

Abstract
Excitotoxicity is thought to play key roles in brain neurodegeneration and stroke. Here we show that neuroprotection against excitotoxicity by trophic factors EFNB1 and brain-derived neurotrophic factor (called here factors) requires de novo formation of ‘survival complexes’ which are factor-stimulated complexes of N-methyl-d-aspartate receptor with factor receptor and presenilin 1. Absence of presenilin 1 reduces the formation of survival complexes and abolishes neuroprotection. EPH receptor B2- and N-methyl-d-aspartate receptor-derived peptides designed to disrupt formation of survival complexes also decrease the factor-stimulated neuroprotection. Strikingly, factor-dependent neuroprotection and levels of the de novo factor-stimulated survival complexes decrease dramatically in neurons expressing presenilin 1 familial Alzheimer disease mutants. Mouse neurons and brains expressing presenilin 1 familial Alzheimer disease mutants contain increased amounts of constitutive presenilin 1–N-methyl-d-aspartate receptor complexes unresponsive to factors. Interestingly, the stability of the familial Alzheimer disease presenilin 1–N-methyl-d-aspartate receptor complexes differs from that of wild type complexes and neurons of mutant-expressing brains are more vulnerable to cerebral ischaemia than neurons of wild type brains. Furthermore, N-methyl-d-aspartate receptor-mediated excitatory post-synaptic currents at CA1 synapses are altered by presenilin 1 familial Alzheimer disease mutants. Importantly, high levels of presenilin 1–N-methyl-d-aspartate receptor complexes are also found in post-mortem brains of Alzheimer disease patients expressing presenilin 1 familial Alzheimer disease mutants. Together, our data identify a novel presenilin 1-dependent neuroprotective mechanism against excitotoxicity and indicate a pathway by which presenilin 1 familial Alzheimer disease mutants decrease factor-depended neuroprotection against excitotoxicity and ischaemia in the absence of Alzheimer disease neuropathological hallmarks which may form downstream of neuronal damage. These findings have implications for the pathogenic effects of familial Alzheimer disease mutants and therapeutic strategies.
Keywords: post-synaptic currents, neuronal survival, survival complexes, trophic factors, excitotoxicity
We describe a presenilin 1-dependent mechanism by which neurotrophic factors protect neurons rom excitotoxicity, a condition with crucial roles in neurodegeneration and stroke. The mechanism includes presenilin1-dependent N-methyl-d-aspartate receptor complexes with factor receptors. Familial Alzheimer disease mutants alter N-methyl-d-aspartate receptor-presenilin1 associations and block factor-dependent neuroprotection in absence of Alzheimer disease hallmarks.
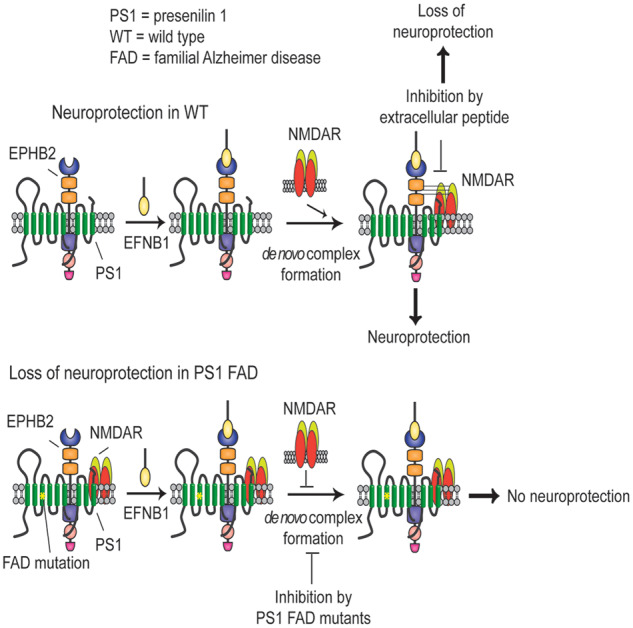
Graphical Abstract
Graphical Abstract.
Introduction
Neurodegenerative disorders, including Alzheimer disease, are characterized by progressive loss of specific neuronal populations (Price et al., 2001), but the mechanisms underlying this selective vulnerability are not fully understood. Neuronal exposure to toxic insults, including glutamate excitotoxicity, oxidative stress and ischaemia, however, is believed to play crucial roles in chronic neurodegenerative disorders such as Alzheimer disease and Huntington diseases (Choi, 1988, 1995; Mattson, 2003; Lipton, 2006). N-methyl-d-aspartate receptors (NMDARs) play central roles in synaptic transmission, plasticity, learning and memory but paradoxically, NMDAR-associated excitotoxicity is also believed to play central roles in neuronal death associated with neurodegenerative diseases, stroke, ischaemia and traumatic brain injury (Arundine and Tymianski, 2003, Hardingham, 2009, Parsons and Raymond, 2014). Thus, it is generally accepted that proper activation of NMDARs promotes neuronal survival while impaired or excessive NMDAR activation leads to excitotoxicity, pathological outcomes and neurodegeneration (Paoletti et al., 2013; Parsons and Raymond, 2014). To counter toxic insults, the brain develops neuroprotective mechanisms some regulated by neuroprotective factors such as brain-derived neurotrophic factor (BDNF) and EFNs (ephrins) (Calo et al., 2006; Nagahara and Tuszynski, 2011; Barthet et al., 2013; Theus et al., 2014).
Mutations of the gene encoding the transmembrane protein presenilin 1 (PS1) are responsible for most cases of early onset autosomal dominant familial Alzheimer disease (FAD). Although the autosomal dominant pattern of FAD transmission suggests these mutants may cause gain of neurotoxic functions or dominant loss of essential functions (Shen and Kelleher, 2007; Wolfe, 2007), the mechanisms by which PS1 FAD mutants cause dominant neurodegeneration remain poorly understood. PS1 has been shown to have diverse functions including a crucial role in the γ-secretase complex that processes transmembrane proteins and mediates production of the Aβ peptides (Barthet et al., 2012; Haapasalo and Kovacs, 2011), and in γ-secretase-independent functions such as autophagy (Lee et al., 2010), calcium homeostasis (Tu et al., 2006), neurotransmitter release (Zhang et al., 2009) and neuroprotection (Barthet et al., 2013). Of particular significance to neuronal survival is the role of PS1 in mediating the neuroprotective functions of EFNB1 and BDNF against excitotoxicity (Barthet et al., 2013). It is known that BDNF and EFNs function in axon guidance, neuronal survival and synaptic activity and that they initiate cellular signalling cascades by stimulating formation of dynamic complexes of their cognate receptors with multiple proteins including the subunits of the NMDAR (Suen et al., 1997; Dalva et al., 2000; Husi et al., 2000; Henderson et al., 2001; Takasu et al., 2002; Wang et al., 2011). Furthermore, literature reports in the last two decades show that BDNF- and EFNB-induced interactions of their cognate receptors with the NMDAR regulate its functions in long-term potentiation, cognition and disease (Minichiello, 2009; Zhang et al., 2012; Cisse and Checler, 2015; Dines and Lamprecht, 2016). Here we show that PS1 is necessary for de novo formation of EFNB1 and BDNF (called here factors)-stimulated complexes of factor receptors (FRs) with the NMDAR and that formation of these complexes is crucial to factor-dependent neuroprotection. In contrast, PS1 FAD mutants inhibit factor-dependent neuroprotection, increase neuronal vulnerability to ischaemia, and reduce the NMDAR-mediated excitatory post-synaptic currents at CA1 synapses. Furthermore, expression of PS1 FAD mutants increases the constitutive levels of the NMDAR–PS1 association but renders it unresponsive to neuroprotective factors.
Materials and methods
Materials and antibodies
Antibodies used in our studies were as follows: for IP experiments; anti-PS1 (rabbit polyclonal, R222) (Georgakopoulos et al., 1999), anti-EPHB2 (rabbit polyclonal, R407) (Barthet et al., 2013), anti-GLUN1 (mouse monoclonal; Novus Biologicals, Littleton, CO, USA) and anti-TRKB (rabbit polyclonal, Abcam; Cambridge, MA, USA); for western blot (WB) detection; anti-PS1 (rabbit polyclonal, R222), anti-PS1 (mouse monoclonal, 33B10) (Huang et al., 2018), anti-EPHB2 (rabbit polyclonal, Zymed; San Francisco, CA, USA), anti-GLUN1 (mouse monoclonal; BD Biosciences, San Jose, CA, USA), anti-GLUN2B (mouse monoclonal; BD Biosciences, San Jose, CA, USA), anti-ACTIN (mouse monoclonal; Abcam, Cambridge, MA, USA), anti-TRKB (rabbit polyclonal, Abcam; Cambridge, MA, USA); for immunofluorescent experiments; anti-EPHB2 (rabbit polyclonal, Abcam; Cambridge, MA, USA), anti-GLUN1 (mouse monoclonal; Biolegend, San Diego, CA, USA) and anti-PS1 (rabbit polyclonal, R222, affinity purified). Anti PS1-CTF and anti PS1-NTF: antibodies against the C- or N-terminals of PS1 respectively. EFNB1-Fc (eB1) ligands prepared as described (Barthet et al., 2013). Accell non-targeting and small interfering RNA (siRNA) SMARTpools against mouse PS1 mRNAs were from Dharmacon (Lafayette, CO, USA); γ-secretase inhibitor (L-685,458) from EMD Millipore (Billerica, MA, USA); EFNB1-Fc from R & D Systems (Minneapolis, MN, USA); and BDNF from Sigma (St. Louis, MO, USA). Clustering of recombinant mouse EFNB1-Fc ligands and anti-Fc was performed as described (Barthet et al., 2013).
Primary cortical neuronal cultures
All procedures involving animals were performed in accordance with the National Institutes of Health (NIH) guide for the Care and Use of Laboratory Animals and were approved by the Icahn School of Medicine at Mount Sinai (New York, NY, USA) Animal Care and Use Committee. Cortical neuronal cultures from mouse brains at embryonic day 15.5 were prepared as described previously (Barthet et al., 2013; Bruban et al., 2015). Dissociated brain cells were plated onto poly-d-lysine-coated plates at a density of ∼1 × 105 cells/cm2 and maintained in neurobasal medium that was supplemented with 2% B27 (Thermo Fisher Scientific), l-glutamine (0.5 mM) and penicillin/streptomycin (1% vol/vol), and used at 10–12 days in vitro (DIV). Under these conditions, post-mitotic neurons represent more than 98% of cultured cells (Huang et al., 2018).
Protein down-regulation by siRNA
Down-regulation of neuronal PS1 by using anti-PS1 small interfering RNA (siRNA), and control siRNAs (Dharmacon) was performed as previously described (Huang et al., 2018). For down-regulation, neurons were treated at 7 DIV with 1 µM of specific siRNA or control siRNA, and cultures were used for experiments at DIV 10–12. Down-regulation of the proteins of interest was examined by WB.
Preparation of mouse brain tissue lysates
Whole tissue lysates were prepared from cortical hemispheres or hippocampi of 4–8 weeks old mice. Briefly, brain tissues were homogenized on ice in 1% Triton X-100 lysis buffer (10 mM HEPES, 2 mM CaCl2, 150 mM NaCl, 0.02% NaN3; pH 7.4) containing protease and phosphatase inhibitors. The homogenate was centrifuged at 14 000 g for 20 min at 4°C, and the supernatant was used as brain tissue lysate for immunoprecipitation experiments, as described below.
Preparation of synaptosomal fractions from post-mortem human brains
We received frontal cortices of post-mortem human brain carrying wild type (WT) PS1 and PS1 S170F mutation from the DIAN Human Brain Tissue Bank at Washington University. Frontal cortices were cut into smaller pieces on ice. Then, equal amount of tissue was used to prepare synaptosomal fractions by biochemical fractionation. Briefly, tissue chunks were homogenized on ice in 0.32 M sucrose, 4 mM HEPES, pH 7.4, containing protease and phosphatase inhibitors. After removing the nuclear fraction by centrifuging at 1000 g for 15 min at 4°C, non-synaptic fractions were further centrifuged at 12 000 g at 4 °C to obtain the crude synaptosomal fraction. Crude synaptosomal pellets were resuspended and solubilized in lysis buffer (50 mM Tris, 140 mM NaCl, 0.5% Triton X-100; pH 7.4) containing protease and phosphatase inhibitors. The resulting solubilized fractions were used for immunoprecipitation and WB experiments.
Cell survival assay
Briefly, mature cortical neuronal cultures were prepared and kept in Neurobasal medium supplemented as described previously; the medium was changed to Hanks’ balanced salt solution (HBSS) (Invitrogen) for 30 min, and then clustered EFNB1-Fc or BDNF was added for another 30 min. Glutamate was then added for 3 h before measurements. For nuclei staining, cells were fixed in 4% paraformaldehyde for 20 min at room temperature and stained with Hoechst 33342 for 10 minutes. Stained nuclei were then observed under a fluorescence microscope using ultraviolet illumination and counted according to manufacturer’s instructions (Sigma). Cells were kept and maintained, and numbers of viable neurons were determined as described (Barthet et al., 2013). Clustered EFNB1-Fc was prepared as reported (Barthet et al., 2013).
Immunoprecipitations
After exposure of the neuronal cultures to the conditions as indicated in the figure legends, total cellular extracts from were prepared in 1% Triton X-100 lysis buffer (10 mM HEPES, 2 mM CaCl2,150 mM NaCl, 0.02% NaN3; pH 7.4) containing protease and phosphatase inhibitors following the method as described previously (Al Rahim et al., 2013). Fixed amount of proteins were subjected to immunoprecipitation by incubating overnight with indicated antibodies at 4°C. Then, 25 μl of protein A/G-agarose conjugated beads was added to each sample and incubated for 2–4 more hours at 4°C. Beads were washed two times with lysis buffer and once with HEPES buffer containing protease and phosphatase inhibitors. Proteins were subsequently eluted from the beads by boiling in 1× SDS sample buffer for 5 min. Eluted proteins were separated on SDS-PAGE gels, transferred to polyvinylidene difluoride (PVDF) membrane (Millipore), blocked with 5% milk, labelled with the indicated primary and horseradish peroxidase (HRP)-conjugated secondary antibodies, and visualized with West Pico or Dura Chemiluminescent reagents (Thermo Scientific). We performed densitometric analysis to determine the level of co-immunoprecipitated protein.
Immunocytochemistry and immunohistochemistry
For detection of surface EPHB2/GLUN1 clusters, neurons were treated with EFNB1-Fc or Fc for 1 h prior to incubation with primary antibodies against EPHB2 (1:100, Rabbit polyclonal) and GLUN1 (1:50, Mouse monoclonal) for 8 min in the incubator. After washing with PBS gently once, neurons were fixed with 4% paraformaldehyde and 2% sucrose for 8 min. After washing with PBS three times, cells were blocked with 1% bovine albumin and 0.2% cold-water fish gelatine for 1 h before incubation with secondary antibodies (Goat anti-rabbit IgG with Alexa Fluor 488 conjugates, and goat anti-mouse IgG with Alexa Fluor 568 conjugates) for 45 min at room temperature. After washing with PBS three times, the coverslips were mounted on slides with Vectashield mounting media with DAPI (Vector Laboratories). For immunohistochemistry, adult mice were euthanized and perfused transcardially with PBS followed by 4% PFA. The brains were removed and post-fixed overnight in 4% PFA, soaked in 30% sucrose for 3 days at 4°C. The brains were later embedded in optimal cutting temperature (OCT) compound and sliced at 20 μm sections on a cryostat. The brain slices were mounted on slides and blocked with 10% heat inactivated goat serum (HIGS) in PBS for 1 h, stained with anti-PS1 and aniti-GLUN1 antibodies overnight at 4°C. The following day, the slides were washed, stained with corresponding secondary antibodies, and mounted.
Electrophysiology
For slice preparation, WT and PS1 FAD (M146V) adolescent mice of either sex, 21–25 days old, were deeply anaesthetized with isoflurane and decapitated. Brains were rapidly removed and placed into ice-cold sucrose-artificial cerebrospinal fluid (sucrose-aCSF) consisting of (in mM): 233.7 sucrose, 26 NaHCO3, 3 KCl, 8 MgCl2, 0.5 CaCl2, 20 glucose and 0.4 ascorbic acid. Coronal hippocampal slices (350 µm) were cut with a Leica VT1000S vibratome and allowed to equilibrate in recording aCSF at room temperature for ∼1 h before being transferred to the recording chamber. The aCSF was composed of (in mM): 117 NaCl, 2.5 KCl, 1.2 MgSO4, 2.5 CaCl2, 1.2 NaH2PO4, 24.9 NaHCO3 and 11.5 glucose. During recording, slices were maintained at 31°C and perfused (1.5 ml/min) with oxygenated aCSF (95% O2–5% CO2) in an immersion chamber containing the GABAA receptor antagonist gabazine (GBZ; 10 µM) and an (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) AMPA receptor antagonist NBQX (10 µM).
Whole-cell patch-clamp recordings
An upright epifluorescence microscope (BX50WI; Olympus) was used to visualize CA1 pyramidal neurons in hippocampus. Whole-cell recordings were performed using glass borosilicate capillaries pulled on a P-87 micropipette puller (Sutter Instruments). The resistance of pipettes used for recordings was 3–5 MΩ. To record pharmacologically isolated NMDA receptor (NMDAR) EPSCs, electrodes were filled with caesium based intracellular solution containing (in mM): 120 Cs-methanesulfonate, 10 HEPES, 0.5 EGTA, 8 NaCl, 5 TEA-Cl, 4 Mg-ATP, 0.4 Na-GTP and 10 phosphocreatine. Osmolarity was adjusted to ∼285 mOsm and pH to 7.3. All recordings were made in voltage clamp mode using a Multiclamp 700B amplifier (Molecular Devices). Analogue signals were low-pass filtered at 2 kHz and digitized at 5 kHz with the use of a Digidata-1440A interface and pClamp10 software (Molecular Devices). Gigaseal and additional access to the intracellular neuronal compartment was achieved in voltage clamp where each cell was held at the holding potential of -70 mV. Cells with series resistance >25 MΩ were discarded. To study isolated NMDA currents, the holding potential was slowly changed to +40 mV without compensating series resistance in the presence of 10 µM NBQX and 10 µM GBZ. Pharmacologically isolated NMDAR EPSCs were then evoked at Schaffer collateral-CA1 synapses by presynaptic stimuli of increasing intensity (50-500 µA). We applied five stimuli (10 s apart) per step of current intensity. Each current step increased by 50 µA. All drugs used in these experiments were purchased from Tocris.
Middle cerebral artery occlusion (MCAO), and measuring neuronal viability by stereology
Experiments were carried out in adult male mice weighing 18–33 g. Animals were housed under controlled diurnal lighting conditions and allowed access to food and water ad libitum until the day of the experiment. Anaesthesia was induced by intraperitoneal injection of a mixture of ketamine (120 mg/kg) and xylazine (20 mg/kg). Depth of anaesthesia was assessed by toe pinch. Focal cerebral ischaemia was induced using an occluding silicon-coated intraluminal suture as previously described (Yenari et al., 1996; Kim et al., 2004). Briefly, following an adapted Koizumi MCAO method, after midline neck incision, the common and external carotid arteries were isolated and ligated with silk suture (Brain tree Inc.). Silk sutures were temporarily placed on the internal carotid artery and common carotid artery. After 50 min, the suture was withdrawn, and surgical incisions were closed. Deltaphase isothermal pads (Brain tree Inc.) were used to keep the animals’ body temperature stable post-anaesthesia, during the surgical procedure and until fully recovered. Successful application of MCAO was determined by Laser Doppler flowmetry. Only mice in which cerebral blood flow was dropped to >70% of the baseline (before MCAO) after the occlusion were used for experiments. An additional criterion was that the infarcted area volume should be 25–45% of the ipsilateral hemisphere volume. After occluding for transient focal cerebral ischaemia for 50 min, brains were isolated and sectioned 30 days later. Brain sections were stained with anti-NeuN antibody (abcam), and NeuN+ cells were counted with stereo investigator software. Number of NeuN+ neurons in the MCAO-induced cortex lesion area was normalized to number of NeuN+ neurons in the contralateral side of each section.
Imaging and analysis
Fluorescent images of neurons in culture were taken using Leica SP5 DMI Confocal microscope with the same background and parameters. Neurons (two per field) were selected at random and analysed in each microscopic field (three fields in each condition). Single channel Images were analysed using MetaMorph Image Analysis software. Co-localization was analysed using ImageJ software with JACoP plugin (Dunn et al., 2011) using Manders’ co-localization coefficients, M1 and M2. M1 is defined as the fraction of overlapping pixels in total pixels of EPHB2, while M2 is defined as the fraction of overlapping pixels in total pixels of GLUN1. Images of the diffuse background fluorescence were thresholded, and clusters were defined as regions with a higher than 2-fold intensity increase in diffuse intensity. Each experimental manipulation was performed in each genotype at least three times. The brain sections were imaged on a Leica SP5 DMI confocal microscope under 100× oil lens with stack images using optimal step. For analysis of PS1/GLUN1 co-localization, Metamorph software available in Mount Sinai imaging core was used. In each batch of the images, background fluorescence was first subtracted by measuring a blank area, and then a threshold was set for each channel (consistent among all images in one batch), after which the area of overlap (co-localization) between two channels was calculated.
Statistical analyses
Data are expressed as means ± SEM. Statistical significance of differences among groups was determined by one-way analysis of variance followed by the post hoc test described in individual figure legends, or with a two-tailed paired t-test. Distribution of the data was assumed to be normal, but this was not formally tested. No statistical methods were used to predetermine sample sizes. To evaluate statistical significance of treatments, two-tailed paired t-tests were performed against the value of the untreated basal condition (*P < 0.05; **P < 0.01; ***P < 0.001). For EPHB2/GLUN1 clusters, statistics were analysed using two-tailed unequal variance unpaired Student’s t-test for 2-group comparison affected by 1 factor. Statistical significance was performed with GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). For electrophysiology data, offline analysis was performed using Clampfit software (Molecular Devices). Comparisons between groups were carried out by two-way ANOVA, with Bonferroni post hoc testing for multiple comparisons using OriginPro10.1 software. Differences were considered significant at values of P ≤ 0.05. All results are presented as means ± SEM.
Data availability
The authors confirm that all the data supporting the findings of this study are available within the article and its Supplementary material. Raw data will be shared by the corresponding author upon request.
Results
The EFNB1-stimulated association of EPHB2 with GLUN1 depends on PS1 and is crucial to neuroprotection
Literature shows that EFNB1 and BDNF (factors) stimulate the association of their receptors, EPHB2 and TRKB respectively, with the GLUN1 subunit of the NMDA receptor (Dalva et al., 2000; Suen et al., 1997; Stucky et al., 2016), and we obtained data that these factors stimulate similar complexes in mouse cortical neuronal cultures (Supplementary Figs 1, 2). In addition, we reported that the neuroprotective functions of both EFNB1 and BDNF depend on PS1 (Barthet et al., 2013), a protein known to play crucial roles in Alzheimer disease neurodegeneration (Russo et al., 2000). To examine whether PS1 plays a role in the association of EPHB2 with NMDAR, we treated cortical neuronal cultures from WT (PS1+/+) mice or from PS1 hemizygous (PS1+/−) or homozygous (PS1−/−) knockout (KO) mice with EFNB1 (eB1) and probed for the formation of EPHB2–GLUN1 complexes. We found that the eB1-dependent stimulation of the EPHB2–GLUN1 complex was strongly inhibited in PS1 hemizygous (PS1+/−) and was undetectable in PS1 null (PS1−/−) neurons (Fig. 1A and B). Furthermore, acute down-regulation of PS1 by means of anti-PS1 siRNA (Bruban et al., 2015), also decreased the eB1-induced association of EPHB2 with GLUN1 (Fig. 1C). Genetic reduction or deletion of the PS1 gene had no effect on the cellular levels of EPHB2 and GLUN1 proteins (Fig. 1A and C, lower panels). Together, our data show that PS1 is necessary for the eB1-induced association of EPHB2 with GLUN1.
Figure 1.

Role of PS1 on EFNB1-stimulated EPHB2-GLUN1 association and neuroprotection. Primary cortical neuronal cultures prepared from WT (PS1 +/+), PS1 hemizygous (PS1 +/−) r PS1 homozygous knock out (PS1 −/−) mouse embryonic brains, were stimulated at 10 DIV with EFNB1-Fc (eB1, 2 µg/ml) or Fc for 60 min, lysed and immunoprecipitated (IPed) with anti-EPHB2 antibody. (A) Obtained immunoprecipitates (IPs) were then probed on WBs with anti-GLUN1 or anti-EPHB2 antibodies (upper panel). Input: neuronal lysates used in this IP experiment were immunoblotted with antibodies against GLUN1, EPHB2, PS1-CTF and ACTIN as shown in figure (lower panel). NIS = non-immune serum. (B) Graphs show quantification of EPHB2-GLUN1 complexes IPed as above. Data are mean ± SEM. *P < 0.05 versus Fc, two-tailed paired t-test, n = 3. (C) Down-regulation of PS1 using PS1 siRNA decreases the EFNB1-dependent complex between EPHB2 and GLUN1. Scrambled siRNA was used as control. (D) ExEPHB2 inhibits the EFNB1-induced association of EPHB2 with GLUN1. Neuronal cultures were pretreated for 1 h with either exEPHB2 or BSA (50 nM) followed by EFNB1 (eB1) stimulation for 1 h. Neurons were lysed afterwards, and extracts were IPed with anti-GLUN1 followed by WB with anti-EPHB2 and anti-GLUN1 antibodies. All detected antigens of co-IPs and Inputs are indicated at left of figures. (E) Extracellular peptide, exEPHB2, abolishes EFNB1 mediated neuroprotection from glutamate excitotoxicity. WT primary cortical neuronal cultures were pretreated for 1 h as indicated in the figure, and then challenged with glutamate (G) at 50 μM for 3 h. Neuronal survival was then determined as described in Materials and methods section. Percent survival is normalized to the survival of the culture treated with glutamate (Glu) alone (dotted horizontal line). EFNB1 rescues neurons from excitotoxicity in the absence (third bar from left) but not in the presence of peptide ex-EPHB2 (50 nM; 4th bar). BSA = bovine serum albumin. Data are mean ± SEM. *P < 0.05, **P < 0.01; one-way ANOVA with Tukey’s post hoc test, n = 3–5. Cell survival in the presence of glutamate is set at 100%.
It is known that PS1 has γ-secretase-dependent and independent functions (Wolfe et al., 1999; Baki et al., 2001; Kallhoff-Munoz et al., 2008) and that EPHB2 is proteolytically processed by γ-secretase (Litterst et al., 2007). We thus asked whether the EPHB2 association with GLUN1 is regulated by this aspartyl protease. Our data (Supplementary Fig. 3A) show that γ-secretase inhibitors have no significant effect on the eB1-induced association of EPHB2 and GLUN1, a result in agreement with an earlier report that γ-secretase does not modulate the eB1-dependent neuroprotection (Barthet et al., 2013). Since PS1 is required for both, the eB1-dependent neuroprotection (Barthet et al., 2013) and the eB1-induced EPHB2–GLUN1 association described here, we asked whether the eB1 stimulation of the EPHB2–GLUN1 complex is critical to the neuroprotective activity of eB1. Based on reports that the eB1-stimulated complex of EPHB2 and GLUN1 is mediated by the extracellular domains of these proteins (Dalva et al., 2000), we constructed peptide exEPHB2 containing the extracellular region of EPHB2 (residues Ser212-Lys540) minus the N-terminal eB1-binding domain. We reasoned that, following eB1 treatment, exEPHB2 would compete with endogenous EPHB2 for binding to GLUN1 leading to reduced stimulation of the neuronal EPHB2–NMDAR complex and attenuated neuroprotection. Indeed, Fig. 1D shows that exEPHB2 strongly reduces the eB1-induced association of EPHB2 with GLUN1. Furthermore, in agreement with our hypothesis, exEPHB2 inhibits the eB1-dependent neuroprotection against glutamate excitotoxicity, while bovine serum albumin (BSA), a protein of comparable size to that of exEPHB2, had no effect on neuroprotection (Fig. 1E). Together, our data show that inhibition of the eB1-stimulated association of EPHB2 with GLUN1 reduces the eB1-stimulated neuroprotection, indicating that the inducible EPHB2–GLUN1 complex plays pivotal roles in the eB1-dependent neuroprotection.
PS1 FAD mutants dominantly block the EFNB1-stimulated EPHB2–GLUN1 association and neuroprotection
PS1 mutations are the most common cause of FAD (Sherrington et al., 1995), (http://www.alzforum.org/mutations), and we showed that PS1 is also essential to the neuroprotective functions of the EFNB1/EPHB2 and BDNF/TRKB ligand–receptor systems (Barthet et al., 2013). We thus asked whether PS1 FAD mutants might affect the neuroprotective functions of EFNB1 (eB1) and BDNF. To test this, we used cortical neuronal cultures from two independently constructed PS1-FAD mutant knock-in (KI) mice, one expressing human FAD mutant allele PS1 M146V (Guo et al., 1999; Nikolakopoulou et al., 2016) and the other expressing FAD allele I213T (Nakano et al., 1999; Baki et al., 2004; Nikolakopoulou et al., 2016). In these models, an endogenous PS1 allele is replaced by an exogenous mutant PS1 allele that remains under the control of endogenous gene expression promoters and shows physiological levels of expression (see input panels of Figs 2B, 4A and 7D). Importantly, these KI mice have a similar genotype as PS1 FAD patients and should be free of potential artefacts reported in protein overexpression Alzheimer disease models (Saito et al., 2016). We observed that expression of PS1 FAD mutant alleles in either heterozygous (KI/WT) or homozygous (KI/KI) state inhibits the neuroprotective functions of both eB1 and BDNF following glutamate excitotoxicity (Fig. 2A). Neuronal cultures from both KI mouse models yielded similar results suggesting that PS1 FAD mutations may have dominant negative effects on the neuroprotective activity of brain neurotrophins. In contrast, the neuroprotective functions of basic fibroblast growth factor (bFGF), were not affected by FAD mutant PS1M146V (Supplementary Fig. 4) or by mutant PS1I213T (data not shown). Together, our data indicate that FAD mutants compromise the neuroprotective activity of specific neuroprotective factors. Importantly, our data that the neuroprotective activities of BDNF and eB1 decrease in neurons expressing FAD mutations support the theory that FAD mutants may contribute to neurodegeneration by reducing neuroprotective activities of brain neurotrophic factors.
Figure 2.

Effects of PS1 FAD mutants on factor-dependent neuroprotection, and EPHB2-GLUN1 association. (A) Cortical neuronal cultures prepared from E15 mouse embryos wild-type (WT), heterozygous (M146V/WT and I213T/WT) or homozygous (M146V/M146V and I213T/I213T) for PS1 FAD alleles M146V or I123T were cultured in 24-well plates and 10–12 days later were treated with either EFNB1-Fc (eB1, 2 µg/ml) or BDNF (50 ng/mL) for 30 min. Cultures were then treated with 50 μM glutamate (Glu) for 3 h. Neuronal viability was quantified by counting healthy nuclei stained with Hoechst kit 33342 as described (Barthet et al., 2013). Percent survival of each neuronal genotype is normalized to the survival of the same culture treated with glutamate alone (dotted horizontal line). Data are mean ± SEM. *P < 0.05, two-tailed paired t-test, n = 4. Cell survival in the presence of glutamate is set at 100%. (B) Cortical neuronal cultures from WT, heterozygous (KI/WT) and homozygous (KI/KI) for PS1 FAD KI mutant M146V or I213T mouse embryonic brains were treated with eB1 or Fc for 60 min, lysed and IPed with anti-EPHB2 antibody, then immunoblotted with anti-GLUN1 and anti-EPHB2 antibodies, respectively. The input of the IP experiment is also shown in the lower panel. (C) Graphs illustrate fold change of GLUN1 IPed with EPHB2 following eB1 stimulation. Experiments were repeated four times. Data are mean ± SEM. *P < 0.05, eB1 versus Fc in WT; #P < 0.05 and ##P < 0.01 FAD Fc control versus WT Fc control, two-tailed paired t-test, n = 4.
Based on our data that the eB1-induced association of EPHB2 and GLUN1 plays crucial roles in the eB1-dependent neuroprotection and that PS1 FAD mutants reduce this activity, we next asked whether FAD mutants also affect the eB1-induced association between EPHB2 and GLUN1. We found that unlike WT neurons that respond to eB1 by increasing the EPHB2–GLUN1 complex (Dalva et al., 2000; also Fig. 1A, PS1+/+, and Supplementary Fig. 1), neurons expressing PS1 FAD mutant M146V or I213T in either heterozygous (KI/WT) or homozygous (KI/KI) states failed to respond to eB1 treatment by stimulating the EPHB2–GLUN1 complex (Fig. 2B). To our surprise, however, we observed that neurons expressing PS1 FAD mutants contained increased levels of the constitutive EPHB2–GLUN1 complex compared to WT neurons (Fig. 2B and C), although total expression of the interacting proteins were comparable among different genotypes (Fig. 2B, lower panel). It has been shown that the eB1-stimulated association of EPHB2 with GLUN1 causes increased co-clustering of these two proteins on neuronal surfaces (Dalva et al., 2000). We thus employed quantitative immunocytochemistry to ask whether PS1 FAD mutants also inhibit the eB1-stimulated co-clustering of these two proteins at the cell surface while increasing the constitutive levels of co-clustered EPHB2 and GLUN1. Figure 3 shows that compared to WT neurons, neurons expressing PS1 FAD mutant M146V in heterozygous (KI/WT) or homozygous (KI/KI) state, had increased levels of constitutive EPHB2–GLUN1 co-clustering. However, in contrast to WT neurons, eB1 treatment failed to stimulate further the association of these proteins on the surface of PS1 FAD mutant-expressing neurons (Fig. 3A and B). These results are in agreement with our co-IP data and support the conclusion that PS1 FAD mutants upregulate the constitutive EPHB2–GLUN1 complex while rendering it unresponsive to eB1. To further test our in vitro findings, the EPHB2–GLUN1 complex was immunoprecipitated from mouse brain hippocampi. In agreement with the data obtained in primary neuronal cultures, brains heterozygous (KI/WT) or homozygous (KI/KI) for PS1 FAD mutant M146V or I213T contained significantly higher amounts of the EPHB2–GLUN1 complex than brains expressing WT PS1 although total amounts of these proteins were comparable in all brains (Fig. 4A and B).
Figure 3.

PS1 FAD mutants modulate EPHB2 co-clustering with GLUN1 on neuronal surface. (A) Cortical neuronal cultures from WT and PS1 FAD (M146V) mouse embryonic brains either heterozygous (M146V KI/WT) or homozygous (M146V KI/KI) were stimulated with eB1 or Fc for 60 min, followed by live-labelling with anti-EPHB2 and anti-GLUN1 antibodies to detect clusters of the receptors on cell surface and their co-localization. In all cases, green indicates EPHB2 staining, red indicates GLUN1 staining, and blue is DAPI nuclear staining. White arrow indicates both single channel immunostaining and co-localized clusters. Scale bar, 5 µm. (B) Graphs showing percent change of co-localization of EPHB2 and GLUN1 over Fc treated WT group by following Mander’s co-localization coefficients. Data are mean ± SEM. **P < 0.01, eB1 versus Fc in WT; ###P < 0.001 FAD Fc control versus WT Fc control, Mander’s co-localization coefficients, n = 9–7.
Figure 4.

PS1 FAD mutants affect EPHB2 association with GLUN1 in mouse brains. (A) Four-week-old WT, heterozygous (KI/WT) and homozygous (KI/KI) for PS1 FAD mutant M146V or I213T mouse brain hippocampi were homogenized in Hepes buffer containing 1% Triton-X 100. After centrifugation, total lysates were IPed with anti-EPHB2 antibody, then immunoblotted with anti-GLUN1 or anti-EPHB2 antibodies, respectively. The neuronal lysates used in the IP experiment were immunoblotted with antibodies against GLUN1, EPHB2 and ACTIN (input, lower panel). (B) Graphs display fold of GLUN1 immunoprecipitated with EPHB2. Data are mean ± SEM. *P < 0.05 versus WT, one-way ANOVA with Tukey’s post hoc test, n = 3.
PS1 FAD mutations decrease the EFNB1-stimulated association of GLUN1 with PS1
Since PS1 has been found in complexes with GLUN1 (Saura et al., 2004), our finding that the EFNB1-stimulated EPHB2–GLUN1 complex depends on PS1 (Fig. 1A) suggested that EFNB1 (eB1) may also stimulate the association of PS1 with GLUN1. Indeed, Fig. 5A shows that eB1 treatment significantly increases the PS1–GLUN1 complex in primary neuronal cultures from WT mice. We also found that eB1 stimulates the PS1–GLUN2B complex supporting the conclusion that eB1 stimulates the PS1 association with the NMDAR. Furthermore, cortical neuronal cultures expressing FAD mutants in either heterozygous or homozygous state show no significant stimulation of the PS1–GLUN1 complex in response to eB1 (Fig. 5B), suggesting dominant negative effects of FAD mutants on the eB1-dependent stimulation of this complex. In addition, neuronal cultures expressing PS1 FAD mutants (KI/WT or KI/KI) showed higher constitutive levels of the PS1–GLUN1 complex than WT neurons (Fig. 5B). Similarly, mouse brains expressing mutant PS1 had increased amounts of the PS1–GLUN1 complexes (Fig. 5C) and immunohistochemistry experiments indicated increased co-localization of PS1 with GLUN1 in mouse brain cortex of PS1 FAD mutant-expressing brains compared to WT brains (Fig. 6A and B). Together, our data indicate higher levels of constitutive PS1–GLUN1 association in FAD mouse brains compared to WT brains. We next asked whether increased association of mutant PS1 with GLUN1 in mouse brains could be reproduced in a non-neuronal system. To this end, HEK293 cells were co-transfected with plasmids expressing GLUN1 and WT or mutant PS1 (M146V or I213T). We found that GLUN1 and PS1 coimmunoprecipitate, indicating that GLUN1 and PS1 form a complex when expressed in HEK293 cells. While both WT and mutant PS1 associated with GLUN1, we observed a clear trend of mutant PS1 being associated more with GLUN1 (Supplementary Fig. 5, left upper panel). However, the increase in the GLUN1 association with PS1 mutants compared to WT proteins failed to reach significance due to variations associated with the overexpressions of WT and mutant proteins (Supplementary Fig. 5, right panel). In addition, we cannot exclude the possibility that neuronal factors absent from HEK cells may affect the stability of the mutant PS1–GLUN1 complex.
Figure 5.

Effects of EFNB1 and PS1 FAD mutants on NMDAR interactions with PS1. (A) Cortical neuronal cultures were stimulated with EFNB1 (eB1) or Fc for 60 min. After stimulation, cells were lysed and IPed with anti-PS1 NTF antibody, then immunoblotted with anti-GLUN1 and anti-GLUN2B antibodies. PS1 NTF (PS1) was detected under the same condition with unboiled samples. Graphs show fold change of GLUN1 immunoprecipitated with PS1 following eB1 stimulation (right panel). Experiments were repeated at least three times. Data are mean ± SEM. *P < 0.05 versus Fc, two-tailed paired t-test, n = 3. The inputs are shown in the lower panel. (B) Cortical neuronal cultures from WT, heterozygous (KI/WT) and homozygous (KI/KI) for PS1 FAD KI mutant M146V or I213T mouse embryonic brains were treated with eB1 or Fc for 60 min, lysed and IPed with anti-PS1 NTF antibody, then immunoblotted with anti-GLUN1. PS1 was detected under the same condition with unboiled samples. PIS = pre-immune serum. (C) Three-month-old WT and PS1 FAD (M146V and I213T) KI heterozygous (KI/WT) or homozygous (KI/KI) mouse brain cortices were homogenized in Hepes buffer containing 1% Triton-X 100. After centrifugation, total lysates were IPed with anti-PS1 NTF antibody, then immunoblotted with anti-GLUN1 and anti-GLUN2B antibodies. PS1 was detected under the same condition with unboiled samples.
Figure 6.

PS1 FAD mutants affect GLUN1 co-localization with PS1, and modulate PS1-GLUN1 complex stability. (A, B) Co-localization of PS1 and GLUN1 in mouse cortex was detected using double fluorescence staining with antibodies against PS1 (green) and GLUN1 (red), followed by analysis with metamorph. Increased overlay of PS1 and GLUN1 staining (yellow) were detected in PS1 FAD mutant M146V heterozygous (KI/WT) and homozygous (KI/KI) mice. White arrow indicates both single channel immunostaining and co-localization. Scale bar 1 µm. One-way ANOVA followed by Tukey’s multiple comparison test was used for statistical analyses. **P < 0.01, ***P < 0.001 versus WT, n = 7–9. (C, D) Cortical neuronal cultures from WT, and heterozygous for PS1 M146V mutant (M146V/WT) mouse embryonic brains were treated with eB1 for 1 h in neurobasal media followed by withdrawal of the ligand, and replacing the media with fresh neurobasal. Cells were thereafter collected and lysed at post-eB1 withdrawal hours as indicated. Neuronal lysates were used for IP experiments as depicted in the figure. No withdrawal means eB1 ligand remained in the media the entire experimental period. (E) Frontal cortices from normal (control) and FAD human brains carrying PS1 S170F mutant (PS1 FAD) were used to isolate crude synaptosomal membrane fractions. The fractions were IPed with anti-PS1 NTF antibody. WB experiments showed constitutively higher amount of PS1-GLUN1 complexes in FAD brains than control brains.
To examine any differences in the stability of the PS1–GLUN1 complex formed in WT neurons and in neurons expressing PS1 FAD mutants, we tested the rate of dissociation of these complexes in neuronal cultures. As expected, eB1 increased the association of GLUN1 with WT PS1 (Fig. 6C) and the levels of the stimulated PS1–GLUN1 complex decreased following withdrawal of eB1. In contrast, consistent with data in Fig. 3B, eB1 was unable to stimulate the PS1–GLUN1 complex in neurons expressing mutant PS1, and the levels of the constitutive complex did not decline following withdrawal of eB1 (Fig. 6D). Together, these data indicate that PS1 FAD mutations stabilize the PS1–GLUN1 association independent of eB1.
Next, we examined the GLUN1–PS1 protein complexes in human post-mortem brains. We obtained frontal cortices from non-demented post-mortem human brains carrying WT PS1 (control brains) and from two individuals each afflicted by FAD and each heterozygous for mutant PS1S170F (mutant brains) from the DIAN Human Brain Tissue Bank at Washington University. Initial efforts to co-IP the PS1–GLUN1 complex using total homogenates of post-mortem brain samples were unsuccessful. We then fractionated our samples to prepare crude synaptosomal membrane fractions that were then used to co-precipitate PS1 and GLUN1 from control and mutant brains. In agreement with our data in mouse brain, we found that brain tissue from the two individuals each expressing mutant PS1S170F showed higher levels of the GLUN1–PS1 complex compared to control brain tissue from non-carriers (Fig. 6E). It is important to note that the two brain tissue samples each carrying mutant PS1S170F are from a male and a female sibling. Additional brain samples from FAD patients carrying different PS1 FAD mutations, including A79V and T245P, did not show increased level of this protein complex. It is unclear whether our inability to detect increased PS1–GLUN1 complex in other human tissues expressing different FAD mutants is related to the quality of the post-mortem brain tissue or an indication that the increase of the PS1–GLUN1 complex is mutation specific.
BDNF increases neuroprotection by inducing a PS1-dependent TRKB–GLUN1 complex
Previous studies show that exogenous BDNF protects neurons against glutamate excitotoxicity (Mattson et al., 2002; Almeida et al., 2005; Barthet et al., 2013), and that, similar to EFNB1 (eB1), the ability of BDNF to protect neurons from excitotoxicity depends on PS1 (Barthet et al., 2013). Furthermore, BDNF stimulates the association of its receptor TRKB with the GLUN1 subunit of NMDAR (Stucky et al., 2016), and we obtained similar data that BDNF stimulates the TRKB–GLUN1 interaction in primary neurons (Supplementary Fig. 2). We thus asked whether PS1 is also required for the BDNF-stimulated association between TRKB and NMDAR. Figure 7A shows that genetic deletion of PS1 inhibits the BDNF-induced TRKB–GLUN1 complex but has no effect on the total amounts of TRKB or GLUN1 (Fig. 7A, lower panel). Similarly, acute down-regulation of PS1 with siRNA as in Fig. 1C also caused a dramatic reduction of the BDNF-induced association of TRKB with GLUN1 (Fig. 7B). Together, our data show that PS1 is required for BDNF-stimulated association of TRKB with GLUN1.
Figure 7.

Role of PS1 on BDNF-stimulated TRKB-GLUN1 association and neuroprotection; and effects of PS1 FAD mutants on GLUN1 interactions with TRKB and PS1. Primary cortical neurons prepared from WT (PS1 +/+), PS1 hemizygous (PS1 +/−) or PS1 homozygous knock out (PS1 −/−) mouse embryonic brains, were stimulated with BDNF (50 ng/ml) or no treatment (NT) for 30 min, lysed and IPed with anti-TRKB antibody. (A) Obtained IPs were then probed on WB with anti-GLUN1 or anti-TRKB antibodies (upper panel). Graphs show quantification of TRKB-GLUN1 complexes IPed as above. Data are mean ± SEM. **P < 0.01, one-way ANOVA with Tukey’s post hoc test, n = 3 (right panel). Input: Neuronal lysates used in this IP experiment were immunoblotted with antibodies against GLUN1, TRKB, PS1-CTF and ACTIN as shown in figure (lower panel). NI IgG = non-immune IgG. (B) Down-regulation of PS1 using PS1 siRNA decreases the BDNF-stimulated complex between TRKB and GLUN1. Scrambled siRNA was used as control. (C) (upper panel) Extracellular peptide, exGLUN1, abolishes BDNF mediated neuroprotection from glutamate excitotoxicity. Primary cortical neuronal cultures were pretreated for one hour as indicated in the figure, and then challenged with glutamate (Glu) at 50 μM for 3 h. Neuronal survival was then determined as described in Materials and methods section. BDNF rescues neurons from excitotoxicity in the absence (3rd bar from left), but not in the presence of peptide ex-GLUN1 (50 nM; 4th bar). Peptide exEPHB2, however, does not abolish BDNF neuroprotection (5th bar). Data are mean ± SEM. **P < 0.01; one-way ANOVA with Tukey’s post hoc test, n = 3–5. Cell survival in the presence of glutamate is set at 100%. (upper panel). ExGLUN1 reduces the BDNF-stimulated association of TRKB with GLUN1. Neuronal cultures were pretreated for 1 h with either exGLUN1 or BSA (50 nM) followed by BDNF stimulation for 30 min. Neurons were lysed afterwards, and extracts were IPed with anti-TRKB followed by WB with anti-GLUN1 or anti-TRKB antibodies. (D) Cortical neuronal cultures from WT, heterozygous (KI/WT) and homozygous (KI/KI) for PS1 FAD (M146V) KI mouse embryonic brains were treated with BDNF for 30 min, lysed and IPed with anti-TRKB antibody, then immunoblotted with anti-GLUN1 and anti-TRKB antibodies, respectively. The input of the IP experiment is also shown in the lower panel. (E) Cortical neuronal cultures from WT, heterozygous (KI/WT) and homozygous (KI/KI) for PS1 FAD (I213T) KI mouse embryonic brains were treated with BDNF for 30 min, lysed and IPed with anti-PS1 NTF (PS1) antibody, then immunoblotted with anti-GluN1. PS1 was detected under the same condition with unboiled samples.
Since PS1 is also necessary for the BDNF-dependent neuroprotection (Barthet et al., 2013), we asked whether the BDNF-induction of the TRKB–GLUN1 complex is critical to its neuroprotection activity. To test this question, we used methods similar to the one we applied to EFNB1–EPHB2 system above. Since it is currently unclear what TRKB sequence mediates the TRKB–GLUN1 interaction, we reasoned that, similar to the eB1-stimlated EPHB2–GLUN1 complex, the extracellular regions of TRKB and GLUN1 are involved in the BDNF-stimulated TRKB–GLUN1 association. This possibility is also supported by data that these two proteins form a complex stimulated by exogenous BDNF (Stucky et al., 2016, and Fig. 7A). To test this hypothesis, we constructed a peptide comprising the N-terminal extracellular region of mouse GLUN1 (exGLUN1; Arg19-Ser560). We reasoned that following BDNF treatment; exGLUN1 would compete with endogenous GLUN1 for the TRKB binding site(s) leading to decreased association of TRKB with GLUN1 and attenuated neuroprotection. In agreement with our hypothesis, exGLUN1 caused a significant decrease in the neuroprotective activity of BDNF against excitotoxicity; but exEPHB2, a peptide that inhibits the eB1 neuroprotection, had no effect on the BDNF neuroprotection (Fig. 7C, upper panel), supporting the specificity of exGLUN1 in attenuating BDNF neuroprotection. Furthermore, Fig. 7C (lower panel), shows that exGLUN1 also decreases the BDNF-stimulated association of TRKB and GLUN1. Thus, attenuation of neuroprotection by peptide exGLUN1 was concomitant with the reduction of the inducible TRKB–GLUN1 complex in neurons treated with this peptide, suggesting that interference with the de novo complex formation has important consequences for BDNF neuroprotection. Together, our data indicate that the BDNF-induced association of TRKB and GLUN1 is crucial to the BDNF-dependent neuroprotection.
PS1 FAD mutants decrease BDNF-stimulated association of GLUN1 with TRKB and PS1
The BDNF–TRKB ligand–receptor system has been implicated in Alzheimer disease (Salehi et al., 1996, Holsinger et al., 2000), and we asked whether PS1 FAD mutants which decrease BDNF neuroprotection (Fig. 2A), might also interfere with the ability of BDNF to stimulate the TRKB-GLUN1 complex. Figure 7D shows that although BDNF increases this complex in WT neurons, it has a reduced effect on the TRKB–GLUN1 complex in neurons expressing PS1 FAD mutants. However, unlike the constitutive EPHB2–GLUN1 complex which increases in FAD mutants (Figs 2B and C, 3 and 4), there was no significant difference in the constitutive amounts of the TRKB–GLUN1 complex between WT and PS1 FAD neuronal cultures (Fig. 7D; NT lanes), suggesting that FAD mutations affect differently these two complexes. We next examined the effects of BDNF on the PS1–GLUN1 complex in WT and FAD mutant-expressing neurons. We observed that BDNF increased the PS1–GLUN1 complexes in WT neuronal cultures (Supplementary Fig. 6) but, similar to eB1, neurons expressing PS1 FAD mutants in heterozygous or homozygous states showed no significant stimulation of the PS1–GLUN1 complex in response to BDNF (Fig 7E), indicating dominant negative effects of FAD mutants on the BDNF-dependent stimulation of this complex.
PS1 FAD mutants alter evoked NMDAR EPSCs and increase neuronal vulnerability to ischaemia in vivo
Since PS1 FAD mutants change the GLUN1 interactions with EPHB2 and PS1, we asked whether these mutations affect NMDAR-mediated synaptic responses. We prepared acute hippocampal slices from PS1 FAD (M146V) or WT control mice and used whole-cell patch-clamp recordings from CA1 pyramidal cells to measure pharmacologically isolated NMDAR-mediated EPSCs at Schaffer collateral-CA1 synapses. NMDAR–EPSCs were elicited by applying stimuli of increasing intensity to presynaptic Schaffer collaterals in the presence of bath-applied AMPA receptor antagonist NBQX (10 μM), and GABAA receptor antagonist Gabazine (GBZ; 10 μM), at a holding potential of +40 mV. Activation of Schaffer collaterals evoked NMDAR EPSCs in CA1 pyramidal neurons in both WT and FAD mutant brain slices (Fig. 8A). However, PS1 FAD mutant M146V significantly reduced the magnitude of the evoked NMDAR-mediated EPSCs in mutant CA1 neurons compared to WT CA1 neurons, as shown by sample current traces (Fig. 8A) and the input–output plots (Fig. 8B). Together, these results show that PS1 FAD mutants alter the NMDAR-mediated synaptic currents probably by affecting the PS1 association with the NMDAR.
Figure 8.

PS1 FAD mutant decreases NMDAR EPSCs in the CA1 region of mouse hippocampal slices and increases neuronal vulnerability after ischaemia in vivo. (A) Representative EPSC traces from WT (black) and heterozygous for PS1 FAD mutant M146V (M146V/WT) (red) CA1 neurons. Traces are averages of 5 NMDAR EPSCs recorded in the presence of 10 µM NBQX and 10 µM GBZ. (B) Input–output plot showing significantly reduced amplitudes of NMDAR EPSCs in PS1 FAD mutant-expressing neurons in comparison with WT neurons at higher stimulation intensities. The values in parentheses indicate the number of neurons/mice used in the analysis. Data are expressed as mean + SEM; P = 0.013, F = 6.36, two-way ANOVA. (C) WT (WT/WT), M146V (WT/M146V) and I213T (WT/I213T) heterozygous mice were subjected to MCAO. Brains were isolated and sectioned 30 days later, followed by counting viable neurons with stereo investigator software. Number of live neurons in the MCAO lesioned cortical area was normalized to number of neurons in the contralateral side of each section. *P < 0.05, two-tailed unpaired t-test versus WT, n = 3 (WT), n = 6 (WT/M146V), n = 6 (WT/I213T).
We next asked whether PS1 FAD mutants increase neuronal vulnerability to ischaemia in vivo by subjecting both wild-type and PS1 FAD mutant-expressing mice (M146V and I213T) to middle cerebral artery occlusion (MCAO) followed by reperfusion. No differences in physiological parameters were observed at various stages of the MCAO between WT and PS1 FAD mutants. Animals were sacrificed 30 days after the onset of reperfusion, and number of NeuN-positive neurons was manually counted (see Materials and methods section). The number of viable neurons was significantly reduced in PS1 mutant mice compared to WT mice (Fig. 8C), suggesting increased neuronal vulnerability to ischaemia of brains expressing PS1 FAD mutants.
Discussion
PS1 promotes neuroprotection by mediating factor-stimulated association of FR with GLUN1
The underlying causes of neuronal dysfunction and death in neurodegenerative disorders, including Alzheimer disease, are imperfectly understood thus contributing to the lack of effective therapeutic methods (Neve and Robakis, 1998). Neurotrophic factors are known to bind cognate receptors and protect neurons from toxic insults (Sampaio et al., 2017) and recently we reported that the ability of neuroprotective factors such as EFNB1 (eB1) and BDNF to protect cortical neurons from glutamate excitotoxicity depends on PS1 (Barthet et al., 2013), a protein with crucial roles in neurodegeneration as PS1 mutants cause most cases of early onset autosomal dominant FAD (Sherrington et al., 1995, http://www.alzforum.org/mutations). Despite its central roles in synaptic transmission, plasticity, learning and memory, over-activation of NMDAR is associated with excitotoxicity and neuronal cell death (Arundine and Tymianski, 2003; Hardingham, 2009; Parsons and Raymond, 2014) suggesting a potential NMDAR involvement in neurodegenerative disorders including Alzheimer disease (Choi, 1988, 1994). It is known that treatment of cell cultures with exogenous BDNF or eB1 stimulate formation of dynamic complexes of their cognate receptors, TRKB and EPHB2 respectively, with the NMDAR receptor, a process involving the binding of the extracellular domains of EPHB2 or TRKB to the GLUN1 subunit of the NMDAR (Suen et al., 1997; Dalva et al., 2000; Husi et al., 2000; Henderson et al., 2001; Wang et al., 2011; Stucky et al., 2016). However, the importance of the factor-stimulated NMDAR–FR complexes in neuroprotection and potential roles of PS1 in their formation are unclear.
Here, we tested the role of the eB1-stimulated EPHB2–NMDAR association in neuroprotection by using peptides containing extracellular sequence of EPHB2 to block the eB1-stimulated association of EPHB2 with NMDAR. Our data show that preventing the de novo formation of the eB1-stimulated EPHB2–GLUN1 complex abolishes the eB1-dependent neuroprotection, showing this complex plays central roles in the eB1-dependent neuronal survival against toxic insults. We also found that the eB1-stimulated association of EPHB2 with GLUN1 depends on PS1 and that eB1 also increases the association of PS1 with the NMDAR, a finding consistent with reports that PS1 physically interacts with the NMDAR (Saura et al., 2004). Similarly, we found that PS1 is needed for the BDNF-stimulated complex of TRKB with GLUN1 and that artificial peptides that prevent formation of this complex also abolish the BDNF-dependent neuroprotection indicating that the association of TRKB with the NMDAR regulates neuroprotective activities of BDNF. Together with previous reports that PS1 is required for the eB1- and BDNF-dependent neuroprotection against excitotoxicity (Barthet et al., 2013), our data support a mechanism by which PS1 promotes neuroprotection by mediating factor-stimulated associations of NMDAR with FR, an association critical to neuroprotection against neurotoxicity. It has also been suggested that the synaptic localization of NMDARs is regulated through physical associations with PS1 (Saura et al., 2004). Combined with evidence that activation of synaptic NMDAR is pro-survival (Hardingham and Bading, 2010; Parsons and Raymond, 2014) our data suggests that PS1-regulated neuroprotective mechanism may also dependent, at least in part, on the activation of NMDARs being complexed with PS1 in the synaptic compartment.
It is known that PS1 interacts with EPHB2 (Litterst et al., 2007) and we show here that eB1, a ligand of EPHB2, stimulates the association of EPHB2 and PS1 with the GLUN1 subunit of the NMDAR (Figs 1A and 5A) but has no effect on the PS1–EPHB2 association (Supplementary Fig. 7). Combined with our findings that PS1 is necessary for the eB1-stimulated association of EPHB2 with GLUN1, our data support a model where binding of eB1 to constitutive EPHB2–PS1 complexes stimulates recruitment of both proteins to GLUN1, a process that leads to de novo formation of the neuroprotective PS1–EPHB2–NMDAR association (see graphical abstract). Our data also show that the role of PS1 in the eB1-stimulated association of EPHB2 with GLUN1 is independent of γ-secretase activity. Similarly, γ-secretase inhibitors had no effect on the BDNF-stimulated association of TRKB with GLUN1. These findings are in agreement with reports that the neuroprotective activities of eB1 and BDNF are independent of γ-secretase (Barthet et al., 2013).
PS1 FAD mutants decrease factors-stimulated associations of GLUN1 with FRs and block neuroprotection
PS1 mutants cause FAD, a condition that shares clinical and neuropathological phenotypes with the more common sporadic Alzheimer disease (SAD) suggesting that both Alzheimer disease forms share overlapping mechanisms (Lippa et al., 1996). We thus explored potential effects of PS1 FAD mutants on the neuroprotective activities of EFNB1 (eB1) and BDNF. To minimize potential interference or toxic effects found in protein overexpression mouse models (Saito et al., 2016), we used neuronal cultures and brain tissue from KI mouse models where exogenous PS1 FAD mutants show similar expression levels as endogenous genes (see Results section). We discovered that in contrast to WT neurons, neurons expressing PS1 FAD mutant M146V or I213T in either heterozygous or homozygous state are rescued from excitotoxicity by neither eB1 nor BDNF. These data show that PS1 FAD mutants have dominant negative effects on the neuroprotective activities of BDNF and eB1 and suggest that FAD mutants interfere with factors-dependent neuroprotective mechanisms. Importantly, FGF-dependent neuroprotection is not affected by PS1 FAD mutants (Supplementary Fig. 4), indicating that these mutants selectively interfere with the activity of a limited number of neuroprotective factors. In agreement with our data that the factor-stimulated association of FR with GLUN1 is crucial to neuroprotection, we found that the ability of eB1 to stimulate the association of EPHB2 with GLUN1 deceases dramatically in neurons expressing PS1 FAD mutants. These data support the theory that PS1 FAD mutants reduce the eB1-dependent neuroprotection by decreasing the eB1-stimulated association of its receptor EPHB2 with GLUN1 (see graphical abstract). Similarly, FAD mutants inhibit the BDNF-dependent neuroprotection and the BDNF-stimulated association of TRKB with GLUN1 indicating that these mutants block the neuroprotective activity of BDNF by inhibiting the BDNF-dependent association of TRKB with GLUN1.
Using co-IP and immunostaining experiments we made the unexpected observation that compared to WT controls, cortical neuronal cultures and mouse brain tissue expressing PS1 FAD mutants have increased amounts of constitutive GLUN1 complexes with EPHB2 and PS1 and show increased co-localization of GLUN1 with EPHB2 and PS1 (see Results section). Importantly, in contrast to WT complexes which are stimulated in response to eB1 treatment, the PS1–GLUN1 and EPHB2–GLUN1 complexes of FAD mutant-expressing neurons form independent of eB1 and do not change in response to eB1 treatment. Furthermore, although the inducible WT PS1–GLUN1 complex dissociates rapidly following eB1 withdrawal, the PS1–GLUN1 complex of FAD mutant-expressing neurons show little or no dissociation following withdrawal of eB1 (Fig. 6C and D) supporting the conclusion that PS1 FAD mutants over-stabilize the PS1–GLUN1 complex independent of eB1. It is tempting to speculate that over-stabilization of the PS1–GLUN1 complex changes the dynamic nature of the interactions of its protein components affecting its neuroprotective functions. Together, our data show that the PS1–GLUN1 complex of WT neurons differs from that formed in neurons expressing FAD mutants. We explored further the physiological relevance of our findings by asking whether human brains expressing PS1 FAD mutants have increased levels of the GLUN1–PS1 complex. We found that post-mortem brain tissue from two siblings carrying the PS1 FAD mutant S170F had increased levels of the GLUN1–PS1 complex. Additional brain samples however, carrying distinct PS1 FAD mutants showed no increased levels of this complex. Our inability to detect increased PS1–GLUN1 complex in brain tissue from these FAD mutant carriers may be related to the quality of the post-mortem brain tissue or an indication that the increase of the PS1–GLUN1 complex is mutant-specific.
It has been proposed that glutamate excitotoxicity and deficiency in neurotrophic support by BDNF play important roles in neurodegenerative disorders such as Alzheimer disease (Almeida et al., 2005; Zuccato and Cattaneo, 2009) and that BDNF promotes neuroprotection by decreasing the toxic signalling of NMDAR (Lau et al., 2015). Our findings that eB1 and BDNF increase neuroprotection by stimulating interactions of their respective receptors with NMDAR suggest that these interactions decrease NMDAR-mediated excitotoxicity. Furthermore, that PS1 FAD mutants impair the factor-stimulated neuroprotective interactions of FR with GLUN1 suggests that restoring these interactions might be beneficial to Alzheimer disease patients.
PS1 FAD mutants affect NMDAR EPSCs and increase neuronal vulnerability to ischaemia
In hippocampus, NMDARs are required for the induction of long-term potentiation and formation of long-term memory (Bliss and Collingridge, 1993; Tsien et al., 1996). Using acute hippocampal slices from PS1 M146V mutant mice, we found that the magnitude of evoked NMDAR-mediated synaptic currents in CA1 across a range of Schaffer-collateral stimulation strengths, is significantly reduced compared to WT, suggesting impaired NMDAR synaptic neurotransmission in brain of mice expressing FAD mutants. Such a deficit in synaptic NMDAR signal strength may underlie the reported spatial memory deficits displayed by these PS1 FAD mutant mice (Sun et al., 2005). Our finding that the FAD mutants also change the constitutive association of PS1 with NMDAR supports the suggestion that this association plays important roles in the NMDAR synaptic transmission. Furthermore, in agreement with our in vitro results of Fig. 2A, the in vivo data of Fig. 8C show that brain neurons of mice expressing PS1 FAD mutants also show increased vulnerability to ischaemic lesions suggesting that such mutants render neurons vulnerable to excitotoxicity in vivo, presumably by reducing the activity of factors-dependent brain neuroprotective mechanisms. In addition, that these mutants also change the association of PS1 with NMDAR and reduce its synaptic currents which is believed to be neuroprotective (Ikonomidou et al., 1999; Hetman and Kharebava, 2006; Hardingham and Bading, 2010; Parsons and Raymond, 2014), supports the theory that the PS1–NMDAR interactions may affect additional neuronal survival mechanisms by modulating the synaptic activity of the NMDAR.
In summary, our study reveals novel PS1-dependent neuroprotective mechanisms of neurotrophic factors BDNF and EFNB1. These mechanisms are perturbed by PS1 FAD mutants leading to decreased neuroprotection against neurotoxicity. Our data suggest a mechanism by which PS1 FAD mutants may increase neurodegeneration by inactivating factor-dependent neuroprotection rendering neurons vulnerable to commonly occurring brain toxicities such as excitotoxicity, oxidative stress, ischaemia and traumatic brain injuries (Choi, 1988; Mattson, 2003). It is tempting to speculate that over many years, reduced neuroprotection to toxic episodes will cause significant neurodegeneration that can lead to dementia. It is also known that mature KI transgenic mice expressing the PS1 FAD variants used in this study show little or no neuropathological hallmarks of Alzheimer disease (Guo et al., 1999; Nakano et al., 1999). Thus, PS1 FAD mutants can increase neuronal vulnerability to toxic insults and cause electrophysiological abnormalities in the absence of Alzheimer disease neuropathology, supporting the suggestion that in Alzheimer disease, neuronal abnormalities may be upstream of the neuropathological hallmarks (Robakis, 2011). Combined with evidence that neuronal damage by excitotoxicity and oxidative stress leads to neuropathology similar to that of Alzheimer disease (Misonou et al., 2000; Lesne et al., 2005; Melov et al., 2007; Liang et al., 2009; Bordji et al., 2010), chronic reduction of neuroprotection against neurotoxicity may lead to dementia of the Alzheimer disease type. The underlying mechanisms by which FAD mutants affect the survival complexes leading to decreased neuroprotection, may offer novel targets for therapeutic intervention.
Supplementary Material
Acknowledgements
We thank Dr. Georgios Voloudakis for expert help with statistical analysis of survival experiments.
Funding
This work was supported by National Institutes of Health grants 2RF1AG008200-29; 2R01-NS047229; P50AG05138; and by Grant AARF-17-531426 of the Alzheimer’s Association.
Competing interests
The authors report no competing interests.
Glossary
- BDNF =
brain-derived neurotrophic factor
- EFN =
ephrin
- eB1 =
EFNB1
- EPHB2 =
EPH receptor B2
- FAD =
familial Alzheimer disease
- FR =
factor receptor
- KI =
knock-in
- NMDA =
N-methyl-d-aspartate
- NMDAR =
N-methyl-d-aspartate receptor
- PS1 =
presenilin 1
- WT =
wild type
References.
- Al Rahim M, Thatipamula S, Hossain MA. Critical role of neuronal pentraxin 1 in mitochondria-mediated hypoxic-ischemic neuronal injury. Neurobiol Dis 2013; 50: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Graos MM, et al. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ 2005; 12: 1329–43. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 2003; 34: 325–37. [DOI] [PubMed] [Google Scholar]
- Baki L, Marambaud P, Efthimiopoulos S, Georgakopoulos A, Wen P, Cui W, et al. Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc Natl Acad Sci USA 2001; 98: 2381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, et al. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J 2004; 23: 2586–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthet G, Dunys J, Shao Z, Xuan Z, Ren Y, Xu J, et al. Presenilin mediates neuroprotective functions of ephrinB and brain-derived neurotrophic factor and regulates ligand-induced internalization and metabolism of EphB2 and TrkB receptors. Neurobiol Aging 2013; 34: 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthet G, Georgakopoulos A, Robakis NK. Cellular mechanisms of gamma-secretase substrate selection, processing and toxicity. Prog Neurobiol 2012; 98: 166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 1993; 361: 31–9. [DOI] [PubMed] [Google Scholar]
- Bordji K, Becerril-Ortega J, Nicole O, Buisson A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid production. J Neurosci 2010; 30: 15927–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruban J, Voloudakis G, Huang Q, Kajiwara Y, Al Rahim M, Yoon Y, et al. Presenilin 1 is necessary for neuronal, but not glial, EGFR expression and neuroprotection via gamma-secretase-independent transcriptional mechanisms. FASEB J 2015; 29: 3702–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo L, Cinque C, Patane M, Schillaci D, Battaglia G, Melchiorri D, et al. Interaction between ephrins/Eph receptors and excitatory amino acid receptors: possible relevance in the regulation of synaptic plasticity and in the pathophysiology of neuronal degeneration. J Neurochem 2006; 98: 1–10. [DOI] [PubMed] [Google Scholar]
- Choi BH. Oxidative stress and Alzheimer’s disease. Neurobiol Aging 1995; 16: 675–8. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and disease of the nervous system. Neuron 1988; 1: 623–34. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res 1994; 100: 47–51. [DOI] [PubMed] [Google Scholar]
- Cisse M, Checler F. Eph receptors: new players in Alzheimer's disease pathogenesis. Neurobiol Dis 2015; 73: 137–49. [DOI] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, et al. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 2000; 103: 945–56. [DOI] [PubMed] [Google Scholar]
- Dines M, Lamprecht R. The role of Ephs and Ephrins in memory formation. Int J Neuropsychopharmacol 2016; 19: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 2011; 300: C723–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakopoulos A, Marambaud P, Efthimiopoulos S, Shioi J, Cui W, Li HC, et al. Presenilin-1 forms complexes with the cadherin/catenin cell-cell adhesion system and is recruited to intercellular and synaptic contacts. Mol Cell 1999; 4: 893–902. [DOI] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, et al. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med 1999; 5: 101–6. [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J Alzheimers Dis 2011; 25: 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans 2009; 37: 1147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signaling: implications for neurodegenerative disorders. Nat Rev Neurosci 2010; 11: 682–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson JT, Georgiou J, Jia Z, Robertson J, Elowe S, Roder JC, et al. The receptor tyrosine kinase EphB2 regulates NMDA-dependent synaptic function. Neuron 2001; 32: 1041–56. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kharebava G. Survival signaling pathways activated by NMDA receptors. Curr Top Med Chem 2006; 6: 787–99. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, Schnarr J, Henry P, Castelo VT, Fahnestock M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer's disease. Brain Res Mol Brain Res 2000; 76: 347–54. [DOI] [PubMed] [Google Scholar]
- Huang Q, Voloudakis G, Ren Y, Yoon Y, Zhang E, Kajiwara Y, et al. Presenilin1/gamma-secretase protects neurons from glucose deprivation-induced death by regulating miR-212 and PEA15. FASEB J 2018; 32: 243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci 2000; 3: 661–9. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999; 283: 70–4. [DOI] [PubMed] [Google Scholar]
- Kallhoff-Munoz V, Hu L, Chen X, Pautler RG, Zheng H. Genetic dissection of gamma-secretase-dependent and -independent functions of presenilin in regulating neuronal cell cycle and cell death. J Neurosci 2008; 28: 11421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Yenari MA, Giffard RG, Cho SW, Park KA, Lee JE. Agmatine reduces infarct area in a mouse model of transient focal cerebral ischemia and protects cultured neurons from ischemia-like injury. Exp Neurol 2004; 189: 122–30. [DOI] [PubMed] [Google Scholar]
- Lau D, Bengtson CP, Buchthal B, Bading H. BDNF reduces toxic extrasynaptic NMDA receptor signaling via synaptic NMDA receptors and nuclear-calcium-induced transcription of inhba/activin A. Cell Rep 2015; 12: 1353–66. [DOI] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141: 1146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Ali C, Gabriel C, Croci N, MacKenzie ET, Glabe CG, et al. NMDA receptor activation inhibits secretase and promotes neuronal amyloid production. J Neurosci 2005; 25: 9367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Dysregulation of tau phosphorylation in mouse brain during excitotoxic damage. JAD 2009; 17: 531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa CF, Saunders AM, Smith TW, Swearer JM, Drachman DA, Ghetti B, et al. Familial and sporadic Alzheimer's disease: neuropathology cannot exclude a final common pathway. Neurology 1996; 46: 406–12. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov 2006; 5: 160–70. [DOI] [PubMed] [Google Scholar]
- Litterst C, Georgakopoulos A, Shioi J, Ghersi E, Wisniewski T, Wang R, et al. Ligand binding and calcium influx induce distinct ectodomain/gamma-secretase-processing pathways of EphB2 receptor. J Biol Chem 2007; 282: 16155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med 2003; 3: 65–94. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Lovell MA, Furukawa K, Markesbery WR. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem 2002; 65: 1740–51. [DOI] [PubMed] [Google Scholar]
- Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2007; 2: e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 2009; 10: 850–60. [DOI] [PubMed] [Google Scholar]
- Misonou H, Morishima-Kawashima M, Ihara Y. Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 2000; 39: 6951–9. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov 2011; 10: 209–19. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Kondoh G, Kudo T, Imaizumi K, Kato M, Miyazaki JI, et al. Accumulation of murine amyloidbeta42 in a gene-dosage-dependent manner in PS1 ‘knock-in’ mice. Eur J Neurosci 1999; 11: 2577–81. [DOI] [PubMed] [Google Scholar]
- Neve RL, Robakis NK. Alzheimer’s disease: a re-examination of the amyloid hypothesis. Trends Neurosci 1998; 21: 15–9. [DOI] [PubMed] [Google Scholar]
- Nikolakopoulou AM, Georgakopoulos A, Robakis NK. Presenilin 1 promotes trypsin-induced neuroprotection via the PAR2/ERK signaling pathway. Effects of presenilin 1 FAD mutations. Neurobiol Aging 2016; 42: 41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 2013; 14: 383–400. [DOI] [PubMed] [Google Scholar]
- Parsons MP, Raymond LA. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014; 82: 279–93. [DOI] [PubMed] [Google Scholar]
- Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol 2001; 58: 1395–402. [DOI] [PubMed] [Google Scholar]
- Robakis NK. Mechanisms of AD neurodegeneration may be independent of Abeta and its derivatives. Neurobiol Aging 2011; 32: 372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C, Schettini G, Saido TC, Hulette C, Lippa C, Lannfelt L, et al. Presenilin-1 mutations in Alzheimer's disease. Nature 2000; 405: 531–2. [DOI] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Yamazaki N, Hashimoto S, Saido TC. Calpain activation in Alzheimer's model mice is an artifact of APP and presenilin overexpression. J Neurosci 2016; 36: 9933–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Verhaagen J, Dijkhuizen PA, Swaab DF. Co-localization of high-affinity neurotrophin receptors in nucleus basalis of Meynert neurons and their differential reduction in Alzheimer's disease. Neuroscience 1996; 75: 373–87. [DOI] [PubMed] [Google Scholar]
- Sampaio TB, Savall AS, Gutierrez MEZ, Pinton S. Neurotrophic factors in Alzheimer's and Parkinson's diseases: implications for pathogenesis and therapy. Neural Regen Res 2017; 12: 549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 2004; 42: 23–36. [DOI] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ. The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA 2007; 104: 403–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995; 375: 754–60. [DOI] [PubMed] [Google Scholar]
- Stucky A, Bakshi KP, Friedman E, Wang HY. Prenatal cocaine exposure upregulates BDNF-TrkB signaling. PLoS One 2016; 11: e0160585. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, et al. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-D-aspartate receptor subunit 1. Proc Natl Acad Sci USA 1997; 94: 8191–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Beglopoulos V, Mattson MP, Shen J. Hippocampal spatial memory impairments caused by the familial Alzheimer’s disease-linked presenilin 1 M146V mutation. Neurodegener Dis 2005; 2: 6–15. [DOI] [PubMed] [Google Scholar]
- Takasu MA, Dalva MB, Zigmond RE, Greenberg ME. Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science 2002; 295: 491–5. [DOI] [PubMed] [Google Scholar]
- Theus MH, Ricard J, Glass SJ, Travieso LG, Liebl DJ. EphrinB3 blocks EphB3 dependence receptor functions to prevent cell death following traumatic brain injury. Cell Death Dis 2014; 5: e1207–e1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 1996; 87: 1327–38. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell 2006; 126: 981–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Crupi D, Liu J, Stucky A, Cruciata G, Di Rocco A, et al. Repetitive transcranial magnetic stimulation enhances BDNF-TrkB signaling in both brain and lymphocyte. J Neurosci 2011; 31: 11044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS. When loss is gain: reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep 2007; 8: 136–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999; 398: 513–7. [DOI] [PubMed] [Google Scholar]
- Yenari MA, Palmer JT, Guo HS, de Crespigny A, Moseley ME, Steinberg GK. Time-course and treatment response with SNX-111, an N-type calcium channel blocker, in a rodent model of focal cerebral ischemia using diffusion-weighted MRI. Brain Res 1996; 739: 36–45. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, et al. Presenilins are essential for regulating neurotransmitter release. Nature 2009; 460: 632–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Kang Z, Li W, Xiao Z, Zhou X. Roles of brain-derived neurotrophic factor/tropomyosin-related kinase B (BDNF/TrkB) signalling in Alzheimer's disease. J Clin Neurosci 2012; 19: 946–9. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 2009; 5: 311–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that all the data supporting the findings of this study are available within the article and its Supplementary material. Raw data will be shared by the corresponding author upon request.