

Abstract
Background
The incidence of colorectal cancer (CRC) among patients <50 years of age has increased dramatically over the last decades. At the same time, the growing proportion of obese children and adolescents and the increasing proportion of young and obese patients with CRC suggests an association between metabolic dysfunction and carcinogenesis. Tumor‐associated macrophages (TAMs) are able to orchestrate tumor promoting and suppressing mechanisms in CRC. The aim of this review was to discuss the different roles of TAMs in CRC and their phenotype‐specific metabolic pathways to identify potential new targets for CRC treatment.
Methods
A literature search was performed using PubMed, Cochrane and Embase to identify studies on TAMs and their metabolism in CRC. The following search terms were used in various combinations: (obesity OR adiposity OR obese) AND (macrophage OR polarization OR macrophage metabolism) AND ((colon cancer*) OR (colon carcinoma) OR (colonic tumor*) OR (colonic neoplasm[MeSH]) OR (rectal cancer*) OR (rectal carcinoma) OR (rectal tumor*) OR (rectal neoplasm[MeSH]) OR (colorectal cancer*) OR (colorectal carcinoma) OR (colorectal tumor*) OR (colorectal neoplasm[MeSH])). Studies including data on the phenotype and metabolism of TAMs in CRC were analyzed.
Results
Evidence for the prognostic utility of macrophage markers in CRC is currently evolving, with a particular role of stage‐dependent cellular metabolism profiles of TAMs. Itaconate is one of the metabolites produced by proinflammatory subtypes of TAMs and it is known to have tumor promoting effects. Metabolic pathways that are involved in macrophage activation and reprogramming play a role in a chronic inflammatory setting, consequently affecting the onset and development of CRC.
Conclusions
Tumor‐promoting metabolites, such as itaconate, are directly regulating these mechanisms, thereby triggering carcinogenesis. Metabolic reprogramming in TAMs can build a bridge between metabolic dysfunction and the onset and progression of CRC through inflammatory pathways, particularly in younger patients with early‐onset CRC.
Keywords: adiposity, colonic neoplasms, colorectal neoplasms, obesity, rectal neoplasms, tumor‐associated macrophages
The incidence of colorectal cancer (CRC) among patients <50 years of age has increased dramatically over the last decades. At the same time, the growing proportion of obese children and adolescents and the increasing proportion of young and obese patients with CRC suggests an association between metabolic dysfunction and carcinogenesis. Metabolic reprogramming in TAMs can build a bridge between metabolic dysfunction and the onset and progression of CRC through inflammatory pathways, particularly in younger patients with early‐onset CRC.
![]()
1. INTRODUCTION
Colorectal cancer (CRC) is one of the most common causes of cancer‐related death in the USA and its incidence among young adults has increased over the last several decades.(1, 2, 3, 4) Simultaneously, the proportion of overweight and obese children and adolescents is rising, suggesting an association between metabolic dysfunction and carcinogenic mechanisms.(4, 5, 6, 7) A direct association between obesity and early‐onset CRC was found among young women as part of the The Nurses' Health Study II.(4) Comparing obese and non‐obese individuals among different cancer types, CRC was the only type of cancer that showed an increase in young adults with obesity undergoing CRC resections.(8)
Most cases of CRC are either sporadic in etiology, based on a somatic mutation involving Adenomatous polyposis coli gene (APC) function, or less often hereditary, as in hereditary nonpolyposis colorectal cancer (HNPCC) or familial adenomatous polyposis (FAP).(9, 10) HNPCC is the most common cause of hereditary CRC.(11) While the proportion of other hereditary cancer syndromes in patients with early‐onset CRC is widely unknown, HNPCC represents 4%‐13.5% of cancers in patients with early‐onset CRC.(11) Only a small percentage of CRC is actually associated with inflammatory bowel disease such as ulcerative colitis or Crohn's disease.(12, 13) Compared to 0.4% of patients with a late onset of CRC, young patients with CRC showed an increased prevalence of inflammatory bowel disease in 3% of cases.(14) There is, however, strong evidence that anti‐inflammatory medication can reduce the risk of CRC, even in the absence of an underlying inflammatory bowel disease.(15, 16) Furthermore, low‐dose aspirin therapy is associated with a reduced risk of advanced stage disease, which might indicate that anti‐inflammatory medication also suppresses CRC progression.(17) This suggests that inflammatory processes may have a greater role in the onset and development of CRC than previously assumed.
As essential components of the immune inflammatory response, macrophages are able to orchestrate inflammatory mechanisms and therefore tumorigenesis. The tumor microenvironment (TME) is a dynamic environment surrounding the tumor and a critical part of these regulatory processes. Tumor cells, immune cells and the blood and lymphatic vascular networks interact with stromal cells and their extracellular matrix, coordinating cancer establishment, tumor growth and metastasis.(18) Current evidence shows that tumor‐associated macrophages (TAMs) play a central role in the dynamic processes within the TME, contributing to tumor promoting effects as well as contributing to tumor suppressing mechanisms in CRC.(18) Various functions require dynamic switching between different TAM phenotypes. These phenotypes depend upon specific metabolic pathways within TAMs, which provide a source for functional metabolites and facilitate phenotype‐specific inflammatory or anti‐inflammatory activities during cancer development.(19, 20) The impact of cell interactions within the TME on a patient's outcome, including risk of CRC recurrence or effectiveness of cytoreductive chemotherapy, radiotherapy, or immunotherapy, is poorly understood.
The aim of this review was to highlight current publications and trendsetting approaches regarding metabolic dysfunction and TAMs in CRC, and to discuss their phenotype‐specific metabolic pathways in order to identify pathogenetic mechanisms as potential new targets for early‐onset CRC treatment.
2. SYSTEMIC METABOLIC DYSFUNCTION AND ITS LINK TO COLORECTAL CANCER
The incidence of overweight and obesity in the general population is increasing worldwide with a remarkable rising trend in children and young adolescents.(21) Simultaneously, an increase in CRC rates among younger adults <50 years of age is reported.(1, 2, 4) Regular screening for CRC is recommended by the U.S. Preventive Services Task Force (USPSTF) beginning at age 50.(22) Due to the large‐scale screening programs in these patients >50 years of age with removal of precancerous polyps during colonoscopy and earlier detection of CRC, the incidence and mortality of CRC has declined.(23, 24) The increasing number of colon cancer cases among younger adults has, however, led to the recommendation of an earlier start of regular screening by the American Cancer Society (ACS) in 2018, beginning at age 45.(25) The parallel increase in the incidence of obesity in the young and early‐onset CRC indicates that obesity‐related inflammatory mechanisms may play a greater role in the development of CRC than assumed.
Overweight and obesity are defined as an abnormal fat accumulation with a respective Body Mass Index (BMI) of ≥25 and ≥30 kg/m2 by the World Health Organization (WHO).(21) Obesity is an established risk factor for type 2 diabetes mellitus and its associated complications.(26) The underlying metabolic dysfunction is due to chronic systemic inflammation that can lead to insulin‐resistance.(27) During the last three decades, epidemiological data and several cohort and case‐control studies have shown an association between obesity and CRC.(28, 29, 30) The simultaneously rising incidence of obesity and CRC in patients younger than 50 years of age indicates a particular contributing role of metabolic dysfunction in the development early‐onset CRC.(4) Liu et al prospectively analyzed a patient cohort of more than 85 000 women aged 25 to 42 years, that were part of the The Nurses' Health Study II.(4) An association between obesity and early‐onset CRC was found among this patient collective. The recent analysis of Hussan et al showed an increasing trend in CRC among young patients with obesity, that could not be demonstrated in other types of gastrointestinal cancer.(8) Focusing on the age of diagnosis in patients with CRC, an increased cancer risk was shown after a diagnosis of type 2 diabetes, especially in men younger than 55 years.(31)
A recent study on the molecular characteristics of early‐onset CRC showed that inflammatory mechanisms, such as deregulated redox homeostasis as one of the hallmarks of CRC in young patients, play a distinct role.(32) The major pathways that are involved in these mechanisms are altered Nuclear factor erythroid 2‐related factor 2 (NRF)‐mediated oxidative stress response, glutathione metabolism, and the chemokine (C‐X‐C motif) ligand 12 ‐ C‐X‐C motif chemokine receptor 4 (CXCL12‐CXCR4) signaling axis.(32) These findings suggest that metabolic dysfunction and obesity represent an important contributing factor in CRC development in young patients.
A chronic inflammatory environment is caused by the proinflammatory endocrine activity of adipose tissue, affecting energy homeostasis and glucose metabolism.(33) Inflammatory macrophages can accumulate within adipose tissue in obese patients and trigger inflammation, which leads to systemic metabolic dysfunction, including insulin resistance.(34) The presence of macrophages is a hallmark of proinflammatory adipose tissue. They form crown‐like structures in subcutaneous and visceral fat deposits.(34) Furthermore, adipose tissue‐derived inflammatory mediators have been shown to induce macrophage polarization toward a proinflammatory phenotype in an in vitro model.(35) In other obesity‐related comorbidities, such as nonalcoholic fatty liver disease (NFLD) or steatohepatitis (NASH), inflamed adipose tissue has been associated with activation of liver macrophages as a determinant for liver fibrosis.(36) Proinflammatory macrophage polarization in tissue macrophages can provide a link between the proinflammatory systemic state in obesity and a chronic inflammatory environment in colon tissue, which in turn can trigger carcinogenic mechanisms in colon epithelium through inflammatory stress.
Itaconate is a macrophage‐specific metabolite, which is produced in proinflammatory macrophages, and which is known to have tumor promoting effects.(37) TAMs in tumor‐bearing mice as well as monocytes isolated from patients with ovarian cancer showed increased itaconate production.(37)
Identifying the role of macrophage metabolism and itaconate in a chronic inflammatory state due to metabolic dysfunction and obesity, could lead to innovative approaches to screening diagnosis and treatment of CRC.
2.1. Chronic Inflammation in colorectal cancer
Chronic inflammation is closely linked to two systems of the human body that have major roles for survival: the immune system with the ability to fight infection and the metabolic system that can provide stored energy during a period of low nutrition.(27) Immunity and metabolism are therefore in a continuous state of interplay through inflammatory pathways. Both systems share several mediators, including hormones, cytokines, transcription factors, signaling proteins, and lipids. A chronic inflammatory state functions as a stressor and promotes tissue damage that can lead to neoplasia. Once a genetic mutation leads to oncogene activation, inflammation will contribute to cell proliferation, tumor establishment, growth, and metastasis. CRC is a cancer type known to be closely associated with chronic inflammation. Even though less than 2% of CRC is colitis‐associated, sporadic CRC shows similar mutations in genes and signaling pathways, such as the Wnt/β‐catenin pathway, K‐Ras or B‐Raf activation, adenomatous polyposis coli (APC) inactivation, transforming growth factor(TGF)‐β, P53, and the DNA mismatch repair (MMR) proteins.(38, 39, 40, 41) The pathogenesis of both CRC types differs in the histological sequence that is followed during development of neoplasia and the initiation of cancer formation.
Numerous clinical and epidemiological studies have shown that the use of aspirin or nonsteroidal anti‐inflammatory drugs (NSAIDs) is associated with a reduced risk of CRC or recurrent adenomatous polyps as well as decreased CRC mortality.(17, 42, 43, 44, 45, 46) Furthermore, low‐dose aspirin therapy seems to slow progression of a tumor that is already established. A recent cohort study of more than 300 000 patients in the United Kingdom demonstrated that new use of low dose aspirin was associated with a reduced risk of advanced stage CRC (Duke's B‐D) at diagnosis.(17) In 2015, the USPSTF started recommending low‐dose aspirin for chemoprevention of CRC in patients with increased cardiovascular risk aged 50‐59 years.(47)
Independent of its pathogenesis, CRC is infiltrated by immune cells such as macrophages, neutrophils or lymphocytes, that induce and maintain cancer‐related inflammation.(48)
In colon adenomas, the precursor lesions of sporadic CRC, TAMs with low major histocompatibility complex class 2 (MHC II) expression were observed, and the density of these macrophages correlates with tumor progression.(49) This suggests that mechanisms within the TME lead to macrophage polarization toward an anti‐inflammatory phenotype during the development of cancer. Furthermore, high‐grade adenomas have been shown to consist of a higher fraction of anti‐inflammatory macrophages than low‐grade adenomas.(50) This leads to the conclusion that macrophages of an anti‐inflammatory type seem to have a role in malignant transformation of colorectal adenomas toward CRC.
The link between immunity and cancer through inflammation was observed as early as the 19th century by the German pathologist Rudolf Virchow, when he described white blood cells as part of the tumor mass. In 1986, the American pathologist Harold Dvorak investigated angiogenesis within tumors, considered these mechanisms similar to those in wounds and depicted tumors as ‘wounds that do not heal’.(51) Inflammatory tissue injury causes chemotactic signaling that attracts immune cells to repair damage, and TAMs are the major cell type orchestrating the pathways within the TME, to either promote or suppress tumor development in CRC.(52) These opposing functions of TAMs are characterized by a respective dominating metabolic pathway of the macrophage that can be affected by extracellular signals within the tumor environment. This polarization into different functional subsets can be affected by proinflammatory cytokines,(53, 54) leading to the conclusion, that there is a direct connection between metabolism, inflammation and macrophage differentiation affecting tumor behavior.
Itaconate is a metabolite within inflammatory macrophages, and a regulator of cellular metabolism as well. It regulates glycolysis and leads to succinate accumulation through inhibition of succinate dehydrogenase.(55) This can lead to decreased production of reactive oxygen species (ROS) and altered activation of numerous transcription factors, such as nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB), hypoxia‐inducible factor 1α (HIF1α), signal transducer and activator of transcription 3 (STAT3), and activator protein 1 (AP‐1).(37, 55) NRF2 is a superordinate regulator of these anti‐inflammatory functions, that is affected by itaconate.(55)
In anti‐inflammatory macrophages, itaconate can further boost anti‐inflammatory functions.(55) Since anti‐inflammatory macrophages play a role in tumor progression in CRC, this suggests that itaconate affects CRC growth.
3. TUMOR‐ASSOCIATED MACROPHAGES IN COLORECTAL CANCER—BASICS FROM THE BENCH
The ability of macrophages to adapt to various environments and to provide a wide variety of functions in tissue is due to dynamic adjustments of their cellular metabolism. These metabolic pathways can be affected by the particular TME inducing the metabolic reprogramming, which in turn leads to different cell phenotypes.
3.1. Cellular metabolism and different phenotypes of tumor‐associated macrophages
Metabolic reprogramming can occur as a result of different stimuli on TAMs, for example, mediators secreted by cancer cells, signals from cells within the tumor microenvironment, self‐secretion or indirect stimuli such as hypoxia. Although switching between phenotypes is a continuous transition with intermediate types present, two main macrophage phenotypes have been described: an M1‐subtype with primarily inflammatory functions and an M2‐subtype, with predominantly anti‐inflammatory and immunosuppressive activity (Figure 1). This simplified classification is an attempt to distinguish between subsets of macrophages that have a primarily—but not exclusively—inflammatory or anti‐inflammatory function. The ‘waterfall model’ illustrates specific characteristics of TAMs during their development from a monocyte to an anti‐inflammatory macrophage subtype.(56) During this process, monocytes that initially present markers, such as C‐C chemokine receptor type 2 (CCR2) and lymphocyte antigen 6 complex (Ly6C), undergo functional and therefore phenotypical changes, losing Ly6C, and gaining MHC II expression.(56) This demonstrates the continuous transition of monocytes and macrophages with overlapping cell surface markers during all stages of development.
Figure 1.
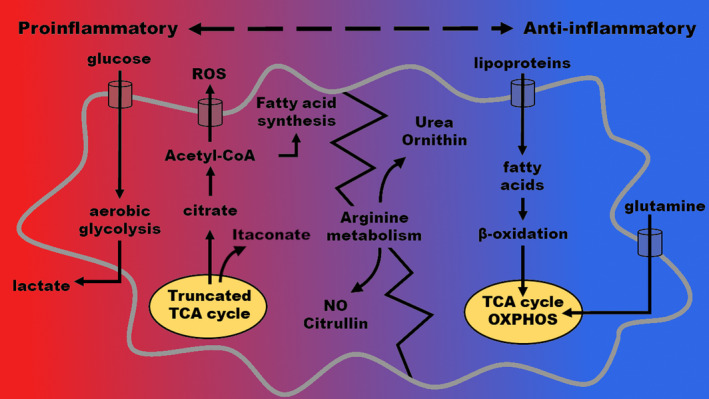
Metabolic pathways in proinflammatory and anti‐inflammatory phenotypes of tumor‐associated macrophages. Simplified model showing the dominating metabolic pathways in both extremes of phenotypes in tumor‐associated macrophages. As macrophages can switch between proinflammatory and anti‐inflammatory phenotypes continuously by changing their cellular metabolism, metabolic pathways can overlap between both types. Proinflammatory macrophages focus on aerobic glycolysis, truncated tricarboxylic acid cycle (TCA cycle) and fatty acid synthesis for energy homeostasis of the cell. Anti‐inflammatory macrophages use the TCA cycle, oxidative phosphorylation and β‐oxidation as their major energy sources. ROS: reactive oxygen species; TCA cycle: tricarboxylic acid cycle
Depending upon their phenotype, macrophages prefer specific metabolic pathways for their energy homeostasis. The characteristic metabolic profiles of inflammatory and anti‐inflammatory macrophages lead to distinct phenotypes with respect to cellular metabolism, which can be studied instead of targeting cell surface markers (Table 1). While aerobic glycolysis is the main pathway in proinflammatory macrophages receiving M1 stimuli, anti‐inflammatory M2 macrophages are characterized by slower rates of aerobic glycolysis and primarily fatty acid oxidation.(57, 58) The classically activated inflammatory M1 macrophages show induction of glycolysis through the AKT/mTOR/HIF pathway.(37) Aerobic glycolysis is an inefficient pathway with a high rate of glucose consumption, but it is essential for rapid energy production and biosynthesis. M1 macrophages utilize this pathway for host‐defense against pathogens, including the production of ROS to kill bacteria or tumor cells. A slower rate of aerobic glycolysis within M2 macrophages is necessary for the production of cytokines.(58) In contrast to M1 macrophages, the M2 subset macrophages show increased oxidative phosphorylation (OXPHOS).(37) As shown in hepatocellular carcinoma, cancer cells can promote glycolysis in M2 macrophages through soluble mediators, increasing the gene expression of the glycolytic enzyme PFKFB3.(59) Therefore, glycolysis plays a role in both macrophage phenotypes, but the respective energy production focuses on different glycolysis‐associated pathways.
Table 1.
Human macrophage characteristics depending on their metabolic phenotype (inflammatory versus anti‐inflammatory)
| Phenotype | Proinflammatory (M1‐like subtype) | Anti‐inflammatory (M2‐like subtype) |
|---|---|---|
| Cell surface markers | CD11c, CD16, CD80, CD86, MHC II | CD163, CD206, CD209 |
| Factors inducing differentiation | IFN‐γ, TNF, LPS, ATP | IL‐4, IL‐10, IL‐13, TGF‐β |
| Metabolic pathways | Aerobic glycolysis, truncated TCA cycle (Itaconate production), fatty acid synthesis | β‐oxidation, oxidative TCA cycle |
| Secreted factors | IL‐1β, IL‐6, IL‐8, IL‐12, IL‐23, IL‐27, TNF‐α, CXCL1, CXCL9, CXCL10, CXCL11, CCL2, CCL5, RNI, ROI, COX2 | IL‐10, IL‐13, IL‐1RA, TGF‐β, CCL17, CCL18, CCL22, CCL24, Arg1, COX1, VEGF, PDGF |
The listed cell surface markers, factors and metabolic pathways are not exclusively present in only one of these macrophage phenotypes. Since macrophages can switch between phenotypes showing fluent transitions, these characteristics might overlap. However, the characteristics that are shown in this table are more likely to be present in the respective phenotype.
Abbreviations: Arg1: arginase 1; ATP: adenosine thiotriphosphate; CCL: CC‐chemokine ligand; CD: cluster of differentiation; COX: cyclooxygenase; CXCL: chemokine (C‐X‐C motif) ligand; IFN: interferone; IL: interleukin; LPS: lipopolysaccharides; MHC II: major histocompatibility complex class 2; PDGF: platelet‐derived growth factor; RNI: reactive nitrogen intermediates; ROI: reactive oxygen intermediates; TCA cycle: tricarboxylic acid cycle; TGF: transforming growth factor; TNF: tumor‐necrosis factor; VEGF: vascular endothelial growth factor.
3.2. The dual role of tumor‐associated macrophages in colorectal cancer
In contrast to other solid human cancers, TAMs in colorectal cancer seem to have the ability to both support and suppress tumor growth. Tumor‐promoting mechanisms are known to result from an interplay between cancer cells, the tumor microenvironment and TAMs. It is hypothesized, that tumor initiation is fostered by mutagenic mechanisms from a chronic inflammatory environment in the subepithelial stroma.(60) Proinflammatory M1 macrophages that produce reactive oxygen and nitrogen species, are able to potentiate this effect, triggering oncogenic mutations in the adjacent epithelial layer (Figure 2). Once neoplasia is initiated, the tumor recruits additional bone marrow‐derived monocytes from the bloodstream and stimulates myelopoiesis by releasing growth factors and chemotactic signals such as CC‐chemokine ligands 2 and 5 (CCL2, CCL5), vascular endothelial growth factor (VEGF) and transforming growth factor beta (TGF‐β).(60, 61, 62) In adipose tissue, a similar mechanism is described, where CCL2 expression leads to increased macrophage infiltration and inflammation, which in turn is associated with insulin resistance.(63) Macrophage colony stimulating factor‐1 (M‐CSF or CSF‐1) has been shown to be produced by colon cancer cells in order to attract and ‘re‐educate’ macrophages.(62) During the early stages of tumor development, neoplastic cells seem to first attract monocytes and ensure their maturation to macrophages within the TME. After their differentiation to TAMs, cancer cells take these macrophages hostage by manipulating their metabolism through multiple signaling pathways and use these TAMs to support further tumor growth and progression. Overexpression of the chemoattractant CCL2 has been associated with advanced tumor stages, metastatic disease and poor prognosis in CRC.(64, 65) Furthermore, CRC cells produce lactic acid as a by‐product of predominantly aerobic glycolysis.(66) Proliferating cancer cells switch their metabolism toward aerobic glycolysis, which is known as the ‘Warburg effect’. Irrespective of the availability of oxygen, they metabolize glucose to lactate, which is also secreted to induce VEGF and arginase 1 (ARG1) expression in TAMs.(66) VEGF expression in macrophages was shown to be upregulated by a pathway described in hypoxia, even under normoxic conditions.(66) This mechanism leads to macrophage recruitment and polarization toward the tumor promoting M2 macrophage phenotype and is therefore associated with metabolic reprogramming in TAMs. Another key mechanism for the alternative activation of tissue macrophages is the peroxisome proliferator activated receptor‐γ (PPARγ) pathway.(67) In animal studies, the disruption of this pathway also was associated with diet‐induced obesity, insulin resistance, and glucose intolerance.(67) PPARγ deficiency can also lead to increased itaconate production, which suggests that itaconate acts as an alternative regulator of M2‐like polarization.(55)
Figure 2.
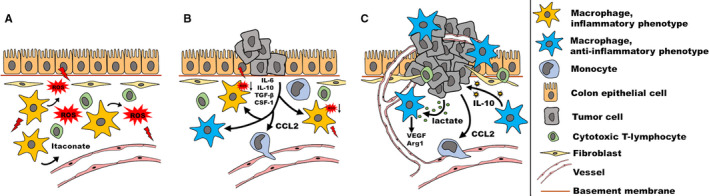
Tumor‐associated macrophages (TAMs) in chronic inflammation and colorectal cancer (CRC) development. (A) Tissue‐resident macrophages with a proinflammatory phenotype might be able to trigger the onset of CRC in the presence of a mutagenic activation of oncogenes in colon epithelial cells due to inflammatory stress and itaconate production. (B) During early cancer development colon cancer cells produce chemokines (CCL2) to attract bone marrow‐derived monocytes and induce macrophage differentiation releasing cytokines and growth factors such as IL‐6, IL‐10, TGF‐β, and M‐CSF (CSF‐1). (C) Colon cancer cells release mediators such as lactate to induce TAM polarization into an anti‐inflammatory phenotype. Reprogrammed macrophages show an increased expression of vascular endothelial growth factor (VEGF) and Arginase 1 (Arg 1), promoting angiogenesis and tumor growth. Furthermore, anti‐inflammatory TAMs promote tumor development by inducing IL‐10 production in colon cancer cells. ROS: reactive oxygen species; IL: interleukin; TGF: transforming growth factor; M‐CSF: Macrophage colony‐stimulating factor
Furthermore, signal transducer and activator of transcription 3 (STAT3) activation leads to M2 polarization of macrophages.(68) This pathway can be induced by glucagon‐like peptide 1 (GLP‐1), a postprandially secreted hormone that improves insulin resistance.(68) TAMs also promote tumor development by inducing interleukin 10 (IL‐10) production in CRC cells through a STAT3 pathway(69, 70) and produce cytokines such as VEGF to induce tumor angiogenesis and tumor growth.(71)
3.3. Tumor‐associated macrophages as prognostic predictors in colorectal cancer
The density of recruited macrophages and their metabolic phenotype were found to be associated with different clinical outcomes in CRC patients. Despite the heterogeneity among study methods used to investigate the degree of TAM infiltration, a high TAM density within the primary tumor is associated with an improved prognosis in CRC patients.(72) In other solid tumors, such as gastric, urogenital and head and neck cancers, a high TAM density is accompanied by worse overall survival.(72) A higher degree of infiltrating macrophages in the invasive front of CRC, in particular those with an M1 phenotype, is associated with a better prognosis in a stage‐dependent manner.(73, 74) Furthermore, it is inversely correlated to lymph node and liver metastases.(75, 76) While M2 macrophages seem to be more prevalent in stage II CRC, M1 macrophages are predominant in less invasive T1 tumors.(77) This indicates that M1 macrophages are primarily responsible for tumor initiation because of inflammatory mechanisms increasing oncogenic potential. Further in the course of the tumor, cancer cells recruit additional bone marrow‐derived blood monocytes and reprogram their metabolism to induce M2‐polarization.(60)
Investigating the different functions and phenotypes of TAMs during tumor development, which in turn promote and suppress tumor growth, is the basis for developing new diagnostic and therapeutic targets, especially in early‐onset CRC. The specific role of itaconate, that can regulate macrophage polarization in tumors, is currently unknown in CRC. Table 2 provides an overview of current studies investigating TAM phenotypes and therefore indirectly TAM metabolism in CRC.
Table 2.
Studies on colorectal cancer investigating cancer‐related mechanisms related to the phenotype and metabolism of tumor‐associated macrophages
| Author | Year of publication | Study model | Aims & objectives | Results | Conclusions regarding TAM metabolism |
|---|---|---|---|---|---|
| Colegio et al [66] | 2014 | Murine/murine cell line |
|
|
|
| Deng et al [90] | 2010 | Murine |
|
|
|
| Edin et al [73] | 2012 | Human tissue |
|
|
|
| Feng et al [86] | 2019 | Human tissue |
|
|
|
| Herbeuval et al [69] | 2004 | Human cell lines |
|
|
|
| Koelzer et al [85] | 2016 | Human tissue |
|
|
|
| Malesci et al [87] | 2017 | Human tissue/human cell lines |
|
|
|
| Nandi et al [91] | 2016 | Murine |
|
|
|
| Oosterling et al [92] | 2005 | Murine |
|
|
|
| Pinto et al [77] | 2019 | Human tissue |
|
|
|
| Umemura et al [93] | 2008 | Murine/murine cell line |
|
|
|
| Zhou et al [75] | 2010 | Human tissue |
|
|
|
Abbreviations: BRAF: B‐Raf proto‐oncogene; CCL: CC‐chemokine ligand; CCR: C‐C Motif Chemokine Receptor; CCR6: C‐C Motif Chemokine Receptor 6; CD: cluster of differentiation; CRC: colorectal cancer; CXCL: chemokine (C‐X‐C motif) ligand; FIZZ1: found in inflammatory zone 1; HIF: hypoxia‐inducible factor; HLA‐DR: human leukocyte antigen‐DR; IL: interleukin; iNOS: inducible nitric oxide synthase; KRAS: Kirsten rat sarcoma viral oncogene homolog gene; LLC: Lewis Lung Carcinoma; MGL: macrophage galactose‐type lectin‐1; MHC II: major histocompatibility complex II; RELM α; STAT3: Signal transducer and activator of transcription 3; TGF‐β1: Transforming growth factor beta 1; TNF: tumor‐necrosis factor; VEGF: vascular endothelial growth factor.
4. PERSPECTIVES FOR TARGETING TUMOR‐ASSOCIATED MACROPHAGES IN CLINICAL PRACTICE
4.1. Tumor‐associated macrophages as diagnostic markers
TAMs have the potential to be used as diagnostic and prognostic markers in CRC and possibly as therapeutic targets. Previous studies have shown that circulating TAMs and the chemokines that they produce could serve as markers in cancer diagnosis.(78, 79, 80) Current research has focused on the identification of circulating TAMs in blood samples by profiling their cell surface markers in different types of cancer as a basis for developing a noninvasive screening tool. Relevant markers are cluster of differentiation (CD) 14, CD163, CD68 or hypoxia‐inducible factor 2α (HIF‐2α).(78, 79, 80) A combination of analyzes of cell surface markers, cytokines secreted by TAMs and soluble factors produced by other cells within the TME could be useful to determine specific cell expression profiles in CRC. Serum levels of neutrophil elastase within the TME have been shown to play a potential role as a diagnostic biomarker in CRC.(81) While serum matrix metalloproteinase‐9 (MMP‐9) was not considered to be an appropriate screening parameter for CRC,(82) tissue inhibitor of metalloproteinase‐1 (TIMP‐1) seems to have a potential diagnostic value.(83) Targeting related factors that are expressed by TAMs or neighboring cells within the TME and circulatory markers may further contribute to the overall diagnostic capacity.
4.2. Tumor‐associated macrophages as prognostic markers
Evidence for prognostic utility of markers in CRC, with a particular role of TAM phenotypes in different tumor stages, is currently evolving. The findings with respect to the association between specific TAM phenotypes and prognosis are inconclusive, suggesting a different role of TAMs during tumor progression. This could also be caused by the fact, that not only the total cell count of either M1 or M2 macrophages seems to be relevant for tumor progression, but also the distribution of these cells within the tumor environment.(73) A low density of TAMs in general, as investigated by CD68+ cell infiltration in tumor tissue, was associated with worse outcome in patients with different stages of CRC.(84) A high proportion of CD163+ macrophages was associated with lower tumor grade and less lymph node metastasis.(85) Other studies report advanced tumor stages and worse prognosis positively correlating with high TAM density.(77) An investigation of the prognostic effect of TAMs in patients with CRC undergoing postoperative chemotherapy recently revealed, that the CD206/CD68 ratio of TAMs can predict high risk of recurrence in patients with stage II colon cancer.(86) As adjuvant chemotherapy is not routinely recommended in these patients, identifying those patients with poor prognosis is leading to targeted and more accurate administration of chemotherapy. The presence of a high density of TAMs in primary tumor tissue and metastatic lymph nodes of stage III CRC can identify patients that benefit from 5‐fluorouracil.(87) In‐vitro results indicating synergistic effects of TAMs and fluoropyrimidines have, however, yet to be proven in an in‐vivo setting.(87)
Since different TAM phenotypes are associated with tumor behavior, the metabolic reprogramming of TAMs to an ‘antitumor’ phenotype is a major aim of ongoing research. In TAMs, the NF‐κB pathway is the main pathway for polarization into an antitumor phenotype. This pathway is affected by Toll‐like receptors, Dectin‐1 receptors and SIGN‐related 1 receptors.(88, 89) Activation of these receptors causes an adaptive immune response enhancing phagocytosis and the release of inflammatory cytokines, such as tumor‐necrosis factor α (TNFα), IL‐2, IL‐10, and IL‐12.(89) The yeast‐derived polysaccharide β‐glucan can act on these membrane receptors, thereby inducing macrophage polarization into a proinflammatory anticancer phenotype.(89) Apart from NF‐κB, other transcription factors can also be regulated to induce M1‐like polarization or to inhibit M2 polarization in macrophages, such as interferon‐regulatory factor (IRF), STAT protein, HIFα and several microRNAs.(54)
Pathways that are known to be involved in macrophage activation and reprogramming in the acute immune response could also play a role in a chronic inflammatory setting, consequently affecting the onset and development of CRC. Identifying inflammatory mediators in obesity that support the polarization of tumor‐promoting macrophages could not only help identify patients at high risk of CRC due to metabolic dysfunction, but also serve as a basis for targeting these mediators in patients with obesity or type 2 diabetes mellitus. The effects of obesity and its associated inflammatory stressors on macrophage polarization,
TAM metabolism and therefore tumor behavior in patients with CRC, need further elucidation.
5. CONCLUSIONS
Tumor‐promoting inflammation is one of the hallmarks of cancer and TAMs are able to orchestrate these mechanisms based on their cellular metabolism. Interactions between TAMs, tumor cells and other components within the TME regulate cancer establishment, tumor growth and metastasis. CRC is closely related to chronic tissue inflammation. Metabolic dysfunction in patients with obesity has the potential to induce reprogramming in TAMs through inflammatory mechanisms. The macrophage metabolite itaconate is produced during TAM polarization and it is known to have tumor promoting effects. Investigating the role of itaconate and other metabolites in TAMs can elucidate processes specific for the onset and progression of CRC on the basis of inflammatory pathways, particularly in early‐onset CRC. There is a potential to detect new diagnostic and prognostic targets for the improvement of neoadjuvant and/or adjuvant therapies in CRC.
CONFLICTS OF INTEREST
K.M.S., A.T.B., S.J.O., and S.G. declare no potential conflicts of interest.
AUTHOR CONTRIBUTION
K.M.S. and S.J.O. performed the literature search (data curation), K.M.S. and A.T.B. identified relevant studies (formal analysis), K.M.S. and S.G. wrote and edited the paper and all authors were involved in final draft changes. K.M.S. and S.G. developed the study and were in charge of overall direction and planning (project administration).
Scheurlen KM, Billeter AT, O'Brien SJ, Galandiuk S. Metabolic dysfunction and early‐onset colorectal cancer – how macrophages build the bridge. Cancer Med. 2020;9:6679–6693. 10.1002/cam4.3315
FUNDING INFORMATION
The Price Institute of Surgical Research, University of Louisville, is financially supported by the John W. Price and Barbara Thruston Atwood Price Trust. The funding sources had no role in the design and conduct of the study as well as on the collection, management, analysis, and interpretation of the data.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article.
REFERENCES
- 1. Brenner DR, Heer E, Sutherland RL, et al. National trends in colorectal cancer incidence among older and younger adults in Canada. JAMA Netw Open. 2019;2(7):e198090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ullah MF, Fleming CA, Mealy K. Changing trends in age and stage of colorectal cancer presentation in Ireland ‐ from the nineties to noughties and beyond. Surgeon. 2018;16(6):350–4. [DOI] [PubMed] [Google Scholar]
- 3. Vuik FE, Nieuwenburg SA, Bardou M, et al. Increasing incidence of colorectal cancer in young adults in Europe over the last 25 years. Gut. 2019;68(10):1820–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu PH, Wu K, Ng K, et al. Association of obesity with risk of early‐onset colorectal cancer among women. JAMA Oncol. 2019;5(1):37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hales CM, Fryar CD, Carroll MD, Freedman DS, Ogden CL. Trends in obesity and severe obesity prevalence in US youth and adults by sex and age, 2007–2008 to 2015–2016. JAMA. 2018;319(16):1723–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosato V, Bosetti C, Levi F, et al. Risk factors for young‐onset colorectal cancer. Cancer Cause Control. 2013;24(2):335–41. [DOI] [PubMed] [Google Scholar]
- 7. Kim JY, Jung YS, Park JH, et al. Different risk factors for advanced colorectal neoplasm in young adults. World J Gastroenterol. 2016;22(13):3611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hussan H, Patel A, Le Roux M, et al. Rising incidence of colorectal cancer in young adults corresponds with increasing surgical resections in obese patients. Clin Transl Gastroenterol. 2020;11(4):e00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rowan AJ, Lamlum H, Ilyas M, et al. APC mutations in sporadic colorectal tumors: a mutational "hotspot" and interdependence of the “two hits”. Proc Natl Acad Sci USA. 2000;97(7):3352–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kanth P, Grimmett J, Champine M, Burt R, Samadder NJ. Hereditary colorectal polyposis and cancer syndromes: a primer on diagnosis and management. Am J Gastroenterol. 2017;112(10):1509–25. [DOI] [PubMed] [Google Scholar]
- 11. Pearlman R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early‐onset colorectal cancer. JAMA Oncol. 2017;3(4):464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jess T, Frisch M, Simonsen J. Trends in overall and cause‐specific mortality among patients with inflammatory bowel disease from 1982 to 2010. Clin Gastroenterol Hepatol. 2013;11(1):43–8. [DOI] [PubMed] [Google Scholar]
- 13. Kanaan Z, Rai SN, Eichenberger MR, et al. Differential microRNA expression tracks neoplastic progression in inflammatory bowel disease‐associated colorectal cancer. Hum Mutat. 2012;33(3):551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gausman V, Dornblaser D, Anand S, et al. Risk factors associated with early‐onset colorectal cancer. Clin Gastroenterol Hepatol. 2019;S1542‐3565(19)31108‐5 10.1016/j.cgh.2019.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burn J, Gerdes AM, Macrae F, et al. Long‐term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378(9809):2081–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cea Soriano L, Soriano‐Gabarro M, Garcia Rodriguez LA. The protective effect of low‐dose aspirin against colorectal cancer is unlikely explained by selection bias: results from three different study designs in clinical practice. PLoS ONE. 2016;11(7):e0159179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garcia Rodriguez LA, Soriano‐Gabarro M, Bromley S, Lanas A, Cea Soriano L. New use of low‐dose aspirin and risk of colorectal cancer by stage at diagnosis: a nested case‐control study in UK general practice. BMC Cancer. 2017;17(1):637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yahaya MAF, Lila MAM, Ismail S, Zainol M, Afizan N. Tumour‐associated macrophages (TAMs) in colon cancer and how to reeducate them. J Immunol Res. 2019;2019:2368249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Viola A, Munari F, Sanchez‐Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. 2019;10:1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mazzone M, Menga A, Castegna A. Metabolism and TAM functions‐it takes two to tango. FEBS J. 2018;285(4):700–16. [DOI] [PubMed] [Google Scholar]
- 21. World Health Organization . Obesity and overweight. https://www.who.int/en/news‐room/fact‐sheets/detail/obesity‐and‐overweight. Accessed May 05, 2020
- 22. US Preventive Services Task Force , Bibbins‐Domingo K, Grossman DC, Curry SJ, et al. Screening for colorectal cancer: US preventive services task force recommendation statement. JAMA. 2016;315(23):2564–75. [DOI] [PubMed] [Google Scholar]
- 23. Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin. 2009;59(6):366–78. [DOI] [PubMed] [Google Scholar]
- 24. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66(4):683–91. [DOI] [PubMed] [Google Scholar]
- 25. Wolf AMD, Fontham ETH, Church TR, et al. Colorectal cancer screening for average‐risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J Clin. 2018;68(4):250–81. [DOI] [PubMed] [Google Scholar]
- 26. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–6. [DOI] [PubMed] [Google Scholar]
- 27. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma Y, Yang Y, Wang F, et al. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS ONE. 2013;8(1):e53916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moghaddam AA, Woodward M, Huxley R. Obesity and risk of colorectal cancer: a meta‐analysis of 31 studies with 70,000 events. Cancer Epidemiol Biomarkers Prev. 2007;16(12):2533–47. [DOI] [PubMed] [Google Scholar]
- 30. Peeters PJ, Bazelier MT, Leufkens HG, de Vries F, De Bruin ML. The risk of colorectal cancer in patients with type 2 diabetes: associations with treatment stage and obesity. Diabetes Care. 2015;38(3):495–502. [DOI] [PubMed] [Google Scholar]
- 31. de Kort S, Masclee AAM, Sanduleanu S, et al. Higher risk of colorectal cancer in patients with newly diagnosed diabetes mellitus before the age of colorectal cancer screening initiation. Sci Rep. 2017;7:46527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holowatyj AN, Gigic B, Herpel E, et al. Distinct molecular phenotype of sporadic colorectal cancers among young patients based on multiomics analysis. Gastroenterology. 2020;158(4):1155–1158.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89(6):2548–56. [DOI] [PubMed] [Google Scholar]
- 34. Bigornia SJ, Farb MG, Mott MM, et al. Relation of depot‐specific adipose inflammation to insulin resistance in human obesity. Nutr Diabetes. 2012;2:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. 2018;61(4):942–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosso C, Kazankov K, Younes R, et al. Crosstalk between adipose tissue insulin resistance and liver macrophages in non‐alcoholic fatty liver disease. J Hepatol. 2019;71(5):1012–21. [DOI] [PubMed] [Google Scholar]
- 37. Weiss JM, Davies LC, Karwan M, et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Invest. 2018;128(9):3794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bopanna S, Ananthakrishnan AN, Kedia S, Yajnik V, Ahuja V. Risk of colorectal cancer in Asian patients with ulcerative colitis: a systematic review and meta‐analysis. Lancet Gastroenterol Hepatol. 2017;2(4):269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta‐analysis. Gut. 2001;48(4):526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol. 2008;14(25):3937–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sheng H, Shao J, Williams CS, et al. Nuclear translocation of beta‐catenin in hereditary and carcinogen‐induced intestinal adenomas. Carcinogenesis. 1998;19(4):543–9. [DOI] [PubMed] [Google Scholar]
- 42. Rothwell PM, Wilson M, Elwin CE, et al. Long‐term effect of aspirin on colorectal cancer incidence and mortality: 20‐year follow‐up of five randomised trials. Lancet. 2010;376(9754):1741–50. [DOI] [PubMed] [Google Scholar]
- 43. Flossmann E, Rothwell PM; British Doctors Aspirin Trial and the UK‐TIA Aspirin Trial . Effect of aspirin on long‐term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369(9573):1603–13. [DOI] [PubMed] [Google Scholar]
- 44. Nan H, Hutter CM, Lin Y, et al. Association of aspirin and NSAID use with risk of colorectal cancer according to genetic variants. JAMA. 2015;313(11):1133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jacobs EJ, Thun MJ, Bain EB, Rodriguez C, Henley SJ, Calle EE. A large cohort study of long‐term daily use of adult‐strength aspirin and cancer incidence. J Natl Cancer Inst. 2007;99(8):608–15. [DOI] [PubMed] [Google Scholar]
- 46. Cole BF, Logan RF, Halabi S, et al. Aspirin for the chemoprevention of colorectal adenomas: meta‐analysis of the randomized trials. J Natl Cancer Inst. 2009;101(4):256–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Force USPST . Recommendation statement: Aspirin to prevent cardiovascular disease and cancer U.S. Preventive Services Task Force. 2015.
- 48. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer‐related inflammation. Nature. 2008;454(7203):436–44. [DOI] [PubMed] [Google Scholar]
- 49. Soncin I, Sheng J, Chen Q, et al. The tumour microenvironment creates a niche for the self‐renewal of tumour‐promoting macrophages in colon adenoma. Nat Commun. 2018;9(1):582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taniyama D, Taniyama K, Kuraoka K, et al. CD204‐positive tumor‐associated macrophages relate to malignant transformation of colorectal adenoma. Anticancer Res. 2019;39(6):2767–75. [DOI] [PubMed] [Google Scholar]
- 51. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–9. [DOI] [PubMed] [Google Scholar]
- 52. Erreni M, Mantovani A, Allavena P. Tumor‐associated macrophages (TAM) and inflammation in colorectal cancer. Cancer Microenviron. 2011;4(2):141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Italiani P, Mazza EM, Lucchesi D, et al. Transcriptomic profiling of the development of the inflammatory response in human monocytes in vitro. PLoS ONE. 2014;9(2):e87680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96. [DOI] [PubMed] [Google Scholar]
- 55. O'Neill LAJ, Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol. 2019;19(5):273–81. [DOI] [PubMed] [Google Scholar]
- 56. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 2015;36(4):229–39. [DOI] [PubMed] [Google Scholar]
- 57. Galvan‐Pena S, O'Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mehla K, Singh PK. Metabolic regulation of macrophage polarization in cancer. Trends Cancer. 2019;5(12):822–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen DP, Ning WR, Jiang ZZ, et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3‐PD‐L1 axis in human hepatocellular carcinoma. J Hepatol. 2019;71(2):333–43. [DOI] [PubMed] [Google Scholar]
- 60. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chun E, Lavoie S, Michaud M, et al. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid‐derived suppressor cell population and function. Cell Rep. 2015;12(2):244–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang H, Shao Q, Sun J, et al. Interactions between colon cancer cells and tumor‐infiltrated macrophages depending on cancer cell‐derived colony stimulating factor 1. Oncoimmunology. 2016;5(4):e1122157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee YS, Park MS, Choung JS, et al. Glucagon‐like peptide‐1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia. 2012;55(9):2456–68. [DOI] [PubMed] [Google Scholar]
- 64. Bailey C, Negus R, Morris A, et al. Chemokine expression is associated with the accumulation of tumour associated macrophages (TAMs) and progression in human colorectal cancer. Clin Exp Metastasis. 2007;24(2):121–30. [DOI] [PubMed] [Google Scholar]
- 65. Hu H, Sun L, Guo C, et al. Tumor cell‐microenvironment interaction models coupled with clinical validation reveal CCL2 and SNCG as two predictors of colorectal cancer hepatic metastasis. Clin Cancer Res. 2009;15(17):5485–93. [DOI] [PubMed] [Google Scholar]
- 66. Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour‐associated macrophages by tumour‐derived lactic acid. Nature. 2014;513(7519):559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Odegaard JI, Ricardo‐Gonzalez RR, Goforth MH, et al. Macrophage‐specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shiraishi D, Fujiwara Y, Komohara Y, Mizuta H, Takeya M. Glucagon‐like peptide‐1 (GLP‐1) induces M2 polarization of human macrophages via STAT3 activation. Biochem Biophys Res Commun. 2012;425(2):304–8. [DOI] [PubMed] [Google Scholar]
- 69. Herbeuval JP, Lelievre E, Lambert C, Dy M, Genin C. Recruitment of STAT3 for production of IL‐10 by colon carcinoma cells induced by macrophage‐derived IL‐6. J Immunol. 2004;172(7):4630–4636. [DOI] [PubMed] [Google Scholar]
- 70. Bollrath J, Phesse TJ, von Burstin VA, et al. gp130‐mediated Stat3 activation in enterocytes regulates cell survival and cell‐cycle progression during colitis‐associated tumorigenesis. Cancer Cell. 2009;15(2):91–102. [DOI] [PubMed] [Google Scholar]
- 71. Barbera‐Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW. Vascular endothelial growth factor secretion by tumor‐infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res. 2002;62(23):7042–9. [PubMed] [Google Scholar]
- 72. Zhang QW, Liu L, Gong CY, et al. Prognostic significance of tumor‐associated macrophages in solid tumor: a meta‐analysis of the literature. PLoS ONE. 2012;7(12):e50946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Edin S, Wikberg ML, Dahlin AM, et al. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS ONE. 2012;7(10):e47045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. 2007;13(5):1472–9. [DOI] [PubMed] [Google Scholar]
- 75. Zhou Q, Peng RQ, Wu XJ, et al. The density of macrophages in the invasive front is inversely correlated to liver metastasis in colon cancer. J Transl Med. 2010;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bacman D, Merkel S, Croner R, Papadopoulos T, Brueckl W, Dimmler A. TGF‐beta receptor 2 downregulation in tumour‐associated stroma worsens prognosis and high‐grade tumours show more tumour‐associated macrophages and lower TGF‐beta1 expression in colon carcinoma: a retrospective study. BMC Cancer. 2007;7:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pinto ML, Rios E, Duraes C, et al. The two faces of tumor‐associated macrophages and their clinical significance in colorectal cancer. Front Immunol. 2019;10:1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Andersen MN, Abildgaard N, Maniecki MB, Moller HJ, Andersen NF. Monocyte/macrophage‐derived soluble CD163: a novel biomarker in multiple myeloma. Eur J Haematol. 2014;93(1):41–7. [DOI] [PubMed] [Google Scholar]
- 79. Tang X. Tumor‐associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 2013;332(1):3–10. [DOI] [PubMed] [Google Scholar]
- 80. Yang L, Zhang Y. Tumor‐associated macrophages: from basic research to clinical application. J Hematol Oncol. 2017;10(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ho AS, Chen CH, Cheng CC, et al. Neutrophil elastase as a diagnostic marker and therapeutic target in colorectal cancers. Oncotarget. 2014;5(2):473–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Otero‐Estevez O, De Chiara L, Rodriguez‐Girondo M, et al. Serum matrix metalloproteinase‐9 in colorectal cancer family‐risk population screening. Sci Rep. 2015;5:13030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Meng C, Yin X, Liu J, Tang K, Tang H, Liao J. TIMP‐1 is a novel serum biomarker for the diagnosis of colorectal cancer: a meta‐analysis. PLoS ONE. 2018;13(11):e0207039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chaput N, Svrcek M, Auperin A, et al. Tumour‐infiltrating CD68+ and CD57+ cells predict patient outcome in stage II‐III colorectal cancer. Br J Cancer. 2013;109(4):1013–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Koelzer VH, Canonica K, Dawson H, et al. Phenotyping of tumor‐associated macrophages in colorectal cancer: Impact on single cell invasion (tumor budding) and clinicopathological outcome. Oncoimmunology. 2016;5(4):e1106677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Feng Q, Chang W, Mao Y, et al. Tumor‐associated macrophages as prognostic and predictive biomarkers for postoperative adjuvant chemotherapy in patients with stage II colon cancer. Clin Cancer Res. 2019;25(13):3896–907. [DOI] [PubMed] [Google Scholar]
- 87. Malesci A, Bianchi P, Celesti G, et al. Tumor‐associated macrophages and response to 5‐fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology. 2017;6(12):e1342918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Albeituni SH, Ding C, Liu M, et al. Yeast‐derived particulate beta‐glucan treatment subverts the suppression of myeloid‐derived suppressor cells (MDSC) by inducing polymorphonuclear MDSC apoptosis and monocytic MDSC differentiation to APC in cancer. J Immunol. 2016;196(5):2167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chan GC, Chan WK, Sze DM. The effects of beta‐glucan on human immune and cancer cells. J Hematol Oncol. 2009;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Deng L, Zhou JF, Sellers RS, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin‐dependent hyperproliferation of colonic epithelium to inflammation‐associated tumorigenesis. Am J Pathol. 2010;176(2):952–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nandi B, Shapiro M, Samur MK, et al. Stromal CCR6 drives tumor growth in a murine transplantable colon cancer through recruitment of tumor‐promoting macrophages. Oncoimmunology. 2016;5(8):e1189052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Oosterling SJ, van der Bij GJ, Meijer GA, et al. Macrophages direct tumour histology and clinical outcome in a colon cancer model. J Pathol. 2005;207(2):147–155. [DOI] [PubMed] [Google Scholar]
- 93. Umemura N, Saio M, Suwa T, et al. Tumor‐infiltrating myeloid‐derived suppressor cells are pleiotropic‐inflamed monocytes/macrophages that bear M1‐ and M2‐type characteristics. J Leukoc Biol. 2008;83(5):1136–1144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article.