacterial clearance from human macrophages by enhancing generation of mitochondrial reactive oxygen species improving bacterial killing [51].
Figure 1.
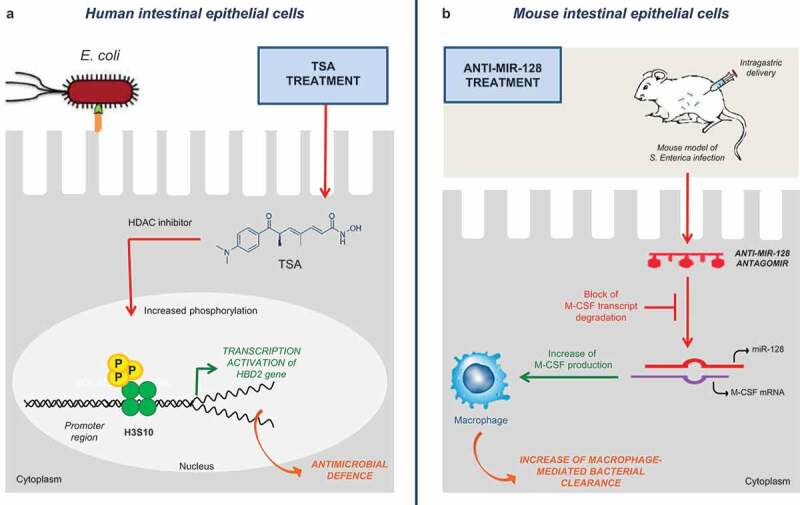
Putative epidrugs to modulate bacterial-host interactions. We illustrate two examples of putative epidrugs and their molecular mechanisms in the treatment of multidrug-resistant bacterial infections. In the panel (a) TSA treatment can enhance antimicrobial defence in human intestinal cells upon Escherichia coli infection by up-regulating the HBD2 gene. Moreover, in the panel (b) Administration of anti-MIR-128 can increase macrophage-mediated bacterial clearance in intestinal epithelial cells of mice infected with Salmonella enterica (see the text for the details).
Abbreviations: HBD2: human beta-defensin-2; HDACi: histone deacetylase inhibitor; M-CSF: macrophage-colony stimulating factor 1; TSA: trichostatin; miR: micro-RNA.
Thus, this might represent a useful therapeutic strategy to treat MDR infections in humans at a time of increasing incidence of antibiotic resistance.
A further promising therapeutic application arises from antago-miRNAs and their immunomodulatory functions [48]. After infection with Salmonella enterica, upregulation of miR-128 promoted the escape of pathogens from clearance exerted by macrophages in human intestinal epithelial cells (HT29) [52]. In detail, increased miR-128 levels correlated with decreased amount of circulating macrophage-colony stimulating factor (M-CSF) leading to impaired M-CSF-related macrophage recruitment [52]. Interestingly, intragastric delivery of anti-miR-128 could reverse this phenomenon and suppress infection in mice (Figure 1(b)) [52].
Epigenetics of MDR infections and critical care
Bacterial infections significantly increase length stay and mortality in the ICU. In 2009, the EPIC II study revealed an incidence of 51% of patients developing infections while in the ICU and 80% of these infections was due to MDR organisms, known as the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) [53]. Bacterial resistance is increasing around the world, especially where antibiotic stewardship is suboptimal and antibiotic medications are over-used [54]. For example, in China, where antibiotics are easily accessible, a new resistance in Enterobacteriaceae involving the plasmid-mediated colistin resistance mechanism (MCR-1) has been discovered as emerging. Also, carbapenem resistance is increasing around the world due to bacterial acquisition of carbapenemase genes (e.g., Pseudomonas aeruginosa and Acinetobacter baumannii in South America and Klebsiella pneumoniae in Greece, Israel, China, South America, and the United States of America) [54]. In addition to alteration of the genetic code, epigenetic modifications may also be responsible for MDR infections in the ICU [55]. For example, resistance of XDR Pseudomonas aeruginosa can involve increased methylation of 30 S RNA leading to reduced antibiotic binding (relevant for aminoglycosides) [56]. Moreover, resistance in Acinetobacterbaumannii includes both increased methylation of the 30 S rRNA and histone lysine acetylation leading to decreased binding of antibiotics [57]. These are potential sites of target for manipulation of MDR bacteria gene expression in the ICU.
Risk evaluation of MDR infections in cerebral death and organ donation: a new challenge for epigenetics
A potential complication of solid organ transplantation is the transmission of latent or active infection from donor to recipient. This occurrence is rare but associated with high morbidity and mortality due to the effects on graft preservability and functionality and the systemic immunosuppressive state after transplantation [58]. Given the limited number of organ donors compared to the patients waiting on the transplantation list, the use of marginal donor is allowed after a careful analysis of the risks-benefits [59]. Bacteraemia and focal infections, such as bacterial meningitis and endocarditis, do not preclude organ donation provided that donors and recipients receive adequate antibiotic treatment and infection in the donor is controlled [59]. Donors with bacteraemia from Staphylococcus aureus and Pseudomonas aeruginosa carry the highest risks of transmission [58]. The colonization or infection of donor with MDR bacteria, such as carbapenem-resistant Pseudomonas aeruginosa (CRPA) and vancomycin-resistant Enterococcus (VRE), poses a great threat to the recipient and no definitive consensus on organ donation in this condition has been reached yet. Moreover, donor infection is often undetected before seroconversion owing to low sensitivity and specificity of routine laboratory tests increasing the risk to recipients of pathogen transmission [60]. Nowadays, there is lack of study evaluating bacterial epigenetics modifications in the clinical scenario of organ donation. However, early and targeted non-invasive epigenetic biomarkers may provide microbiological screening able to recognize MDR pathogens preventing donor-derived infections, followed by customized antibiotic treatments.
MDR infections in the follow-up of organ transplantation
Generally, a life-long immunosuppression therapy required to maintain the allograft function is responsible for frequent post-transplant infection, also [61]. Moreover, epidemiological studies reported that a high percentage of patients can develop a pre-transplant infection causing a six-fold higher risk to be removed from waiting list with respect to uninfected patients [61]. Thus, a pre-transplant evaluation would be relevant to care by identifying the risk for pre- and post-transplant infections [61]. The re-purposing of existing drugs for use as antimicrobial agents could help us in developing useful antibacterial strategies. For example, the UVI5008, as gyrase inhibitor with inhibitory effects on the growth of Staphylococcus aureus, can further act by inhibiting both DNMT and SIRT enzymes leading to structural modifications of the bacterial cell wall [62]. Moreover, during the prospective phase 1/2 trial (NCT00810602) vorinostat (HDACi) in combination with standard therapy did not appear to increase the risk for infectious complications in patients with graft-versus-host disease after haemopoietic stem-cell transplantation [63]. Further studies should focus their attention on the role of these epidrugs as useful antibacterial strategy to prevent or counteract infections in pre- and post-transplantation setting.
Key research questions and key unknown of value to pursue
In this section, we highlight the main research points and strategies to fight MDR bacteria. The first key research question regards the clarification of which molecular networks are early manipulated by MDR pathogens to survive in the host and resist to antibiotics. Indeed, the early interactions between a specific pathogen and the innate immune system are crucial in modulating the individual susceptibility to onset of critical illness [64]. Advanced next-generation sequencing platforms and bioinformatic analysis are strongly supporting the bacterial remodelling of the host epigenome providing cell- and stimulus-specific sites of aberrant DNA methylation, histone modifications, and non-coding RNAs in patients affected by critical illness (Table 1). However, how these molecular networks are spatio-temporally coordinated during the different phases of infectious remain key unresolved issues. Preclinical evidence demonstrated that the innate immune system can mount a non-specific immunological memory (trained innate immunity) by using a complex epigenetic reprogramming (Table 2) [65–68]. This evidence provides the potential to protect from reinfections and reverse immunotolerant states (upregulation of trained responses) as well as to reduce excessive immune activation in chronic inflammatory diseases (downregulation of trained immunity) (Table 2). Two large unexplored aspects of the trained immunity are: 1) the contribution of DNA methylation, miRNAs, and lncRNAs; 2) the duration of protection provided by trained immunity. Moreover, evidence in humans is less than in experimental models [69,70]. Successful therapeutic strategies able to manipulate the innate immune memory of infectious will emerge from a comprehensive understanding of key molecular networks and epigenetic interactions [62]. However, untangling the direction of effect and the causal pathway is challenging. In Figure 2, we report three examples among the abovementioned clinical studies to delineate a diagram of cause–effect relationship by highlighting current gaps in the experimental procedures and what should be addressed in future studies. In detail, we illustrate two experimental flow-charts examining which points have been clarified in the bacterial-induced signalling pathway (blu solid line) in human host cells. In the first example, the writer of DNA hypermethylation in host macrophages upon infection is still unknown. In the second example, we highlight that authors did not validate epigenetic-sensitive networks in animal models or in humans. In contrast, in the third example, we indicate as the plasma miRNA profiling of patients did not provide functional information on the bacterial-host interaction but only a putative useful use as clinical biomarker. Moreover, in this type of studies, it is needed to validate data provided from bioinformatic prediction. Thus, much efforts should be done in clarifying a detailed molecular map of MDR-host interactions. One of the most ambitious lines of research to pursue is represented by the transgenerational epigenetic inheritance providing a mechanistic link between environmental signals and foetal reprogramming with huge implication for primary prevention [31–34]. Indeed, modelling the early epigenetic trajectories may provide useful predictive biomarkers and drug targets. In this direction, advanced liquid-based assays could be useful to: 1) assess molecular changes at different time points, 2) establish direct causal effects, and 3) clarify their putative clinical relevance in risk evaluation, diagnosis, and drug management [71]. Despite encouraging results in biomarker discovery, however, there are significant challenges to implement the epigenetic pharmacotherapy in clinics: (1) to develop tissue-specific delivery systems avoiding unwanted side-effects; and (2) to test single or combined epidrugs in large prospective clinical trials [31–34,72]. To date, network medicine is one of the most useful integrated approaches combining heterogeneous big data, bioinformatics, and clinical information which might reach precision medicine and personalized therapy of critical illness. Combining network medicine approach with transgenerational effects may identify a chain of epigenetic changes perturbing the human interactome over time as well as across multiple generations, rather than a single modification [71,73,74]. Thus, large clinical trials should investigate if these findings can be translated from bench to bedside in ICU.
Table 2.
Some examples of epigenetics coming from training responses in experimental models.
| Sample source | Primary stimulus/ disease | Admistration route | Omic platform | Epigenetic change | Localization | Results | Ref. |
| Infection | |||||||
| Macrophage cultures from bone marrows mice | LPS | Intraperitoneal injection | ChIP-seq | Stable H3K4me1 (24h) Transient H3K27Ac (4h) |
Latent enhancer (unmarked in the basal state) |
LPS can activate 514 latent enhancers at 4 h and 1,002 at 24 h leading to a short-term transcriptional memory | [65] |
| Peritoneal macrophages from mice | Candida albicans (β-glucans) |
Intraperitoneal injection | Chip-seq | Increase in H3K4me3 | Promoters | Protection against reinfection in a monocyte-dependent manner by enhancing dectin-1, C-type lectin receptors, pro-inflammatory cytokines such as TNF-α, IL-6 and IL-18, TLRs and TLR adaptor protein Myd88 | [66] |
| Immune system diseases | |||||||
| Mononuclear cells from mice |
arthritis | // | Chip-seq | Elevated levels of phosphorylated H3 |
Promoters | Upregulation of the AURKA and AURKB genes can lead to promotes proliferation of T lymphocytes. Treatment with the VX-680 attenuated inflammatory reactions and promoted the apoptosis of B cells |
[67] |
| Mouse model of transient hyperglycemia and bovine aortic endothelial cells | Hyperglycemia | // | Chip-seq | Enhanced H3K4 and reduced H3K9 methylation | Promoters | High-glucose-mediated NFKB-p65 gene upregulation that is refractory to glycemic correction | [68] |
Abbreviations: AURKA: aurora kinases A; AURKB: aurora kinases B; VX-680; aurora kinase-specific inhibitor.
Figure 2.
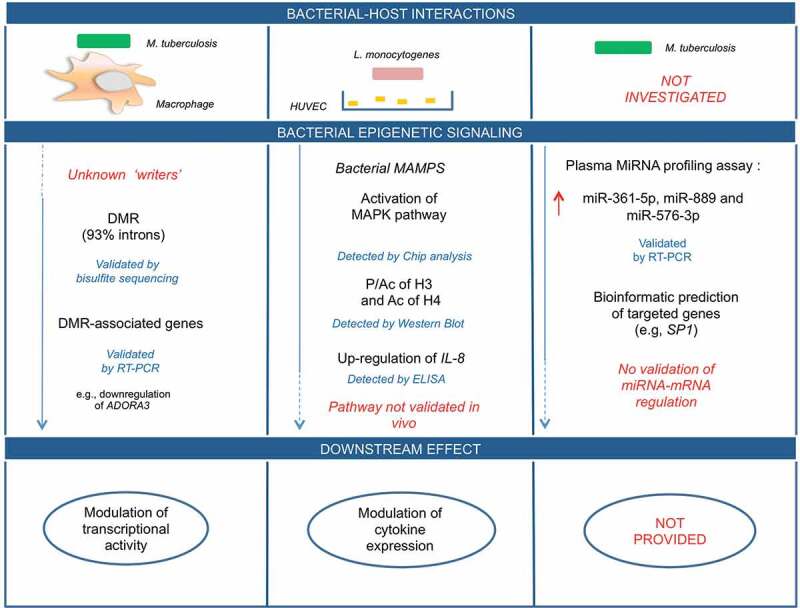
Host-bacterial interactions in humans: experimental gaps in causal-effect pathways. In this diagram, we illustrate three examples for which the cause-effect pathway is not fully detailed at molecular level in bacterial-host interactions. Mainly, the lack of knowledge is represented in red and by blu dashed lines. Blu solid lines represent what has been validated through experimental procedures (see the text for the details).
Abbreviations: DMR: differentially methylated regions; ELISA: enzyme-linked immunoassay; IL-8: interleukin 8; MAMPS: microbe-associated molecular patterns; MAPK: Mitogen-activated protein kinase; RT-PCR: real time-polymerase chain reaction; miR: micro-RNA; SP1: transcription factor SP1.
Acknowledgments
We would like to thank Dr. Concetta Schiano for her dedication to artwork of this manuscript.
Funding Statement
This work was supported by PRIN2017F8ZB89 from Italian Ministry of Research (PI Prof Napoli). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Dr. Giuditta Benincasa is a PhD student of Translational Medicine awarded ESC Congress 2019 and she is supported by Educational Grant from the University of Campania, Naples, Italy.
Disclaimer
This research was supported (in whole or in part) by HCA and/or HCA affiliated entity. The views expressed in this publication represent those of the author(s) and do not necessarily represent the official views of HCA or any of its affiliated entities.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Kollef MH, Bassetti M, Francois B, et al. The intensive care medicine research agenda on multidrug-resistant bacteria, antibiotics, and stewardship. Intensive Care Med. 2017;43:1187–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jasovský D, Littmann J, Zorzet A, et al. Antimicrobial resistance-a threat to the world’s sustainable development. Ups J Med Sci. 2016;121:159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Magiorakos AP, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18:268–281. [DOI] [PubMed] [Google Scholar]
- [4].Bassetti M, Poulakou G, Timsit JF.. Focus on antimicrobial use in the era of increasing antimicrobial resistance in ICU. Intensive Care Med. 2016;42:955–958. [DOI] [PubMed] [Google Scholar]
- [5].Blair JM, Webber MA, Baylay AJ, et al. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 2015;13:42–51. [DOI] [PubMed] [Google Scholar]
- [6].Motta SS, Cluzel P, Aldana M.. Adaptive resistance in bacteria requires epigenetic inheritance, genetic noise, and cost of efflux pumps. PLoS One. 2015;10:e0118464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cohen NR, Lobritz MA, Collins JJ. Microbial persistence and the road to drug resistance. Cell Host Microbe. 2013;13:632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kauffman SA. The epigenetic landscape and clinical implications. J Crit Care. 2011;26:e15. [Google Scholar]
- [9].Casadesús J, Low DA. Programmed heterogeneity: epigenetic mechanisms in bacteria. J Biol Chem. 2013;288:13929–13935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Day T. Interpreting phenotypic antibiotic tolerance and persister cells as evolution via epigenetic inheritance. Mol Ecol. 2016;25:1869–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fernández L, Breidenstein EB, Hancock RE. Creeping baselines and adaptive resistance to antibiotics. Drug Resist Updat. 2011;14:1–21. [DOI] [PubMed] [Google Scholar]
- [12].Netea MG, Joosten LA, Latz E, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Crimi E, Cirri S, Benincasa G, et al. Epigenetics mechanisms in multiorgan dysfunction syndrome. Anesth Analg. 2019;129:1422–1432. doi: 10.1213/ANE.0000000000004331 [DOI] [PubMed] [Google Scholar]
- [14].van der Wijst MG, Venkiteswaran M, Chen H, et al. Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics. 2015;10:671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590–607. [DOI] [PubMed] [Google Scholar]
- [16].Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. [DOI] [PubMed] [Google Scholar]
- [17].Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. [DOI] [PubMed] [Google Scholar]
- [18].Fritz KS. Chemical acetylation and deacetylation. Methods Mol Biol. 2013;1077:191–201. [DOI] [PubMed] [Google Scholar]
- [19].Taniguchi Y. BET Bromodomain as a target of epigenetic therapy. Chem Pharm Bull (Tokyo). 2016;64:540–547. [DOI] [PubMed] [Google Scholar]
- [20].Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6:a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chan AO, Lam SK, Wong BC, et al. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut. 2003;52:502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bussiere FI, Michel V, Memet S, et al. H. pylori-induced promoter hypermethylation downregulates USF1 and USF2 transcription factor gene expression. Cell Microbiol. 2010;12:1124–1133. [DOI] [PubMed] [Google Scholar]
- [25].Yan J, Zhang M, Zhang J, et al. Helicobacter pylori infection promotes methylation of WWOX gene in human gastric cancer. Biochem Biophys Res Commun. 2011;408:99–102. [DOI] [PubMed] [Google Scholar]
- [26].Yao Y, Tao H, Park DI, et al. Demonstration and characterization of mutations induced by helicobacter pylori organisms in gastric epithelial cells. Helicobacter. 2006;11:272–286. [DOI] [PubMed] [Google Scholar]
- [27].Ando T, Yoshida T, Enomoto S, et al. DNA methylation of microRNA genes in gastric mucosae of gastric cancer patients: its possible involvement in the formation of epigenetic field defect. Int J Cancer. 2009;124:2367–2374. [DOI] [PubMed] [Google Scholar]
- [28].Tolg C, Sabha N, Cortese R, et al. Uropathogenic E. coli infection provokes epigenetic downregulation of CDKN2A (p16INK4A) in uroepithelial cells. Lab Invest. 2011;91:825–836. [DOI] [PubMed] [Google Scholar]
- [29].Pacis A, Tailleux L, Morin AM, et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. 2015;25:1801–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sharma G, Sowpati DT, Singh P, et al. Genome-wide non-CpG methylation of the host genome during M. tuberculosis infection. Sci Rep. 2016;6:25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Napoli C, Benincasa G. Loscalzo J epigenetic inheritance underlying pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2019;39:653–664. doi: 10.1213/ANE.0000000000004331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Napoli C, Benincasa G, Schiano C, et al. Differential epigenetic factors in the prediction of cardiovascular risk in diabetic patients. Eur Heart J Cardiovasc Pharmacother. 2019. DOI: 10.1093/ehjcvp/pvz062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].de Nigris F, Cacciatore F, Mancini FP, et al. Epigenetic hallmarks of fetal early atherosclerotic lesions in humans. JAMA Cardiol. 2018;3:1184–1191. doi: 10.1001/jamacardio.2018.3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Napoli C, Crudele V, Soricelli A, et al. Primary prevention of atherosclerosis: a clinical challenge for the reversal of epigenetic mechanisms? Circulation. 2012;125:2363–2373. doi: 10.1161/CIRCULATIONAHA.111.085787 [DOI] [PubMed] [Google Scholar]
- [35].Bobetsis YA, Barros SP, Lin DM, et al. Bacterial infection promotes DNA hypermethylation. J Dent Res. 2007;86:169–174. [DOI] [PubMed] [Google Scholar]
- [36].Grabiec M, Potempa J. Epigenetic regulation in bacterial infections: targeting histone deacetylases. Crit Rev Microbiol. 2018;44:336–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bierne H, Hamon M, Cossart P. Epigenetics and bacterial infections. Cold Spring Harb Perspect Med. 2012;2:a010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schmeck B, Beermann W, van Laak V, et al. Intracellular bacteria differentially regulated endothelial cytokine release by MAPK-dependent histone modification. J Immunol. 2005;175:2843–2850. [DOI] [PubMed] [Google Scholar]
- [39].Bandyopadhaya A, Tsurumi A, Maura D, et al. A quorum-sensing signal promotes host tolerance training through HDAC1-mediated epigenetic reprogramming. Nat Microbiol. 2016;1:16174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Descamps HC, Herrmann B, Wiredu D, et al. The path toward using microbial metabolites as therapies. EBioMedicine. 2019;44:747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Saeed S, Quintin J, Kerstens HH, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rolando M, Sanulli S, Rusniok C, et al. Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe. 2013;13:395–405. [DOI] [PubMed] [Google Scholar]
- [43].Qi Y, Cui L, Ge Y, et al. Altered serum microRNAs as biomarkers for the early diagnosis of pulmonary tuberculosis infection. BMC Infect Dis. 2012;12:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cui JY, Liang HW, Pan XL, et al. Characterization of a novel panel of plasma microRNAs that discriminates between mycobacterium tuberculosis infection and healthy individuals. PLoS One. 2017;12:e0184113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Alipoor SD, Tabarsi P, Varahram M, et al. Serum exosomal miRNAs are associated with active pulmonary tuberculosis. Dis Markers. 2019;2019:1907426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang Y, Wang C. MicroRNA-211-3p has a role in the effects of lipopolysaccharide on endoplasmic reticulum stress in cultured human skin fibroblasts. Med Sci Monit Basic Res. 2019;25:164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yan H, Xu R, Zhang X, et al. Identifying differentially expressed long non-coding RNAs in PBMCs in response to the infection of multidrug-resistant tuberculosis. Infect Drug Resist. 2018;11:945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Parmeciano Di Noto G, Molina MC, Quiroga C. Insights into non-coding RNAs as novel antimicrobial drugs. Front Genet. 2019;10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li W, Sun Z. Mechanism of action for HDAC inhibitors-insights from omics approaches. Int J Mol Sci. 2019;20:1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fischer N, Sechet E, Friedman R, et al. Histone deacetylase inhibition enhances antimicrobial peptide but not inflammatory cytokine expression upon bacterial challenge. Proc Natl Acad Sci U S A. 2016;113:2993–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ariffin JK, Das Gupta K, Kapetanovic R, et al. Histone deacetylase inhibitors promote mitochondrial reactive oxygen species production and bacterial clearance by human macrophages. Antimicrob Agents Chemother. 2015;60:1521–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang T, Yu J, Zhang Y, et al. Salmonella enterica serovar enteritidis modulates intestinal epithelial miR-128 levels to decrease macrophage recruitment via macrophage colony-stimulating factor. J Infect Dis. 2014;209:2000–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Vincent JL, Rello J, Marshall J, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302:2323–2329. [DOI] [PubMed] [Google Scholar]
- [54].Zilahi G, Artigas A, Martin-Loeches I. What’s new in multidrug-resistant pathogens in the ICU? Ann Intensive Care. 2016;6:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Eichenberger EM, Thaden JT. Epidemiology and mechanisms of resistance of extensively drug resistant gram-negative bacteria. Antibiotics (Basel). 2019;8. DOI: 10.3390/antibiotics8020037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lioy VS, Goussard S, Guerineau V, et al. Aminoglycoside resistance 16S rRNA methyltransferases block endogenous methylation, affect translation efficiency and fitness of the host. RNA. 2014;20:382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liao J, Tsai C, Patel S, et al. Acetylome of Acinetobacter baumanni SK17 reveals a highly-conserved modification of histone-like protein HU. Front Mol Biosci. 2017;4:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Len O, Garzoni C, Lumbreras C, et al. Recommendations for screening of donor and recipient prior to solid organ transplantation and to minimize transmission of donor-derived infections. Clin Microbiol Infect. 2014;20:10–18. [DOI] [PubMed] [Google Scholar]
- [59].Westphal GA, Garcia VD, Souza RL, et al. Guidelines for the assessment and acceptance of potential brain-dead organ donors. Rev Bras Ter Intensiva. 2016;28:220–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Grossi PA. Donor-derived infections, lessons learnt from the past, and what is the future going to bring us. Curr Opin Organ Transplant. 2018;23:417–422. [DOI] [PubMed] [Google Scholar]
- [61].Fishman JA. Infection in organ transplantation. Am J Transplant. 2017;17:856. [DOI] [PubMed] [Google Scholar]
- [62].Franci G, Folliero V, Cammarota M, et al. Epigenetic modulator UVI5008 inhibits MRSA by interfering with bacterial gyrase. Sci Rep. 2018;8:13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Choi SW, Braun T, Chang L, et al. Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host disease after related-donor reduced-intensity conditioning allogeneic haemopoietic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. 2014;15:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cole J, Morris P, Dickman MJ, et al. The therapeutic potential of epigenetic manipulation during infectious diseases. Pharmacol Ther. 2016;167:85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ostuni R, Piccolo V, Barozzi I, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. [DOI] [PubMed] [Google Scholar]
- [66].Quintin J, Saeed S, Martens JHA, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Glant TT, Besenyei T, Kádár A, et al. Differentially expressed epigenome modifiers, including aurora kinases A and B, in immune cells in rheumatoid arthritis in humans and mouse models. Arthritis Rheum. 2013;65:1725–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Brasacchio D, Okabe J, Tikellis C, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009;58:1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kleinnijenhuis J, Quintin J, Preijers F, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A. 2012;109:17537–17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].van Splunter M, van Osch TLJ, Brugman S, et al. Induction of trained innate immunity in human monocytes by bovine milk and milk-derived immunoglobulin G. Nutrients. 2018;10. DOI: 10.3390/nu10101378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Benincasa G, Mansueto G, Napoli C. Fluid-based assays and precision medicine of cardiovascular diseases: the ‘hope’ for Pandora’s box? J Clin Pathol. 2019;72:785–799. doi: 10.1136/jclinpath-2019-206178 [DOI] [PubMed] [Google Scholar]
- [72].Sommese L, Zullo A, Mancini FP, et al. Clinical relevance of epigenetics in the onset and management of type 2 diabetes mellitus. Epigenetics. 2017;12:401–415. doi: 10.1080/15592294.2016.1278097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Benincasa G, Marfella R, Della Mura N, et al. Strengths and opportunities of network medicine in cardiovascular diseases. Circ J. 2020;84:144–152. doi: 10.1253/circj.CJ-19-0879 [DOI] [PubMed] [Google Scholar]
- [74].Miryala SK, Ramaiah S. Exploring the multi-drug resistance in Escherichia coli O157: h7by gene interaction network: a systems biology approach. Genomics. 2019;111:958–965. [DOI] [PubMed] [Google Scholar]