

Abstract
What biological factors account for resilience to pain or resilience to behavioral stress? We discuss examples of cellular and molecular mechanisms within disparate parts of the nervous system as contributors to such resilience. In some especially well-studied humans, it is possible to identify particular neuronal cell-types in the peripheral nervous system and pinpoint specific genes that are major contributors to pain resilience. We also discuss far more complex factors that operate within the central nervous system to confer resilience to behavioral stress. We propose that genetic and neurobiological substrates for resilience are discoverable and suggest more generally that neurology and psychiatry hold lessons for each other as investigators search for actionable, biological underpinnings of disease.
What is Resilience?
A spectrum of human responses is seen with neuropathic or inflammatory pain, with some individuals feeling severe discomfort, while others experience it as moderate or minimal, even in the context of similar nervous system injuries or inflammatory lesions. The latter patients can be viewed as exhibiting a form of resilience. Similarly, when confronted with threatening or stressful situations, some people develop behavioral abnormalities defined as a psychiatric syndrome (e.g., depression, anxiety, posttraumatic stress disorder [PTSD]), while most can maintain normal psychological and physiological functioning. The latter again is referred to as “resilient.”
The nervous system, where resilience resides at least in large part, has a tiered organization, which includes higher associative cortex fed by primary sensory cortices, which sit upon thalamic, midbrain and brainstem structures that, in turn, are superimposed upon spinal circuitry and peripheral neurons. Here, we discuss cellular and molecular mechanisms of resilience that operate at cerebral, sub-cerebral, spinal, or peripheral levels. A focus on resilience to pain and to behavioral stress, which currently comprise largely separate lines of investigation, provides important comparisons and contrasts for neurological versus psychiatric syndromes, and underscores the need to define both at multiple levels of the nervous system.
There is a strong link between chronic pain, stress and affective disorders (see Clinician’s Corner). Expectations and social and cultural contexts play important roles in pain and stress resilience, but these are difficult to understand at a fundamental biological level. Here, we take a reductionist approach and highlight neurobiological and genetic factors at the cellular and molecular level as contributors to pain and stress resilience. We conclude with directions for future research on neurological and psychiatric disorders.
Clinicians’ Corner.
Clinicians commonly encounter patients in whom quality of life is impacted by chronic pain or by behavioral stress. Thus far the majority of research has focused on understanding and treating susceptibility to these issues.
Some individuals, however, appear to be resilient to pain or to behavioral stress. Recent research is beginning to identify some of the cellular and molecular factors that contribute to resilience.
Resilience to pain can, at least in some human subjects, be attributed to a specific gene and to properties of a specific group of neurons. Inherited erythromelalgia (IEM) provides a genetic model of pain with 100% heritability. Outlier IEM patients carrying the same pain-producing mutation of peripheral sodium channel NaV1.7 experience less pain. An additional mutation in KV7.2 in one of the patients dampened the phenotype and provides a mechanistic basis for pain resilience.
The translation from studying resilience to pain versus resilience to stress is hampered by the lack of objective biological measures or biomarkers of stress susceptibility versus resilience. Notably, it is difficult to test neurobiological mechanisms of psychiatric endpoints in humans.
Heritability of depression and PTSD is ~35%, which provides a rough indication of the heritability of stress vulnerability. However, there are no known single gene mutations of strong effect and high penetrance that determine stress susceptibility
Deep brain stimulation of selected limbic targets has been shown to exert antidepressant effects in individuals with severe, treatment-resistant depression. There are also reports that deep brain stimulation can attenuate the overall experience of pain.
Retigabine, a small molecule potentiator of K+ channels in VTA promotes behavioral resilience in mice and can also reverse signs of susceptibility induced by prior stress. Inducing mechanisms of natural resilience represents a novel strategy for antidepressant drug discovery.
While much more research is needed, it appears likely that improved understanding of resilience to these conditions will in the future lead to the development of fundamentally novel treatments that would remain unknown by a sole focus on susceptibility.
Cellular and Molecular Contributors to Pain Resilience
Inter-individual differences in pain are well-documented, with some individuals reporting more severe pain and others reporting less severe pain in response to similar noxious insults and tissue damage. Recent studies have demonstrated that resilience to pain can, at least in some human subjects, be attributed to a specific gene and to properties of a specific group of neurons. These studies have capitalized on the availability of a genetic model of neuropathic pain, termed inherited erythromelalgia (IEM, see Glossary). IEM is an autosomal dominant disorder characterized by episodes of excruciating burning pain triggered by mild warmth which is usually innocuous. IEM is caused at the cellular level by hyperexcitability of dorsal root ganglion (DRG) neurons. Microneurographic recordings from patients with IEM point to abnormal firing of DRG neurons as the cause of pain [1, 2], consistent with the notion that increased firing of these peripheral neurons is a common source of neuropathic pain [3–9].
IEM is caused by gain-of-function mutations in SCN9A, which encodes sodium (Na+) channel NaV1.7. NaV1.7 is preferentially expressed in pain-signaling DRG neurons where its presence controls neuronal excitability: NaV1.7 acts to set the gain, like a volume knob within DRG neurons. Mutations that cause IEM enhance activation of NaV1.7 and slow its deactivation [10]. As a result of increased overlap between activation and steady-state inactivation, IEM mutations also depolarize DRG neurons [11]. Together, these biophysical changes in mutant Nav1.7 channels in IEM produce profound DRG neuron hyperexcitability that underlies the dramatic pain that characterizes this disorder.
Because of its well-established genetic and molecular basis and highly characteristic phenotype, IEM is regarded as a genetic model of neuropathic pain. Most patients with IEM exhibit a relatively uniform picture of very intense pain that limits their activities [12]. However, there are rare kindreds that contain outlier individuals, exemplified by individuals within the same family carrying the same pain-driving NaV1.7 mutation, but with very different pain profiles [13, 14]. It might have been expected that pain resilience in these outliers would arise from personality traits like will-power, courage, optimism or faith, and this cannot be ruled out. Yet recent studies indicate that, in the case of pain, peripheral mechanisms can be large contributors to pain resilience. Mis et al. studied one such family [15] composed of two IEM subjects (a mother and her son), both carrying the same pain-causing NaV1.7-S241T mutation. This muation enhances NaV1.7 channel activation [16], thereby hyperexciting DRG neurons [17]. The son complained of severe pain as is characteristic of IEM, while the mother described an unusual clinical picture of much less pain. The difference in clinical phenotypes [13, 14] showed that, despite carrying the same NaV1.7-S241T mutation, the son and mother had strikingly different pain profiles (Figure 1A,B). Interestingly, the mother’s resilience was manifested primarily in temporal domains, and was reflected by a remarkably reduced attack duration, small number of pain attacks, and other temporal measures like time in pain per day [13].
Figure 1. Strategy for identifying and functionally validating a pain resilience gene.
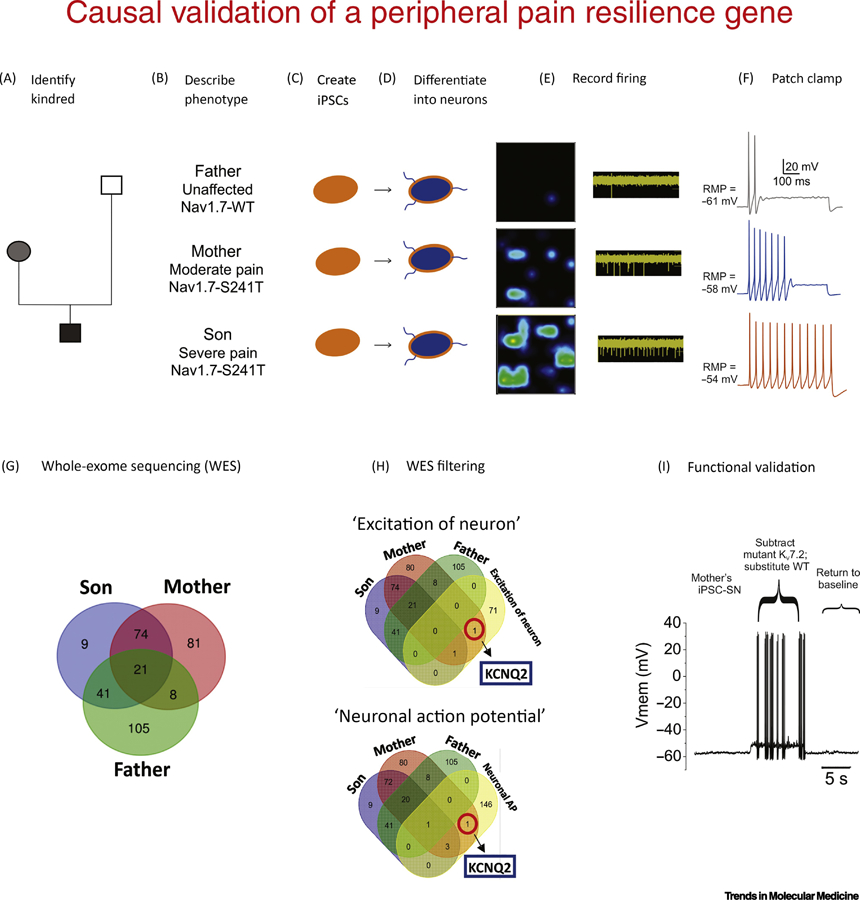
(A,B) In inherited erythromelalgia (IEM) with underlying NaV17 241T mutation the son exhibited severe pain while the mother was less effected. The unaffected father did not carry a NaV1.7 mutation. (C-D) iPSCs from blood or fibroblasts can be differentiated into nociceptive sensory neurons. (E) Heatmaps show representative multi-electrode array recordings from cells exposed to mildly increased temperature. The firing frequency of each active electrode is color-coded: white/green = high firing frequency; blue/black = low firing frequency. (F) Patch-clamp recordings display differences in resting potential, threshold for action potential generation, and frequency of firing paralleled the pain profile. (G) Whole exome sequencing (WES) to find gene variants that might underlie the different excitability in iPSC-SNs from the pain-resilient mother. Venn diagram showing the numbers of detected variants in samples from the three subjects. (H) Targeted Gene Ontology analysis can then be used to filter the results. (I) Dynamic-clamp analysis permits comparison of the functional effect of a mutant ion channel, versus the wild-type, within single cells. In this case, substitution of the mutant T730A IM current with WT IM in the mother’s iPSC-SNs increases the excitability of these cells, as indicated by reduced threshold. Panels E,F,G.H,I dapted from Mis et al. [15]
iPSC-derived neurons and WES can pinpoint pain resilience genes
Induced pluripotent stem cell (iPSC) modeling, whole exome sequencing (WES) and dynamic clamp analysis provided insights on the difference in pain between these related individuals who shared one-half of their genes and carried the same pain-causing mutation [15]. iPSCs from the affected son (severe pain, S241T Nav1.7 mutation) and mother (mild pain; same S241T NaV1.7 mutation), and the unaffected father were differentiated into peripheral sensory neuron-like cells (iPSC-SNs) (Figure 1C,D). Consistent with the fact that pain in IEM is triggered by increased temperature, mild warmth increased the mean firing rate and number of active iPSC-SNs from both the son and mother compared with the unaffected father. However, cells from the mother showed substantially less hyperexcitability compared to those from the son (Figure 1E). Further parameters that reflect neuronal excitability (resting membrane potential (RMP), current threshold, induced firing and percentage of repetitively firing neurons) in iPSC-SNs from the son, mother and father paralleled their pain profiles (Figure 1F). Differences in resting potential between iPSC-SNs from the mother and son were a major contributor to the differences in excitability of their derived neurons.
To pinpoint the genetic basis for these differences in pain, WES on samples from this kindred were filtered to focus on variants of genes expressed in DRG neurons (Figure 1G). Specific Gene Ontology (GO) terms related to neuronal excitability identified a variant in KCNQ2 encoding for potassium (K+) channel KV7.2, in the pain-resilient mother (Figure 1H). KV7.2 contributes to the non-inactivating M current (IM) in DRG neurons, and is known to be a major determinant of RMP of DRG neurons, where it regulates excitability [18, 19]. The KCNQ2 missense variant T730A in the mother might indeed contribute to her pain phenotype as the KV7.2-T730A mutation hyperpolarizes KV7.2 activation and thus enhances IM close to resting potential, which would be expected to dampen excitability.
Dynamic clamp revealed that substituting the KV7.2-T730A IM with an equivalent amount of WT conductance caused RMP depolarization. The KV7.2-T730A variant also had a strong effect in increasing current threshold and decreasing firing frequency (Figure 1H) of iPSC-SNs from the mother. These results show that a gain-of-function variant in KV7.2 significantly reduces the excitability of iPSC-SNs produced by another gain-of-function variant in NaV1.7, and provides a mechanistic basis for pain resilience in a human subject. Genetic variants in Na+ channels also contribute to pain sensitivity and thus, by implication, to resilience (Box 1). There are undoubtedly additional mechanisms, operating at higher levels of the neuraxis that can attenuate the pain experience even in the presence of unaltered hyperactivity of peripheral pain signaling neurons. Descending modulatory systems at the spinal level, and endogenous pain control circuitry at higher levels, can almost certainly modulate the experience of pain. Courage, faith, hope and will-power probably play important roles in some individuals as well. Elucidation of the molecular, cellular, and circuit mechanisms that underlie these central nervous system (CNS) functions is a major challenge at the interface between neurology and psychiatry.
Box 1: Genetic variants of sodium channels also contribute to pain resilience.
An example is provided by the R1150W polymorphism of NaV1.7 which occurs in about 10–14% of chromosomes from control populations. Although the effect of this amino acid substitution on NaV1.7 channel function remains to be resolved [20, 21], it is known that this channel variant depolarizes the RMP and increases excitability of DRG neurons [20]. The minor (“W”) allele (rs575030) was associated with a reduced pain threshold in response to experimental stimuli in normal individuals [21, 22]. The minor allele was also associated with higher pain scores in patients with osteoarthritis, sciatica, postamputation phantom pain and spinal disc herniation, although no correlation was seen with severity of pain in pancreatitis [21, 23, 24]. While some other studies have not found an association of the minor allele with increased pain [25]—which underscores the challenge of measuring pain and studying its genetic associations in large populations—this common missense variant may confer susceptibility to pain. In another way, the major allele present in the majority of individuals, can be viewed as conferring resilience.
Variants in SCN10A, which encodes Na+ channel NaV1.8, also modulate pain sensitivity [26]. The minor allele has multiple effects, including shifting channel activation and accelerating channel inactivation, with the net effect being reduced DRG neuron excitability, consistent with lower mechanical pain sensitivity.
Lessons From Pain Resilience For Studying Stress-Induced Psychiatric Syndromes
It might have been hoped that pain resilience would hold immediate lessons for resilience to behavioral stress. Yet, pain resilience can be conferred by mechanisms that operate in peripheral neurons, and this is not the case for behavioral stress or resilience to it. Repeated stress across the lifespan is the strongest known risk factor for human depression and related syndromes (anxiety, PTSD) [27]. Yet, it is well-established that most humans, despite exposure to extreme levels of psychological or physical stress, remain resilient, they avoid developing a psychiatric syndrome [28]. However, going from this observation to defining a molecular-cellular basis of stress resilience is far more difficult than for pain in IEM. First, there are no known single gene mutations of strong effect and high penetrance that determine stress susceptibility versus resilience as seen for NaV1.7 in IEM. Second, no single cell-type plays the same type of defining role in stress susceptibility versus resilience as played by KV7.2 within DRG neurons in pain. Third, there are no objective biomarkers of stress susceptibility versus resilience. This makes it impossible to ascertain whether functional differences in induced neurons from susceptible versus resilient individuals contributes to behavioral phenotypes. In fact, it is not even clear what type of cell-types one would want to study for the molecular basis of stress susceptibility versus resilience, since numerous brain regions and cell-types are implicated.
Cellular and Molecular Contributors to Stress Resilience
Increasing evidence in humans shows that resilience represents an active, adaptive process and not simply the absence of pathological responses that occur in more susceptible individuals [29, 30]. Resilient individuals tend to exhibit more optimism, possess stronger belief system and are part of stronger social communities [30]. The study of human resilience is still reflected by a mostly phenomenological literature, given the difficulty of testing neurobiological mechanisms of psychiatric endpoints in humans. Most studies have focused on peripheral neuroendocrine changes that are predictive of resilience [29]. Brain imaging studies have likewise implicated broad limbic regions of brain in controlling mood and anxiety states in general as well as susceptibility or resilient responses to severe stress. These include regions of prefrontal cortex (PFC), hippocampus and amygdala in the temporal lobe, key reward circuitry composed of the ventral tegmental area (VTA) and nucleus accumbens (NAc), and hypothalamus, among other regions [31, 32]. However, no known measure can predict where on the spectrum between stress susceptibility versus resilience any given individual will fall when exposed to stress.
Stress susceptibility versus resilience is determined by a combination of genetic factors and environmental exposures. Heritability of depression and PTSD is ~35%, which provides a rough indication of the heritability of stress vulnerability [33, 34]. This is in contrast to IEM, where the heritability is 100%. Importantly, this heritability for depression and PTSD is highly complex, with many hundreds of genes likely involved; such genes are only now beginning to be identified, work that has required analysis of hundreds of thousands of individuals [33, 34].
The most widely used model to understand the neurobiology of resilience is chronic social defeat stress (CSDS), where a test C57BL/6J mouse is placed into the home cage of a larger, more aggressive CD1 mouse [35]. This induces a broad behavioral syndrome in two-thirds of the test mice—termed susceptible—which is reminiscent of human depression, anxiety, and PTSD: mice exhibit anhedonia, social avoidance, reduced exploratory behavior, overeating and obesity, increased self-administration of drugs of abuse, and disrupted sleep and circadian rhythms [35, 36]. The remaining third of test mice - termed resilient - show similar deficits in exploratory behavior, but avoid the other behavioral abnormalities [36, 37].
Circuit mechanisms
This model of stress susceptibility versus resilience made it possible to identify multiple underlying brain circuits. VTA-NAc reward circuit is particularly important (Figure 2): susceptible but not resilient mice display increased excitability of VTA dopamine neurons that project to NAc, but reduced excitability of those that project to PFC [32, 41]. Optogenetic control over these two subpopulations of VTA dopamine neurons established that both responses causally contribute to behavioral susceptibility versus resilience [41]. Likewise, optogenetic control over activity of principal neurons in NAc, PFC, hippocampus and thalamus also influenced susceptible versus resilient responses to CSDS (for review, see [31, 32]).
Figure 2. Strategy to mine mechanisms of natural stress resilience for novel antidepressant drug discovery.
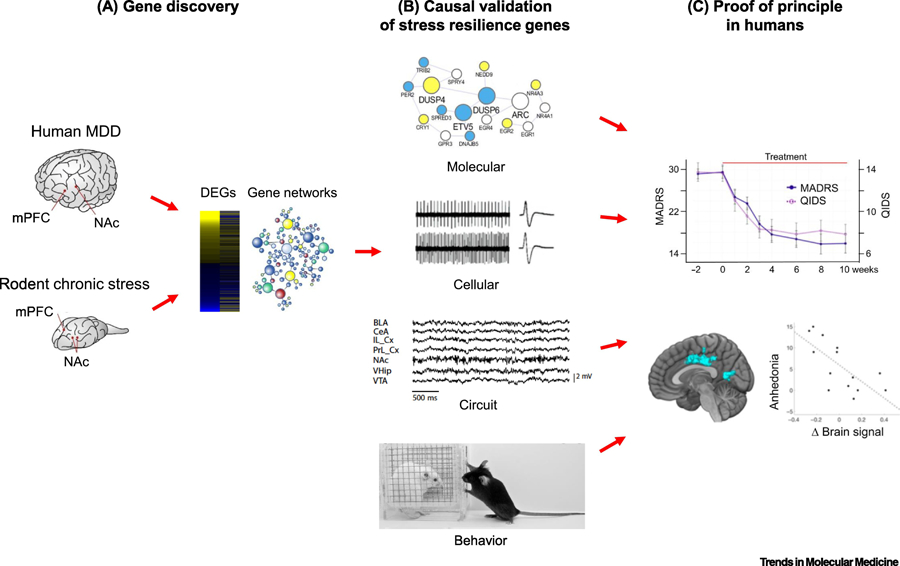
(A) Gene discovery involves using RNA-sequencing or other unbiased, genome-wide approaches to obtain an open-ended view of gene expression changes found in postmortem brains of humans with Major depressive disorder (MDD) or other stress-related disorders and in rodent chronic stress models. The latter include chronic social defeat stress, chronic variable stress, and paradigms of early life stress (e.g., maternal separation). Differentially expressed genes (DEGs) and key driver genes in gene networks are identified and correlated with resilience in rodents, and opposite effects in depressed humans. (B) A causal role of these key drivers in mediating resilience is then validated across molecular, cellular, circuit, and behavioral levels of analysis in rodent models. (C) Small molecules that affect a key driver can be studied in humans as a proof of principle, combining behavioral analyses with brain imaging measures. In the example shown, ezogabine decreases symptoms of depression in adult depressed humans based on the MADRS and QIDS behavioral rating scales: anhedonia measures within these rating scales correlate with a change in BOLD signal by functional MRI in the patients’s nucleus accumbens. (Data from [56]).
More recent work has indicated even greater complexity of mechanisms contributing to stress resilience, involving the hypothalamic-pituitary-adrenal (HPA) axis, which has long been known as a major feature of mammalian stress responses [42].
Immune mechanisms
Susceptible animals also display evidence of a hyperinflammatory state as indicated by increased levels of certain proinflammatory cytokines (e.g., interleukin 6 [IL6]) in blood. Transplantation of bone marrow from susceptible C57BL/6J mice into irradiated control mice render those control mice more susceptible to CSDS, an effect not seen when bone marrow from IL6 knockout mice is used [43]. These findings may relate to hyperinflammation observed in a subset of depressed patients. CSDS also induces changes in endothelial cells in specific brain regions of susceptible but not resilient mice that result in disruption of the blood-brain barrier, an effect observed in depressed humans as well [44]. This might enable circulating factors like IL6 to penetrate the brain and contribute to deleterious effects of stress in susceptible individuals. Abnormal patterns of gene expression and cellular function of brain microglia, oligodendrocytes, and astrocytes have also been observed in susceptible mice [45]. The mechanisms by which resilient mice avoid these abnormalities requires further analysis.
Molecular and cellular mechanisms
Open-ended studies of global changes in gene expression within specific brain regions have identified dramatic transcriptional reorganization across limbic brain regions in both susceptible and resilient animals [37, 45, 46]. These studies highlight that—in most brain regions—many more genes show altered expression levels in resilient mice than susceptible mice (see Figure 2A). This further suggests that susceptibility could represent a failure of the adaptive plasticity mounted by resilient individuals. Sequencing of dissected brain regions in CSDS and other chronic stress models has revealed regulated transcripts expressed by several neuronal, glial, or endothelial cells. This further emphasizes the challenges of unraveling the cellular complexity of susceptible versus resilient responses [e.g., 44, 64].
Large genome-wide datasets helped to identify key genes and enabled the demonstration of their causal role in mediating behavioral resilience (Figure 2B). Unique upregulation of several K+ channel subunits was found in VTA of resilient mice, an effect not seen in susceptible mice [37]. Interestingly, several Kv7 subtypes—including those implicated in pain responses in IEM, are among these upregulated K+ channels in the resilient VTA [37]. Overexpression of these K+ channels in VTA, or systemic administration of small molecule potentiators of these channels (e.g., retigabine), promotes an animal’s behavioral resilience and can also reverse signs of susceptibility induced by prior stress [37, 47, 48]. Inducing mechanisms of natural resilience could represent a novel strategy for antidepressant drug discovery, a possibility discussed below. Altered activity of HCN (hyperpolarization and cyclic nucleotide activated) channels in VTA dopamine neurons also contributes to responses to CSDS [48].
In addition to ion channels, several other proteins have been linked causally to behavioral resilience. Prominent among these are the transcription factors ∆FOSB, ß-catenin, ESR1, and ZFP189, each of which promotes stress resilience through actions in a specific neuronal cell type within a given limbic brain region (Table 1) [49–52]. Identifying target genes through which these resilience-promoting transcription factors exert their effects can be used to advance therapeutic discovery for stress-related illnesses by boosting mechanisms of natural resilience.
Table 1.
Examples of Pro-Resilient Transcription Factors for Behavioral Stress1
| Transcription Factor | Brain Region | Cell Type | Ref. |
|---|---|---|---|
| ∆FOSB | Nucleus accumbens | D1-type medium spiny neuron | 49,50 |
| ß-Catenin | Nucleus accumbens | D2-type medium spiny neuron | 51 |
| ESR1 (estrogen receptor-α) | Nucleus accumbens | N/A | 65 |
| ZFP189 | Prefrontal cortex | Pyramidal neurons | 64 |
The pro-resilient role of each of these factors was established primarily in the CSDS model.
Epigenetic mechanisms
Why do genetically inbred mice display such dramatically different behavioral responses to the same social stress? Inbred mouse lines differ in their inherent rates of susceptibility versus resilience, indicating the importance of genetic factors which is consistent with human data. Environmental exposures also influence a mouse’s behavioral response to stress later in life [53]. The divergent responses to CSDS by inbred C57BL/6J mice bred and raised in the same facility and presumably exposed to nearly identical environmental conditions implicates random events during development which are mediated by epigenetic mechanisms. Indeed, numerous epigenetic changes have been shown to mediate behavioral resilience to stress acting in several limbic brain regions [54–56]. Ongoing research investigates the mechanisms by which these epigenetic factors—in concert with pro-resilient transcription factors—mediate stress resilience.
Effects of behavioral stress on pain perception
CSDS and other forms of chronic stress have been shown to alter an animal’s sensitivity to acute painful stimuli; some studies report stress-induced analgesia, while others find stress-induced hyperalgesia [57–60]. These differences may relate to the severity and duration of the different chronic stress paradigms used. Endogenous opioid pathways have been implicated in these responses [57, 58], but further work is needed to define the effects of behavioral stress on pain perception and chronic pain syndromes. Of interest, a recent study found shared transcriptional responses in several limbic brain regions induced in mice by chronic variable stress on the one hand and a chronic pain (nerve injury) model on the other [61].
Translating Insights from Animal Models to Humans
It has been difficult to translate findings from animal models to the treatment of pain and psychiatric disorders. One obstacle is the paucity of tool compounds to test specific hypotheses arising from the basic literature. Open-ended, unbiased transcriptomic and epigenomic mapping has implicated several factors playing roles in mediating resilience, yet it is not straightforward to test whether replicating such mechanisms (e.g., activation of pro-resilient transcription or epigenetic factors) in humans might boost resilience.
Nevertheless, one example of a putative pro-resilient protein that can already be targeted today is KV7 channels and ovservations on retigabine: Induction of several KV7 subunits occurs in VTA dopamine neurons of resilient mice [37] and experimental potentiation of KV7 channel function in VTA promotes resilience and antidepressant-like effects in stressed mice [37, 47, 48]. This provided the rationale for an open-label study of a small number of depression patients with retigabine. Retigabine produced a statistically significant improvement in depressive symptoms which correlated with correction of abnormal activity in brain reward circuits as assessed by magnetic resonance imaging (Figure 2C) [62]. Development of derivative drugs that selectively target specific KV7 subtypes implicated in stress resilience, with a more acceptable side-effect profile, might be an important step forward. While results of this pilot retigabine study must be viewed with considerable caution, they do support the validity of the general approach: of mining large genome-wide datasets to identify putative targets for inducing resilience (treatment of depression or chronic pain) in humans.
Concluding Remarks
While resilience to pain and resilience to stress are complex and only partially understood, there has been progress in unraveling their neurobiological and genetic underpinnings. The findings of Mis et al. [15] show that inter-individual differences in pain can be modeled in vitro using subject-specific iPSC-SNs and provides proof-of-concept to pinpoint mechanisms and identify specific gene variants that contribute to pain resilience.
By contrast, our discussion of stress resilience highlights dimensions of far greater complexity. Despite these obstacles, multiple leads at the molecular, cellular and circuit levels contribute to deciphering central mechanisms of stress and pain resilience and are even beginning to be translated toward the clinic.
Caveats and complexities
Among the complexities in the study of resilience to pain and stress are the many levels within the hierarchical nervous system (i.e., peripheral, spinal, subcortical, cortical). One of the challenges is also that resilience is impacted by expectation, environmental influences and social and cultural factors. Many elements, at the molecular and cellular levels, can interact in complex ways, as exemplified by NaV1.7 and KV7.2 in the pain-resilient patient with IEM. Even a single factor can have multivalent actions, for example the NaV1.8 variant [26] which has both pro- and anti-excitatory biophysical attributes, requiring assessment of the net effect of multiple offsetting actions. Importantly, intronic as well as exonic variants of protein-coding genes, and non-coding RNAs, also probably contribute to pain and stress resilience, but these remain more difficult to study.
Adding another level of complexity, resilience is not necessarily fixed or static over time. On the contrary, it almost certainly varies across the lifetime of any given individual, increasing with age in some individuals and decreasing in others. Within the nervous system, this is probably paralleled by changes in synaptic efficiency, connectivity and intrinsic neuronal excitability that occur over time.
Functional imaging has demonstrated abnormal patterns of brain activation that occur in association with chronic neuropathic pain [63]. Effective pain treatment can be accompanied, within weeks, by a shift toward a more normal pattern of brain activation, suggesting that the changes are not necessarily hard-wired within the brain [14]. Likewise, numerous inter-connected limbic brain regions are implicated in susceptibility versus resilience to chronic stress and depression [64] and contribute to the overall pain experience as well [65, 66]. Deep brain stimulation of selected limbic targets has been shown to exert antidepressant effects in roughly one-half of individuals with severe, treatment-resistant depression [67]. Such stimulation corrects some of the circuit abnormalities found in depression, providing direct demonstration that modulation of specific brain circuits can control behavioral susceptibility and resilience. There are also reports that deep brain stimulation can attenuate the overall experience of pain [68, 69].
These complexities represent challenges for our understanding of resilience. Nonetheless, it has been possible in selected individuals to pinpoint particular cell-types within the nervous system and to identify the genes responsible for resilience [15]. Advances have also been made in revealing the molecular and cellular mechanisms that contribute to stress resilience, albeit with far less precision and certainty. New technologies, including iPSC modeling of disease, CRISPR gene editing, next-generation sequencing and dynamic clamp, among many others, will help to unravel the mysteries of resilience.
Lessons for the search for resilience factors
The search for neurobiological and genetic factors that confer resilience to pain, stress and other related conditions is still at early stages. Nonetheless, several lessons are emerging. First, it is important not to confuse complexity with intractability. Dissection of resilience at the level of peripheral neurons is more straightforward than for central mechanisms of resilience, involving numerous brain regions with multiple neuronal and nonneuronal cell-types; and by many peripheral organs which interact bidirectionally with the brain. Still, we propose that the phenomenon of resilience is tractable and that, while the path of discovery will be long and arduous, tangible progress with direct medical implications will be made. In this way, the field of psychiatry, which has focused for decades on deciphering such complex mechanisms, can help guide neurology, just as neurology has led the way in establishing the genetic underpinnings of brain diseases.
WES and whole genome sequencing, combined with very large cohorts is now under way. Furthermore, epigenetic and transcriptional analyses at single cell level, and gene network analyses, such as weighted gene co-expression network analysis, make it possible to integrate these large and complex datasets to identify targets, in an open-ended, unbiased manner. Examples of this network approach have appeared for pain and stress models [45, 70], including a focus on resilience [71, 72]. While big data approaches involving thousands of patients are being used widely to study the genetic basis of pain and stress vulnerability, smaller kindreds of well-characterized individuals can still teach important lessons about resilience. The principle of mutations in specific ion channel genes in mediating resilience to may be true for psychiatric syndromes. Deep phenotyping of large pedigrees might find rare genetic variations of large effect and high penetrance that control stress susceptibility versus resilience.
Furthermore, we may need to discard pre-conceived notions. At least in some individuals, a component of pain resilience arises not within the CNS, but within peripheral neurons. Likewise, stress resilience can arise not only within the CNS, but also within lymphoid cells in bone marrow and their release of pro-inflammatory cytokines in peripheral blood. Additional neuronal and nonneuronal cell types and genes will be identified as contributors to pain and stress resilience. Unraveling the complexity of resilience will reveal additional surprises and previously unexplored territory.
Finally, temporal aspects of a disease’s presentation and course may hold lessons for the study of stress. In the kindred studied by Mis et al. [15] temporal features of pain provided greater insight than other measures. Similar lessons derive from clinical trials on trigeminal neuralgia [73] and migraine [74, 75], where the number of pain attacks per day/month can be quantitated. It would be interesting to devise similar longitudinal scales to measure stress susceptibility versus resilience, which might offer some advantages over sole reliance on cross-sectional data.
Beyond pain and stress, the phenomenon of resilience is also being explored for other neuropsychiatric conditions, such as Alzheimer’s disease [76]. Overall, our expectation is that an improved understanding of resilience will lead to the developmental of fundamentally novel treatments that would remain unknown by a sole focus on disease susceptibility.
Figure 3. Multiscale mechanisms of pain and stress resilience.
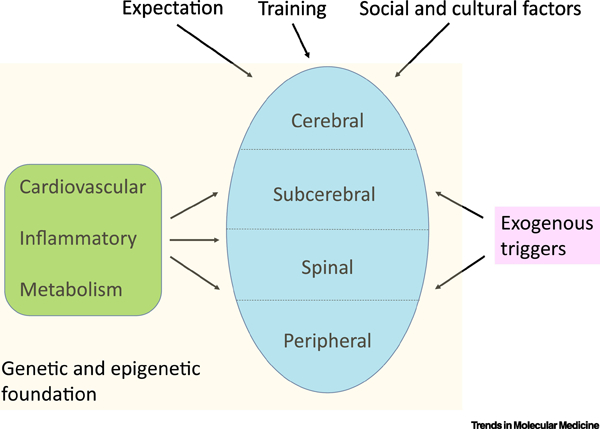
The figure illustrates the involvement of the entire neuraxis, from peripheral neurons (all excitatory) to more complex spinal, sub-cerebral, and cerebral circuits which reflect a combination of excitatory and inhibitory neurons and their regulation by several glia cell-types. Nervous system function in turn is controlled by peripheral factors such as those from the cardiovascular, inflammatory, and metabolic systems, among others. The functioning of the nervous system and peripheral organs is driven by a foundation of genomic sequence and its regulation by epigenetic factors, which mediate in part the influence of a host of external stimuli on the organism over the life cycle. These combinations of factors result ultimately in an individual’s inherent level of resilience versus susceptibility to pain, stress, and many other challenges.
OUTSTANDING QUESTIONS.
Can additional cell-types be identified as contributors to pain resilience?
Can specific genes or molecules be pinpointed as significant contributors to pain resilience in these cells?
Can environmental, epigenetic, hormonal or other contributors to pain resilience be identified?
Can these discoveries be translated into new therapies that will confer resilience to pain in patients?
Can additional cell-types be identified as contributors to behavioral stress resilience?
Can specific genes or molecules be pinpointed as significant contributors to stress resilience in these cells?
Can environmental, epigenetic, hormonal or other contributors to resilience to behavioral stress be identified?
Can these discoveries be translated into new therapies that will confer resilience to behavioral stress in patients?
HIGHLIGHTS.
Chronic pain and behavioral stress represent major unmet medical needs
Some individuals are relatively resilient to pain or to behavioral stress
In some especially well-studied humans, it has been possible to identify particular neuronal cell-types in the peripheral nervous system and pinpoint particular genes that are major contributors to pain resilience
Recent research is beginning to illuminate the far more complex factors that operate within the CNS to confer resilience to behavioral stress and pain
Improved understanding of resilience to pain and behavioral stress is likely to inform the development of novel treatments that would remain unknown by a sole focus on disease susceptibility.
ACKNOWLEDGEMENTS
Work in the laboratory of EJN is supported by P01MH096890 and R01MH051399. Work in the laboratory of SGW is supported by Center Grant B9253-C from the U.S. Department of Veterans Affairs Rehabilitation Research and Development Service, by a grant from The Erythromelalgia Association, and by the Regenerative Medicine Research Fund (RMRF) of CT Innovations. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University.
Glossary
- Anhedonia
reduced interest in pleasurable stimuli such as food, drink, sex, and exercise
- Chronic social defeat stress (CSDS)
an experimental model where a test C57BL/6J mouse is placed into the home cage of a larger, more aggressive CD1 mouse [35]. Fighting ensues quickly but is restricted to 5–10 min to limit physical injury, after which time the two mice are separated by a divider which prevents further physical contact but allows the aggressive cues of the larger mouse to be perceived. Over 10 days the test mouse is exposed to a different aggressive mouse each day. CSDS induces similar rates of susceptibility and resilience in male and female C57BL/6J mice, although different experimental procedures are used for the two sexes [38, 39]. Moreover, development of susceptible versus resilient responses to CSDS does not require physical defeat per se: C57BL/6J mice that simply witness another C57BL/6J mouse being defeated develop a similar ratio of susceptibility versus resilience [38–40]
- Dorsal root ganglion (DRG)
a cluster of primary sensory neurons that generate (pain) signals from the periphery and transmit them to the spinal cord
- Dynamic clamp
a powerful electrophysiological method to compare the effects of wild-type and variant ion channels within the same cell. It can be used to assess the role of mutant channels in modulating excitability of neurons
- Epigenome
mediates the interactions between environmental exposures and an individual’s genome sequence. Environmentally mediated alterations are for example changes in histone-modifying enzymes, chromatin remodeling proteins, DNA methylation and microRNAs
- Hypothalamic-pituitary-adrenal (HPA) axis
controls the body’s secretion of glucocorticoids, has long been known as a major feature of mammalian stress responses and more recently implicated in stress resilience
- Inherited erythromelalgia (IEM)
also called the “Man on Fire” syndrome, is an autosomal dominant disorder characterized by episodes of excruciating burning pain triggered by mild warmth which is usually innocuous, such as wearing socks or a sweater, or entering a room at 70°F. The feeling is described “as if hot lava has been poured into the body”. People with IEM tend to immerse their limbs in cold water or even ice to alleviate pain, which might imply abnormal impulse generation in axon terminals within the skin
- Pain scores
There are different pain assessment scales for different patient types. Generally higher numbers indicate more severe pain
- Retigabine (also known as ezogabine)
A KV7 channel potentiator that has been marketed for the treatment of epilepsy but rarely used due to side-effects; Retigabine selectively targets specific KV7 subtypes implicated in stress resilience
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
Eric Nestler is a founder and advisor for PsychoGenics and a Director of BERG. Stephen Waxman reports serving as an advisor or consultant to Amgen, Biogen, GlaxoSmithKline, Chromocell, RedPin Therapeutics and SiteOne Therapeutics.
REFERENCES
- 1.Orstavik K, et al. , Pathological C-fibres in patients with a chronic painful condition. Brain, 2003. 126(Pt 3): p. 567–78. [DOI] [PubMed] [Google Scholar]
- 2.Namer B, et al. , Specific changes in conduction velocity recovery cycles of single nociceptors in a patient with erythromelalgia with the I848T gain-of-function mutation of Nav1.7. Pain, 2015. 156(9): p. 1637–46. [DOI] [PubMed] [Google Scholar]
- 3.Zhang XY, et al. , Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet, 2013. 93(5): p. 957–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ochoa J and Torebjork E, Sensations evoked by intraneural microstimulation of C nociceptor fibres in human skin nerves. J Physiol, 1989. 415: p. 583–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleggetveit IP, et al. , High spontaneous activity of C-nociceptors in painful polyneuropathy. Pain, 2012. 153(10): p. 2040–7. [DOI] [PubMed] [Google Scholar]
- 6.Haroutounian S, et al. , Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain, 2014. 155(7): p. 1272–9. [DOI] [PubMed] [Google Scholar]
- 7.Vaso A, et al. , Peripheral nervous system origin of phantom limb pain. Pain, 2014. 155(7): p. 1384–91. [DOI] [PubMed] [Google Scholar]
- 8.Devor M, Neuropathic pain: pathophysiological response of nerves to injury, in Wall and Melzack’s Textbook of Pain., McMahon SL, Koltzenburg M, Tracey I and Turk DC, Editor. 2013, Churchill Livingstone: London: p. pp861–888. [Google Scholar]
- 9.Waxman SG, et al. , Sodium channels and pain. Proc Natl Acad Sci U S A, 1999. 96(14): p. 7635–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dib-Hajj SD, et al. , The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci, 2013. 14(1): p. 49–62. [DOI] [PubMed] [Google Scholar]
- 11.Vasylyev DV, et al. , Dynamic-clamp analysis of wild-type human Nav1.7 and erythromelalgia mutant channel L858H. J Neurophysiol, 2014. 111(7): p. 1429–43. [DOI] [PubMed] [Google Scholar]
- 12.Drenth JP and Waxman SG, Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest, 2007. 117(12): p. 3603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonnell A, et al. , Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain, 2016. 139(Pt 4): p. 1052–65. [DOI] [PubMed] [Google Scholar]
- 14.Geha P, et al. , Pharmacotherapy for Pain in a Family With Inherited Erythromelalgia Guided by Genomic Analysis and Functional Profiling. JAMA Neurol, 2016. 73(6): p. 659–67. [DOI] [PubMed] [Google Scholar]
- 15.Mis MA, et al. , Resilience to Pain: A Peripheral Component Identified Using Induced Pluripotent Stem Cells and Dynamic Clamp. J Neurosci, 2019. 39(3): p. 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lampert A, et al. , Size matters: Erythromelalgia mutation S241T in Nav1.7 alters channel gating. J Biol Chem, 2006. 281(47): p. 36029–35. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, et al. , Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Na(V)1.7 mutant channel. Nat Commun, 2012. 3: p. 1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du X, et al. , Control of somatic membrane potential in nociceptive neurons and its implications for peripheral nociceptive transmission. Pain, 2014. 155(11): p. 2306–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Passmore GM, et al. , KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci, 2003. 23(18): p. 7227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Estacion M, et al. , A sodium channel gene SCN9A polymorphism that increases nociceptor excitability. Ann Neurol, 2009. 66(6): p. 862–6. [DOI] [PubMed] [Google Scholar]
- 21.Reimann F, et al. , Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc Natl Acad Sci U S A, 2010. 107(11): p. 5148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan G, et al. , The Effect of SCN9A Variation on Basal Pain Sensitivity in the General Population: An Experimental Study in Young Women. J Pain, 2015. 16(10): p. 971–80. [DOI] [PubMed] [Google Scholar]
- 23.Kurzawski M, et al. , Common Missense Variant of SCN9A Gene Is Associated with Pain Intensity in Patients with Chronic Pain from Disc Herniation. Pain Med, 2018. 19(5): p. 1010–1014. [DOI] [PubMed] [Google Scholar]
- 24.Valdes AM, et al. , Role of the Nav1.7 R1150W amino acid change in susceptibility to symptomatic knee osteoarthritis and multiple regional pain. Arthritis Care Res (Hoboken), 2011. 63(3): p. 440–4. [DOI] [PubMed] [Google Scholar]
- 25.Holliday KL, et al. , The non-synonymous SNP, R1150W, in SCN9A is not associated with chronic widespread pain susceptibility. Mol Pain, 2012. 8: p. 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duan G, et al. , A SCN10A SNP biases human pain sensitivity. Mol Pain, 2016. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melchers MC, et al. , Differentiating Burnout from Depression: Personality Matters! Front Psychiatry, 2015. 6: p. 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feder A, Nestler EJ, and Charney DS, Psychobiology and molecular genetics of resilience. Nat Rev Neurosci, 2009. 10(6): p. 446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russo SJ, et al. , Neurobiology of resilience. Nat Neurosci, 2012. 15(11): p. 1475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Southwick SMC, D.S., Resilience: The Science of Mastering Life’s Greatest Challenges. 2018.
- 31.Cathomas F, et al. , Neurobiology of Resilience: Interface Between Mind and Body. Biol Psychiatry, 2019. 86(6): p. 410–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han MH and Nestler EJ, Neural Substrates of Depression and Resilience. Neurotherapeutics, 2017. 14(3): p. 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maul S, et al. , Genetics of resilience: Implications from genome-wide association studies and candidate genes of the stress response system in posttraumatic stress disorder and depression. Am J Med Genet B Neuropsychiatr Genet, 2019. [DOI] [PubMed]
- 34.McIntosh AM, Sullivan PF, and Lewis CM, Uncovering the Genetic Architecture of Major Depression. Neuron, 2019. 102(1): p. 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berton O, et al. , Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science, 2006. 311(5762): p. 864–8. [DOI] [PubMed] [Google Scholar]
- 36.Golden SA, et al. , A standardized protocol for repeated social defeat stress in mice. Nat Protoc, 2011. 6(8): p. 1183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krishnan V, et al. , Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell, 2007. 131(2): p. 391–404. [DOI] [PubMed] [Google Scholar]
- 38.Harris AZ, et al. , A Novel Method for Chronic Social Defeat Stress in Female Mice. Neuropsychopharmacology, 2018. 43(6): p. 1276–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi A, et al. , Establishment of a repeated social defeat stress model in female mice. Sci Rep, 2017. 7(1): p. 12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warren BL, et al. , Neurobiological sequelae of witnessing stressful events in adult mice. Biol Psychiatry, 2013. 73(1): p. 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaudhury D, et al. , Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature, 2013. 493(7433): p. 532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinlein SA and Karatsoreos IN, The hypothalamic-pituitary-adrenal axis as a substrate for stress resilience: Interactions with the circadian clock. Front Neuroendocrinol, 2020. 56: p. 100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodes GE, et al. , Neuroimmune mechanisms of depression. Nat Neurosci, 2015. 18(10): p. 1386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menard C, et al. , Social stress induces neurovascular pathology promoting depression. Nat Neurosci, 2017. 20(12): p. 1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bagot RC, et al. , Circuit-wide Transcriptional Profiling Reveals Brain Region-Specific Gene Networks Regulating Depression Susceptibility. Neuron, 2016. 90(5): p. 969–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bagot RC, et al. , Ketamine and Imipramine Reverse Transcriptional Signatures of Susceptibility and Induce Resilience-Specific Gene Expression Profiles. Biol Psychiatry, 2017. 81(4): p. 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedman AK, et al. , KCNQ channel openers reverse depressive symptoms via an active resilience mechanism. Nat Commun, 2016. 7: p. 11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Friedman AK, et al. , Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science, 2014. 344(6181): p. 313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamilton PJ, et al. , Cell-Type-Specific Epigenetic Editing at the Fosb Gene Controls Susceptibility to Social Defeat Stress. Neuropsychopharmacology, 2018. 43(2): p. 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lobo MK, et al. , DeltaFosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. J Neurosci, 2013. 33(47): p. 18381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vialou V, et al. , DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci, 2010. 13(6): p. 745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dias C, et al. , beta-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature, 2014. 516(7529): p. 51–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pena CJ, et al. , Early life stress confers lifelong stress susceptibility in mice via ventral tegmental area OTX2. Science, 2017. 356(6343): p. 1185–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Covington HE 3rd, et al. , Antidepressant actions of histone deacetylase inhibitors. J Neurosci, 2009. 29(37): p. 11451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pena CJ and Nestler EJ, Progress in Epigenetics of Depression. Prog Mol Biol Transl Sci, 2018. 157: p. 41–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun H, et al. , BAZ1B in Nucleus Accumbens Regulates Reward-Related Behaviors in Response to Distinct Emotional Stimuli. J Neurosci, 2016. 36(14): p. 3954–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McLaughlin JP, et al. , Social defeat stress-induced behavioral responses are mediated by the endogenous kappa opioid system. Neuropsychopharmacology, 2006. 31(6): p. 1241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miczek KA, Thompson ML, and Shuster L, Opioid-like analgesia in defeated mice. Science, 1982. 215(4539): p. 1520–2. [DOI] [PubMed] [Google Scholar]
- 59.Pagliusi M Jr., et al. , Therapeutic and Preventive Effect of Voluntary Running Wheel Exercise on Social Defeat Stress (SDS)-induced Depressive-like Behavior and Chronic Pain in Mice. Neuroscience, 2020. 428: p. 165–177. [DOI] [PubMed] [Google Scholar]
- 60.Sawicki CM, et al. , Microglia Promote Increased Pain Behavior through Enhanced Inflammation in the Spinal Cord during Repeated Social Defeat Stress. J Neurosci, 2019. 39(7): p. 1139–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Descalzi G, et al. , Neuropathic pain promotes adaptive changes in gene expression in brain networks involved in stress and depression. Sci Signal, 2017. 10(471). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan A, et al. , Effects of the KCNQ channel opener ezogabine on functional connectivity of the ventral striatum and clinical symptoms in patients with major depressive disorder. Mol Psychiatry, 2019: p. in press. [DOI] [PMC free article] [PubMed]
- 63.Hashmi JA, et al. , Shape shifting pain: chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain, 2013. 136(Pt 9): p. 2751–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dunlop BW and Mayberg HS, Neuroimaging-based biomarkers for treatment selection in major depressive disorder. Dialogues Clin Neurosci, 2014. 16(4): p. 479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baliki MN, et al. , Predicting value of pain and analgesia: nucleus accumbens response to noxious stimuli changes in the presence of chronic pain. Neuron, 2010. 66(1): p. 149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bushnell MC, Ceko M, and Low LA, Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci, 2013. 14(7): p. 502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holtzheimer PE and Mayberg HS, Deep brain stimulation for psychiatric disorders. Annu Rev Neurosci, 2011. 34: p. 289–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Russo JF and Sheth SA, Deep brain stimulation of the dorsal anterior cingulate cortex for the treatment of chronic neuropathic pain. Neurosurg Focus, 2015. 38(6): p. E11. [DOI] [PubMed] [Google Scholar]
- 69.Farrell SM, Green A, and Aziz T, The Current State of Deep Brain Stimulation for Chronic Pain and Its Context in Other Forms of Neuromodulation. Brain Sci, 2018. 8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng N, et al. , Weighted gene co-expression network analysis reveals specific modules and hub genes related to neuropathic pain in dorsal root ganglions. Biosci Rep, 2019. 39(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lorsch ZS, et al. , Stress resilience is promoted by a Zfp189-driven transcriptional network in prefrontal cortex. Nat Neurosci, 2019. 22(9): p. 1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lorsch ZS, et al. , Estrogen receptor alpha drives pro-resilient transcription in mouse models of depression. Nat Commun, 2018. 9(1): p. 1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zakrzewska JM, et al. , Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol, 2017. 16(4): p. 291–300. [DOI] [PubMed] [Google Scholar]
- 74.Dodick DW, et al. , Effect of Fremanezumab Compared With Placebo for Prevention of Episodic Migraine: A Randomized Clinical Trial. JAMA, 2018. 319(19): p. 1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goadsby PJ, et al. , A Controlled Trial of Erenumab for Episodic Migraine. N Engl J Med, 2017. 377(22): p. 2123–2132. [DOI] [PubMed] [Google Scholar]
- 76.Ridge PG, et al. , Linkage, whole genome sequence, and biological data implicate variants in RAB10 in Alzheimer’s disease resilience. Genome Med, 2017. 9(1): p. 100. [DOI] [PMC free article] [PubMed] [Google Scholar]