

Abstract
Huntington’s disease is characterized by a triad of motor, cognitive and psychiatric impairments, as well as unintended weight loss. Although much of the research has focused on cognitive, motor and psychiatric symptoms, the extent of peripheral pathology and the relationship between these factors, and the core symptoms of Huntington’s disease, are relatively unknown. Gut microbiota are key modulators of communication between the brain and gut, and alterations in microbiota composition (dysbiosis) can negatively affect cognition, behaviour and affective function, and may be implicated in disease progression. Furthermore, gut dysbiosis was recently reported in Huntington’s disease transgenic mice. Our main objective was to characterize the gut microbiome in people with Huntington’s disease and determine whether the composition of gut microbiota are significantly related to clinical indicators of disease progression. We compared 42 Huntington’s disease gene expansion carriers, including 19 people who were diagnosed with Huntington’s disease (Total Functional Capacity > 6) and 23 in the premanifest stage, with 36 age- and gender-matched healthy controls. Participants were characterized clinically using a battery of cognitive tests and using results from 16S V3 to V4 rRNA sequencing of faecal samples to characterize the gut microbiome. For gut microbiome measures, we found significant differences in the microbial communities (beta diversity) based on unweighted UniFrac distance (P = 0.001), as well as significantly lower alpha diversity (species richness and evenness) between our combined Huntington’s disease gene expansion carrier group and healthy controls (P = 0.001). We also found major shifts in the microbial community structure at Phylum and Family levels, and identified functional pathways and enzymes affected in our Huntington’s disease gene expansion carrier group. Within the Huntington’s disease gene expansion carrier group, we also discovered associations among gut bacteria, cognitive performance and clinical outcomes. Overall, our findings suggest an altered gut microbiome in Huntington’s disease gene expansion carriers. These results highlight the importance of gut biomarkers and raise interesting questions regarding the role of the gut in Huntington’s disease, and whether it may be a potential target for future therapeutic intervention.
Keywords: Huntington’s disease, neurodegenerative disease, gut microbiome, cognitive impairment, gut–brain axis
Emerging evidence links the gut–brain axis to neurodegenerative diseases. Wasser et al. report the first evidence of gut dysbiosis in people with Huntington’s disease. Their results highlight the importance of gut biomarkers and the prospect of future therapeutic interventions in Huntington’s disease.
Graphical Abstract
Graphical Abstract.

Introduction
Huntington’s disease is an inherited neurodegenerative disease that causes progressive motor decline, cognitive dysfunction and neuropsychiatric symptoms (Walker, 2007). Huntington’s disease is caused by age-dependent penetrance of an expanded sequence of cytosine–adenine–guanine (CAG) repeats in the huntingtin gene (Tabrizi et al., 2011). Larger expansions of CAG repeats are associated with earlier disease onset and more rapid disease progression (Andrew et al., 1993). Across the disease course, Huntington’s disease affects a wide range of functions, including communication, awareness, executive functioning, organization, memory and visuospatial abilities, all of which decline over time (Craufurd et al., 2001; Berrios et al., 2002; Paulsen et al., 2008; Ross and Tabrizi, 2011). Cognitive decline eventually progresses to dementia, and furthermore, depression and suicide are estimated to be 5–10 times more common in Huntington’s disease than in the general population (Berrios et al., 2002; Walker, 2007). In the brain, Huntington’s disease affects the basal ganglia early and severely, but the brain effects of Huntington’s disease progress and generalize to a range of cortical and subcortical structures, affecting both grey and white matter (Tabrizi et al., 2009, 2011; Domínguez et al., 2016). These brain changes have been assumed to account for the clinical manifestations seen in Huntington’s disease (Ross and Tabrizi, 2011), however, peripheral pathology is an underexplored area of investigation.
In addition to the cognitive, motor and neuropsychiatric symptoms, which likely relate to brain changes, people with Huntington’s disease also experience a range of gastrointestinal disturbances, including diarrhoea, nutrient deficiencies, gastritis and unintended weight-loss, which are considered clinical features of Huntington’s disease (Djousse et al., 2002; Robbins et al., 2006; van der Burg et al., 2017). Some evidence suggests that these disturbances are a manifestation of dysfunction in the gastrointestinal tract (van der Burg et al., 2011). In addition, gastrointestinal disturbances may be indirect effects of other progressive disease features, such as cognitive decline and decreased mobility. Whilst the majority of research investigating the pathogenesis of Huntington’s disease has focused on brain atrophy and the accompanying cognitive, behavioural and psychiatric symptoms, the extent to which peripheral pathology, and in particular the gut, is related to core symptoms of Huntington’s disease remains relatively unknown (Kong et al., 2018).
Adding to this picture, extensive evidence highlights the relationship among the enteric nervous system, the gut microbiome and the brain, which are collectively referred to as the gut–brain axis (Carabotti et al., 2015). The gut–brain axis is a bidirectional chemical signalling system between the gastrointestinal system and the central nervous system (Burokas et al., 2015). Furthermore, alterations of the gut microbiome influence mood regulation, cognition and sleep (Cryan and Dinan, 2012), and increasing evidence suggests that diversity in the gut microbiome is essential for brain function (Dinan and Cryan, 2017). The recognition of the functional significance of the gut–brain axis is increasingly influential, and has even been argued to be a paradigm shift in neuroscience (Mayer et al., 2014). With the rise in research activity on this topic, a substantial, growing body of evidence now links the gut–brain axis to neurodegenerative, neurodevelopmental and neuropsychiatric diseases (Marques et al., 2016; Sarkar and Banerjee, 2019).
Altered gut microbiome profiles have been documented in several neurodegenerative diseases, such as multiple sclerosis (Newland et al., 2016), Parkinson’s disease (Scheperjans et al., 2015), amyotrophic lateral sclerosis (Fang et al., 2016) and Alzheimer’s disease (Vogt et al., 2017; for a full review refer to Sarkar and Banerjee, 2019). The most abundant evidence of gut dysbiosis is in Parkinson’s disease (Keshavarzian et al., 2015; Scheperjans et al., 2015; Fitzgerald et al., 2019). In Parkinson’s disease, alterations in gut microbiota are thought to promote α-synuclein-mediated motor deficits and neuroinflammation, which specifically connects the gut–brain axis to disease pathogenesis (Sampson et al., 2016; Fitzgerald et al., 2019). Furthermore, transplants of gut microbiota from humans with Parkinson’s disease into mice, exacerbate motor deficits, underlining the interplay between gut microbiota and the progression of disease (Sampson et al., 2016). People with Parkinson’s disease also exhibit dysbiosis, and a greater abundance of Enterobacteriaceae correlates with severity of motor signs, including postural instability and gait abnormalities (Scheperjans et al., 2015). Parkinson’s disease, as well as other neurodegenerative diseases, is linked to lower than normal levels of Eubacterium hallii and Prevotella (Louis et al., 2010; Engels et al., 2016; Gerhardt and Mohajeri, 2018). These bacteria are involved in the production of short-chain fatty acids, which play a major role in intestinal homeostasis, gut and colon function (Bourassa et al., 2016), and may be a mechanism underlying some of the gut signs and symptoms observed in Parkinson’s disease.
Compared to the extensive evidence emerging in Parkinson’s disease, no gut microbiome studies have been reported in people with Huntington’s disease, despite intriguing findings from Huntington’s disease animal models (Kong et al., 2018; Radulescu et al., 2019). For example, studies in Huntington’s disease mouse models show gut dysfunction, along with evidence of endocrine dysfunction, including hormonal abnormalities, decreased enteric neuropeptides, decreased mucosal thickness and villus length (Ferrante et al., 2000; van der Burg et al., 2011; McCourt et al., 2015). Furthermore, deficient energy metabolism, as well as serum and cerebrospinal fluid metabolic differences, has been observed in presymptomatic transgenic Huntington’s disease rats (Verwaest et al., 2011). More recently, in R6/1 transgenic mice, Kong et al. (2018) observed the first specific evidence of gut dysbiosis in a Huntington’s disease model, finding increases in Bacteroidetes and a proportional decrease in Firmicutes. In this study, gut dysbiosis was also associated with body weight impairment and motor deficits.
Given the preclinical evidence suggesting gut dysbiosis in Huntington’s disease models and clinical evidence of gut symptoms in Huntington’s disease, together with the emerging picture of gut dysbiosis in neurodegenerative conditions, in this study we used faecal samples to investigate whether the gut microbiome in Huntington’s disease gene expansion carriers (HDGECs) differs from age- and gender-matched healthy controls (HCs). We sought to investigate differences in terms of the richness and evenness of diversity of the gut, which confers resilience of gut function, the composition of microbial communities, as indicated by taxonomic analysis and the various Phyla and Families of bacteria constituting compositional differences, and the functional pathways and enzymes associated with these gut profiles. We also examined the relationship among Phyla, Family and certain species of bacteria, with clinical measures of motor and cognitive function.
Materials and methods
Participants
We studied 42 HDGECs, including 19 people who were diagnosed with Huntington’s disease, 23 people in the premanifest stage and 36 HCs. All premanifest HDGECs had been genetically confirmed to have the Huntington’s disease CAG expansion (i.e. CAG ≥ 39; except for one premanifest HDGEC close to manifest diagnosis). For the Huntington's disease sample, diagnosis was made by a neurological examination and the neurologist’s classification of the participant as clinically symptomatic, based on unequivocal motor signs of Huntington’s disease using the Unified Huntington’s Disease Rating Scale (UHDRS; Kieburtz et al., 2001). We used scaled CAG-age product (CAPS) scores to classify our premanifest HDGEC participants based on a 5-year probability of symptomatic diagnosis into cut-offs of low probability, medium probability and high probability, based on the optimization algorithm from the PREDICT-HD study (Epping et al., 2013). CAPS is calculated by multiplying the age at time of testing (Age0) by scaling the CAG repeat length, where CAPS = Age0 × (CAG - 33.66)/432.3326 (Zhang et al., 2011). We had seven premanifest participants in the low cut-off (CAPS < 0.67), seven participants in the medium cut-off (CAPS between 0.67 and 0.85), and seven participants in the high cut-off (CAPS > 0.85). We could not calculate CAPS for two participants because of missing genetic data.
We recruited participants from the Statewide Progressive Neurological Disease Service at Calvary Health Care Bethlehem Hospital and the Experimental Neuropsychology Research Unit and Clinical Cognitive Neuroscience Huntington’s disease research participant database at Monash University, Melbourne, Australia. Exclusion criteria for all participants included diagnosis by a general practitioner or medical professional of irritable bowel syndrome, coeliac disease, diabetes or history of any diagnosed condition that may likely influence general gut health (e.g. Crohn’s disease and ulcerative colitis). Furthermore, participants were excluded if they had psychiatric illness (except for psychiatric symptoms attributable to Huntington’s disease), neurological illness (except for Huntington’s disease), a history of traumatic brain injury, recent antibiotic or anti-inflammatory medication use (past 2 months) or current recreational substance use.
Sample characterization measures
Depression is prevalent in Huntington’s disease (Walker, 2007), and can negatively affect cognition and the gut microbiome. We therefore screened participants using the Center for Epidemiologic Studies Depression Scale Revised (Eaton et al., 2004). Center for Epidemiologic Studies Depression Scale Revised scores range from 0 to 80, with higher scores indicative of greater depressive symptoms. As the focus of this study was on the gut microbiome, we wanted to consider gastrointestinal symptoms. Therefore, we included a clinically used measure of gut health, the self-report Gastrointestinal Health Appraisal Questionnaire (Metagenics®, 1992). The Gastrointestinal Health Appraisal Questionnaire includes 35 questions relating to gastric functioning, gastrointestinal inflammation, functioning of the small intestine and pancreas and colon function. Gastrointestinal Health Appraisal Questionnaire scores range from 0 to 280, with higher scores indicative of higher gastrointestinal symptoms. We administered the Wechsler Test of Adult Reading (Holdnack, 2001) to estimate participants’ premorbid intelligence. The Wechsler Test of Adult Reading comprises a list of 50 words that do not follow typical pronunciation rules, and participants are required to read the words aloud. A higher score (i.e. larger number of correctly pronounced items) reflects a higher estimated premorbid intelligence quotient score. On these measures, groups did not statistically differ on depressive symptoms, self-reported gastrointestinal symptoms or estimated premorbid intelligence.
For the HDGECs, the UHDRS (Kieburtz et al., 2001) Total Motor Score (TMS) and Total Functional Capacity (TFC) components were used to characterize severity of motor signs and the stage of disease. Higher TMS indicates more severe motor signs. The TFC is a clinician rating scale including items related to capacity to work, handle finances, perform domestic chores, self-care and ability to live independently (Shoulson and Fahn, 1979). These ratings yield a TFC score, with higher scores indicating better functioning and greater independence. The TFC and TMSs were obtained from each HDGEC participant’s treating neurologist/clinician. A TMS for three participants was unavailable. Demographic and clinical features of the participant groups are presented in Table 1. Supplementary Table 1 contains demographic and clinical information of HDGEC participants separated by disease status (i.e. premanifest or manifest).
Table 1.
Demographic and clinical profile of HDGEC and HC participants
| HDGEC = 42 | Control = 36 | P/χ2 value | ||
|---|---|---|---|---|
| Gender | M:F | 22:20 | 15:21 | 0.345 |
| Age (years) | M (SD) | 50.16 (12.14) | 50.55 (13.90) | 0.948 |
| Range | 26–75 | 24–69 | ||
| UHDRS–TMS | M (SD) | 9.21 (12.71) | ||
| Range | 0–41 | |||
| UHDRS–TFC | M (SD) | 10.90 (2.64) | ||
| Range | 6–13 | |||
| Disease duration | M (SD) | 5.06 (3.23) | ||
| (years after diagnosis) | Range | 1–13 | ||
| CAG length | M (SD) | 41.26 (1.80) | ||
| Range | 38–46 | |||
| CAPS | M (SD) | 0.88 (0.25) | ||
| Range | 0.32–1.40 | |||
| Years of education | M (SD) | 14.61 (2.94) | 14.83 (3.20) | 0.461 |
| Range | 8–21 | 9–23 | ||
| Estimated IQ | M (SD) | 104.04 (6.99) | 106.14 (6.43) | 0.171 |
| Range | 87–115 | 89–117 | ||
| GIHAQ | M (SD) | 21.47 (25.44) | 15.81 (21.33) | 0.294 |
| Range | 0–119 | 0–116 | ||
| CESD-R | M (SD) | 10.92 (10.45) | 7.06 (7.75) | 0.071 |
| Range | 0–43 | 0–31 |
CAPS: scaled CAG-age product score (for premanifest HDGECs only); estimated intelligence quotient (IQ) calculated by Wechsler Test of Adult Reading; CESD-R: Center for Epidemiologic Studies Depression Scale-Revised (range: 0–80); F: female; GIHAQ: Gastrointestinal Health Appraisal Questionnaire (range: 0–280); M: male; TFC: Total Functional Capacity (range: 0–13); disease duration is for manifest HDGECs only; TMS: Total Motor Score (range: 0–124). P/χ2 value signifies probability value between groups, using independent samples t-tests or Chi-squared test (χ2 for gender).
Ethical approval
Monash University Human Research Ethics Committee approved this study (MUHREC: 8031). All participants provided written informed consent in accordance with the Declaration of Helsinki (World Medical Association, 2013).
Procedure
The data reported in this study were collected as baseline data for a probiotic clinical trial, which was ongoing when this manuscript was prepared. Participants were screened for eligibility by phone, and then scheduled for a 90-min assessment session in the lab. To enable the collection of a faecal sample, participants were mailed a stool collection kit in advance. The kit included instructions on collecting and storing the stool sample, as well as collection tubes, latex gloves, plastic containers and sealable plastic bags. To enable the assessment of a panel of gut bacteria, participants provided a faecal sample within 24 h of their in-lab testing session, which they stored on ice until attending the testing session. Once participants arrived at the testing session, we stored their stool sample at −80°C for later processing. We then administered a battery of neuropsychological tasks and a measure used to estimate premorbid intelligence. Participants also filled out self-report measures of depression and gastrointestinal function. We reimbursed participants for their time at the end of the testing session.
Measures
Cognitive measures
We used cognitive measures from the Huntington’s Disease Cognitive Assessment Battery (HD-CAB), which is a battery specifically designed for cognitive assessment in Huntington’s disease clinical trials (Stout et al., 2014). HD-CAB includes six cognitive tests. We describe each task below; for more details refer to Stout et al. (2014).
The Hopkins Verbal Learning Test-Revised (Benedict et al., 1998) is a paper and pencil task that assesses verbal learning and memory (immediate and delayed recall). On this task, participants must recall as many items as they can from a word list, during three learning trials and the delayed recall trial. The main outcome measure is total number of words recalled across the three trials summed with the number of words recalled on the delayed trial.
The Symbol Digit Modalities Test measures processing speed and visual attention (Smith, 1982). Participants must refer to a key and fill in each empty box with a number that correctly corresponds to each symbol, as quickly as they are able to within 90 s. The outcome measure is the total numbers of boxes correctly completed.
The Trail Making Test (Reitan, 1958) parts A and B measures processing and manual movement speed, visuospatial attention and executive functioning. The outcome measure we used for this task is the time taken (in seconds) to complete Trail Making Test part B, where participants are required to draw a continuous line connecting numbers and letters in alteration (1 to A, A to 2, 2 to B, etc.). Longer times are indicative of poorer performance.
Paced tapping (Rowe et al., 2010; Stout et al., 2014) is a computerized task used to measure psychomotor coordination and timing. Participants are instructed to tap a mouse using alternating thumbs at a consistent rhythm. They complete four identical trials. The outcome measure for this task is the reciprocal of the standard deviation of the inter-tap interval, which measures the overall consistency of the tapping rate. Higher values indicate better performance.
The Emotion Recognition Task (Johnson et al., 2007) is a computerized measure of participants’ ability to recognize emotions in facial expressions. Participants are presented with black and white photographed faces that depict seven emotional expressions: anger, disgust, fear, happiness, sadness, surprise or neutral. The outcome measure is the correct number of negative emotions identified (anger, disgust, fear and sadness). Higher scores are indicative of greater accuracy.
The One Touch Stockings of Cambridge (OTS) is a modified, shortened version (6Touch10) of the CANTAB® OTS task. OTS is a computerized task that measures working memory and spatial planning (Robbins et al., 1994; Watkins et al., 2000). Participants have to imagine moving balls (one at a time), to make the bottom arrangement of balls match the top, in as few moves as possible. During each trial, participants are instructed to select the minimum numbers of moves (from one to six) required to make the bottom set match the top set. The outcome measure is the mean time to reach a correct response (seconds), averaged across all trials. Higher values are indicative of poorer performance.
Using the six tests included in the HD-CAB, we computed a composite score for a cognitive outcome measure. We first rescaled Trail Making Test part B and OTS so that higher scores represented better performance. For these variables, all values were multiplied by −1 and then a constant value (highest value for each variable) was added to all values. We then computed an equal-weighting composite score by adding the average standardized Z-score for each individual test (Hopkins Verbal Learning Test-Revised, Symbol Digit Modalities Test, Trail Making Test part B, Paced tapping, Emotion Recognition and OTS). We also computed separate equal-weighting cognitive composite scores, standardized from only our HDGEC group, for certain analyses restricted to the HDGEC group.
Faecal DNA extraction, sequencing and pre-processing
We extracted a 200–250 mg aliquot from each frozen faecal sample and placed them in 2 ml bead extraction tubes. The bead extraction tubes were stored on dry ice and couriered to the Australian Genomic Research Facility in Adelaide, South Australia, for bacterial genomic DNA extraction. All samples included in this report, were extracted at a single time-point to eliminate variation in results because of possible batch effects. The 16S V3–V4 rRNA gene (341F–806 R), which is a hypervariable region of bacterial rRNA for tracing phylogeny, was amplified and the final concentrations were determined by fluorometry. All aliquots included had > 0.50 ng/µl of usable amplicon, and were deemed sufficient for the sequencing stage. Amplicon was pooled and sequenced using an Illumina MiSeq next-generation sequencer. The reads were then demultiplexed and quality trimmed before denoizing using default parameters on DADA2 v1.12 in R (Callahan et al., 2016) to obtain the amplicon sequence variants (ASVs) table. Samples that did not have at least 1000 reads were not included in our analyses. The ASVs were mapped against SILVA 132_99 database (Quast et al., 2012) to obtain taxonomic identity of the reads. Results from this process were provided as a tabulation of the relative abundance of bacteria at the Phylum and Family levels. The full dataset of results received from the analyses included bacteria from seven Phyla, 32 Families and 79 Genera. The majority of the bacterial Phyla across the samples were Firmicutes (83%), Actinobacteria (9%), Bacteroidetes (4%) and Verrucomicrobia (1.1%).
Determination of key outcome measures from faecal samples
To characterize richness, evenness and composition of the microbiome environment, we assessed alpha and beta diversity. We used the number of ASVs observed, as well as the Fisher index, to determine richness and evenness, i.e. alpha diversity of each participant’s microbial community, and unweighted UniFrac distances for group environment composition, i.e. beta diversity. For alpha diversity measures, the reads were rarefied to 1984 reads, which is the global minimum number of reads in the cohort. For beta diversity, unweighted UniFrac distances were calculated and ordinated in principal coordinated analysis to visualize the variation in the data. The unweighted UniFrac distance accounts for phylogenetic relationship between ASVs. Adonis (Permutation multivariate ANOVA) from the ‘vegan’ v2.5.6 R package was performed with 999 permutations (Dixon, 2003). Adonis provides an R2 value on a scale of 0 to 1, indicating the amount of variance explained by the factors tested. Both alpha and beta diversity were analysed using the ‘Phyloseq’ v1.28 R package (McMurdie and Holmes, 2013). For examining differences which describe observed differences in the composition of the microbial environment, we used taxonomic analysis at the Phylum and Family level. Rare bacterial families were filtered using the cut-off of minimum 10 reads per sample, resulting in 29 bacterial Families altogether and 7 Phyla.
Gut function analysis using PICRUSt2 to identify functional pathways and enzymes
Raw sequences were passed through PICRUSt2 workflow to obtain enzyme commission numbers and Kyoto Encyclopedia of Genes and Genomes Ortholog abundances, which describe potential disturbance in gut microbiome enzymes and functional pathways, respectively (Douglas et al., 2020). As with the microbiome composition analysis, alpha and beta diversity of Kyoto Encyclopedia of Genes and Genomes Ortholog abundances and enzyme commission numbers were analysed using Phyloseq in R.
Statistical analysis
We used the Kruskal–Wallis test to compare species richness and alpha-diversity measurements between groups, and Adonis Permutation multivariate ANOVA to compare microbial composition (beta diversity indices) between groups. For examining taxonomies, we used analysis of composition of microbiomes, implemented in QIIME2 v2019.7 (Caporaso et al., 2010) to test for differential abundances of bacteria between the HDGEC and HC groups in the Phylum and Family levels (Mandal et al., 2015). Analysis of composition of microbiomes is shown to control for false-discovery rates while maintaining high power and is much better suited for analysing microbiome relative abundance data, compared to other standard statistical tests such as t-test and ANOVA (Mandal et al., 2015). The W-statistic value outputted by analysis of composition of microbiomes indicates the number of times the null hypothesis has been rejected for each ASV, with a false-discovery rate of < 0.05. For all analyses of group differences (HDGECs versus HC) we controlled for gender effects. To compare functional pathways and enzymes between groups, we used multivariate sparse partial least square discriminant analysis, a supervised learning method implemented in mixOmics v6.8.5 R package, (Le Cao et al., 2016), to identify the signature discriminating the HDGEC from HC group.
Exploratory analyses
Clinical outcomes and correlation with gut microbiome
We used an independent samples t-test to examine differences in cognitive performance (HD-CAB composite score) between our HDGEC and HC groups. Next, using only the HDGECs, we examined associations among the gut microbiome, cognitive outcomes (HD-CAB composite score) and clinical outcomes related to Huntington’s disease (UHDRS–TMS, UHDRS–TFC and CAPS), using Spearman’s rank correlation coefficients, given the unevenness in distribution of our gut variables. For the associations among UHDRS–TMS, UHDRS–TFC and bacterial data, it was necessary to limit analysis to only manifest HDGECs, because these clinical measures, which were developed for use in symptomatic HD, exhibit substantial range restrictions (ceiling effects) in the premanifest group, where motor symptoms and functional decline are not yet readily apparent. Furthermore, we investigated the association between bacterial data and CAPS scores only for premanifest HDGECs, given that CAPS is a predictive measure of symptomatic disease onset. Gender and age variables did not significantly correlate with these clinical outcomes.
16S rRNA sequencing produces thousands of gut variables, hence it is essential to restrict analyses of these variables to limit Type 1 error. Thus, we selected bacteria outcomes based on previous research in Parkinson’s disease and other conditions, which has documented associations among particular bacteria, disease progression and cognition (Bajaj et al., 2012; Bruce-Keller et al., 2015; Manderino et al., 2017; Parashar and Udayabanu, 2017; Gerhardt and Mohajeri, 2018; Zhan et al., 2018; Weis et al., 2019). For these analyses, at the Phylum level we examined Bacteroidetes, Firmicutes, Verrucomicrobia and Proteobacteria; at the Family level, we examined Akkermansiacea, Bacteroidaceae, Bifidobacteriaceae, Christensenellaceae, Lachnospiraceae, Ruminococcaceae and Streptococcaceae. We also examined a single species, E. hallii.
Data availability
Raw data from these analyses are available upon reasonable request.
Results
In terms of the richness and evenness of the microbial environment, which is typically associated with resilience of the gut environment, HDGECs showed less alpha diversity compared to HCs. Specifically, for alpha diversity, Observed and Fisher indices were lower (P = 0.001), indicating fewer species present and lower evenness, in HDGECs than in HCs; Fig. 1A. Beta diversity, indicating microbial community structure, significantly differed in HDGECs compared to HCs, based on unweighted UniFrac distances, P = 0.001; Fig. 1B.
Figure 1.

Alpha and beta diversity differences in the gut microbiome in HDGEC and HC groups. (A) Alpha diversity analysis investigating richness (Observed) and evenness (Fisher) in HDGEC versus HC groups. (B) Beta diversity analysis showing variation in the composition of the samples based on their phylogenetic distance (unweighted UniFrac distance) in HDGEC versus HC groups.
Next, using analysis of composition of microbiomes to examine differences observed in gut microbiome composition at the Phylum and Family levels, we found a significant gender by group interaction, with several differences identified in males. Specifically, for males, at the Phylum level, Euryarchaeota, Firmicutes and Verrucomicrobia differed significantly between groups. At the Family level, many Families differed significantly by gender between groups, including Acidaminococcaceae, Akkermansiaceae, Bacteroidaceae, Bifidobacteriaceae, Christensenellaceae, Clostridiaceae, Coriobacteriaceae, Eggerthellaceae, Enterobacteriaceae, Erysipelotrichaceae, Flavobacteriaceae, Lachnospiraceae, Methanobacteriaceae, Peptococcaceae, Peptostreptococcaceae and Rikenellaceae for males. For females, neither the Phylum nor the Family level revealed significant differences.
In terms of functional gut pathways, Kyoto Encyclopedia of Genes and Genomes Ortholog abundances differed significantly between HDGECs and HCs in both alpha and beta diversity. Specifically, for alpha diversity, the HDGEC group showed significantly less richness and evenness in functional pathways (Observed and Fisher) compared to HCs (P = 0.001). HDGECs and HCs also differed significantly in composition of gut pathways (beta diversity, P = 0.001). Additional analyses on predicted pathways using sparse partial least square discriminant analysis revealed specific functional gut pathways affected in the HDGEC group, including the Super pathway of serine and glycine biosynthesis, Starch degradation V pathway, Methylerythritol phosphate pathway I, Methylerythritol phosphate pathway II and the NAD biosynthesis I pathway (Fig. 2). At the enzyme level, enzymes of glutathione transferase and homoserine O-succinyltransferase were significantly different in the HDGEC group compared to HCs (Fig. 3).
Figure 2.

Functional pathways differences in the gut microbiome between HDGEC and HC groups.
Figure 3.

Enzyme differences in the gut microbiome between HDGEC and HC groups.
Although the samples within the HDGEC group were small, we explored whether the manifest HDGECs (n = 19) differed from the premanifest HDGECs (n = 23). We found no statistically significant differences in richness, evenness and in group microbial composition in the gut, from alpha and beta diversity measures. Differences describing microbial composition, as measured by taxonomic analysis, also showed no differences between these groups, apart from a significant difference in the abundance of Ruminococcaceae family, for male HDGECs only. Given these findings, we limited our examination of the gut results presented above, to comparisons between the combined HDGEC group (manifest and premanifest HD) and HCs.
Interestingly, in our exploratory analyses linking the gut measures with clinically relevant characteristics in the HDGECs, we found several associations, see Fig. 4. In particular, in manifest HDGECs, lower abundance of E. hallii correlated with more severe motor signs (UHDRS–TMS; rs = −0.512, n = 16, P = 0.043), however, we did not find associations between E. hallii abundance and our clinical measure of functional capacity (UHDRS–TFC; rs= 0.329, n = 19, P = 0.183). In premanifest HDGECs, we observed a significant negative relationship between the abundance of E. hallii and estimated proximity to disease onset (CAPS), rs = −0.568, n = 21, P = 0.007.
Figure 4.

Relationship among E. hallii, clinical measures of disease progression in manifest HDGECs (TMS & TFC) and estimated disease onset (CAPS) in premanifest HDGECs.
Prior to undertaking an evaluation of the cognitive measures in the context of the gut outcomes, we examined group differences in overall cognition to establish that as expected, the HDGEC group was cognitively impaired compared to the HC group. Group analysis confirmed this expected difference. Overall, the HDGEC group performed more poorly than controls on the HD-CAB composite measure of overall function [t(76) = 3.940, P < 0.001], with a large effect size (Cohen's d = 0.91), see Fig. 5.
Figure 5.
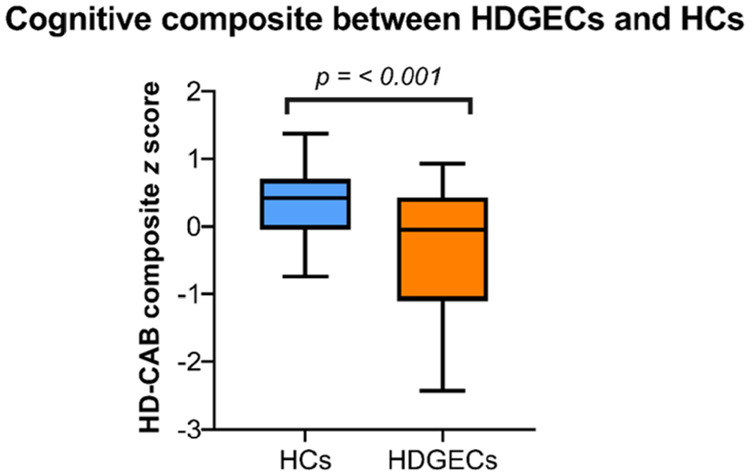
Overall performance between groups (HDGEC and HC) on cognitive outcomes (HD-CAB composite score).
In terms of associations between cognition and gut outcomes, E. hallii was associated with memory performance on the Hopkins Verbal Learning Test-Revised, rs = 0.315, n = 42, P = 0.042, and a similar trend was observed for a measure of processing speed and attention, Symbol Digit Modalities Test, rs = 0.278, n = 42, P = 0.074. These associations, however, appeared in the context of many other non-significant correlations, and therefore must be interpreted with caution as they may be spurious. Supplementary Tables 2–4 show the full extent of these correlational analyses among gut bacteria data, clinical measures and cognitive performance in HDGECs. Supplementary Table 5 details correlations between self-reported gastrointestinal symptoms (Gastrointestinal Health Appraisal Questionnaire) and alpha diversity indices, which were non-significant.
Discussion
The rationale for examining the gut in Huntington’s disease is supported by several lines of evidence linking differences in gut microbiota changes across a variety of diseases (Dinan and Cryan, 2017; Kowalski and Mulak, 2019; Sarkar and Banerjee, 2019), clinical indicators of gut dysfunction in Huntington’s disease (e.g. weight loss, nutrition intake abnormalities and gastrointestinal issues), and recent evidence of gut dysfunction in Huntington’s disease mice (Kong et al., 2018). Using 16S rRNA gene sequencing, our study provides the first evidence of gut dysbiosis in people with Huntington’s disease. These results supported our primary hypothesis of an altered gut microbiome in HDGECs compared to age- and gender-matched HCs, in terms of differences in richness, evenness and overall microbial composition of the gut, as indicated by alpha and beta diversity, and further described by taxonomic changes and functional pathway and enzyme differences. We also discovered consistent trends among Huntington’s disease progression, gut microbiota and cognitive outcomes, although these must be interpreted with caution given our small sample size and the need to conduct several statistical tests.
Consistent with our current findings in Huntington’s disease of lower alpha diversity and beta diversity differences, indicating less than normal richness, evenness and altered microbial gut composition, others have reported similar observations in neurodegenerative conditions such as Parkinson’s disease, Alzheimer’s disease and amyotrophic lateral sclerosis (Keshavarzian et al., 2015; Rowin et al., 2017; Vogt et al., 2017). Whilst the bacteria responsible for observed changes in diversity metrics vary across the spectrum of diseases, altered diversity of the gut microbiota across these diseases indicates that gut dysbiosis appears to be a common feature of at least some neurodegenerative diseases.
Homeostasis of the gut microbiome is important for human health, and influences behaviour and brain function via the gut–brain axis (Dinan and Cryan, 2017). Thus, some of the clinical manifestations or brain changes evident in Huntington’s disease may be related to gut-driven modulation of brain inflammatory pathways, via communication among the gut, endocrine, immune and neural pathways (Burokas et al., 2015). Studies in Alzheimer’s disease and Parkinson’s disease support a link between the gut and neuropathology, with evidence that microbiota influence amyloid development in Alzheimer’s disease (Kowalski and Mulak, 2019), and α-synuclein pathology in Parkinson’s disease (Sampson et al., 2016; Fitzgerald et al., 2019). Further studies of Huntington’s disease will be needed to determine associations between the gut microbiota and Huntington’s disease-specific neuropathology.
Although we observed differences between HDGECs and HCs at the taxonomy level, including gender effects, we did not observe an increase in Bacteroidetes and a proportional decrease in Firmicutes, such as that reported in the preclinical Huntington’s disease study by Kong et al. (2018). In our sample, the differences observed in Phyla and Families had some, but not complete overlap with published results from other neurodegenerative diseases, which is not surprising given the discrepancy in the literature both amongst and between different clinical populations (for a full review see: Gerhardt and Mohajeri, 2018). Our finding of significantly lower abundances of Firmicutes, Lachnospiraceae and Akkermansiaceae in HDGEC males is interesting, because it links to inflammatory processes, given the role of Akkermansiaceae in the maintenance of the gut barrier (Chelakkot et al., 2018), and Lachnospiraceaes’ and Firmicutes’ role in the production of butyric acid, reducing inflammation (Louis et al., 2010; Stilling et al., 2016; Truax et al., 2018). Previously, reductions in these bacteria have been shown to be related to depression (Huang et al., 2018), but we found no such significant associations when controlling for depressive symptoms or age. The finding of a gender effect from taxonomic analyses indicates that the gut is affected not only by Huntington’s disease, but also by other factors such as sex, which could interact with the relationship between gut microbiota and disease progression. Whilst differences in gut composition are interesting, particularly in the early phase of our understanding of the gut microbiome in conditions such as Huntington’s disease, ongoing research must determine whether observed changes in the relative abundances of bacteria are a cause, or a product of abnormal gut–brain function.
Differences in gut composition in Huntington’s disease compared to HCs do not necessarily translate into gastrointestinal problems. In contrast, differences in gut function have, by definition, consequences that may be clinically relevant. For example, the functional pathway and enzyme differences observed in our study related to serine, methionine (including enzyme differences in homoserine) and glycine biosynthesis. These changes may be important, because methionine links to oxidative stress (Campbell et al., 2016), and these pathways are related to glutamate release, tryptophan metabolism and synthesis of serotonin and melatonin production (O'Mahony et al., 2015; Yu and Lau, 2018; Hoglund et al., 2019). The finding of differences in these pathways in our Huntington’s disease sample supports previous observations showing abnormal handling of tryptophan metabolism in Huntington’s disease, resulting in higher levels of oxidative stress, which may contribute to neuroinflammation and brain dysfunction, as well as depressive symptoms and memory difficulties (Stoy et al., 2005; Mendelsohn et al., 2009; Bourassa et al., 2016; Kałużna-Czaplińska et al., 2019; Gao et al., 2020). Furthermore, at the enzyme level, significantly lower levels of glutathione transferase have been found in Parkinson’s disease and Alzheimer’s disease, and these changes are linked to increased oxidative stress and neuroinflammation (Mazzetti et al., 2015). Thus, our finding in the HDGEC group of functional pathway and enzyme differences raises interesting questions about the possibility of future development of gut biomarkers.
Published evidence relates cognition to bacteria across a range of taxonomic levels, but previous studies in other neurodegenerative samples have yielded mixed findings of relative higher or lower abundance of specific bacteria and their associations with cognition and disease progression (Keshavarzian et al., 2015; Vogt et al., 2017; Fitzgerald et al., 2019; Sarkar and Banerjee, 2019). Although speculative, the consistent trend we observed among E. hallii, Huntington’s disease progression and cognition is also interesting given this bacterium’s role in short-chain fatty acid production, tryptophan metabolism and inflammation. We did not see any association among Verrucomicrobia, Bacteroidetes, Proteobacteria and cognitive outcomes, which has been reported in other studies such as Manderino et al. (2017).
This study provides the first evidence of gut dysbiosis in people with Huntington’s disease, adding to the growing number of studies that have demonstrated an altered gut microbiome profile in different neurological, and particularly neurodegenerative populations. Understanding the interplay among the gut–brain axis, gut microbiome and the pathogenesis of disease progression remains an ongoing challenge, particularly given the large number of variables to analyse, and the inherent challenge of establishing a large enough sample size in humans to reliably establish meaningful differences. This is particularly relevant in studies such as ours, which are limited by small sample sizes, a cross-sectional design, and other confounding variables that make assessing healthy gut microbiota challenging, given the influence of many factors such as age, medication, diet, drugs, environment and lifestyle factors. Nonetheless, from a Huntington’s disease perspective, given the clinical evidence of gut symptoms in Huntington’s disease, recent evidence of an altered gut microbiome in Huntington’s disease mice (Kong et al., 2018), and now our study describing altered gut microbiota in humans with Huntington’s disease, these initial results highlight the importance of understanding how the gut fits into the progression of Huntington’s disease. To fully understand this key aspect of the disease, larger and longitudinal studies are essential. Furthermore, our results raise the prospect of identifying gut biomarkers in Huntington’s disease, which will be enabled by further examination and characterization of the gut microbiome in relation to other phenotypic and possibly genotypic information. Lastly, these results raise the tantalizing proposition of whether the gut may be a potential target for future therapeutic intervention to improve outcomes in Huntington’s disease and other neurodegenerative diseases.
Supplementary Material
Acknowledgements
The authors thank all participants who voluntarily donated their time to participate in this research study.
Funding
J.S. is an Australian Government National Health and Medical Research Council (NHRMC) Leadership Fellow and A.H. is an NHMRC Principal Research Fellow. This project was partly supported by an NHMRC- Australian Research Council Dementia Research Development Fellowship (grant number 1100862) awarded to Y. G-J. We greatly appreciate further generous donations received from The Fox Family Foundation, Torquay Coasters Cycling Group, and a private donation from Tacie Fox.
Competing interests
J.S. is director of a research company, Zindametrix, which supports clinical trial sponsors in the use of the HD-CAB and other cognitive testing in clinical trials, and provides consulting services regarding cognitive assessment in Huntington’s disease to clinical trial sponsors via Stout Neuropsych Pty Ltd. The remaining authors have no competing interests to report.
Glossary
- ASVs =
amplicon sequence variants
- CAG =
cytosine–adenine–guanine
- CAPS =
scaled CAG-age product score
- HC =
healthy control
- HD-CAB =
Huntington’s Disease Cognitive Assessment Battery
- HDGECs =
Huntington’s disease gene expansion carriers
- OTS =
One Touch Stockings of Cambridge
- TFC =
Total Functional Capacity
- TMS =
Total Motor Score
- UHDRS =
Unified Huntington’s Disease Rating Scale
References
- Andrew SE, Goldberg PY, Kremer B, Telenius H, Theilmann J, Adam S, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet 1993; 4: 398–403. [DOI] [PubMed] [Google Scholar]
- Association WM. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013; 310: 2191. [DOI] [PubMed] [Google Scholar]
- Bajaj JS, Ridlon JM, Hylemon PB, Thacker LR, Heuman DM, Smith S, et al. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am J Physiol Gastrointestin Liver Physiol 2012; 302: G168–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict RH, Schretlen D, Groninger L, Brandt J.. Hopkins Verbal Learning Test-Revised: normative data and analysis of inter-form and test–retest reliability. Clin Neuropsychol 1998; 12: 43–55. [Google Scholar]
- Berrios G, Wagle A, Markova I, Wagle S, Rosser A, Hodges J.. Psychiatric symptoms in neurologically asymptomatic Huntington's disease gene carriers: a comparison with gene negative at risk subjects. Acta Psychiatr Scand 2002; 105: 224–30. [DOI] [PubMed] [Google Scholar]
- Bourassa MW, Alim I, Bultman SJ, Ratan RR.. Butyrate, neuroepigenetics and the gut microbiome: can a high fiber diet improve brain health? Neurosci Lett 2016; 625: 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Salbaum JM, Luo M, Blanchard E, Taylor CM, Welsh DA, et al. Obese-type gut microbiota induce neurobehavioral changes in the absence of obesity. Biol Psychiatry 2015; 77: 607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burokas A, Moloney RD, Dinan TG, Cryan JF.. Microbiota regulation of the Mammalian gut–brain axis. Adv Appl Microbiol 2015; 91: 1–62. [DOI] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP.. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 2016; 13: 581–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K, Vowinckel J, Keller MA, Ralser M.. Methionine metabolism alters oxidative stress resistance via the pentose phosphate pathway. Antioxid Redox Signal 2016; 24: 543–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7: 335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabotti M, Scirocco A, Maselli MA, Severi C.. The gut–brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 2015; 28: 1–7. [PMC free article] [PubMed] [Google Scholar]
- Chelakkot C, Choi Y, Kim DK, Park HT, Ghim J, Kwon Y, et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp Mol Med 2018; 50: e450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craufurd D, Thompson JC, Snowden JS.. Behavioral changes in Huntington disease. Cogn Behav Neurol 2001; 14: 219–26. [PubMed] [Google Scholar]
- Cryan JF, Dinan TG.. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 2012; 13: 701–12. [DOI] [PubMed] [Google Scholar]
- Dinan TG, Cryan JF.. The microbiome–gut–brain axis in health and disease. Gastroenterol Clin N Am 2017; 46: 77–89. [DOI] [PubMed] [Google Scholar]
- Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci 2003; 14: 927–30. [Google Scholar]
- Djousse L, Knowlton B, Cupples L, Marder K, Shoulson I, Myers R.. Weight loss in early stage of Huntington’s disease. Neurology 2002; 59: 1325–30. [DOI] [PubMed] [Google Scholar]
- Domínguez JF, Stout JC, Poudel G, Churchyard A, Chua P, Egan GF, et al. Multimodal imaging biomarkers in premanifest and early Huntington's disease: 30-month IMAGE-HD data. Br J Psychiatry 2016; 208: 571–8. [DOI] [PubMed] [Google Scholar]
- Douglas GM, Maffei VJ, Zaneveld J, Yurgel S, Brown JR, Taylor CM, et al. PICRUSt2: an improved and extensible approach for metagenome inference. BioRxiv 2020; 672295.
- Eaton WW, Muntaner C, Tien A, Ybarra M. Center for epidemiologic studies depression scale: Review and revision (CESD and CESD-R). In: Maruish ME, editor. The use of psychological testing for treatment planning and outcomes assessment. 3rd ed. Mahwah, NJ: Lawrence Erlbaum; 2004. p. 363–377.
- Engels C, Ruscheweyh HJ, Beerenwinkel N, Lacroix C, Schwab C.. The common gut microbe Eubacterium hallii also contributes to intestinal propionate formation. Front Microbiol 2016; 7: 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epping EA, Mills JA, Beglinger LJ, Fiedorowicz JG, Craufurd D, Smith MM, et al. Characterization of depression in prodromal Huntington disease in the neurobiological predictors of HD (PREDICT-HD) study. J Psychiatr Res 2013; 47: 1423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Wang X, Yang S, Meng F, Wang X, Wei H, et al. Evaluation of the microbial diversity in amyotrophic lateral sclerosis using high-throughput sequencing. Front Microbiol 2016; 7: 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, et al. Neuroprotective effects of creatine in a transgenic mouse model of Huntington's disease. J Neurosci 2000; 20: 4389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald E, Murphy S, Martinson HA.. Alpha-synuclein pathology and the role of the microbiota in Parkinson’s disease. Front Neurosci 2019; 13: 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K, Mu CL, Farzi A, Zhu WY.. Tryptophan metabolism: A link between the gut microbiota and brain. Adv Nutr 2020; 11: 709–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt S, Mohajeri MH.. Changes of colonic bacterial composition in Parkinson's disease and other neurodegenerative diseases. Nutrients 2018; 10: 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund E, Overli O, Winberg S.. Tryptophan metabolic pathways and brain serotonergic activity: a comparative review. Front Endocrinol 2019; 10: 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdnack H, Wechsler Test of Adult Reading: WTAR San Antonio, TX: The Psychological Corporation; 2001. [Google Scholar]
- Huang Y, Shi X, Li Z, Shen Y, Shi X, Wang L, et al. Possible association of Firmicutes in the gut microbiota of patients with major depressive disorder. NDT 2018; 14: 3329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Stout JC, Solomon AC, Langbehn DR, Aylward EH, Cruce CB, et al. Beyond disgust: impaired recognition of negative emotions prior to diagnosis in Huntington's disease. Brain 2007; 130: 1732–44. [DOI] [PubMed] [Google Scholar]
- Kałużna-Czaplińska J, Gątarek P, Chirumbolo S, Chartrand MS, Bjørklund G.. How important is tryptophan in human health? Crit Rev Food Sci Nutr 2019; 59: 72–88. [DOI] [PubMed] [Google Scholar]
- Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, et al. Colonic bacterial composition in Parkinson's disease. Mov Disord 2015; 30: 1351–60. [DOI] [PubMed] [Google Scholar]
- Kieburtz K, Penney JB, Corno P, Ranen N, Shoulson I, Feigin A, et al. Unified Huntington’s Disease Rating Scale: reliability and consistency. Neurology 2001; 11: 136–42. [Google Scholar]
- Kong G, Cao KL, Judd LM, Li S, Renoir T, Hannan AJ.. Microbiome profiling reveals gut dysbiosis in a transgenic mouse model of Huntington's disease. Neurobiol Dis 2018; 135: 104268. [DOI] [PubMed] [Google Scholar]
- Kowalski K, Mulak A.. Brain–gut–microbiota axis in Alzheimer's disease. J Neurogastroenterol Motil 2019; 25: 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Cao K-A, Costello M-E, Lakis VA, Bartolo F, Chua X-Y, Brazeilles R, et al. MixMC: a multivariate statistical framework to gain insight into microbial communities. PLoS One 2016; 11: e0160169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis P, Young P, Holtrop G, Flint HJ.. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA: acetate CoA-transferase gene. Environ Microbiol 2010; 12: 304–14. [DOI] [PubMed] [Google Scholar]
- Mandal S, Van Treuren W, White RA, Eggesbo M, Knight R, Peddada SD.. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis 2015; 26: 27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderino L, Carroll I, Azcarate-Peril MA, Rochette A, Heinberg L, Peat C, et al. Preliminary evidence for an association between the composition of the gut microbiome and cognitive function in neurologically healthy older adults. J Int Neuropsychol Soc 2017; 23: 700–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques T, Holster S, Wall R, König J, Brummer R, de Vos WM, Correlating the gut microbiome to health and disease In: Hyland N, Stanton C, editors, The gut–brain axis. Academic Press; 2016. p. 261–91. [Google Scholar]
- Mayer EA, Knight R, Mazmanian SK, Cryan JF, Tillisch K.. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci 2014; 34: 15490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzetti AP, Fiorile MC, Primavera A, Lo Bello M.. Glutathione transferases and neurodegenerative diseases. Neurochem Int 2015; 82: 10–8. [DOI] [PubMed] [Google Scholar]
- McCourt AC, O'Donovan KL, Ekblad E, Sand E, Craufurd D, Rosser A, et al. Characterization of gastric mucosa biopsies reveals alterations in Huntington's disease. PLoS Curr 2015; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S.. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 2013; 8: e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn D, Riedel WJ, Sambeth A.. Effects of acute tryptophan depletion on memory, attention and executive functions: a systematic review. Neurosci Biobehav Rev 2009; 33: 926–52. [DOI] [PubMed] [Google Scholar]
- Newland PK, Heitkemper M, Zhou Y.. The emerging role of the gut microbiome in adult patients with multiple sclerosis. J Neurosci Nurs 2016; 48: 358–64. [DOI] [PubMed] [Google Scholar]
- O’Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF.. Serotonin, tryptophan metabolism and the brain–gut–microbiome axis. Behav Brain Res 2015; 277: 32–48. [DOI] [PubMed] [Google Scholar]
- Parashar A, Udayabanu M.. Gut microbiota: implications in Parkinson's disease. Parkinsonism Relat Disord 2017; 38: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, et al. Detection of Huntington's disease decades before diagnosis: the predict-HD study. J Neurol Neurosurg Psychiatry 2008; 79: 874–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 2012; 41: D590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulescu CI, Garcia-Miralles M, Sidik H, Bardile CF, Yusof N, Lee HU, et al. Manipulation of microbiota reveals altered callosal myelination and white matter plasticity in a model of Huntington disease. Neurobiol Dis 2019; 127: 65–75. [DOI] [PubMed] [Google Scholar]
- Reitan RM. Validity of the Trail Making Test as an indicator of organic brain damage. Percept Mot Skills 1958; 8: 271–6. [Google Scholar]
- Robbins A, Ho A, Barker R.. Weight changes in Huntington's disease. Eur J Neurol 2006; 13: e7. [DOI] [PubMed] [Google Scholar]
- Robbins TW, James M, Owen AM, Sahakian BJ, McInnes L, Rabbitt P.. Cambridge Neuropsychological Test Automated Battery (CANTAB): a factor analytic study of a large sample of normal elderly volunteers. Dement Geriatr Cogn Disord 1994; 5: 266–81. [DOI] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ.. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011; 10: 83–98. [DOI] [PubMed] [Google Scholar]
- Rowe KC, Paulsen JS, Langbehn DR, Duff K, Beglinger LJ, Wang C, et al. Self-paced timing detects and tracks change in prodromal Huntington disease. Neuropsychology 2010; 24: 435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowin J, Xia Y, Jung B, Sun J.. Gut inflammation and dysbiosis in human motor neuron disease. Physiol Rep 2017; 5: e13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell 2016; 167: 1469–80.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SR, Banerjee S.. Gut microbiota in neurodegenerative disorders. J Neuroimmunol 2019; 328: 98–104. [DOI] [PubMed] [Google Scholar]
- Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov Disord 2015; 30: 350–8. [DOI] [PubMed] [Google Scholar]
- Shoulson I, Fahn S.. Huntington disease clinical care and evaluation. Neurology 1979; 29: 1. [DOI] [PubMed] [Google Scholar]
- Smith A. Symbol digits modalities test. Los Angeles, CA: Western Psychological Services; 1982.
- Stilling RM, van de Wouw M, Clarke G, Stanton C, Dinan TG, Cryan JF.. The neuropharmacology of butyrate: the bread and butter of the microbiota–gut–brain axis? Neurochem Int 2016; 99: 110–32. [DOI] [PubMed] [Google Scholar]
- Stout JC, Queller S, Baker KN, Cowlishaw S, Sampaio C, Fitzer‐Attas C, et al. HD‐CAB: a cognitive assessment battery for clinical trials in Huntington's disease 1,2,3. Mov Disord 2014; 29: 1281–8. [DOI] [PubMed] [Google Scholar]
- Stoy N, Mackay GM, Forrest CM, Christofides J, Egerton M, Stone TW, et al. Tryptophan metabolism and oxidative stress in patients with Huntington's disease. J Neurochem 2005; 93: 611–23. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, et al. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol 2009; 8: 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol 2011; 10: 31–42. [DOI] [PubMed] [Google Scholar]
- Truax AD, Chen L, Tam JW, Cheng N, Guo H, Koblansky AA, et al. The inhibitory innate immune sensor NLRP12 maintains a threshold against obesity by regulating gut microbiota homeostasis. Cell Host Microbe 2018; 24: 364–78.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg JM, Gardiner SL, Ludolph AC, Landwehrmeyer GB, Roos RA, Aziz NA.. Body weight is a robust predictor of clinical progression in Huntington disease. Ann Neurol 2017; 82: 479–83. [DOI] [PubMed] [Google Scholar]
- van der Burg JM, Winqvist A, Aziz NA, Maat-Schieman ML, Roos RA, Bates GP, et al. Gastrointestinal dysfunction contributes to weight loss in Huntington's disease mice. Neurobiol Dis 2011; 44: 1–8. [DOI] [PubMed] [Google Scholar]
- Verwaest KA, Vu TN, Laukens K, Clemens LE, Nguyen HP, Van Gasse B, et al. (1)H NMR based metabolomics of CSF and blood serum: a metabolic profile for a transgenic rat model of Huntington disease. Biochim Biophys Acta 2011; 1812: 1371–9. [DOI] [PubMed] [Google Scholar]
- Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep 2017; 7: 13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FO. Huntington's disease. Lancet 2007; 369: 218–28. [DOI] [PubMed] [Google Scholar]
- Watkins L, Rogers R, Lawrence A, Sahakian B, Rosser A, Robbins T.. Impaired planning but intact decision making in early Huntington’s disease: implications for specific fronto-striatal pathology. Neuropsychologia 2000; 38: 1112–25. [DOI] [PubMed] [Google Scholar]
- Weis S, Schwiertz A, Unger MM, Becker A, Faßbender K, Ratering S, et al. Effect of Parkinson's disease and related medications on the composition of the fecal bacterial microbiota. NPJ Parkinsons Dis 2019; 5: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Lau AY.. Glutamate and glycine binding to the NMDA receptor. Structure 2018; 26: 1035–43.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan G, Yang N, Li S, Huang N, Fang X, Zhang J, et al. Abnormal gut microbiota composition contributes to cognitive dysfunction in SAMP8 mice. Aging 2018; 10: 1257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Long JD, Mills JA, Warner JH, Lu W, Paulsen JS, et al. Indexing disease progression at study entry with individuals at-risk for Huntington disease. Am J Med Genet B 2011; 156: 751–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data from these analyses are available upon reasonable request.