

Abstract
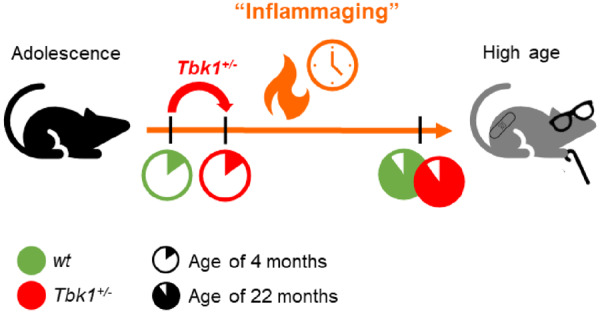
Loss-of-function mutations in TANK-binding kinase 1 cause genetic amyotrophic lateral sclerosis and frontotemporal dementia. Consistent with incomplete penetrance in humans, haploinsufficiency of TANK-binding kinase 1 did not cause motor symptoms in mice up to 7 months of age in a previous study. Ageing is the strongest risk factor for neurodegenerative diseases. Hypothesizing that age-dependent processes together with haploinsufficiency of TANK-binding kinase 1 could create a double hit situation that may trigger neurodegeneration, we examined mice with hemizygous deletion of Tbk1 (Tbk1+/− mice) and wild-type siblings up to 22 months. Compared to 4-month old mice, aged, 22-month old mice showed glial activation, deposition of motoneuronal p62 aggregates, muscular denervation and profound transcriptomic alterations in a set of 800 immune-related genes upon ageing. However, we did not observe differences regarding these measures between aged Tbk1+/− and wild-type siblings. High age did also not precipitate TAR DNA-binding protein 43 aggregation, neurodegeneration or a neurological phenotype in Tbk1+/− mice. In young Tbk1+/− mice, however, we found the CNS immune gene expression pattern shifted towards the age-dependent immune system dysregulation observed in old mice. Conclusively, ageing is not sufficient to precipitate an amyotrophic lateral sclerosis or frontotemporal dementia phenotype or spinal or cortical neurodegeneration in a model of Tbk1 haploinsufficiency. We hypothesize that the consequences of Tbk1 haploinsufficiency may be highly context-dependent and require a specific synergistic stress stimulus to be uncovered.
Keywords: ALS, ageing, inflammation, TBK1, neurogenetics
We found that hemizygous Tbk1 loss does not lead to motor neuron disease or an altered neuroinflammation in mice studied up to an age of 22 months. However, transcriptomic data suggest that Tbk1 haploinsufficiency prepones immune ageing in young mice.
Graphical Abstract
Graphical Abstract.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are fatal age-related neurodegenerative diseases affecting predominantly motor neurons (MNs) or neurons of the frontal and temporal cortex. This leads to peripheral and central palsy or cognitive deficits and personality changes, respectively. We have recently discovered that mutations in TANK-binding kinase 1 (TBK1) can cause both conditions (Freischmidt et al., 2015). The association between ALS/FTD and TBK1 is almost exclusively based on deleterious loss-of-function mutations. TBK1 loss-of-function mutations have been found evenly distributed over the TBK1 coding sequence, and consistently result in a loss of expression of the mutant TBK1 in both post-mortem brain tissue and patient-derived cell lines (Pottier et al., 2015; Catanese et al., 2019). Thus, TBK1 is haploinsufficient, and loss of one of the two TBK1 gene copies causes ALS. TBK1 is a regulator of selective autophagy (Wild et al., 2011; Weidberg and Elazar, 2011; Pilli et al., 2012), innate and adaptive immune responses (Hemmi et al., 2004, Jin et al., 2012; Yu et al., 2015), energy metabolism (Reilly et al., 2013, 2015) and tumorigenesis (Ou et al., 2011). Long before its discovery as an ALS gene the role of TBK1 in immune signalling has been studied in mice. Mice on a C57BL/6 background heterozygous for Tbk1 are viable while full knockouts die at E 14.5 due to liver necrosis (Bonnard et al., 2000). In contrast, mice carrying both Tbk1 alleles mutated to encode a protein with no catalytic activity on the 129S5 background are viable but develop severe immune cell infiltrates in multiple organs (Marchlik et al., 2010). Furthermore, mice with a heterozygous endogenous knock-in of the Tbk1G217R variant (almost completely abolished protein expression), as well as homozygous and heterozygous mice with a knock-in of Tbk1R228H (reduced kinase activity with normal protein expression) are viable and do not develop clinical or histological signs of motor symptoms up to 2 years of age (Gerbino et al., 2020). We and others have recently shown that heterozygous Tbk1 deletion prepones muscular denervation and tremor but extends survival in SOD1G93A transgenic mice, which are characterized by dysregulation of proteostasis and pronounced neuroinflammation.
Consequently, as TBK1 mutations display incomplete penetrance in patients and Tbk1+/− mice did not develop a neurological phenotype during the study period of 7 months (Brenner et al., 2019), we hypothesized that additional factors besides the Tbk1 loss-of-function mutation alone are required to cause signs of neurodegeneration or neuromuscular junction (NMJ) denervation.
Ageing is the strongest risk factor for ALS and FTD (Niccoli et al., 2017). To refer to the pro-inflammatory phenotype accompanying ageing in mammals the term ‘inflamm-ageing’ has been coined in 2000 (Franceschi et al., 2006). Moreover, age-related effects in the spinal cord, which are highly relevant for the modelling of ALS in rodents, can contrast with the respective alterations in the brain (Holtman et al., 2015; Galatro et al., 2017) and are poorly characterized to date.
Therefore, we set out to test the hypothesis that a double hit situation of heterozygous Tbk1 deletion and ageing could synergize to cause a manifest a subclinical ALS/FTD phenotype.
Materials and methods
Animals
Heterozygous B6.129P2-Tbk1tm1Aki mice (Tbk1+/− mice from now on; http://www.informatics.jax.org/allele/key/29752, accessed 25 August 2020; Hemmi et al., 2004) were kindly provided by Shizuo Akira. Mice were maintained at 22°C with a 14-/10-h light/dark cycle with food and water ad libitum. All animal experiments were performed in accordance with institutional guidelines of the University of Ulm and were approved by the local authority (Regierungspräsidium Tübingen, Germany; animal permission no. 1242).
Motor and behavioural testing
All animal behavioural tests were performed at Ulm University. The Viewer 3 software from ‘Biobserve’ was used to track and record animal movements automatically. Biweekly, male mice were subjected to weighing, motor testing and behavioural testing. Detailed test descriptions can be found in the Supplementary section.
Gene expression analysis and immunostaining
All immunostainings (including western blots) and Nanostring© methods were adapted from Brenner et al. (2019). Further information can be found in the Supplementary section of this manuscript. All male mice were sacrificed at the age of 4 or 22 months.
Immunostainings
Spinal cord and brain sections were stained with combinations of mouse anti-p62 (1:500, ab56416, Abcam), goat anti-ChAT (1:100, AB114P, Millipore), rabbit anti-GABARAPL1 (1:1000, 11010 AP, Proteintech), rat anti-Clec7a (1:30, mabg-mdect, InvivoGen), rabbit anti-Iba1 (1:500), goat anti-Iba1 (1:1000, 01919741, Wako), chicken anti-GFAP (1:1000, ab4674, Abcam), goat anti-Sox9 (1:100, AF3075-SP, R&D) and rabbit anti-PU.1 (1:100, 2258S, Abcam). APC anti mouse-CD31 (PECAM-1, 1:50, 102510, BioLegend), PE anti mouse-LY6-A/E (1:50, 108108, BioLegend) and rabbit anti-ITGA7 (1:100, ab203254, Abcam) were used for the representative pictures in the Supplementary section. Muscles were stained with α-bungarotoxin 488 (1:1000, B13422, Invitrogen) and anti-Synaptophysin (1:1000, ab32594, Abcam). Brain slices were further stained for TAR DNA-binding protein 43(TDP-43) (Rabbit-Anti-TDP-43, T1580, Sigma). Antibodies were diluted in TBS containing 0.25% (v/v) Triton X-100 and 5% (v/v) horse serum. Sections were incubated with the primary antibody overnight at 4°C, washed three times with TBS for 20 min and incubated with the secondary antibody for 1 h at RT protected from light. Secondary antibodies used for immunofluorescence were donkey anti-Rat/-Rabbit/-Mouse/-Goat as well as goat anti-Chicken/-Rabbit/-Mouse Alexa Fluor 488/546/647 (1:750, Invitrogen). NeuroTrace™ (NT) 530/615 Red and 640/660 Deep red from Thermo Fisher, 1:100, was used to mark neurons in the spinal cord and in the brain cortex. Slides were mounted with medium containing DAPI. Rabbit anti-LC3B (1:1000, 2775, Cell Signaling), mouse anti-SQSTM1/p62 (1:1000, ab56416, Abcam), rabbit anti-GAPDH (1:5000, 10494-1-AP, Proteintech) and secondary goat anti-mouse-HRP (1:1000, Life Technologies) and goat anti-rabbit-HRP (1:1000, Life Technologies) were used for staining of western blot membranes accordingly to Brenner et al. (2019).
Image and data analysis
An Axio Observer.A1 microscope (Zeiss) was only used to acquire pictures for MNs count. For all other stainings a TCS SP8 (Leica) confocal laser scanning microscope was used. All stainings were analysed using ImageJ software and plug-ins and by blinded investigators.
Statistical analysis
For comparison of multiple groups, the statistical significance of endpoints was evaluated by paired t-test or two-way ANOVA followed by Tukey’s multiple comparisons post hoc test. Data are presented as means ± SEM in bar graphs. Statistical significance is reported by the P-value of the statistical test procedures and was assessed as significant (*P < 0.05), strongly significant (**P < 0.01) or highly significant (***P < 0.001; ****P < 0.0001). All statistical analyses were performed with Prism software.
Data availability statement
Raw data were generated at Ulm University and are available upon request.
Results
Hemizygous loss of Tbk1 does not cause a behavioural phenotype in aged mice
Mice with heterozygous Tbk1 deletion lacked a phenotype until the age of 7 months in a previous study (Brenner et al., 2019). To test whether a double hit situation of hemizygous Tbk1 deletion and high age precipitates an ALS- or FTD-like phenotype in Tbk1+/− mice, we analysed Tbk1+/− mice and wild-type (wt) siblings for gross abnormalities, motor, social or cognitive deficits until the age of 22 months. Morphologically, Tbk1+/− mice were normal during the whole observation period (Fig. 1A). The weight curves (Fig. 1B) and rotarod performance (Fig. 1C) did not differ between Tbk1+/− and wt mice when tested on a weekly basis between 9 and 22 months. Moreover, as assessed by a test battery consisting of open field, Y-maze and three-chamber tests, Tbk1+/− mice did not show altered exploration behaviour, memory deficits or altered social interaction (Fig. 1D–F), respectively. Tbk1+/− mice did not exhibit a premature lethality (Fig. 1G).
Figure 1.

Mice lacking one Tbk1 allele do not show morphological or behavioural changes. (A) Photographs showing a wt mouse and a Tbk1+/− sibling. (B) Weight development is similar in Tbk1+/− mice and wt siblings during the study period. (C) Tbk1+/− mice and wt siblings show a similar performance in the rotarod test during the study period. (D) Tbk1+/− and wt mice show similar lengths of stay in the centre of the open field arena. (E) Spontaneous alternation performance value does not differ between Tbk1+/− and wt mice in the Y-maze test. (F) The lengths of stay in the social areas in the three-chamber test is not different between Tbk1+/− and wt mice. (G) Tbk1+/− and wt mice do not display premature lethality during the study period. N = 18–20 males per genotype. Data in B–F are shown as mean ± SEM and were analysed by paired t-test. Kaplan–Meier plot was analysed using the Log-rank (Mantel–Cox) test.
Decline in muscular innervation and autophagy depends on age but not Tbk1 haploinsufficiency
Tbk1+/ − and wt mice were sacrificed at an early- and a late-adult time-point (4 and 22 months) and were histopathologically and biochemically analysed to assess subclinical parameters potentially altered by age and/or Tbk1 haploinsufficiency. As shown in Fig. 2A–D, similar numbers of spinal MNs and cortical neurons in layer V were counted in Tbk1+/− and wt animals at both time points. Regarding NMJ integrity, proximal muscles (m. quadriceps) showed only a slight trend towards denervation at the higher age compared to the earlier time point (Fig. 2E and F), while a significant increase in partially or fully denervated motor endplates was found in distal muscles (m. lumbricalis) in old mice compared to young animals (Fig. 2G and H) consistent with age-related NMJ defects observed by a previous study (Valdez et al., 2012). This effect, however, was not modified by the Tbk1 genotype. Furthermore, as analysed by electron microscopy, the g-ratio (axon diameter/total diameter of the axon plus the myelin sheath as a measure for myelination), the amount of axon abnormalities and the diameter distribution did not differ between both genotypes at the age of 22 months (Fig. 2I–L).
Figure 2.

Hemizygous deletion of Tbk1 does not affect MN viability and muscular innervation. (A) Representative pictures of LSC anterior horn sections of both genotypes at 4 and 22 months stained for NT and choline acetyltransferase. Scale bar = 100 µm. (B) Quantification of LSC MNs shows similar numbers in Tbk1+/− and wt mice at either age. N = 6 mice. (C) Representative pictures of motor cortex sections of 4- and 22-month old Tbk1+/− and wt mice stained with NT. Scale bar = 50 µm. (D) Quantification of NT-positive neurons per mm2 in the V layer of the motor cortex shows similar numbers in Tbk1+/− when compared to aged matched wt mice. N = 6 mice. (E) Representative pictures of NMJs of the m. vastus medialis and m. quadriceps of 4- and 22-month old Tbk1+/− and wt mice stained for α-Bungarotoxin and synaptophysin. Scale bar = 50 µm. (F) Quantification of partially and fully denervated NMJs of the m. vastus medialis/m. quadriceps. N = 6–7 mice. (G) Representative pictures of NMJ of the m. lumbricalis of both genotypes at 4 and 22 months stained for α-Bungarotoxin and synaptophysin). White arrows indicate denervated NMJs. Scale bar = 50 µm. (H) Quantification of fully and partially denervated NMJs of the m. lumbricalis shows a genotype-independent age-related muscular denervation. N = 6–11 mice. (I) Representative pictures of transmission electron microscopy of the corticospinal tract in the LSC of 22-month old Tbk1+/− and wt mice. Scale bar = 20 µm. Lower higher magnification images show from left to right, examples of axons with myelin shield containing inner protrusion, detached axon, myelin out folding and an axon with degenerated myelin. Scale bar = 5 µm. (J) Graphs showing the correlation between the diameter and the g-ratio (axon diameter/total diameter of the axon plus the myelin sheath) per axon. N = 5 mice. (K) Quantification of different axon abnormalities. N = 4 mice. (L) Analysis of the axon diameters distribution shows no differences between 22-month old Tbk1+/− and wt mice. N = 5 mice. Data in B, D, F and H have been analysed by two-way ANOVA followed by Tukey’s multiple comparisons correction. **P < 0.01; *P < 0.05. Data in K and L have been analysed by t-test. All bar graphs are shown as mean ± SEM.
The regulation of selective autophagy is a well-characterized function of TBK1. Hemizygous deletion of TBK1, respectively, leads to accumulation of p62 positive intracytoplasmic protein inclusions in iPSC-derived MNs and spinal MNs of SOD1G93A mutant mice (Brenner et al., 2019; Catanese et al., 2019). However, hemizygous Tbk1 knock-out alone, i.e. without an additional burden of the proteostatic system by mutant SOD1G93A overexpression, did not lead to an accumulation of p62 inclusions in spinal MNs of young (4 months old) mice (Brenner et al., 2019). Ageing has been suggested to be associated with a decline of neuronal autophagic capacity (Stavoe et al., 2019). Consequently, we speculated whether Tbk1 haploinsufficiency would cooperate with ageing to impair autophagic function. In an immunohistological analysis, the number of spinal MNs containing tiny p62+ inclusions tended to be non-significantly higher in old compared to young mice, while we did not observe such a trend by heterozygous Tbk1 deletion (Fig. 3A and C). Since GABARAPL1, an ATG8 homologue, has been shown to be associated with autophagic vesicles (Chakrama et al., 2010) and we observed a co-localization with p62 (data not shown), we decided to further validate our results by quantification of the amount of GABARAPL1+ aggregates. We observed an age-dependent trend towards more GABARAPL1+ aggregates in the lumbar spinal cord (LSC), independent of the Tbk1 deletion (Fig. 3B and D). We further performed the same quantification for p62 and GABARAPL1 in the motor cortex of young and aged mice. Again, we could detect only a small trend towards more aggregates in the aged cohort, without any effect due to heterozygous Tbk1 deletion (Fig. 3E–H). Western blotting showed similar levels of the autophagy markers LC3-I, LC3-II and p62 in spinal cord and cortex lysates from young and old mice of both genotypes (Fig. 4A–G). As cytoplasmatic aggregates of (hyperphosphorylated) TDP-43 protein are the main neuropathological marker for ALS, we performed also TDP-43 staining in the motor cortex of 22-month old mice. However, we detected only nuclear TDP-43 staining but no cytoplasmatic TDP-43 aggregates in either genotype (Supplementary Fig. 1A).
Figure 3.

Autophagy in spinal cord and motor cortex is not influenced by hemizygous loss of Tbk1. (A) Representative pictures of LSC sections stained for NT and the autophagy marker p62. Scale bar = 100 µm. (B) Representative pictures of LSC sections stained for NT and the autophagy marker GABARAPL1. Scale bar = 100 µm. (C) and (D) Bar graphs showing the percentage of neurons containing p62 and GABARAPL1 positive inclusions in spinal cord of 4- and 22-month old mice from both genotypes. In both graphs a trend can be seen towards the aged cohorts. (E) and (F) Representative pictures of brain motor cortex sections stained for NT and p62 and NT and GABARAPL1. respectively. Scale bar = 50 µm. (G) and (H) Bar graphs showing the percentage of neurons containing p62 and GABARAPL1 positive inclusions in brain cortex of 4- and 22-month old mice from both genotypes. A genotype-independent age-related accumulation of GABARAPL1 inclusions is observed in the cortex of the transgene mice. All bar graphs are shown as mean ± SEM. All data analysed by two-way ANOVA followed by Tukey’s multiple comparisons. *P < 0.05. N = 5–6.
Figure 4.

Autophagic markers stay unchanged upon ageing and Tbk1 reduction in vivo. (A) Representative western blot picture of spinal cord and brain cortex lysates. Protein expression levels of LC3-I/-II and p62 have been analysed; 30 µg of protein have been loaded per lane. GAPDH has been used as loading control. (B), (D) and (F) bar graphs showing the quantifications of p62 and LC3-I/-II protein expression in mice spinal cord lysate while (C), (E) and (G) show quantifications of the same protein expression in brain cortex lysates. All bar graphs are shown as mean ± SEM. All data analysed by two-way ANOVA followed by Tukey’s multiple comparisons. N = 4.
Age-dependent but Tbk1-independent activation of glial cells
TBK1 is an important regulator of inflammatory responses, mainly by controlling the type I interferon response (Trinchieri, 2010; Ahmad et al., 2016; Brenner et al., 2019). Hemizygous deletion of Tbk1 substantially alters neuroinflammation in the CNS of SOD1G93A transgenic mice (Brenner et al., 2019), and mice carrying two Tbk1 mutated alleles (leading to a reduced and inactive protein) develop a pro-inflammatory auto-immune phenotype on the S129 background (Marchlik et al., 2010). Also, normal ageing has been repeatedly shown to be associated with a low-grade inflammation in higher organisms (Chung et al., 2019). Consequently, to determine whether Tbk1 haploinsufficiency would interact with age-associated inflammation, we analysed microglial and astrocytic markers in the CNS of young and aged Tbk1+/− and wt mice. We detected a robust increase in the number of cells double positive for the microglial/myeloid cell marker Iba1 and PU.1 in both the spinal cord and cortical grey matter of 22-month old mice compared to 4-month old mice (Fig. 5A–F). We further quantified the mean cell surface of double positive microglia in both tissues, and found a significant increase in cell size in the aged cohort in the spinal cord, but not in the motor cortex, where only the aged Tbk1+/− cohort had a significantly greater cell surface (Fig. 5F). However, heterozygous deletion of Tbk1 had no effect on glial cell numbers at either age.
Figure 5.

Tbk1 heterozygous loss does not influence age-related microglia activation in mice spinal cord and motor cortex. (A) Representative pictures of LSC sections stained for the microglia markers Iba1, Clec7a (Clc7) and PU.1. Enlargements show that only microglia of 22-month old mice are Clec7a-positive. Scale bar = 100 µm. (B) Quantification of Iba1+/PU.1+ cells in the grey matter of the LSC shows a genotype-independent increase in microglia numbers in 22-month old mice. (C) The mean cell size of Iba1+/PU.1+ microglial cells in the LSC differs between young and old but not between Tbk1+/− and wt mice. (D) Representative pictures of motor cortex sections stained for PU.1 and Iba1. Scale bar = 50 µm. (E) Age-dependent but genotype-independent increase in the numbers of Iba1+/PU.1+ microglia in the motor cortex. (F) The mean cell size of Iba1+/PU.1+ microglial cells in the motor cortex differs significantly only between young and old Tbk1+/− mice. (G) LSC sections stained for the astrocytic markers GFAP and Sox9. Enlargements show GFAP+/Sox9+ astrocytes. Scale bar = 100 µm. (H) Quantification of GFAP+/Sox9+ astrocytes in the LSC reveals similar numbers, independently of genotype and age. (I) The mean cell size of GFAP+/Sox9+ astrocytes in the LSC does not differ between Tbk1+/− and wt and young and old mice. (J) Representative pictures of motor cortex sections stained for GFAP and Sox9. Scale bar = 50 µm. (K) Quantification of GFAP+/Sox9+ astrocytes in motor cortex shows higher numbers only in aged wt mice compared to young ones. N = 6 for all analyses. (L) Volcano plot of RNA sequencing analysis of laser micro-dissected LSC MNs from wt and Tbk1+/− mice at the age of 22 months. Horizontal line indicates threshold for FDR (0.1). After FDR correction for multiple testing only four genes remain significantly altered (including Tbk1). (M)–(O) qPCR results of mRNA analysis from whole LSC lysates for validation of Avpr1a, Stat6 and Mon2 levels. N = 5. All bar graphs are shown as mean ± SEM. All data have been analysed by two-way ANOVA followed by Tukey’s multiple comparisons. *P < 0.05; **P < 0.01;***P < 0.001.
We additionally performed immunohistochemical staining of Clec7a. Clec7a is a prominent component of the disease-associated microglial signature downstream of Trem2, which has previously been described in various mouse models of neurodegeneration (Holtman et al., 2015; Krasemann et al., 2017). While Clec7a+ microglial cells were abundant in the LSC of aged mice, they were virtually absent in young mice spinal cords (Supplementary Fig. 1B). Again, we did not observe a Tbk1-dependent effect at either age.
Moreover, the number and the cell size of cells double positive for the astrocytic markers GFAP and Sox9 (Sun et al., 2017) was not affected by age or Tbk1 haploinsufficiency in the spinal cord (Fig. 5G–I). In contrast, the abundance of GFAP/Sox9 positive cells was mildly, but significantly increased in the motor cortex of aged wt compared to young wt mice. Again, there was no difference between Tbk1+/− and wt mice at either age (Fig. 5J and K).
Hemizygous deletion of Tbk1 causes only minor changes in the transcriptome of aged spinal cord MNs
In absence of behavioural or histopathological abnormalities in the motor system of Tbk1+/− mice we aimed to detect changes in gene expression that might reflect with high sensitivity possible consequences of the Tbk1 deletion. To that end, MNs were captured by laser microdissection from the LSCs of 22-month old Tbk1+/− and wt mice. Subsequent RNA sequencing detected a total of 11.095 transcripts per sample. After correction for multiple testing, apart from the expected downregulation of Tbk1 only the abundance of three RNA transcripts remained significantly different between Tbk1+/− and wt mice: Mon2, Stat6 (both upregulated) and Avpr1a (downregulated) (Fig. 5L). RT-qPCR quantification of these transcripts from whole spinal cord lysates showed similar levels in both genotypes (Fig. 5M–O). Moreover, both genotypes were not separated in a principal component analysis of the MN transcriptomes (data not shown). Conclusively, even a transcriptomic approach at the single cell level did not reveal evidence for major molecular alterations of aged Tbk1+/− mice compared to wt mice.
Haploinsufficiency of TBK1 shifts the spinal inflammatory transcriptome towards ageing in young mice
To corroborate our immunohistochemical analysis of neuroinflammation, we performed a quantitative NanoString ncounter© mRNA expression profiling of a large inflammation-related gene panel (800 genes) in total LSC lysates of mice of all four groups. Ageing significantly changed 219 mRNA transcripts in wt mice (Fig. 6A and Supplementary data). While the heterozygous Tbk1 deletion significantly altered five mRNA transcripts in 4-month old mice compared to age-matched siblings (Tbk1, Esam, Ly6a, Setd1a and Pecam1; Fig. 6F), we did not find significant differences between wt and Tbk1+/− siblings beyond Tbk1 at 22 months. Three of the five transcripts altered in young Tbk1+/− mice are predominantly expressed by endothelial cells (Newman, 1991; Hirata et al., 2001; Zhang et al., 2014). Consistently, analysis of cell type-specific marker gene signatures according to Danaher et al. (2017) showed a significant enrichment of endothelial markers in young Tbk1+/− mice compared to wt siblings (Fig. 6E). To identify the specific expression of these endothelial markers we also performed IHC staining for their protein product, LY6A and PECAM-1 and we could not observe higher extracellular expression other than at the endothelial level (Supplementary Fig. 1C and D). Furthermore, the microglia gene score was robustly increased in aged mice, while we did not observe differences between wt and Tbk1+/− mice (Fig. 6B), in line with our immunohistochemical analysis of microglial activation. Similarly, the astrocyte gene score was increased in aged wt mice compared to young ones. The astrocyte gene score did not differ between wt and Tbk1+/− siblings at high age, but we observed an increase in the astrocyte gene score due to Tbk1 loss at 4 months of age (Fig. 6C). The astroglial marker gene score is also composed of the genes Itga7, Nwd1 and Egfr. With a fold change of 1.15 Itga7 has the biggest impact on this score. Immunofluorescent staining for ITGA7 confirmed astrocytic expression (Supplementary Fig. 1). The oligodendrocyte marker score was unaltered in all comparisons (Fig. 6D).
Figure 6.

Haploinsufficiency of TBK1 shifts the spinal inflammatory transcriptome towards ageing in young mice. (A) Unbiased hierarchical cluster analysis (average linkage) on all 800 genes of the neuroinflammation panel in the spinal cord of young and aged wt and Tbk1+/− mice. Cluster analysis completely separates young from old mice and wt from Tbk1+/− mice at the age of 4 months. (B) The microglia-specific gene score shows an age-dependent increase that is independent of Tbk1. (C) The astrocyte-specific gene score is increased in Tbk1+/− mice compared to wt siblings at 4 but not at 22 months. The astroglia gene score is age-dependently increased in wt mice. (D) The oligodendrocyte-specific gene score is not affected by genotype or age. (E) The endothelial-specific gene score is increased in Tbk1+/− mice compared to wt siblings at 4 but not at 22 months. (F) Volcano plot showing 5 (including Tbk1) out of 800 genes after FDR correction for multiple testing differently regulated when comparing both genotypes at age 4 months. Horizontal line indicates threshold for FDR (0.05). (G) A principal component analysis of the same 800 gene panel shows a shift of the transcriptomic inflammatory profile of young Tbk1+/− mice towards that of old ones. (H) Positive correlation of age-associated changes with changes caused by Tbk1 haploinsufficiency in young mice, based on the 219 genes significantly altered by ageing. (B)–(E) Graphs have been analysed by two-way ANOVA followed by Tukey’s multiple comparisons. All bar graphs are shown as mean ± SEM. *P < 0.05; **P < 0.01;***P < 0.001. N = 5.
In an unbiased hierarchical cluster analysis based on the whole gene panel young mice were completely separated from old mice, and wt were incompletely distinguished from Tbk1+/− siblings at 4 months but not at 22 months of age (Fig. 6A). The impact of Tbk1 deletion in young animals was further corroborated by a principal component analysis, which showed that transcriptomic profile of young Tbk1+/− mice is separated from the wt mice and shifted towards the aged groups, while Tbk1 haploinsufficiency did not have an impact in old mice (Fig. 6G). We found further evidence for a mild shift in the immune transcriptome due to Tbk1 haploinsufficiency in young animals, in that the nominal expression changes induced by Tbk1 haploinsufficiency at young age correlated with the gene expression alterations induced by ageing (r = 0.4571; P < 0.0001; based on the 219 genes significantly regulated by age after false discovery rate (FDR) correction for multiple testing; Fig. 6H).
Discussion
Here, we provide a behavioural, histological and molecular in-depth analysis of mouse motor cortex and spinal cord with heterozygous Tbk1 knock-out at young and old age, to decipher whether and how the ALS/FTD-causing TBK1 mutation interacts with ageing. We observed age-dependent effects on glial activation, trends to altered markers of proteostasis and NMJ innervation of distal muscles that were independent of the Tbk1 genotype.
Employing RNA sequencing of laser-captured MNs of aged mice we found only very few mRNAs significantly altered by the heterozygous Tbk1 knock-out despite the presumed high sensitivity of this approach to detect possible cellular consequences of Tbk1 deletion. The alteration of these few transcript could not be validated by RT-qPCR from whole spinal cord tissue, suggesting that the observed alterations in Mon2, Stat6 and Avpr1 are restricted to one or few cell types (and thus, not detected in the whole tissue, as compared to laser microdissected MNs).
The results from our transcriptomic panel analysis of neuroinflammatory markers in the spinal cord are in line with previous reports of an age-dependent ‘inflamm-ageing’ in the mouse and brain (Franceschi et al., 2006; Norden and Godbout, 2013; Koellhoffer et al., 2017; Lin et al., 2018). The robust age-related alterations described in our analyses make it less likely that the complete lack of an ALS phenotype in old Tbk1+/− mice was due to a lack of biological ageing. Moreover, the fact that deletion of just one Tbk1 allele robustly modified markers of autophagy, NMJ denervation, neuroinflammation and even the motor phenotype in SOD1G93A transgenic mice (Brenner et al., 2019), show that a heterozygous Tbk1 knock-out (using the same mutation and mouse line as in this study) is principally sufficient to result in detectable biological alterations in the mouse CNS. However, these mutant Tbk1-linked effects seem to usually require a very specific context (or second hit) to be uncovered in the CNS. Tbk1 reduction modulates the disease in SOD1G93A mice by also slowing the progression at later stages, indicating that the loss of this kinase activity could actually be beneficial, in a disease model such as the SOD1G93A mouse, by reducing the innate immune response (Gerbino et al., 2020).
Although in none of the previously published studies the Tbk1+/− mice were as systematically investigated as in ours, the results obtained by us are in line with the recent study of Gerbino et al. In this study, Tbk1G217R heterozygous mice (largely reduced expression and therefore not viable in homozygous state) show no sign of motor deficits and MNs loss until 2 years of age and only age-related NMJs changes independent of the Tbk1 mutation (Gerbino et al., 2020). In the study presented here, Tbk1 heterozygous knockouts were thoroughly and deeply characterized by the use of a large battery of socio-cognitive and motor tests detailed RNA and protein analysis and ultrastructure analysis of spinal cord axons. No subclinical differences were detected even at very high age.
Surprisingly, however, we found evidence for a mild effect of the heterozygous Tbk1 knock-out in young mice, with a mild premature shift of the immune transcriptome towards ‘inflamm-ageing’, which may be relevant for ALS pathogenesis. The premature ‘inflamm-ageing’ in young Tbk1+/− mice is mainly based on an altered astrocyte and endothelial marker gene scores. In fact, of the four transcripts differently regulated between wt and Tbk1+/− at 4 months, three are associated with endothelial cells, making of them the element that contributes the most to the premature ‘inflamm-ageing’ in young Tbk1+/− mice.
In our analysis we found an upregulation of Ly6a, Esam and Pecam1 that are expressed by endothelial cells and are associated with the blood–brain barrier (BBB) function. While a stabilizing function for PECAM-1 was described in in vitro models of the BBB, the functions of LY6A and ESAM are still largely unknown in BBB function (Wimmer et al., 2019). The role of the BBB in ALS is the topic of active research. Initial studies using different superoxide dismutase 1 models provide evidence that disruption of the BBB precedes MN loss (Zhong et al., 2008; Miyazaki et al., 2011). The specific role of TBK1 in the regulation of endothelial cells and the BBB remains to be examined by further studies.
Taken together, our study provides valuable data on the robust age-dependent alterations in the mouse CNS, which are of importance for both ageing and ALS research. Moreover, we show that TBK1 mutations associated with ALS or FTD in humans do not in a simple way synergize with ageing, but rather prepone immune ageing in young-adult mice. Although age clearly is a risk factor for neurodegeneration, additional and more specific second hits that cooperate with Tbk1 mutations to cause ALS/FTD are probably waiting to be identified.
Supplementary Material
Acknowledgements
We would like to thank A. Sage, N. Todt and A. Jesionek for their technical support and the core facilities ‘Konfokale und Multiphotonen Mikroskopie’ as well as ‘Zentrale Einrichtung Elektronenmikroskopie’ of the University of Ulm. We thank Christine V. Möser and Ellen Niederberger (Pharmazentrum Frankfurt/Zentrum für Arzneimittelforschung, Entwicklung und Sicherheit, Institut für Klinische Pharmakologie, Klinikum der Goethe-Universität, Frankfurt, Germany) for sharing of mice and protocols.
Funding
This work was funded by the Baustein-Programm of the Medical Faculty of the University of Ulm (LSBR.0030 to D.B.); the Bruno and Ilse Frick Foundation for ALS Research (award 2015 to J.H.W.) and the Association pour la Recherche sur la Sclérose latérale amyotrophique et autres maladies du motoneurone (ARSla, France) and l’Aide à la Recherche des Maladies du Cerveau (ARMC, France) (to C.S.L.). This work benefited from equipment and services from the iGenSeq (RNA sequencing) and iCONICS (RNAseq analysis) core facilities at the ICM (Institut du Cerveau et de la Moelle épinière, Hôpital Pitié-Salpêtrière, PARIS, France), which received funding from the program ‘Investissements d’avenir’ ANR-10-IAIHU-06.
Supplementary material
Supplementary material is available at Brain Communications online.
Competing interests
The authors report no competing interests.
Glossary
- ALS =
amyotrophic lateral sclerosis
- BBB =
blood–brain barrier
- FDR =
false discovery rate
- FTD =
frontotemporal dementia
- LSC =
lumbar spinal cord
- MN =
motor neuron
- NMJ =
neuromuscular junction
- NT =
NeuroTrace™
- TBK1 =
TANK-binding kinase 1
- TDP-43/TARDBP =
TAR DNA-binding protein 43
- wt =
wild-type
References
- Ahmad L, Zhang SY, Casanova JL, Sancho-Shimizu V. Human TBK1: a gatekeeper of neuroinflammation [Review]. Trends Mol Med 2016; 22: 511–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnard M, Mirtsos C, Suzuki S, et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J 2000; 19: 4976–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner D, Sieverding K, Bruno C, Lüningschrör P, Buck E, Mungwa S, et al. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J Exp Med 2019; 216: 267–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanese A, Olde Heuvel F, Mulaw M, Demestre M, Higelin J, Barbi G, et al. Retinoic acid worsens ATG10-dependent autophagy impairment in TBK1-mutant hiPSC-derived motoneurons through SQSTM1/p62 accumulation. Autophagy 2019; 15: 1719–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrama FZ, Seguin-Py S, Le Grand JN, Fraichard A, Delage-Mourroux R, Despouy G, et al. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy 2010; 6: 495–505. [DOI] [PubMed] [Google Scholar]
- Chung HY, Kim DH, Lee EK, Chung KW, Chung S, Lee S, et al. Redefining chronic inflammation in aging and age-related diseases: proposal of the senoinflammation concept. Aging Dis 2019; 10: 367–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaher P, Warren S, Dennis L, D’Amico L, White A, Disis ML, et al. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer 2017; 5: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Bonafé M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging: an evolutionary perspective on immunosenescence. Ann N Y Acad Sci 2006; 908: 244–54. [DOI] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 2015; 18: 631–6. [DOI] [PubMed] [Google Scholar]
- Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci 2017; 20: 1162–71. [DOI] [PubMed] [Google Scholar]
- Gerbino V, Kaunga E, Ye J, Canzio D, O’Keeffe S, Rudnick ND, et al. The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 2020; 106: 789–805.e5. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 2004; 199: 1641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata K-I, Ishida T, Penta K, Rezaee M, Yang E, Wohlgemuth J, et al. Cloning of an immunoglobulin family adhesion molecule selectively expressed by endothelial cells. J Biol Chem 2001; 276: 16223–31. [DOI] [PubMed] [Google Scholar]
- Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun 2015; 3: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Xiao Y, Chang JH, Yu J, Hu H, Starr R, et al. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-κB signaling. Nat Immunol 2012; 13: 1101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koellhoffer EC, McCullough LD, Ritzel RM. Old maids: aging and its impact on microglia function. IJMS 2017; 18: 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017; 47: 566–81.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Liu Gene A, Perez E, Rainer RD, Febo M, Cruz-Almeida Y, et al. Systemic inflammation mediates age-related cognitive deficits. Front Aging Neurosci 2018; 10: 1663–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchlik E, Thakker P, Carlson T, Jiang Z, Ryan M, Marusic S, et al. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J Leukoc Biol 2010; 88: 1171–80. [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Ohta Y, Nagai M, Morimoto N, Kurata T, Takehisa Y, et al. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J Neurosci Res 2011; 89: 718–28. [DOI] [PubMed] [Google Scholar]
- Newman PJ. Switched at birth: a new family for PECAM-1. J Clin Invest 1999; 103: 5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccoli T, Partridge L, Isaacs AM. Ageing as a risk factor for ALS/FTD [Review]. Hum Mol Genet 2017; 26: R105–13. [DOI] [PubMed] [Google Scholar]
- Norden DM, Godbout JP. Microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 2013; 39: 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell 2011; 41: 458–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012; 37: 223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 2015; 130: 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SM, Ahmadian M, Zamarron BF, Chang L, Uhm M, Poirier B, et al. A subcutaneous adipose tissue-liver signalling axis controls hepatic gluconeogenesis. Nat Commun 2015; 6: 6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, et al. An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat Med 2013; 19: 313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavoe AK, Gopal P, Gubas A, Tooze SA, Holzbaur EL. Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. eLife 2019; 8: e44219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Cornwell A, Li J, Peng S, Osorio MJ, Aalling N, et al. SOX9 is an astrocyte-specific nuclear marker in the adult brain outside the neurogenic regions. J Neurosci 2017; 37: 4493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. Type I interferon: friend or foe? J Exp Med 2010; 207: 2053–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR. Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 2012; 7: e34640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Elazar Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci Signal 2011; 4: pe39. [DOI] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011; 333: 228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer I, Tietz S, Nishihara H, Deutsch U, Sallusto F, Gosselet F, et al. PECAM-1 Stabilizes blood-brain barrier integrity and favors paracellular T-cell diapedesis across the blood-brain barrier during neuroinflammation. Front Immunol 2019; 10: 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhou X, Chang M, Nakaya M, Chang JH, Xiao Y, et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat Commun 2015; 6: 6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex [published correction appears in J Neurosci 2015; 35: 846–6]. J Neurosci 2014; 34: 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O'Banion MK, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci 2008; 11: 420–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data were generated at Ulm University and are available upon request.