

Abstract
Multiple sclerosis is a complex autoimmune disease caused by a combination of genetic and environmental factors. Translation of Genome-Wide Association Study findings into therapeutics and effective preventive strategies has been limited to date. We used summary-data-based Mendelian randomization to synthesize findings from public expression quantitative trait locus, methylation quantitative trait locus and Multiple Sclerosis Genome-Wide Association Study datasets. By correlating the effects of methylation on multiple sclerosis, methylation on expression and expression on multiple sclerosis susceptibility, we prioritize genetic loci with evidence of influencing multiple sclerosis susceptibility. We overlay these findings onto a list of ‘druggable’ genes, i.e. genes which are currently, or could theoretically, be targeted by therapeutic compounds. We use GeNets and search tool for the retrieval of interacting genes/proteins to identify protein–protein interactions and druggable pathways enriched in our results. We extend these findings to a model of Epstein-Barr virus-infected B cells, lymphoblastoid cell lines. We conducted a systematic review of prioritized genes using the Open Targets platform to identify completed and planned trials targeting prioritized genes in multiple sclerosis and related disease areas. Expression of 45 genes in peripheral blood was strongly associated with multiple sclerosis susceptibility (False discovery rate 0.05). Of these 45 genes, 20 encode a protein which is currently targeted by an existing therapeutic compound. These genes were enriched for Gene Ontology terms pertaining to immune system function and leucocyte signalling. We refined this prioritized gene list by restricting to loci where CpG site methylation was associated with multiple sclerosis susceptibility, with gene expression and where expression was associated with multiple sclerosis susceptibility. This approach yielded a list of 15 prioritized druggable target genes for which there was evidence of a pathway linking methylation, expression and multiple sclerosis. Five of these 15 genes are targeted by existing drugs and three were replicated in a smaller expression Quantitative Trait Loci dataset (CD40, MERTK and PARP1). In lymphoblastoid cell lines, this approach prioritized 7 druggable gene targets, of which only one was prioritized by the multi-omic approach in peripheral blood (FCRL3). Systematic review of Open Targets revealed multiple early-phase trials targeting 13/20 prioritized genes in disorders related to multiple sclerosis. We use public datasets and summary-data-based Mendelian randomization to identify a list of prioritized druggable genetic targets in multiple sclerosis. We hope our findings could be translated into a platform for developing targeted preventive therapies.
Keywords: multiple sclerosis, Mendelian randomization, therapeutics
The genetic architecture of multiple sclerosis is complex, spanning across >200 loci in the genome. Here, we integrate genomic, expression and methylation datasets to prioritize potentially druggable target genes which could be modulated to influence susceptibility to multiple sclerosis.
Graphical Abstract
Graphical Abstract.

Introduction
Genome-wide association studies (GWAS) in multiple sclerosis have revealed over 200 risk loci associated with an increased risk of developing the disease (International Multiple Sclerosis Genetics Consortium, 2019b). However, translating findings from GWAS into therapeutic strategies have proved challenging for several reasons. GWAS provide insights into potential genomic risk loci that are likely to harbour single or multiple causal variants. Despite the advent of analytical techniques such as fine-mapping, advanced annotation tools, co-localization and Mendelian randomization for inferring causal variants from risk loci, difficulties remain in inferring with certainty which variants are truly causal. Understanding how these variants mechanistically influence disease phenotypes provides additional challenges (Wu et al., 2018). These challenges arise from factors such as complex linkage disequilibrium and potential effects on distant (i.e. trans) genes. In addition, dynamic, context-specific effects of variants are likely to vary depending on time, cell type and context.
Multiple sclerosis is a complex autoimmune disease, which is a leading cause of disability in young people. There are currently neither effective cures nor preventive measures for multiple sclerosis. Licenced therapies for multiple sclerosis include rationally designed monoclonal antibodies, such as ocrelizumab (anti-CD20), natalizumab (anti-alpha4 integrin) and alemtuzumab (anti-CD52) and fingolimod and other sphingosine-1-phosphate-receptor modulators, in addition to drugs with multiple mechanisms of action such as cladribine, glatiramer acetate and beta-interferon (Tintore et al., 2019). Autologous haematopoietic stem cell transplant has an increasing evidence base for use in multiple sclerosis. Existing highly active therapies are effective at controlling inflammatory disease activity, but have several serious side-effects; alongside this, prognostication at the time of diagnosis remains imperfect. There is thus an unmet clinical need for highly effective therapies with a better safety profile, which is even more important when considering preventive drugs for individuals at high-risk of disease.
Many GWAS hits reside in intronic or intergenic regions, or within genes that do not make attractive druggable targets, for example due to conformational considerations, cellular localization or concerns about off-target effects. Outside of rare, missense or nonsense coding variants, moving from GWAS hits into druggable targets has had limited success. Expression quantitative trait loci (eQTLs) studies provide a tissue-specific measure of genetic expression and enable a deeper understanding of the influence of transcription levels alongside genetic variation. Summary-data-based Mendelian randomization (SMR) provides a data-driven approach for integrating GWAS data with gene expression data from eQTL studies to prioritize potentially causal genes from GWAS hits (Zhu et al., 2016). SMR extends the concept of Mendelian randomization, enabling testing of the hypothesis that genetically determined levels of gene expression are associated with a disease phenotype.
In this paper, we focus on a previously curated list of 4479 genes that were identified as ‘druggable’ on the basis of various considerations (Finan et al., 2017). We apply SMR to recent multiple sclerosis GWAS data, complemented by pathway analysis to provide a data-driven prioritization of druggable genes in multiple sclerosis.
Materials and methods
Datasets
Multiple sclerosis GWAS data were provided by the International MS Genetics Consortium (IMSGC) from the latest IMSGC discovery-stage summary statistics (International Multiple Sclerosis Genetics Consortium, 2019b). These data originate from 14 802 individuals with multiple sclerosis and 26 703 controls of European descent. Allele frequencies for single nucleotide polymorphisms (SNPs) in the multiple sclerosis GWAS discovery data were obtained from the 1000 genomes samples of European ancestry (n GWA03) (Consortium, T. 1000 G. P., & The 1000 Genomes Project Consortium, 2015).
For SMR analyses, we used several eQTLs datasets. The primary analysis used cis-eQTL data from the eQTLgen consortium. This dataset contains cis-eQTLs for all 19 250 genes expressed in whole blood obtained from 31 684 individuals. Each SNP–gene pair for which data was available in ≥2 cohorts and with a SNP–gene distance of ≤1 MB was tested (Võsa et al., 2018). Data were downloaded from the eQTLgen consortium website (https://www.eqtlgen.org/cis-eqtls.html) (Võsa et al., 2018). Whole blood results obtained with the eQTLgen dataset were replicated using the Consortium for the Architecture of Gene Expression (CAGE) consortium eQTL data obtained from peripheral blood of 2765 individuals (Lloyd-Jones et al., 2017). CAGE data were downloaded from the SMR website (https://cnsgenomics.com/software/smr/#DataResource). Data on eQTLs in lymphoblastoid cell lines (LCLs)—Epstein-Barr virus (EBV)-immortalized B cells—were obtained from the Geuvadis consortium results available from the SMR website (Lappalainen et al., 2013).
For multi-omic SMR, we used peripheral blood methylation quantitative trait locus (mQTL) data from a meta-analysis of the Lothian Birth Cohort and the Brisbane Systems Genetics Study (McRae et al., 2018; Wu et al., 2018). These data are limited to DNA methylation probes with ≥1 cis-mQTL associated at P ass × 10−0 and SNPs ≤2 MB from each DNA methylation probe. mQTL data were downloaded from the SMR website.
Druggable genome
The druggable genome can be defined as the set of protein-coding genes for which the gene products could potentially be modulated by therapeutic compounds. This includes protein products which are already targeted by existing drugs and proteins with structural and functional properties suggestive of druggability but which are not currently targeted by existing compounds. The list of druggable genes used in this work was taken from Supplementary Table 1 of the paper by Finan et al. (2017). The final list was developed from a list of protein-coding genes, T-cell receptor genes, immunoglobulins, polymorphic pseudogenes and selected non-protein-coding genes believed to have functional consequences. Genes were classified into three tiers based on their druggability. Genes were classified as ‘Tier 1’ if they were already being targeted by compounds in clinical use or clinical development. ‘Tier 2’ genes were not currently targeted by existing compounds but have a peptide sequence product with high sequence homology to ‘Tier 1’ druggable genes. ‘Tier 3’ genes incorporated gene products with a degree of peptide sequence homology to targets of existing compounds, genes encoding major classes of druggable protein (kinases, ion channels, G-protein-coupled receptors, nuclear hormone receptors and phosphodiesterases), genes encoding extracellular proteins (either secreted or membrane-bound) and cluster of differentiation (CD) antigen genes. Tier 3 was divided into 3A and 3B based on proximity to GWAS hits for various common diseases, with genes ≤50 KB from a GWAS hit deemed more likely to be druggable (3A).
Summary-data-based Mendelian randomization
SMR is a technique used to determine associations between genetically determined traits, such as gene expression and methylation, and outcomes of interest, such as disease phenotypes (Zhu et al., 2016). eQTLs refer to genetic variants which are associated with levels of expression of a particular transcript. These are derived from the measurement of gene expression, most commonly using RNA sequencing, which is then correlated with genotyping data. Importantly, eQTLs are time and tissue-specific: the genetic regulation of gene expression varies widely between tissues and depends on context.
In SMR, SNP association statistics with an outcome (e.g. a disease phenotype) are regressed on SNP association statistics with expression of a particular transcript to determine an estimate approximating the effect on the disease phenotype for a genetically determined increase in the expression of that transcript. If βE is the per-allele beta for a genetically determined increase in gene expression, and βD is the per-allele log odds ratio for a binary disease phenotype, then the SMR estimate for the effect of genetically determined increased expression of that transcript βSMR is:
A key assumption of this approach is that the same underlying causal variant determines both gene expression and the disease phenotype. Due to LD, it is possible that βSMR could be non-zero even when this assumption is violated, i.e. if SNP i is in linkage with SNP j, which determines the expression of transcript t, and is also in linkage with SNP k, which directly influences the disease phenotype, then βSMR may be non-zero despite no direct causal pathway from SNP to transcript to disease [see Fig. 1 in Wu et al. (2018)]. This is different to the ‘vertical pleiotropy’ situation upon which instrumental variable analysis and MR are based, which assumes a direct causal pathway among genetic variant, gene expression and disease phenotype. Importantly, SMR cannot distinguish between vertical pleiotropy—the situation in which variant influences phenotype via gene expression, and horizontal pleiotropy, the situation in which variant influences phenotype and gene expression, but influences the phenotype at least partly independently of gene expression.
Figure 1.
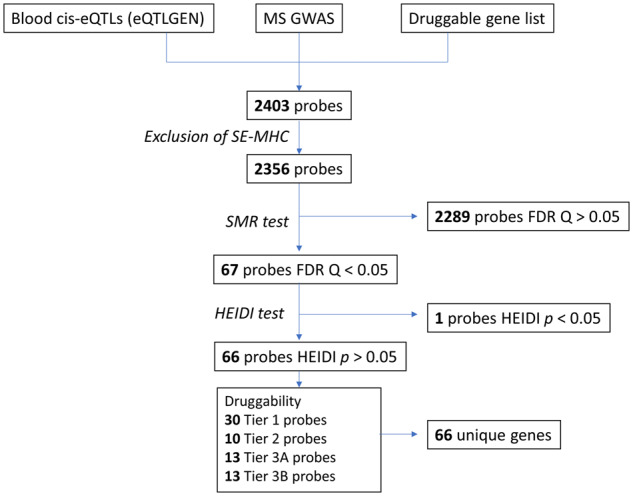
Flow diagram of numbers of probes included in the analysis of eQTLgen data. SMR was used to determine the causal effect of perturbations in genetically determined gene expression in peripheral blood on multiple sclerosis susceptibility.
To distinguish pleiotropy from linkage, Zhu et al. (2016) developed the heterogeneity in dependent instruments (HEIDI) test, which exploits the observation that if gene expression and disease phenotype are in vertical pleiotropy with the same causal variant, βSMR is identical for any variant in linkage disequilibrium with the causal variant. Thus greater heterogeneity among βSMR statistics calculated for all significant cis-eQTLs implies a greater likelihood that linkage, rather than causality/vertical pleiotropy, explains the observed βSMR. The heterogeneity statistic, the ‘HEIDI’ statistic, tests the hypothesis HEIDI = 0. This provides a formal test of heterogeneity, with P-values < 0.05 suggestive of linkage, rather than pleiotropy, as the underlying biological model (Zhu et al., 2016).
In this work, we performed SMR using the SMR software tool (SMR v1.0.2) in the command line using default options (Zhu et al., 2016). Default options are as follows: cis-eQTLs selected based on minimum P = 5 × 10−8, eQTLs included for the HEIDI test based on minimum P = 1.57 × 10−3, eQTLs included for the HEIDI test if R2 with the top cis-eQTL was between 0.05 and 0.9, minimum number of SNPs included in the HEIDI test = 3, maximum number of SNPs included in the HEIDI test = 20 and physical window around probe within which the top cis-eQTL was selected = 2 MB.
P-values were adjusted in R (v3.6.1) to control the false discovery rate at ɑ = 0.05 using the Benjamini–Hochberg procedure. Associations with pHEIDI<0.01—the cutoff used by Wu et al. (2018)—were considered likely due to linkage and thus discarded from the analysis. Probes were excluded if any of the transcript or the top eQTL resided within the super-extended major histocompatibility complex (hg19 6:25 000 000–35 000 000) given the complex linkage disequilibrium structures within this region. Linkage disequilibrium estimation was performed using reference genomes obtained from the 1000 genomes samples of European ancestry (n = 503) (Consortium, T. 1000 G. P., & The 1000 Genomes Project Consortium, 2015).
Multi-omic SMR
To further refine the list of plausible druggable targets developed using the techniques above, we performed multi-omic SMR using the methods described in Wu et al. (2018). This approach prioritizes genes by layering SNP associations with CpG methylation sites, gene expression and the phenotype of interest. As the majority of GWAS hits are in non-coding regions, they are likely to influence disease through regulation of gene expression (Wu et al., 2018). As CpG methylation sites are partly genetically determined, methylome-wide association study data can be exploited to determine possible causal relationships between CpG methylation and other traits, including molecular traits such as gene expression. We applied the following steps to prioritize druggable genes in multiple sclerosis:
Use SMR to determine associations between CpG methylation sites and gene expression
Use SMR to determine associations between CpG methylation sites and multiple sclerosis
Use SMR to determine associations between gene expression and multiple sclerosis
If there is strong evidence of causal association (i.e. pSMR < threshold and pHEIDI > 0.01) at each of the above three steps (associations between CpG methylation sites and gene expression, associations between CpG methylation sites and MS and associations between gene expression and MS), this provides evidence of a causal pathway linking genetic variation, methylation, gene expression and multiple sclerosis at that genetic locus.
Custom gene tracks for locus plots were downloaded from the University of California at Santa Cruz Genome Browser (https://genome.ucsc.edu/cgi-bin/hgTables). All genomic coordinates specified are genome build hg19 (GRCh37).
Pathway analysis, functional annotation and prediction of protein interactors
Pathway analysis and annotation was conducted using the search tool for the retrieval of interacting genes/proteins (STRING) database of protein–protein interactions (https://string-db.org/). For data visualization, we used default settings to map protein–protein interactions between the prioritized druggable targets. STRING database interaction scores are curated from multiple sources (experimental data, co-expression, text-mining and predictions from peptide sequences) and reflect the probability that two proteins are linked in the same Kyoto encyclopedia of genes and genomes metabolic pathway. By default, interactions with medium confidence (>0.4) are shown (von Mering et al., 2005; Szklarczyk et al., 2019). For functional enrichment analysis, we examined the list of SMR-prioritized genes (not restricted to the druggable genome).
Enrichment for Gene Ontology (GO) terms and biological pathways was determined by querying GO terms, Kyoto encyclopedia of genes and genomes pathways and REACTOME pathways using STRING (Franceschini et al., 2012). Enrichment P-values are computed using hypergeometric tests and corrected using the Benjamini–Hochberg procedure. The hypergeometric test for enrichment is based on the observation that under the null hypothesis (no enrichment), the number of genes from the gene list which have a particular GO term attached follows an approximately binomial distribution for large n-values (Rivals et al., 2007).
To identify additional druggable targets not prioritized in the primary analysis but likely to interact with prioritized genes, we used the GeNets tool (http://apps.broadinstitute.org/genets). GeNets uses pre-trained random forest classifiers to predict the likelihood that any given protein interacts functionally with another. In addition to calculating interaction strength between given gene products, GeNets can also predict likely interacting partners given a gene set. We ran GeNets using the online tool, with the SMR-prioritized gene list from our primary analysis (eQTLgen SMR, Supplementary Table 3) as input, and with default settings (Li et al., 2018).
Systematic review of clinical trials
Finally, we performed a systematic evaluation of the clinical trial literature for all Tier 1 druggable targets associated with multiple sclerosis from our primary analysis (eQTL SMR). We searched the Open Targets Platform (https://www.targetvalidation.org/) for each prioritized gene candidate. For each gene, we collated all trials targeting the gene in autoimmune diseases (e.g. multiple sclerosis, Rheumatoid Arthritis, Inflammatory Bowel Disease, Psoriasis, Ankylosing Spondylitis, Systemic Lupus Erythematosus, Primary Biliary Cholangitis, Type 1 Diabetes Mellitus, Sjogren’s Syndrome and Primary Sclerosing Cholangitis) and haematological malignancies. These diseases areas were chosen given their potentially shared aetiology with multiple sclerosis; successful trials in these diseases may suggest possible therapeutic benefit in multiple sclerosis.
Statistical analysis, software and computing
All analyses were conducted using SMR and R v3.6.1. This research was supported by the High-Performance Cluster computing network hosted by Queen Mary University of London (King et al., 2017). SMR locus plots were made using the online tool hosted at the SMR website (https://cnsgenomics.com/software/smr/omicsplot/).
Data availability
All data used in this study are publicly available at the URLs given in the methods. The SMR programme is available at the URL given in the methods. SMR was run using default command line options.
All code is available here: https://github.com/benjacobs123456/MS_SMR_druggable_targets.
Results
Expression of druggable gene targets is modulated by multiple sclerosis risk loci
We used SMR to test the association of genetically determined expression of druggable genes in peripheral blood with multiple sclerosis. Expression of 45 tested probes (mapping to 45 unique genes; 2356 probes tested in total) was associated with multiple sclerosis (FDRSMR < 0.05, pHEIDI > 0.01, Fig. 1). Twenty of these genes were Tier 1 druggable targets (Figs 1–4 and Table 1). Seventeen of these 45 signals were replicated in the CAGE dataset (Supplementary Tables 1 and 2, Fig. 1). For these replicated genes, the association statistics (betaSMR) with multiple sclerosis were highly correlated between the two datasets (Pearson's correlation coefficient 0.93, 95% CI 0.83–0.98, P = 4.20 × 10−8, Supplementary Tables 1 and 2, Fig. 2).
Figure 2.

Number of SMR-prioritized druggable targets from the analysis of eQTLgen data. Druggability tiers refer to the extent to which a gene target can or could be targeted by drugs, as expanded in the Materials and methods section.
Figure 3.

Top eQTL associations with both expression of the gene and multiple sclerosis susceptibility for prioritized druggable genes. For prioritized druggable genes, the top eQTL associations with both expression of the gene (x) and multiple sclerosis susceptibility (y) are shown. Both associations are expressed as betas, i.e. the log(OR) of multiple sclerosis susceptibility.
Figure 4.

Top eQTL associations with both expression of the gene and multiple sclerosis susceptibility for prioritized druggable genes with rsID. As per Fig. 3, but each top eQTL is labelled with rsID.
Table 1.
SMR-prioritized druggable genes associated with multiple sclerosis
| Gene | CHR | Probe BP | Top eQTL | A1 | Freq | Beta SMR | P SMR | P HEIDI | Druggability tier | FDR Q-value |
|---|---|---|---|---|---|---|---|---|---|---|
| CD5 | 11 | 60882595 | rs4939489 | T | 0.409543 | −0.67129 | 7.37E−14 | 0.026639 | Tier 1 | 5.78E−11 |
| CD40 | 20 | 44752706 | rs13037326 | T | 0.261431 | −0.49717 | 1.89E−12 | 0.023461 | Tier 1 | 1.11E−09 |
| MERTK | 2 | 1.13E+08 | rs55812028 | T | 0.256461 | 0.192908 | 4.92E−08 | 0.047402 | Tier 1 | 8.27E−06 |
| CD37 | 19 | 49842510 | rs1320302 | T | 0.259443 | −0.34411 | 1.29E−07 | 0.056106 | Tier 1 | 1.89E−05 |
| KIF11 | 10 | 94384096 | rs10882098 | T | 0.422465 | −1.15164 | 4.37E−06 | 0.082185 | Tier 1 | 4.89E−04 |
| TYK2 | 19 | 10476280 | rs8101195 | G | 0.152087 | −0.43108 | 8.30E−06 | 0.078946 | Tier 1 | 8.13E−04 |
| MAPK3 | 16 | 30130126 | rs55732507 | C | 0.437376 | −0.15962 | 1.01E−05 | 0.125369 | Tier 1 | 9.15E−04 |
| S1PR1 | 1 | 1.02E+08 | rs1922987 | C | 0.392644 | −0.66394 | 3.88E−05 | 0.781547 | Tier 1 | 0.003146 |
| SLAMF7 | 1 | 1.61E+08 | rs489286 | A | 0.32008 | 0.396246 | 6.93E−05 | 0.241083 | Tier 1 | 0.00509 |
| MAP3K11 | 11 | 65374039 | rs2004649 | A | 0.440358 | 0.147259 | 7.31E−05 | 0.07623 | Tier 1 | 0.005213 |
| VEGFB | 11 | 64004134 | rs7943988 | A | 0.298211 | −0.64641 | 1.23E−04 | 0.746483 | Tier 1 | 0.008243 |
| CHEK2 | 22 | 29111070 | rs134547 | G | 0.111332 | −1.08044 | 1.96E−04 | 0.06031 | Tier 1 | 0.01215 |
| NCSTN | 1 | 1.6E+08 | rs6668576 | C | 0.486083 | 0.115574 | 1.96E−04 | 0.013308 | Tier 1 | 0.01215 |
| ERBB2 | 17 | 37865423 | rs1453559 | T | 0.481113 | −1.06676 | 2.53E−04 | 0.012659 | Tier 1 | 0.013813 |
| PARP1 | 1 | 2.27E+08 | rs2793379 | A | 0.164016 | 0.260005 | 2.83E−04 | 0.159161 | Tier 1 | 0.014781 |
| IFNGR2 | 21 | 34813428 | rs17879003 | T | 0.141153 | 0.127669 | 4.31E−04 | 0.776412 | Tier 1 | 0.0207 |
| MPO | 17 | 56352756 | rs917016 | C | 0.225646 | −0.24996 | 5.21E−04 | 0.023533 | Tier 1 | 0.023098 |
| CXCR4 | 2 | 1.37E+08 | rs11897084 | A | 0.477137 | −1.87471 | 5.39E−04 | 0.891624 | Tier 1 | 0.023468 |
| HDAC3 | 5 | 1.41E+08 | rs1421896 | T | 0.417495 | 0.473559 | 0.001156 | 0.707142 | Tier 1 | 0.041839 |
| ABCC2 | 10 | 1.02E+08 | rs2756109 | G | 0.429423 | 0.370597 | 0.001244 | 0.666814 | Tier 1 | 0.044326 |
| TYMP | 22 | 50966333 | rs140522 | T | 0.32008 | −0.17244 | 3.49E−10 | 0.962601 | Tier 2 | 1.17E−07 |
| STAT5A | 17 | 40451763 | rs6503694 | C | 0.449304 | −1.34492 | 1.91E−08 | 0.019111 | Tier 2 | 3.75E−06 |
| NR1H3 | 11 | 47280123 | rs326222 | T | 0.302187 | −0.22802 | 2.91E−06 | 0.112627 | Tier 2 | 3.42E−04 |
| MANBA | 4 | 1.04E+08 | rs6533034 | C | 0.332008 | 0.328618 | 1.62E−04 | 0.011624 | Tier 2 | 0.010557 |
| PHOSPHO1 | 17 | 47304426 | rs6504606 | A | 0.232604 | 0.420936 | 2.14E−04 | 0.835836 | Tier 2 | 0.012581 |
| EIF2AK3 | 2 | 88891676 | rs867529 | C | 0.298211 | 0.708537 | 7.88E−04 | 0.193829 | Tier 2 | 0.031402 |
| GALK1 | 17 | 73754733 | rs7209235 | G | 0.294235 | −0.29782 | 8.17E−04 | 0.011387 | Tier 2 | 0.032039 |
| CDK11B | 1 | 1580538 | rs11486028 | C | 0.44334 | 0.361917 | 9.55E−04 | 0.090301 | Tier 2 | 0.035664 |
| MMEL1 | 1 | 2543279 | rs10752747 | T | 0.310139 | 0.283971 | 2.08E−14 | 0.113363 | Tier 3A | 3.73E−11 |
| FCRL3 | 1 | 1.58E+08 | rs2210913 | T | 0.474155 | −0.12963 | 1.15E−08 | 0.094284 | Tier 3A | 2.46E−06 |
| IL7 | 8 | 79652868 | rs894221 | C | 0.285288 | −1.52297 | 7.12E−06 | 0.115854 | Tier 3A | 7.28E−04 |
| ACP2 | 11 | 47265655 | rs2957873 | G | 0.191849 | −0.14897 | 1.60E−05 | 0.513203 | Tier 3A | 0.001345 |
| CD27 | 12 | 6557458 | rs1059501 | G | 0.45328 | −0.69764 | 2.21E−04 | 0.014897 | Tier 3A | 0.012686 |
| PIK3IP1 | 22 | 31683049 | rs4820963 | C | 0.274354 | 0.119862 | 7.09E−04 | 0.154287 | Tier 3A | 0.029239 |
| AMICA1 | 11 | 1.18E+08 | rs7939622 | A | 0.200795 | −0.15402 | 9.11E−04 | 0.068043 | Tier 3A | 0.03511 |
| GPR25 | 1 | 2.01E+08 | rs296545 | A | 0.292247 | −0.83806 | 1.54E−09 | 0.065113 | Tier 3B | 4.52E−07 |
| CATSPER1 | 11 | 65789105 | rs56154109 | C | 0.446322 | 0.898415 | 1.75E−06 | 0.0853 | Tier 3B | 2.16E−04 |
| CD3D | 11 | 1.18E+08 | rs17540708 | G | 0.306163 | −0.66788 | 2.14E−04 | 0.080303 | Tier 3B | 0.012581 |
| SLC12A7 | 5 | 1081324 | rs35188965 | C | 0.442346 | −0.10226 | 2.43E−04 | 0.03212 | Tier 3B | 0.013622 |
| KLHL8 | 4 | 88111507 | rs10026594 | G | 0.260437 | 0.506605 | 3.65E−04 | 0.034025 | Tier 3B | 0.018648 |
| PPIL2 | 22 | 22029562 | rs2236643 | G | 0.416501 | −0.41586 | 3.81E−04 | 0.32388 | Tier 3B | 0.019028 |
| SLC6A16 | 19 | 49810688 | rs181344 | T | 0.296223 | 0.363643 | 3.88E−04 | 0.019443 | Tier 3B | 0.019028 |
| PGLYRP1 | 19 | 46524367 | rs11669048 | G | 0.380716 | −0.18954 | 5.05E−04 | 0.391924 | Tier 3B | 0.022837 |
| PLXNC1 | 12 | 94621975 | rs2306664 | T | 0.414513 | 0.729647 | 7.37E−04 | 0.069415 | Tier 3B | 0.029874 |
| PKD2L2 | 5 | 1.37E+08 | rs6871007 | C | 0.49503 | 0.560794 | 0.001109 | 0.17167 | Tier 3B | 0.040769 |
eQTL data are from the eQTLgen consortium. Values <0.01 indicate that the SMR association is driven by linkage rather than by an underlying shared causal variant.
A1: effect allele; Beta SMR: causal estimate for effect of each unit increase in gene expression on multiple sclerosis susceptibility; CHR: chromosome; Druggability tier: tier derived from Finan et al.; FDR: false discovery rate; Freq: A1 frequency; P HEIDI: P-value for the HEIDI test; Probe BP: position (hg19); P SMR: P-value for SMR association; Q-value: false-discovery rate corrected P-value; Top eQTL: cis-eQTL for gene with smallest P-value for association.
We found evidence for multiple protein–protein interactions within this list of prioritized druggable genes (Fig. 5). Extending the SMR approach beyond the druggable genome, expression of 235 genes was associated with multiple sclerosis risk (FDRSMR < 0.05, pHEIDI >0.01, Supplementary Table 3 and Fig. 6). This full set of SMR-prioritized genes was enriched for multiple GO terms and biological pathways implicated in immune system function and dysfunction (Supplementary Tables 4–6). No significant enrichment was found for REACTOME pathways nor for GO cellular components.
Figure 5.

Protein–protein interactions among druggable SMR-prioritized genes derived from the STRING database. The strength of the line indicates the confidence of the interaction.
Figure 6.

Protein–protein interactions among druggable SMR-prioritized genes derived from the STRING database As per Fig. 5, but including all SMR-prioritized genes, i.e. not restricted to druggable targets.
Methylation at CpG sites is modulated by multiple sclerosis risk loci
Next, to determine CpG methylation sites associated with multiple sclerosis risk, we applied SMR to CpG mQTL data from a meta-analysis of the Lothian Birth Cohort and Brisbane Systems Genetics Study cohorts (see Materials and methods section). We found evidence for an association of 574 CpG methylation probes with multiple sclerosis risk (Supplementary Table 12).
Multi-omic SMR to prioritize druggable gene candidates in multiple sclerosis
To determine the associations between CpG methylation sites and gene expression, we performed SMR using the Lothian Birth Cohort and Brisbane Systems Genetics Study mQTL meta-analysis and the eQTLgen consortium eQTL data. We restricted this analysis to druggable gene transcripts and excluded the major histocompatibility complex. We identified 23 731 associated pairs of CpG probes and transcripts (FDRSMR < 0.05, pHEIDI > 0.01) mapping to 2548 unique genes (Supplementary Table 7).
We then overlaid SMR evidence for causal relationships among associations between CpG methylation sites and gene expression, associations between CpG methylation sites and multiple sclerosis and associations between gene expression and multiple sclerosis for the druggable genome. We found 35 druggable loci mapping to 15 unique genes (Table 2). Five of these genes were Tier 1 (highly druggable) genes: CD40, ERBB2, VEGFB, MERTK and PARP1. Eight of these 15 unique genes were replicated using the CAGE dataset, including three of the five Tier 1 genes (CD40, MERTK and PARP1). In addition, another Tier 1 gene, IL12RB1, was associated with multiple sclerosis in the CAGE but not the eQTLgen dataset (Table 2 and Supplementary Table 8). Locus plots with chromatin state annotations from the Roadmap Epigenomics Projects are shown for the three replicated Tier 1 genes (Supplementary Fig. 6).
Table 2.
Druggable genetic targets associated with multiple sclerosis prioritized by multi-omic SMR
| Chromosome | Methylation probe | Ensembl gene ID | Gene | Druggability tier | Replicated in CAGE |
|---|---|---|---|---|---|
| 20 | cg01943874 | ENSG00000101017 | CD40 | Tier 1 | Yes |
| 20 | cg17929951 | ENSG00000101017 | CD40 | Tier 1 | Yes |
| 20 | cg19785066 | ENSG00000101017 | CD40 | Tier 1 | Yes |
| 20 | cg21601405 | ENSG00000101017 | CD40 | Tier 1 | Yes |
| 20 | cg25239996 | ENSG00000101017 | CD40 | Tier 1 | Yes |
| 2 | cg04202892 | ENSG00000153208 | MERTK | Tier 1 | Yes |
| 2 | cg08443563 | ENSG00000153208 | MERTK | Tier 1 | Yes |
| 2 | cg18646521 | ENSG00000153208 | MERTK | Tier 1 | Yes |
| 1 | cg23712594 | ENSG00000143799 | PARP1 | Tier 1 | Yes |
| 11 | cg23844623 | ENSG00000025434 | NR1H3 | Tier 2 | Yes |
| 11 | cg25783544 | ENSG00000025434 | NR1H3 | Tier 2 | Yes |
| 11 | cg26365553 | ENSG00000025434 | NR1H3 | Tier 2 | Yes |
| 11 | cg23844623 | ENSG00000134575 | ACP2 | Tier 3A | Yes |
| 11 | cg25783544 | ENSG00000134575 | ACP2 | Tier 3A | Yes |
| 11 | cg26365553 | ENSG00000134575 | ACP2 | Tier 3A | Yes |
| 1 | cg08786003 | ENSG00000160856 | FCRL3 | Tier 3A | Yes |
| 1 | cg19602479 | ENSG00000160856 | FCRL3 | Tier 3A | Yes |
| 1 | cg25259754 | ENSG00000160856 | FCRL3 | Tier 3A | Yes |
| 1 | cg09808235 | ENSG00000142606 | MMEL1 | Tier 3A | Yes |
| 1 | cg12750103 | ENSG00000142606 | MMEL1 | Tier 3A | Yes |
| 1 | cg26783079 | ENSG00000142606 | MMEL1 | Tier 3A | Yes |
| 11 | cg01637289 | ENSG00000167286 | CD3D | Tier 3B | Yes |
| 17 | cg09639931 | ENSG00000141736 | ERBB2 | Tier 1 | No |
| 11 | cg08904394 | ENSG00000173511 | VEGFB | Tier 1 | No |
| 22 | cg05329888 | ENSG00000025708 | TYMP | Tier 2 | No |
| 22 | cg09435190 | ENSG00000025708 | TYMP | Tier 2 | No |
| 22 | cg11654620 | ENSG00000025708 | TYMP | Tier 2 | No |
| 22 | cg16367976 | ENSG00000025708 | TYMP | Tier 2 | No |
| 22 | cg22927510 | ENSG00000025708 | TYMP | Tier 2 | No |
| 11 | cg01637289 | ENSG00000160593 | AMICA1 | Tier 3A | No |
| 8 | cg01069256 | ENSG00000104432 | IL7 | Tier 3A | No |
| 8 | cg05575058 | ENSG00000104432 | IL7 | Tier 3A | No |
| 11 | cg19088912 | ENSG00000175294 | CATSPER1 | Tier 3B | No |
| 22 | cg03321319 | ENSG00000100023 | PPIL2 | Tier 3B | No |
| 22 | cg25898577 | ENSG00000100023 | PPIL2 | Tier 3B | No |
If the association was replicated at all three stages in the CAGE dataset, this is indicated in the final column.
CAGE: Consortium for the Architecture of Gene Expression.
Prediction of novel interactions
To extend the list of functionally prioritized druggable targets, we used GeNets to predict which proteins interact with the targets identified in our analysis. As input, we used the full list of 235 SMR-prioritized genes (including non-druggable targets) from the primary analysis (Supplementary Table 3). This approach yielded an additional 75 predicted protein interaction partners and multiple novel and established therapeutic compounds targeting our list of prioritized protein networks (Supplementary Fig. 4 and Table 10).
Druggable targets in lymphoblastoid cell lines
EBV-transformed cell lines, LCLs, provide a model for studying gene expression on B cells in vitro. These cells are immortalized using EBV, which appears to play a significant role in multiple sclerosis risk. We attempted to identify druggable gene targets in LCLs using LCL–eQTL data from the Geuvadis consortium (Lappalainen et al., 2013). SMR identified seven prioritized druggable gene targets for multiple sclerosis in LCLs (Supplementary Table 10 and Fig. 5). Expression of 3/7 of these transcripts (SLC12A7, FCRL3 and SLAMF7) was also associated with multiple sclerosis susceptibility in blood, with concordant directions of effects for all three genes. The FCRL3 gene was the only gene that overlapped with our list of genes prioritized using multi-omic SMR, although notably the TNFRSF14 gene prioritized from the LCL analysis is adjacent to MMEL1, which was nominated by the multi-omic method as a likely causal gene in peripheral blood. Comparison of SMR causal estimates for gene expression of the 1215 overlapping genes which passed the HEIDI test (P > 0.01) revealed a weak but precisely estimated positive correlation between association statistics with multiple sclerosis in blood and LCLs (Pearson’s rho = 0.27, 95% CI 0.21–0.32, P < 2 × 10−16).
Systematic review of clinical trials
We conducted a systematic review of clinical trials targeting our 20 Tier 1 prioritized genes from the primary analysis (eQTL SMR with eQTLgen data). Using the Open Targets platform, we retrieved trial data for trials in autoimmune diseases (including multiple sclerosis) and in haematological malignancies for each druggable target. Full results are presented in Supplementary Table 9. Of the 20 genes queried, we found drug trials in related disease areas for 13/20. Gene targets without any trials in relevant areas were MPO, ABCC2, CD5, MERTK, NCSTN, MAP3K11 and VEGFB. Only one gene target, S1PR1, was being currently targeted in multiple sclerosis. Phase III trial data have demonstrated efficacy of S1PR1 modulators (fingolimod, siponimod, ozanimod and ponesimod) in reducing relapse rate for relapsing disease (Cohen et al., 2010; Kappos et al., 2010; Calabresi et al., 2014) and slowing disability progression for secondary-progressive disease (Kappos et al., 2018). Four drug targets had been targeted in trials for at least one autoimmune disease (excluding multiple sclerosis): IFNGR2, HDAC3, TYK2 and CD40. Of these targets, only compounds targeting TYK2 had progressed into phase III/IV trial development. Completed phase III trials have shown efficacy of TYK2 inhibitors (e.g. tofacitinib) in Rheumatoid Arthritis (van Vollenhoven et al., 2012), psoriasis/psoriatic arthropathy (Gladman et al., 2017) and Ulcerative Colitis (Sandborn et al., 2017). The other genes prioritized and replicated by the multi-omic SMR approach—CD40 and PARP1—were not being targeted by any compounds beyond phase II development. CD40 inhibitors (lucatumumab and dacetuzumab) and PARP1 inhibitors (veliparib, niraparib, talazoparib) were in multiple phase I and II trials for haematological malignancies. An anti-CD40L mAb, INX-021, has entered phase I testing in multiple sclerosis.
Discussion
In this report, we combine the latest IMSGC GWAS data with large mQTL and eQTL datasets and a list of curated ‘druggable genes’ to provide a data-driven list of 45 prioritized gene products which are possible druggable targets to mitigate the risk of multiple sclerosis. Three of these targets—CD40, MERTK and PARP1—have strong evidence to support a pleiotropic pathway from genetic variation to CpG site methylation to gene expression to disease, and are already being targeted by existing compounds at various stages of clinical development. Several are being investigated in clinical or preclinical studies for multiple sclerosis and/or autoimmune disorders. It is important to note that the phenotype examined in the GWAS used in this study is multiple sclerosis susceptibility (i.e. a binary disease trait), rather than multiple sclerosis severity. Thus, our findings should be interpreted as pointing to biological targets and pathways which could, in principle, be modulated to affect the risk of developing multiple sclerosis. While at least some of the pathways uncovered by this approach may play a role in determining multiple sclerosis severity, this assumption remains to be proven.
CD40 is a member of the Tumour Necrosis Factor superfamily which is constitutively expressed on the cell surface of specialized antigen-presenting cells such as B cells and dendritic cells (Aarts et al., 2017). Binding of CD40 by its ligand, CD40L, initiates diverse signalling cascades via Tumour Necrosis Factor Receptor-Associated-Factors culminating in B-cell differentiation, activation, proliferation and germinal centre formation (Aarts et al., 2017). CD40L is primarily expressed on activated T cells, and the CD40L–CD40 interaction provides an antigen-specific co-stimulatory signal for propagating the humoral immune response. Several observations support a pathogenic role for CD40 signalling in multiple sclerosis: CD40L is upregulated in CD4+ T cells in multiple sclerosis lesions, soluble CD40L (a secreted ligand for CD40) is upregulated in serum and CSF of people with active multiple sclerosis, CD40-expressing mononuclear cells infiltrate the CNS in rodent experimental autoimmune encephalomyelitis, the density of CD40 appears higher on B cells from people with multiple sclerosis (Aarts et al., 2017), and variation at the CD40 locus is associated with multiple sclerosis susceptibility (International Multiple Sclerosis Genetics Consortium, 2019b). The precise mechanism by which genetic variation at the CD40 locus influences multiple sclerosis risk is unclear—perhaps counter-intuitively, multiple sclerosis-associated SNPs are associated with decreased expression of CD40 mRNA in peripheral blood and B lymphocyte subsets (Field et al., 2015; Smets et al., 2018), an effect which is replicated in our study (BetaSMR −0.50, pSMR = 1.89 × 10−12). In a study of peripheral blood mononuclear cells from people with multiple sclerosis and healthy controls, the susceptibility SNP rs4810485 was associated with both alternative splicing of CD40 transcripts—with relative upregulation of the splice isoform lacking exon 5 leading to lower cell-surface expression—and with lower levels of the anti-inflammatory cytokine IL-10 in peripheral blood (Smets et al., 2018). These data suggest that genetically determined modulation of CD40 mRNA expression may promote multiple sclerosis risk via effects of alternatively spliced transcripts on IL-10 production, although the exact mechanism remains to be clarified. Monoclonal antibodies blocking the interaction between CD40L and CD40 have shown efficacy in rodent and primate experimental autoimmune encephalomyelitis (Aarts et al., 2017).
Drug trials of anti-CD40 therapies in humans have been limited by safety concerns. Early phase II data showed promising efficacy of an anti-CD40L mAb in lupus nephritis (Boumpas et al., 2003). Despite phase I safety of the anti-CD40L antibody, IDEC-131, in multiple sclerosis, the phase II trial was halted due to increased risk of thromboembolic disease caused by inhibition of the thrombus-stabilizing interaction between CD40L and platelet alpha2, beta3 integrins (André et al., 2002; Aarts et al., 2017). Subsequently, various strategies have been deployed to overcome this safety concern. The thrombogenicity of CD40L antagonists is dependent on an Fc-FcγRIIa interaction leading to platelet cross-linking (Robles-Carrillo et al., 2010); development of a PEGylated monovalent anti-CD40L Fab fragment lacking an Fc portion appears to have overcome this problem (Tocoian et al., 2015). A placebo-controlled phase II trial of dapirolizumab pegol for individuals with Systemic Lupus Erythematosus has been completed (results pending; NCT02804763). An anti-CD40L antibody-like molecule based on a Tn3 protein scaffold has shown phase I safety and preliminary efficacy in individuals with Rheumatoid Arthritis (Karnell et al., 2019).
In addition to directly targeting the CD40L–CD40 interaction, it may be beneficial to target downstream signalling. Upon activation by CD40L, CD40 signals via nuclear factor kappa-light-chain-enhancer of activated B cells, phosphoinositide 3-kinases and the mitogen-activated protein kinases (MAPKs), c-Jun N-terminal kinase and p38 MAPKs, depending on cellular context (Michel et al., 2017). The potential utility of drugging the MAPK signalling pathway is clear from our study, with MAPK3, MAPK1 and MAP3K11 all prioritized in the primary analysis. Phosphoproteomic scans of differences in protein phosphorylation in people with multiple sclerosis have revealed constitutive high levels of activation of MAPK signalling in immune cells from people with multiple sclerosis (Kotelnikova et al., 2019). Inhibitors of MEK1, a downstream target of MAPK signalling, have been trialled in Rheumatoid Arthritis, Crohn’s disease, psoriasis and multiple myeloma (Arthur and Ley, 2013).
MERTK is a member of the TAM (Tyro3, Axl and Mer) family of receptor tyrosine kinases which have multiple endogenous ligands, and exist in both membrane-bound and circulating (decoy receptor) forms (Wium et al., 2018). Upon ligand binding, MERTK signals via phosphoinositide 3-kinases/Akt, MAPK and nuclear factor kappa-light-chain-enhancer of activated B cells pathways, in addition to several others (Wium et al., 2018). Physiological roles of MERTK include regulation of platelet function, inflammation and phagocytosis (Scott et al., 2001). Disruption of phagocytic clearance of dead and dying cells is thought to contribute to aberrant exposure of Damage-Associated Molecular Patterns, such as dsDNA, which may act as a substrate for loss of immune tolerance to self-antigens and subsequent autoimmunity (Wium et al., 2018). In addition, MERTK appears to facilitate astrocytic clearance of synaptic debris in the CNS (Chung et al., 2013). MERTK is a risk locus in the IMSGC GWAS (International Multiple Sclerosis Genetics Consortium, 2019b), with the multiple sclerosis risk-increasing alleles associated with increased MERTK expression (BetaSMR 0.19, pSMR 4.92 × 10−8). In cuprizone-treated rodents, knockout of the MERTK ligand, Growth Arrest Specific 6 (Gas6), delays remyelination and recovery, suggesting that the Gas6-MERTK interaction facilitates remyelination (Binder et al., 2008, 2011). In post-mortem tissue, soluble and membrane-bound MERTK were upregulated in chronic multiple sclerosis lesions; soluble MERTK was negatively correlated with lesional Gas6 (Weinger et al., 2009). It is plausible that upregulation of MERTK leads to increased decoy receptor shedding (depending on metalloproteinase action) and subsequent inhibition of Gas6 by soluble MERTK. One study suggested that CSF Gas6 levels may be elevated in early relapse, but do not differ between controls and people with multiple sclerosis outside of an acute relapse (Sainaghi et al., 2013), which may reflect a homeostatic response to acute demyelination. Small molecule inhibitors of MERTK have shown efficacy in mouse models of ALL (Lee-Sherick et al., 2018).
Poly(ADP-ribose) polymerase 1(PARP1) is an intracellular enzyme which catalyses the transfer of ADP-ribose groups from NAD to targets such as histones, DNA repair proteins and transcription factors. PARP1 is involved in DNA repair and the regulation of gene expression. It also promotes inflammation and innate immune responses via nuclear factor kappa-light-chain-enhancer of activated B cells signalling, and may be involved in B-cell differentiation and activation (Rosado et al., 2013). There is some evidence linking increased PARP1 activity to autoimmunity. Specific PARP1 haplotypes are associated with Rheumatoid Arthritis (Pascual et al., 2003). Some, but not all, experimental autoimmune encephalomyelitis studies have demonstrated efficacy of PARP1 inhibitors in limiting disease severity (Rosado et al., 2013). In our study, surprisingly, the direction of SMR effect implies that increased PARP1 expression may be protective against multiple sclerosis (BetaSMR −0.31, 2.95 × 10−195).
Data-driven efforts to prioritize drug targets in multiple sclerosis using SMR have been limited to date. One study used older GWAS data and smaller eQTL datasets (Westra et al., 2013 and CAGE) to prioritize 10 non-major histocompatibility complex genes and 20 non-major histocompatibility complex methylation loci associated with multiple sclerosis in peripheral blood (Mo et al., 2019). Our use of larger, more recent datasets, a more lenient HEIDI threshold, and a false discovery rate cutoff rather than a Bonferroni correction is likely to explain our discovery of a greater number of genes. To our knowledge, ours is the first attempt to synthesize SMR with a curated list of druggable targets to prioritize genes for preventive therapy in multiple sclerosis. Reassuringly, our list of prioritized druggable genes shares several candidates with studies which examined eQTLs in people with multiple sclerosis, strengthening the argument that eQTLs are not disease-specific (James et al., 2018). Furthermore, all of our prioritized ‘Tier 1’ genes were identified by the IMSGC as likely implicated in disease using a variety of annotation methods (International Multiple Sclerosis Genetics Consortium, 2019b).
An important limitation is that our analyses focus on whole blood, partly because this is the tissue for which the largest eQTL and mQTL datasets exist. As the genetic determinants of gene expression vary between tissues, this paper focuses on identifying differentially expressed genes in blood potentially suitable for preventive interventions. Not only does this miss possible tissue-specific effects of other genes in multiple sclerosis, it also does not distinguish between different types of immune cells. Although multiple sclerosis GWAS hits are primarily enriched for eQTLs and epigenomic marks of active chromatin in various types of immune cells, which are included in eQTL datasets from peripheral blood, there is also enrichment in CNS-resident microglia and, at the tissue level, in the thymus (International Multiple Sclerosis Genetics Consortium, 2019a, b). Our analysis would not detect cell type-specific effects acting in other tissues (e.g. CNS and thymus), and cell type-specific effects within particular leucocyte subsets which are not reflected in ‘averaged’ datasets across multiple cell types. It is likely that the pathogenesis of multiple sclerosis involves dysregulated signalling in specific subsets of immune cells, and our work misses this complexity. A useful extension of our work would be to apply this approach to eQTL datasets garnered from specific immune cell types. DNA methylation, like gene expression, is a dynamically regulated process which varies substantially depending on cellular context, and thus we would urge a cautious interpretation of our methylation results, which exploit ‘snapshot’ mQTL data from healthy individuals to make inferences about methylation in multiple sclerosis.
A further cell type we explore using eQTL data (mQTL data are not available) is LCLs, an in vitro model of EBV-infected B cells. This analysis allows us to make inferences about genes which may govern multiple sclerosis susceptibility within EBV-infected B cells. We prioritize seven genes from this analysis, several of which have biologically plausible mechanisms linking their expression to multiple sclerosis susceptibility. The clearest of these is TNFRSF14: a herpes virus entry mediator, which facilitates infection of B lymphocytes by herpes viruses (e.g. EBV and HHV6) and lies within an multiple sclerosis risk locus(Blanco-Kelly et al., 2011; International Multiple Sclerosis Genetics Consortium, 2019b). The relatively weak correlation between SMR signals in LCLs and whole blood emphasizes the diversity of relationships between gene expression and phenotype between different cell types. The importance of multiple sclerosis risk loci acting within EBV-infected cells is reinforced by evidence that multiple sclerosis susceptibility SNPs are enriched for loci influencing EBV copy number and EBV micro-RNA expression within LCLs (Afrasiabi et al., 2020).
A further limitation of our work is that SMR identifies genes in which the level of expression is associated with disease. This is consistent with both vertical pleiotropy—i.e. a causal pathway—and with horizontal pleiotropy, whereby the variant may influence disease susceptibility via a mechanism independent of affecting transcription of the prioritized gene: SMR is unable to distinguish these possibilities, and thus we would caution against overinterpretation of our results as causal (Zhu et al., 2016). Moreover, there are many ways whereby gene and protein function may influence phenotype independently of levels of gene expression, such as through changes in subcellular localization and post-translational modifications. In addition, the effect of expression of a single gene is likely to depend on the overall transcriptional and translational state of the cell at that point; this complexity is not captured by considering the expression of individual genes separately. Furthermore, the curated list of druggable genes does not imply that a gene is necessarily a sensible target; some prioritized genes with multiple roles in cell signalling, such as CD40, may be challenging to target in practice without unacceptable off-target effects. Lastly, in our analyses we examine the overall effect on gene expression, not considering individual transcripts. This approach does not capture the subtle effects of alternative transcripts, which recent evidence suggests may be an important mechanisms by which multiple sclerosis GWAS loci influence disease susceptibility (Ban et al., 2020).
In summary, using data-driven Mendelian randomization and publicly available datasets, we highlight several possible drug targets which could be modulated to affect susceptibility to multiple sclerosis. In particular, we highlight a central role for the CD40 signalling pathway, which could be targeted with small molecule compounds already being trialled for other indications.
Supplementary Material
Acknowledgements
We would like to thank the IMSGC, CAGE, eQTLgen, GEUVADIS, LBC and BSGS investigators for making their data publicly available. We would also like to the Complex Traits Genomics group who developed the SMR software and have made their code publicly available.
Funding
This work was funded via a grant from Barts Charity (grant ref MGU0365).
Supplementary material
Supplementary material is available at Brain Communications online.
Competing interests
B.J., T.T. and A.A. have no competing interests to declare. D.B. reports consultancies with Canbex therapeutics Japan Tobacco, Merck, Novartis and Roche. G.G. reports receiving research grant support from Bayer-Schering Healthcare, Biogen, GW Pharma, Ironwood, Merck, Merck-Serono, Merz, Novartis, Roche, Sanofi-Genzyme, Takeda and Teva. G.G. has received personal compensation for participating on advisory boards, trial steering committees and trial data and safety monitoring boards from: AbbVie, Actelion, Atara Bio, Almirall, Bayer-Schering Healthcare, Biogen, Biogen-Idec, Canbex, Celgene, Eisai, Elan, Fiveprime, Sanofi-Genzyme, Genentech, GSK, GW Pharma, Ironwood, Merck, Merck-Serono, Novartis, Pfizer, Roche, Serono, Synthon BV, Takeda, Teva, UCB Pharma and Vertex. A.J.N. reports support from Parkinson’s UK, Virginia Kieley benefaction, GE Healthcare, Roche, Profile, Biogen, BIAL and Britannia. R.D. reports receiving support from Biogen, Merck, Teva, Sanofi and Celgene.
Glossary
- CAGE =
Consortium for the Architecture of Gene Expression
- CD =
cluster of differentiation
- EBV =
Epstein-Barr virus
- eQTLs =
expression quantitative trait loci
- GO =
Gene Ontology
- GWAS =
genome-wide association studies
- HEIDI =
heterogeneity in dependent instruments
- IMSGC =
International MS Genetics Consortium
- LCLs =
lymphoblastoid cell lines
- MAPKs =
mitogen-activated protein kinases
- mQTL =
methylation quantitative trait locus
- SMR =
summary-data-based Mendelian randomization
- SNP =
single nucleotide polymorphism
- STRING =
search tool for the retrieval of interacting genes/proteins
- UCSC =
University of California at Santa Cruz
References
- Aarts SABM, Seijkens TTP, van Dorst KJF, Dijkstra CD, Kooij G, Lutgens E. The CD40-CD40L dyad in experimental autoimmune encephalomyelitis and multiple sclerosis. Front Immunol 2017; 8: 1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afrasiabi A, Parnell GP, Swaminathan S, Stewart GJ, Booth DR. The interaction of multiple sclerosis risk loci with Epstein-Barr virus phenotypes implicates the virus in pathogenesis. Sci Rep 2020; 10: 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André P, Prasad KSS, Denis CV, He M, Papalia JM, Hynes RO, et al. CD40L stabilizes arterial thrombi by a beta3 integrin-dependent mechanism. Nat Med 2002; 8: 247–52. [DOI] [PubMed] [Google Scholar]
- Arthur JSC, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol 2013; 13: 679–92. [DOI] [PubMed] [Google Scholar]
- Ban M, Liao W, Baker A, Compston A, Thorpe J, Molyneux P, et al. Transcript specific regulation of expression influences susceptibility to multiple sclerosis. Eur J Hum Genet 2020; 28: 826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder MD, Cate HS, Prieto AL, Kemper D, Butzkueven H, Gresle MM, et al. Gas6 deficiency increases oligodendrocyte loss and microglial activation in response to cuprizone-induced demyelination. J Neurosci 2008; 28: 5195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder MD, Xiao J, Kemper D, Ma GZM, Murray SS, Kilpatrick TJ. Gas6 increases myelination by oligodendrocytes and its deficiency delays recovery following cuprizone-induced demyelination. PLoS One 2011; 6: e17727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Kelly F, Alvarez-Lafuente R, Alcina A, Abad-Grau MM, de Las Heras V, Lucas M, et al. Members 6B and 14 of the TNF receptor superfamily in multiple sclerosis predisposition. Genes Immun 2011; 12: 145–8. [DOI] [PubMed] [Google Scholar]
- Boumpas DT, Furie R, Manzi S, Illei GG, Wallace DJ, Balow JE, et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum 2003; 48: 719–27. [DOI] [PubMed] [Google Scholar]
- Calabresi PA, Radue E-W, Goodin D, Jeffery D, Rammohan KW, Reder AT, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol 2014; 13: 545–56. [DOI] [PubMed] [Google Scholar]
- Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013; 504: 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JA, Barkhof F, Comi G, Hartung H-P, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010; 362: 402–15. [DOI] [PubMed] [Google Scholar]
- Consortium T. 1000 G. P., & The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015; 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J, Shahijanian F, Schibeci S, Johnson L, Gresle M, Laverick L, et al. The MS risk allele of CD40 is associated with reduced cell-membrane bound expression in antigen presenting cells: implications for gene function. PLoS One 2015; 10: e0127080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med 2017; 9: eaag1166. 10.1126/scitranslmed.aag1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2012; 41: D808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladman D, Rigby W, Azevedo VF, Behrens F, Blanco R, Kaszuba A, et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med 2017; 377: 1525–36. [DOI] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. A systems biology approach uncovers cell-specific gene regulatory effects of genetic associations in multiple sclerosis. Nat Commun 2019. a; 10: 2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019. b; 365: eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James T, Lindén M, Morikawa H, Fernandes SJ, Ruhrmann S, Huss M, et al. Impact of genetic risk loci for multiple sclerosis on expression of proximal genes in patients. Hum Mol Genet 2018; 27: 912–28. [DOI] [PubMed] [Google Scholar]
- Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 2018; 391: 1263–73. [DOI] [PubMed] [Google Scholar]
- Kappos L, Radue E-W, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010; 362: 387–401. [DOI] [PubMed] [Google Scholar]
- Karnell JL, Albulescu M, Drabic S, Wang L, Moate R, Baca M, et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci Transl Med 2019; 11: eaar6584. [DOI] [PubMed] [Google Scholar]
- King T, Butcher S, Zalewski L. Apocrita-high performance computing cluster for Queen Mary University of London. 2017. 10.5281/zenodo.438045. [DOI]
- Kotelnikova E, Kiani NA, Messinis D, Pertsovskaya I, Pliaka V, Bernardo-Faura M, et al. MAPK pathway and B cells overactivation in multiple sclerosis revealed by phosphoproteomics and genomic analysis. Proc Natl Acad Sci USA 2019; 116: 9671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ‘T Hoen PAC, Monlong J, Rivas MA, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013; 501: 506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Sherick AB, Jacobsen KM, Henry CJ, Huey MG, Parker RE, Page LS, et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight 2018; 3: e97941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Kim A, Rosenbluh J, Horn H, Greenfeld L, An D, et al. GeNets: a unified web platform for network-based genomic analyses. Nat Methods 2018; 15: 543–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Jones LR, Holloway A, McRae A, Yang J, Small K, Zhao J, et al. The genetic architecture of gene expression in peripheral blood. Am J Hum Genet 2017; 100: 228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae AF, Marioni RE, Shah S, Yang J, Powell JE, Harris SE, et al. Identification of 55,000 replicated DNA methylation QTL. Sci Rep 2018; 8: 17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel NA, Zirlik A, Wolf D. CD40L and its receptors in Atherothrombosis—an Update. Front Cardiovasc Med 2017; 4: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo X-B, Lei S-F, Qian Q-Y, Guo Y-F, Zhang Y-H, Zhang H. Integrative analysis revealed potential causal genetic and epigenetic factors for multiple sclerosis. J Neurol 2019; 266: 2699–709. [DOI] [PubMed] [Google Scholar]
- Pascual M, López-Nevot MA, Cáliz R, Ferrer MA, Balsa A, Pascual-Salcedo D, et al. A poly(ADP-ribose) polymerase haplotype spanning the promoter region confers susceptibility to rheumatoid arthritis: PARP-1 gene promoter polymorphism and RA susceptibility. Arthritis Rheum 2003; 48: 638–41. [DOI] [PubMed] [Google Scholar]
- Rivals I, Personnaz L, Taing L, Potier M-C. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics 2007; 23: 401–7. [DOI] [PubMed] [Google Scholar]
- Robles-Carrillo L, Meyer T, Hatfield M, Desai H, Dávila M, Langer F, et al. Anti-CD40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. J Immunol 2010; 185: 1577–83. [DOI] [PubMed] [Google Scholar]
- Rosado MM, Bennici E, Novelli F, Pioli C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013; 139: 428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainaghi PP, Collimedaglia L, Alciato F, Molinari R, Sola D, Ranza E, et al. Growth arrest specific gene 6 protein concentration in cerebrospinal fluid correlates with relapse severity in multiple sclerosis. Mediators Inflamm 2013; 2013: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandborn WJ, Su C, Sands BE, D’Haens GR, Vermeire S, Schreiber S, et al. Tofacitinib as induction and maintenance therapy for Ulcerative Colitis. N Engl J Med 2017; 376: 1723–36. [DOI] [PubMed] [Google Scholar]
- Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001; 411: 207–11. [DOI] [PubMed] [Google Scholar]
- Smets I, Fiddes B, Garcia-Perez JE, He D, Mallants K, Liao W, et al. Multiple sclerosis risk variants alter expression of co-stimulatory genes in B cells. Brain 2018; 141: 786–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019; 47: D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintore M, Vidal-Jordana A, Sastre-Garriga J. Treatment of multiple sclerosis—success from bench to bedside. Nat Rev Neurol 2019; 15: 53–8. [DOI] [PubMed] [Google Scholar]
- Tocoian A, Buchan P, Kirby H, Soranson J, Zamacona M, Walley R, et al. First-in-human trial of the safety, pharmacokinetics and immunogenicity of a PEGylated anti-CD40L antibody fragment (CDP7657) in healthy individuals and patients with systemic lupus erythematosus. Lupus 2015; 24: 1045–56. [DOI] [PubMed] [Google Scholar]
- van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 2012; 367: 508–19. [DOI] [PubMed] [Google Scholar]
- von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, et al. STRING: known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Res 2005; 33: D433–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Võsa U, Claringbould A, Westra HJ, Bonder MJ. Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis. bioRxiv2018. https://www.biorxiv.org/content/10.1101/447367v1.abstract.
- Weinger JG, Omari KM, Marsden K, Raine CS, Shafit-Zagardo B. Up-regulation of soluble Axl and Mer receptor tyrosine kinases negatively correlates with Gas6 in established multiple sclerosis lesions. Am J Pathol 2009; 175: 283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra H-J, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 2013; 45: 1238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wium M, Paccez JD, Zerbini LF. The dual role of TAM receptors in autoimmune diseases and cancer: an overview. Cells 2018; 7: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zeng J, Zhang F, Zhu Z, Qi T, Zheng Z, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun 2018; 9: 918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016; 48: 481–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this study are publicly available at the URLs given in the methods. The SMR programme is available at the URL given in the methods. SMR was run using default command line options.
All code is available here: https://github.com/benjacobs123456/MS_SMR_druggable_targets.