

Many genes associate with the adaptive diversification of red warning coloration in neotropical Heliconius butterflies.
Abstract
Characterizing the genetic complexity of adaptation and trait evolution is a major emphasis of evolutionary biology and genetics. Incongruent findings from genetic studies have resulted in conceptual models ranging from a few large-effect loci to massively polygenic architectures. Here, we combine chromatin immunoprecipitation sequencing, Hi-C, RNA sequencing, and 40 whole-genome sequences from Heliconius butterflies to show that red color pattern diversification occurred via many genomic loci. We find that the red wing pattern master regulatory transcription factor Optix binds dozens of loci also under selection, which frequently form three-dimensional adaptive hubs with selection acting on multiple physically interacting genes. Many Optix-bound genes under selection are tied to pigmentation and wing development, and these loci collectively maintain separation between adaptive red color pattern phenotypes in natural populations. We propose a model of trait evolution where functional connections between loci may resolve much of the disparity between large-effect and polygenic evolutionary models.
INTRODUCTION
Identifying the genetic basis of both the origin and maintenance of adaptive traits is essential for the study of diversification. Recent studies disagree, however, on the complexity of adaptive trait architecture, largely because approaches used for genetic mapping are often inconsistent. Quantitative trait loci (QTL) studies investigating the genetic basis of adaptive traits have repeatedly mapped natural variation to a few Mendelian loci (1, 2). Similarly, studies of the loci differentiating adaptive phenotypes in hybrid zones frequently find a handful of genes that strongly segregate by phenotype (3–5). Yet, these observations are increasingly incongruent with genome-wide association studies (GWAS), selection associated with trait variation, and studies incorporating artificial selection, which all regularly find many natural genetic variants of differing effect sizes that associate with trait variation (6–8). This has led to several proposals that many traits may be highly or massively polygenic (9, 10). An oligogenic or polygenic architecture would, in turn, suggest that a larger subset of causal variants might also contribute to maintaining adaptive traits between hybridizing taxa. The discrepancy between QTL studies, known for bias toward a few loci of large effect, and the often multigenic composition of natural trait-associated variation is not new, yet reconciliation of these disparate findings has been a challenge (9).
Here, we study hybridizing populations of Heliconius erato butterflies as a natural model for resolving this conflict. In Ecuador and Peru, evolutionarily derived Radiate morphs of H. erato form hybrid zones along the Andes with several Postman morphs (Fig. 1A) (5). In this region, genome-wide divergence is low, effective population sizes are high (11), gene flow is frequent between hybridizing morphs (12), and populations are only strongly differentiated by three known color pattern–associated loci around WntA, cortex, and optix (Fig. 1B). The master regulator gene optix acts as a classic Mendelian gene of large effect, and both QTL studies and scans for genomic differentiation in hybrid zones have mapped control of adaptive red mimicry color pattern variation to optix (3, 13–16). Recent study of the Radiate phenotype in H. erato found that wing patterns evolved via selection on at least five pleiotropic cis-regulatory loci and that these elements were shared by all Radiate morphs through adaptive introgression (17). Yet, despite the singular importance of optix for red wing pattern evolution, studies in Heliconius have found that quantitative variation in red wing patterns maps to at least 15 of the 21 chromosomes in Heliconius (shown in Fig. 1C), indicating that many more loci may be important contributors to red color pattern adaptation in the genus (18–21). In this study, we investigate the population genetic signatures of additional loci associated with adaptive diversification and assess the extent to which these may be functionally connected to optix.
Fig. 1. Genomic differentiation and selection in H. erato metapopulations.
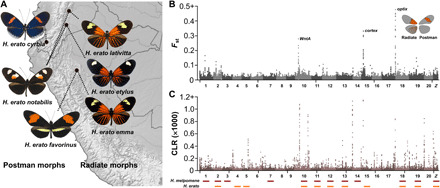
(A) Postman (western) and Radiate (eastern) metapopulations show Andean hybrid zone between morphs. (B) Genomic differentiation between Radiate and Postman metapopulations with three major color pattern loci annotated. (C) Signatures of selective sweeps in the Radiate metapopulation shows many loci under selection. Bars mark chromosomes significantly associated with red pattern variation in H. melpomene (red) and H. erato (orange).
RESULTS
Adaptive evolution of Optix-bound loci in Radiate morph butterflies
We predicted that additional loci in the optix gene network may have contributed to the adaptive diversification of the Radiate morphology. Because Optix is a transcription factor that binds cis-regulatory elements (CREs; fig. S1A), focusing on Optix-bound loci limited our analysis to genes directly regulated by the master regulator of red wing coloration in butterflies. To test for additional adaptive evolution causally linked to red color pattern variation in H. erato, we intersected a whole genome scan for selective sweeps using SweepFinder2 (22) in a metapopulation of Radiate morphs (Fig. 1C) with downstream targets of the Optix protein in midpupal wing tissue. Consistent with the hypothesis that much of the Optix-associated gene network may be under selection, Optix-bound CREs showed significantly greater evidence of selective sweeps than expected from CREs drawn randomly from wing assay for transposase-accessible chromatin sequencing (ATAC-seq) data at the same developmental stage (Fig. 2A). The composite likelihood ratio produced by SweepFinder2 is capable of detecting hard sweeps (22), introgressed sweeps (23), and, to a lesser extent, soft sweeps [e.g., (24)]. Thus, much of the enriched composite likelihood ratio (CLR) signal observed at Optix-bound CREs is likely due to selection on alleles with varying origins and evolutionary histories, and some additional soft sweeps may be identified in future studies.
Fig. 2. Optix binding sites mark loci of adaptation in the Radiate metapopulation.
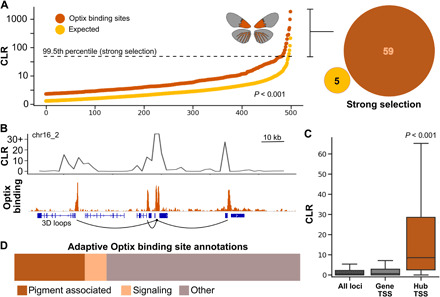
(A) Optix binding sites show significantly greater signatures of selective sweeps than expected from annotated CREs chosen randomly from the same developmental stage. The 99.5th percentile line shows the cutoff threshold for strong signals of selection, and bracket highlights the number of Optix-bound and control loci above this threshold. (B) Optix-bound loci with strong signals of selection often form three-dimensional (3D) adaptive hubs. (C) Adaptive hub transcriptional start sites (TSSs) show elevated selection compared to other regulatory elements. (D) Functional characterization of known genes at loci with strong signals of selection and adaptive hubs are often pigment associated.
We next used physical, Hi-C–based evidence of CRE-to-gene interactions to link Optix binding sites to gene promoters and selected CREs intersecting with selective sweep values (CLR) in the top 0.5% of all CLR scores as loci displaying strong signals of positive selection (fig. S1, B and C). Fifty-nine loci met the criteria of Optix binding and high CLR values and were designated as additional loci of putative adaptive evolution driven by changing optix expression domains (Fig. 2B). Targets of the Optix protein displaying a strong signal of selection were found on 15 of 21 chromosomes, including all but two chromosomes implicated in red color pattern variation (Fig. 1C and fig. S2A) (18–21). Together, evidence of increased selection on Optix binding sites and numerous loci with strong signals of selection in the Radiate metapopulation provide compelling support for a multigenic adaptive process such as the two-step model proposed by Sheppard et al. (13) and Baxter et al. (20).
Our finding that Optix binding sites are generally under increased selection pressure led us to investigate the three-dimensional landscape of adaptation at loci with strong signatures of selection. To accomplish this, we compared our results to RNA sequencing (RNA-seq) assays of differential gene expression between wing tissue of Radiate and Postman butterflies at the same developmental stage (25). We found that 65% of Optix binding sites that display strong signals of selection formed cis-acting adaptive hubs, characterized by a single CRE in the top 0.5% of CLR values that physically connects with two or more genes that are differentially expressed between Radiate and Postman butterflies (Fig. 2C). Adaptive hubs are defined locally by the presence of an Optix binding site with a strong signal of selection in the Radiate metapopulation, with Hi-C evidence of physical loops to at least two additional transcriptional start sites (TSS) of genes differentially expressed between wing pattern morphs. Thus, adaptive hubs are cis-acting networks of genes in close physical proximity that are regulated by Optix and associated with color pattern variation between morphs. This differs from developmental hubs derived from statistical analysis of network connectivity that focus primarily on identifying large-scale genetic interaction networks with both cis and trans interactions [e.g., (26, 27)]. Adaptive hub genes, excluding the hub center with a strong signal of selection, displayed significantly greater signatures of selection than expected when compared to both all genomic loci tested (all CLR values) and selection at all annotated gene TSSs (Fig. 2D). The median CLR value of hub genes indicated a moderate degree of selection and was greater than the 95th percentile of either control dataset. Consistent with our previous results showing increased selection on Optix-bound loci, two-thirds of adaptive hub genes were also bound by Optix, suggesting that selection can favor specific three-dimensional Optix-associated landscapes in addition to individual gene alleles.
We next annotated the function of known genes at adaptive hubs and loci with strong signatures of selection to determine the composition of putative adaptive loci. In total, 160 genes with known functions were linked to strong signals of selection on the Radiate optix expression domain, as determined by selection on Optix-bound TSSs or selection on distal CREs looping to the relevant TSS. Of these, 25% (39) had annotated pigmentation or wing pattern–associated gene functions, including some well-characterized pigmentation genes, while an additional 8% (13) of genes performed a role in important signaling pathways such as the Toll or Dpp pathways (Fig. 2D and fig. S2B). Incorporation of pigmentation, patterning, and signaling genes with genes of other, less obvious functions points to a complex adaptive process targeting wing color–patterning genes, biochemical processes, and potentially even compensatory mutations to account for maladaptive effects of the changing optix expression pattern.
Optix-bound loci under selection differentiate Postman from Radiate butterflies
Multiple lines of evidence implicate numerous loci in the adaptive diversification of the Radiate morph. However, the extent to which multiple loci also assist in maintaining adaptive divergence in the Radiate metapopulation remains unclear. Current models of diversification within H. erato suggest that the Radiate phenotype evolved in the western Amazon region about 300,000 years ago and became the model for several comimetic species in the Heliconius and Eueides genera (17, 28). Yet, despite strong ecological selection favoring the Radiate morphology, extensive hybridization with non-Radiate morphs of H. erato occurs along the Andes (3). The strongest signature of genomic differentiation between hybridizing red color pattern morphs occurs around optix, and this remains true with a fixation index (Fst) of 0.66 when comparing the Radiate metapopulation data to 22 whole-genome datasets from the Postman-like metapopulation along the western side of the Andes (Fig. 1B). Examination of loci with strong signals of selection also bound by Optix, however, makes it clear that many of these regions also act as a significant engine of genomic differentiation between metapopulations.
As a case study, we looked more closely at the Optix-bound promoter of the cotranscribed domeless/washout (dome/wash) genes, which is 500 kb away from the major color-patterning gene cortex and displays the strongest signature of selection within the Radiate metapopulation (Fig. 3A). The Optix-bound dome/wash promoter was not classified as an adaptive hub, indicating that while many loci associated with selection on Optix binding sites form adaptive hubs, strong candidate loci for color pattern–associated divergence are not limited to hub loci. dome has been previously implicated in multiple butterfly wing patterning roles, including yellow mimicry patterns in Heliconius (29, 30). Specifically, the study of the presence or absence of a yellow hindwing bar phenotype in H. melpomene found a significant association between single-nucleotide polymorphisms (SNPs) at the dome/wash locus and the yellow bar pattern. In crosses of Peruvian H. melpomene Radiate and Postman morph butterflies, homozygotes for the yellow hindwing bar allele were dominant over the Radiate hindwing phenotype. In H. erato, this relationship was more complicated and dependent on alleles at multiple Mendelian loci (31). Evidence of Optix binding at the dome/wash promoter and prior association with mimicry phenotypes thus suggests that optix interacts epistatically with dome/wash, although additional study of this locus will be necessary to confirm this function.
Fig. 3. Optix-bound loci with strong signatures of selection drive genomic differentiation between Radiate and Postman metapopulations.
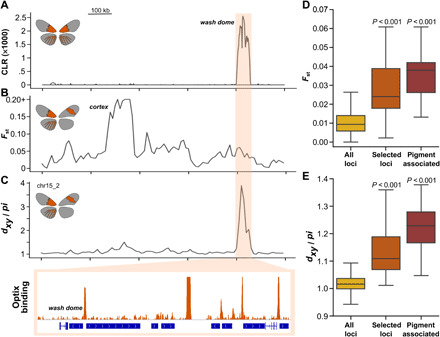
The dome/wash color-patterning locus exemplifies a strong signal of selection (A) on an Optix binding site that displays elevated population differentiation (B) and population divergence (C). Loci with strong signals of selection (selected loci) and the “pigment-associated” subset show increasingly significant population differentiation (D) and divergence (E) between Radiate and Postman metapopulations relative to the genome-wide distributions.
A sliding window scan of Fst, which measures within- versus between-population differentiation, showed a high degree of differentiation at the cortex locus as expected, and that differentiation decreases with distance. Fst between the Radiate and Postman metapopulations elevates again around dome/wash, with differentiation well above the 99th percentile (Fig. 3B). While selection will often lower nucleotide diversity, which, in turn, reduces the average pairwise genomic divergence between neighboring populations [e.g., (32)], any accumulated SNPs should differ between metapopulations at loci that maintain genomic separation across hybrid zones (see Supplementary Text). Therefore, we next sought to determine the extent to which the available nucleotide polymorphisms drove divergence between the Radiate and Postman metapopulations independent of within-population diversity (33). To accomplish this, we measured the nucleotide divergence between the Radiate and Postman metapopulations, normalized to the available nucleotide diversity (dxy/pi; Fig. 3C). Confirming our Fst-derived observation implicating dome/wash as a locus separating metapopulations, dxy/pi was extremely elevated (99.99th percentile) with no evidence of haplotype exchange between the metapopulations.
To investigate the genome-wide significance of this observation, we extended our analysis of metapopulation differentiation to all Optix-bound loci with strong signals of selection. Fst values were significantly greater (P < 0.001) at putative adaptive loci compared to the genome-wide distribution, with the median value in the 93rd percentile and an upper bound of 0.083 (Fig. 3D and fig. S3). dxy/pi was also strongly elevated (P < 0.001) and showed an even greater increase over the genome-wide distribution (with a median in the 97th percentile) than the relative measure of genomic differentiation (Fig. 3E). Consistent with these loci separating metapopulations, rather than solely reflecting selection, neither Fst nor dxy/pi was strongly dependent on CLR at loci with strong signatures of selection (fig. S4). Together, our results clearly demonstrate that downstream targets of Optix regulation under recent positive selection also act as moderate to strong barriers to gene flow between hybridizing morphs.
A multigenic model of adaptive diversification of the Radiate morphology
Updating the previous hypothesis for red color pattern evolution in Heliconius (34), our results now suggest a two-component model of adaptive diversification that captures the distinct processes that underlie the evolution and maintenance of red mimicry color patterns in the Radiate H. erato metapopulation: First, multigenic adaptation occurs via selection on optix, a gene with complete developmental control of red pigmentation in butterflies, and many additional loci directly regulated by changing optix expression domains (Fig. 4A). The genes associated with adaptive evolution of the Radiate morphology indicate selection on pigment composition (e.g., black and UCH), wing morphology and patterning (nkd and osa), and apparent epistatic interactions with other color pattern networks (dome/wash and Wnt signaling). In the Radiate metapopulation, selection will be a product of the relative frequency of a variant (e.g., a novel mutation versus standing genetic variation), the effect size of selected variants, and the ecological significance of the resulting phenotype. Given the very large effective population size of most Heliconius species, selection can easily act on relatively weak-effect variants to optimize even seemingly minor aspects of the Radiate morphology. While our data-driven approach cannot identify all genes associated with red color pattern evolution, we nonetheless found considerable evidence that adaptation relies on many genes in the optix network.
Fig. 4. A model of adaptation and maintenance of the Radiate morphology.
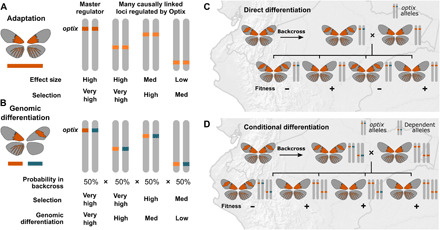
(A) Model showing adaptation occurs at many loci, and selection is a product of population size, the effect of a mutation, and the significance of the trait. (B) Genomic differentiation in hybrid zones is determined by both selection and the probability that a conditionally dependent allele will be functional in backcrosses. (C and D) Cartoons show phenotypes and genotypes for hybrid and pure butterflies, and potential backcross offspring broods under two scenarios of differentiation. Direct differentiation (C) of a dominant allele occurs when a hybrid individual backcrosses into the source population. Any offspring with the maladaptive allele from the hybrid parent is less fit. In conditional differentiation (D), offspring with the maladaptive dependent allele are only less fit when that individual also has the maladapted optix allele.
The Radiate phenotype is then maintained by varying degrees of genomic isolation from the Postman metapopulation (Fig. 4B) at many loci through both direct and conditional (i.e., dependent on trans-allelic effects) differentiation at loci under selection (Fig. 4, C and D). Along the Andean hybrid zone between the Radiate and Postman metapopulations, genomic differentiation begins when a hybrid individual (F1 generation) backcrosses with one of the two source populations (F0 genotypes). The backcross produces maladapted offspring subject to selection in the Radiate metapopulation if the F1 Postman color pattern alleles are transferred to the F2 offspring. Allowing for recombination to narrow maladaptive haplotypes, this results in a relatively small region that fails to pass through the hybrid zone. Similar to selection on beneficial variants, the strength of differentiation is determined by the degree of selection on the resulting phenotype but also by the likelihood of a functional allele. In the case of optix, which is functionally independent of developmentally downstream variants, 50% of F2 offspring will have the maladaptive optix allele (18, 31, 35) and strong, direct differentiation occurs (Fig. 4C).
Our results suggest, however, that conditional differentiation is also prevalent in the Radiate metapopulation along the hybrid zone. Adaptive alleles that are regulated by and functionally dependent on optix will continue to segregate at a roughly 50:50 ratio. But because these loci mostly appear on other chromosomes, maladaptive Optix-dependent haplotypes will only appear (and be subject to selection) alongside a maladaptive optix allele in 25% of F2 offspring (Fig. 4D). Genomic differentiation at Optix-dependent loci is thus conditional on allelic variation at optix itself, when in the presence of a maladaptive optix haplotype, additional downstream genetic variants from the adjacent morph will also be maladaptive. For example, genetic variants relying on Optix activation in the hindwing will have no effect in individuals without the hindwing optix expression domain. The conditional effect on differentiation is compounded in cases where dependent variants may also interact, such as implied by the synergistic effect of HDAC4 and lesswright mutant alleles on Drosophila eye pigmentation (36). Since the degree of genomic differentiation at a locus is a function of the strength of selection and likelihood of a functional allele, the combination of selection and independent segregation of alleles can produce a wide range of differentiation at adaptive loci causally linked to optix expression. This can nonetheless be a strong force for maintaining genomic separation between phenotypes.
Our current data provide some additional findings consistent with the expectations of our adaptive diversification model. Consistent with our proposal of selection on loci of varying effect size and ecological significance, selection coefficients for Optix-bound loci with strong signals of selection were generally 4- to 10-fold lower than observed at the optix locus (Fig. 5A). Similarly, our data support the view that adaptive loci that separate morphs through the process of conditional differentiation will display signatures of genomic differentiation significantly elevated above background. In our analysis, we found that Fst at 17 of 59 Optix-bound loci with strong signals of selection was at or above the 97.5th percentile (the third SD) and often much greater (Fig. 5B). Ultimately, however, additional studies of developmental function and gene flow will be necessary to rigorously test our model of adaptive diversification of the Radiate morphology.
Fig. 5. Selection coefficients and conditional differentiation at Optix-bound CREs.
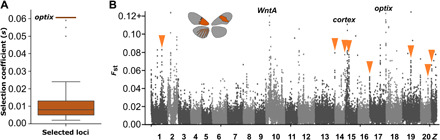
(A) Plot shows the estimated selection coefficients derived from alpha produced by SweepFinder2; outlier from the dome/wash locus is not shown (s = 0.082). Red bar indicates the largest selection coefficient in the optix locus. (B) Evidence of moderately strong genomic differentiation at Optix-bound loci provides support for a model incorporating conditional differentiation. Orange arrowheads highlight Optix-bound loci with Fst greater than 0.04.
DISCUSSION
In this study, we show how signatures of adaptation can manifest across a gene network. To what extent might future studies expect to uncover additional genetic variation underlying trait adaptation? The mode of multigenic evolution will partly determine the propensity for both additional genetic variation and conditional differentiation. Models of classic polygenic adaptation suggest that evolution from standing genetic variation at many independent loci is a common feature of multigenic adaptive processes (37). Adaptive alleles will often be difficult to detect for classic polygenic adaptation, but none are likely to be “hidden” from genetic studies of trait architecture. Multiple-step adaptation, such as the proposed two-step model for Heliconius wing pattern evolution (13, 20) supported here, diverges from classic polygenic models by implicating sequential selection at multiple loci over time and is thus less limited to only ancestral variants. That is, in multistep processes, novel variants that further modify a trait might undergo a classic hard sweep, and the multigenic trait architecture would consist of both novel and ancestral alleles. In either scenario, dependency of some alleles on another locus of large effect will likely produce an effect similar to our findings here.
The ability to detect multiple loci of adaptation will also depend on the biological context of a study system. Hard sweeps are often detected by CLR methods, while soft sweeps and minor frequency shifts proposed in some polygenic models can be more difficult to detect (38, 39). Soft sweeps on rare variants, such as deleterious alleles or variants that arose just preceding the adaptive event, will often appear similar to hard sweeps (40) and can be detected with CLR methods. Multistep adaptation along a hybrid zone, as proposed here, will likely favor soft sweeps on rare variants if the subsequent selection occurs early in the diversification process. In this scenario, the initial adaptation will create a founder-like effect as the populations split and neutral variants are slowly reintroduced by gene flow. Population size variation, such as seasonal fluctuation in population size, will also increase the “hardness” of a sweep (39). Where adaptation occurs from older, neutral polymorphisms in response to an environmental shift, alternative methods of detecting selective sweeps may be necessary (38). Subtle shifts in allele frequencies at multiple loci is the most problematic scenario for detecting multigenic adaptation, although this is only expected when mutation rates are extremely high or the loci under selection are highly redundant with one another (41). For taxa where adaptive introgression is likely, the ability to detect introgressed sweeps will generally fall somewhere between hard and soft selective sweeps, depending on the age of the adaptive allele in the source population, the migration rate, and the origin of the new allele. In sum, tests for selection more suited to partial sweeps from neutral variation may identify additional loci associated with diversification in Heliconius butterflies and other taxa.
Ultimately, our multigenic model of adaptive diversification in Heliconius butterflies provides some resolution to the disparity between a few QTLs of large effect and the multigenic trait architecture often seen in GWAS. QTL studies from a few source individuals and scans for genomic outliers separating phenotypes will favor a few loci of large effect when additional trait-associated variants are functionally dependent on a single gene. Understanding the molecular and developmental mechanisms of adaptation can, however, bring hidden, conditional loci of adaptation to light. Thus, more detailed analysis of the mechanisms of large-effect loci will be necessary to develop a complete theory for the processes of adaptation and diversification. Future studies incorporating many more individuals will be important for understanding the complex genetic architecture of wing polymorphisms in Heliconius. Our findings, however, show that a few functional assays can substitute for hundreds or thousands of genomic samples to identify adaptive network components (42).
MATERIALS AND METHODS
Optix binding site analysis
Genome-wide targets of Optix binding were determined using chromatin immunoprecipitation sequencing (ChIP-seq) for the Optix protein, as previously described (17). Forewing and hindwing tissue from four to six Radiate H. erato individuals of mixed sex at 3 days after pupation were used to perform the ChIP assay. Samples were taken from a laboratory colony of H. erato derived from Ecuadorian stock and kept pure for the Radiate phenotype. Samples were dissected in phosphate-buffered saline, fixed for 7 min with fresh formaldehyde, and flash-frozen on dry ice. Samples were then combined by tissue for two replicates of both forewing and hindwing assays. Chromatin from dounced and lysed samples was sheared to approximately 250 base pairs (bp) using a Bioruptor, and IP was performed overnight with a homemade Heliconius-derived Optix antibody. Input samples were taken before adding the antibody as a control. Library preparation was performed using the NEBNext Ultra II DNA library preparation kit, and both input and control libraries were sequenced on a NextSeq 500.
ChIP-seq and input control libraries were aligned to the Radiate H. erato lativitta genome assembly (43) using Bowtie 2 (44). Nonuniquely aligning reads were removed using a custom script, and data were deduplicated using picard tools. Peak calling was performed with MACS2 (45) on the paired BAM file using default settings, a genome size estimate of 3.6 × 108, and the input sequencing data as a control. Peak calls from forewing and hindwing samples showed a fivefold median increase over input, and 95% of ChIP peaks overlapped previously annotated sites of accessible chromatin from ATAC-seq assays of the same developmental stage (25). Comparison of forewing and hindwing samples found that, as expected, 95.4% of peaks overlapped between wing tissues, so we generated a single, high-quality ChIP peak set of 5051 Optix binding sites that were used in subsequent analyses.
Hi-C processing
Hi-C libraries used to detect Optix binding site interactions were generated using the in situ Hi-C protocol from Rao et al. (46) with minor modifications as previously described (17). Wing tissues from forewing and hindwing Radiate H. erato butterflies were prepared as described for the ChIP-seq assays. Four to five mixed-sex individuals were combined for library preparation, wing tissue was briefly dounced, and nuclei were permeabilized using 0.1% SDS. Samples were centrifuged after ligation to remove any fragments not contained within permeabilized nuclei before reverse cross-linking, and an additional step was included to reduce fragments with unligated biotin in the final libraries. Samples were sheared on a Covaris S2 sonicator, and size was selected with 1.2× AMPure beads before library preparation. Sequencing libraries were prepared with the NEBNext Ultra II DNA library preparation kit. Samples were sequenced on a NextSeq 500.
Hi-C data alignment and analysis were performed as previously described: Hi-C libraries were aligned to the H. erato lativitta genome assembly using Juicer (47). Matching our ChIP-seq analysis, alignments from all replicates were then merged to produce a final merged_nodups.txt file containing all Hi-C fragments for subsequent analyses.
Hi-C interaction calls
To link Optix binding sites to gene TSSs, we used a two-component pipeline. All TSS-proximal ChIP-seq peaks of less than 3 kb from a potential target (generally promoter peaks) were assigned to the proximal gene TSS. We then used the hicContactCaller pipeline from Ray et al. (48) to determine the loci that interact significantly at distances greater than 3 kb compared to an empirical background model. This method calculates the observed number of reads connecting 3-kb windows around an Optix binding site (bait) to TSSs within a predesignated distance (300 kb, prey window). An empirical expected value is determined from the local Hi-C alignments, and a Fisher’s exact test is calculated using observed and expected read counts connecting the bait and prey loci. Interaction between the bait and prey loci were considered significant for P values of less than 0.05, as this threshold was found to be well below the 10% false discovery rate threshold in the previous study (48).
SNP calling
SNP calling for H. erato samples was performed as follows: Eighteen whole genome resequencing samples from three geographically adjacent H. erato Radiate populations (Radiate metapopulation: H. erato lativitta, H. erato etylus, and H. erato emma) and 22 samples from three geographically adjacent H. erato Postman populations (Postman metapopulation: H. erato favorinus, H. erato cyrbia, and H. erato notabilis) (3) were realigned to the Radiate H. erato lativitta genome assembly (43) to produce gVCF (genomic variant call format) files using GATK HaplotypeCaller with default settings (49). Populations were categorized as either “Radiate” or “Postman,” consistent with previous analyses (3). Samples included all Radiate and Postman individuals collected along the Andean hybrid zone region; Radiate and Postman individuals from geographically distant populations, such as H. erato demophoon in Panama, were excluded. SNPs were filtered using VariantFiltration with the filter “MQRankSum < -12.5 || FS > 60.0 || MQ < 20.0” and called using GenotypeGVCFs. The VCF file was then additionally filtered to include only biallelic SNPs using VCFtools (50).
Detecting selective sweeps
SweepFinder2 (51) was used to detect signatures of selective sweeps in the Radiate H. erato metapopulation. Allele counts for all biallelic Radiate sample SNPs were generated using VCFtools. SNPs were polarized using H. erato phyllis [outgroup for East Andean Radiate and Postman metapopulations (3)] whole-genome sequencing data aligned to the H. erato lativitta genome assembly (3). The ancestral allele was marked using the VCFtools fill-aa script. A custom script was then used to convert VCFtools allele counts output to the SweepFinder2 input format. SweepFinder2 was run using default settings and set to test SNPs every 2000 bp (-sg 2000).
Tests for selection on Optix binding sites
To test for the increased selection on all Optix binding sites, the highest CLR values from the SweepFinder2 output within 2000 bp of Optix ChIP peaks were determined using bedtools (52). To prepare a control dataset, we randomly selected the same number of unbound peaks from H. erato lativitta ATAC-seq data using “shuf” and determined the highest CLR value within 2000 bp. This random sample was performed 10 times, then all samples were ranked from lowest to highest, and the values were averaged for each rank position to estimate the random expected value of the same number of CREs sampled from the CRE distribution in the genome. A Kolmogorov-Smirnov test was used to test for the significant difference between the expected and Optix-bound CRE distributions. The top 500 data points for each category were plotted to increase figure resolution.
To identify putatively adaptive Optix-bound loci, we designated the top 0.5% of all CLR values, equal to or greater than 63, as sites under with a strong signal of selection. We then selected all Optix-bound loci with a CLR value at or above 63 as our initial dataset. These were predominantly CRE loci but did include one gene that showed a significant selection in the gene body and was bound by Optix at the promoter. Because some highly repetitive regions show very high CLR values and were likely regions of new repeats found only in the Radiate clade, we manually curated selected loci to discard any sites where a CLR window overlapped repetitive sequences. The three major color pattern loci identified in scans of genomic differentiation were excluded from all tests for the selection on Optix-bound loci to preclude evidence of selection on the optix locus and other known loci of large effect.
Adaptive hub analysis
To test for evidence of three-dimensional evolution associated with selection on Optix binding sites, we combined our Hi-C data with 12 RNA-seq datasets from wing tissue at 3 days after pupation (25). RNA-seq samples were collected from laboratory colonies of a Radiate morph (H. erato lativitta) and a Postman morph (H. erato petiverana). To reduce the impact of nonadaptive evolution and nonevolving loci on our results, we limited our analysis to the loci with evidence of strong selection and differentially expressed genes between Radiate and Postman phenotypes. Thus, adaptive hubs were characterized by two requirements: (i) an Optix-bound “hub center” meeting our definition of a strong signal of selection and (ii) Hi-C–based evidence of physical interaction with two or more hub gene TSSs that were differentially expressed between Radiate and Postman samples. All CLR values were used to determine the genome-wide distribution of CLR values, while all TSS and adaptive hub gene loci were characterized by the highest CLR value within 2 kb, as described above. Hub centers with strong signals of selection were excluded from our analysis of selection at hub gene loci except for the few cases where two loci displaying strong signatures of selection interacted, in which case, the hub center was excluded but the interacting locus was included. Tests for significance between adaptive hub gene CLR values and control datasets were performed with a Mann-Whitney U test.
Gene function annotation
Adaptive hub genes and genes at loci with strong signals of selection were annotated using Lepbase and FlyBase. For all genes with characterized proteins or which showed some similarity to characterized proteins, gene function was manually curated. Gene annotations were categorized as “pigmentation associated” given (i) evidence of a known effect on pigmentation or color patterning, (ii) the gene was annotated as a regulator of known color-patterning genes in Lepidoptera (e.g., regulation of Wnt signaling), or (iii) the gene had known effects on wing morphology in Drosophila. Gene annotations were categorized as “signaling,” given evidence of an important role in major signaling pathways and lacking evidence of a functional role in pigmentation. All other genes were categorized as “other.” Detailed gene annotations are available in the Supplementary Materials.
Estimating population differentiation and divergence
We calculated Fst and dxy/pi to determine the extent to which loci with strong signals of selection may associate with differentiation between the Radiate and Postman metapopulations. Sliding window Fst analysis between the Radiate and Postman metapopulation samples was calculated for 20-kb intervals with 10-kb steps using VCFtools. dxy and pi were calculated for the same intervals and step size using the Genomics-General Python library (53). Statistical tests for significant deviation from the genome-wide distribution of both statistics were performed using Mann-Whitney U tests.
Estimating selection coefficients
In addition to detecting selective sweeps, SweepFinder2 can be used to estimate selection coefficients (23, 54). To do this, we used the formula s = (r*ln(2N)) / a to estimate selection coefficients for loci with strong signals of selection. We used the previously calculated average recombination rate (r) of 1.343 × 10−7 and estimated the Radiate metapopulation size (N) at 10M or roughly twice the size of the metapopulation effective population size (23). Alpha was taken for each locus from the SweepFinder2 output. We verified our parameters by comparing our resulting selection coefficient at optix (0.0605) with a previously calculated coefficient using the same method (0.0595).
Butterfly stocks
All animals were reared in accordance with institutional guidelines.
Supplementary Material
Acknowledgments
We thank the Cornell University Insect Collection for use of the specimens to produce butterfly images. We thank K. van der Burg, L. Campagna, V. Lewis, and two anonymous reviewers for helpful comments on the manuscript. Funding: J.J.L. was supported by NASA 17-EXO-17-2-0112, NSF DEB-1546049, and NSF IOS-1656514. S.M.V.B. was supported by NSF EPSCoR RII Track-2 FEC (OIA 1736026) and, in part, by the National Institutes of Health, NIGMS COBRE Phase 2 Award, Center for Neuroplasticity at the University of Puerto Rico (grant no. 1P20GM103642). S.M.V.B. and R.P. acknowledge support from the Puerto Rico Science, Technology & Research Trust catalyzer award (grant no. 2020-00142) and NSF IOS-1656389. C.G.D. acknowledges support from the National Institutes of Health Common Fund 4D Nucleome Program (grant U01HL129958). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Author contributions: J.J.L. conceived and designed the study. J.J.L. and S.M.V.B. produced and analyzed data. C.G.D., R.D.R., and R.P. provided materials and resources. J.J.L. and S.M.V.B. interpreted the data, and all authors wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: ChIP-seq, Hi-C, ATAC-seq, and RNA-seq data used in this study are available at NCBI-GEO GSE123700, GSE123701, GSE123703, GSE109889, and GSE111022. Whole-genome resequencing data are available at SRA SAMN05224096-SAMN05224211. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/39/eabb8617/DC1
REFERENCES AND NOTES
- 1.Kratochwil C. F., Liang Y., Gerwin J., Woltering J. M., Urban S., Henning F., Machado-Schiaffino G., Hulsey C. D., Meyer A., Agouti-related peptide 2 facilitates convergent evolution of stripe patterns across cichlid fish radiations. Science 362, 457–460 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Cresko W. A., Amores A., Wilson C., Murphy J., Currey M., Phillips P., Bell M. A., Kimmel C. B., Postlethwait J. H., Parallel genetic basis for repeated evolution of armor loss in Alaskan threespine stickleback populations. Proc. Natl. Acad. Sci. U.S.A. 101, 6050–6055 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Belleghem S. M., Rastas P., Papanicolaou A., Martin S. H., Arias C. F., Supple M. A., Hanly J. J., Mallet J., Lewis J. J., Hines H. M., Ruiz M., Salazar C., Linares M., Moreira G. R. P., Jiggins C. D., Counterman B. A., McMillan W. O., Papa R., Complex modular architecture around a simple toolkit of wing pattern genes. Nat. Ecol. Evol. 1, 0052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campagna L., Repenning M., Silveira L. F., Fontana C. S., Tubaro P. L., Lovette I. J., Repeated divergent selection on pigmentation genes in a rapid finch radiation. Sci. Adv. 3, e1602404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toews D. P. L., Taylor S. A., Vallender R., Brelsford A., Butcher B. G., Messer P. W., Lovette I. J., Plumage genes and little else distinguish the genomes of hybridizing warblers. Curr. Biol. 26, 2313–2318 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Lee J. J., Wedow R., Okbay A., Kong E., Maghzian O., Zacher M., Nguyen-Viet T. A., Bowers P., Sidorenko J., Linnér R. K., Fontana M. A., Kundu T., Lee C., Li H., Li R., Royer R., Timshel P. N., Walters R. K., Willoughby E. A., Yengo L.; 23andMe Research Team; COGENT (Cognitive Genomics Consortium; Social Science Genetic Association Consortium, Alver M., Bao Y., Clark D. W., Day F. R., Furlotte N. A., Joshi P. K., Kemper K. E., Kleinman A., Langenberg C., Mägi R., Trampush J. W., Verma S. S., Wu Y., Lam M., Zhao J. H., Zheng Z., Boardman J. D., Campbell H., Freese J., Harris K. M., Hayward C., Herd P., Kumari M., Lencz T., Luan J., Malhotra A. K., Metspalu A., Milani L., Ong K. K., Perry J. R. B., Porteous D. J., Ritchie M. D., Smart M. C., Smith B. H., Tung J. Y., Wareham N. J., Wilson J. F., Beauchamp J. P., Conley D. C., Esko T., Lehrer S. F., Magnusson P. K. E., Oskarsson S., Pers T. H., Robinson M. R., Thom K., Watson C., Chabris C. F., Meyer M. N., Laibson D. I., Yang J., Johannesson M., Koellinger P. D., Turley P., Visscher P. M., Benjamin D. J., Cesarini D., Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergey C. M., Lopez M., Harrison G. F., Patin E., Cohen J. A., Quintana-Murci L., Barreiro L. B., Perry G. H., Polygenic adaptation and convergent evolution on growth and cardiac genetic pathways in African and Asian rainforest hunter-gatherers. Proc. Natl. Acad. Sci. U.S.A. 115, E11256–E11263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castro J. P. L., Yancoskie M. N., Marchini M., Belohlavy S., Hiramatsu L., Kučka M., Beluch W. H., Naumann R., Skuplik I., Cobb J., Barton N. H., Rolian C., Chan Y. F., An integrative genomic analysis of the Longshanks selection experiment for longer limbs in mice. eLife 8, e42014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rockman M. V., The QTN program and the alleles that matter for evolution: All that’s gold does not glitter. Evolution 66, 1–17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyle E. A., Li Y. I., Pritchard J. K., An expanded view of complex traits: From polygenic to omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Belleghem S. M., Baquero M., Papa R., Salazar C., McMillan W. O., Counterman B. A., Jiggins C. D., Martin S. H., Patterns of Z chromosome divergence among Heliconius species highlight the importance of historical demography. Mol. Ecol. 27, 3852–3872 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Supple M. A., Papa R., Hines H. M., McMillan W. O., Counterman B. A., Divergence with gene flow across a speciation continuum of Heliconius butterflies. BMC Evol. Biol. 15, 204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheppard P. M., Turner J. R. G., Brown K. S., Benson W. W., Singer M. C., Genetics and the evolution of Muellerian mimicry in Heliconius butterflies. Phil. Trans. R Soc. Lond. B 308, 433–610 (1985). [Google Scholar]
- 14.Morris J., Navarro N., Rastas P., Rawlins L. D., Sammy J., Mallet J., Dasmahapatra K. K., The genetic architecture of adaptation: Convergence and pleiotropy in Heliconius wing pattern evolution. Heredity 123, 138–152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L., Mazo-Vargas A., Reed R. D., Single master regulatory gene coordinates the evolution and development of butterfly color and iridescence. Proc. Natl. Acad. Sci. U.S.A. 114, 10707–10712 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Supple M. A., Hines H. M., Dasmahapatra K. K., Lewis J. J., Nielsen D. M., Lavoie C., Ray D. A., Salazar C., McMillan W. O., Counterman B. A., Genomic architecture of adaptive color pattern divergence and convergence in Heliconius butterflies. Genome Res. 23, 1248–1257 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis J. J., Geltman R. C., Pollak P. C., Rondem K. E., Van Belleghem S. M., Hubisz M. J., Munn P. R., Zhang L., Benson C., Mazo-Vargas A., Danko C. G., Counterman B. A., Papa R., Reed R. D., Parallel evolution of ancient, pleiotropic enhancers underlies butterfly wing pattern mimicry. Proc. Natl. Acad. Sci. U.S.A. 116, 24174–24183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papa R., Kapan D. D., Counterman B. A., Maldonado K., Lindstrom D. P., Reed R. D., Nijhout H. F., Hrbek T., McMillan W. O., Multi-allelic major effect genes interact with minor effect QTLs to control adaptive color pattern variation in Heliconius erato. PLOS ONE 8, e57033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nadeau N. J., Ruiz M., Salazar P., Counterman B., Medina J. A., Ortiz-Zuazaga H., Morrison A., McMillan W. O., Jiggins C. D., Papa R., Population genomics of parallel hybrid zones in the mimetic butterflies. H. melpomene and H. erato. Genome Res. 24, 1316–1333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baxter S. W., Johnston S. E., Jiggins C. D., Butterfly speciation and the distribution of gene effect sizes fixed during adaptation. Heredity 102, 57–65 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Bainbridge H. E., Brien M. N., Morochz C., Salazar P. A., Rastas P., Nadeau N. J., Limited genetic parallels underlie convergent evolution of quantitative pattern variation in mimetic butterflies. bioRxiv 10.1101/2020.06.15.151613 , (2020). [DOI] [PubMed] [Google Scholar]
- 22.DeGiorgio M., Huber C. D., Hubisz M. J., Hellmann I., Nielsen R., S weep F inder 2: Increased sensitivity, robustness and flexibility. Bioinformatics 32, 1895–1897 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Moest M., Van Belleghem S. M., James J. E., Salazar C., Martin S. H., Barker S. L., Moreira G. R. P., Mérot C., Joron M., Nadeau N. J., Steiner F. M., Jiggins C. D., Selective sweeps on novel and introgressed variation shape mimicry loci in a butterfly adaptive radiation. PLOS Biol. 18, e3000597 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schrider D. R., Kern A. D., Soft sweeps are the dominant mode of adaptation in the human genome. Mol. Biol. Evol. 34, 1863–1877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis J. J., Reed R. D., Genome-wide regulatory adaptation shapes population-level genomic landscapes in Heliconius. Mol. Biol. Evol. 36, 159–173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swings T., Weytjens B., Schalck T., Bonte C., Verstraeten N., Michiels J., Marchal K., Network-based identification of adaptive pathways in evolved ethanol-tolerant bacterial populations. Mol. Biol. Evol. 34, 2927–2943 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helsen J., Frickel J., Jelier R., Verstrepen K. J., Network hubs affect evolvability. PLOS Biol. 17, e3000111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoyal Cuthill J. F., Charleston M., Wing patterning genes and coevolution of Müllerian mimicry in Heliconius butterflies: Support from phylogeography, cophylogeny, and divergence times. Evolution 69, 3082–3096 (2015). [DOI] [PubMed] [Google Scholar]
- 29.L. Livraghi, J. J. Hanly, L. S. Loh, A. Ren, I. A. Warren, C. Concha, C. Wright, J. M. Walker, J. Foley, H. Arenas-Castro, L. R. Brenes, A. Martin, W. O. McMillan, C. D. Jiggins, The gene cortex controls scale colour identity in Heliconius. bioRxiv:2020.05.26.116533 (2020).
- 30.Nadeau N. J., Pardo-Diaz C., Whibley A., Supple M. A., Saenko S. V., Wallbank R. W. R., Wu G. C., Maroja L., Ferguson L., Hanly J. J., Hines H., Salazar C., Merrill R. M., Dowling A. J., ffrench-Constant R. H., Llaurens V., Joron M., Millan W. O. M., Jiggins C. D., The gene cortex controls mimicry and crypsis in butterflies and moths. Nature 534, 106–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mallet J., Clarke C., The genetics of warning colour in Peruvian hybrid zones of Heliconius erato and H. melpomene. Proc. R Soc. Lond. 236, 163–185 (1989). [Google Scholar]
- 32.Irwin D. E., Alcaide M., Delmore K. E., Irwin J. H., Owens G. L., Recurrent selection explains parallel evolution of genomic regions of high relative but low absolute differentiation in a ring species. Mol. Ecol. 25, 4488–4507 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Cruickshank T. E., Hahn M. W., Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23, 3133–3157 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Reed R. D., Papa R., Martin A., Hines H. M., Counterman B. A., Pardo-Diaz C., Jiggins C. D., Chamberlain N. L., Kronforst M. R., Chen R., Halder G., Nijhout H. F., McMillan W. O., optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 333, 1137–1141 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Mallet J., Barton N., Lamas G., Santisteban J., Muedas M., Eeley H., Estimates of selection and gene flow from measures of cline width and linkage disequilibrium in heliconius hybrid zones. Genetics 124, 921–936 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwartz S., Truglio M., Scott M. J., Fitzsimons H. L., Long-term memory in Drosophila is influenced by histone deacetylase HDAC4 interacting with sumo-conjugating enzyme Ubc9. Genetics 203, 1249–1264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pritchard J. K., Di Rienzo A., Adaptation – not by sweeps alone. Nat. Rev. Genet. 11, 665–667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pennings P. S., Hermisson J., Soft sweeps III: The signature of positive selection from recurrent mutation. PLOS Genet. 2, e186 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Messer P. W., Petrov D. A., Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol. Evol. 28, 659–669 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hermisson J., Pennings P. S., Soft sweeps and beyond: Understanding the patterns and probabilities of selection footprints under rapid adaptation. Methods Ecol. Evol. 8, 700–716 (2017). [Google Scholar]
- 41.Höllinger I., Pennings P. S., Hermisson J., Polygenic adaptation: From sweeps to subtle frequency shifts. PLOS Genet. 15, e1008035 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dudbridge F., Power and predictive accuracy of polygenic risk scores. PLOS Genet. 9, e1003348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis J. J., van der Burg K. R. L., Mazo-Vargas A., Reed R. D., ChIP-seq-annotated Heliconius erato genome highlights patterns of cis-regulatory evolution in lepidoptera. Cell Rep. 16, 2855–2863 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao S. S. P., Huntley M. H., Durand N. C., Stamenova E. K., Bochkov I. D., Robinson J. T., Sanborn A. L., Machol I., Omer A. D., Lander E. S., Aiden E. L., A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Durand N. C., Shamim M. S., Machol I., Rao S. S. P., Huntley M. H., Lander E. S., Aiden E. L., Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ray J., Munn P. R., Vihervaara A., Lewis J. J., Ozer A., Danko C. G., Lis J. T., Chromatin conformation remains stable upon extensive transcriptional changes driven by heat shock. Proc. Natl. Acad. Sci. U.S.A. 116, 19431–19439 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van der Auwera G. A., Carneiro M. O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., Banks E., Garimella K. V., Altshuler D., Gabriel S., DePristo M. A., From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R.; Group 1000 Genomes Project Analysis , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huber C. D., DeGiorgio M., Hellmann I., Nielsen R., Detecting recent selective sweeps while controlling for mutation rate and background selection. Mol. Ecol. 25, 142–156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin S. H., Möst M., Palmer W. J., Salazar C., McMillan W. O., Jiggins F. M., Jiggins C. D., Natural selection and genetic diversity in the butterfly Heliconius melpomene. Genetics 203, 525–541 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nielsen R., Williamson S., Kim Y., Hubisz M. J., Clark A. G., Bustamante C., Genomic scans for selective sweeps using SNP data. Genome Res. 15, 1566–1575 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dinwiddie A., Null R., Pizzano M., Chuong L., Leigh Krup A., Ee Tan H., Patel N. H., Dynamics of F-actin prefigure the structure of butterfly wing scales. Dev. Biol. 392, 404–418 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Rousset R., Mack J. A., Wharton K. A. Jr., Axelrod J. D., Cadigan K. M., Fish M. P., Nusse R., Scott M. P., naked cuticle targets dishevelled to antagonize Wnt signal transduction. Genes Dev. 15, 658–671 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lv W.-W., Wei H.-M., Wang D.-L., Ni J.-Q., Sun F.-L., Depletion of histone deacetylase 3 antagonizes PI3K-mediated overgrowth of Drosophila organs through the acetylation of histone H4 at lysine 16. J. Cell Sci. 125, 5369–5378 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Drelon C., Rogers M. F., Belalcazar H. M., Secombe J., The histone demethylase KDM5 controls developmental timing in Drosophila by promoting prothoracic gland endocycles. Development 146, dev182568 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collins R. T., Treisman J. E., Osa-containing Brahma chromatin remodeling complexes are required for the repression of Wingless target genes. Genes Dev. 14, 3140–3152 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sowa A. S., Martin E., Martins I. M., Schmidt J., Depping R., Weber J. J., Rother F., Hartmann E., Bader M., Riess O., Tricoire H., Schmidt T., Karyopherin α-3 is a key protein in the pathogenesis of spinocerebellar ataxia type 3 controlling the nuclear localization of ataxin-3. Proc. Natl. Acad. Sci. U.S.A. 115, E2624–E2633 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Byers H. R., Yaar M., Eller M. S., Jalbert N. L., Gilchrest B. A., Role of cytoplasmic dynein in melanosome transport in human melanocytes. J. Invest. Dermatol. 114, 990–997 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Lakatos Z., Lőrincz P., Szabó Z., Benkő P., Kenéz L. A., Csizmadia T., Juhász G., Sec20 is required for autophagic and endocytic degradation independent of Golgi-ER retrograde transport. Cell 8, 768 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seo E. Y., Jin S.-P., Sohn K.-C., Park C.-H., Lee D. H., Chung J. H., UCHL1 regulates melanogenesis through controlling MITF stability in human melanocytes. J. Invest. Dermatol. 137, 1757–1765 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Ooi C. E., Dell’Angelica E. C., Bonifacino J. S., ADP-ribosylation factor 1 (ARF1) regulates recruitment of the ap-3 adaptor complex to membranes. J. Cell Biol. 142, 391–402 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sasaki M., Horikoshi T., Uchiwa H., Miyachi Y., Up-regulation of Tyrosinase gene by nitric oxide in human melanocytes. Pigment Cell Res. 13, 248–252 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Hayashi H., Fujii R., Possible involvement of nitric oxide in signaling pigment dispersion in teleostean melanophores. Zoolog. Sci. 18, 1207–1215 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Zhou L., Liang H., Zhou X., Jia J., Ye C., Hu Q., Xu S., Yu Y., Zou G., Hu G., Genetic characteristic and RNA-seq analysis in transparent mutant of carp–goldfish nucleocytoplasmic hybrid. Genes 10, 704 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Protiva P., Gong J., Sreekumar B., Torres R., Zhang X., Belinsky G. S., Cornwell M., Crawford S. E., Iwakiri Y., Chung C., Pigment epithelium-derived factor (PEDF) inhibits Wnt/β-catenin signaling in the liver. Cell. Mol. Gastroenterol. Hepatol. 1, 535–549.e14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang H., Kambris Z., Lemaitre B., Hashimoto C., Two proteases defining a melanization cascade in the immune system of Drosophila. J. Biol. Chem. 281, 28097–28104 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Abrisqueta M., Herraiz C., Pérez Oliva A. B., Sanchez-Laorden B. L., Olivares C., Jiménez-Cervantes C., García-Borrón J. C., Differential and competitive regulation of human melanocortin 1 receptor signaling by β-arrestin isoforms. J. Cell Sci. 126, 3724–3737 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Tare M., Sarkar A., Bedi S., Kango-Singh M., Singh A., Cullin-4 regulates Wingless and JNK signaling-mediated cell death in the Drosophila eye. Cell Death Dis. 7, e2566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sembajwe L. F., Katta K., Grønning M., Kusche-Gullberg M., The exostosin family of glycosyltransferases: mRNA expression profiles and heparan sulphate structure in human breast carcinoma cell lines. Biosci. Rep. 38, BSR20180770 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee H., Cheong S.-M., Han W., Koo Y., Jo S.-B., Cho G.-S., Yang J.-S., Kim S., Han J.-K., Head formation requires Dishevelled degradation that is mediated by March2 in concert with Dapper1. Development 145, dev143107 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Court H., Ahearn I. M., Amoyel M., Bach E. A., Philips M. R., Regulation of NOTCH signaling by RAB7 and RAB8 requires carboxyl methylation by ICMT. J. Cell Biol. 216, 4165–4182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hariharan I. K., Hu K. Q., Asha H., Quintanilla A., Ezzell R. M., Settleman J., Characterization of rho GTPase family homologues in Drosophila melanogaster: Overexpressing Rho1 in retinal cells causes a late developmental defect. EMBO J. 14, 292–302 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ooi C. E., Moreira J. E., Dell’Angelica E. C., Poy G., Wassarman D. A., Bonifacino J. S., Altered expression of a novel adaptin leads to defective pigment granule biogenesis in the Drosophila eye color mutant garnet. EMBO J. 16, 4508–4518 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jin N., Chow C. Y., Liu L., Zolov S. N., Bronson R., Davisson M., Petersen J. L., Zhang Y., Park S., Duex J. E., Goldowitz D., Meisler M. H., Weisman L. S., VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P2 in yeast and mouse. EMBO J. 27, 3221–3234 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fu W., Sun J., Huang G., Liu J. C., Kaufman A., Ryan R. J. H., Ramanathan S. Y., Venkatesh T., Singh B., Squamous cell carcinoma-related oncogene (SCCRO) family members regulate cell growth and proliferation through their cooperative and antagonistic effects on cullin neddylation. J. Biol. Chem. 291, 6200–6217 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mummery-Widmer J. L., Yamazaki M., Stoeger T., Novatchkova M., Bhalerao S., Chen D., Dietzl G., Dickson B. J., Knoblich J. A., Genome-wide analysis of Notch signalling in Drosophila by transgenic RNAi. Nature 458, 987–992 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsuda M., Seong K.-H., Aigaki T., POSH, a scaffold protein for JNK signaling, binds to ALG-2 and ALIX in Drosophila. FEBS Lett. 580, 3296–3300 (2006). [DOI] [PubMed] [Google Scholar]
- 81.Lee N. G., Hong Y. K., Yu S. Y., Han S. Y., Geum D., Cho K. S., dXNP, a Drosophila homolog of XNP/ATRX, induces apoptosis via Jun-N-terminal kinase activation. FEBS Lett. 581, 2625–2632 (2007). [DOI] [PubMed] [Google Scholar]
- 82.Ketosugbo K. F., Bushnell H. L., Johnson R. I., A screen for E3 ubiquitination ligases that genetically interact with the adaptor protein Cindr during Drosophila eye patterning. PLOS ONE 12, e0187571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Holmes R. S., Comparative studies of vertebrate iduronate 2-sulfatase (IDS) genes and proteins: Evolution of A mammalian X-linked gene. 3 Biotech 7, 22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aznar N., Midde K. K., Dunkel Y., Lopez-Sanchez I., Pavlova Y., Marivin A., Barbazán J., Murray F., Nitsche U., Janssen K.-P., Willert K., Goel A., Abal M., Garcia-Marcos M., Ghosh P., Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. eLife 4, e07091 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rosengren Pielberg G., Golovko A., Sundström E., Curik I., Lennartsson J., Seltenhammer M. H., Druml T., Binns M., Fitzsimmons C., Lindgren G., Sandberg K., Baumung R., Vetterlein M., Strömberg S., Grabherr M., Wade C., Lindblad-Toh K., Pontén F., Heldin C.-H., Sölkner J., Andersson L., A cis-acting regulatory mutation causes premature hair graying and susceptibility to melanoma in the horse. Nat. Genet. 40, 1004–1009 (2008). [DOI] [PubMed] [Google Scholar]
- 86.Kanuka H., Kuranaga E., Hiratou T., Igaki T., Nelson B., Okano H., Miura M., Cytosol-endoplasmic reticulum interplay by Sec61α translocon in polyglutamine-mediated neurotoxicity in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 100, 11723–11728 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bikkavilli R. K., Malbon C. C., Arginine methylation of G3BP1 in response to Wnt3a regulates β-catenin mRNA. J. Cell Sci. 124, 2310–2320 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McLean C. A., Lutz A., Rankin K. J., Elliott A., Moussalli A., Stuart-Fox D., Red carotenoids and associated gene expression explain colour variation in frillneck lizards. Proc. Biol. Sci. 286, 20191172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/39/eabb8617/DC1