

Abstract
Importance
Pulmonary inflammatory myofibroblastic tumors (PIMTs) are primary lung tumors in children. Misdiagnosis easily occurs because of the nonspecific clinical manifestations, laboratory examination results, and imaging findings in affected patients.
Objective
To summarize the clinical characteristics, diagnosis, and prognosis of children with PIMTs.
Methods
This retrospective analysis included 23 children with PIMTs who received treatment in our hospital from January 2008 to January 2019. The clinical manifestations, laboratory examination results, and computed tomography findings were retrospectively analyzed.
Results
The population included 13 boys and 10 girls, with a median age at onset of 78 months (range, 10–126 months). Fourteen patients had PIMT lesions in the right lung and nine patients had PIMT lesions in the left lung. The surgical procedures included pulmonary wedge resection, pulmonary lobectomy, and total pneumonectomy. The median operation time was 115 min (range, 45–235 min); the median intraoperative blood loss volume was 30 mL (range, 3–500 mL). During the operation, one patient each had pulmonary hemorrhage, vena cava hemorrhage, and thoracic duct injury. Postoperative complications included pulmonary embolism in one patient and tumor recurrence in two patients; neither of these complications recurred after reoperation. The median follow‐up period was 49 months (range, 2–127 months).
Interpretation
Children with PIMTs exhibited good prognoses and primarily underwent surgical resection as treatment. Complete preoperative evaluation should be performed in affected patients.
Keywords: Pulmonary inflammatory myofibroblastic tumors, Surgery, Children
Pulmonary inflammatory myofibroblastic tumor (PIMT) is relatively rare in the clinical setting, but it is still common among pulmonary primary tumors in children. The prognosis of PIMT is good, and the main therapy of PIMT is surgical resection, thus complete preoperative evaluation should be performed. New treatment options, such as targeted therapy, may reduce the risk of surgery.
![]()
INTRODUCTION
Inflammatory myofibroblastic tumors (IMTs) are mesenchymal tumors composed of differentiated myofibroblast spindle cells; they are often accompanied by large amounts of plasma cells and/or lymphocytes. IMTs exhibit variable histological morphology, characterized by low potential malignancy or borderline biological behavior. IMTs can occur at any age, but are more common in children and adolescents. They also occur at any site, 1 however, they are most commonly observed in the lung. Pulmonary IMTs (PIMTs) rarely occur in children; to the best of our knowledge, there have been few large sample studies regarding the characteristics and treatment of pediatric patients with PIMTs. Here, we investigated the clinical characteristics, diagnosis, treatment, and prognosis of PIMTs in children admitted to our hospital, with the aim of improving clinical knowledge regarding diagnosis and treatment of these tumors.
METHODS
Ethical approval
This study was approved by the Ethics Committee of Beijing Children’s Hospital. Since this was a retrospective study and the data analysis were performed anonymously, the study was exempt from informed consent from patients’ guardians.
Inclusion criteria
This study included all 23 children with pathologically confirmed PIMTs who were treated from January 2008 to January 2019 in Beijing Children’s Hospital.
Analysis of clinical characteristics
The clinical manifestations, laboratory examination results, and computed tomography (CT) findings of the patients at the initial visit were retrospectively analyzed to summarize their clinical features, treatment, and prognosis.
Statistical analysis
Statistical analysis was performed using SPSS version 20.0 (IBM Corp., Armonk, N Y, USA). Count data were expressed as frequencies and proportions, while measurement data were expressed as medians. The event free survival (EFS) and overall survival (OS) are estimated with the Kaplan–Meier method.
RESULTS
Clinical characteristics of patients with PIMT
Contemporaneous cases were reviewed, 85 children with lung tumors were admitted to our hospital between 2008 and 2019. Following exclusion of 31 patients with pulmonary metastasis, 23 (43%) patients with PIMTs were identified; thus, PIMTs were the most common primary lung tumors in our population. In 81 patients with IMTs who were admitted to our hospital, 23 had IMTs in the lung, which constituted the largest subgroup of patients with IMTs.
The 23 children in this study included 14 boys and nine girls and median age at onset of 78 months (range, 10–126 months). The primary tumor sites were the right lung in 14 patients (60.9%) and left lung in 9 patients (39.1%). In 18 patients, tumors were detected during imaging examinations because of respiratory tract symptoms such as pyrexia, cough, and expectoration; the remaining five patients were asymptomatic and their tumors were incidentally discovered during clinical examinations (e.g., the presence of pulmonary space on a routine physical examination in a child with no obvious clinical symptoms). Ten (43.4%) children had elevated preoperative inflammatory indices (e.g., leukocyte count, C‐reactive protein level, and erythrocyte sedimentation rate), 17 (73.9%) had elevated platelet levels, and 17 (73.9%) had reduced hemoglobin levels. Five are positive in ALK immunohistochemical analysis (Table 1).
TABLE 1.
Demographic and clinical data of children with pulmonary inflammatory myofibroblastic tumors
| Items | Number of patients, n (%) |
|---|---|
| Gender | |
| Male | 14 (60.9) |
| Female | 9 (39.1) |
| The primary site of the tumor | |
| Right lung | 14 (60.9) |
| Left lung | 9 (39.1) |
| Symptoms | |
| Fever, cough | 18 (78.3) |
| Asymptomatic | 5 (21.7) |
| Laboratory examination | |
| CRP/leukocyte elevate | 10 (43.5) |
| Platelet elevate | 17 (73.9) |
| Hemoglobin decline | 17 (73.9) |
| Immunohistochemical | |
| ALK(+) | 5 (21.7) |
PIMT, pulmonary inflammatory myofibroblastic tumor.
The results of preoperative imaging examinations (B‐mode ultrasound, CT) suggested a pulmonary space‐occupying mass (Figures 1, 2, 3, 4). CT examinations revealed that most masses were indistinct with uneven density; some masses exhibited calcification. Most masses exhibited uneven enhancement. One patient had multiple lesions, while the remaining 22 patients had focal tumors; notably, 18 of the 23 patients exhibited peripheral organ infiltration, which led to enhanced surgical difficulty.
FIGURE 1.
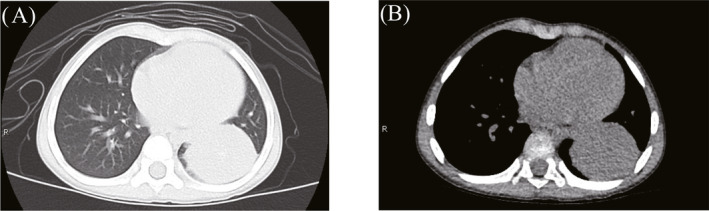
Lesion in the left lower lobe of a patient, detected during preoperative chest computed tomography. (A) Lung window, (B) Mediastinal window.
FIGURE 2.
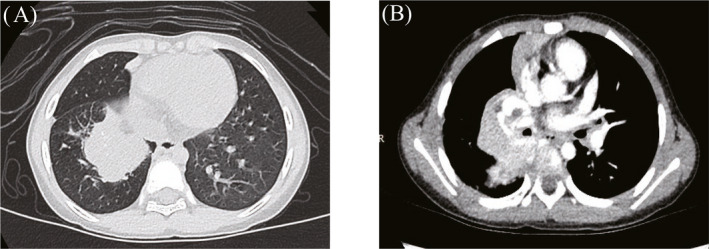
Lesion in the hilum of the right lung of a patient, detected during preoperative chest computed tomography. (A) Lung window, (B) Mediastinal window.
FIGURE 3.
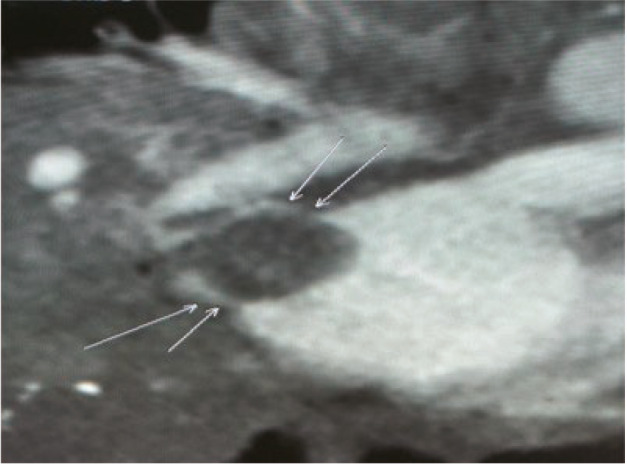
Lesion in the hilum of the right lung invading the left atrium of a patient, detected during preoperative cardiac computed tomography.
FIGURE 4.
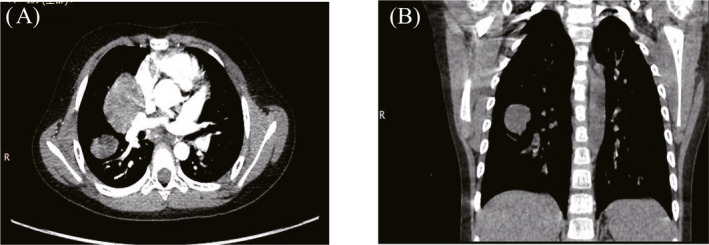
Multiple lesions in the posterior upper lobe of the right lung and dorsal segment of the lower lobe of a patient, detected during preoperative chest CT. (A) Horizontal position, (B) Coronal position.
Surgical procedures and intraoperative complications
All 23 patients underwent surgical treatment; 17 underwent thoracotomy and six underwent thoracoscopic surgery (five patients underwent conversion to thoracotomy during operation). Twenty patients underwent one‐stage complete tumor excision. The surgical procedures were pulmonary wedge resection (n = 5), pulmonary lobectomy (n = 12), and total pneumonectomy (n = 3, including one patient who underwent total pneumonectomy during the initial surgery; one patient who underwent right lower‐middle lobectomy during the initial surgery and emergency right upper lobectomy at 3 days postoperatively because of an embolism in the right upper pulmonary vein; and one patient who underwent right upper‐middle lobectomy during the initial surgery and right lower lobectomy at 17 months postoperatively because of recurrence in the right lower lobe). Three underwent subtotal resection (one underwent resection of an upper‐lobe lesion because of large lesion range and difficult exposure, whereas two underwent major tumor resection because of wide invasion range and extensive adhesion). The median operative time was 115 min (range, 45–235 min). The median blood loss volume was 30 mL (range, 3–500 mL).
The pericardium was opened in four patients because of difficulty dissecting the hilum of the lung during pulmonary lobe resection. During the operation, one patient each exhibited pulmonary hemorrhage, vena cava hemorrhage, and thoracic duct injury. In the patient with pulmonary hemorrhage, the tumor was closely adhered to the pulmonary trunk and the pulmonary trunk was torn; the artery was repaired after distal and proximal ends were blocked. In the patient with vena cava hemorrhage, the tumor was closely adhered to the superior vena cava; local bleeding developed in the superior vena cava during dissection. The superior vena cava was repaired after intraoperative application of local pressure to stop the bleeding. In the patient with thoracic duct injury, the tumor was located in the lower‐middle lobe of the right lung; it was closely adhered to the diaphragm, mediastinum, esophagus, pericardium, and pulmonary vessels. The lymph vessels were damaged during tumor separation; they were repaired and postoperative total parenteral nutrition was administered. All of these complications were discovered intraoperatively; all three patients recovered well after timely treatment. Intraoperative gross examination revealed incomplete resection in three other patients. No perioperative death, postoperative bleeding, or bronchopleural fistula occurred. During the operation, the texture was found to differ among PIMTs (tumor specimens are shown in Figure 5). Three exhibited a soft texture, while most (20/23) exhibited a hard texture.
FIGURE 5.
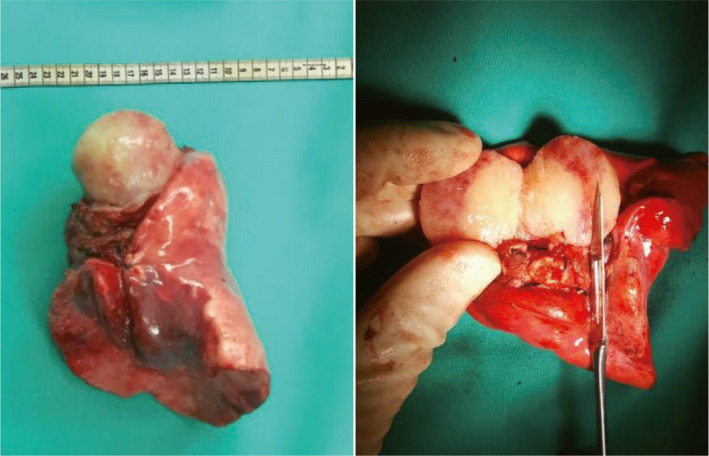
Tumor specimen from total pneumonectomy of a patient.
Prognosis
Twenty‐two patients received periodic re‐examinations for 2 to 127 months postoperatively; one patient was lost to follow‐up. The 5‐year event‐free survival rate was 86%, while the 5‐year overall survival rate was 100%. Two patients developed local recurrence at 17 and 33 months after complete resection, respectively. Reoperations were performed in both patients; disease‐free survival was achieved at 32 and 54 months relative to the second operation, respectively, following the second procedure. Nineteen children achieved disease‐free survival, and three survived with the tumor.
DISCUSSION
Elucidation of the clinical characteristics of IMTs has a relatively long history. In 1939, Brunn 2 first described two patients with spindle cell benign tumors in the lung. In 1954, Uminker and Iverson 3 proposed that such spindle cell proliferation in the lung constituted a post‐inflammatory tumor. Subsequently, inflammatory pseudoneoplasms were identified. At that time, the lesions were widely considered to be non‐neoplastic. Since 1953, similar lesions have been identified in the soft tissue, dura mater, spleen, lymph nodes, heart, bladder, and kidneys; they have been given different names, such as inflammatory fibroblast proliferation, plasma cell granuloma, inflammatory pseudotumor, fibrous histiocytoma, xanthoma pseudotumor, plasma cell/ histiocytoma syndrome, and inflammatory fibrosarcoma. 4 , 5 In 1984, Spencer proposed the possible coexistence of pulmonary plasma cell granuloma with histiocytoma. At that time, it was unclear whether an inflammatory pseudotumor constituted a real tumor. Although some patients exhibit a benign clinical process, others exhibit metastasis, invasion, or recurrence. These lesions have since been evaluated by immunohistochemical and cytogenetic analyses, which led to identification as true neoplasms, rather than simple inflammatory processes. In 2002, the World Health Organization defined these lesions as inflammatory myofibroblastic tumors. The exact etiology and pathogenesis of IMTs remain unknown. They are considered secondary to surgery, trauma, radiotherapy, infection, steroid hormone use, an autoimmune response, and abnormal expression and genetic mutation of the anaplastic lymphoma kinase gene. 6
The incidence of PIMTs is low. IMTs reportedly constituted 0.04% to 1% of all lung tumors. 6 , 7 These data are based on findings in the general population 8 , 9 ; the incidence in children remains unclear. IMTs can occur at any site, including the head and neck, abdomen, pelvis (e.g., gastrointestinal tract, mesentery, greater omentum, posterior peritoneum, hepatobiliary system, and urogenital tract), soft tissue of the torso and extremities, and the heart; they are most commonly observed in the lung. 10 The two common primary lung tumors in children are pleuropulmonary blastomas and PIMTs. The incidences of these two tumors have not been reported in the literature. Our single‐center study showed that PIMTs were most common among all lung tumors, which may have been related to a higher incidence of PIMT; alternatively, it might have resulted from the high degree of malignancy and rapid progression of pleuropulmonary blastomas, such that children could not wait for an accurate diagnosis. IMTs can occur at any age, but are more common in children and teenagers. No significant sex‐related differences have been found in previous studies.
PIMTs have no specific clinical manifestations. Most cases have insidious onset and are incidentally discovered during physical examinations. Some children develop symptoms consistent with respiratory tract infections (e.g., pyrexia, cough, and expectoration). Laboratory examination results regarding PIMTs lack specificity. Some patients exhibit an elevated leukocyte count, C‐reactive protein level, red blood cell count, and platelet count, as well as a reduced hemoglobin level. 11 Anti‐infective therapy was ineffective in our study, but inflammatory indicators returned to normal levels after tumors had been resected. Thus, PIMTs should be considered in children with symptoms of respiratory tract disease; elevated leukocyte count, C‐reactive protein level, erythrocyte sedimentation rate, and platelet count; reduced hemoglobin level; and a poor response to anti‐inflammatory therapy.
Notably, PIMTs also lack characteristic imaging findings. The CT manifestations vary and can be divided into invasive, massive, and nodular types. A single lesion is most common, 7 but multiple lesions are also occasionally reported. 12 In this study, CT examinations revealed masses that were indistinct with uneven density; some exhibited calcification and uneven enhancement (Figures 1, 2, 3, 4). These findings were consistent with literature reports. Focal tumors can be located in any pulmonary lobe; infiltrative tumors can invade the chest wall, diaphragm, trachea, hilum of the lung, esophagus, or heart. The tumor mass can be located in the margin of lung or near the central aspect of the hilum (within bronchi). Peripheral lesions are reportedly more common than central lesions, 13 , 14 but our findings were not consistent with the prior literature.
In PIMTs, large amounts of fibroblasts and myofibroblasts are distributed on a background of inflammatory cells. In 1995, Coffin et al 15 proposed three pathological types based on a pathological analysis of 84 cases of IMTs: mucus blood vessel, spindle cell intensive, and fibrous scar. However, most studies have shown that this typing approach has no clinical significance. In the present study, the pathological diagnosis did not include tumor typing. To exclude other diagnoses, definitive diagnosis of IMTs depends on histopathology and immunohistochemistry for immunotyping of myofibroblasts, such as examination of vimentin, smooth muscle actin, muscle‐specific action, and anaplastic lymphoma kinase.
PIMTs are tumors with low malignancy, but some PIMTs may deteriorate and metastasize. Thus, surgical treatment is needed during early stages of tumor growth. Surgical intervention for patients with PIMTs include pulmonary wedge resection, pulmonary lobectomy, and total pneumonectomy. Resection of PIMTs is relatively difficult, compared with resection of other tumors. In particular, aggressive tumor growth causes enhanced surgical difficulty. Our findings indicated that surgical difficulty was associated with the tumor ’s range, site, blood supply, and texture.
After complete resection, most patients reportedly have a good prognosis. Schweckendiek et al 16 reported a 5‐year survival rate of 99.0% and a 10‐year survival rate of 77.7%. Instances of spontaneous regression have also been reported. 6 PIMTs often relapse in patients who have undergone incomplete resection; 5% of patients with PIMTs develop distant metastasis. 16 However, these findings were from studies of adult patients with PIMTs; there have been few reports regarding children with PIMTs. In the present study, 22 of 23 patients underwent follow‐up for 2 to 127 months; all patients survived until the end of the study period. Two patients developed recurrence at 17 and 33 months after the initial surgery, respectively. Both patients exhibited ALK‐negative tumors and had developed local recurrence after complete resection in the initial surgery. Neither patient developed recurrence after the second surgery; both achieved disease‐free survival. Three patients with a residual tumor survived with their tumors; they did not exhibit tumor growth, shrinking, or regression during re‐examination. The pathogenesis of PIMT recurrence remains unclear and requires further investigation.
The role of targeted therapy is unclear based upon our limited experience. Recent studies have reported that targeted therapy (i.e., use of ALK inhibitors) efficacious and safe in trials of children with IMT in the United States, Europe, and other regions. They put forward this heterogeneous disease as being largely a kinase fusion driven neoplasm, and targeted therapeutic may be rational strategies. 17 Several ALK rearrangements have been identified in IMTs, including TPM‐3/4, ATIC, CLTC, CARS, RANBP2, and several novel ALK gene fusions (such as LMNA‐ALK and PRKAR1A‐ALK, the latter of which was detected in a congenital IMT), in addition, the possibility of targeted therapy with an ALK inhibitor would have been missed for patients with ALK testing by IHC alone. 18 Other actionable targets, such as ROS1 and PDGFb, have been identified in ALK‐negative IMTs. 19 Thus, consideration could be given to the concept of reducing the risk of surgical morbidity in large, difficult, multi‐focal tumors by treatment with an ALK inhibitor (e.g., crizotinib) prior to definitive surgical resection of in ALK‐positive tumors. This approach could facilitate maintenance of excellent outcomes with definitive surgery, while reducing surgical risk in patients with the most difficult tumors.
In conclusion, the diagnosis of PIMTs requires histopathological examination, immunohistochemistry, and (when possible) NGS analysis. Patients with PIMTs have good prognoses; however, the difficulty of surgical intervention varies widely. Thus, complete preoperative evaluation ensures the success of surgery. New molecular diagnosis, targeted therapies, or combination therapies may reduce the extent of surgery and related risks; these might facilitate better outcomes in future treatment.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Zhang N, Zeng Q, Chen C, et al. Clinical characteristics and prognosis of pulmonary inflammatory myofibroblastic tumor: An over 10‐year retrospective analysis. Pediatr Invest. 2020;4:192–197. 10.1002/ped4.12218
Funding source
Beijing Hospital Administration pediatric discipline collaborative development center pediatrics special project (XTZD20180105).
REFERENCES
- 1. Hedlund GL, Navoy JF, Galliani CA, Johnson WH Jr. Aggressive manifestations of inflammatory pulmonary pseudotumor in children. Pediatr Radiol. 1999;29:112–116. [DOI] [PubMed] [Google Scholar]
- 2. Brunn H. Two interesting benign lung tumors of contradictory histopathology. J Thorac Surg. 1939;9:119–131. [Google Scholar]
- 3. Umiker WO, Iverson LC. Post inflammatory tumor of the lung: report of four cases simulating xanthoma, fibroma or plasma cell granuloma. J Thorac Surg. 1954;28:55–62. [PubMed] [Google Scholar]
- 4. Coffin CM, Fletcher JA. Inflammatory myofibroblastic tumor In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. World Health Organization classification of tumors of soft tissue and bone. Lyon: IARC Press; 2013:83–84. [Google Scholar]
- 5. Gaudichon J, Jeanne‐Pasquier C, Deparis M, Veyssière A, Heyndrickx M, Minckes O, et al. Complete and repeated response of a metastatic ALK‐rearranged inflammatory myofibroblastic tumor to crizotinib in a teenage girl. J Pediatr Hematol Oncol. 2016;38:308–311. [DOI] [PubMed] [Google Scholar]
- 6. Surabhi VR, Chua S, Patel RP, Takahashi N, Lalwani N, Prasad SR. Inflammatory myofibroblastic tumors: Current update. Radiol Clin North Am. 2016;54:553–563. [DOI] [PubMed] [Google Scholar]
- 7. Maurya V, Gupta UA, Dewan RK, Jain S, Shah A. Spontaneous resolution of an inflammatory pseudotumour of the lung subsequent to wedge biopsy. Arch Bronconeumol. 2013;49:31–34. (in Spanish) [DOI] [PubMed] [Google Scholar]
- 8. Li X, Li J, Rao X, Ao Q, Cao X, Huang Y, et al. A case report of tracheal inflammatory myofibroblastic tumor in a 34‐week pregnant woman misdiagnosed with asthma. Medicine (Baltimore). 2017;96:e7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Camela F, Gallucci M, di Palmo E, Cazzato S, Lima M, Ricci G, et al. Pulmonary inflammatory myofibroblastic tumor in children: A case report and brief review of literature. Front Pediatr. 2018;6:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu CH, Huang HY, Chen HK, Chuang JH, Ng SH, Ko SF. Huge pelvi‐abdominal malignant inflammatory myofibroblastic tumor with rapid recurrence in a 14‐year‐old boy. World J Gastroenterol. 2010;16:2698–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mergan F, Jaubert F, Sauvat F, Hartmann O, Lortat‐Jacob S, Révillon Y, et al. Inflammatory myofibroblastic tumor in children: Clinical review with anaplastic lymphoma kinase, Epstein‐Barr virus, and human herpesvirus 8 detection analysis. J Pediatr Surg. 2005;40:1581–1586. [DOI] [PubMed] [Google Scholar]
- 12. Swain RS, Tihan T, Horvai AE, Di Vizio D, Loda M, Burger PC, et al. Inflammatory myofibroblastic tumor of the central nervous system and its relationship to inflammatory pseudotumor. Hum Pathol. 2008;39:410–419. [DOI] [PubMed] [Google Scholar]
- 13. Hammas N, Chbani L, Rami M, Boubbou M, Benmiloud S, Bouabdellah Y, et al. A rare tumor of the lung: Inflammatory myofibroblastic tumor. Diagn Pathol. 2012;7:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dhouib A, Barrazzone C, Reverdin A, Anooshiravani M, Hanquinet S. Inflammatory myofibroblastic tumor of the lung: A rare cause of atelectasis in children. Pediatr Radiol. 2013;43:381–384. [DOI] [PubMed] [Google Scholar]
- 15. Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995;19:859–872. [DOI] [PubMed] [Google Scholar]
- 16. Schweckendiek D, Inci I, Schneiter D, Weder W. Inflammatory myofibroblastic tumor of the lung: Two progressing pulmonary nodules in a 25‐year‐old adult with a Moraxella catharalis infection. Ann Thorac Surg. 2015;100:e123–e124. [DOI] [PubMed] [Google Scholar]
- 17. Trahair T, Gifford AJ, Fordham A, Mayoh C, Fadia M, Lukeis R, et al. Crizotinib and surgery for long‐term disease control in children and adolescents with ALK‐positive inflammatory myofibroblastic tumors. JCO Precis Oncol. 2019;3:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kovach SJ, Fischer AC, Katzman PJ, Salloum RM, Ettinghausen SE, Madeb R, et al. Inflammatory myofibroblastic tumors. J Surg Oncol. 2006;94:385–391. [DOI] [PubMed] [Google Scholar]
- 19. Lovly CM, Gupta A, Lipson D, Otto G, Brennan T, Chung CT, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]