

Abstract
Motivation
Intercellular communication plays an essential role in multicellular organisms and several algorithms to analyze it from single-cell transcriptional data have been recently published, but the results are often hard to visualize and interpret.
Results
We developed Cell cOmmunication exploration with MUltiplex NETworks (COMUNET), a tool that streamlines the interpretation of the results from cell–cell communication analyses. COMUNET uses multiplex networks to represent and cluster all potential communication patterns between cell types. The algorithm also enables the search for specific patterns of communication and can perform comparative analysis between two biological conditions. To exemplify its use, here we apply COMUNET to investigate cell communication patterns in single-cell transcriptomic datasets from mouse embryos and from an acute myeloid leukemia patient at diagnosis and after treatment.
Availability and implementation
Our algorithm is implemented in an R package available from https://github.com/ScialdoneLab/COMUNET, along with all the code to perform the analyses reported here.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Single-cell RNA sequencing (scRNA-seq) has been extensively used in the past few years to analyze intercellular communication in tissues (see e.g. Bonnardel et al., 2019; Camp et al., 2017; Caruso et al., 2019; Cohen et al., 2018; Halpern et al., 2018; Kumar et al., 2018; Puram et al., 2017; Schiebinger et al., 2019; Sheikh et al., 2019; Skelly et al., 2018; Vento-Tormo et al., 2018; Wang, 2020; Zepp et al., 2017; Zhou et al., 2017). Several algorithms to perform these analyses have been published (for instance, Boisset et al., 2018; Caruso et al., 2019; Efremova et al., 2020; Ramilowski et al., 2015; Rieckmann et al., 2017; Vento-Tormo et al., 2018; Wang, 2020) and they all start from a database of interacting molecular partners (e.g. ligand and receptor pairs) to infer, from their expression patterns, a list of potential communication pathways between cell types.
While the results of these analyses can reveal important insights into the functioning of complex tissues composed of many different cell types, their interpretation can still be challenging with the existing algorithms.
For example, the standard visualization strategies based on graphs or heatmaps, are often busy, poorly interpretable and can hinder data-driven hypothesis generation. Given the sheer number of potentially communicating cell types and interacting partners, it can become challenging to identify all the molecules that could mediate the communication between a given set of cell types. Moreover, to understand the biological relevance of specific interacting molecular partners and the cellular communication they generate, it is desirable to be able to quantitatively compare cellular communication across different datasets.
Here, we present Cell cOmmunication exploration with MUltiplex NETworks (COMUNET), a new tool to visualize and interpret cell–cell communication that is based on multiplex networks and addresses all the challenges mentioned above. More specifically, COMUNET allows (i) unsupervised clustering of interacting partners (e.g. ligand–receptor pairs), (ii) search for specific patterns of communication and (iii) comparison between two biological conditions, aiding the interpretability of the results and the identification of promising candidate molecules to follow-up on. In this article, we show how COMUNET can perform these tasks on two scRNA-seq datasets from mouse embryos and from the bone marrow of an acute myeloid leukemia patient. COMUNET can be easily installed and run, since it is available as an R package from https://github.com/ScialdoneLab/COMUNET.
2 Materials and methods
2.1 Multiplex networks
As a first step, COMUNET uses a multiplex network to represent all significant interacting partners (consisting of, e.g. pairs of ligands and receptors) that can mediate cell–cell communication identified by an algorithm of choice. For the sake of definiteness, in the examples below we use CellPhoneDB (Efremova et al., 2020) to find significant interacting partners. However, COMUNET can work with any algorithm [e.g. ImmProt (Rieckmann et al., 2017) or any of the other algorithms cited in Section 1] that provides a quantification of the strength/statistical significance of the communication between cell types mediated by a given pair of interacting partners. This information can be represented in the form of a matrix (see Fig. 1), which from now on we will refer to as the weight matrix. The input of COMUNET is a set of weight matrices, one for each pair of interacting partners (Fig. 1).
Fig. 1.
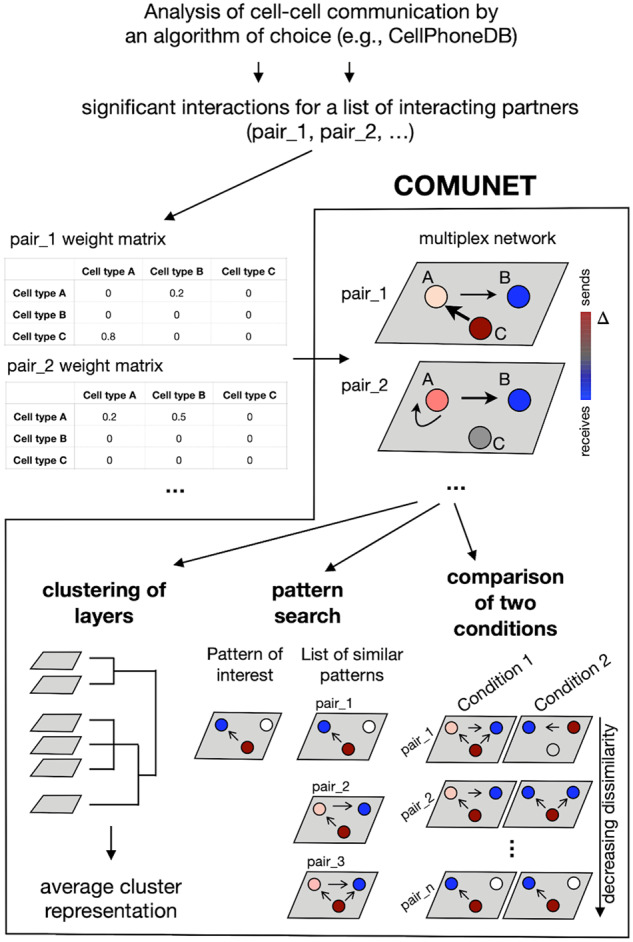
Schematic representation of COMUNET workflow. Once the weight matrices for a set of interacting partners (e.g. ligands–receptor pairs) are estimated with an algorithm of choice (e.g. CellPhoneDB), COMUNET represents them as layers in a multiplex network, where nodes are cell types (indicated with A, B and C). Each node is colored based on the difference between the weighted in- and out-degree (indicated with ), in such a way that, in the case of ligands/receptors, the nodes that preferentially send signals are red, whereas the nodes that preferentially receive signals are blue. Next, COMUNET calculates pairwise dissimilarities between layers in the multiplex network and can perform: clustering of layers, to reveal interacting partners sharing similar communication patterns; search of interacting partners showing a specific communication pattern; comparison of communication patterns between two biological conditions.
Each weight matrix is interpreted as an adjacency matrix of a directed weighted graph, where the entries are the edge weights. For each node (representing cell types), the difference between the weighted in- and out-degree, , is calculated and encoded in the color of the node.
If the pair of interacting partners is a ligand–receptor pair, a positive value of indicates that the node (cell type) is mostly communicating by producing the ligand (‘sending’ node); conversely, a negative marks nodes that communicate mainly by receiving signals through the receptor (‘receiving node’). The arrows of the graph start at the sending nodes (which express the ligand) and point to the receiving nodes (which express the receptor), whereas the thickness of an edge indicates the edge weight.
In case, no directionality is specified for the pair of interacting partners P1 and P2 (i.e. as for adhesion molecules), the arrows start at the node expressing partner P1 and points to the node expressing partner P2. The graphs built from the matrices are then stacked together as layers of a multiplex network (Fig. 1).
2.2 Dissimilarity measure
Once the pairs of interacting partners are represented as layers in a multiplex network, COMUNET calculates pairwise dissimilarities between them.
Given two layers and , their dissimilarity is defined as:
with
where is the weight of the directed edge between nodes and in the layer , is the total number of nodes, is the set of edges in the layer , is the cardinality of the union of all edges in layers and . This definition of dissimilarity can be seen as a modified version of the Jaccard similarity index (Jaccard, 1912) between the sets of edges in the two layers that also takes into account the weights and directionality of the edges.
2.3 Clustering of interacting partners
In this step, COMUNET identifies pairs of interacting partners with similar patterns of intercellular communication and organizes them into clusters (Fig. 1).
This is done by performing hierarchical clustering of the dissimilarity matrix of layers (by default, the ‘hclust’ R function with the ‘average’ agglomeration method is used) and the results can be visualized as a heatmap or a UMAP plot (McInnes et al., 2018). The number of clusters is estimated using the ‘cutreeHybrid’ R function [package dynamicTreeCut, version 1.63-1(Langfelder et al., 2008)] with ‘deepSplit’ equal to 0 and default ‘minClusterSize’ equal to 6. For each cluster, a graph that represents the ‘average’ pattern is built by averaging the adjacency matrices and the (see Section 2.1) of the nodes of all the graphs in the cluster (Figs 1 and 2A).
2.4 Cell–cell communication pattern search
For a more supervised analysis, it is useful to retrieve all pairs of interacting partners that show a specific communication pattern. To this aim, our algorithm classifies the pairs of interacting partners based on their similarity to a user-specified intercellular communication pattern.
A pattern of interest is defined with a binary adjacency matrix, 1 indicating the presence of an edge and 0 the absence. The adjacency matrices of the layers are also binarized based on the presence/absence of the edges and then the dissimilarities with the user-specified adjacency matrix are calculated (see Section 2.2). The output is a list of pairs of interacting partners sorted by increasing dissimilarity with the user-specified pattern (Figs 1 and 2B, Supplementary Table S2).
2.5 Comparative analysis
Given the results of a cell–cell communication analysis of two datasets including the same cell types, COMUNET can estimate the differences in the intercellular communication patterns between corresponding interacting partners in the two datasets. First, the results are represented as multiplex networks, as described above; then, pairwise dissimilarities between the shared layers (e.g. layers representing the same pairs of interacting partners) of the two datasets are calculated (see Section 2.2). The pairs of interacting partners are then sorted by decreasing dissimilarity, i.e. from those that change the most across the two datasets to those that remain unaltered (Figs 1 and 2C, Supplementary Table S3).
3 Results
Below, we apply COMUNET to scRNA-seq datasets from two different publications to show how the clustering, the pattern search and the comparative analysis can help extract biologically relevant information from a cell–cell communication analysis performed with CellPhoneDB (Efremova et al., 2020; Vento-Tormo et al., 2018). In the first example, we used COMUNET to characterize the cellular communication patterns in a mouse embryo dataset, and in the second example, we found the changes in cellular communication in the bone marrow of a leukemia patient between time of diagnosis and after treatment.
3.1 E6.5 mouse embryo
We took scRNA-seq data from a mouse embryo at the E6.5 stage from Nowotschin et al. (2019) (Fig. 2A, Supplementary Fig. S1). Around this stage gastrulation is starting, with signaling between cell types that begin to define where and when the first organs will arise (Tam and Loebel, 2007). As an example, the external layer of cells called visceral endoderm, in addition to being responsible for nutrient uptake and transport, determines the direction of the anterior–posterior axis and the region of epiblast where mesodermal cells will differentiate, through mechanisms and signaling pathways that are still not fully understood (Stower and Srinivas, 2018). On this dataset, including five different cell types, we ran CellPhoneDB and then the clustering algorithm of COMUNET, which finds eight clusters (see Fig. 2A, Supplementary Table S1). These clusters classify the interacting partners according to the pattern of communication they might generate between the different cell types and offer an overview of the main communication patterns present in the data.
Fig. 2.

Application of COMUNET to datasets from mouse embryos and cancer. (A) We used COMUNET to identify clusters of interacting partners in published scRNA-seq data from E6.5 mouse embryo. The dataset includes five cell types: extraembryonic visceral endoderm (exVE), trophectoderm (TE), mesoderm (Mes), embryonic visceral endoderm (emVE) and epiblast (EPI). Eight clusters of interacting partners were identified, each corresponding to a specific communication pattern, whose average representation is depicted in the squares. In particular, Cluster 3 represents communication from the emVE to EPI and Mes, Lefty1:Tdgf1 being a representative ligand:receptor pair included in this cluster. (B) As an example of pattern search, using the same scRNA-seq data of panel A, we searched for the interacting partners showing the pattern of communication depicted in the top left: i.e. a signal originating from the extraembryonic visceral endoderm and received by all other embryonic tissues, which corresponds to the adjacency matrix shown in the top right. COMUNET returned a list of interacting partners sorted by increasing dissimilarity with the specified pattern (bottom left). The bottom right panel illustrates the graphs corresponding to some selected pairs of interacting partners. (C) We applied COMUNET to a published scRNA-seq dataset from the bone marrow of an AML patient at diagnosis (d0) and after treatment (d29) to find differences in communication patterns between the two time points. TNFSF13:FAS and CCL5:CCR5 are examples of interacting pairs with a dramatic change of communication patterns between d0 and d29.
As an example, if we consider the ligands/receptor pairs in Cluster 3, they are responsible for signaling that originates preferentially from the embryonic visceral endoderm (emVE) and is directed to other cell types (Fig. 2A, Supplementary Fig. S1). Indeed, this cluster includes LEFTY1-TDGF1 (Fig. 2A;Supplementary Fig. S1 and Supplementary Table S1), which are part of the nodal signaling that regulates expression of mesodermal genes in the epiblast (Tam and Loebel, 2007).
In addition to this unsupervised analysis, all interacting partners having a specific communication pattern can be identified. For example, in Figure 2B and Supplementary Table S2, we show how a list of all interacting partners that might be responsible for signaling from the extraembryonic visceral endoderm to all the other cell types can be easily obtained. At the top, we found Igf2, which is known to promote embryonic growth (DeChiara et al., 1991), and its receptor Igf2r, which attenuates Igf2 signaling (Brown et al., 2009).
3.2 Human acute myeloid leukemia dataset
There is growing evidence that communication between different cell types in a tissue is altered in diseases like cancer. For instance, in healthy bone marrow, hematopoietic stem cells stay in close contact with niche cells, including endothelial cells, megakaryocytes and adipocytes (Asada et al., 2017; Behrmann et al., 2018), which regulate their proliferation state (Ambrosi et al., 2017; Bruns et al., 2014; Itkin et al., 2016). In hematological malignancies as well as solid cancers, the normal control mechanisms executed by the niche cells are disrupted, such that a novel tumor-specific communicational landscape dominates the tissue. Tumor cells can actively recruit the niche cells from their environment (e.g. fibroblasts, mesenchymal stem cells, T cells or macrophages) and receive growth factors secreted by the niche, to evade from the immune response or to metastasize (Behrmann et al., 2018; Yuan et al., 2016). To investigate the alteration of communication patterns in cancer, it is crucial to have a tool that can perform the communication comparison between conditions, such as before and after treatment.
In this example, we used the data from (van Galen et al., 2019) to show how COMUNET can find differences in communication patterns between two datasets. This publication includes a scRNA-seq data of a bone marrow sample of a 74 years old newly diagnosed female patient with acute myeloid leukemia (AML) (#328) at diagnosis (day 0) and after Azacitidine-Venetoclax-C1D27 treatment (day 29). At the two timepoints, the bone marrow of this patient contains both normal hematopoietic cells, as well as tumor cells (HSC-like, progenitor-like, monocyte-like and cDC-like) (Supplementary Fig. S2). We compared the communication patterns in the two samples and revealed that six pairs of interacting partners dramatically change their pattern of communication upon treatment. Among those we found TNFSF13: FAS and CCL5: CCR5, two pairs of ligands/receptors that are known to play a role in AML and other cancers (Aldinucci and Colombatti, 2014; Brenner et al., 2016; Chapellier et al., 2019; Gmeiner et al., 2015; Mollica Poeta et al., 2019) (Fig. 2C, Supplementary Fig. S2 and Table S3). In AML, TNFSF13, secreted by normal myeloid cells, supports proliferation and apoptosis resistance of tumor cells (Chapellier et al., 2019). In solid cancers, inflammatory chemokines such as CCL5 together with their receptors have been shown to attract neutrophils and monocytes at the tumor site and reprogram them, leading to altered immune response (Mollica Poeta et al., 2019). This signaling might be relevant for the immune response and survival of a subset of tumor cells in this patient after treatment.
4 Discussion
In this article, we introduced COMUNET, a powerful tool for visualization and analysis of intercellular communication. Starting from the output generated by any algorithm that estimates the likelihood or strength of cellular communication, COMUNET represents the pattern of cell communication mediated by each pair of interacting molecules as layers in a multiplex network and calculates the dissimilarity between them. Based on this dissimilarity, COMUNET clusters the pairs of interacting partners based on the pattern of intercellular communication that they can mediate. This facilitates the visualization of the results and gives an overview of the cell types that are involved in the communication, in addition to the molecules that could be responsible for it. Furthermore, COMUNET can prioritize the molecules that are more likely to mediate the communication between a set of specific cell types.
We showed how COMUNET can perform these two tasks in a dataset from mouse embryos, where we classified the communication patterns in eight different clusters and we were able to easily retrieve a list of potential mediators for signaling originating from extraembryonic visceral endoderm.
A comparison of cellular communication between two different conditions is often necessary to understand the role of signaling molecules. COMUNET can perform such a comparison to identify the changes in communication and the molecules that are responsible for them. We showed this in a dataset from an AML bone marrow before and after treatment and we were able to single out molecules that were previously linked with important processes like apoptosis resistance and immune response in cancer.
These examples show how COMUNET significantly increases our ability to go beyond a simple descriptive study of cellular communication by helping to generate hypotheses on the role of communication and to identify candidate genes for validation.
Another feature of COMUNET is the possibility to use it on the output generated by any algorithm that quantifies intercellular communication. This is particularly important given the rapid pace at which new, refined algorithms for cellular communication are being developed and holds true irrespective of the kind of data used to infer communication, including, for instance, single-cell proteomics data (Labib and Kelley, 2020), which can be even more informative on cellular communication than RNA-seq.
5 Data and code availability
COMUNET is available as an R package from https://github.com/ScialdoneLab/COMUNET. In this GitHub repository, a comprehensive tutorial is provided with all the code and the data to reproduce the analyses shown in this article.
Supplementary Material
Acknowledgements
The authors thank Jonathan Fiorentino, Gabriele Lubatti and Frank Ziemann for useful discussions and comments on the tutorials.
Author contributions
M.S. and A.S. designed research; M.S. implemented the method; M.S. and A.S. interpreted the results; M.S. and A.S. wrote the article; A.S. supervised the study.
Financial Support: none declared.
Conflict of Interest: none declared.
References
- Aldinucci D., Colombatti A. (2014) The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm., 2014, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosi T.H. et al. (2017) Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell, 20, 771–784.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada N. et al. (2017) Complexity of bone marrow hematopoietic stem cell niche. Int. J. Hematol., 106, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrmann L. et al. (2018) Acute myeloid leukemia and the bone marrow niche-take a closer look. Front. Oncol., 8, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisset J.-C. et al. (2018) Mapping the physical network of cellular interactions. Nat. Methods, 15, 547–553. [DOI] [PubMed] [Google Scholar]
- Bonnardel J. et al. (2019) Stellate cells, hepatocytes, and endothelial cells imprint the Kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity, 51, 638–654.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner A.K. et al. (2016) A subset of patients with acute myeloid leukemia has leukemia cells characterized by chemokine responsiveness and altered expression of transcriptional as well as angiogenic regulators. Front. Immunol., 7, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. et al. (2009) Keeping IGF-II under control: lessons from the IGF-II-IGF2R crystal structure. Trends Biochem. Sci., 34, 612–619. [DOI] [PubMed] [Google Scholar]
- Bruns I. et al. (2014) Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med., 20, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp J.G. et al. (2017) Multilineage communication regulates human liver bud development from pluripotency. Nature, 546, 533–538. [DOI] [PubMed] [Google Scholar]
- Caruso F.P. et al. (2019) A MAP of tumor–host interactions in glioma at single cell resolution. bioRxiv, 827758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapellier M. et al. (2019) Arrayed molecular barcoding identifies TNFSF13 as a positive regulator of acute myeloid leukemia-initiating cells. Haematologica, 104, 2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. et al. (2018) Lung single-cell signaling interaction map reveals basophil role in macrophage imprinting. Cell, 175, 1031–1044.e18. [DOI] [PubMed] [Google Scholar]
- DeChiara T.M. et al. (1991) Parental imprinting of the mouse insulin-like growth factor II gene. Cell, 64, 849–859. [DOI] [PubMed] [Google Scholar]
- Efremova M. et al. (2020) CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc., 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- van Galen P. et al. (2019) Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell, 176, 1265–1281.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gmeiner W.H. et al. (2015) Thymineless death in F10-treated AML cells occurs via lipid raft depletion and Fas/FasL co-localization in the plasma membrane with activation of the extrinsic apoptotic pathway. Leuk. Res., 39, 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern K.B. et al. (2018) Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat. Biotechnol., 36, 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itkin T. et al. (2016) Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature, 532, 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaccard P. (1912) The distribution of the flora in the alpine zone.1. New Phytol., 11, 37–50. [Google Scholar]
- Kumar M.P. et al. (2018) Analysis of single-cell RNA-seq identifies cell–cell communication associated with tumor characteristics. Cell Rep., 25, 1458–1468.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib M., Kelley S.O.. et al. (2020) Single-cell analysis targeting the proteome. Nat. Rev. Chem., 4, 143–158. [DOI] [PubMed] [Google Scholar]
- Langfelder P. et al. (2008) Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics, 24, 719–720. [DOI] [PubMed] [Google Scholar]
- McInnes L. et al. (2018) UMAP: uniform manifold approximation and projection. JOSS, 3, 861. [Google Scholar]
- Mollica Poeta V. et al. (2019) Chemokines and chemokine receptors: new targets for cancer immunotherapy. Front. Immunol., 10, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S. et al. (2019) The emergent landscape of the mouse gut endoderm at single-cell resolution. Nature, 569, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram S.V. et al. (2017) Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell, 171, 1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilowski J.A. et al. (2015) A draft network of ligand–receptor-mediated multicellular signalling in human. Nat. Commun., 6, 7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieckmann J.C. et al. (2017) Social network architecture of human immune cells unveiled by quantitative proteomics. Nat. Immunol., 18, 583–593. [DOI] [PubMed] [Google Scholar]
- Schiebinger G. et al. (2019) Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell, 176, 928–943.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh B.N. et al. (2019) Systematic identification of cell–cell communication networks in the developing brain. iScience, 21, 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly D.A. et al. (2018) Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep., 22, 600–610. [DOI] [PubMed] [Google Scholar]
- Stower M.J., Srinivas S. (2018) Chapter fourteen – The Head’s Tale: anterior–posterior axis formation in the mouse embryo In: Plusa B., Hadjantonakis A.-K. (eds) Current Topics in Developmental Biology. Academic Press, Oxford, UK, pp. 365–390. [DOI] [PubMed] [Google Scholar]
- Tam P.P.L., Loebel D.A.F. (2007) Gene function in mouse embryogenesis: get set for gastrulation. Nat. Rev. Genet., 8, 368–381. [DOI] [PubMed] [Google Scholar]
- Vento-Tormo R. et al. (2018) Single-cell reconstruction of the early maternal–fetal interface in humans. Nature, 563, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. (2020) talklr uncovers ligand–receptor mediated intercellular crosstalk. bioRxiv, 2020.02.01.930602.
- Yuan Y. et al. (2016) Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep., 35, 2499–2515. [DOI] [PubMed] [Google Scholar]
- Zepp J.A. et al. (2017) Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell, 170, 1134–1148.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.X. et al. (2017) Extracting intercellular signaling network of cancer tissues using ligand–receptor expression patterns from whole-tumor and single-cell transcriptomes. Sci. Rep., 7, 8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
COMUNET is available as an R package from https://github.com/ScialdoneLab/COMUNET. In this GitHub repository, a comprehensive tutorial is provided with all the code and the data to reproduce the analyses shown in this article.