

Abstract
The glioblastoma multiforme (GBM) is one of the deadliest tumors. It has been speculated that virus plays a role in GBM but the evidences are controversy. Published researches are mainly limited to studies on the presence of human cytomegalovirus (HCMV) in GBM. No comprehensive assessment of the brain virome, the collection of viral material in the brain, based on recently sequenced data has been performed. Here, we characterized the virome from 111 GBM samples and 57 normal brain samples from eight projects in the SRA database by a tested and comprehensive assembly approach. The annotation of the assembled contigs showed that most viral sequences in the brain belong to the viral family Retroviridae. In some GBM samples, we also detected full genome sequence of a novel picornavirus recently discovered in invertebrates. Unlike previous reports, our study did not detect herpes virus such as HCMV in GBM from the data we used. However, some contigs that cannot be annotated with any known genes exhibited antibody epitopes in their sequences. These findings provide several avenues for potential cancer therapy: the newly discovered picornavirus could be a starting point to engineer novel oncolytic virus; and the exhibited antibody epitopes could be a source to explore potential drug targets for immune cancer therapy. By characterizing the virosphere in GBM and normal brain at a global level, the results from this study strengthen the link between GBM and viral infection which warrants the further investigation.
Keywords: assembly, GBM, metagenomics., virosphere
We employed a data driven approach to identify virus species from the GBM NGS sequencing data. These virus could be potential novel therapeutic targets for cancer therapy.

1. BACKGROUND
In 2019, an estimated 86 970 new cases of brain and other central nervous system (CNS) tumors are expected to be diagnosed in the United States alone. 1 It was projected that 47.7% of primary malignant brain tumors are glioblastoma multiforme (GBM)—one of the most killing tumors with a 5‐year survival rate less than 6% and a 12‐15 months median survival time even with the most advanced treatment. 2 , 3 , 4 , 5 , 6 Although there is rapid advancement in cancer research and therapies, outcomes for GBM patients remain dismal due to the lack of knowledge of GBM etiology. GBM is not usually inherited 7 and the causes of GBM have always been a topic of controversy. Hypothesized causes of GBM include exposure to ionizing radiation, 8 use of electronics, 8 , 9 , 10 , 11 or viral infections. 12 , 13 , 14
Viruses have been identified as important factors in the incidence of various cancers. 15 , 16 Many efforts have been devoted to detect the cancer causing virus or design oncolytic virus for tumor treatment. 17 , 18 For example, a novel Merkel cell polyomarvirus was discovered in Merkel cell carcinoma, 19 , 20 and the herpes virus Epstein‐Barr virus (EBV) was identified from the large B‐cell lymphomas, 21 Burkitt's lymphomas, 22 and gastric carcinoma. 23 In addition, the human papillomaviruses (HPV) have been proven to play essential roles in promoting oncogenesis in cervical carcinoma. 16 The Hepatitis B virus (HBV) and its integrations were also identified as a major risk factors for the development of hepatocellular carcinoma. 24 , 25 , 26 , 27 Furthermore, there have been studies focusing on identifying insertion sites of viruses in the human genome from next‐generation sequencing data in the Cancer Genome Atlas (TCGA). 16 , 28 These studies clearly demonstrate the importance of investigating the association between viruses and cancer development.
Since 2002, there have been significant efforts to investigate the correlation between human cytomegalovirus (HCMV) 12 and GBM occurrence by different methods such as polymerase chain reaction, in situ hybridization, immunohistochemistry, and next‐generation sequencing. Despite of these efforts, the presence of HCMV as well as other herpes virus in brain and their correlation with the development of GBM remains an area of controversy. 12 , 14 , 16 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53
In addition to HCMV, some studies observed the presence of human papillomavirus (HPV) and hepatitis B in low‐grade gliomas (LGG) 52 from next‐generation sequencing data. In these studies, short sequence reads were aligned to the reference viral genome sequences to identify these viruses. One limitation of such approach is the high false positive results due to the congregation of short reads in highly repetitive regions, or in the regions that contain artificial sequences in some of the reference genomes. 54 In addition, traditional approaches had only identified 4021 characterized virus species according to Baltimore virus classification, 55 which only represent a tiny fraction of the virome diversity. Furthermore, a large number of unknown reads that cannot be mapped to any reference genome are discarded. Therefore, current approach does not provide a full depiction of the landscape of the virome in the brain, and a comprehensive assessment of the virome and its correlation to GBM is needed.
The assembly of the metagenomics is vitally important to the quality of viral detection. However, assembly of the viral genome has always been challenging due to the fast evolving and fragmented nature of the viral genome. 56 , 57 In recent decades, several metagenomic assemblers have been designed for the assembly of different sequencing data. 58 , 59 , 60 The assembly software with long k‐mer length can generate contigs more accurately by reducing chimeric sequences. 61 In addition, the annotation of the assembly directly against a reference sequence database via BLAST is an easy and effective approach to characterize sequences. 62
In this study, we applied metagenomics approach to characterize the virosphere of GBM at a global scale and observed some novel viruses previously isolated only from nonhuman organisms. We also observed that the contigs matching the genome sequences of the herpes virus only make up a small portion of the whole viral genome. In addition, we identified some novel sequences with no known annotations. Further analysis showed that these sequences have the signature for antibody epitopes. These findings will provide novel avenues toward future GBM research and therapies.
2. METHODS
2.1. Data source and availability
We searched the NCBI Sequence Read Archive (SRA: https://www.ncbi.nlm.nih.gov/sra), Gene Expression Omnibus (GEO: https://www.ncbi.nlm.nih.gov/geo/), and PubMed literature to collect NGS studies relating to GBM and normal brain tissues. We also identified a set of samples infected with known viruses as our “positive controls” to test if our assembly approaches can detect these viruses from the sequencing data. We limited our study to the data generated from Illumina sequencing platform and the RNA‐seq data were downloaded from SRA database. The list of accessions for the source data are shown in Supplemental File 1.
2.2. Positive controls and brain sample assembly
The raw reads in each study were first trimmed and checked using Trimmomatic (version 0.36) 63 and fastqc. 64 Ambiguous nucleotides (N’s), extreme short reads (<30 nt), and low‐quality bases were trimmed with a sliding window size of 4. The reads were then mapped to the human genome (GRCh38.p13) via STAR. 65 Reads that cannot be mapped to human genome were collected for further analysis.
For the samples with known virus infections, the MEGAHIT was used for contig assembly, and the resulting contigs were compared with the reference viral genomes (Figure 1). For brain RNA‐seq data, viral sequences were detected by the pathogen discovery program, READSCAN. 66 A read is considered as a viral sequence if it covers at least 10% of the reference genome of the virus. The assembly of the viral sequences was conducted with MEGAHIT and Trinity, and their assembly results are compared and evaluated (Figure 2). The pair‐end and single‐end reads were pooled and assembled by MEGAHIT. 67 , 68 Trinity is also an efficient and robust software for de novo assembly of transcriptomes from RNA‐seq data, and was also used for the assembly. The pair‐end and single‐end reads were assembled separately. The longest isoform for each gene assembled was selected using get_longest_isoform_seq_per_trinity_gene.pl. In order to reduce redundancy, the assembly was then processed by CD‐Hit (version 4.5.4) to remove duplicated contigs. 69 The threshold of sequence identity was set at 1.0, with the alignment coverage greater than 90% of the shorter sequence, and word length of 5.
FIGURE 1.
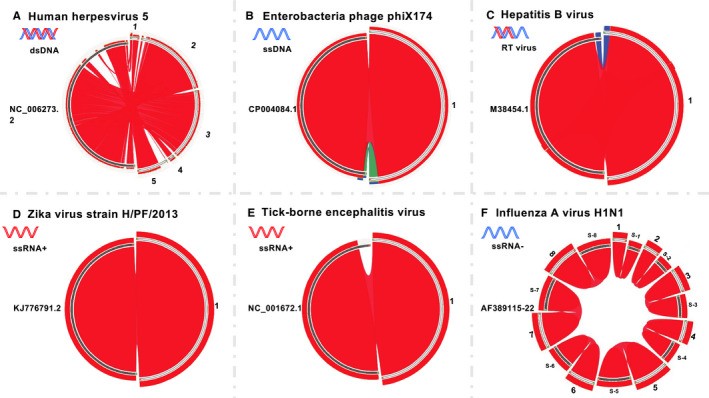
The assembled contigs from known viral infections and synteny analysis with their reference genomes. A, Human herpesvirus 5, reference genome accession: NC_006273 contigs: 1. k89_1468; 2. k89_1723; 3. k89_1974; 4. k89_821; 5. k89_887. B, Enterobacteria phage phiX174 reference genome accession: CP004084.1 contigs: 1. k141.3724. C, Hepatitis B virus reference genome accession: M38454.1 contigs: 1. k141.13661. D, Zika virus strain H/PF/2013 reference genome accession: KJ776791.2 contigs: k95.45717. E, Tick‐borne encephalitis virus reference genome accession: NC.001672.1 contigs: k79.90. F, Influenza A virus (A/Puerto Rico/8/34/Mount Sinai(H1N1)) reference genome accession: S‐1: ENA.AF389122.AF38912; S‐2: ENA.AF389121.AF38912; S‐3: ENA.AF389119.AF38911; S‐4: ENA.AF389120.AF38912; S‐5: ENA.AF389115.AF38911; S‐6: ENA.AF389118.AF38911; S‐7: ENA.AF389116.AF38911; S‐8: ENA.AF389117.AF38911; contigs: 1. k59.54; 2. k59.36; 3. k59.42; 4. k59.46; 5. k59.58; 6. k59.53; 7. k59.41; 8. k59.56
FIGURE 2.
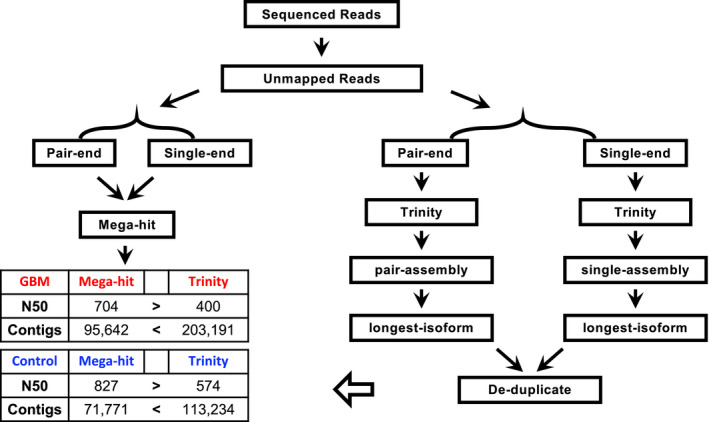
The assembly approach used for GBM and normal brain RNA‐seq dataset
2.3. Viral contig annotation with RefSeq database
The contigs with length over 500 bp were annotated to known viruses references in both protein and nucleotide databases at NCBI via BLAST 70 and Diamond 71 with the cutoff of e‐value < 1e‐10. For “positive controls,” the annotated virus contigs and its synteny with the virus genome were visualized with Circos using tBLASTN. Ribbons are colored based on the E‐value, with red represents the best hit. 72
The number of reads contributed to the assembly of each “viral” contig from each sample was calculated to ensure the assembly quality (Figure 4) by mapping to the “viral” contigs using Bowtie2 73 and viewed by Tabular. 74 The charts were generated using the R ggplot package. 75
FIGURE 4.
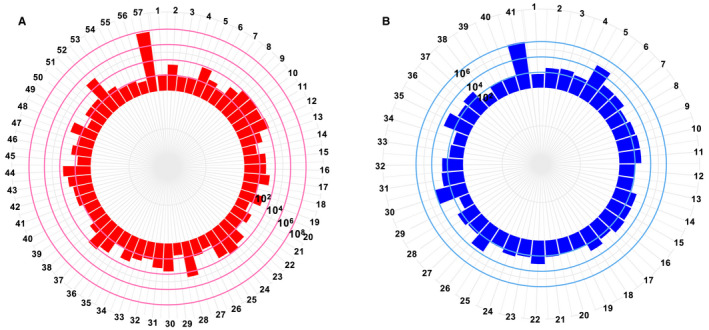
The reads abundance for the annotated contigs from Table 1. A, The GBM and B, normal brain. The X‐axis represents the name of contigs (Table 2), the Y‐axis represents the number of reads that can be mapped to the contigs, in log10 scale
2.4. Novel contigs annotation and characterization
The shared contigs that have no annotation from the above analysis are view in Venn diagram (Figure 3). 76 The unknown contigs are extracted and the phylogenetic tree was built using Fast tree (version 1.0.1). 77 The potential viral open reading frames (ORFs) were predicted by ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/). The minimal ORF length was set as 75, with any sense codon and standard genetic code applied. For each of the putative protein‐coding contigs, we applied TMHMM Server v. 2.0 to predict transmembrane domains. 78 Antibody epitope prediction was conducted by Bepipred Linear Epitope Prediction method in Immune Epitope Database (IEDB) (https://www.iedb.org/home_v3.php). 79 , 80 , 81 , 82 In order to ensure the quality of each contig, we calculated the reads coverage for each sample in the samtools, 83 and only kept those contigs with the coverage over 60% of its entire length for analysis.
FIGURE 3.
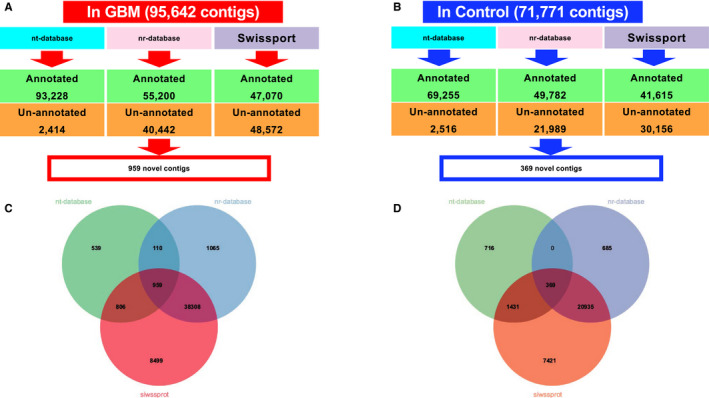
The annotation results from nt‐database, nr‐database, and Swiss‐Prot databases. The annotation results for A, GBM and B, normal brain. The overlap of the unknown annotations in C, GBM and D, normal brain
3. RESULTS
3.1. Assembly of positive controls
To validate our approach, we tested six samples with known viral infections as positive controls to evaluate our methods for viral sequence assembly. These six samples include Human herpesvirus 5 (double stranded DNA virus), Enterobacteria phage phiX174 (single stranded DNA virus), HBV (double stranded DNA virus with reverse transcription), Zika virus (single‐stranded, positive‐sense RNA virus), Tick‐borne Encephalitis virus (single‐stranded, positive‐sense RNA virus), and Influenza A virus H1N1 (fragmented, single‐stranded, negative‐sense RNA virus). These viruses cover major categories of different types of viruses to ensure the validity of our approach.
After trimming and mapping the reads to human genome, the unmapped high‐quality reads from the positive controls were assembled via MEGAHIT. The assembly results from each virus infected samples were compared to its corresponding reference sequences. We observed that for each positive control, the assembled contigs can cover over 90% of the reference genome of the corresponding virus (Figure 1). Five assembled contigs from the human herpesvirus 5 virus sample cover more than 90% of the viral genome (Figure 1A). One assembled contig from phage X174, HBV, zika, and Encephalitis samples each covers more than 90% of the corresponding viral genome (Figure 1B‐E). Furthermore, eight contigs from Influenza A virus infected sample can cover the eight segments of the influenza A H1N1 reference genome, respectively (Figure 1F). These results showed that our assembly approach is suitable and reliable for the metagenomic studies.
3.2. Brain RNA‐Seq reads assembly
We collected 111 GBM and 57 healthy brain data sets from eight different projects. This large number of datasets ensures the quality of contig assembly (Supplemental 2). In total, there are 6609 M (Million) raw sequencing reads for GBM and 2681 M for healthy brain. The low‐quality reads and reads that map to human genome were then removed to yield 210.0 M high quality reads for GBM and 115.4 M for health brain. For each group, reads were pooled together and assembled with MEGAHIT and Trinity, respectively. Using N50, N90 and the number of contigs as a criteria, MEGAHIT performed better than Trinity in assembling the GBM RNA‐Seq reads (Figure 2): MEGAHIT generated 95 642 contigs with N50 = 704 bp while Trinity generated 203 191 contigs with N50 = 400bp. In healthy brain, MEGAHIT generated 71 771 contigs with N50 = 827bp while Trinity generated 113 234 contigs with N50 = 574bp. Therefore, MEGAHIT results were used for further analysis. In total, GBM assembly contains 39 000 contigs longer than 500 nt and the largest contig is 14kb. The normal brain assembly contains 33 640 contigs longer than 500 nt, with largest contig reaching 37.5 kb.
3.3. Assembly annotation
The assembled contigs were annotated with the nucleotide collection database for Blast (nr/nt) at NCBI as well as Swiss‐Prot. Among the 95 642 contigs assembled from GBM samples, 93 228 can be annotated by nt database, 55 200 are annotated by nr database, and 47 070 contigs are annotated by Swiss‐Prot database. Only 959 contigs cannot be annotated by neither of the three databases (Figure 3A,C). Out of 71 771 contigs assembled from healthy brain samples, 69 255 can be annotated by nt database, 49 782 are annotated by nr database and 41 615 are annotated by Swiss‐Prot database, with only 369 contigs cannot be annotated (Figure 3B,D).
Of the annotated contigs over 500 bp long, 57 from GBM and 42 from healthy brain were identified as putative viral sequences of nonhuman origin (Table 1). Most of these contigs have a minimum read depth of 100 over the entire contig (Figure 4A,B, Table 2). Figure 5 shows the detailed information about these contigs. Most of these viral annotations can be characterized as retroviridae. Surprisingly, five contigs were annotated as a novel picornavirus previously identified from invertebrates. 84 These viral contigs were detected in five GBM but none of the healthy brain samples. The synteny analysis shows that these five contigs can match up to more than 90% of the picorna‐like virus 2 reference genome (Figure 6A). This result suggests a possible cross species transmission of the virus.
TABLE 1.
The annotated contigs with length > 500 bp by nr, nt, and Swiss‐Prot in GBM and normal brain. (A) The annotated virus from nr, nt, Swiss‐Prot in GBM. (B) The annotated virus from nr, nt, Swiss‐Prot in normal brain
|
Swiss‐Prot >333 AA |
Swiss‐Prot 167‐333 AA |
NR >333AA |
NR 167‐333AA |
NT >1000 |
NT 500‐1000 |
|---|---|---|---|---|---|
| (A) | |||||
| k141_1966 | k141_17176 | k141_17176 | k141_21057 | k141_17176 | k141_14851 |
| k141_20413 | k141_21082 | k141_1966 | k141_22611 | k141_20413 | k141_19740 |
| k141_22611 | k141_22611 | k141_20413 | k141_26285 | k141_33111 | k141_22611 |
| k141_24342 | k141_25917 | k141_22611 | k141_31289 | k141_34506 | k141_26285 |
| k141_31655 | k141_31289 | k141_65527 | k141_32573 | k141_50766 | k141_34855 |
| k141_32066 | k141_32573 | k141_83074 | k141_33111 | k141_5616 | k141_41235 |
| k141_33111 | k141_33111 | k141_85368 | k141_54776 | k141_83074 | k141_47897 |
| k141_34506 | k141_34506 | k141_90342 | k141_72529 | k141_50766 | |
| k141_37037 | k141_37401 | k141_9526 | k141_80924 | k141_54776 | |
| k141_50766 | k141_40368 | k141_58075 | |||
| k141_52202 | k141_40862 | k141_78048 | |||
| k141_67072 | k141_44479 | k141_9526 | |||
| k141_79178 | k141_48992 | ||||
| k141_83074 | k141_50917 | ||||
| k141_84281 | k141_53095 | ||||
| k141_85368 | k141_54776 | ||||
| k141_8782 | k141_5936 | ||||
| k141_59436 | |||||
| k141_59727 | |||||
| k141_60933 | |||||
| k141_6834 | |||||
| k141_7441 | |||||
| k141_74451 | |||||
| k141_77374 | |||||
| k141_77641 | |||||
| k141_83074 | |||||
| k141_85368 | |||||
| k141_86666 | |||||
| k141_9065 | |||||
| k141_91608 | |||||
| k141_9526 | |||||
| (B) | |||||
| k119_16633 | k119_11170 | k119_11210 | k119_12208 | ||
| k119_20176 | k119_12162 | k119_12208 | k119_33960 | ||
| k119_23522 | k119_12208 | k119_17584 | k119_54092 | ||
| k119_31731 | k119_12631 | k119_20176 | |||
| k119_47334 | k119_13303 | k119_23522 | |||
| k119_56431 | k119_16853 | k119_66335 | |||
| k119_58902 | k119_17584 | k119_7909 | |||
| k119_60013 | k119_18222 | ||||
| k119_18699 | |||||
| k119_20176 | |||||
| k119_21423 | |||||
| k119_21825 | |||||
| k119_25761 | |||||
| k119_26055 | |||||
| k119_37222 | |||||
| k119_37745 | |||||
| k119_3825 | |||||
| k119_43612 | |||||
| k119_44406 | |||||
| k119_45310 | |||||
| k119_46849 | |||||
| k119_47632 | |||||
| k119_48152 | |||||
| k119_48193 | |||||
| k119_50374 | |||||
| k119_5065 | |||||
| k119_55758 | |||||
| k119_56955 | |||||
| k119_59983 | |||||
| k119_60013 | |||||
| k119_7909 | |||||
| k119_7937 | |||||
TABLE 2.
The assembled contigs annotated as viral origin with number of mapped reads and labels presented in Figure 4
| GBM assembly | Normal brain assembly | ||||
|---|---|---|---|---|---|
| Label | Contigs | Total reads | Label | Contigs | Total reads |
| 1 | k141_41235 | 128 | 1 | k119_12208 | 60 |
| 2 | k141_59727 | 2354 | 2 | k119_60013 | 379 |
| 3 | k141_21082 | 78 | 3 | k119_33960 | 531 |
| 4 | k141_22611 | 126 | 4 | k119_55758 | 244 |
| 5 | k141_83074 | 6389 | 5 | k119_54092 | 58 879 |
| 6 | k141_85368 | 442 | 6 | k119_3825 | 1115 |
| 7 | k141_9526 | 133 | 7 | k119_11170 | 1076 |
| 8 | k141_59436 | 1824 | 8 | k119_25761 | 219 |
| 9 | k141_14851 | 9209 | 9 | k119_26055 | 443 |
| 10 | k141_19740 | 11 153 | 10 | k119_46849 | 151 |
| 11 | k141_34855 | 6415 | 11 | k119_5065 | 632 |
| 12 | k141_47897 | 14 070 | 12 | k119_50374 | 107 |
| 13 | k141_77374 | 584 | 13 | k119_16633 | 122 |
| 14 | k141_65527 | 58 | 14 | k119_37745 | 651 |
| 15 | k141_74451 | 643 | 15 | k119_20176 | 734 |
| 16 | k141_17176 | 646 | 16 | k119_23522 | 1084 |
| 17 | k141_40368 | 2165 | 17 | k119_21825 | 251 |
| 18 | k141_44479 | 104 | 18 | k119_16853 | 1018 |
| 19 | k141_5616 | 1184 | 19 | k119_45310 | 90 |
| 20 | k141_5936 | 42 | 20 | k119_48193 | 108 |
| 21 | k141_86666 | 377 | 21 | k119_37222 | 138 |
| 22 | k141_1966 | 113 | 22 | k119_44406 | 1131 |
| 23 | k141_20413 | 3011 | 23 | k119_58902 | 174 |
| 24 | k141_24342 | 8541 | 24 | k119_7909 | 329 |
| 25 | k141_25917 | 4573 | 25 | k119_18222 | 124 |
| 26 | k141_31289 | 50 | 26 | k119_47632 | 4153 |
| 27 | k141_31655 | 151 | 27 | k119_31731 | 172 |
| 28 | k141_32066 | 41 520 | 28 | k119_47334 | 342 |
| 29 | k141_32573 | 39 | 29 | k119_13303 | 23 |
| 30 | k141_33111 | 4324 | 30 | k119_48152 | 9792 |
| 31 | k141_34506 | 1697 | 31 | k119_12631 | 608 |
| 32 | k141_37037 | 183 | 32 | k119_43612 | 461 |
| 33 | k141_37401 | 512 | 33 | k119_56955 | 61 |
| 34 | k141_40862 | 2007 | 34 | k119_56431 | 38 |
| 35 | k141_48992 | 110 | 35 | k119_17584 | 2603 |
| 36 | k141_50766 | 3945 | 36 | k119_18699 | 246 |
| 37 | k141_50917 | 15 136 | 37 | k119_21423 | 734 |
| 38 | k141_52202 | 472 | 38 | k119_7937 | 21 |
| 39 | k141_53095 | 108 | 39 | k119_59983 | 114 |
| 40 | k141_54776 | 116 | 40 | k119_11210 | 82 |
| 41 | k141_58075 | 338 | 41 | k119_66335 | 1 280 118 |
| 42 | k141_60933 | 342 | |||
| 43 | k141_67072 | 1032 | |||
| 44 | k141_6834 | 3787 | |||
| 45 | k141_72529 | 54 | |||
| 46 | k141_7441 | 418 | |||
| 47 | k141_77641 | 147 | |||
| 48 | k141_79178 | 2654 | |||
| 49 | k141_80924 | 43 | |||
| 50 | k141_84281 | 481 | |||
| 51 | k141_8782 | 475 | |||
| 52 | k141_9065 | 95 447 | |||
| 53 | k141_91608 | 348 | |||
| 54 | k141_26285 | 77 | |||
| 55 | k141_78048 | 83 | |||
| 56 | k141_21057 | 40 | |||
| 57 | k141_90342 | 64 234 471 | |||
FIGURE 5.
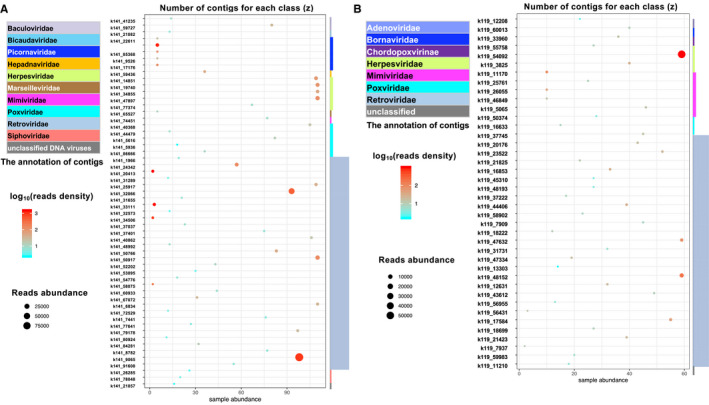
The distribution of virus contigs in different samples, (phage excluded). A, The GBM and B, normal brain. The X‐axis represents the number of samples that harbor these contigs. The Y‐axis list the individual contigs; the reads abundance is represented by the size of the dot; the color represents the reads density (reads number/sample numbers) in log 10 scale; the taxonomy of the annotated virus is presented on the right of the chart, with the z‐axis for the number of contigs for each order
FIGURE 6.
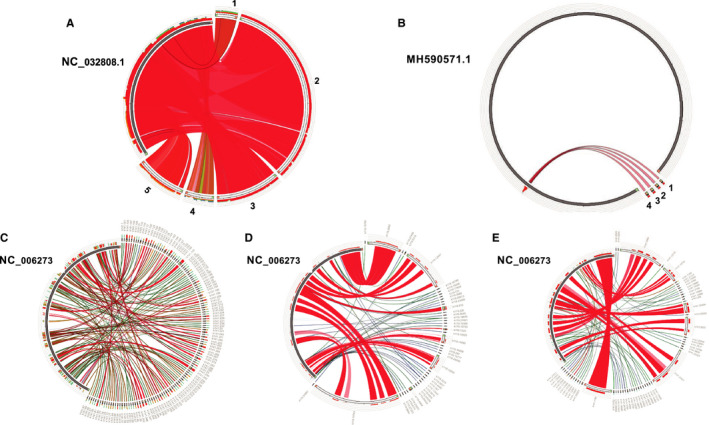
The assembled contigs from known viral infections and synteny analysis with their reference genomes. A, Wenzhou picorna‐like virus 2 strain. Contigs: 1: k141.22611 2: k141.83074; 3: k141.85368; 4: k141.9526; 5. k141.17176. B, Human gammaherpesvirus 4, reference genome accession: MH590571.1 contigs: 1: k141.19740 2: k141.34855 3: k141.14851 4: k141.47897. C, HCMV from seropositive healthy human samples D, HCMV from fetal lung fibroblast cells from naturally infection E, latent HCMV from hematopoietic cell
We also identified four contigs (k141_19740 (length = 664); k141_34855 (length = 739); k141_14851 (length = 753); k141_47897 (length = 501)) that were annotated as EBV, the only herpes virus to be found with moderate length of contigs. However, the synteny analysis showed that they are mapped to the same small region of the EBV reference genome (Figure 6B). In contrast, the synteny analysis of the presence of herpes virus in positive control showed significant number of contigs homologous to the HCMV reference genome (Figure 1). Significant homology over large genomic area is also observed in HCMV contigs from CMV seropositive healthy human samples (Figure 6C), fetal lung fibroblast cells from naturally infected people (Figure 6D), and HCMV latent hematopoietic cell (Figure 6E). In addition, READSCAN analysis of GBM virome does not support the presence of herpesviruses in GBM despite of few reads in few samples appeared to be mapped to a small proportion of the viral genome (Supplemental 3). 64 Therefore, both the contig assembly and sequence reads mapping from our analysis do not support the presence of EVB and other herpesviruses in GBM. However, our analysis cannot rule out the presence of latent herps virus whose genomic DNA is inserted into the genome of GBM tumor cells.
3.4. Novel contig antigen prediction
For unannotated 959 contigs from GBM and 369 from healthy brain (Figure 3C,D), we performed phylogenetic analysis to group them into three major clusters (Supplemental 4A, 4B). ORF was predicted for each contig longer than 500bp. The resulting protein sequences from these predicted ORF were subject to TMHMM v2.0 (http://www.cbs.dtu.dk/services/TMHMM/) analysis to predict the transmembrane domains. Significant transmembrane domains were found in 31 unknown contigs from GBM and three unknown contigs from health brain. Among these transmembrane contigs, we found that the linear B‐cell epitopes were enriched and analyzed. Some of the contigs, such as k141_31618 assembled from 22 out of 110 GBM samples and k141_77976 from 33 of GBM samples, contains putative antigen epitopes (Figure 7). If real and validated by experiments, these contigs can potentially be recognized by immune system and used as targets for drug development.
FIGURE 7.
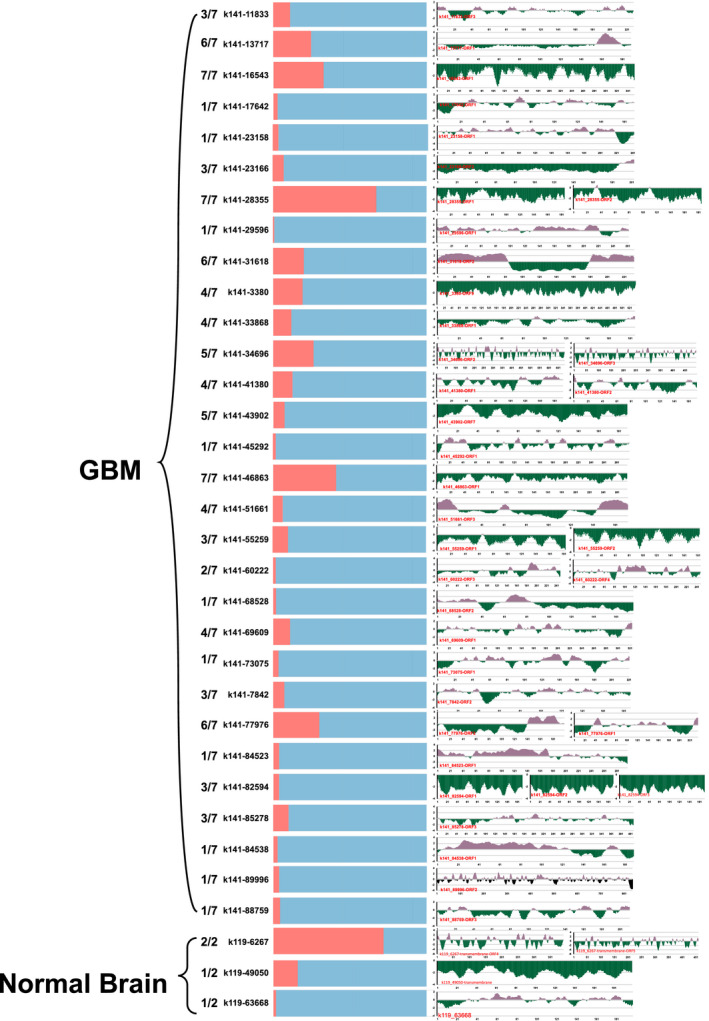
The antibody epitope prediction and sample distribution. The antibody epitope prediction results are on the right, Y‐axis represents the score of the antigen prediction and the X‐axis represents the position of the predicted open reading frame. On the left are the proportion of samples (blue) that harbor this contig out of 110 GBM and 57 normal brain tissues. The number represents the proportion of projects that harbor the contigs
4. DISCUSSION
As the most lethal type of cancer, GBM kills thousands every year. Although many studies have investigated the risk factors of GBM, our knowledge of their etiology is still lacking. 9 , 11 , 12 , 13 , 14 , 85 Emerging evidence suggests that viral infection can cause tumors. For GBM, the main focus was on HCMV, with a small number of studies on other viruses such as EBV 86 or HPV 87 by amplifying viral genome segments. However, the presence and association of virus with GBM is not firmly established and an un‐biased data‐driven approach to investigate the virome in human brain is needed. Analyzing virome in GBM can provide insight on etiology of GBM, and maybe it's unexplainable relationship with other neurological disorder such as Alzheimer's disease.
Next‐generation sequencing technologies had been successfully applied to characterize the virome in various human tissues such as skin and blood. 88 , 89 , 90 Traditional methods for viral detection are based on aligning short sequence reads to the reference viral genome sequences with commonly used software such as PathSequation 91 or RINs. 92 However, these methods could suffer from false positive results where short sequence reads can be congregated in highly repetitive regions. Besides, some reference viral genomes may also contain artificial sequences. 54 Our approach avoided this drawback by first mapping sequence reads to the human genome to filter out human protein‐coding genes and other highly repetitive elements such as human endogenous retrovirus or transposable element sequences. The unmapped reads containing viral sequences were then assembled into relative longer contigs. Our study is the first to explore the GBM virome in an assembly annotation approach, and indeed we identified contigs that match viral sequences. Among them, most were retrovirus sequences, probably due to the close relationship of the retrovirus with human transposable elements. 93 We also found extensive presence of phage sequences in both GBM and healthy brain. Even though it is possible that they come from the gut, 94 previous studies often consider them from bacterial infections contaminated by the commercial phiX174. 88 , 95 , 96
It is surprising to find the sequences of a Picornavirus in five GBM samples (Supplemental 5), as this virus was first reported in invertebrate. 9 , 10 , 11 , 85 However, it is unlikely due to sample contamination or sequence mismatches as the five assembled contigs cover more than 90% of the reference genome of the virus. Picornaviruses are small, single‐stranded positive RNA viruses infecting a wide range of hosts. Given that some viruses infect their hosts ranging from plants to animals, 97 the ubiquitous presence of the Picornaviruses suggests a complex nature of virosphere and an extensive horizontal genetic exchanges of viral genomics. 98 Our finding also indicates that this virus could be a new candidate for oncolytic viral therapy since several other picornaviruses had been proven to have the oncolytic potentials. For example, a recombinant oncolytic poliovirus, PVSRIPO has demonstrated to be oncolytic in a wide range of brain cancer cell lines such as GBM cell lines 99 or astrocytomas cancer cell lines. 100 , 101 Other attenuated polioviruses such as incompetent poliovirus 1 (PV1) replicons have also shown cytotoxicity against various tumors and promising results in prolong survival of GBM mouse models. 102 Taken together, the detection of picornavirus in the GBM but not healthy samples suggests the potential of the discovered picornavirus as a candidate to engineer future oncolytic virus. 103
The presence of EBV in gliomas has always been controversy. 86 Consistent with some of the previous studies, 34 , 35 , 39 , 52 , 104 our results suggest that EBV is absent from gliomas. In addition, contig segments matching herpes virus sequences may come from homologous sequences. However, one possibility we cannot rule out is that the herpes virus is in latent in GBM or inserted into the human genome in various tissues that cannot be captured by RNA‐seq.
A number of contigs cannot be annotated by any databases we used. It is possible that those contigs are artificial or formed from artificial sequences such as vectors or contaminations. However, we observed that various samples from different projects have reads that can cover more than 60% of the contig. For example, over 60% of the length of contig k141‐31618 can be covered by the reads originated from 22 studies from six out of seven projects in the GBM group, making it evident that contigs like this are not contaminations but rather originated from a valid source. Transmembrane analysis and antibody epitope prediction show that significant amount of those contig sequences has antibody epitope sequence signature, suggesting a potential to be used as drug targets for cancer immune therapy.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
ZY, WJZ, and ZA conceived and designed the study, ZY and LZ performed the analysis, XY, LZ, NZ, ZA and WJZ made revisions, ZA and WJZ supervised the project. All authors support the publication of the manuscript.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Yuan Z, Ye X, Zhu L, Zhang N, An Z, Jim Zheng W. Virome assembly and annotation in brain tissue based on next-generation sequencing. Cancer Med. 2020;9:6776–6790. 10.1002/cam4.3325
Funding information
The authors used computing resources from the Texas Advanced Computing Center at The University of Texas at Austin, and the Data Science and Informatics Core for Cancer Research at the School of Biomedical Informatics, University of Texas Health Science Center at Houston. This work is partly supported by the Cancer Prevention and Research Institute of Texas grant RP170668 (Zheng), PR150551& RP190561 (An), the National Institutes of Health (NIH) through grants 1UL1TR003167 and R01AG066749 (Zheng) and the Welch Foundation AU‐0042‐20030616 (An).
Contributor Information
Zhiqiang An, Email: zhiqiang.an@uth.tmc.edu.
W. Jim Zheng, Email: zhiqiang.an@uth.tmc.edu, Email: wenjin.j.zheng@uth.tmc.edu.
DATA AVAILABILITY STATEMENT
We searched the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra), Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/), and literatures to collect the next‐generation sequencing (NGS) studies relating to GBM and normal brain tissues, as well as samples infected with known virus as “positive controls” to test our assembly approaches. The raw RNA‐seq fastq files from Illumina platform were downloaded from SRA database, and the list of accessions for the source data is shown in the Supplemental File 1.
REFERENCES
- 1. Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C et al CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2009–2013. Neuro‐oncology. 2016;18(suppl_5):v1–v75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C et al CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro‐Oncol. 2015;17(suppl 4):iv1–iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz‐Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro‐oncology. 2018;20(suppl_4):iv1–iv86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathologica. 2005;109(1):93–108. [DOI] [PubMed] [Google Scholar]
- 5. ACS . Cancer facts and figures. 2015.
- 6. Meyer MA. Malignant gliomas in adults. N Engl J Med. 2008;359(17):1850. [DOI] [PubMed] [Google Scholar]
- 7. Alexander BM, Cloughesy TF. Adult glioblastoma. J Clin Oncol. 2017;35(21):2402–9. [DOI] [PubMed] [Google Scholar]
- 8. Fisher JL, Schwartzbaum JA, Wrensch M, Wiemels JL. Epidemiology of brain tumors. Neurologic Clinics. 2007;25(4):867–90. [DOI] [PubMed] [Google Scholar]
- 9. Barchana M, Margaliot M, Liphshitz I. Changes in brain glioma incidence and laterality correlates with use of mobile phones‐a nationwide population based study in Israel. Asian Pacific J Cancer Prevention. 2012;13(11):5857–63. [DOI] [PubMed] [Google Scholar]
- 10. Deltour I, Auvinen A, Feychting M, Johansen C, Klaeboe L, Sankila R et al Mobile phone use and incidence of glioma in the nordic countries 1979–2008: consistency check. Epidemiology. 2012;23(2):301–307. [DOI] [PubMed] [Google Scholar]
- 11. Little MP, Azizova TV, Bazyka D, Bouffler SD, Cardis E, Chekin S et al Systematic review and meta‐analysis of circulatory disease from exposure to low‐level ionizing radiation and estimates of potential population mortality risks. Environ Health Perspectives. 2012;120(11):1503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH et al Human cytomegalovirus infection and expression in human malignant glioma. Cancer research. 2002;62(12):3347–50. [PubMed] [Google Scholar]
- 13. Wrensch M, Minn Y, Chew T, Bondy M, Berger MS. Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro‐oncology. 2002;4(4):278–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saddawi‐Konefka R, Crawford JR. Chronic viral infection and primary central nervous system malignancy. J Neuroimmune Pharmacol. 2010;5(3):387–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nature Rev Cancer. 2010;10(12):878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang K‐W, Alaei‐Mahabadi B, Samuelsson T, Lindh M, Larsson E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nature Communications. 2013;4:2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Strong K, Mathers C, Epping‐Jordan J, Resnikoff S, Ullrich A. Preventing cancer through tobacco and infection control: how many lives can we save in the next 10 years? Eur J Cancer Prevention. 2008;17(2):153–61. [DOI] [PubMed] [Google Scholar]
- 18. Isakov O, Modai S, Shomron N. Pathogen detection using short‐RNA deep sequencing subtraction and assembly. Bioinformatics. 2011;27(15):2027–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arora R, Chang Y, Moore PS. MCV and Merkel cell carcinoma: a molecular success story. Curr Opinion Virol. 2012;2(4):489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Strong MJ, O'Grady T, Lin Z, Xu G, Baddoo M, Parsons C et al Epstein‐Barr virus and human herpesvirus 6 detection in a non‐Hodgkin's diffuse large B‐cell lymphoma cohort by using RNA sequencing. J Virol. 2013;87(23):13059–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nonoyama M, Huang C, Pagano J, Klein G, Singh S. DNA of Epstein‐Barr virus detected in tissue of Burkitt's lymphoma and nasopharyngeal carcinoma. Proc Natl Acad Sci. 1973;70(11):3265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strong MJ, Xu G, Coco J, Baribault C, Vinay DS, Lacey MR et al Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: implications for possible immune adjuvant therapy. PLoS Pathogens. 2013;9(5):e1003341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sung W‐K, Zheng H, Li S, Chen R, Liu X, Li Y et al Genome‐wide survey of recurrent HBV integration in hepatocellular carcinoma. Nature Genetics. 2012;44(7):765. [DOI] [PubMed] [Google Scholar]
- 25. Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y et al The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012;22(4):593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams R. Global challenges in liver disease. Hepatology. 2006;44(3):521–6. [DOI] [PubMed] [Google Scholar]
- 27. Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV‐and HCV‐associated hepatocellular carcinoma. Nature Rev Cancer. 2013;13(2):123. [DOI] [PubMed] [Google Scholar]
- 28. Chen Y, Yao H, Thompson EJ, Tannir NM, Weinstein JN, Su X. VirusSeq: software to identify viruses and their integration sites using next‐generation sequencing of human cancer tissue. Bioinformatics. 2012;29(2):266–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amirian ES, Bondy ML, Mo Q, Bainbridge MN, Scheurer ME. Presence of viral DNA in whole‐genome sequencing of brain tumor tissues from the cancer genome atlas. J Virol. 2014;88(1):774‐. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baumgarten P, Michaelis M, Rothweiler F, Starzetz T, Rabenau HF, Berger A et al Human cytomegalovirus infection in tumor cells of the nervous system is not detectable with standardized pathologico‐virological diagnostics. Neuro‐Oncol. 2014;16(11):1469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lehrer S, Labombardi V, Green S, Pessin‐Minsley MS, Germano IM, Rosenzweig KE. No circulating cytomegalovirus in five patients with glioblastoma multiforme. Anticancer Res. 2011;31(3):959–60. [PubMed] [Google Scholar]
- 32. Bhattacharjee B, Renzette N, Kowalik TF. Genetic analysis of cytomegalovirus in malignant gliomas. J Virol. 2012;86(12):6815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bianchi E, Roncarati P, Hougrand O, Guérin‐El Khourouj V, Boreux R, Kroonen J et al Human cytomegalovirus and primary intracranial tumours: frequency of tumour infection and lack of correlation with systemic immune anti‐viral responses. Neuropathol Appl Neurobiol. 2015;41(2):e29–e40. [DOI] [PubMed] [Google Scholar]
- 34. Cimino PJ, Zhao G, Wang D, Sehn JK, Lewis JS Jr, Duncavage EJ. Detection of viral pathogens in high grade gliomas from unmapped next‐generation sequencing data. Exp Molecular Pathol. 2014;96(3):310–5. [DOI] [PubMed] [Google Scholar]
- 35. Cosset É, Petty TJ, Dutoit V, Cordey S, Padioleau I, Otten‐Hernandez P et al Comprehensive metagenomic analysis of glioblastoma reveals absence of known virus despite antiviral‐like type I interferon gene response. Int J Cancer. 2014;135(6):1381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ding D, Han S, Wang Z, Guo Z, Wu A. Does the existence of HCMV components predict poor prognosis in glioma? J Neuro‐Oncol. 2014;116(3):515–22. [DOI] [PubMed] [Google Scholar]
- 37. dos Santos CJ, Stangherlin LM, Figueiredo EG, Corrêa C, Teixeira MJ, da Silva MCC. High prevalence of HCMV and viral load in tumor tissues and peripheral blood of glioblastoma multiforme patients. J Med Virol. 2014;86(11):1953–61. [DOI] [PubMed] [Google Scholar]
- 38. Fonseca RF, Kawamura MT, Oliveira JA, Teixeira A, Alves G, Carvalho MdGdC. The prevalence of human cytomegalovirus DNA in gliomas of Brazilian patients. Memórias do Instituto Oswaldo Cruz. 2012;107(7):953–4. [DOI] [PubMed] [Google Scholar]
- 39. Khoury JD, Tannir NM, Williams MD, Chen Y, Yao H, Zhang J et al Landscape of DNA virus associations across human malignant cancers: analysis of 3,775 cases using RNA‐Seq. J Virol. 2013;87(16):8916–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lau SK, Chen Y‐Y, Chen W‐G, Diamond DJ, Mamelak AN, Zaia JA et al Lack of association of cytomegalovirus with human brain tumors. Modern Pathol. 2005;18(6):838. [DOI] [PubMed] [Google Scholar]
- 41. Lucas KG, Bao L, Bruggeman R, Dunham K, Specht C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J Neuro‐Oncol. 2011;103(2):231–8. [DOI] [PubMed] [Google Scholar]
- 42. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE et al Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro‐Oncol. 2008;10(1):10–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Poltermann S, Schlehofer B, Steindorf K, Schnitzler P, Geletneky K, Schlehofer J. Lack of association of herpesviruses with brain tumors. J Neurovirol. 2006;12(2):90–9. [DOI] [PubMed] [Google Scholar]
- 44. Rahbar A, Orrego A, Peredo I, Dzabic M, Wolmer‐Solberg N, Strååt K et al Human cytomegalovirus infection levels in glioblastoma multiforme are of prognostic value for survival. J Clin Virol. 2013;57(1):36–42. [DOI] [PubMed] [Google Scholar]
- 45. Rahbar A, Stragliotto G, Orrego A, Peredo I, Taher C, Willems J et al Low levels of Human Cytomegalovirus Infection in Glioblastoma multiforme associates with patient survival;‐a case‐control study. Herpesviridae. 2012;3(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J Virol. 2012;86(2):854–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sabatier J, Uro‐Coste E, Pommepuy I, Labrousse F, Allart S, Tremoulet M et al Detection of human cytomegalovirus genome and gene products in central nervous system tumours. Br J Cancer. 2005;92(4):747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El‐Zein R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathologica. 2008;116(1):79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Slinger E, Maussang D, Schreiber A, Siderius M, Rahbar A, Fraile‐Ramos A et al HCMV‐encoded chemokine receptor US28 mediates proliferative signaling through the IL‐6–STAT3 axis. Sci Signal. 2010;3(133):ra58. [DOI] [PubMed] [Google Scholar]
- 50. Tang KW, Hellstrand K, Larsson E. Absence of cytomegalovirus in high‐coverage DNA sequencing of human glioblastoma multiforme. Int J Cancer. 2015;136(4):977–81. [DOI] [PubMed] [Google Scholar]
- 51. Yamashita Y, Ito Y, Isomura H, Takemura N, Okamoto A, Motomura K et al Lack of presence of the human cytomegalovirus in human glioblastoma. Modern Pathol. 2014;27(7):922. [DOI] [PubMed] [Google Scholar]
- 52. Strong MJ, Blanchard E, Lin Z, Morris CA, Baddoo M, Taylor CM et al A comprehensive next generation sequencing‐based virome assessment in brain tissue suggests no major virus‐tumor association. Acta Neuropathologica Commun. 2016;4(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Holdhoff M, Guner G, Rodriguez FJ, Hicks JL, Zheng Q, Forman MS et al Absence of cytomegalovirus in glioblastoma and other high‐grade gliomas by real‐time PCR, immunohistochemistry, and in situ hybridization. Clin Cancer Res. 2017;23(12):3150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chou H‐H, Holmes MH. DNA sequence quality trimming and vector removal. Bioinformatics. 2001;17(12):1093–104. [DOI] [PubMed] [Google Scholar]
- 55. Li Y, Tian K, Yin C, He RL, Yau SS‐T. Virus classification in 60‐dimensional protein space. Molecular Phylogenetics Evolution. 2016;99:53–62. [DOI] [PubMed] [Google Scholar]
- 56. García‐López R, Vázquez‐Castellanos JF, Moya A. Fragmentation and coverage variation in viral metagenome assemblies, and their effect in diversity calculations. Front Bioeng Biotechnol. 2015;3:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hatfull GF. Bacteriophage genomics. Curr Opinion Microbiol. 2008;11(5):447–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Peng Y, Leung HC, Yiu S‐M, Chin FY. Meta‐IDBA: a de Novo assembler for metagenomic data. Bioinformatics. 2011;27(13):i94–i101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS et al SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. J Computational Biol. 2012;19(5):455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boisvert S, Raymond F, Godzaridis É, Laviolette F, Corbeil J. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 2012;13(12):R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gruninger RJ, Nguyen TTM, Reid ID, Yanke J, Wang P, Abbott DW et al Application of transcriptomics to compare the carbohydrate active enzymes that are expressed by diverse genera of anaerobic fungi to degrade plant cell wall carbohydrates. Front MICROBIOL. 2018;9:1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hesse U, Van Heusden P, Kirby BM, Olonade I, van Zyl LJ, Trindade M. Virome assembly and annotation: a surprise in the Namib Desert. Front Microbiol. 2017;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Andrews S. FastQC: a quality control tool for high throughput sequence data. Cambridge, UK: Babraham Bioinformatics, Babraham Institute; 2010. [Google Scholar]
- 65. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Naeem R, Rashid M, Pain A. READSCAN: a fast and scalable pathogen discovery program with accurate genome relative abundance estimation. Bioinformatics. 2013;29(3):391–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Roux S, Emerson JB, Eloe‐Fadrosh EA, Sullivan MB. Benchmarking viromics: an in silico evaluation of metagenome‐enabled estimates of viral community composition and diversity. PeerJ. 2017;5:e3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sutton TD, Clooney AG, Ryan FJ, Ross RP, Hill C. Choice of assembly software has a critical impact on virome characterisation. Microbiome. 2019;7(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li W, Godzik A. Cd‐hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–9. [DOI] [PubMed] [Google Scholar]
- 70. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Molecular Biol. 1990;215(3):403–10. [DOI] [PubMed] [Google Scholar]
- 71. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nature Methods. 2015;12(1):59. [DOI] [PubMed] [Google Scholar]
- 72. Darzentas N. Circoletto: visualizing sequence similarity with Circos. Bioinformatics. 2010;26(20):2620–2621. [DOI] [PubMed] [Google Scholar]
- 73. Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nature Methods. 2012;9(4):357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L et al Using Tablet for visual exploration of second‐generation sequencing data. Briefings Bioinformatics. 2012;14(2):193–202. [DOI] [PubMed] [Google Scholar]
- 75. Wickham H. Ggplot2: Elegant graphics for data analysis. New York, NY: Springer‐Verlag; 2016. [Google Scholar]
- 76. Bardou P, Mariette J, Escudié F, Djemiel C, Klopp C. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics. 2014;15(1):293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular Biol Evolution. 2009;26(7):1641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sonnhammer EL, Von Heijne G, Krogh A, editors. A hidden Markov model for predicting transmembrane helices in protein sequences. Ismb. 1998;6:175–182. [PubMed] [Google Scholar]
- 79. Larsen JEP, Lund O, Nielsen M. Improved method for predicting linear B‐cell epitopes. Immunome Res. 2006;2(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ponomarenko JV, Bourne PE. Antibody‐protein interactions: benchmark datasets and prediction tools evaluation. BMC Struct Biol. 2007;7(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Haste Andersen P, Nielsen M, Lund O. Prediction of residues in discontinuous B‐cell epitopes using protein 3D structures. Protein Sci. 2006;15(11):2558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR et al The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2018;47(D1):D339–D43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shi M, Lin X‐D, Tian J‐H, Chen L‐J, Chen X, Li C‐X et al Redefining the invertebrate RNA virosphere. Nature. 2016;540(7634):539. [DOI] [PubMed] [Google Scholar]
- 85. Fisher JL, Schwartzbaum JA, Wrensch M, Berger MS. Evaluation of epidemiologic evidence for primary adult brain tumor risk factors using evidence‐based medicine. Guiding Neurosurgery by Evidence. 19. Karger Publishers; 2006;p. 54–79. [DOI] [PubMed] [Google Scholar]
- 86. Akhtar S, Vranic S, Cyprian FS, Moustafa A. Epstein‐Barr virus in gliomas: cause, association, or artifact? Front Oncol. 2018;8:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vidone M, Alessandrini F, Marucci G, Farnedi A, de Biase D, Ricceri F et al Evidence of association of human papillomavirus with prognosis worsening in glioblastoma multiforme. Neuro‐Oncol. 2013;16(2):298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Moustafa A, Xie C, Kirkness E, Biggs W, Wong E, Turpaz Y et al The blood DNA virome in 8,000 humans. PLoS Pathogens. 2017;13(3):e1006292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hannigan GD, Meisel JS, Tyldsley AS, Zheng Q, Hodkinson BP, SanMiguel AJ et al The human skin double‐stranded DNA virome: topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome. MBio. 2015;6(5):e01578‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tang K‐W, Larsson E. Tumour virology in the era of high‐throughput genomics. Philos Transac R Soc B: Biol Sci. 2017;372(1732):20160265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, Getz G et al PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nature Biotechnol. 2011;29(5):393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bhaduri A, Qu K, Lee CS, Ungewickell A, Khavari PA. Rapid identification of non‐human sequences in high‐throughput sequencing datasets. Bioinformatics. 2012;28(8):1174–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cloyd MW. Human retroviruses. Medical Microbiology 4th edition. University of Texas Medical Branch at Galveston; 1996. [PubMed] [Google Scholar]
- 94. Górski A, Ważna E, Dąbrowska B‐W, Dąbrowska K, Świtała‐Jeleń K, Międzybrodzki R. Bacteriophage translocation. FEMS Immunol Med Microbiol. 2006;46(3):313–9. [DOI] [PubMed] [Google Scholar]
- 95. Glassing A, Dowd SE, Galandiuk S, Davis B, Chiodini RJ. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathogens. 2016;8(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Strong MJ, Xu G, Morici L, Bon‐Durant SS, Baddoo M, Lin Z et al Microbial contamination in next generation sequencing: implications for sequence‐based analysis of clinical samples. PLoS Pathogens. 2014;10(11):e1004437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shi M, Lin X‐D, Chen X, Tian J‐H, Chen L‐J, Li K et al The evolutionary history of vertebrate RNA viruses. Nature. 2018;556(7700):197. [DOI] [PubMed] [Google Scholar]
- 98. Baranowski E, Ruiz‐Jarabo CM, Domingo E. Evolution of cell recognition by viruses. Science. 2001;292(5519):1102–5. [DOI] [PubMed] [Google Scholar]
- 99. Ochiai H, Campbell SA, Archer GE, Chewning TA, Dragunsky E, Ivanov A et al Targeted therapy for glioblastoma multiforme neoplastic meningitis with intrathecal delivery of an oncolytic recombinant poliovirus. Clinical Cancer Res. 2006;12(4):1349–54. [DOI] [PubMed] [Google Scholar]
- 100. Yang X, Chen E, Jiang H, Muszynski K, Harris RD, Giardina SL et al Evaluation of IRES‐mediated, cell‐type‐specific cytotoxicity of poliovirus using a colorimetric cell proliferation assay. J Virological Methods. 2009;155(1):44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dobrikova EY, Broadt T, Poiley‐Nelson J, Yang X, Soman G, Giardina S et al Recombinant oncolytic poliovirus eliminates glioma in vivo without genetic adaptation to a pathogenic phenotype. Molecular Ther. 2008;16(11):1865–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ansardi DC, Porter DC, Jackson CA, Gillespie GY, Morrow CD. RNA replicons derived from poliovirus are directly oncolytic for human tumor cells of diverse origins. Cancer Res. 2001;61(23):8470–9. [PubMed] [Google Scholar]
- 103. McCarthy C, Jayawardena N, Burga LN, Bostina M. Developing picornaviruses for cancer therapy. Cancers. 2019;11(5):685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hashida Y, Taniguchi A, Yawata T, Hosokawa S, Murakami M, Hiroi M et al Prevalence of human cytomegalovirus, polyomaviruses, and oncogenic viruses in glioblastoma among Japanese subjects. Infect Agents Cancer. 2015;10(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Data Availability Statement
We searched the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra), Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/), and literatures to collect the next‐generation sequencing (NGS) studies relating to GBM and normal brain tissues, as well as samples infected with known virus as “positive controls” to test our assembly approaches. The raw RNA‐seq fastq files from Illumina platform were downloaded from SRA database, and the list of accessions for the source data is shown in the Supplemental File 1.