

Abstract
Familial hypokalaemic periodic paralysis is a rare skeletal muscle disease caused by the dysregulation of sarcolemmal excitability. Hypokalaemic periodic paralysis is characterized by repeated episodes of paralytic attacks with hypokalaemia, and several variants in CACNA1S coding for CaV1.1 and SCN4A coding for NaV1.4 have been established as causative mutations. Most of the mutations are substitutions to a non-charged residue, from the positively charged arginine (R) in transmembrane segment 4 (S4) of a voltage sensor in either CaV1.1 or NaV1.4. Mutant channels have aberrant leak currents called ‘gating pore currents’, and the widely accepted consensus is that this current is the essential pathological mechanism that produces susceptibility to anomalous depolarization and failure of muscle excitability during a paralytic attack. Here, we have identified five hypokalaemic periodic paralysis cases from two different ethnic backgrounds, Japanese and French, with charge-preserving substitutions in S4 from arginine, R, to lysine, K. An R to K substitution has not previously been reported for any other hypokalaemic periodic paralysis families. One case is R219K in NaV1.4, which is located at the first charge in S4 of Domain I. The other four cases all have R897K in CaV1.1, which is located at the first charge in S4 of Domain III. Gating pore currents were not detected in expression studies of CaV1.1-R897K. NaV1.4-R219K mutant channels revealed a distinct, but small, gating pore current. Simulation studies indicated that the small-amplitude gating pore current conducted by NaV1.4-R219K is not likely to be sufficient to be a risk factor for depolarization-induced paralytic attacks. Our rare cases with typical hypokalaemic periodic paralysis phenotypes do not fit the canonical view that the essential defect in hypokalaemic periodic paralysis mutant channels is the gating pore current and raise the possibility that hypokalaemic periodic paralysis pathogenesis might be heterogeneous and diverse.
Keywords: hypokalaemic periodic paralysis, voltage sensing domain, NaV, CaV, gating pore current
We have identified typical hypokalaemic periodic paralysis cases from two different ethnic backgrounds, Japanese and French, with charge-preserving substitutions in S4 from arginine, R, to lysine, K. One case has a mutation, R219K, in SCN4A, and other the four cases have a mutation, R897K, in CACNA1S gene.
Graphical Abstract
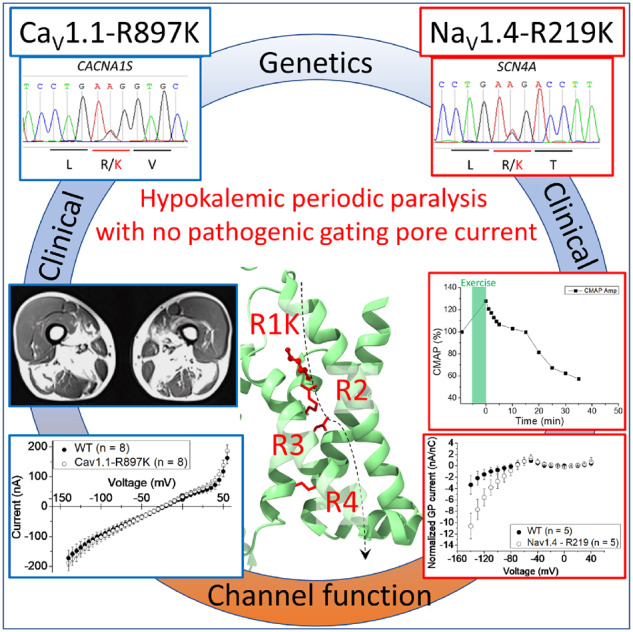
Graphical Abstract.
Introduction
Familial hypokalaemic periodic paralysis (HypoPP) is a rare skeletal muscle disease caused by the dysregulation of sarcolemmal excitability (Cannon, 2015). HypoPP is characterized by repeated episodes of weakness in the setting of hypokalaemia, and several mutations in CACNA1S encoding CaV1.1 and SCN4A encoding NaV1.4, have been identified as causative (Venance et al., 2006). Most mutations are substitutions of a positively charged arginine (R) in the fourth transmembrane segment (S4) within a voltage sensor domain (VSD) of either CaV1.1 or NaV1.4 (Cannon, 2010). The reason why mutations in two different channels can cause identical clinical manifestations had been a mystery for a long time. The explanation came when the biophysical experiments using Shaker K+ channel and voltage-gated Na+ channels demonstrated that missense mutations of arginine residues in the S4 segments create a leakage current, called the ‘omega current’ or ‘gating pore current’ (Starace and Bezanilla, 2004; Sokolov et al., 2007). Subsequently, similar leak currents were detected in muscle fibres isolated from HypoPP mouse models with CaV1.1-R528H and NaV1.4-R663H, S4 arginine to histidine knock-in mutations (Wu et al., 2011, 2012). Computational models of fibre excitability show that although the gating pore leak is small (about 5% of the total resting conductance of the fibre), the current is sufficient to cause paradoxical depolarization of the resting potential in low extracellular K+, which is the hallmark of HypoPP. This ‘gating pore current theory’ has been postulated to be the common pathogenic feature in HypoPP (Matthews et al., 2009; Cannon, 2010). Interestingly, a mutation of the α2 subunit of the Na+/K+-ATPase encoded by ATP1A2 has been recently reported to be responsible for HypoPP by producing anomalous leak current, which further strengthens the unifying pathomechanism (Sampedro Castañeda et al., 2018).
For 19 of 20 reported HypoPP mutations, the genetic lesion causes substitution of arginine to a non-charged residue in the S4 segment of a VSD. Functional expression studies have demonstrated gating pore currents in all 11 HypoPP R/X mutations in S4 segments of NaV1.4 channels (Sokolov et al., 2007; Struyk and Cannon, 2007; Struyk et al., 2008; Francis et al., 2011; Bayless-Edwards et al., 2018) and all 4 of the S4 R/X mutants tested in CaV1.1 (Wu et al., 2012, 2018b; Fuster et al., 2017a). For the one exception, p. Val876Glu in the S3 segment of domain III in CaV1.1, expression studies also detected a gating pore current (Fuster et al., 2017a). All available data are consistent with the notion that the gating pore current, usually as the result of a non-charged R/X missense mutation in the S4 segment of a VSD, is the essential anomaly that produces susceptibility to episodic loss of fibre excitability during attacks of weakness in HypoPP. Moreover, from the biophysical point of view on the structure–function relations for the VSDs in NaV1.4 or CaV1.1, it is plausible that the loss of a positively charged residue might be necessary for the generation of gating pore currents, by disrupting the interactions with the counter negatively charged residues, glutamate (E) or aspartate (D), in S2 and S3 (Tao et al., 2010; Pan et al., 2018).
Here, we report that five patients with typical HypoPP clinical phenotypes from four pedigrees (two Japanese and two French), harbouring missense from arginine to lysine (R/K) mutations, which preserves the positive charge in the voltage sensor in either NaV1.4 or CaV1.1. We detected a gating pore current for the R219K mutation in NaV1.4, albeit with a very small amplitude that was about 1/17th of that seen for other non-charge preserving R/X HypoPP mutations. Conversely, we were unable to detect a gating pore current for the R897K mutation in CaV1.1. Our results suggest that alternative mechanisms, besides the canonical gating pore current, may also be a cause of intermittent loss of fibre excitability in HypoPP.
Materials and methods
Patient consent and genetic analysis
We reported clinical information of Japanese patients under the approval of the ethical committee in Osaka university medical hospital. We conducted the genetic analysis after obtaining written form of the informed consent which has been approved by the ethical committee in Osaka university graduate school of medicine. Genetic diagnosis in French patients was performed in accordance with French Bioethics Laws.
Genomic DNA was extracted from the patients’ blood lymphocytes. The region comprising all exons of SCN4A, CACNA1S and KCNJ2 gene were amplified by PCR, and the purified fragments were sequenced using an automated DNA sequencer (Big Dye Terminator v 3.1 and ABI310; PE Applied Biosystems, Foster City, CA, USA).
Molecular biology
The α and β1 subunits of the rat skeletal muscle voltage-gated sodium channel (rNaV1.4) were cloned into pBSTA vectors for expression studies in Xenopus oocytes. For patch-clamp experiments using HEK293t cells, the human skeletal muscle NaV1.4 channel (hNaV1.4) and β1 subunit were cloned into the mammalian expression construct, pRc/CMV. The R291K missense mutation of NaV1.4 was introduced with the site-directed mutagenesis kit (KOD-Plus mutagenesis kit; TOYOBO, Osaka, Japan) and verified by Sanger sequencing.
The human α1S subunit of the calcium channel (hCaV1.1) was co-expressed with rat α2δ, rabbit β1 and mouse Stac3, as previously described (Wu et al., 2018b). The CaV1.1, Stac3 and β1 constructs were subcloned into an oocyte-optimized expression vector, pGEMHE (Liman et al., 1992) and the α2δ cDNA was subcloned into pcDNA3. Site-directed mutagenesis of CaV1.1 to create CaV1.1-R897K was performed with the QuikChange II mutagenesis kit (Agilent) and verified by sequencing the entire cDNA insert and flanking regions of pGEMHE.
Expression of rNaV1.4, rNaVβ1 and hCaV1.1 in Xenopus oocytes
For expression studies of rNaV1.4 in oocytes, cRNAs for the α and β1 subunits were transcribed in vitro using the T7 mMESSAGE cRNA kit (Ambion). Freshly isolated oocytes from Xenopus laevis were injected with 75 ng of a 1:1 molar ratio of cRNAs and maintained in standard oocyte saline solution: 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 200 mg/L sodium pyruvate, pH 7.4, for 1–4 days at 18°C. Expression studies of CaV1.1 were performed similarly, except 100 ng of the cRNA mixture (CaV1.1, α2δ, β1 and Stac3) was injected and oocytes were stored in 0.5× Leibovitz’s L-15 medium (HyClone) supplemented with 1% horse serum, 100 U/ml penicillin, 100 µg/ml streptomycin and 100 µg/ml amikacin. All experiments were performed within the guidelines established by the Institutional Animal Care and Use Committees at the University of Chicago and the University of California, Los Angeles.
Expression of hNaV1.4 and hβ1 in HEK293t cells
Expression studies of hNaV1.4 in mammalian cells were performed by transient transfection in HEK293t cells as previously described (Takahashi and Cannon, 1999). Briefly, plasmid cDNAs encoding wild type and mutant human NaV1.4 α-subunits (0.42 μg/35-mm dish), the human Nav channel β1-subunit (0.42 μg/35-mm dish) and a CD8 marker (0.08 μg/35-mm dish) were transfected by calcium phosphate precipitation.
Electrophysiology
Sodium ionic currents were measured using a conventional whole-cell patch-clamp technique as described previously (Takahashi and Cannon, 1999). Briefly, at 2–3 days after transfection, the Human embryonic kidney (HEK) cells were trypsinized briefly and passaged onto 12 mm diameter round glass coverslips for electrophysiological recording. Individual transfection-positive cells were identified by labelling with anti-CD8 antibody crosslinked to microbeads. Currents were recorded with an Axopatch 200B amplified (Molecular Devices, San Jose, CA, USA). The tip of the electrode was heat-polished, and the resistance was 1.8–2.5 MΩ. Series resistance was 2–6 MΩ, and 75–80% of the series resistance was compensated by the analog circuitry of the amplifier. Cells with peak currents of <1 or >10 nA upon step depolarization from –120 mV to –10 mV were excluded. The pipette (internal) solution consisted of (mM): 105 CsF, 35 NaCl, 10 ethylene-glycol tetraacetic acid and 10 Cs-HEPES (pH 7.4). The bath solution consisted of (mM): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5 glucose and 10 Na-HEPES (pH 7.4).
Charge-displacement currents and gating pore currents were recorded using a cut-open oocyte voltage clamp setup (Stefani and Bezanilla, 1998). For charge-displacement measurements, the external solution contained 115 mM N-methylglucamine-methylsulfonate (MS), 10 mM HEPES, and 2 mM Ca-MS, pH 7.4. The internal solution contained 115 mM N-methylglucamine-methylsulfonate, 10 mM HEPES and 2 mM ethylene-glycol tetraacetic acid, pH 7.4. For recording rNaV1.4 currents, oocytes were voltage-clamped at −100 mV for at least 5 min to remove slow inactivation. After measurements of gating currents, the solution in the chambers was replaced into the pair of solutions; the external solution contained 115 mM Na-MS, 10 mM HEPES and 2 mM Ca-MS, pH 7.4 and the internal solution contained 115 mM N-methylglucamine-methylsulfonate, 10 mM HEPES and 2 mM ethylene-glycol tetraacetic acid, pH 7.4. To block ionic currents, 10 μM tetrodotoxin was applied to the upper and guard chambers. After replacing solutions, the oocyte membrane was held at −100 mV, again, for at least 5 min, then the gating pore currents were recorded. For recording Ca2+ currents in oocytes expressing hCaV1.1 channels, the external solution contained 96 mM Na-MS, 6 mM Ca2+ acetate, 10 mM HEPES, pH 7.0 with methanesulfonic acid. In some gating pore experiments, the Na-MS was reduced to 60 mM and the solution was supplemented with 60 mM guanidinium acetate. The ‘early current’ 3–4 ms after a test depolarization was measured to test for the presence of a gating pore current. This time window is after the charge-displacement current has settled, but too early for activation of the Ca2+ current through the pore, and thereby provides an optimal interval isolating the gating pore current (Wu et al., 2018b). Blockers were not used to suppress the Ca2+ current to avoid the possibility of also blocking a gating pore current.
Data analysis
The currents from Xenopus oocytes were recorded with a PC44 board and digitized on the 16-bit A/D converter, and were sampled at 10 ms/point. The data acquisition program was developed in house. For gating currents measurements, linear leak and membrane capacitive currents were subtracted manually using currents obtained from a subtracting holding potential of +20 mV. All data were obtained at 20°C. Gating pore currents were obtained without any subtraction.
The ionic currents obtained from HEK293t cells were acquired by Digidata 1440 A (Molecular Devices, San Jose, CA, USA) and pClamp 10 software (Molecular devices, San Jose, CA, USA) was used for the data collection. Curve fitting was manually performed using Origin software (Microcal LLC, Northampton, MA, USA) as stated previously (Takahashi and Cannon, 1999).
Statistical analysis
All data indicate the mean values ± standard error of the mean. Statistical significance was determined in cases where P-values were less than 0.05 by unpaired t-test.
Data availability
The data which support the findings in this study are available from the corresponding authors when we receive the reasonable requests.
Results
Cases
Case 1
The proband is a 16-year-old Japanese boy. One day, the patient felt muscle pains after Judo class. On the next day, the weakness in upper limbs appeared, spreading to lower limbs on the following day. On the fourth day after the judo class, the patient was brought to the emergency department because of severe weakness in all limbs. Upon arrival, he had tetraparesis with a Medical Research Council (MRC) Grade 2/5 in upper limbs and the Grade 1/5 in the lower limbs. There is no family history of neuromuscular disorders. The laboratory data showed hypokalaemia (1.5 mmol/l) and hyperCKemia (603 U/l), but no abnormality in thyroid function or adrenal function. The ECG revealed abnormal U waves and prolonged Q-Tc, most likely due to hypokalaemia. Potassium chloride was administered intravenously, with monitoring of ECG and serum potassium. After a few days of treatment, his tetraparesis recovered completely, and he showed no neurological deficit. In order to confirm a suspected diagnosis of HypoPP, clinical neurophysiological assessment was performed. Nerve conduction study showed no abnormalities. Needle electromyography did not reveal myotonic discharges or spontaneous activity in all muscles examined including biceps brachii, first dorsalis interosseus, vastus lateralis and tibialis anterior muscles. The compound muscle action potential (CMAP) during the short exercise test showed no apparent changes (Supplementary Fig. 1). The prolonged exercise test, performed after potassium chloride administration, revealed a marked decrement of the CMAP amplitude (−30%) compared to the reference before the exercise task (Fig. 1B) (Fournier et al., 2004; Tan et al., 2011). Genetic analysis using the DNA extracted from the patient’s and both parents’ lymphocytes was performed. The proband had a heterozygous substitution (c. 2690G>A) in CACNA1S gene resulting in p. R897K mutation in CaV1.1, which was not present in either parent. The clinical, CMAP exercise, and genetic data support a diagnosis of de novo HypoPP type 1, with R897K missense mutation of CaV1.1 (Fig. 1A).
Figure 1.

Clinical information of HypoPP patients with the R/K substitution in either CACNA1S or SCN4A. (A) Schematic representation of R897K location in CaV1.1 channel (upper panel) and the result of the Sanger sequencing obtained from Case 1. (B) Prolonged exercise test in Case 1 elicited an early increase and a late decrease of ∼30% for the CMAP. (C) MRI images of skeletal muscles in Case 2-2. T1 weighted images (upper panel) and short tau inversion recovery images (lower panel) revealed fatty infiltration in soleus and medial gastrocnemius muscles. Quadriceps, gracilis, sartorius, semi-membranosus and semi-tendinosus muscles are grossly respected. (D) CT scan images of skeletal muscle in Case 3. CT scan image of lumbar portion (upper panel) showed low density area in paraspinal muscles, indicating fatty infiltration. CT scan image of thigh (lower panel) revealed low density area in bilateral hamstrings indicating fatty infiltration. (E) Schematic representation of the R219K location in NaV1.4 (upper panel) and the results of Sanger sequencing obtained from Case 4. (F) Prolonged exercise test in Case 4 revealed a decrement of the CMAP by 40%, which supports a diagnosis of periodic paralysis.
Cases 2-1 and 2-2
The probands were two French brothers. The elder brother, Case 2-1, experience repeated episodes of tetraparesis with hypokalaemia since adolescence. He developed a myopathy at age 55, at which time a muscle biopsy showed vacuoles and tubular aggregates. The patient died from liver cancer. A clinical electrophysiological assessment was never performed. The younger brother, Case 2-2, showed repeated episodes of tetraparesis since age 16. Serum potassium concentration during the paralytic attack was measured only once, which showed around 1 mmol/l. He reported 3–4 attacks per year over a period of 10 years that were triggered by rest after strenuous exercise, alcohol or carbohydrates-rich meals. The prolonged exercise test at the age of 57 showed a significant decrement of CMAP (−31%) representing the pattern V according to Fournier’s classification. The frequency of the paralytic attacks decreased with the administration of acetazolamide and potassium chloride supplementation. Beginning at age 59, permanent weakness of the pelvic girdle muscle was noticed. The patient complained of limitations with running, climbing stairs and getting up from the ground. The lower-extremity MRI scan performed at age 64 showed muscle atrophy, fatty changes and the short tau inversion recovery hyperintensity in the posterior compartment of the lower leg (thigh and calve) (Fig. 1C, Supplementary Fig. 2). The genetic analysis of the two brothers showed a heterozygous R897K mutation in CACNA1S gene. Their father also had similar symptoms. They had four sisters who were asymptomatic and never had genetic testing.
Case 3
The proband was a French boy who presented with a first episode of tetraparesis at age 20, following a football match. The blood potassium level measured during the paralytic attack was 2 mmol/l. He experienced episodes of tetraparesis once or twice a month for about 10 years. Triggering factors were rest after sustained exercise, cold weather and carbohydrate-rich meals. Treatment with potassium chloride supplementation and potassium-sparing diuretics (spironolactone) significantly reduced the frequency of the paralytic attacks. Since the age of 46, he developed progressive permanent weakness of the pelvic girdle with difficulties climbing stairs or rising from squatting positions. A CT scan showed paravertebral and pelvic muscle atrophy with fatty infiltration (Fig. 1D). The prolonged exercise test revealed a 25% decrement of the CMAP amplitude, whereas a 40% decrement is considered diagnostic of periodic paralysis (Fournier et al., 2004; Tan et al., 2011). The genetic analysis showed a heterozygous R897K mutation in CACNA1S gene. He has three unaffected siblings who have never been genetically tested, and there is no family history of neuromuscular disorders.
Case 4
The proband was a 36-year-old Japanese male with no family history of neuromuscular disorders. Since the age of 30, he experienced transient muscle weakness in the lower limbs, twice a month on average, but never visited the clinic. At the age of 31, he was transferred to the emergency department and hospitalized because of severe tetraparesis. The laboratory data, while still weak, showed hypokalaemia (2.1 mmol/l), hyperCKemia (541 U/L), normal thyroid function and normal adrenal function. The ECG showed low amplitude T wave in aVF and III, and U waves in V2–V5, most likely due to hypokalaemia. By giving potassium chloride, the hypokalaemia normalized, followed by the recovery of tetraparesis. At age 34, the patient experienced similar attacks of severe tetraparesis again and consulted a neurologist for the diagnosis. When he was evaluated in our outpatient clinic, there were no abnormal findings in the neurological examination. The needle electromyography examination did not reveal myotonic discharges, but the prolonged exercise test elicited a large decrement of CMAP amplitude, more than 40% (Fig. 1F) (Fournier et al., 2004; Tan et al., 2011). The genetic analysis revealed the heterozygous substitution (c.656G>A) in the SCN4A gene resulting in p. R219K in NaV1.4 (Fig. 1E). The material from other family members was not available for genetic analysis.
In addition to the specific variants identified for each of the cases above, we performed comprehensive whole-exon screening in all five patients for genes associated with familial periodic paralysis, including SCN4A (NM_000334.4), CACNA1S (NM_000069.3) and KCNJ2 (NM_000891.2). No other variants, except for known single nucleotide polymorphisms, were detected. Neither p. R219K in NaV1.4 nor p. R897K in CaV1.1 has been registered in the database of Tohoku Medical Megabank Organization (ToMMo), which has genotypes of 47 000 Japanese individuals (4.7KJPN), nor in the ExAC database. PolyPhen-2 (Adzhubei et al., 2010) showed that R219K in NaV1.4 is a ‘probably damaging’ mutation with a score of 1.000, whereas R897K in CaV1.1 is a ‘benign’ mutation with a score of 0.013. By Mutation Taster (Schwarz et al., 2010), both R219K in NaV1.4 and R897K in CaV1.1 are predicted as ‘disease-causing’ mutations. Sorting Intolerant From Tolerant (SIFT) (Sim et al., 2012) displayed that both R219K in NaV1.4 and R897K in CaV1.1 are ‘intolerant’. Clinical information for all cases is summarized in Table 1.
Table 1.
Clinical information summary
| Case | Pedigree (race) | 1 | 2 |
3 | 4 | |
|---|---|---|---|---|---|---|
| (Japanese) | (French) |
(French) | (Japanese) | |||
| 2-1 | 2-2 | |||||
| Sex | Male | Male | Male | Male | Male | |
| Genetic diagnosis | R897K in CACNA1S | R219K in SCN4A | ||||
| Onset | 16 years old | Adolescence | 16 years old | 20 years old | 30 years old | |
| Family history | None (de novo) | + (brothers and their father) | None | None | ||
| LET | +(30% decrement) | N/A | +(31% decrement) | +(25% decrement) | +(43% decrement) | |
| Serum potassium during attack | Hypokalaemia (1.5 mmol/l) | Hypokalaemia | Hypokalaemia | Hypokalaemia | Hypokalaemia (2.1 mmol/l) | |
| Other features | HyperCKemia(603 U/l) during PP attack | Myopathy (+) (vacuoles and tubular aggregates),Died of liver cancer | Acetazolamide effective | Myopathy (+) | HyperCKemia (541 U/l) during PP attack | |
LET indicates the prolonged exercise test.
Functional analysis of NaV1.4-R219K and CaV1.1-R897K
Functional expression studies of ion channel mutations associated with periodic paralysis have revealed derangements in the voltage-dependent gating of NaV1.4, and specifically for HypoPP, the presence of an anomalous gating pore current conducted through a ‘leaky’ VSD of NaV1.4 or CaV1.1 HypoPP mutant channels (Sokolov et al., 2007; Struyk and Cannon, 2007; Cannon, 2010, 2015; Mi et al., 2014; Wu et al., 2018b). The HypoPP mutations previously shown to support gating pore currents are almost all (19 of 20) missense mutations in the first or second arginine residues of S4 transmembrane segments (i.e. near the extracellular end). The charge-preserving R/K mutations reported herein are located at the first arginine of S4 in domain I of NaV1.4 (R219K, Fig. 1E) and at the first arginine of S4 in domain III of CaV1.1 (R897K, Fig. 1A). We, therefore, measured Na+ currents to characterize the voltage-dependence of gating for NaV1.4 WT and R219K channels and tested for the presence of anomalous gating pore currents in NaV1.4-R219K and CaV1.1-R897K by measuring currents at hyperpolarized potentials in the presence of pore blockers.
Voltage-dependence of gating was not disrupted by the NaV1.4-R219K mutation
Sodium currents were measured from HEK293t cells transiently transfected with plasmids encoding WT or R219K NaV1.4 plus the axillary β1 subunit. Expression of WT and R219K mutant channels was comparable with Na+ current densities of −394.9 ± 104.0 A/F for WT (n = 7) and −306.5 ± 68.0 A/F for R219K (n = 8) based on the peak current at a test potential of −10 mV. The voltage-dependence of activation was indistinguishable for WT and R219K channels, as shown by the overlapping data in the conductance–voltage relation (Fig. 2A right axis and Supplementary Fig. 3). Similarly, the inactivation gating behaviour of R219K channels was comparable to WT for steady-state voltage-dependence (Fig. 2A, left axis), as were the kinetics of entry to and recovery from inactivation (Fig. 2B). All gating parameters of Na+ ionic currents are shown in Table 2.
Figure 2.

The functional analysis of NaV1.4-R219K gating. (A) The voltage dependent conductance (G–V relationship) and the voltage dependent steady-state fast inactivation (V-hinf) in wild type NaV1.4 (n = 7) and NaV1.4-R219K (n = 8) are shown. The pulse protocols used are shown as insets, in the right for G-V and in the left for V-hinf. (B) Voltage dependent time constants for fast inactivation (Tau–V relationship) in wild type NaV1.4 (n = 5–7) and NaV1.4-R219K (n = 6–8) are shown. The left upper panel indicates the two-pulse protocol for the entry to fast inactivation from −80 mV to −50 mV, and the left lower for the recovery from fast inactivation from −110 mV to −90 mV, respectively. Error bars indicate standard error of the mean. (C) Representative charge-displacement currents from the wild type of NaV1.4 (WT) and NaV1.4 with R219K mutation (NaV1.4-R219K). The pulse protocol used is shown as inset in the left. (D, E) The relative charge moved by the voltage sensors (integral of charge-displacement current) is shown as a function of test potential in the Q–V relationship (D) and Tau–V relationship (E) for WT (n = 5) and NaV1.4-R219K (n = 5), respectively. The time constant of gating currents were fitted by two-exponential fitting and plotted both fast components and slow ones separately (opened triangles and squares). The mean of time constant was calculated by the weighted average and are shown in filled marks.
Table 2.
Parameters for ionic Na+ currents in patch clamp recording fitted by two state model
| Activation |
Steady-state fast inactivation |
|||
|---|---|---|---|---|
| k (mV/e-fold) | V 1/2 (mV) | k (mV/e-fold) | V 1/2 (mV) | |
| WT (n = 7) | 4.2 ± 0.4 | −25.5 ± 1.4 | 4.6 ± 0.2 | 62.6 ± 0.8 |
| R219K (n = 8) | 5.0 ± 0.4 | −22.5 ± 1.6 | 4.7 ± 0.3 | 63.7 ± 0.9 |
V 1/2 indicates the mid-point value of the steady-state fast inactivation curve (V-hinf) and the voltage dependence of activation (G-V). k is the slope factor. Errors indicate standard error of the mean.
Movement of the channel voltage-sensor in response to a change in membrane potential was measured as the charge-displacement current in the cut-open oocyte preparation (Fig. 2C–E), where the channel expression is higher and the clamp speed is faster. The voltage for the mid-point of charge displacement, as well as the steepness with voltage, were indistinguishable for WT and R219K channels, which is consistent with the expectations for a charge-conserving mutation of the voltage-sensor domain.
Gating pore current is small for NaV1.4-R219K and undetectable for CaV1.1-R897K
The oocyte expression system was used to test for the presence of anomalous gating pore currents because the sensitivity is increased by the higher membrane expression of channel density compared to HEK293t cells. The Na-conducting pore was blocked by tetrodotoxin, and steady-state currents were measured for a series of test depolarizations from a holding value of −100 mV. The current amplitude was divided by the maximal charge-displacement, to normalize for the expression level of NaV1.4 channels in each oocyte, and plotted as a function of test potential (Fig. 3A). An excess of inward current (negative) was detected at voltages of −80 mV and more negative, for oocytes expressing NaV1.4-R219K channels compared to WT. This difference is consistent with a gating pore current. Moreover, the inward rectification of this current shows the gating pore current is activated by hyperpolarization, which is expected for a mutation in the outermost arginine of S4 (R219K). The inward displacement of the S4 segment with hyperpolarization brings the R219K mutation into the hydrophobic charge transfer centre, where a tight interaction with R219 normally prevents the leakage of ions. The amplitude of the R219K gating pore current, however, is much smaller than those observed in other HypoPP-2 mutant NaV1.4 channels (Struyk and Cannon, 2007; Struyk et al., 2008; Francis et al., 2011).
Figure 3.

Gating pore current is small in NaV1.4-R219K and undetectable in CaV1.1-R897K. (A) Gating pore current was observed for test potentials < −80 mV (n = 5, respectively). Leak currents recorded were subtracted by the linear fitted line from −20 mV to +20 mV in each oocyte. Error bars indicate standard error of the mean. No gating pore current was detected for oocytes expression CaV1.1-R897K in two different conditions: (B) external Na+ or (C) a mixture of 60 mM N-methylglucamine and 60 mM guanidine. For (B), n = 8 oocytes, and for (C), n = 6 oocytes.
The WT or R897K CaV1.1 channels were co-expressed with the α2δ, β1 and Stac3 subunits in oocytes. Membrane expression of WT channels was higher than for R897K mutant channels, based on the maximum charge displacement, 0.60 ± 0.074 nC (n = 8) and 0.29 ± 0.018 nC (n = 8), respectively. We are confident that the expression level of R897K channels was sufficient to detect a gating pore current because we had previously demonstrated robust gating pore currents for the HypoPP mutation CaV1.1-R528H when the maximum charge displacement was comparable (0.25 nC) (Wu et al., 2018b). In an external Na+ solution, the early current for oocytes expressing CaV1.1-R897K was not different from those expressing WT CaV1.1 (Fig. 3B). For many, but not all, anomalous gating pore conductances created by R/X missense mutations in S4, including CaV1.1-R528G (Wu et al., 2018b), the gating pore current amplitude is dramatically enhanced in the presence of external guanidinium. We, therefore, tested CaV1.1-R897K in 60 mM external guanidinium, but again the steady-state current was identical to that observed in WT CaV1.1 (Fig. 3C). We conclude that under the expression conditions used in this study, there was no detectable gating pore current for CaV1.1-R897K channels.
Discussion
In this study, we report five cases of HypoPP from four different pedigrees in which a charge-preserving missense mutation (R/K) in S4 of a VSD was identified in CaV1.1 or NaV1.4. Expression studies revealed a small gating pore current for NaV1.4-R219K channels, but no detectable gating pore current for CaV1.1-R897K channels.
The clinical phenotypes of these R/K cases are typical for HypoPP
Recurrent episodic attacks of severe paresis with ictal hypokalaemia (<3.0 mmol/l) was observed in all five patients. Episodes were triggered by rest after exercise, ethanol or carbohydrate-rich meals, and clinical improvement occurred with acetazolamide, K supplementation or K-sparing diuretics. Missense variants were identified in all cases, with four having CaV1.1-R897K and one with NaV1.4-R219K. These variants were not found in unaffected family members or in gene databases of subjects without neuromuscular disease. The familial transmission was demonstrated in one family with CaV1.1-R897K, and the others were de novo. Moreover, another missense mutation at this residue, CaV1.1-R897S, has been associated with a severe HypoPP phenotype (Chabrier et al., 2008; Hanchard et al., 2013). The prolonged CMAP exercise test was consistent with HypoPP [pattern V, with ≥40% decrement (Fournier et al., 2004; Tan et al., 2011)] for the NaV1.4-R219K case and for one of the three CaV1.1-R897K patients who were tested, with the other two being borderline (25–31% decrement). Among the CaV1.1-R897K cases, 3 of 4 had clinical and imaging evidence of proximal myopathy, and in one a muscle biopsy showed vacuolar changes and T-tubular aggregates. The one case with NaV1.4-R219K did not have fixed myopathy, but he is younger than the others at age 34. Taken together, the clinical data provide convincing evidence of typical HypoPP. The genetic variants segregate with affected individuals, and all occur at arginine residues in S4 segments, adjacent to established R/X HypoPP mutations. Therefore, we propose that the CaV1.1-R897K and NaV1.4-R219K variants are disease-causing mutations.
The anomalous gating pore current typically found for HypoPP mutant channels was not detected in the R/K mutants
The gating pore current arising from non-conserved missense mutations at the first or second arginine in the S4 segments of CaV1.1 or NaV1.4 has been a consistent finding in HypoPP mutant channels. Gating pore currents are present for all 11 HypoPP R/X mutations of NaV1.4 reported to date (Sokolov et al., 2007; Struyk and Cannon, 2007; Struyk et al., 2008; Francis et al., 2011; Bayless-Edwards et al., 2018). Expression studies have been performed on 7 of the 9 HypoPP mutations identified in CaV1.1. Gating pore currents were observed for six of these mutant constructs: one in a knock-in mutant mouse (Wu et al., 2012), four expressed in oocytes (Wu et al., 2018a, b) which included the same mutant construct in the knock-in mouse, and another two by transient expression in mouse skeletal muscle (Fuster et al., 2017a, b). This latter group includes the one atypical HypoPP mutation that is not located at an arginine in S4 (V876E). We did not detect a gating pore current for the HypoPP mutation CaV1.1-R900S expressed in oocytes, but another HypoPP mutation at this same R residue caused by substitution with a smaller amino acid, CaV1.1-R900G, did support a typical gating pore current (Wu et al., 2018a). In aggregate, these multiple studies provide compelling evidence that the gating pore current is an essential contributor to the pathogenesis of HypoPP.
A gating pore current was detected for NaV1.4-R219K (Fig. 3A), but the amplitude was considerably small than those currents observed in other NaV1.4 HypoPP mutant channels. For example, at the typical resting potential of skeletal muscle of −90 mV, the gating pore current for R219K was −2.8 nA/nC (Fig. 3A), whereas for our prior studies of HypoPP-2 mutations at R669, R672, or R1132, the gating pore current amplitude was −48.5 ± 13 nA/nC (range −17.3 to −99.7 nA/nC). A contributing factor to the small amplitude of the R219K gating pore current at −90 mV was the left-shifted (more negative) voltage for inward rectification which was about −80 mV. Similarly, we found a relatively negative rectification voltage (−45 mV) for the R669H channel, which consequently also had the smallest amplitude gating pore current (−17.3 nA/nC) amongst the HypoPP mutant channels in S4 of domain II. Both R219 and R669 are the outermost arginines in the S4 segments of domain I and II, respectively, and so a more negative membrane hyperpolarization is required to sufficiently shift the S4 segment far enough inward such that the mutant residue misaligns with the hydrophobic charge transfer centre to produce the leak. By comparison, the rectification voltage of the gating pore current for the R672 HypoPP mutant channels (second arginine from outside) is −25 mV (Struyk et al., 2008).
We used our model simulation of fibre excitability (Cannon, 2018) to test whether the small-amplitude gating pore current observed for NaV1.4-R219K is sufficient to cause the paradoxical depolarization that occurs in an attack of HypoPP. Figure 4 shows the membrane potential of a simulated fibre as the extracellular K is varied. In normal muscle for which there is no gating pore current (Fig. 4, solid line in black), the membrane potential becomes more negative (hyperpolarized) as K is reduced because of the Nernst potential for K shifts to more negative potentials (e.g. 4.5 mM to 3.5 mM). In extremely low [K+], 2.25 mM in this example, a paradoxical depolarization to −50 mV occurs because a reduction of the inward rectifier K+ current is no longer able to maintain the normal resting potential. At this depolarized potential, the fibre is chronically refractory and inexcitable leading to flaccid paralysis. Such low values of [K+] do not normally occur in vivo, and so we do not have attacks of weakness. The addition of the gating pore current, however, shifts this catastrophic depolarization to higher [K+] that may overlap with the low physiologic range. The question is whether the small gating pore current observed for NaV1.4-R219K is sufficient to cause susceptibility to HypoPP. As described above, the amplitude of the gating pore current at −90 mV for a typical NaV1.4 HypoPP mutant channel expressed in a Xenopus oocyte is −48 nA/nC. This current density is equivalent to a slope conductance of 12 µS/cm2 in a muscle fibre that is heterozygous for the HypoPP mutation, as in patients (Mi et al., 2014). The addition of this gating pore leakage current now causes paradoxical depolarization in 3.5 mM [K+], which simulates the HypoPP phenotype (Fig. 4, solid line in red). If the amplitude of the gating pore current is reduced to 50% of the prior value, then depolarization is predicted to occur for [K+] = 2.9 mM, a low value that rarely occurs and so an attack of HypoPP is expected to be very infrequent. If the gating pore current is further reduced to one-fourth the typical value in HypoPP, then [K+] must be reduced to 2.6 mM for the fibre to depolarize. Similarly, with one-eighth the gating pore current, [K+] must be 2.4 mM for depolarization. The gating pore current for NaV1.4-R219K at −90 mV was one-seventeenth the amplitude of a typical HypoPP mutant channel. According to our model simulations (Fig. 4), such a small leakage current is not predicted substantially shift the point of catastrophic depolarization to a [K+] value different from normal muscle. Therefore, we conclude the small gating pore current observed for NaV1.4-R219K is not sufficient to cause a risk for HypoPP.
Figure 4.
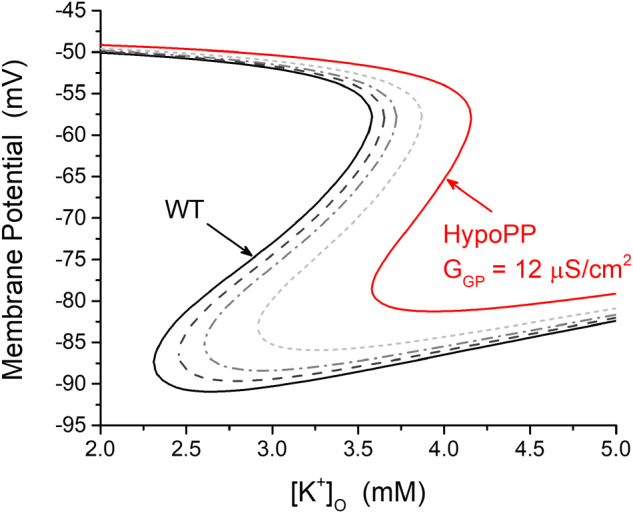
Simulation shows the small gating pore current for NaV1.4-R219K is not predicted to cause susceptibility to HypoPP. Curves show membrane potential as the extracellular [K+] is varied, for a simulated muscle fibre. Black trace indicates the response for a normal fibre, without gating pore currents. In this case, paradoxical depolarization occurs only for extremely low [K+]O, < 2.25 mM. The red trace shows the response for a representative HypoPP fibre with a gating pore conductance, GGP = 12 µS/cm2, set at the mean value observed for S4 R/X HypoPP mutant channels of domains II and III. The gating pore current causes the right shift of the curve, such that paradoxical depolarization now occurs for [K+]O in the low physiological range (e.g. 3.5 mM). The additional curves show the response of the membrane potential if the gating pore conductance is reduced to half (short-dashed line in light grey), one-fourth (dash-dot line in grey) or one-eighth (dashed line in dark grey) of the typical value for a HypoPP mutation (solid line in red). By comparison, the gating pore current for NaV1.4-R219K at −90 mV is 17 times smaller than for other HypoPP mutant channels, which is insufficient to be a risk for HypoPP by the mechanism in this simulation.
We were unable to detect a gating pore current for CaV1.1-R897K in the oocyte expression system. This failure is not because of limited expression of the mutant channel in the membrane because the charge-displacement current (a functional measure of voltage-sensor activity in the membrane) was comparable to the value we achieved with CaV1.1-R528H HypoPP channels that had robust gating pore currents (Wu et al.., 2018b). Moreover, we used a physiological extracellular solution (primarily Na+) and did not use CaV1.1 pore blockers (e.g. Co2+) that might have inadvertently blocked a gating pore conductance. Chloride-free solutions were used to greatly reduce the contribution of endogenous Cl− currents in oocytes, which interfere with the detection of a gating pore current. The charge-conserving R/K substitution raises the interesting theoretical possibility of a Cl−-selective gating pore leak. Although we did not test for such a possibility, a small leakage Cl− current is not predicted to alter the excitability of a muscle fibre because the resting Cl− conductance via ClC-1 channels is enormous. Other data clearly show that a missense mutation at CaV1.1-R897 is capable of supporting a gating pore current. The CaV1.1-R897S mutation has been identified in two de novo HypoPP cases, both of which had severe clinical phenotypes (Chabrier et al., 2008; Hanchard et al., 2013), and expression studies in oocytes revealed gating pore currents about 2× larger than values observed for the most common HypoPP mutation, CaV1.1-R528H (Wu et al., 2018a). Another possibility is that the oocyte expression system is inadequate to reveal the creation of an anomalous gating pore conductance for CaV1.1-R897K channels. Many potential causes could be proposed: an interacting protein is missing, post-translational modification is different in the oocyte or an endogenous substance in the oocyte occludes the gating pore conductance. Expression studies in mammalian muscle fibres are needed to explore these possibilities.
Other hypotheses for pathological mechanisms of HypoPP
In addition to the anomalous gating pore current, other functional defects of HypoPP mutant channels have been reported. For HypoPP mutations of NaV1.4 with typical clinical features, several loss-of-function defects have been reported. Inactivation (fast or slow) may be enhanced (Jurkat-Rott et al., 2000; Struyk et al., 2000), current density may be reduced, or the coupling of voltage-sensor movement to channel opening may be impaired (Mi et al., 2014). While these loss-of-function changes would exacerbate a depolarization-induced inhibition of Na channel availability arising from the gating pore current, these loss-of-function defects alone do not cause depolarization-induced weakness in low potassium. Because the NaV1.4-R219K variant was identified in a single proband and no genetic information is available for the parents, we cannot exclude the possibility R219K is a benign variant. If this were the case, then we propose the HypoPP phenotype may be from another gene (one-third of clinically definite HypoPP patients do not have an identified mutation in CACNA1S or SCN4) or in a non-coding region of CACNA1S or SCN4A. Atypical forms of HypoPP that are unusual because of coexisting myotonia or HyperPP have mixed gain-of-function and loss-of-function defects (Sugiura et al., 2003; Kokunai et al., 2018; Luo et al., 2018). Alternative pathomechanisms are less apparent for HypoPP mutations of CaV1.1 without gating pore currents. The fundamental challenge is that CaV1.1 normally has little or no influence on the membrane resting potential, and so altered gating or expression level of mutant CaV1.1 is not expected to destabilize the resting potential. Two CaV1.1 mutations with convincing clinical phenotypes of typical HypoPP do not have detectable gating pore currents in the oocyte expression system: the charge-conserving CaV1.1-R897K herein and the non-conserved CaV1.1-R900S (Wu et al., 2018a). These exceptions raise the possibility that another class of functional defect remains to be discovered for CaV1.1 HypoPP mutant channels.
Conclusion
This study describes five cases with typical clinical features of HypoPP, but with previously unreported charge-preserving amino acid substitutions at the outermost arginines in the voltage sensor of domain III in CaV1.1 or domain I in NaV1.4. No gating pore current was detectable for CaV1.1-R897K, and only a small gating pore current was observed for NaV1.4-R219K. Simulation studies suggest the small gating pore current for NaV1.4-R219K is not sufficient to cause paradoxical depolarization in low K+. These findings suggest additional functional defects, besides the gating pore leakage current, may cause susceptibility to HypoPP.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
All authors appreciate Dr. Francisco Bezanilla and Dr. Ana M. Correa for kind equipment support of cut-open voltage clamp set-up and providing the rNav1.4 clone and rNavβ1 clone. We thank Ms. Kimie Hayashi for her technical supports.
Funding
This work is supported by Japan Society for the Promotion of Science (JSPS) KAKENHI (JP18K07524) and by Takeda Science Foundation Grant for medical science research to T.K., by the Research Grant for Intractable Disease from the Ministry of Health, Labour and Welfare (H29-Nanchitou(Nan)-Ippan-030 and 20FC1036), the Japan Agency for Medical Research and Development (JP19ek0109230 and JP19bm0804005) and JSPS KAKENHI (JP15K09314) to M.P.T., by grants from the Institut national de la santé et de la recherche médicale (INSERM), Sorbonne University and the French Muscular Dystrophy Association (AFM-Telethon) (strategic grant to B.F.). Support was provided by National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health (AR063182 to S.C.C.).
Competing interests
The authors report no competing interests.
Glossary
- CMAP =
compound muscle action potential
- HypoPP =
hypokalaemic periodic paralysis
- MRC =
Medical Research Council
- VSD =
voltage sensor domain
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayless-Edwards L, Winston V, Lehmann-Horn F, Arinze P, Groome JR, Jurkat-Rott K.. NaV1.4 DI-S4 periodic paralysis mutation R222W enhances inactivation and promotes leak current to attenuate action potentials and depolarize muscle fibers. Sci Rep 2018; 8: 10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC. Voltage-sensor mutations in channelopathies of skeletal muscle. J Physiol 2010; 588: 1887–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC. Channelopathies of skeletal muscle excitability. Compr Physiol 2015; 5: 761–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC. Sodium channelopathies of skeletal muscle. Handb Exp Pharmacol 2018; 246: 309–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrier S, Monnier N, Lunardi J.. Early onset of hypokalaemic periodic paralysis caused by a novel mutation of the CACNA1S gene. J Med Genet 2008; 45: 686–8. [DOI] [PubMed] [Google Scholar]
- Fournier E, Arzel M, Sternberg D, Vicart S, Laforet P, Eymard B, et al. Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol 2004; 56: 650–61. [DOI] [PubMed] [Google Scholar]
- Francis DG, Rybalchenko V, Struyk A, Cannon SC.. Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia. Neurology 2011; 76: 1635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster C, Perrot J, Berthier C, Jacquemond V, Allard B.. Elevated resting H + current in the R1239H type 1 hypokalaemic periodic paralysis mutated Ca 2+ channel. J Physiol 2017. a; 595: 6417–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster C, Perrot J, Berthier C, Jacquemond V, Charnet P, Allard B.. Na leak with gating pore properties in hypokalemic periodic paralysis V876E mutant muscle Ca channel. J Gen Physiol 2017. b; 149: 1139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchard NA, Murdock DR, Magoulas PL, Bainbridge M, Muzny D, Wu YQ, et al. Exploring the utility of whole-exome sequencing as a diagnostic tool in a child with atypical episodic muscle weakness. Clin Genet 2013; 83: 457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkat-Rott K, Mitrovic N, Hang C, Kouzmenkine A, Iaizzo P, Herzog J, et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA 2000; 97: 9549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokunai Y, Dalle C, Vicart S, Sternberg D, Pouliot V, Bendahhou S, et al. A204E mutation in Nav1.4 DIS3 exerts gain- and loss-of-function effects that lead to periodic paralysis combining hyper- with hypo-kalaemic signs. Sci Rep 2018; 8: 16681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P.. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 1992; 9: 861–71. [DOI] [PubMed] [Google Scholar]
- Luo S, Sampedro Castañeda M, Matthews E, Sud R, Hanna MG, Sun J, et al. Hypokalaemic periodic paralysis and myotonia in a patient with homozygous mutation p.R1451L in NaV1.4. Sci Rep 2018; 8: 9714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology 2009; 72: 1544–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi W, Rybalchenko V, Cannon SC.. Disrupted coupling of gating charge displacement to Na+ current activation for DIIS4 mutations in hypokalemic periodic paralysis. J Gen Physiol 2014; 144: 137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Li Z, Zhou Q, Shen H, Wu K, Huang X, et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with β1. Science 2018; 362: 10.1126/science.aau2486. [DOI] [PubMed] [Google Scholar]
- Sampedro Castañeda M, Zanoteli E, Scalco RS, Scaramuzzi V, Marques Caldas V, Conti Reed U, et al. A novel ATP1A2 mutation in a patient with hypokalaemic periodic paralysis and CNS symptoms. Brain 2018; 141: 3308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D.. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–6. [DOI] [PubMed] [Google Scholar]
- Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC.. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012; 40: W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T, Catterall WA.. Gating pore current in an inherited ion channelopathy. Nature 2007; 446: 76–8. [DOI] [PubMed] [Google Scholar]
- Starace DM, Bezanilla F.. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature 2004; 427: 548–53. [DOI] [PubMed] [Google Scholar]
- Stefani E, Bezanilla F.. Cut-open oocyte voltage-clamp technique. Methods Enzymol 1998; 293: 300–18. [DOI] [PubMed] [Google Scholar]
- Struyk AF, Cannon SC.. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol 2007; 130: 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struyk AF, Markin VS, Francis D, Cannon SC.. Gating pore currents in DIIS4 mutations of NaV1.4 associated with periodic paralysis: saturation of ion flux and implications for disease pathogenesis. J Gen Physiol 2008; 132: 447–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struyk AF, Scoggan KA, Bulman DE, Cannon SC.. The human skeletal muscle Na channel mutation R669H associated with hypokalemic periodic paralysis enhances slow inactivation. J Neurosci 2000; 20: 8610–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, Makita N, Li L, Noble PJ, Kimura J, Kumagai Y, et al. Cold induces shifts of voltage dependence in mutant SCN4A, causing hypokalemic periodic paralysis. Neurology 2003; 61: 914–8., [DOI] [PubMed] [Google Scholar]
- Takahashi MP, Cannon SC.. Enhanced slow inactivation by V445M: a sodium channel mutation associated with myotonia. Biophys J 1999; 76: 861–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SV, Matthews E, Barber M, Burge JA, Rajakulendran S, Fialho D, et al. Refined exercise testing can aid DNA-based diagnosis in muscle channelopathies. Ann Neurol 2011; 69: 328–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Lee A, Limapichat W, Dougherty DA, MacKinnon R.. A gating charge transfer center in voltage sensors. Science 2010; 328: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain 2006; 129: 8–17. [DOI] [PubMed] [Google Scholar]
- Wu F, Mi W, Burns DK, Fu Y, Gray HF, Struyk AF, et al. A sodium channel knockin mutant (NaV1.4-R669H) mouse model of hypokalemic periodic paralysis. J Clin Invest 2011; 121: 4082–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Mi W, Hernández-Ochoa EO, Burns DK, Fu Y, Gray HF, et al. A calcium channel mutant mouse model of hypokalemic periodic paralysis. J Clin Invest 2012; 122: 4580–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Quinonez M, Cannon SC.. Gating Pore Currents in DIII Hypopp Mutations of Ca V 1.1 (Conference abstract). Biophys J 2018. a; 114: 639a. [Google Scholar]
- Wu F, Quinonez M, DiFranco M, Cannon SC.. Stac3 enhances expression of human CaV1.1 in Xenopus oocytes and reveals gating pore currents in HypoPP mutant channels. J Gen Physiol 2018. b; 150: 475–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data which support the findings in this study are available from the corresponding authors when we receive the reasonable requests.