

Abstract
Summary
As the importance of microbiome research continues to become more prevalent and essential to understanding a wide variety of ecosystems (e.g. marine, built, host associated, etc.), there is a need for researchers to be able to perform highly reproducible and quality analysis of microbial genomes. MetaSanity incorporates analyses from 11 existing and widely used genome evaluation and annotation suites into a single, distributable workflow, thereby decreasing the workload of microbiologists by allowing for a flexible, expansive data analysis pipeline. MetaSanity has been designed to provide separate, reproducible workflows that (i) can determine the overall quality of a microbial genome, while providing a putative phylogenetic assignment, and (ii) can assign structural and functional gene annotations with varying degrees of specificity to suit the needs of the researcher. The software suite combines the results from several tools to provide broad insights into overall metabolic function. Importantly, this software provides built-in optimization for ‘big data’ analysis by storing all relevant outputs in an SQL database, allowing users to query all the results for the elements that will most impact their research.
Availability and implementation
MetaSanity is provided under the GNU General Public License v.3.0 and is available for download at https://github.com/cjneely10/MetaSanity. This application is distributed as a Docker image. MetaSanity is implemented in Python3/Cython and C++. Instructions for its installation and use are available within the GitHub wiki page at https://github.com/cjneely10/MetaSanity/wiki, and additional instructions are available at https://cjneely10.github.io/year-archive/. MetaSanity is optimized for users with limited programing experience.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The analysis of microbial genomes has become an increasingly common task for many fields of biology and geochemistry. Researchers can routinely generate hundreds/thousands of environmentally derived microbial genomes using methodologies, such as metagenomics (Tully et al., 2018), high-throughput culturing (Thrash et al., 2015) and single-cell sorting (Stepanauskas et al., 2017). However, analyzing the data can be problematic, as data analysis is computationally intensive and requires a knowledge of software that is constantly changing and may be difficult to install or execute. For the average researcher, the task of evaluating and annotating a set of microbial genomes may be time intensive and computationally rigorous. Here, we present MetaSanity, a comprehensive solution for generating evaluation and annotation pipelines for bacterial and archaeal isolate genomes, metagenome-assembled genomes (MAGs) and single-amplified genomes (SAGs). MetaSanity provides genome quality evaluation, phylogenetic assignment, as well as structural and functional annotation through a variety of integrated programs based on the procedure described in Tully (2019). By providing users with the ability to create annotation pipelines using multiple annotation suites, MetaSanity achieves a broad level of functional annotation that may be missed by implementing a single annotation program. MetaSanity provides a workflow that combines all outputs into a single queryable database that operates easily from the command line. Installation can be performed at the user level, limiting the need for intervention by system administrators, and, except for certain memory intensive programs, can be run locally on high-end personal computers.
2 Materials and methods
MetaSanity consists of two smaller workflows (Fig. 1): (i) PhyloSanity, to evaluate the completion, contamination, redundancy and phylogeny of each genome in a dataset, and (ii) FuncSanity, to provide structural and functional annotations of each genome. Each component consists of several optional applications that can be modified to specific research needs. While each component contained within the two pipelines runs independently and generates component-specific outputs, MetaSanity combines all outputs into a single queryable SQL database that allows fast and easy retrieval of data—in this case, gene annotations and other related genomic data. MetaSanity focuses on allowing users the ability to fine tune and adjust their data analysis pipelines with minimal effort and maximize computational and storage efficiency (Supplementary Table S1). MetaSanity is distributed as a Docker image (Merkel, 2014) and is implemented using a combination of Python3 (Python Software Foundation (2014)/Cython (Bradshaw et al., 2011) and C++ (ISO/IEC 2014). MetaSanity is also available as a source code installation for systems that do not support Docker.
Fig. 1.
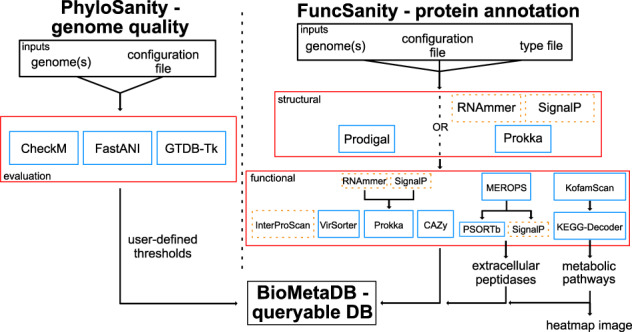
MetaSanity pipeline schema. Programs and databases that are part of the MetaSanity installation are in blue boxes. Programs in the dotted orange boxes must be installed separately by the user due to licensing agreements. DB, database. (Color version of this figure is available at Bioinformatics online.)
2.1 Phylosanity
PhyloSanity is designed to provide metrics of genome quality and to filter genomes for downstream analysis based on user-defined quality metrics. The workflow integrates CheckM v1.0.18 (Parks et al., 2015), GTDB-Tk v.0.3.2 (Parks et al., 2018) and FastANI (Jain et al., 2018) as part of its evaluation pipeline. CheckM estimates the completion and contamination of each genome (Parks et al., 2015). Next, FastANI compares each genome in a pairwise fashion against all other genomes to determine the average nucleotide identify (ANI) for each genome pair (Jain et al., 2018). For any set of genomes that shares an ANI above a user-defined value, a non-redundant genome representative will be selected from the set that is the most complete and least contaminated. This allows users the option to exclude redundant genomes from further analysis. Differentiating genomes as non-redundant versus redundant can be useful for researchers working with MAGs or SAGs that are generated from replicate samples and may not have biological meaning when working with isolates or strain level differences. All genomes can undergo phylogenetic assignment based on relative evolutionary distance (Parks et al., 2018) through GTDB-Tk (Pierre-Alain et al., 2020), which will replace the CheckM-returned taxonomic assignment.
2.2 Funcsanity
FuncSanity provides structural and functional annotation of microbial genomes. The workflow incorporates annotation suites from eight existing and widely used programs. The use of multiple annotation programs has the advantage of capturing functional predictions that may not have been detected due to database or search limitations. Specialized annotation programs, such as VirSorter v1.0.5 (Roux et al., 2015), use custom tools and/or databases to return relevant annotations that are not captured by other programs in MetaSanity. Open reading frames (ORFs) are predicted using Prodigal v2.6.3 (Hyatt et al., 2010); however, users may opt to use the putative coding DNA sequences (CDS) generated by Prokka v1.13.3 (Seemann, 2014). From here, putative ORFs are processed by a set of annotation tools that can be selected by the user with user-defined filtering and cut-off values.
2.2.1 Kyoto Encyclopedia of Genes and Genomes annotation
Putative ORFs can be searched against the KofamKOALA database using KofamScan v.1.1.0 (Aramaki et al., 2019). Default parameters are used, and the ‘mapper’ tab-delimited output option is generated, linking ORF IDs to Kyoto Encyclopedia of Genes and Genomes (KEGG) Ontology (KO) IDs. Users can query any KO ID to generate specific functional search results in BioMetaDB.
2.2.2 KEGG-Decoder
KEGG annotations can be used to estimate the completeness of various biogeochemically relevant metabolic pathways in a genome using KEGG-Decoder v.1.0.10 (Graham et al., 2018; https://github.com/bjtully/BioData/tree/master/KEGGDecoder). Users can search genomes based on completeness of a pathway or function of interest. An additional heatmap summary visualization is generated.
2.2.3 Virsorter
VirSorter v1.0.5 (Roux et al., 2015) can be implemented to identify phage and prophage signatures in each genome using default parameters. Users can search for matches to each of the phage and prophage categories returned by VirSorter and generate lists of contigs and/or genomes with the assignments (Supplementary Table S1).
2.2.4 Interproscan
InterProScan 5.36-75.0 (Jones et al., 2014) is an optional installation and can be used for domain prediction on putative ORFs. Users have the option of downloading all of the InterProScan databases, including TIGRfam (Haft et al., 2003), Pfam (Finn et al., 2016), CDD (Marchler-Bauer et al., 2010, Zhang et al., 2017) and PANTHER (Mi et al., 2019). Each InterProScan database result is indexed separately in BioMetaDB and can be used to return matching genomes using database specific IDs (e.g. PF01036 would return putative rhodopsin ORFs from a Pfam result).
2.2.5 Prokka annotation
If not chosen as the option for structural annotation, genomes can be annotated using Prokka and its associated databases with the parameters –addgenes (adds the ‘gene’ feature to each CDS in the GenBank output format), –addmrna (adds the ‘mRNA’ feature to each CDS in the GenBank output format), –usegenus (use the genus-specific databases), –metagenome (improve gene predictions for fragmented genomes) and –rnammer [sets RNAmmer as the preferred rRNA prediction tool instead of Barrnap (http://www.vicbioinformatics.com/software.barrnap.shtml)]. rRNA identification with RNAmmer v.1.2 (Lagesen et al., 2007) and signal peptide detection with SignalP v.4.1 (Nielsen, 2017) are optional installations. Users are given the option to use Prokka-derived putative CDS, which take into consideration the locations of RNA gene sequences (tRNA, rRNA, tmRNA and ncRNA) in their downstream analysis. These putative proteins are used in further analysis instead of the Prodigal-derived CDS, which are not aware of RNA boundaries.
2.2.6 Carbohydrate-active enzyme annotation
Putative ORFs can be assigned a putative carbohydrate-active enzyme (CAZy) functionality (Cantarel et al., 2009) based on the dbCANv2 database (Zhang et al., 2018). ORFs are searched against dbCANv2 using HMMER v3.1b2 (Eddy, 2011) with the minimum score threshold set to 75 (-T parameter).
2.2.7 Peptidase annotation
Putative ORFs can be assigned to a peptidase family using a set of HMMs that represent the MEROPS database (Rawlings et al., 2014). PSORTb v.3.0 (Yu et al., 2010) and SignalP can be optionally performed on MEROPS matches to determine if a putative enzyme is predicted to be extracellular. An extracellular assignment is made if PSORTb predicts ‘extracellular’ or ‘outer membrane’ localization or if PSORTb returns ‘unknown’ localization and SignalP predicts the presence of a signal peptide. Users can search for genes and genomes based on overall MEROPS annotations or by searching for specific designations (e.g. GT41 for glycosyl transferase family 41).
InterProScan, SignalP and RNAmmer are not automatically distributed with MetaSanity and require users to download their binaries separately and agree to their individual license requirements.
2.2.8 Configuration adjustments
Each run of MetaSanity can be adjusted to the needs of the genome(s) being analyzed. One of the required parameters for MetaSanity if a configuration (config) file that sets which analyses will be performed on a genome or set of genomes, including optional settings (e.g. number of CPU threads, etc.). Config files allow users to determine specific pipelines of analyses, as needed, and provide a record which can be used for future genome sets for rapid re-deployment of analyses.
2.3 Biometadb
MetaSanity outputs consist of tab-delimited descriptions of genomic data, which can easily be analyzed by a variety of external tools. We additionally provide BioMetaDB, a specialized relational database management system tool that integrates modularized storage and retrieval of FASTA records with the metadata describing them. This application uses tab-delimited data files to generate table relation schemas via Python3. Based on SQLAlchemy v.1.3.7 (Bayer, 2012), BioMetaDB allows researchers to efficiently manage data from the command line by providing operations that include: (i) the ability to store information from any valid tab-delimited data file and to quickly retrieve FASTA records or annotations related to these datasets by using SQL-optimized command-line queries; and, (ii) the ability to run all CRUD operations (create, read, update, delete) from the command line and from python scripts. Output from both workflows is stored into a BioMetaDB project, providing users a simple interface to comprehensively examine their data (Supplementary Table S2). Users can query application results used across the entire genome set for specific information that is relevant to their research, allowing the potential to screen genomes based on returned taxonomy, quality, annotation, putative metabolic function or any combination thereof. More information on using BioMetaDB is available via the BioMetaDB GitHub (https://github.com/cjneely10/BioMetaDB).
3 Results
MetaSanity was tested on two separate systems—a personal computer with an Intel core i5-4570 CPU @ 3.20 GHz processor with 4 cores and 32 GB of RAM operating the Ubuntu 18.04.3 LTS Linux distribution, and an academic server with an Intel Xeon E7-4850 v2 @ 2.30 GHz processor with 96 cores and 1 TB of RAM operating the Ubuntu 18.04.3 LTS Linux distribution. Reduced options were calculated on the personal computer using all four available threads and preset parameter flags that skip memory-intensive processes. Complete options were calculated on the academic server using 10 threads and no parameters to reduce memory usage. Runtime results are available in Supplementary Table S3. The current architecture relies on sequential completion of time intensive processes, several of which are optional for users. Ongoing modifications that take advantage of parallelizing these processes should decrease the overall computation time.
Supplementary Material
Acknowledgements
We would like to thank Taylor Reiter, Roth Conrad, Jay Osvatic and Luiz Irber for providing code contributions to KEGG-Decoder as part of the Moore Foundation funded ‘Speeding Up Science’ hackathon. This is C-DEBI Contribution 533 .
Funding
This work was supported by the National Science Foundation Science and Technology Center, the Center for Dark Energy Biosphere Investigations (C-DEBI) [OCE-0939654 to B.J.T.].
Conflict of Interest: none declared.
References
- Aramaki T. et al. (2019) KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics, 36, 2251–2252, 10.1101/602110 [DOI] [PMC free article] [PubMed]
- Bayer M. (2012) SQLAlchemy In: Brown A., Wilson G. (eds) The Architecture of Open Source Applications Volume II: Structure, Scale, and a Few More Fearless Hacks. http://aosabook.org. [Google Scholar]
- Bradshaw R. et al. (2011) The Cython Compiler http://cython.org. (October 2019, date last accessed).
- Camacho C. et al. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel B.L. et al. (2009) The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res., 37, D233–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. (2011) Accelerated profile HMM searches. PLoS Comput. Biol., 7, e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D. et al. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res., 44, D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham E.D. et al. (2018) Potential for primary productivity in a globally-distributed bacterial phototroph. Isme J., 12, 1861–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D.H. et al. (2003) The TIGRFAMs database of protein families. Nucleic Acids Res., 31, 371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. et al. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISO/IEC. (2014) ISO International Standard ISO/IEC 14882:2014(E) – Programming Language C++. [Working Draft]. International Organization for Standardization (ISO), Geneva, Switzerland: https://isocpp.org/std/the-standard. [Google Scholar]
- Jain C. et al. (2018) High-throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun., 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. et al. (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics, 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K. et al. (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res., 35, 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A. et al. (2010) pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics, 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel D. (2014) Docker: lightweight Linux containers for consistent development and deployment. Linux J. [Google Scholar]
- Mi H. et al. (2019) Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc., 14, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H. (2017) Predicting secretory proteins with signalP In: Kihara D. (ed.) Protein Function Prediction. Methods in Molecular Biology. Vol. 1611 Humana Press, New York, NY, pp. 59–73. [DOI] [PubMed] [Google Scholar]
- Parks D.H. et al. (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res., 25, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D.H. et al. (2018) A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol., 36, 996–914. [DOI] [PubMed] [Google Scholar]
- Pierre-Alain C. et al. (2020) GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics., 36, 1925–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Python Software Foundation. (2014) Python Language Reference, Version 3 http://www.python.org.
- Rawlings N.D. et al. (2014) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res., 42, D503–D509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S. et al. (2015) VirSorter: mining viral signal from microbial genomic data. PeerJ., 3, e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics, 30, 2068–2069. [DOI] [PubMed] [Google Scholar]
- Stepanauskas R. et al. (2017) Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles. Nat. Commun., 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash J.C. et al. (2015). Cultivating fastidious microbes In: Hydrocarbon and Lipid Microbiology Protocols. Vol. 39 Springer, Berlin, Heidelberg, pp. 57–78. doi: 10.1007/8623_2015_67. [DOI] [Google Scholar]
- Tully B.J. (2019) Metabolic diversity within the globally abundant Marine Group II Euryarchaea offers insight into ecological patterns. Nat. Commun., 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully B.J. et al. (2018) The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data, 5, 170203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N.Y. et al. (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics, 26, 1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. et al. (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res., 45, D200–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. et al. (2018) dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res., 46, W95–W101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.