

Abstract
Summary
Diverse traits have evolved through cis-regulatory changes in genome sequence that influence the magnitude, timing and cell type-specificity of gene expression. Advances in high-throughput sequencing and regulatory genomics have led to the identification of regulatory elements in individual species, but these genomic regions remain difficult to align across taxonomic orders due to their lack of sequence conservation relative to protein coding genes. The groundwork for tracing the evolution of regulatory elements is provided by the recent assembly of hundreds of genomes, the generation of reference-free Cactus multiple sequence alignments of these genomes, and the development of the halLiftover tool for mapping regions across these alignments. We present halLiftover Post-processing for the Evolution of Regulatory Elements (HALPER), a tool for constructing contiguous regulatory element orthologs from the outputs of halLiftover. We anticipate that this tool will enable users to efficiently identify orthologs of regulatory elements across hundreds of species, providing novel insights into the evolution of traits that have evolved through gene expression.
Availability and implementation
HALPER is implemented in python and available on github: https://github.com/pfenninglab/halLiftover-postprocessing.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The availability of genomes from thousands of species (Gross, 2018) provides us with an unprecedented opportunity to study evolution across species whose most recent common ancestor lived hundreds of millions of years ago. Many traits have evolved at least in part through gene expression (King and Wilson, 1975), making identifying orthologous regulatory elements essential for studying trait evolution. Although high-throughput sequencing technologies have enabled the identification of regulatory elements in a wide range of tissues across diverse species (Giuffra et al., 2019; Mouse ENCODE Consortium et al., 2012; The ENCODE Project Consortium, 2012), identifying these regulatory elements’ orthologs in other species’ genomes remains challenging. Popular methods for ortholog identification involve using pairwise sequence alignments (Kent et al., 2003), which are often less accurate for distantly related species than multi-species alignments are (Blanchette et al., 2004; Paten et al., 2011; Rosenberg, 2005). Alternatively, MultiZ multi-species alignments are anchored to a reference species that might not be closely related to the species in a study and cannot account for certain genomic structural rearrangements, such as inversions (Blanchette et al., 2004). In contrast, new Cactus multi-species alignments are reference-free and can account for a wide range of structural rearrangements, overcoming many limitations of previous methods (Paten et al., 2011). The current Cactus alignment with the most distantly related species contains 600 mammalian and avian genomes (https://www.biorxiv.org/content/10.1101/730531v2), enabling us to map regulatory elements across an unprecedentedly diverse group of species.
The 'Hierarchical Alignment (HAL) Format API' (Hickey et al., 2013) provides a suite of methods for working with Cactus alignments, including a method called halLiftover that identifies putative orthologs of genomic regions from a query species in a target species. Given a region of interest in the query species, the method outputs all regions in the target species that align to any part of the query species region. Thus, if the target species has insertions relative to the query species or if different parts of the region in the query species map to different target genomic regions, the output will consist of many fragmented regions instead of a single contiguous region. In practice, for regulatory element-sized queries, the output of halLiftover often contains over an order of magnitude more regions than the input. This makes the outputs of halLiftover difficult to interpret.
We developed halLiftover Post-processing for the Evolution of Regulatory Elements (HALPER), a tool for creating contiguous putative region orthologs from the outputs of halLiftover (Fig. 1). To identify putative orthologs, HALPER connects mapped fragments surrounding the target species ortholog of a specified focal position from the query species region (e.g. a peak summit), discarding putative orthologs that do not meet user-specified criteria. We anticipate that HALPER will help researchers trace the evolution of regulatory elements across hundreds of genomes, ultimately providing insights into the evolution of many traits.
Fig. 1.
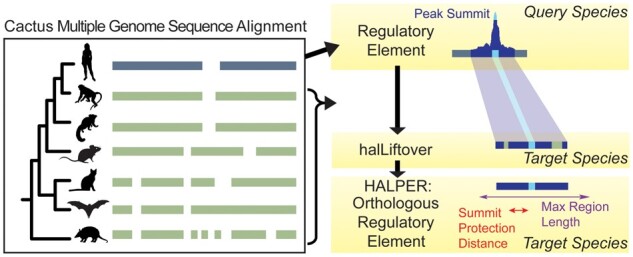
HALPER overview. HALPER constructs contiguous orthologs from the outputs of halLiftover by first identifying the mapped fragment in the target species that contains the ortholog of the query species peak summit. It then extends the region until all mapped fragments on the same chromosome have been included. Putative target species orthologs are excluded if they do not meet user-specified criteria. Animal silhouettes were downloaded from http://phylopic.org/
2 Materials and methods
2.1 Preparing data for HALPER
The intuition behind HALPER is that regulatory elements contain focal positions (such as peak summits) around which functionally important sequences are clustered (Supplementary Material). Thus, any region’s putative ortholog should contain the ortholog of that focal position. To this end, HALPER requires three input BED files: a ‘query file’ of genomic regions of interest in a query species (e.g. the output file from a peak-caller), a ‘target file’ containing the raw outputs of the HAL Format API tool halLiftover (Hickey et al., 2013) run to map regions in the query file to the genome of a target species of interest, and a ‘summits file’. The summits file contains the outputs of halLiftover run to map the focal position (summit) of each query region (peak) to the target species. If the regions of interest in the query file are open chromatin or transcription factor (TF) Chromatin Immunoprecipitation Sequencing (ChIP-seq) peaks, the summits file should be the outputs of halLiftover run on the peak summits. HALPER also comes with guidelines for creating a ‘summits file’ for histone modification ChIP-seq and other genomic regions, for which the position with the greatest alignment depth can be used as the focal position instead of peak summits (Supplementary Material).
2.2 HALPER method
HALPER constructs contiguous orthologs from the typically fragmented outputs of halLiftover (Hickey et al., 2013). For each query species region of interest mapped to a target species, HALPER extends the region with the mapped summit outward to include the neighboring mapped fragments (Fig. 1). HALPER continues this until the constructed region includes all target species-mapped fragments on the same chromosome as the mapped summit. HALPER then discards the putative target species ortholog if it does not meet the following criteria: (i) length is at least a user-specified minimum length, (ii) length does not exceed a user-specified maximum length and (iii) distance from the mapped summit to the putative ortholog’s end in either direction is at least a user-specified ‘Summit protection distance.’ Thus, HALPER enables users to specify the criteria for defining candidate orthologs (Fig. 1).
3 Conclusions
HALPER constructs contiguous orthologs from the outputs of halLiftover, enabling researchers to leverage state-of-the-art Cactus genome alignments when tracing the evolution of regulatory elements. This tool will enable researchers to map orthologs of regulatory elements and other genomic regions across hundreds of genomes. We anticipate that HALPER will help provide insights into the gene regulatory mechanisms underlying traits that have evolved through gene expression.
Supplementary Material
Acknowledgements
We thank the members of the Pfenning Lab for useful discussions and the members of the Paten Lab and Zoonomia Project for sharing Cactus alignments.
Funding
I. M. K. was supported by the Carnegie Mellon University Computational Biology Department (CMU CBD) Lane Fellowship; M. W. by the CMU BrainHub Fellowship; A. R. P. by the Alfred P. Sloan Foundation Research Fellowship and the National Institutes of Health, National Institute on Drug Abuse (NIDA) [DP1DA046585], which also partially supported I. M. K. and M. W.; T. Y. P. by the Center for the Neural Basis of Cognition Undergraduate Research Program in Neuro Computation (CNBC uPNC) and X. Z. by a CMU Small Undergraduate Research Grant (CMU SURG).
Conflict of Interest: none declared.
Contributor Information
Xiaoyu Zhang, Department of Computer Science.
Irene M Kaplow, Department of Computational Biology; Neuroscience Institute.
Morgan Wirthlin, Department of Computational Biology; Neuroscience Institute.
Tae Yoon Park, Department of Biology, Carnegie Mellon University, Pittsburgh, PA 15213, USA.
Andreas R Pfenning, Department of Computational Biology; Neuroscience Institute.
References
- Blanchette M. (2004) Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res., 14, 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffra E., Tuggle C.K.FAANG Consortium. (2019) Functional Annotation of Animal Genomes (FAANG): current achievements and roadmap. Annu. Rev. Anim. Biosci., 7, 65–88. [DOI] [PubMed] [Google Scholar]
- Gross M. (2018) The genome sequence of everything. Curr. Biol., 28, R719–R721. [Google Scholar]
- Hickey G. et al. (2013) HAL: a hierarchical format for storing and analyzing multiple genome alignments. Bioinformatics, 29, 1341–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J. et al. (2003) Evolution’s cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. USA, 100, 11484–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M.C., Wilson A.C. (1975) Evolution at two levels in humans and chimpanzees. Science, 188, 107–116. [DOI] [PubMed] [Google Scholar]
- Mouse ENCODE Consortium. et al. (2012) An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol., 13, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paten B. et al. (2011) Cactus: algorithms for genome multiple sequence alignment. Genome Res., 21, 1512–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg M.S. (2005) Multiple sequence alignment accuracy and evolutionary distance estimation. BMC Bioinformatics, 6, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.