

Abstract
Progranulin (PGRN) is a growth factor with significant biological effects in different types of cancer. However, its role in melanoma progression has not been explored. In this study, we first analyze clinical datasets and show that high PGRN expression levels are correlated with poor prognosis of melanoma patients. Further, we demonstrate in a transplanted murine melanoma model in which the endogenous Grn gene encoding PGRN has been deleted that tumor-derived, not host-derived PGRN, promotes melanoma growth and metastasis. Immunological analyses reveal an enhanced infiltration of natural killer cells, but not T lymphocytes, into PGRN-deficient tumors compared to the wild type control. Antibody-mediated depletion confirms the critical role of NK cells in controlling B16 tumor growth. RNA-seq analysis reveals that several chemokines including CCL5 are strongly upregulated in PGRN-deficient tumor. Silencing CCL5 expression in PGRN-deficient tumor reduces NK cell recruitment and restores tumor growth to the control level. Lastly, we show that PGRN inhibits Ccl5 gene expression at the transcriptional level. This study highlights a novel and critical role of PGRN in melanoma growth and metastasis and suggests that it may represent a potential therapeutic target.
Keywords: Progranulin, melanoma, natural killer cell, CCL5, immune response
1. Introduction
Progranulin (PGRN) is a growth factor also known as acrogranin, granulin/epithelin precursor (GEP), proepithelin (PEPI), PC cell-derived growth factor (PCDGF), and 88-kDa glycoprotein (GP88) [1]. PGRN has activities in various biological processes such as tissue remodeling and repair during embryonic development, cell proliferation, angiogenesis, tumorigenesis, wound repair, and inflammation [2]. Progranulin consists of 593 amino acids, and its precursor’s protein has a secretory signal peptide with seven and a half cysteine-rich ~ 6 kDa repetitions in tandem, each of which is called granulin that can be generated by proteolytic cleavage of PGRN into the individual granulin peptides or bioactive fragments [3–8]. Secretory leukocyte protease inhibitor (SLPI) interacts with PGRN and protects it against elastase digestion [3]. Generally speaking, the role of PGRN is considered anti-inflammatory whereas the granulin peptides have pro-inflammatory activities [9].
Recently, numerous studies have demonstrated the importance of PGRN in tumorigenesis and metastasis [10]. Overexpression of PGRN has been observed in clinical specimens of many types of cancer especially in cancers with high malignancy [11]. He et al. first showed that PGRN overexpression could promote renal epithelial cells’ malignant transformation and increase their tumorigenesis [12]. High PGRN expression is associated with elevated vascular endothelial growth factor (VEGF) expression in breast cancer, esophageal squamous cell carcinoma, and colorectal tumors [13–16]. PGRN promotes tumor cell division, survival, and invasion through the activation of the PI3K and ERK pathways, and induction of cyclin D1 and cyclin B expression [17, 18]. PGRN also increases cell migration and invasion by upregulation of the EMT regulators and activation of matrix metalloproteins 2 and 9 in tumor cells [19–21]. PGRN facilitates breast cancer and multiple myeloma’s resistance to anticancer drugs but the mechanisms are currently unknown [22–24]. Last but not least, PGRN renders hepatocellular carcinoma cells resistant to natural killer cytotoxicity [25].
Melanoma is a cancer that originates from melanocytes which are found predominantly in skin and eye. Skin cancer is the third most common human cancer in the world. Each year, 2–3 million cases of skin cancer are reported across the globe. At the early stage, it is curable by surgical resection however metastatic melanoma has a very poor prognosis and does not respond to current therapies [26]. Thus, new and more effective therapeutic targets and strategies are desired. In this study, we provide the first evidence for the impact of PGRN in melanoma growth and metastasis with human clincal data and in a murine model of melanoma. Our study suggests that PGRN may represent a novel clinical biomarker for diagnosis and prognosis of melanoma, as well as a potential target for therapy.
2. Materials and methods
2.1. Mice
6–8 weeks old WT female C57BL/6 were purchased from Shanghai Jihui Laboratory Animal Care (Shanghai, China) and Grn−/− mice were generated as described previously [27]. Mice from both stains were maintained in the pathogen-free condition. All animal experiments were performed in accordance with approved protocols by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University.
2.2. Cell lines
B16-F10 murine cell line was purchased from the Cell Center of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
To generate the stable B16-F10/Grn−/− cells, we used an online CRISPR Design Tool (http://tools.genome-engineering.org), which provided by Zhang Feng team. Six different single-guide RNA (sgRNA) constructs were used with the following sequences, the locations of PGRN-targeted sequences in the genome of mouse PGRN were shown in Supplementary Fig 1.A.
sgRNA1: ACCATCAACCATAATGCAGC
sgRNA2: AGATGATGGCTCGTTATTCT
sgRNA3: TCTATTTGCAGATAACCCCT
sgRNA4: ATAACGAGCCATCATCTAGA
sgRNA5: GGCTACCAGCCCTGCCGCGA
sgRNA6: GCCGGAACACAGTGTCCAGA
The sgRNA expression constructs were amplified by PCR and were cloned into the LentiCRISPRV2 plasmids. Large-scale extraction of the plasmid was performed by using the QIAGEN Plasmid Plus kit. After sequence verification of the gene of interest in the plasmid, a pair of plasmids were cotransfected to the B16-F10 cells by Lipofectamine 2000 according to the manufacturer’s instructions. 48 hours post-transfection, cells were selected for single-cell clones expanded in 96-well plates. Clones were screened at the DNA (Supplementary Fig 1.B), RNA (Supplementary Fig 1.C) and protein level (Supplementary Fig 1.D) to ensure the obtained clones were successfully knocked out. After evaluation, clone P1+P2 which is simultaneously transfected by two plasmids containing sgRNA1 and sgRNA2 was selected for experiments.
Mouse PGRN reconstitution was obtained by transfecting plasmid pLVX-IRES-Puro with mPGRN cDNA insertion. CCL5 was knockdown by infection of lentiviruses pLKO.1-TRC containing two RANTES shRNA sequences (GTGTGTGCCAACCCAGAGA and CTATTTGGAGAT-GAGCTAG). Scramble shRNA (TTCTCCGAACGTGTCACGT) served as control.
2.3. Tumorigenesis studies
For B16-F10 primary tumor growth, 1×106 cells in 100 ul of PBS were injected subcutaneously in right flank of the mice. Tumor sizes were monitored every other day using an electronic caliper. Tumor volume size was calculated by the equation (length×width×hight×0.52). To produce experimental lung metastasis, 5×105 tumor cells were injected intravenously (IV) into the lateral tail vein. Mice were sacrificed two weeks after injection and the lungs were dissected and performed immunological studies.
2.4. Cell proliferation assay
Cell proliferation was evaluated by using MTS assay (Promega) according to the manufacture’s protocol. B16-F10 cells (1×103 cells/well) were plated in 96 well plates and incubated for 24 hours. Then, the medium was replaced with 100 μl of fresh medium, 20 μl of the MTS reagent (CellTiter 96® AQueous One Solution Reagent) was added per each well. the plate was incubated in the 37 °C with 5% CO2 for 30 minutes. The absorbance was detected at 490 nm wave length with a 96-well plate reader.
2.5. Flow cytometry
The single cell suspensions were obtained followed protocol described previously. In brief, the primary tumors were dissociated in PBS with 1 mg/ml collagenase D and 4 μg/ml DNase I for 1 hour in 37 °C with periodic vortexing, and centrifuged at 500 g for 5–10 minutes, followed by removing red blood cells (RBC) by RBC lysis buffer. The cells were first blocked by anti-CD16/32 (101302, BioLegend) for 10 minutes to reduce nonspecific binding and then stained with following antibodies individually: PerCP/Cyanine5.5 anti-mouse CD45 (103131; BioLegend), PE anti-mouse NK-1.1 (108707; BioLegend), APC anti-mouse CD3 (100236; BioLegend), FITC anti-mouse CD69 (104505; BioLegend), FITC anti-human/mouse Granzyme B (515403; BioLegend), and FITC anti-mouse IFN-γ (505805; BioLegend). Zombie Violet™ Fixable Viability Kit (423113; BioLegend) was used to determine cell viability.
2.6. In vivo NK and CD8 T cells depletion
The depletion of NK cells was performed by utilizing an anti-NK1.1 monoclonal antibody, which was provided by Dr. Joseph sun’s lab [28]. 250 μg of anti-NK1.1 (PK136) antibody per mouse was injected intraperitoneal on days −7, 7, 14 post tumor inoculation. The NK cells population were monitored by flow cytometry on day twenty by using NK1.1+ and CD3− antibodies.
To deplete CD8 T cells, 50 μg of anti-CD8a antibody per mouse were injected intraperitoneally on days −1, 3, 7, 11,15 post tumor cells injection. Control mice received equal amounts of IgG2a or PBS. The depletion efficiency was verified by FACS analysis of PBMC following anti-CD8α treatment.
2.7. Wound healing assay
A total of 5×105 WT or Grn−/− B16-F10 cells were seeded in 6-well plates in DMEM supplemented with 10% FBS. Twenty-four hours later, the plates were scratched by a 200 ul pipette tip to generate a wound in cells monolayer. The closure of wound was monitored under an inverted microscope at different time points.
2.8. Invasion transwell assay
The Transwell assay was performed to determine cell migration and invasion as previously described. 8 um pore size chambers with BD Matrigel bedding in the basement membrane were used to determine cell invasion. 2×104 tumor cells were resuspended in 200 ul of serum-free media in the upper chambers and 750 ul of DMEM containing 10% FBS was placed in the lower chamber. After 12 hours of incubation, the filters were washed and fixed with 4% paraformaldehyde and stained with 0.5% crystal violet.
2.9. Soft agar colony formation assay
Each 6-well plate was coated with 1.5 ml of bottom agar (DMEM with/without 20% FBS and 0.5% agarose (CAS No. 9012-36-6)). 5×103 cells (B16-F10 cell lines) were suspended in top agar (DMEM with/without 20% FBS and 0.3% Agarose) in each well. The plate was incubated in 37 °C for 3 weeks. 100 μl of medium was added twice weekly to prevent desiccation of agar. Then the colonies were visualized by staining with 0.3 ml of 0.1% crystal violet solution (3 mL 0.5% crystal violet + 1.5 mL ethanol + 10.5 mL water).
2.10. Western blot
Cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitor. Total protein concentration was quantified by BCA Protein Assay Kit (23227; Thermofisher). Protein (20–40 μg) was resolved on a 10% SDS/PAGE gel and transferred to the PVDF membrane. The membrane was blocked with 3–5% BSA and probed by the primary antibodies against Granulin (ab191211, Abcam), or β-actin (sc-47778, Santa Cruz Biotechnology).
2.11. ELISA
The quantification of mouse CCL5 was performed by utilizing DuoSet ELISA kit (DY478, R&D system) according to the manufacturer’s protocol.
2.12. RNA extraction and qPCR assay
Total RNA was extracted using E.Z.N.A Total RNA Kit (R6812–02; Omega Bio-Tek), and cDNA was synthesized by using the PrimeScript RT reagent kit (TAKARA, RR047A) according to the manufacturer’s protocol. Real-time PCR analysis was carried out on CFX real-time PCR system (Bio-rad). The primer sets used in quantitation are mPGRN (forward 5’-CTGCCCGTTCTCTAA-GGGTG-3’, reverse 5’-ATCCCCACGAACCATCAACC-3’), mCCL5 (forward 5’-GATGGAC-ATAGAGGACACAACT-3’, reverse 5’-TGGGACGGCAGATCTGAGGG-3’).
2.13. Luciferase reporter assay
B16-F10 cells were transiently transfected with the PGRN expression plasmid and luciferase-reporter vectors containing CCL5 promoter region, and then stimulated by 100 unit/ml of IFN-γ for 16 hours followed by luciferase reporter assay by using Dual-Luciferase system (Promega). The activity from Renilla luciferase were used as internal control. The luciferase activities were calculated based on the ratio of firefly to Renilla luciferase activity in transfected samples.
2.14. Bioinformatics analysis
The GRN mRNA expression data in Figure 1A was retrieved from GSE3189 [29] dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE3189). The expression and clinical data of Figure 1B was from The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov). The clinical survival data of melanoma patients was retrieved according to the pipeline of Liu [30]. Kaplan–Meier analysis of percent survival was performed with R version 3.4.4 (https://www.r-project.org/about.html).
Figure 1. High GRN expression correlates with poor survival of melanoma patients.
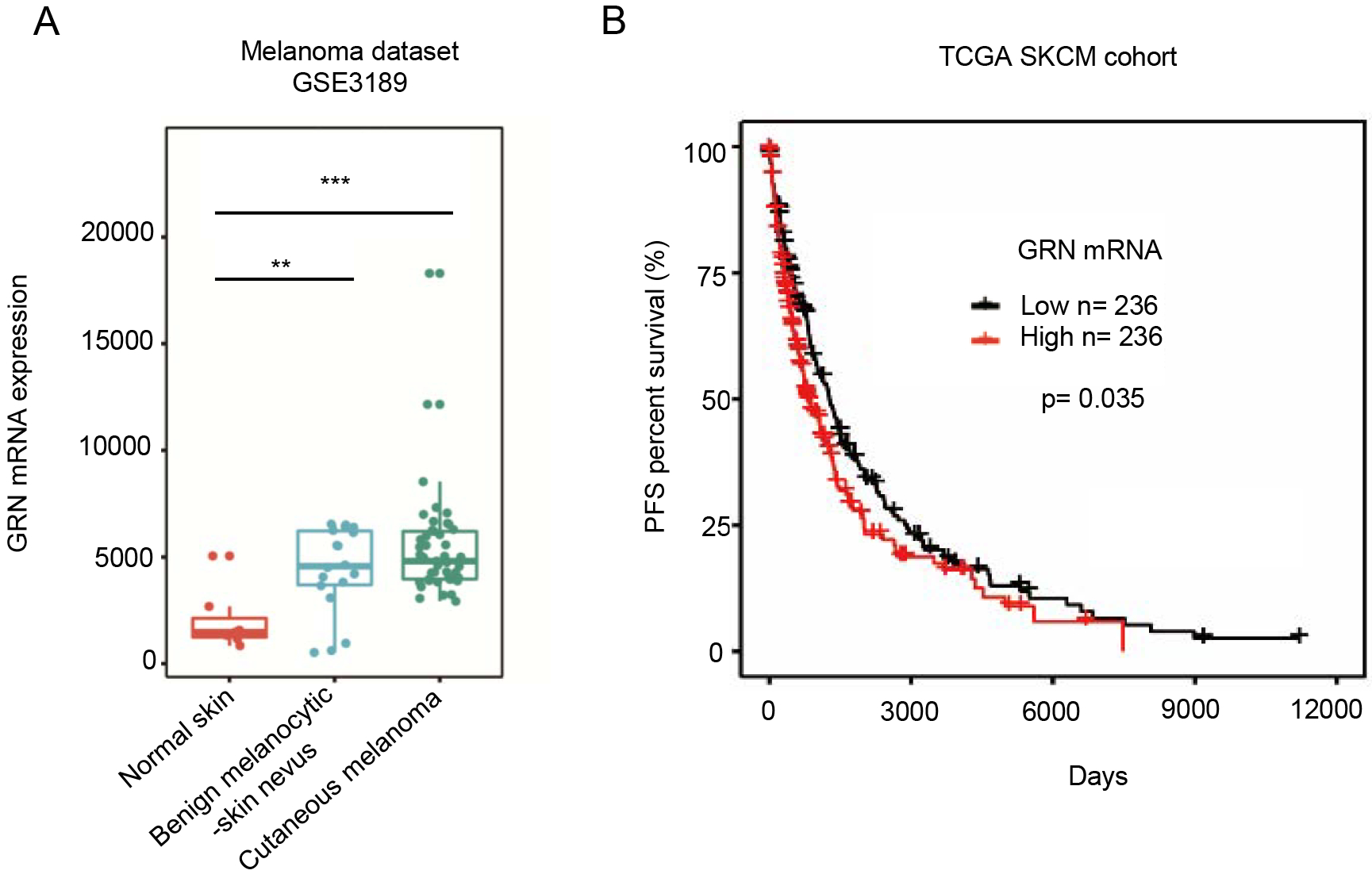
(A) Analysis of GRN mRNA expression across various types of samples based on melanoma dataset GSE3189 from The Cancer Genome Atlas Genomic Common (TCGA-GDC) Data Portal. (B) Kaplan–Meier curve based on TCGA Skin Cutaneous Melanoma (SKCM) dataset showing melanoma patient progression-free survival (PFS) grouped by the median expression of GRN (RNA-seq). (** P < 0.01; *** P < 0.01, two-tailed Student’s t-test).
2.15. Pathway enrichment analysis
The 276 differentially expressed genes between WT and Grn KO B16-F10 cells were subjected to pathway enrichment analysis via Gene Ontology Biological Process of the Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/). The detailed expression profile of the most enriched pathway, immune response, was shown in the heatmap, which was generated with R version 3.4.4 (https://www.r-project.org/about.html).
2.16. Statistics
For data analyzing, a two-tailed Student t-test was used. We considered p-value <0.05 statistically significant and all data were presented as mean ± SEM or SD.
3. Results
3.1. High PGRN expression in human melanoma patients correlates with poor prognosis
To determine the role of PGRN in human melanoma, we used publicly available datasets of melanoma patients in The Cancer Genome Atlas (TCGA) and GEO databases. Bioinformatics analysis with the dataset of GSE3189 revealed that PGRN mRNA was significantly upregulated in malignant melanoma tissues compared with normal tissues (Fig. 1A). Moreover, a Kaplan–Meier analysis based on the TCGA data revealed that high PGRN expression positively correlated with poor survival of melanoma patients in the cohort of cutaneous melanoma (Fig. 1B). According to the GRN expression level, the 472 samples of melanoma patient were allocated into low and high GRN-expressing groups, each group contains 236 samples. The Kaplan-Meier survival plot was grouped by the median expression of GRN in melanoma samples. Taken together, these results illustrate a significant association of PGRN expression with reduced survival in melanoma patients.
3.2. Tumor-derived, not host-derived PGRN regulates melanoma tumor growth and lung metastasis
To determine the role of PGRN in melanoma growth and metastasis, we used the B16-F10 mouse melanoma model. We generated four monoclonal B16-F10 cell lines, sequentially numbered 1 through 4, in which the endogenous Grn gene was deleted via CRISPR/Cas9. The clones exhibited very similar proliferative rates in vitro to the WT B16-F10 cells (Fig. 2A) To further address the question about the cellular source of PGRN important for melanoma tumor progression, i.e., cancer cells, host cells or both, we used PGRN-deficient mice (Grn−/−) and one of the four B16-F10/Grn−/− clones, which were inoculated into the WT and Grn−/− mice. In both WT and Grn−/− mice, B16-F10/Grn−/− tumor cells lost to a significant degree the ability to grow whereas WT B16-F10 tumor cells retained the ability to grow even in Grn−/− mice (Fig. 2B). We then examined the ability of B16-F10/Grn−/− cells to metastasize to the lungs via intravenous injection and metastasis. The result shows that B16-F10/Grn−/− cells were highly defective in lung metastasis in both WT and Grn−/− mice (Fig. 2C). These data indicate that tumor-derived, not host-derived PGRN, plays a dominant role in B16-F10 progression, particularly in metastasis.
Figure 2. Tumor-derived PGRN regulates melanoma tumor growth and lung metastasis.
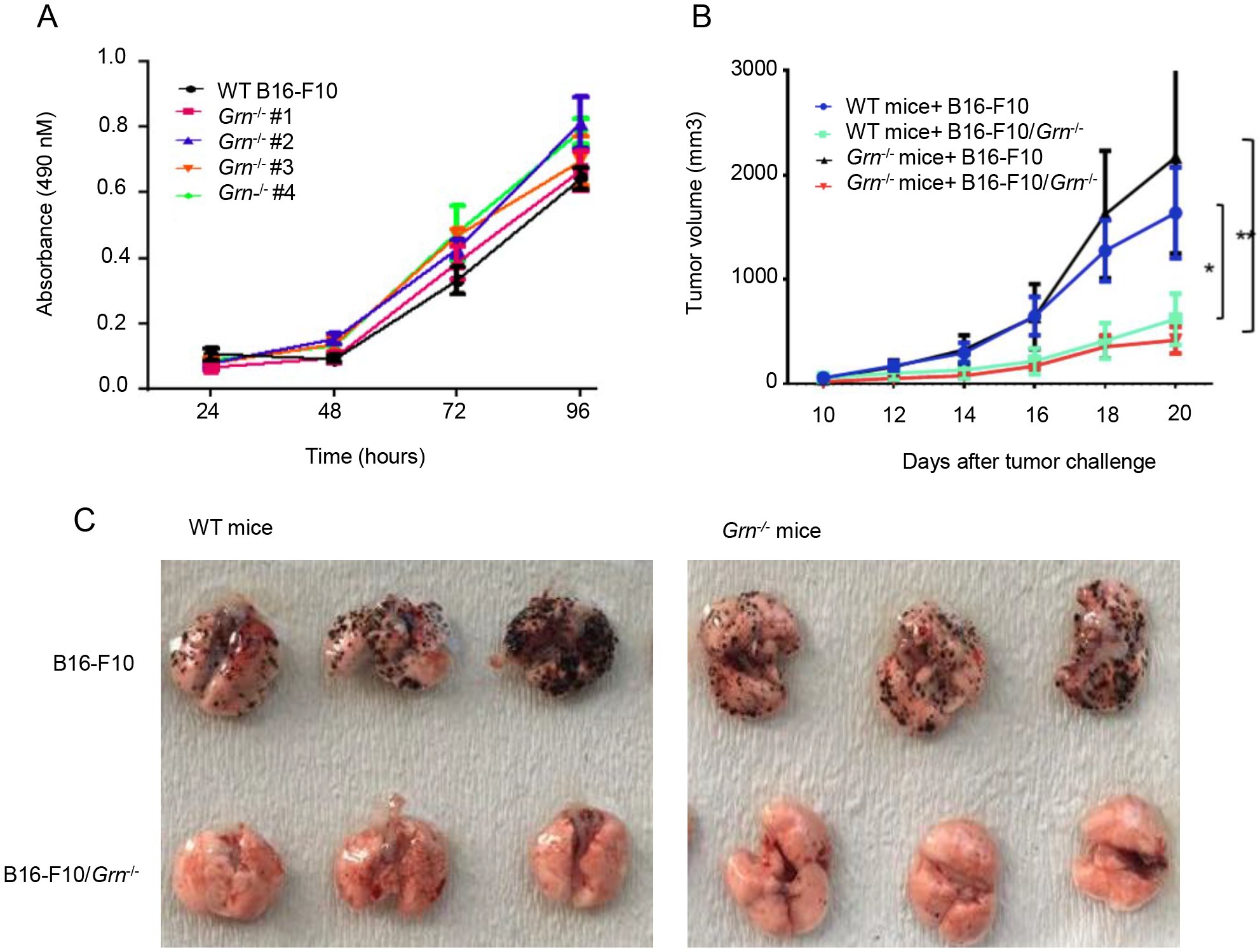
(A) Proliferation of B16-F10 and knocked out clones was measured by MTS assay. Error bars represent standard deviations (n=3). (B) 1×106 WT or Grn−/− B16-F10 tumor cells were injected subcutaneously to the WT or GRN KO mice (n=3) individually. Tumor growth was monitored with caliper every four days with a caliper and the tumor volume was shown as average ± SEM (*P < 0.05, ** P < 0.01, two-tailed Student’s t test). (C) 5×105 WT or Grn−/− B16-F10 tumor cells were injected intravenously through tail vein to the WT or GRN KO mice (n=3 with 3 repeats) individually. After 12 days, mice were sacrificed and lung tissues were dissected. Representative images of whole lung with metastatic tumor nodules from 3 mice each group were shown.
3.3. Reconstitution of PGRN expression restores tumor growth and metastasis
To rule out the off-target effect of the CRISPER/Cas9 method used to edit the Grn gene in B16-F10 melanoma, we reconstituted PGRN expression in B16-F10/Grn−/− cells to the WT level via a lentivirus-mediated expression vector. Real-time PCR (Fig. 3A) and Western blot (Fig. 3B) analyses confirmed the successful reconstitution of mPGRN expression. Upon subcutaneous inoculation of the PGRN reconstituted-PGRN B16-F10/Grn−/− cells in mice, we observed that their growth was completely restored to the WT level (Fig. 3C–D). By i.v. injection of the reconstituted tumor cells, a complete rescue of the severely impaired lung metastasis of B16-F10/Grn−/− cells was also ascertained (Fig. 3E). Together, these data validate the growth-and metastasis-promoting effects of tumor-derived PGRN.
Figure 3. Tumor growth and metastasis of Grn−/− B16-F10 tumor cells was rescued after mPGRN reconstitution.
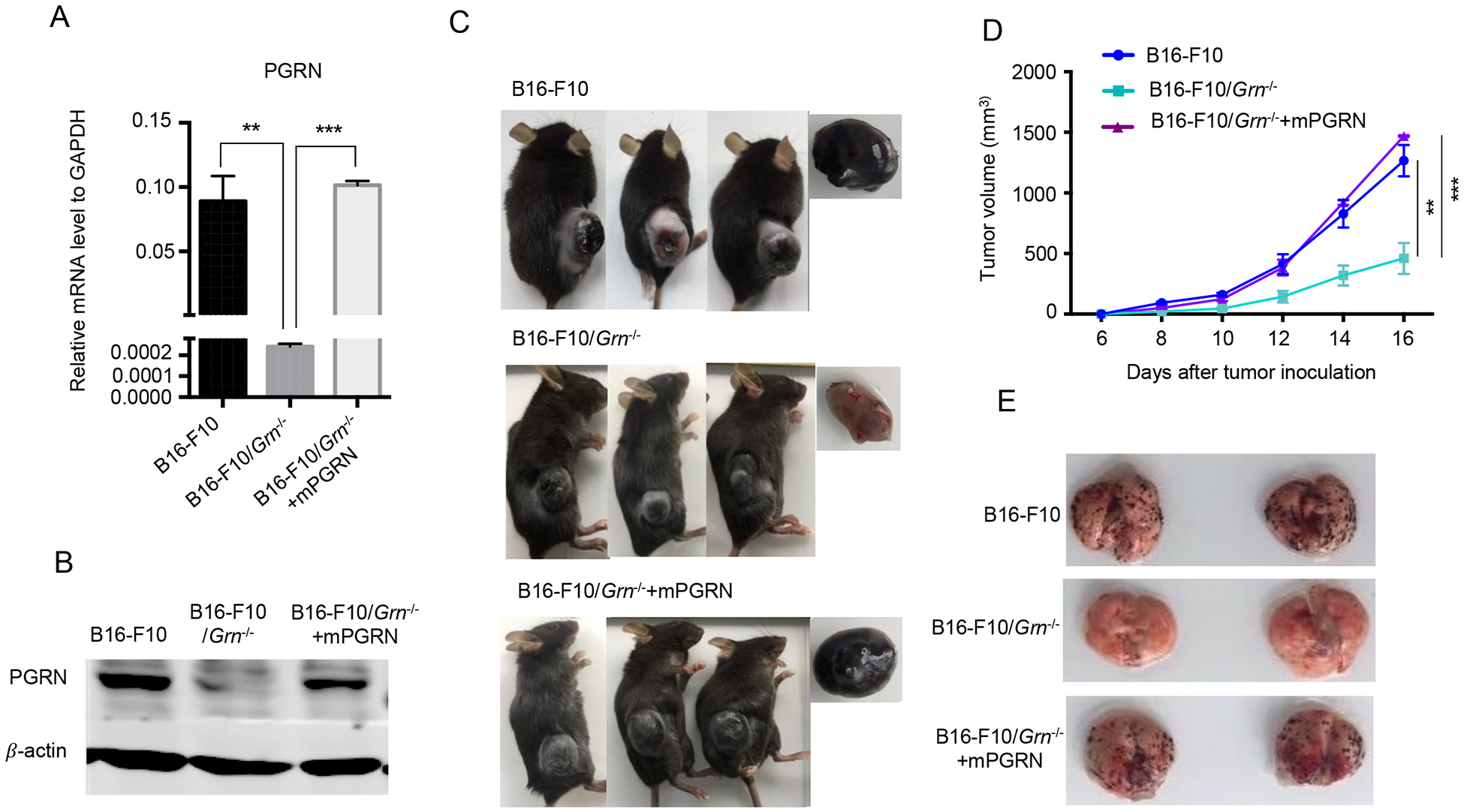
Grn−/− B16-F10 cells were transfected by plasmid encoded mPGRN mRNA, and stable cell lines B16-F10/Grn−/− mPGRN were generated based on puromycin selection. The mRNA (A) and protein (B) levels of PGRN in B16-F10/Grn−/− mPGRN is equivalent the one in WT B16-F10 cells. Quantitative PCR experiments were performed in triplicate and repeated three times. Results were presented as mean ± SEM. (**P < 0.01, *** P < 0.001, two-tailed Student’s t test). (C) 1×106 WT, or Grn−/−, or Grn−/−mPGRN B16-F10 cells were injected subcutaneously in the right flank subcutaneously to C57BL/6 mice (n=3 per group). Representative images of tumor dissected after 16 days are shown in (C) right side. (D) Tumor size was measured at the indicated times. The experiment was repeated two times. Data, presented as mean ± SEM, are representative of the two independent experiments. (**P < 0.01, *** P < 0.001, two-tailed student’s t test). (E) 5×105 WT, or Grn−/−, or Grn−/−mPGRN B16-F10 cells were injected intravenously into the tail vein (n=4 per group) to 6–8 weeks old female C57BL/6 mice. After 12 days, the mice were sacrificed, and lung tissues were dissected. Representative images of whole lungs with metastatic tumor nodules from 2 mice each group are shown.
3.4. PGRN deficiency results in reduced B16-F10 motility
Recently PGRN has been shown to promote tumor cell migration in multiple models such as bladder [31], ovarian [19], and breast cancers [21]. To determine the effect of PGRN on B16-F10 cell migration, we performed wound healing and transwell invasion assays in vitro. The results show that both migratory and invasive abilities of B16-F10/Grn−/− cells were compromised compared to WT B16-F10 cells (Fig. 4A and 4B, respectively). Besides, we evaluated anchorage-independent growth by assaying the colony-forming ability of B16-F10 cells in soft agarose to examine the effect of PGRN on tumorigenicity of B16-F10 melanoma cells. The result showed, B16-F10/Grn−/− almost completely abolished anchorage-independent growth in which the size and the number of colonies were reduced in comparison with control group (Fig. 4C). These data demonstrate that tumor-derived PGRN has an intrinsic ability to influence migration and invasion, which underlies its metastasis-promoting activities in vivo.
Figure 4. PGRN deficiency reduced tumors’ migratory and metastatic capacities.
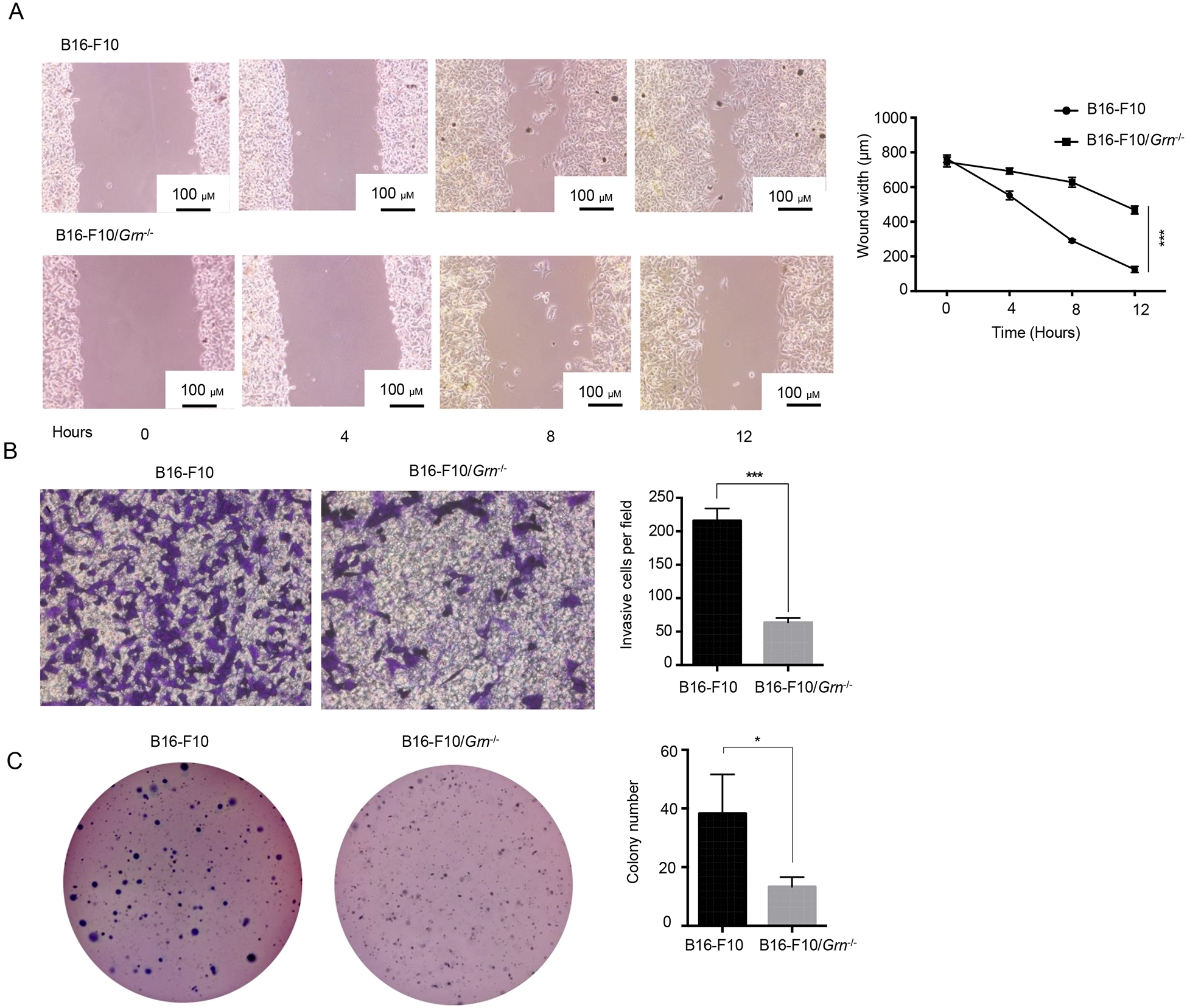
(A) Scratch wound-healing assay of WT and Grn−/− B16-F10 cells. The distance was measured at indicated time points. Data from triplicate experiments were presented as mean ± SD. (*** P < 0.001, two-tailed Student’s t test). (B) Transwell invasion assay of WT and Grn−/− B16-F10 cells. The experiment was performed in triplicate for each group. (C) Grn deletion decreases anchorage-independent growth of B16-F10 cells. WT and Grn−/− B16-F10 cells were subjected to soft-agar colony formation assay. The colonies were stained by crystal violet and counted by using ImageJ software. Data from triplicate experiments were presented as mean ± SD. (*P < 0.05, *** P < 0.001, two-tailed Student’s t test).
3.5. PGRN deficiency causes immunological changes in the tumor microenvironment
We sought to determine the role of immune cells in PGRN-regulated B16-F10 growth by analyzing the infiltrating cells in the tumor microenvironment (TME) by flow cytometry. The results show that several major populations of CD45+ infiltrating cells increased to varying degrees in B16-F10/Grn−/− tumors: CD3−, NK1.1+ cells (Fig. 5A), CD11b+, F4/80+ macrophages (Fig. 5B) and CD11c+ MHC II+ dendritic cells (Fig. 5C), whereas CD4+ T cells (Fig. 5D), CD8+ T cells (Fig. 5E), and Gr1+ cells (further separated into Ly6C+ and Ly6G+ subpopulations) (Supplementary Fig 2. A), were not altered, nor were NK cells, macrophages, CD8+, and CD4+ T cells in the spleen (Supplementary Fig 2. B, C, D, E, respectively). Consistent with an activated state of NK cells, MHC I expression was upregulated on the surface of B16-F10/Grn−/− tumors (Fig. 5F). The tumor sections were analyzed by immunohistochemistry, which confirmed that NK cells were more robustly infiltrated into the B16-F10/Grn−/−, compared with B16-F10 tumors (Fig. 5G). These data imply that NK cells present in TME may play an important role in PGRN-deficiency-caused tumor reduction.
Figure 5. Differential involvement of the immune system in Grn−/− B16-F10 tumor.
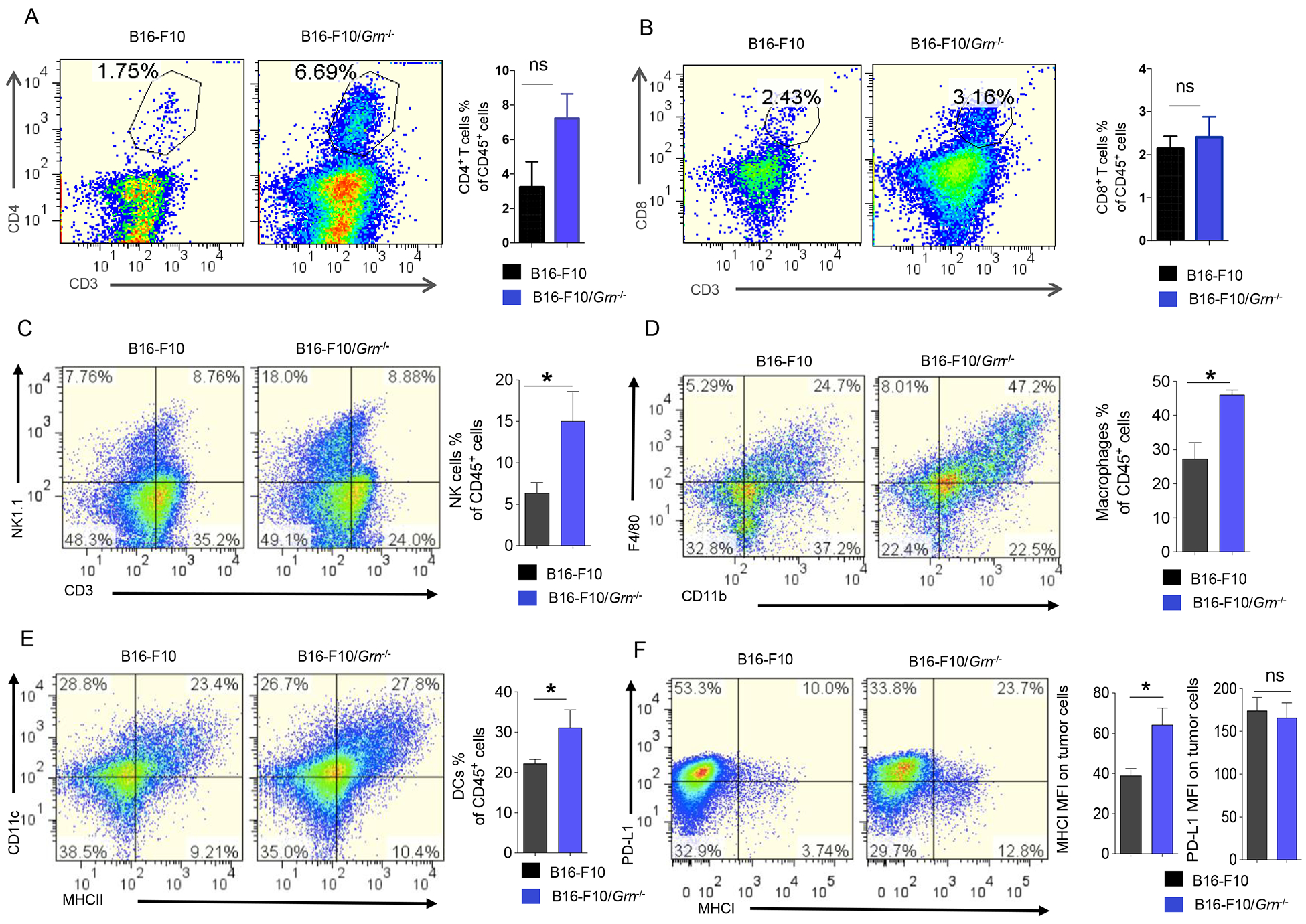
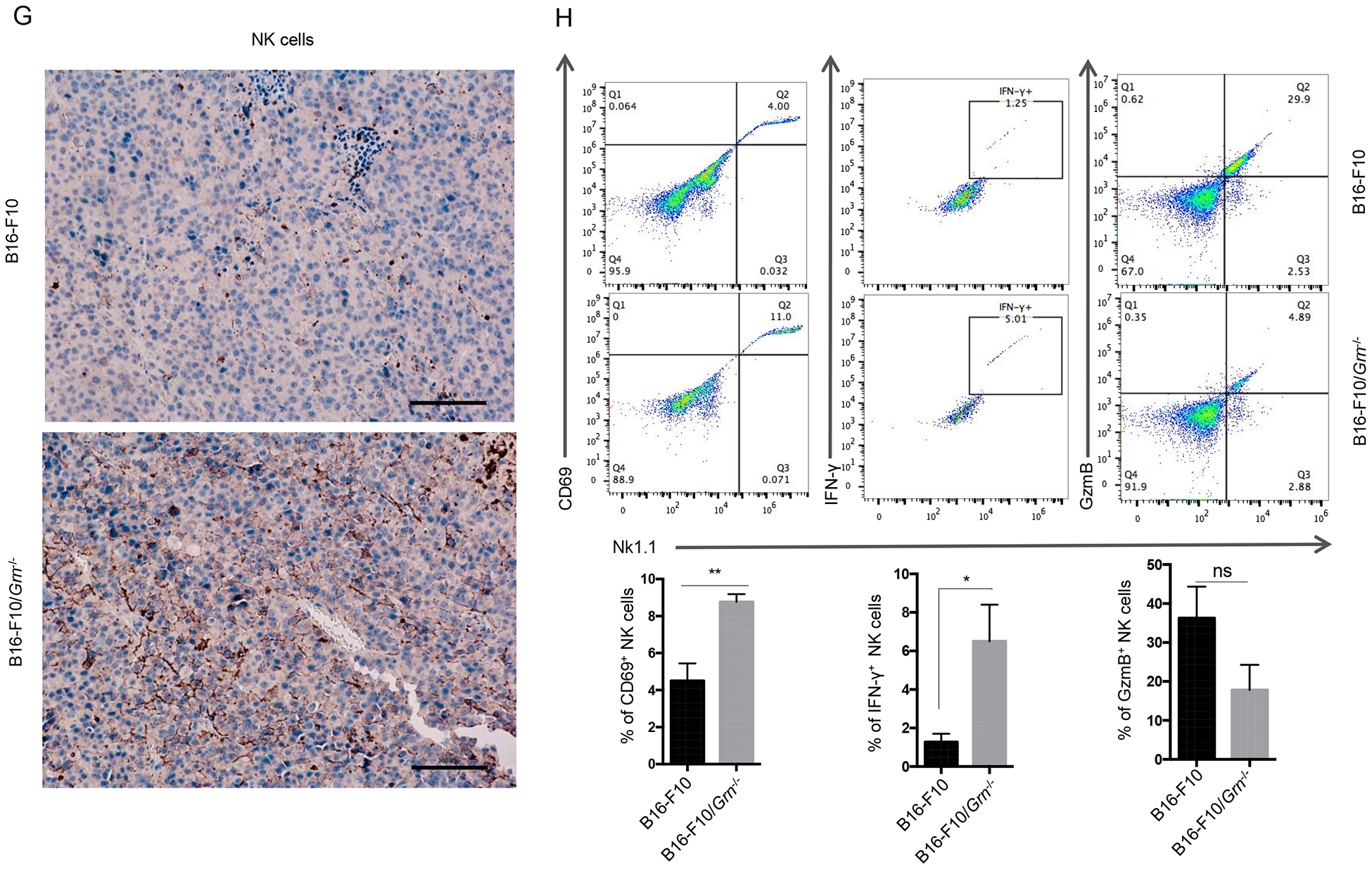
1×106 WT, or Grn−/−B16-F10 cells were injected subcutaneously in the right flank to C57BL/6 mice.18 days later, primary tumors were dissected. Single cell suspensions were obtained from these tissues and stained for (A) CD3− NK1.1+ cells, (B) CD11b+ F4/80+ macrophages, (C) CD11c+ MHC II+ dendritic cells, (D) CD4+ T cells, (E) CD8+ T cells. The percentage of each type cells were calculated based on total CD45+ cells. (F) MHC I and PD-L1 expression on the B16-F10/Grn−/− tumors were analyzed by FCM. Data were presented as mean ± SD (* P < 0.05, two-tailed Student’s t test). n=3 mice per group. (G) Immunohistochemical staining for NK cells in ctrl and B16-F10/Grn−/− tumor section by using anti-NK1.1 antibody (Scale bar, 100 μm). (H) Live cells were gated and the gate including CD45+ cells were analyzed for NK1.1, CD69, IFN-γ, and GzmB. Quantification of active NK cells (NK1.1+CD69+, NK1.1+IFN-γ+, NK1.1+GzmB+) infiltrating to the tumor was reported as a percentage of total NK cells. Data were presented as mean ± SEM (* P < 0.05, *** P < 0.001, two-tailed Student’s t test). n=3 mice per group.
To investigate the effect of PGRN deficiency on cytolytic activity of NK cells, we analyzed CD69 expression as an early activation marker as well as intracellular IFN-γ and Granzyme B (GzmB) levels in tumor infiltrating NK cells from mice bearing B16-F10 and B16-F10/Grn−/− tumors. The result shows that PGRN deficiency caused increased CD69 and IFN-γ levels in NK cells. However, no significant difference was observed for GzmB expression between the ctrl and Grn−/− group (Fig. 5H). Overall, we conclude that PGRN-deficiency-associated tumor reduction is due to enhanced NK cell infiltration and activation in the TME.
3.6. NK but not T cells are crucial in controlling PGRN-regulated B16-F10 growth
To determine if NK cells were required for the B16-F10 tumor growth inhibition relevant to PGRN, they were depleted in vivo using an anti-NK1.1 monoclonal antibody. The depletion efficiency was greater than 50% (Fig. 6A, B), and it resulted in a complete restoration of tumor growth in mice carrying the Grn−/− tumor to and over the WT level (Fig. 6C). In contrast, antibody-mediated depletion of CD8+ T cells had very little impact on WT and B16-F10/Grn−/− tumor growth in vivo (Fig. 6D). Taken together these results demonstrate that NK, but not T cells, are the major immune effectors that control B16-F10 tumor growth and that PGRN intrinsically affects NK infiltration into TME.
Figure 6. NK cells are required to control PGRN-regulated B16-F10 growth.

(A-B) NK cells were depleted in B16-F10 and B16-F10/Grn−/− tumor-bearing mice by using anti-NK1.1 monoclonal antibody. The NK cell depletion in tumor was confirmed by NK1.1+ CD3− staining gated on CD45+ cells. (C) Tumor volume was measured every two days starting day 10. n=5 mice per group. Data are presented as mean ± SD (* P < 0.05, **P < 0.01, and *** P < 0.001 by Student t test). (D) B16-F10 and Grn−/− cells were injected subcutaneously and treated with 50 μg of anti-CD8α antibody. Tumor growth were measured from day 10 onward. Data are presented as mean ± SD (**P < 0.01 by Student t test).
3.7. PGRN inhibits CCL5 expression NK cell recruitment
To understand how tumor-derived PGRN intrinsically affect NK recruitment to TME, we performed RNA-seq analysis of primary B16-F10 and B16-F10/Grn−/− tumors isolated from mice. Bioinformatic analysis of the data shows that 18 pathways were enriched in B16-F10/Grn−/− vs WT tumors. The most differently expressed genes were involved in immune response pathways (Fig. 7A), and a heat map presents 22 genes that were significantly upregulated in the B16-F10/Grn−/− group in two independent tumors (Fig. 7B). Notably, numerous chemokines including CCL5 were among the upregulated genes in this group. High CCL5 expression was associated with recruitment of activated T cells, NK cells and M1 macrophages in human triple negative breast cancer [32] and in experimental tumor models [33]. CCL5 is instrumental in NK-mediated liver injury [34]. Therefore, we focused our attention on CCL5. To determine the role of CCL5 in NK cell infiltration to the tumor and in controlling tumor growth, we “knocked down” CCL5 expression in B16-F10/Grn−/− cells stably by using Lentivirus vectors delivering shRNAs (Grn−/−Ccl5KD). The efficiency of CCL5 expression silencing was verified by ELISA (Fig. 7C) and the effect of silencing CCL5 expression on NK cell recruitment to TME was confirmed by flow cytometric analysis (Fig. 7D). As a result of this manipulation, B16-F10/Grn−/−Ccl5KD tumor fully regained its growth capacity in vivo (Fig. 7E). These data demonstrate that CCL5 produced by B16-F10/Grn−/− tumor is crucial for NK recruitment and control of B16-F10 tumor growth.
Figure 7. CCL5 overexpression pivotally affects the B16-F10/Grn−/− tumor growth.
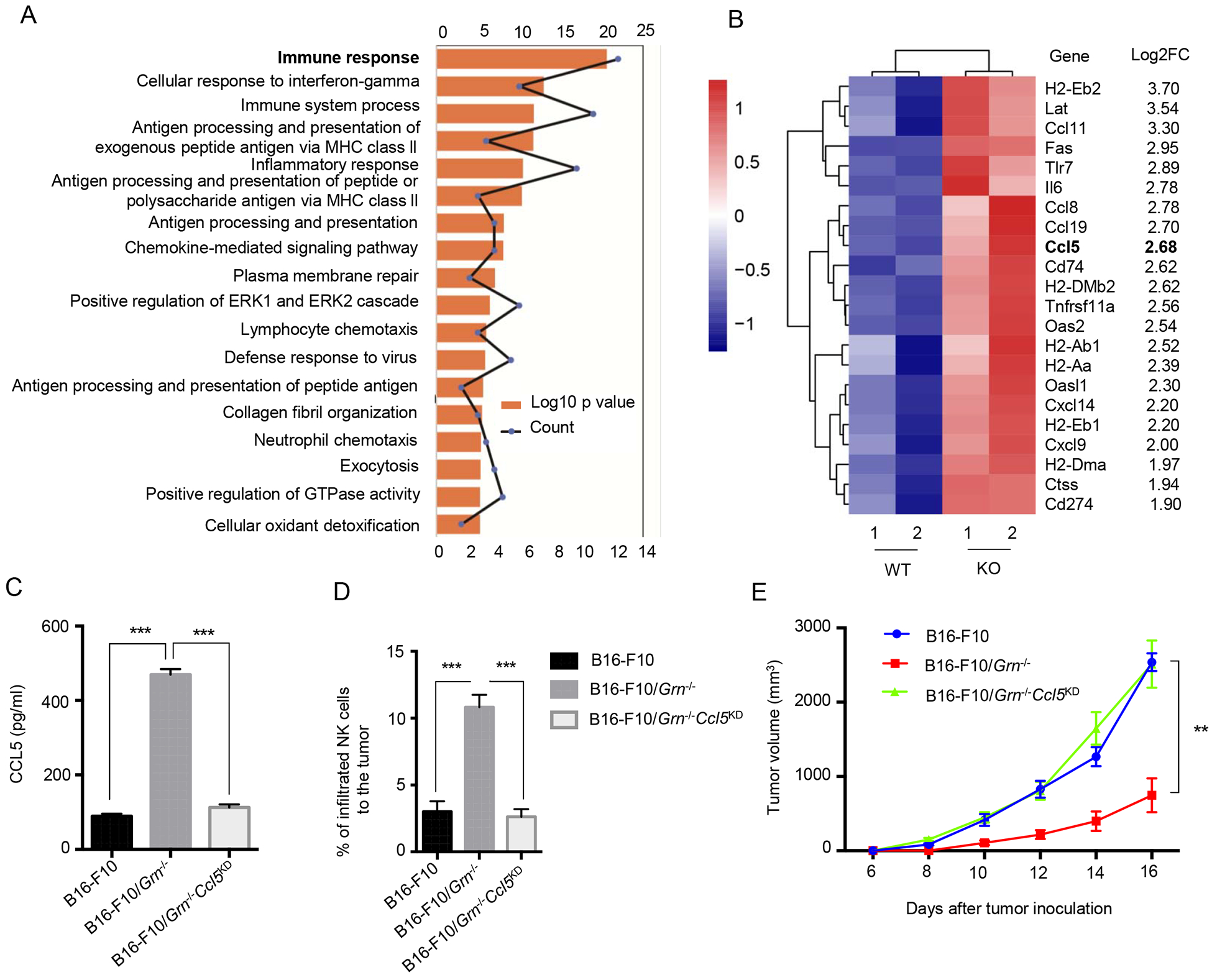
(A) RNAseq was performed for WT, or Grn−/− tumor dissected from mice and the data were analyzed by using DAVID (Database for Annotation, Visualization, and Integrated Discovery) for 276 genes. The top 18 enriched terms are presented as bar graphs. (B) The heat map shows genes with ≥ 4-fold increasing in B16-F10/Grn−/− RNA-Seq data compared to WT tumor. (C) 24 hours after cell seeding, CCL5 secretion into the supernatant of the cells was measured in B16-F10, B16-F10/Grn−/− and B16-F10/Grn−/−Ccl5KD mice by ELISA. Data are presented as the average ± SEM. *** P-value < 0.001 by Student t test. (D) B16-F10, B16-F10/Grn−/− and B16-F10/Grn−/−Ccl5KD tumors cells were injected to the 6–8 weeks old C57BL/6 mice subcutaneously. At day 18, tumors were dissected and infiltrating NK cells were measured by flow cytometry. n=4 per each group. Data were presented as mean ± SEM (*** P < 0.001 by Student t test). (E) Tumor growth was measured after B16-F10, B16-F10/Grn−/− and B16-F10/Grn−/−Ccl5KD cells (1×106 cells/mouse) injection subcutaneously. Data are presented as mean ± SEM. ** P < 0.01 (two-tailed student’s t test). n=4 per each group.
3.8. PGRN inhibits Ccl5 transcription in tumor cells
The fact that B16-F10/Grn−/− tumor produced elevated CCL5 suggests that PGRN is an inhibitor of CCL5 expression. This notion was confirmed by reconstitution of B16-F10/Grn−/− tumor with mPGRN, which inhibited CCL5 expression at mRNA and protein levels with or without IFN-γ stimulation (Fig. 8A, B).
Figure 8. PGRN inhibits Ccl5 transcription in tumor cells.
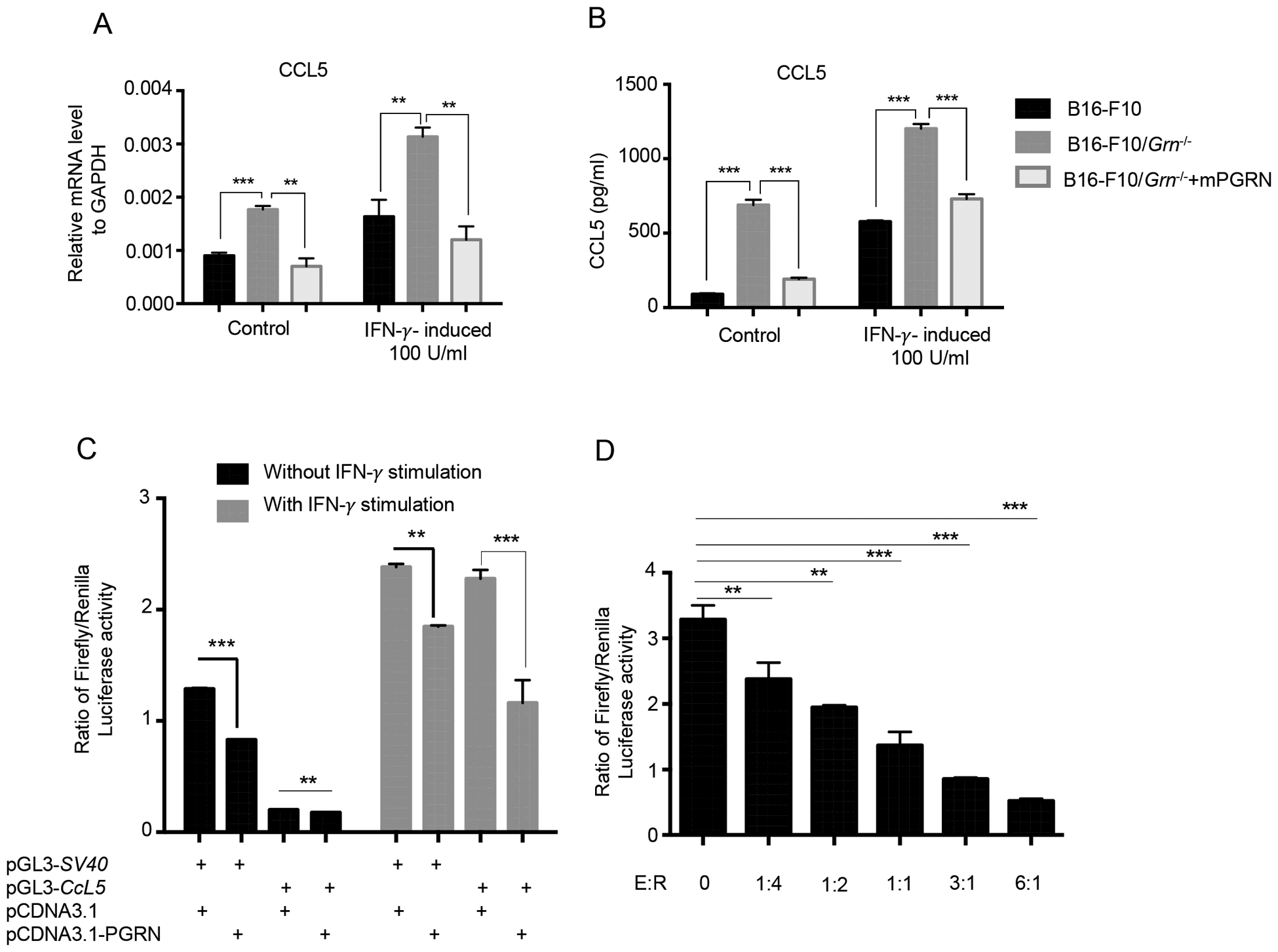
(A) Quantitative PCR measuring mRNA level of CCl5 in B16-F10 cells after stimulation with 100 U/ml IFN-γ. Data (means ± SEM) are representative of two independent experiments with triplicate each group. (* P < 0.05, **P < 0.01, *** P < 0.001 by Student t test). (B) CCL5 protein level was measured by ELISA in the supernatant of WT, or Grn−/−, or Grn−/− mPGRN B16-F10 cells after stimulation with 100 U/ml IFN-γ. Data presented as means ± SEM (* P < 0.05, **P < 0.01, *** P < 0.001 by Student t test). (C) B16-F10 cells were cotransfected with mouse Ccl5 Promoter-Luc and PGRN overexpressing, followed with or without IFN-γ stimulation for 16 h. Luciferase activity was measured from cell lysates by dual luciferase assay. Results are presented as mean ± SEM of three individual experiments. **P-value < 0.01, ***P-value < 0.001 (D) 1 μg of pGL3-CCL5-Luc was cotransfected with increasing concentrations of pCDNA3.1-PGRN. Data were presented as mean ± SEM. ** < 0.01; *** P < 0.001 by student t test.
To further explore the molecular mechanism whereby PGRN inhibits CCL5 expression, we cloned the murine Ccl5 promoter region (−979, +8) upstream of a firefly luciferase reporter gene on the backbone of pGL3. The resulting reporter, pGL3-Ccl5, was transiently cotransfected with pCDNA3.1 expressing mPGRN (effector) into B16-F10 cells. The transfected cells were then stimulated with or without IFN-γ for 16 hours. The cells were lysed and luciferase activity was measured by a luminometer. The result shows that PGRN inhibited Ccl5 promoter-driven luciferase transcriptional activity (Fig. 8C). The inhibitory effect of PGRN on Ccl5 promoter was more pronounced when the cells were stimulated by IFN-γ. The inhibition appeared to be selectively on the Ccl5 promoter as the pGL3-SV40 promoter-driven reporter activity was not significantly inhibited in IFN-γ stimulated cells. In addition, the PGRN-mediated inhibition was dose-dependent (Fig. 8D). These data demonstrate that PGRN inhibits CCL5 expression at the transcriptional level.
4. Discussion
PGRN as a growth factor is critically involved in a wide variety of physiological and pathological processes. Overall, many studies have demonstrated an anti-inflammatory role of PGRN in various pathological situations, as our previous work revealed that PGRN deficiency exacerbated immunological pathogenesis induced during sepsis and endotoxemia by uncontrolled induction of pro-inflammatory cytokines simultaneously by a reduction in IL-10 secretion [2]. PGRN’s activities in cancer are well-established, and numerous studies have pointed to its importance in cell proliferation [10, 35, 36], migration, invasion [20, 21], and angiogenesis [13]. In this study, we show that PGRN expression is associated with early and late stages of melanoma development and poor prognosis in human patients, suggesting that PGRN may be a disease marker for this type of cancer. Indeed, it has been suggested that PGRN concentrations in serum, urine or its expression in tissues can be useful for monitoring the clinical course of tumors and patient prognosis. PGRN expression in breast cancer tumor samples was found to be significantly correlated with tumor size, lymph node metastasis and angiogenesis [37]. Similar observations have been made in ovarian cancer [38, 39], prostate cancer [40], kidney cancer [41], chronic lymphocytic leukemia [42], non-small cell lung carcinoma [43], and malignant lymphoma [44]. More extensive studies are needed to further explore the potential of and authenticate PGRN as a disease marker in melanoma.
It is noted with interest that PGRN levels are high in both benign melanocytic nevi and malignant melanomas, suggesting that PGRN may be required for tumor growth but it is unlikely a driver of melanocytic malignant development per se. The ultimate proof will involve more definitive approaches such as overexpressing PGRN in normal melanocytes to see if it will drive the malignant transformation.
The impact of PGRN on tumor growth has been widely studied [12]. Here we show by editing the Grn gene and silencing its expression in B16-F10 tumor, the cellular motility and invasiveness decreased in vitro and tumor metastasis was impaired in mice. Whether the cell-intrinsic property of PGRN on cellular mobility can account completely for the B16-F10 tumor’s lung metastasis without the involvement of extrinsic factors in TME remains to be formally established. We also demonstrate that tumor-derived PGRN promotes B16-F10 tumor growth and PGRN deficiency enhances the production of CCL5 by tumor cells, which helps recruit immune cells, particularly NK cells, to TME to control tumor growth. NK cells have strong cytolytic activity against microbially infected cells and cancer cells [45]. Improving the infiltration of cytotoxic immune cells such as NK cells to the tumor bed can enhance the therapeutic effects of NK cell-based tumor immunotherapies [46]. Recently, the importance of NK-based immunotherapy in cancers has been extensively discussed. This process in the clinic has some limitations beside its advantage, for instance, NK Cell-based immunotherapy might be dysfunctional in solid tumors, but clinical trial studies have provided credible evidence that this strategy is beneficial for malignant melanoma therapy [47, 48].
CCL5, also known as RANTES, is one of the CC chemokine family members that are expressed in a variety of cells such as T lymphocytes, platelet, macrophages, synovial fibroblasts, tubular epithelium and specific type of tumor cells [49, 50]. CCL5 as a ligand can interact with three receptors: CCR1, CCR3, and CCR5. CCL5 induces chemotaxis in the inflammatory site by recruiting immune cells. In tumors, CCL5 may work as a double-edged sword. In some studies, it has been shown that tumor-derived CCL5 upregulates matrix metalloproteinase (MMP) expression that facilitates tumor invasion and metastasis by degrading the extracellular matrix [51]. Moreover, tumor-derived CCL5 can inhibit T cell responses and induce mammary primary tumor growth in mice [52, 53]. In contrast to the above studies there is some evidence that expression of CCL5 intratumorally induces anti-tumor immunity by affecting tumor-specific and non-specific immune cells, such as DCs, CD4+ Th1 cells, CD8+ T cells, and NK cells and their recruitment to TME [54]. Mrowietz et al. reported that a subset of human melanoma cells express mRNA and secrete CCL5 protein which may be partly responsible for the recruitment of monocytes, T-cells and DCs into the tumors. However, higher levels of CCL5 secretion by six human melanoma cell lines were associated with increased tumor formation upon s.c. injection in nude mice [55]. The apparent “paradox” of this finding with our observation in this study may be squared off by the fact the experiment in question was performed in nude mice in which many types of immune cells are either absent or functionally defective.
There is ample evidence for a critical role of CCL5 in NK cell recruitment to inflamed tissues [56] but its role in NK cell recruitment to TME has not been extensively studied. Our finding is consistent with the recent report of Mgrditchian et al. [46] that tumor-derived CCL5 is a significant factor in recruiting NK cells to TME. The RNA-seq data presented in Fig 7 revealed several chemokines including CCL5 were strongly upregulated in PGRN-deficient B16-F10 tumor. It would be of interest to explore the involvement of other chemokines in immune cell mobilization and tumor control.
After secretion from the cytoplasm, PGRN may translocate to the nucleus and effect on select transcriptional machineries. Hoque et al. reported that PGRN could bind the histidine-rich region of cyclin T1 and inhibits the function of the cellular positive transcription elongation factor b (P-TEFb) in the nucleus [57, 58]. In this study, we identified for the first time PGRN regulates Ccl5 gene transcription through which PGRN can inhibit on-going inflammatory cell mobilization and infiltration. Currently, it remains to be determined if the transcriptional inhibition of Ccl5 by PGRN is direct or indirect. Furthermore, it will be of interest to explore the impact of PGRN on major transcription factors such as NF-κB that drive CCL5 expression during inflammatory responses.
In summary, this study establishes a novel and substantial role for PGRN in melanoma growth and metastasis and suggests that it may represent a tangible therapeutic target for melanoma therapy, in addition to the prospect that PGRN may be a new marker for diagnosis of melanoma at an early stage.
Supplementary Material
Highlights.
Progranulin (PGRN) expression is associated with human melanoma progression.
PGRN promotes B16 experimental melanoma growth and metastasis.
NK cells are crucial in controlling PGRN-facilitated B16 growth.
Tumor-derived CCL5 is responsible for the recruitment of activated NK cells to the microenvironment.
PGRN inhibits CCL5 gene transcription.
Acknowledgements
This work was supported by grants from Natural Science Foundation of China to X.M. (31670913, 81872353).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest Statement
All authors declare no conflict of interests.
References
- [1].He Z, Bateman A, Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis, J Mol Med, 81 (2003) 600–612. [DOI] [PubMed] [Google Scholar]
- [2].Yan W, Ding A, Kim H-J, Zheng H, Wei F, Ma X, Progranulin controls sepsis via C/EBPα-regulated Il10 transcription and ubiquitin ligase/proteasome-mediated protein degradation, The Journal of Immunology, 197 (2016) 3393–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair, Cell, 111 (2002) 867–878. [DOI] [PubMed] [Google Scholar]
- [4].Kessenbrock K, Fröhlich L, Sixt M, Lämmermann T, Pfister H, Bateman A, Belaaouaj A, Ring J, Ollert M, Fässler R, Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin, The Journal of clinical investigation, 118 (2008) 2438–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xu D, Suenaga N, Edelmann MJ, Fridman R, Muschel RJ, Kessler BM, Novel MMP-9 substrates in cancer cells revealed by a label-free quantitative proteomics approach, Molecular & Cellular Proteomics, 7 (2008) 2215–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Suh H-S, Choi N, Tarassishin L, Lee SC, Regulation of progranulin expression in human microglia and proteolysis of progranulin by matrix metalloproteinase-12 (MMP-12), PloS one, 7 (2012) e35115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Butler GS, Dean RA, Tam EM, Overall CM, Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding, Molecular and cellular biology, 28 (2008) 4896–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bai X-H, Wang D-W, Kong L, Zhang Y, Luan Y, Kobayashi T, Kronenberg HM, Yu X-P, C.-j. Liu, ADAMTS-7, a direct target of PTHrP, adversely regulates endochondral bone growth by associating with and inactivating GEP growth factor, Molecular and cellular biology, 29 (2009) 4201–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jian J, Konopka J, Liu C, Insights into the role of progranulin in immunity, infection, and inflammation, Journal of leukocyte biology, 93 (2013) 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Feng T, Zheng L, Liu F, Xu X, Mao S, Wang X, Liu J, Lu Y, Zhao W, Yu X, Growth factor progranulin promotes tumorigenesis of cervical cancer via PI3K/Akt/mTOR signaling pathway, Oncotarget, 7 (2016) 58381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].He Z, Bateman A, Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis, Journal of molecular medicine, 81 (2003) 600–612. [DOI] [PubMed] [Google Scholar]
- [12].He Z, Bateman A, Progranulin gene expression regulates epithelial cell growth and promotes tumor growth in vivo, Cancer research, 59 (1999) 3222–3229. [PubMed] [Google Scholar]
- [13].Eguchi R, Nakano T, Wakabayashi I, Progranulin and granulin-like protein as novel VEGF-independent angiogenic factors derived from human mesothelioma cells, Oncogene, 36 (2017) 714. [DOI] [PubMed] [Google Scholar]
- [14].Yang D, Wang L-L, Dong T-T, Shen Y-H, Guo X-S, Liu C-Y, Liu J, Zhang P, Li J, Sun Y-P, Progranulin promotes colorectal cancer proliferation and angiogenesis through TNFR2/Akt and ERK signaling pathways, American journal of cancer research, 5 (2015) 3085. [PMC free article] [PubMed] [Google Scholar]
- [15].Chen X.-y., Li J.-s., Liang Q.-p., He D.-z., Jing Z, Expression of PC cell-derived growth factor and vascular endothelial growth factor in esophageal squamous cell carcinoma and their clinicopathologic significance, Chinese medical journal, 121 (2008) 881–886. [PubMed] [Google Scholar]
- [16].Li L.-q., Huang H.-l., Ping J.-l., Wang X.-h., Zhong J, Dai L.-c., Clinicopathologic and prognostic implications of progranulin in breast carcinoma, Chinese medical journal, 124 (2011) 2045–2050. [PubMed] [Google Scholar]
- [17].Zanocco-Marani T, Bateman A, Romano G, Valentinis B, He Z-H, Baserga R, Biological activities and signaling pathways of the granulin/epithelin precursor, Cancer Research, 59 (1999) 5331–5340. [PubMed] [Google Scholar]
- [18].Lu R, Serrero G, Mediation of estrogen mitogenic effect in human breast cancer MCF-7 cells by PC-cell-derived growth factor (PCDGF/granulin precursor), Proceedings of the National Academy of Sciences, 98 (2001) 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dong T, Yang D, Li R, Zhang L, Zhao H, Shen Y, Zhang X, Kong B, Wang L, PGRN promotes migration and invasion of epithelial ovarian cancer cells through an epithelial mesenchymal transition program and the activation of cancer associated fibroblasts, Experimental and molecular pathology, 100 (2016) 17–25. [DOI] [PubMed] [Google Scholar]
- [20].Liu Y, Xi L, Liao G, Wang W, Tian X, Wang B, Chen G, Han Z, Wu M, Wang S, Inhibition of PC cell-derived growth factor (PCDGF)/granulin-epithelin precursor (GEP) decreased cell proliferation and invasion through downregulation of cyclin D and CDK 4 and inactivation of MMP-2, BMC cancer, 7 (2007) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tangkeangsirisin W, Serrero G, PC cell-derived growth factor (PCDGF/GP88, progranulin) stimulates migration, invasiveness and VEGF expression in breast cancer cells, Carcinogenesis, 25 (2004) 1587–1592. [DOI] [PubMed] [Google Scholar]
- [22].Tangkeangsirisin W, Hayashi J, Serrero G, PC cell-derived growth factor mediates tamoxifen resistance and promotes tumor growth of human breast cancer cells, Cancer research, 64 (2004) 1737–1743. [DOI] [PubMed] [Google Scholar]
- [23].Wang W, Hayashi J, Serrero G, PC Cell–Derived Growth Factor Confers Resistance to Dexamethasone and Promotes Tumorigenesis in Human Multiple Myeloma, Clinical cancer research, 12 (2006) 49–56. [DOI] [PubMed] [Google Scholar]
- [24].Abrhale T, Brodie A, Sabnis G, Macedo L, Tian C, Yue B, Serrero G, GP88 (PC-Cell Derived Growth Factor, progranulin) stimulates proliferation and confers letrozole resistance to aromatase overexpressing breast cancer cells, Bmc Cancer, 11 (2011) 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cheung PF, Yip CW, Wong NC, Fong DY, Ng LW, Wan AM, Wong CK, Cheung TT, Ng IO, Poon RT, Granulin–epithelin precursor renders hepatocellular carcinoma cells resistant to natural killer cytotoxicity, Cancer immunology research, 2 (2014) 1209–1219. [DOI] [PubMed] [Google Scholar]
- [26].Gray-Schopfer V, Wellbrock C, Marais R, Melanoma biology and new targeted therapy, Nature, 445 (2007) 851. [DOI] [PubMed] [Google Scholar]
- [27].Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice, Journal of Experimental Medicine, 207 (2010) 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gasteiger G, Hemmers S, Firth MA, Le Floc’h A, Huse M, Sun JC, Rudensky AY, IL-2–dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells, Journal of Experimental Medicine, 210 (2013) 1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Talantov D, Mazumder A, Jack XY, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y, Novel genes associated with malignant melanoma but not benign melanocytic lesions, Clinical cancer research, 11 (2005) 7234–7242. [DOI] [PubMed] [Google Scholar]
- [30].Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, Omberg L, Wolf DM, Shriver CD, Thorsson V, Caesar-Johnson SJ, Demchok JA, Felau I, Kasapi M, Ferguson ML, Hutter CM, Sofia HJ, Tarnuzzer R, Wang Z, Yang L, Zenklusen JC, Zhang J, Chudamani S, Liu J, Lolla L, Naresh R, Pihl T, Sun Q, Wan Y, Wu Y, Cho J, DeFreitas T, Frazer S, Gehlenborg N, Getz G, Heiman DI, Kim J, Lawrence MS, Lin P, Meier S, Noble MS, Saksena G, Voet D, Zhang H, Bernard B, Chambwe N, Dhankani V, Knijnenburg T, Kramer R, Leinonen K, Liu Y, Miller M, Reynolds S, Shmulevich I, Thorsson V, Zhang W, Akbani R, Broom BM, Hegde AM, Ju Z, Kanchi RS, Korkut A, Li J, Liang H, Ling S, Liu W, Lu Y, Mills GB, Ng K-S, Rao A, Ryan M, Wang J, Weinstein JN, Zhang J, Abeshouse A, Armenia J, Chakravarty D, Chatila WK, de Bruijn I, Gao J, Gross BE, Heins ZJ, Kundra R, La K, Ladanyi M, Luna A, Nissan MG, Ochoa A, Phillips SM, Reznik E, Sanchez-Vega F, Sander C, Schultz N, Sheridan R, Sumer SO, Sun Y, Taylor BS, Wang J, Zhang H, Anur P, Peto M, Spellman P, Benz C, Stuart JM, Wong CK, Yau C, Hayes DN, Parker JS, Wilkerson MD, Ally A, Balasundaram M, Bowlby R, Brooks D, Carlsen R, Chuah E, Dhalla N, Holt R, Jones SJM, Kasaian K, Lee D, Ma Y, Marra MA, Mayo M, Moore RA, Mungall AJ, Mungall K, Robertson AG, Sadeghi S, Schein JE, Sipahimalani P, Tam A, Thiessen N, Tse K, Wong T, Berger AC, Beroukhim R, Cherniack AD, Cibulskis C, Gabriel SB, Gao GF, Ha G, Meyerson M, Schumacher SE, Shih J, Kucherlapati MH, Kucherlapati RS, Baylin S, Cope L, Danilova L, Bootwalla MS, Lai PH, Maglinte DT, Van Den Berg DJ, Weisenberger DJ, Auman JT, Balu S, Bodenheimer T, Fan C, Hoadley KA, Hoyle AP, Jefferys SR, Jones CD, Meng S, Mieczkowski PA, Mose LE, Perou AH, Perou CM, Roach J, Shi Y, Simons JV, Skelly T, Soloway MG, Tan D, Veluvolu U, Fan H, Hinoue T, Laird PW, Shen H, Zhou W, Bellair M, Chang K, Covington K, Creighton CJ, Dinh H, Doddapaneni H, Donehower LA, Drummond J, Gibbs RA, Glenn R, Hale W, Han Y, Hu J, Korchina V, Lee S, Lewis L, Li W, Liu X, Morgan M, Morton D, Muzny D, Santibanez J, Sheth M, Shinbro E, Wang L, Wang M, Wheeler DA, Xi L, Zhao F, Hess J, Appelbaum EL, Bailey M, Cordes MG, Ding L, Fronick CC, Fulton LA, Fulton RS, Kandoth C, Mardis ER, McLellan MD, Miller CA, Schmidt HK, Wilson RK, Crain D, Curley E, Gardner J, Lau K, Mallery D, Morris S, Paulauskis J, Penny R, Shelton C, Shelton T, Sherman M, Thompson E, Yena P, Bowen J, Gastier-Foster JM, Gerken M, Leraas KM, Lichtenberg TM, Ramirez NC, Wise L, Zmuda E, Corcoran N, Costello T, Hovens C, Carvalho AL, de Carvalho AC, Fregnani JH, Longatto-Filho A, Reis RM, Scapulatempo-Neto C, Silveira HCS, Vidal DO, Burnette A, Eschbacher J, Hermes B, Noss A, Singh R, Anderson ML, Castro PD, Ittmann M, Huntsman D, Kohl B, Le X, Thorp R, Andry C, Duffy ER, Lyadov V, Paklina O, Setdikova G, Shabunin A, Tavobilov M, McPherson C, Warnick R, Berkowitz R, Cramer D, Feltmate C, Horowitz N, Kibel A, Muto M, Raut CP, Malykh A, Barnholtz-Sloan JS, Barrett W, Devine K, Fulop J, Ostrom QT, Shimmel K, Wolinsky Y, Sloan AE, De Rose A, Giuliante F, Goodman M, Karlan BY, Hagedorn CH, Eckman J, Harr J, Myers J, Tucker K, Zach LA, Deyarmin B, Hu H, Kvecher L, Larson C, Mural RJ, Somiari S, Vicha A, Zelinka T, Bennett J, Iacocca M, Rabeno B, Swanson P, Latour M, Lacombe L, Têtu B, Bergeron A, McGraw M, Staugaitis SM, Chabot J, Hibshoosh H, Sepulveda A, Su T, Wang T, Potapova O, Voronina O, Desjardins L, Mariani O, Roman-Roman S, Sastre X, Stern M-H, Cheng F, Signoretti S, Berchuck A, Bigner D, Lipp E, Marks J, McCall S, McLendon R, Secord A, Sharp A, Behera M, Brat DJ, Chen A, Delman K, Force S, Khuri F, Magliocca K, Maithel S, Olson JJ, Owonikoko T, Pickens A, Ramalingam S, Shin DM, Sica G, Van Meir EG, Zhang H, Eijckenboom W, Gillis A, Korpershoek E, Looijenga L, Oosterhuis W, Stoop H, van Kessel KE, Zwarthoff EC, Calatozzolo C, Cuppini L, Cuzzubbo S, DiMeco F, Finocchiaro G, Mattei L, Perin A, Pollo B, Chen C, Houck J, Lohavanichbutr P, Hartmann A, Stoehr C, Stoehr R, Taubert H, Wach S, Wullich B, Kycler W, Murawa D, Wiznerowicz M, Chung K, Edenfield WJ, Martin J, Baudin E, Bubley G, Bueno R, De Rienzo A, Richards WG, Kalkanis S, Mikkelsen T, Noushmehr H, Scarpace L, Girard N, Aymerich M, Campo E, Giné E, Guillermo AL, Van Bang N, Hanh PT, Phu BD, Tang Y, Colman H, Evason K, Dottino PR, Martignetti JA, Gabra H, Juhl H, Akeredolu T, Stepa S, Hoon D, Ahn K, Kang KJ, Beuschlein F, Breggia A, Birrer M, Bell D, Borad M, Bryce AH, Castle E, Chandan V, Cheville J, Copland JA, Farnell M, Flotte T, Giama N, Ho T, Kendrick M, Kocher J-P, Kopp K, Moser C, Nagorney D, O’Brien D, O’Neill BP, Patel T, Petersen G, Que F, Rivera M, Roberts L, Smallridge R, Smyrk T, Stanton M, Thompson RH, Torbenson M, Yang JD, Zhang L, Brimo F, Ajani JA, Angulo Gonzalez AM, Behrens C, Bondaruk J, Broaddus R, Czerniak B, Esmaeli B, Fujimoto J, Gershenwald J, Guo C, Lazar AJ, Logothetis C, Meric-Bernstam F, Moran C, Ramondetta L, Rice D, Sood A, Tamboli P, Thompson T, Troncoso P, Tsao A, Wistuba I, Carter C, Haydu L, Hersey P, Jakrot V, Kakavand H, Kefford R, Lee K, Long G, Mann G, Quinn M, Saw R, Scolyer R, Shannon K, Spillane A, Stretch J, Synott M, Thompson J, Wilmott J, Al-Ahmadie H, Chan TA, Ghossein R, Gopalan A, Levine DA, Reuter V, Singer S, Singh B, Tien NV, Broudy T, Mirsaidi C, Nair P, Drwiega P, Miller J, Smith J, Zaren H, Park J-W, Hung NP, Kebebew E, Linehan WM, Metwalli AR, Pacak K, Pinto PA, Schiffman M, Schmidt LS, Vocke CD, Wentzensen N, Worrell R, Yang H, Moncrieff M, Goparaju C, Melamed J, Pass H, Botnariuc N, Caraman I, Cernat M, Chemencedji I, Clipca A, Doruc S, Gorincioi G, Mura S, Pirtac M, Stancul I, Tcaciuc D, Albert M, Alexopoulou I, Arnaout A, Bartlett J, Engel J, Gilbert S, Parfitt J, Sekhon H, Thomas G, Rassl DM, Rintoul RC, Bifulco C, Tamakawa R, Urba W, Hayward N, Timmers H, Antenucci A, Facciolo F, Grazi G, Marino M, Merola R, de Krijger R, Gimenez-Roqueplo A-P, Piché A, Chevalier S, McKercher G, Birsoy K, Barnett G, Brewer C, Farver C, Naska T, Pennell NA, Raymond D, Schilero C, Smolenski K, Williams F, Morrison C, Borgia JA, Liptay MJ, Pool M, Seder CW, Junker K, Omberg L, Dinkin M, Manikhas G, Alvaro D, Bragazzi MC, Cardinale V, Carpino G, Gaudio E, Chesla D, Cottingham S, Dubina M, Moiseenko F, Dhanasekaran R, Becker K-F, Janssen K-P, Slotta-Huspenina J, Abdel-Rahman MH, Aziz D, Bell S, Cebulla CM, Davis A, Duell R, Elder JB, Hilty J, Kumar B, Lang J, Lehman NL, Mandt R, Nguyen P, Pilarski R, Rai K, Schoenfield L, Senecal K, Wakely P, Hansen P, Lechan R, Powers J, Tischler A, Grizzle WE, Sexton KC, Kastl A, Henderson J, Porten S, Waldmann J, Fassnacht M, Asa SL, Schadendorf D, Couce M, Graefen M, Huland H, Sauter G, Schlomm T, Simon R, Tennstedt P, Olabode O, Nelson M, Bathe O, Carroll PR, Chan JM, Disaia P, Glenn P, Kelley RK, Landen CN, Phillips J, Prados M, Simko J, Smith-McCune K, VandenBerg S, Roggin K, Fehrenbach A, Kendler A, Sifri S, Steele R, Jimeno A, Carey F, Forgie I, Mannelli M, Carney M, Hernandez B, Campos B, Herold-Mende C, Jungk C, Unterberg A, von Deimling A, Bossler A, Galbraith J, Jacobus L, Knudson M, Knutson T, Ma D, Milhem M, Sigmund R, Godwin AK, Madan R, Rosenthal HG, Adebamowo C, Adebamowo SN, Boussioutas A, Beer D, Giordano T, Mes-Masson A-M, Saad F, Bocklage T, Landrum L, Mannel R, Moore K, Moxley K, Postier R, Walker J, Zuna R, Feldman M, Valdivieso F, Dhir R, Luketich J, Mora Pinero EM, Quintero-Aguilo M, Carlotti JCG, Dos Santos JS, Kemp R, Sankarankuty A, Tirapelli D, Catto J, Agnew K, Swisher E, Creaney J, Robinson B, Shelley CS, Godwin EM, Kendall S, Shipman C, Bradford C, Carey T, Haddad A, Moyer J, Peterson L, Prince M, Rozek L, Wolf G, Bowman R, Fong KM, Yang I, Korst R, Rathmell WK, Fantacone-Campbell JL, Hooke JA, Kovatich AJ, Shriver CD, DiPersio J, Drake B, Govindan R, Heath S, Ley T, Van Tine B, Westervelt P, Rubin MA, Lee JI, Aredes ND, Mariamidze A, Hu H, An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics, Cell, 173 (2018) 400–416.e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Monami G, Gonzalez EM, Hellman M, Gomella LG, Baffa R, Iozzo RV, Morrione A, Proepithelin promotes migration and invasion of 5637 bladder cancer cells through the activation of ERK1/2 and the formation of a paxillin/FAK/ERK complex, Cancer research, 66 (2006) 7103–7110. [DOI] [PubMed] [Google Scholar]
- [32].Araujo JM, Gomez AC, Aguilar A, Salgado R, Balko JM, Bravo L, Doimi F, Bretel D, Morante Z, Flores C, Gomez HL, Pinto JA, Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer, Sci Rep, 8 (2018) 4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lavergne E, Combadiere C, Iga M, Boissonnas A, Bonduelle O, Maho M, Debre P, Combadiere B, Intratumoral CC chemokine ligand 5 overexpression delays tumor growth and increases tumor cell infiltration, J Immunol, 173 (2004) 3755–3762. [DOI] [PubMed] [Google Scholar]
- [34].Geng J, Wang X, Wei H, Sun R, Tian Z, Efficient attenuation of NK cell-mediated liver injury through genetically manipulating multiple immunogenes by using a liver-directed vector, J Immunol, 190 (2013) 4821–4829. [DOI] [PubMed] [Google Scholar]
- [35].Wang W, Hayashi J, Kim WE, Serrero G, PC cell-derived growth factor (granulin precursor) expression and action in human multiple myeloma, Clinical cancer research, 9 (2003) 2221–2228. [PubMed] [Google Scholar]
- [36].Kong W-J, Zhang S-L, Chen X, Zhang S, Wang Y-J, Zhang D, Sun Y, PC cell-derived growth factor overexpression promotes proliferation and survival of laryngeal carcinoma, Anti-cancer drugs, 18 (2007) 29–40. [DOI] [PubMed] [Google Scholar]
- [37].Li LQ, Min LS, Jiang Q, Ping JL, Li J, Dai LC, Progranulin expression in breast cancer with different intrinsic subtypes, Pathol Res Pract, 208 (2012) 210–216. [DOI] [PubMed] [Google Scholar]
- [38].Carlson AM, Maurer MJ, Goergen KM, Kalli KR, Erskine CL, Behrens MD, Knutson KL, Block MS, Utility of progranulin and serum leukocyte protease inhibitor as diagnostic and prognostic biomarkers in ovarian cancer, Cancer Epidemiol Biomarkers Prev, 22 (2013) 1730–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Han JJ, Yu M, Houston N, Steinberg SM, Kohn EC, Progranulin is a potential prognostic biomarker in advanced epithelial ovarian cancers, Gynecol Oncol, 120 (2011) 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pan CX, Kinch MS, Kiener PA, Langermann S, Serrero G, Sun L, Corvera J, Sweeney CJ, Li L, Zhang S, Baldridge LA, Jones TD, Koch MO, Ulbright TM, Eble JN, Cheng L, PC cell-derived growth factor expression in prostatic intraepithelial neoplasia and prostatic adenocarcinoma, Clin Cancer Res, 10 (2004) 1333–1337. [DOI] [PubMed] [Google Scholar]
- [41].Donald CD, Laddu A, Chandham P, Lim SD, Cohen C, Amin M, Gerton GL, Marshall FF, Petros JA, Expression of progranulin and the epithelin/granulin precursor acrogranin correlates with neoplastic state in renal epithelium, Anticancer Res, 21 (2001) 3739–3742. [PubMed] [Google Scholar]
- [42].Gobel M, Eisele L, Mollmann M, Huttmann A, Johansson P, Scholtysik R, Bergmann M, Busch R, Dohner H, Hallek M, Seiler T, Stilgenbauer S, Klein-Hitpass L, Duhrsen U, Durig J, Progranulin is a novel independent predictor of disease progression and overall survival in chronic lymphocytic leukemia, PLoS One, 8 (2013) e72107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Edelman MJ, Feliciano J, Yue B, Bejarano P, Ioffe O, Reisman D, Hawkins D, Gai Q, Hicks D, Serrero G, GP88 (progranulin): a novel tissue and circulating biomarker for non-small cell lung carcinoma, Hum Pathol, 45 (2014) 1893–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yamamoto Y, Goto N, Takemura M, Yamasuge W, Yabe K, Takami T, Miyazaki T, Takeuchi T, Shiraki M, Shimizu M, Adachi S, Saito K, Shibata Y, Nakamura N, Hara T, Serrero G, Saito K, Tsurumi H, Association between increased serum GP88 (progranulin) concentrations and prognosis in patients with malignant lymphomas, Clin Chim Acta, 473 (2017) 139–146. [DOI] [PubMed] [Google Scholar]
- [45].Cerwenka A, Lanier LL, Natural killer cells, viruses and cancer, Nature reviews immunology, 1 (2001) 41. [DOI] [PubMed] [Google Scholar]
- [46].Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, Van Moer K, Kreis S, Guerin C, Buart S, Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner, Proceedings of the National Academy of Sciences, 114 (2017) E9271–E9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cheng M, Chen Y, Xiao W, Sun R, Tian Z, NK cell-based immunotherapy for malignant diseases, Cellular & molecular immunology, 10 (2013) 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S, Functions of natural killer cells, Nature immunology, 9 (2008) 503. [DOI] [PubMed] [Google Scholar]
- [49].Lv D, Zhang Y, Kim H-J, Zhang L, Ma X, CCL5 as a potential immunotherapeutic target in triple-negative breast cancer, Cellular & molecular immunology, 10 (2013) 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Soria G, Ben-Baruch A, The inflammatory chemokines CCL2 and CCL5 in breast cancer, Cancer letters, 267 (2008) 271–285. [DOI] [PubMed] [Google Scholar]
- [51].Chuang JY, Yang WH, Chen HT, Huang CY, Tan TW, Lin YT, Hsu CJ, Fong YC, Tang CH, CCL5/CCR5 axis promotes the motility of human oral cancer cells, Journal of cellular physiology, 220 (2009) 418–426. [DOI] [PubMed] [Google Scholar]
- [52].Adler EP, Lemken CA, Katchen NS, Kurt RA, A dual role for tumor-derived chemokine RANTES (CCL5), Immunology letters, 90 (2003) 187–194. [DOI] [PubMed] [Google Scholar]
- [53].Stormes KA, Lemken CA, Lepre JV, Marinucci MN, Kurt RA, Inhibition of metastasis by inhibition of tumor-derived CCL5, Breast cancer research and treatment, 89 (2005) 209–212. [DOI] [PubMed] [Google Scholar]
- [54].Lapteva N, Huang XF, CCL5 as an adjuvant for cancer immunotherapy, Expert opinion on biological therapy, 10 (2010) 725–733. [DOI] [PubMed] [Google Scholar]
- [55].Mrowietz U, Schwenk U, Maune S, Bartels J, Kupper M, Fichtner I, Schroder JM, Schadendorf D, The chemokine RANTES is secreted by human melanoma cells and is associated with enhanced tumour formation in nude mice, Br J Cancer, 79 (1999) 1025–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Campbell JJ, Qin S, Unutmaz D, Soler D, Murphy KE, Hodge MR, Wu L, Butcher EC, Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire, The Journal of Immunology, 166 (2001) 6477–6482. [DOI] [PubMed] [Google Scholar]
- [57].Hoque M, Young TM, Lee C-G, Serrero G, Mathews MB, Pe’ery T, The growth factor granulin interacts with cyclin T1 and modulates P-TEFb-dependent transcription, Molecular and cellular biology, 23 (2003) 1688–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hoque M, Tian B, Mathews MB, Pe’ery T, Granulin and Granulin Repeats Interact with the Tat· P-TEFb Complex and Inhibit Tat Transactivation, Journal of Biological Chemistry, 280 (2005) 13648–13657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.