

Abstract
Background and Purpose
Apolipoprotein A‐I (apoA‐I) mimetic peptides (AAMPs) are short peptides that can mimic the physiological effects of apoA‐I, including the suppression of atherosclerosis by reversely transporting peripheral cholesterol to the liver. As the hydrophobicity of apoA‐I is considered important for its lipid transport, novel AAMPs were designed and synthesized in this study by gradually increasing the hydrophobicity of the parent peptide, and their anti‐atherosclerotic effects were tested.
Experimental Approach
Seventeen new AAMPs (P1–P17) with incrementally increased hydrophobicity were designed and synthesized by replacing the amino acids 221–240 of apoA‐I (VLESFKVSFLSALEEYTKKL). Their effects on cholesterol efflux were evaluated. Their cytotoxicity and haemolytic activity were also measured. The in vitro mechanism of the action of the new peptides was explored. Adult apolipoprotein E−/− mice were used to evaluate the anti‐atherosclerotic activity of the best candidate, and the mechanistic basis of its anti‐atherosclerotic effects was explored.
Key Results
Seventeen new AAMPs (P1–P17) were synthesized, and their cholesterol efflux activity and cytotoxicity were closely related to their hydrophobicity. P12 (FLEKLKELLEHLKELLTKLL) was the best candidate and most strongly promoted cholesterol efflux among the non‐toxic peptides (P1–P12). With its phospholipid affinity, P12 facilitated cholesterol transport through the ATP‐binding cassette transporter A1. In vivo, P12 exhibited prominent anti‐atherosclerotic activity via coupling with HDL.
Conclusion and Implications
P12 featured adequate hydrophobicity, which ensured its efficient binding with cytomembrane phospholipids, cholesterol and HDL, and provided a basis for its ability to reversely transport cholesterol and treat atherosclerosis.
Keywords: apolipoprotein A‐I mimetic peptide, atherosclerosis, cholesterol, hydrophobicity, lipoprotein
Abbreviations
- ABCA1
ATP‐binding cassette transporter A1
- AAMP
ApoA‐I mimetic peptide
- ApoA‐I
apolipoprotein A‐I
- C‐TPP
5‐(4‐carboxyphenyl)‐10,15,20‐triphenylporphyrin
- DMPC
dimyristoyl phosphatidylcholine
- RCT
reverse cholesterol transport
- TC
total cholesterol
- TG
triglyceride
What is already known
Apolipoprotein A‐I (apoA‐1) mimetic peptides have emerged as potentially useful treatments for atherosclerosis.
What this study adds
The contribution of hydrophobicity to the activity of apoA‐I mimetic peptide was important.
The optimal apoA‐I mimetic peptide (P12) was identified.
What is the clinical significance
P12 is a promising candidate for development as a new atheroprotective agent for cardiovascular disease.
1. INTRODUCTION
Reverse cholesterol transport (RCT) has received increasing attention as an anti‐atherosclerotic strategy in recent decades (Lewis & Rader, 2005; Ohashi, Mu, Wang, Yao, & Chen, 2005). However, despite intense efforts, drugs specifically designed to stimulate RCT are barely available in the market (Reddy, Navab, Anantharamaiah, & Fogelman, 2014). In vivo, HDL carries out the RCT, thereby removing cholesterol from peripheral cells (Fisher, Feig, Hewing, Hazen, & Smith, 2012). In the formation of HDL, lipid‐free apolipoprotein A‐I (apoA‐I) is synthesized and secreted by the liver and, then, it binds phospholipids through interactions with the ATP‐binding cassette transporter protein A1 (ABCA1) and forms nascent disc‐shaped HDL (preβ‐HDL) particles. Preβ‐HDL particles constantly encapsulate peripheral intracellular cholesterol esters into its hydrophobic core to generate mature spherical HDL (α‐HDL) and have stronger cholesterol carrying capacity than mature α‐HDL particles. Eventually, cholesterol ester is removed from HDL and transported into hepatocytes via the scavenger receptor BI (SR‐BI) or transferred to LDL by cholesterol ester transfer protein. Thus, cholesterol is mobilized and transferred from the peripheral tissues to the liver through the RCT process (Khera & Rader, 2010).
ApoA‐I, a structural and functional protein of HDL, is the key protein that drives the RCT process (Mei & Atkinson, 2015). ApoA‐I consists of 243 amino acids and is divided into 10 amphipathic α‐helical fragments by proline (Leman, Maryanoff, & Ghadiri, 2014). These 10 fragments share three common structural features (Ingenito et al., 2010; Islam et al., 2018). (i) Their secondary structure is an amphiphilic α‐helix that exhibits, at the same time, a hydrophobic and a hydrophilic surface (ii) The connection points of amphiphilic surfaces are positively charged amino acid residues, such as Lys, Arg, and His. (iii) The middle of the hydrophilic surface contains negatively charged amino acid residues, such as Asp and Glu. These characteristics are closely related to its activity. ApoA‐I has high lipid affinity and markedly increases the efflux of cholesterol from peripheral cells. Unfortunately, the use of apoA‐I as a drug has been restricted by its high production cost and lack of oral bioavailability. Fortunately, shorter apoA‐I mimetic peptides (AAMPs) may represent alternative treatment options. Therefore, based on their sequence characteristics, many AAMPs with short sequences have been derived. The class A and amphipathic α‐helix peptide, 18A (DWLKAFYDKVAEKLKEAF), was designed to mimic apoA‐I, which has hydrophobic amino acids (W, L, A, F, Y, and V) on the hydrophobic face and hydrophilic amino acids (D, E, K, and R) on the hydrophilic face of its α‐helix, as well as a positively charged amino acid (K) at the hydrophobic/hydrophilic interface and a negatively charged amino acid (D or E) at the centre of the hydrophilic face. Peptide 18A was found to associate strongly with liposomes and displace apoA‐I from native HDL and apolipoprotein E (apoE) from native VLDL (Getz & Reardon, 2011). The derived peptide 5A (DWLKAFYDKVAEKLKEAF‐P‐DWAKAAYDKAAEKAKEAA), a proline‐linked dimer of 18A, promoted cholesterol efflux more strongly than 18A. This dimer readily complexed with phospholipids and formed an HDL‐like structure (Amar et al., 2010). Peptide 4F (Ac‐DWFKAFYDKVAEKFKEAF‐NH2), which was produced by replacing the leucine of 18A at Sites 3 and 14 with phenylalanine, is the most representative AAMP. L‐4F (l‐amino acid) and D‐4F (enantiomer of L‐4F) were revealed to have good cholesterol efflux activity in vitro and in vivo. In clinical research, oral and intraperitoneal administration of peptide 4F reduced evolving atherosclerotic lesions, plaque lipids and macrophage activity in vein grafts, but this peptide did not affect established lesions in the aortic sinus. In addition, the doses required and cost of production were major issues in the project (Navab et al., 2002; Navab et al., 2011; Osei‐Hwedieh, Amar, Sviridov, & Remaley, 2011). These AAMPs feature the three common sequence–structure characteristics of the 10 fragments of apoA‐I, as already mentioned.
Regarding the anti‐atherosclerotic mechanism of AAMPs, after binding to HDL, the peptides promote RCT from arteriosclerotic plaques to the liver. AAMPs must have a strong affinity for phospholipids and cholesterol in HDL or cells to transport cholesterol (Stoekenbroek, Stroes, & Hovingh, 2015). Cholesterol efflux is a rate‐limiting step in the process of RCT. The factors affecting cholesterol efflux induced by AAMPs are hydrophobicity, charge, and secondary structure (Leman et al., 2014). Among them, hydrophobicity is the most important and fundamental factor that can both affect the solubility and absorption of the peptide and directly determine its lipid affinity and secondary structure (Sviridov et al., 2013). Adequate hydrophobicity of AAMPs can promote binding between AAMPs and ABCA1 for membrane phospholipid efflux to form preβ‐HDL and induce cellular cholesterol efflux (D'Souza et al., 2010). In short, hydrophobicity has a considerable influence on cholesterol efflux induced by AAMPs, highlighting the need for detailed studies of this variable.
Chroni et al. (2003) found that amino acids 220–231 of apoA‐I form the binding domain for ABCA1. The 10th helix of apoA‐I (containing amino acids 220–231) is the least conserved but most indispensable sequence (Palgunachari et al., 1996; Panagotopulos et al., 2002). Truncation mutants of apoA‐I lacking the 10th helix do not promote cholesterol efflux (Panagotopulos et al., 2002). In the present study, based on apoA‐I221–240, which comprises the active fragment of the apoA‐I C‐terminal (Wool, Reardon, & Getz, 2014), 17 new AAMPs were designed (P1–P17) and synthesized by gradually increasing their hydrophobicity through amino acid replacement. The relationships of hydrophobicity with the secondary structures, anti‐atherosclerotic activity, and toxicity of the new mimetic peptides were evaluated. Of these, P12, a lipid‐free 20‐amino acid AAMP, featuring an amphiphilic α‐helix structure and the best biological function of apoA‐I, was found to have the greatest effects on cholesterol efflux. The anti‐atherosclerotic mechanisms and effect of this candidate AAMP were elucidated in apoE−/− mice.
2. METHODS
2.1. Peptide and fluorescent probe synthesis
ApoA‐I221–240, 17 novel AAMPs (P1–P17), L‐4F, and 5‐(4‐carboxyphenyl)‐10,15,20‐triphenylporphyrin (C‐TPP)‐labelled P12 were synthesized using Wang resin and N‐9‐fluorenyl methoxycarbonyl (Fmoc) based on solid‐phase peptide synthesis. The peptide extension reaction conditions were as follows: threefold excess of 2‐(1H‐benzotriazol‐1‐yl)‐1,1,3,3‐tetramethyluronium hexafluorophosphate (HBTU)/hydroxybenzotriazole (HOBt) as the coupling reagents, N‐dimethylformamide as a solvent, sixfold excess of diisopropylethylamine, and threefold excess of Fmoc protecting group amino acids. The cleavage of the peptide from the resin was performed using a reagent composed of 95% trifluoroacetic acid, 2.5% triisopropylsilane, and 2.5% deionized water (3 h, room temperature). The peptide was precipitated in diethyl ether (1:1, volume ratio).
C‐TPP and C‐TPP‐P12 were generated as described previously (Ushasri et al., 2014). Briefly, 4‐carboxybenzaldehyde and benzaldehyde were mixed with propionic acid. The reaction mixture was heated to 120°C and pyrrole was added dropwise. The obtained solid material was subjected to silica gel column chromatography and eluted with CH2Cl2/CH3OH to obtain pure C‐TPP. Then, 1 equivalent of P12 that was not cleaved from the resin, 10 equivalents of C‐TPP, 10 equivalents of HBTU, and 10 equivalents of HOBt were dissolved in dimethylformamide separately and mixed. This mixture was added to the resin, and 10 equivalents of diisopropylethylamine were added to start the coupling reaction. After 24 h, the resin was dried, and the cleavage of peptide from the resin was achieved by treatment using a solution containing trifluoroacetic acid at room temperature for 10 h (Figures S1 and S2).
The obtained crude peptides were purified by reversed‐phase HPLC (Waters, MA, USA) on a C18 column (19 × 250 mm). The hydrophobicity and purity of the peptides were analysed using reversed‐phase HPLC on a C18 column (4.6 × 250 mm). All peptides prepared were more than 95% pure. Their molecular masses were confirmed via electrospray ionization MS (MaXis 4G, Bruker, USA).
2.2. The design strategy of AAMPs
As shown in Table 1 and Figure 1, 17 novel AAMPs were designed and synthesized using apoA‐I221–240 (VLESFKVSFLSALEEYTKKL) as the parent peptide. To ensure alignment to the amphiphilic surface of apoA‐I221–240, L2, S4, F5, V7, S8, F9, L10, S11, L13, E15, Y16, and K19 of apoA‐I221–240 were replaced with V2, K4, A5, E7, A8, A9, E10, H11, K13, A15, A16, and A19 respectively. Then, the amino acids on the hydrophilic surface of the new peptides were confirmed, The template of the novel AAMPs was X1X2EKX5KEX8X9EHX12KEX15X16TKX19X20 (X represents hydrophobic amino acids), and the hydrophobicity of P1–P17 was gradually increased by replacing the hydrophobic amino acid with Ala, Leu, Val, Phe, or Trp. Hydrophobicity and the hydrophobic moment were calculated using HeliQuest online software.
TABLE 1.
Amino acid sequences and α‐helix content of human apolipoprotein A1 (apoA‐I) fragments and apoA‐I mimetic peptides.
| α‐helix content, % | |||
|---|---|---|---|
| Peptide | Sequence | in H2O | in 50%TFE |
| apoA‐I221–240 | H‐VLESFKVSFLSALEEYTKKL‐OH | 18.97 | 46.62 |
| P1 |

|
10.11 | 65.87 |
| P2 |

|
8.15 | 59.60 |
| P3 |

|
7.11 | 61.11 |
| P4 |

|
6.87 | 59.25 |
| P5 |

|
5.67 | 67.23 |
| P6 |

|
‐0.41 | 72.80 |
| P7 |

|
3.55 | 68.64 |
| P8 |

|
9.71 | 61.36 |
| P9 |

|
9.24 | 61.45 |
| P10 |

|
17.42 | 63.11 |
| P11 |

|
59.23 | 63.85 |
| P12 |

|
70.23 | 62.34 |
| P13 |

|
75.16 | 91.96 |
| P14 |

|
75.84 | 74.80 |
| P15 |

|
30.27 | 42.62 |
| P16 |

|
40.01 | 15.36 |
| P17 |

|
30.78 | 16.41 |
| L‐4F a | Ac‐DWFKAFYDKVAEKFKEAF‐NH2 | 52.77 | 66.25 |
L‐4F was used as a positive control. The amino acids that were substituted in the parent peptide apoA‐I221–240, are shown in bold. The letters in the box represent the unchanged amino acid residues of the hydrophilic face. apoA‐I, apolipoprotein A1; TFE, trifluoroethanol.
FIGURE 1.
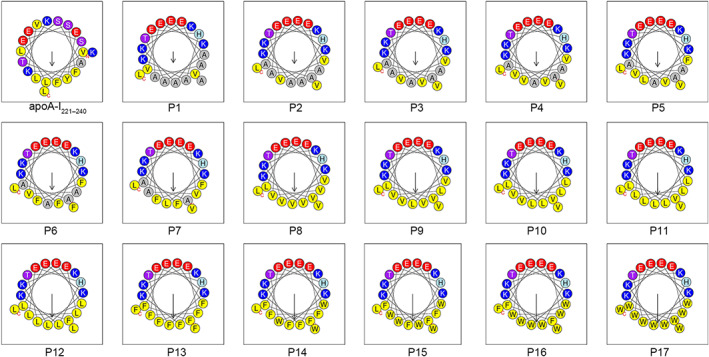
The designed peptides (P1–P17) with cholesterol efflux activities possess an amphipathic α‐helix structure (figures were exported from HeliQuest online software [https://heliquest.ipmc.cnrs.fr/]). The design strategy was as follows: First, the hydrophobic amino acids on the hydrophilic surface of apoA‐I221–240 were replaced with hydrophilic amino acids, and the hydrophilic amino acids on the hydrophobic surface of apoA‐I221–240 were replaced with hydrophobic amino acids. Subsequently, to facilitate the study of the effect of hydrophobicity on the amphiphilic apoA‐I mimetic peptides, the substituted amino acids on the hydrophilic surface were fixed, and the hydrophobic amino acids on the hydrophobic surface were gradually replaced with more hydrophobic amino acids. The difference in the hydrophobicity value between two adjacent numbered peptides was approximately 0.05
2.3. Circular dichroism measurements
To study the relationship between the secondary structures and cholesterol efflux activity of the new peptides, the circular dichroism (CD) spectra of the peptides were measured at 25°C on a J‐810 spectrometer (Jasco, Japan) using a quartz cell with a 1.0‐mm path length. Recording was performed over the range of 190–260 nm with a scanning speed of 50 nm·min−1, bandwidth of 1 nm, and response time of 1 s. The peptide solutions were prepared at a final concentration of 50 μM in deionized water (mimicking an aqueous environment) and 50% trifluoroethanol (TFE, mimicking the hydrophobic environment of the biological membrane). The acquired CD spectra were then converted to the mean residue ellipticity using the following equation:
where θ M is the residue ellipticity (deg·cm2·dmol−1), θ obs is the observed ellipticity corrected for the buffer at a given wavelength (mdeg), c is the peptide concentration (mM), l is the path length (mm), and n is the number of amino acids.
2.4. Dimyristoyl phosphatidylcholine vesicle solubilization assay
Dimyristoyl phosphatidylcholine (DMPC) vesicles (1 mg·ml−1) were prepared by resuspending DMPC in 75°C PBS (pH = 7.4) and vortexing. Then, 5 μl of 1‐mM AAMP in PBS (pH = 7.4) was added to 96‐well plates, followed by 95 μl of 1 mg·ml−1 of DMPC vesicles in PBS. The working concentration of the peptide was 50 μM. Changes in light scattering upon peptide addition were monitored at 23.9°C every 15 s for 90 min at 430 nm, with shaking in a multimode plate reader. Each sample had five parallel duplicates, and the test was repeated in five to six independent experiments.
2.5. Cell culture
RAW264.7 cells (RRID:CVCL_0493) were cultured in DMEM supplemented with 10% FBS, and THP‐1 cells (RRID:CVCL_0006) were cultured in RPMI‐1640 medium supplemented with 10% FBS. Cells were maintained at 37°C in 5% CO2.
2.6. Determination of cellular cholesterol efflux
The cholesterol efflux activity of the new peptides was determined using a previously described method with modifications (Avdulov, Chochina, Igbavboa, & Wood, 2000; Zhang et al., 2017). Briefly, RAW264.7 (4 × 104 per well) or THP‐1 (1 × 104 per well) cells were labelled with 22‐NBD‐cholesterol (2.5 mg·L−1) for 24 h in serum‐free medium, and then cellular cholesterol efflux was examined in the presence of 0‐, 1‐, 2‐, 5‐, 10‐, 20‐, 50‐, 100‐, or 200‐μM AAMPs for 12 h in serum‐free medium. Cells were lysed with 2% Triton X‐100. The fluorescence intensity (FI; Ex = 469 nm, Em = 538 nm) of 22‐NBD‐cholesterol in medium and cell lysates was monitored using a multimode reader. The cholesterol efflux rate was calculated as (FImedium)∕(FImedium + FIcell) × 100%. Each sample had five parallel duplicates, and the test was repeated in five to six independent experiments.
For the further determination of cholesterol efflux using flow cytometry, RAW264.7 and THP‐1 cells were labelled with 22‐NBD‐cholesterol (2.5 mg·L−1) for 24 h in 24‐well plates, and cellular cholesterol efflux was induced by incubation with apoA‐I221–240 (10 μM), L‐4F (10 μM), or P12 (1, 10, and 20 μM) for 12 h. The uptake and efflux of 22‐NBD‐cholesterol (Ex = 488 nm, Em = 550 nm) in cells were determined using a flow cytometer (LSRFortessa, BD). Each sample had five parallel duplicates, and theo test was repeated in five to six independent experiments.
Finally, ABCA1‐dependent cellular cholesterol efflux stimulated by P12 was determined using two methods:
In the first method (Shimizu et al., 2015; Uehara et al., 2013), RAW264.7 or THP‐1 cells were labelled with 22‐NBD‐cholesterol (2.5 mg·L−1) for 12 h in serum‐free medium. Cells were washed with PBS (pH = 7.4). To promote the ABCA1 expression, the labelled cells were treated with 9‐cis‐retinoic acid (retinoid X receptor ligand) and TO‐901317 (liver X receptor agonist) for 10 h in serum‐free medium (each 5 μM). To suppress ABCA1 function, cells were treated with 20‐μM probucol for 2 h at 37°C. Intracellular cholesterol efflux was induced by treatment with P12 (1 and 10 μM) for 4 h, and L‐4F was used as a positive control. FI of 22‐NBD‐cholesterol in medium and cell lysates was monitored using a multimode reader.
In the second method (Carballo‐Jane et al., 2010), RAW264.7 or THP‐1 cells were labelled with 22‐NBD‐cholesterol (2.5 mg·L−1) for 24 h in serum‐free medium, and then cellular cholesterol efflux was induced by exposure to P12 (0, 1, 5, 10, and 20 μM) while providing energy for ABCA1 activity using cAMP (0.3 mM) for 12 h in serum‐free medium. L‐4F was used as a positive control. FI of 22‐NBD‐cholesterol in medium and cell lysates was monitored using a multimode reader.
2.7. Cytotoxicity assay and haemolytic activity
RAW264.7 cells were seeded on 96‐well plates at a density of 1 × 104 per well. After incubation for 6 h to allow adherence, 100 μl of AAMP (0, 1, 2, 5, 10, 20, 50, 100, or 200 μM) in serum‐free medium was added to the plates for 12 h, after which 10 μl of MTT (50 mg·ml−1 in water) was added. After 4 h, the formazan in the cells was dissolved in DMSO. The absorbance was measured using a multimode reader at 490 nm. Each sample had five parallel duplicates, and the test was repeated in five to six independent experiments.
The venous blood of C57BL/6J mice (male, 20 ± 2 g) was collected from the venous plexus of the fundus and centrifuged at 1,000 g for 10 min. The plasma supernatant was removed, and 8% erythrocyte suspension was prepared with PBS (pH = 7.4). Then, 100 μl of 8% erythrocyte suspension and 100 μl of AAMPs (0, 1, 2, 5, 10, 20, 50, 100, or 200 μM) in PBS were added to the 96‐well plates, and 2% Triton X‐100 was used as a positive control. The plates were incubated at 37°C for 1 h and then centrifuged at 12,000 g for 15 min. Plasma supernatants (150 μl) were transferred to new 96‐well plates. The absorbance was measured using a multimode reader at 490 nm. Each sample had five parallel duplicates, and the test was repeated in five to six independent experiments.
2.8. Localization of P12 and 22‐NBD‐cholesterol in macrophages
As described previously, THP‐1 and RAW264.7 cells were loaded with 22‐NBD‐cholesterol (2.5 mg·L−1) for 24 h in laser scanning confocal microscope (LSCM) dishes, and cellular cholesterol efflux was induced by exposure to C‐TPP‐P12 for 12 h. The fluorescent images of cells were photographed in track 1 (22‐NBD‐cholesterol, Ex = 488 nm, Em = 500–550 nm, green), track 2 (C‐TPP‐P12, Ex = 515 nm, Em = 640–730 nm, red), and the bright‐field via LSCM (LSM710, Zeiss).
2.9. Animal experiments
2.9.1. Animal management
All animal care and experimental procedures were performed according to protocols approved by the Animal Care and Use Committee of Lanzhou University. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020).
Mice were purchased from Beijing Charles River Laboratories, maintained in a temperature‐ and light‐controlled facility, and fed with specific pathogen‐free chow. ApoE−/− mice and background wild‐type C57BL/6J mice (RRID:IMSR_JAX:000664; 20 ± 1 g, 10 weeks) were used for the in vivo experiments. Background wild‐type C57BL/6J mice (n = 10, 5 females and 5 males) were fed with a commercial basal diet, and apoE−/− mice (n = 60, 30 females and 30 males) were fed with a high‐fat diet (0.5% cholesterol and 10% fat). After 4 weeks, apoE−/− mice were divided into six groups. Low and high doses (10 and 20 mg·kg−1, respectively) of P12 were injected intraperitoneally into mice three times per week for 12 weeks. Saline, L‐4F (10 mg·kg−1), and apoA‐I221–240 (10 mg·kg−1) were used as background, positive, and negative controls respectively. After 16 weeks, mice were anaesthetized with isoflurane and killed via cervical dislocation, after an overnight fast. Plasma samples, organs, and tissues were then collected.
Other batches of wild‐type C57BL/6J (n = 20, male) and apoE−/− (n = 20, male) mice were fed using the aforementioned method for the following four supplementary experiments: the determination of plasma apoA‐I levels after P12 injection, plasma clearance of P12, the effects of 1 week of P12 exposure on blood lipid levels, and the effect of 2 weeks of P12 on cholesterol excretion (five C57BL/6J and five apoE−/− mice for each experiment).
In all experiments, animals were randomly assigned to treatment groups. The numbers of mice in each group are designated in the individual figures. The sample size in each group was determined according to previous studies with similar experimental protocols. The studies were blinded for treatment assignment and outcome assessment.
2.9.2. Histopathological examination
Paraffinized sections of the aortic arch were stained with haematoxylin and eosin. The images were captured using a CX21 microscope (Olympus, Japan). The atherosclerotic lipid plaque and relative thickness of the arterial wall were quantified via computer image analysis using Adobe Photoshop CC 2017 software (RRID:SCR_014199).
2.9.3. Serum lipid profiles, turbidity, inflammatory factors, and lipid peroxidation
Serum levels of total cholesterol (TC), LDL‐C, HDL‐C, triglyceride (TG), and C‐reactive protein (CRP) were determined using biochemical kits according to the manufacturer's instructions on an automatic biochemical analyzer (Hitachi, Japan). Serum IL‐6, the chemokine CCL2, and TNF‐α levels were measured using elisa assay kits according to the manufacturer's protocol. Serum turbidity was represented as the absorbance as measured using a multimode reader at 430 nm. Serum malondialdehyde (MDA) and SOD levels were determined in all mice using biochemical kits according to the manufacturer's instructions.
2.9.4. Plasma clearance of P12 in C57BL/6J and apoE−/− mice
Both wild‐type C57BL/6J (n = 5) and apoE−/− (n = 5) mice were intravenously injected with 10 mg·kg−1 of P12. P12 levels in plasma were measured using HPLC at 0, 1, 5, 10, 15, 30, 60, 90, 120, 180, 240, 360, 480, 720, 1,440, and 2,880 min after injection. The half‐life (t 1/2) of P12 in plasma was calculated in C57BL/6J and apoE−/− mice using Microsoft Excel 2019 (Microsoft Excel, RRID:SCR_016137).
2.9.5. Effect of P12 on plasma apoA‐I levels
Plasma apoA‐I levels in wild‐type C57BL/6J (n = 5) and apoE−/− (n = 5) mice were measured before and after the intravenous injection of P12 (10 mg·kg−1) using an apoA‐I assay kit.
2.9.6. Regulatory effect of 1 week of P12 exposure on blood lipids
Five new male apoE−/− mice were maintained on their diets for 1 week after 4 months of feeding with the high‐fat diets. Five new male wild‐type C57BL/6J mice were used as the control group. They received intraperitoneal injections of P12 (10 mg·kg−1·day−1). Serum TC, LDL‐C, HDL‐C, and non‐HDL‐C levels were measured in both mouse groups before and after daily P12 administration.
2.9.7. Effect of P12 on cholesterol excretion for 2 weeks
After 4 months of feeding, an additional five male apoE−/− mice fed with the high‐fat diet and five male wild‐type C57BL/6J mice fed with basal diet, were maintained on their respective diets for two additional weeks. In the first week, faeces were collected from mice in each group. In the second week, mice received intraperitoneal injections of P12 (10 mg·kg−1·day−1). Faeces were collected for measuring of cholesterol levels. To extract the cholesterol, 1 g of faeces powder was soaked in 5 ml of chloroform and shaken 10 min on a vortex mixer. After filtering samples, non‐cholesterol impurities in chloroform were extracted using water, and cholesterol in chloroform was measured using a cholesterol kit according to the manufacturer's instructions.
2.10. In vivo mechanism experiments
2.10.1. Lipoprotein analysis by size exclusion chromatography
Fresh blood was drawn from apoE−/− mice into tubes containing potassium EDTA, and plasma was separated immediately by centrifugation at 500 g for 30 min at 4°C. In total, 5 μl of peptides (P12 and C‐TPP‐P12, final concentration = 0.25 mg·ml−1) was mixed with 45 μl of mouse plasma and incubated at 37°C for 2 h. Size exclusion chromatography was performed at ambient temperature using a Superose 6 column (10 × 1,000 mm) on a semi‐preparative HPLC system (Waters). A 200‐μl aliquot of the peptide/plasma mixture was injected per run and eluted in PBS (pH = 7.4) with 1‐mM EDTA at a flow rate of 1 ml·min−1. A total of 60 fractions (1.5 ml per fraction) were directly collected into microtitre plates for further analysis. The TC content in each fraction was measured using a cholesterol kit according to the manufacturer's instructions, and FI represented the C‐TPP‐P12 concentration in each fraction. Saline was used as acontrol.
2.10.2. Western blot quantification of mouse preβ‐HDL levels
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. Peptide solution (5 μl) was mixed with 45 μl of apoE−/− mouse plasma, incubated at 37°C with shaking at 300 r.p.m. for 2 h, and then subjected to one‐dimensional, non‐reducing, anti‐mouse apoA‐I Western blotting. Briefly, samples were diluted 1:1 with Tris‐glycine native sample buffer and 4–20% native polyacrylamide gel electrophoresis as performed at 90 V for 10 min and at 150 V for 1 h. Proteins were transferred to a PVDF membrane (0.45 μm) overnight (200 mA at 4°C). After transfer, the membrane was blocked with 6% lipid‐free milk and washed three times with Tris‐buffered saline with Tween 20 (10‐mM Tris, pH 7.4, 150‐mM NaCl, and 0.1% Tween 20). Rabbit anti‐mouse apoA‐I antibody (1:500) and HRP‐conjugated goat anti‐rabbit antibody (1:1,000) were used as the primary and secondary antibodies respectively. Blots were developed using the HRP substrate luminol reagent and scanned using a Typhoon 9400 Imager (GE Healthcare, Piscataway, NJ, USA). Preβ‐HDL was identified on the basis of its electrophoretic mobility, and the band was quantified using Adobe Photoshop CC 2017 software (Adobe Photoshop, RRID:SCR_014199). The results were expressed as the grey value representing the signal intensity of the band.
2.10. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. All experiments were randomized and blinded. The sample size in each group was determined based on our previous studies with similar experimental protocols. At least five samples per group (n = 5) were included in the statistical analysis, where n is the number of independent values. Data for animals in which relative arterial wall thickness could not be measured because of the rupture of arterial plaque was excluded from the analysis. A normal probability plot was used to examine data distributions. Statistical analyses were performed using GraphPad Prism 5 software (GraphPad Prism, RRID:SCR_002798), and data are presented as mean ± SEM unless otherwise stated. Student's t test was used for comparisons between two groups. For comparisons of three or more groups, one‐way ANOVA followed by Dunnett's post hoc test was used for normal distributions with one variable. The significance level was set at P < .05.
2.11. Materials
22‐NBD‐cholesterol, DMEM, RPMI‐1640, and FBS were purchased from Thermo Fisher Scientific (MA, USA). Serum TC, LDL‐C, HDL‐C, TG, and CRP assay kits were purchased from Maccura Biotechnology Co., Ltd. (Sichuan, China). IL‐6, MCP‐1, and TNF‐α assay kits were purchased from Cloud‐Clone Corp (Beijing, China). 9‐cis‐Retinoic acid, TO‐901317, cAMP, MDA, and SOD were purchased from Sigma‐Aldrich (St. Louis, MO, USA). A cholesterol assay kit was purchased from Beijing Apllygen Gene Technology Co. Ltd. (Beijing, China). Rabbit anti‐mouse apoA‐I antibody (Proteintech Cat# 14427‐1‐AP, RRID:AB_2056524) and HRP‐conjugated goat anti‐rabbit antibody (Proteintech Cat# SA00001‐2, RRID:AB_2722564) were purchased from Proteintech Group, Inc. (IL, USA).
2.12. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY (http://www.guidetopharmacology.org) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Cidlowski et al., 2019; Alexander, Kelly et al., 2019).
3. RESULTS
3.1. Secondary structure and retention time was influenced by hydrophobicity
Changes in the secondary structures of the new AAMPs were observed with the increases in hydrophobicity. In aqueous medium, apoA‐I221–240 and P1–P10 did not form α‐helices, whereas L‐4F and P11–P14 had stable α‐helical conformations. Interestingly, P15–P17 had β‐sheet conformations. In the simulated cell membrane environment of 50% TFE, apoA‐I221–240, L‐4F, and P1–P14 had α‐helical structures, whereas P15–P17 had α‐helical and β‐sheet structures (Figures 2a,b and S3).
FIGURE 2.
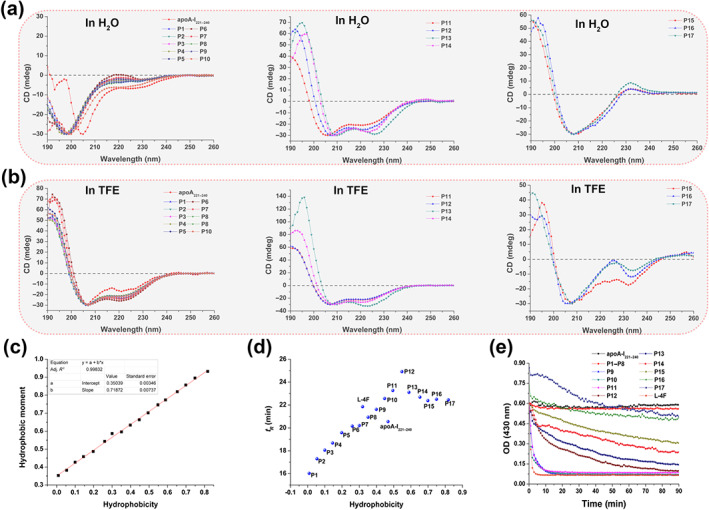
Characterization of circular dichroism (CD) spectroscopy, hydrophobicity, retention time (t R), and lipid affinity of the new peptides. CD of apoA‐I221–240 and P1–P17 (a) in H2O and (b) in 50% trifluoroethanol (TFE). P11–P14 always maintained an α‐helical structure in aqueous and simulated biofilm environment. (c) Hydrophobicity and hydrophobic moment of P1–P17 have a linear relationship (hydrophobic moment = 0.7187 hydrophobicity + 0.3504, R 2 = 0.998). These data were calculated using HeliQuest online software (https://heliquest.ipmc.cnrs.fr/). (d) Effects of hydrophobicity on t R as determined using reversed‐phase HPLC. As the hydrophobicity increased, t R of P1–P12 increased gradually, whereas the opposite pattern was observed for P12–P17. (e) Lipid affinity evaluation of the peptides. Dimyristoyl phosphatidylcholine (DMPC) vesicle solubilization is visually presented as changes in the OD of the mixed system of peptides (50 μM) and DMPC (1 mg·ml−1) at 430 nm. DMPC vesicles were solubilized by apoA‐I221–240, P1–P17, and L‐4F. Similar to apoA‐I221–240, P1–P8 did not solubilize DMPC vesicles. Surprisingly, P9–P11 and L‐4F made DMPC vesicles clear within 10 min. P12–P17 gradually and slowly induced DMPC vesicle solubilization. P12 induced DMPC vesicle solubilization within 90 min. P16 and P17 were precipitated in DMPC vesicles, and thus, their absorbance was initially greater than 0.6
The hydrophobicity and hydrophobic moment of P1–P17 had a linear relationship (Figure 2c). The retention time (t R) of these new peptides was measured using analytical HPLC. The results showed that t R of P1–P12 gradually increased, whereas those of P12–P17 gradually decreased. However, the theoretical value of hydrophobicity was increased from P1 to P17 (Figure 2d).
3.2. The lipid affinity of P12 ensures its cholesterol efflux activity
Macroscopically, the DMPC emulsion is dissolved by sufficiently hydrophobic AAMPs into a clear liquid. Of the studied AAMPs, apoA‐I221–240 and P1–P8 induced no changes of the DMPC emulsion within 90 min. However, P9–P11 and L‐4F quickly dissolved in DMPC within 10 min. Meanwhile, the emulsion became gradually clear in the presence of P12–P17, but the emulsion became completely clear after 90 min only following exposure to P12 (Figure 2e).
3.3. P12, a new peptide with excellent activity and low toxicity, was identified
In RAW264.7 cells, apoA‐I221–240 and P1–P5 did not induce cholesterol efflux, and their intracellular cholesterol efflux rates were less than 2% at concentrations of 1–200 μM. L‐4F and P6–P17 displayed concentration‐dependent cholesterol efflux activity. When the concentration exceeds the safe limit, P13–P17 could damage the membrane of RAW264.7 cells, leading to cholesterol leakage from cells and consequent changes of the dose–effect curves (Table 2 and Figures 3a, S4, and S5).
TABLE 2.
The cholesterol efflux activity (CE), cytotoxicity and haemolytic effects of human apoA‐I fragments and apoA‐I mimetic peptides (AAMPs)
| Peptide | CE (%; 10 μM), 12 h | Cell viability (%; 100 μM), 12 h | Haemolysis (%; 200 μM), 1 h |
|---|---|---|---|
| apoA‐I221–240 | 0.38 ± 0.24 | 84.27 ± 1.91 | 1.34 ± 0.66 |
| P1 | −1.17 ± 0.17 | 90.65 ± 4.93 | −1.84 ± 0.26 |
| P2 | 0.18 ± 0.13 | 89.40 ± 2.48 | 1.25 ± 0.65 |
| P3 | 0.91 ± 0.25 | 90.43 ± 3.40 | −0.28 ± 0.49 |
| P4 | 1.45 ± 0.15 | 84.20 ± 4.40 | 0.99 ± 0.66 |
| P5 | −0.12 ± 0.17 | 93.98 ± 2.71 | 1.20 ± 0.78 |
| P6 | 1.41 ± 0.11 | 97.80 ± 5.20 | −1.78 ± 0.17 |
| P7 | 2.29 ± 0.17 | 85.60 ± 3.20 | −0.74 ± 0.41 |
| P8 | 1.82 ± 0.13 | 87.50 ± 1.60 | −0.52 ± 0.49 |
| P9 | 3.09 ± 0.12 | 89.40 ± 2.20 | −0.37 ± 0.87 |
| P10 | 3.14 ± 0.22 | 106.70 ± 4.80 | −0.41 ± 0.60 |
| P11 | 3.30 ± 0.26 | 95.30 ± 3.90 | 1.64 ± 0.78 |
| P12 | 3.39 ± 0.28 | 106.77 ± 5.07 | 1.71 ± 0.73 |
| P13 | 4.33 ± 0.38 | 68.20 ± 5.30 | 1.99 ± 0.67 |
| P14 | 2.49 ± 0.44 | 66.00 ± 3.00 | 2.63 ± 0.70 |
| P15 | 4.01 ± 0.31 | 52.41 ± 3.16 | 7.06 ± 0.86 |
| P16 | 9.75 ± 0.97 | 42.61 ± 4.00 | 5.94 ± 0.58 |
| P17 | 12.23 ± 0.63 | 36.62 ± 3.40 | 7.86 ± 0.64 |
| L‐4F a | 3.61 ± 0.19 | 47.07 ± 1.89 | 2.10 ± 0.48 |
Note: Data are presented as the mean ± SEM. More detailed data are presented in the Supporting Information.
Abbreviations: apoA‐I, apolipoprotein A1.
L‐4F was used as a positive control.
FIGURE 3.
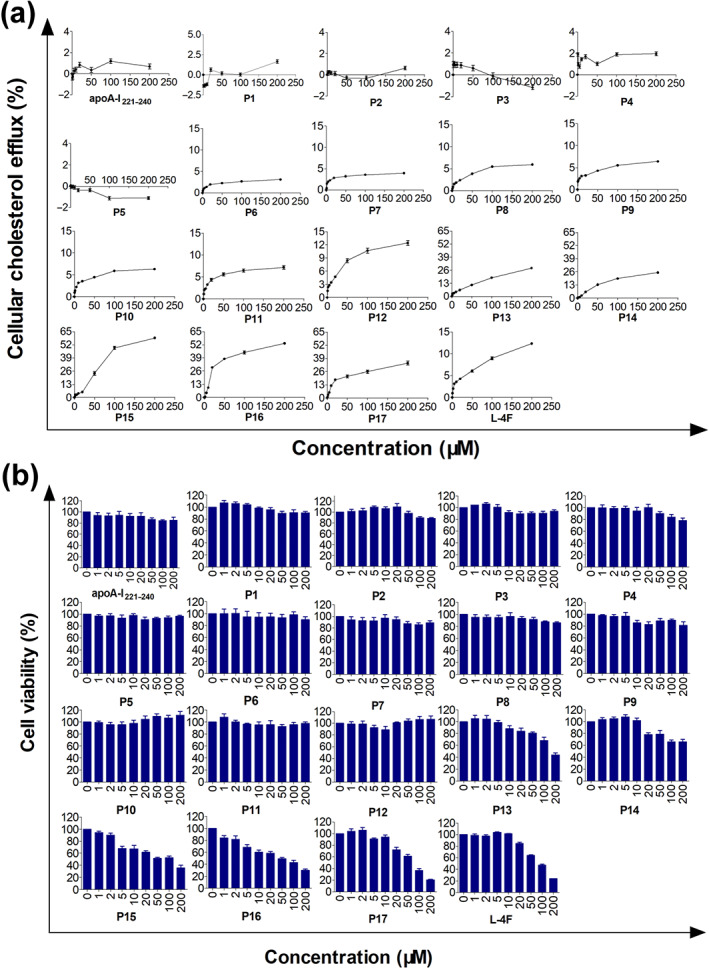
Cholesterol efflux activity and cytotoxicity of the parent peptide apoA‐I221–240, P1–P17, and L‐4F in RAW264.7 cells. (a) Cholesterol efflux activity of apoA‐I mimetic peptides in RAW264.7 cells after 12 h of treatment. ApoA‐I221–240 and P1–P5 had no concentration‐dependent cholesterol efflux activities. P6–P17 and L‐4F had concentration‐dependent cholesterol efflux activities. (b) The cytotoxicity of apoA‐I mimetic peptides in RAW264.7 cells after 12 h of treatment. Over a concentration range from 0‐200μM, the parent peptide ApoA‐I221–240 and P1–P12 showed no cytotoxicity, whereas P13–P17 and L‐4F induced cytotoxicity. n = 5. Each experiment was repeated independently five times
Then, the cytotoxicity of these new mimetic peptides was determined in RAW264.7 cells. The results illustrated that apoA‐I221–240 and P1–P12 had no cytotoxic effects. P13 had significant cytotoxicity at concentrations of 100 and 200 μM, resulting in cell viability rates of less than 80%. P14 and P15 exhibited cytotoxic effects at 20 and 5 μM respectively. P16, P17, and L‐4F had concentration‐dependent cytotoxic effects in RAW264.7 cells (Table 2 and Figure 3b).
The influence of new mimetic peptides on mouse erythrocytes in comparison with their parent peptide and L‐4F is presented in Table 2 and Figure S6. After 12 h, apoA‐I221–240, P1–P13, and L‐4F induced slight haemolysis, and the haemolytic rate was less than 2% at a concentration of less than 200 μM. P14 was haemolytic at concentrations exceeding 200 μM (haemolytic rate >2%). P15–P17 induced obvious haemolysis at 20 μM, and the haemolytic rate approached 8% at 200 μM. Based on these results, P12, with good activity and no toxicity, was screened for further study.
2.11. In vitro mechanism
2.11.1. P12 can promote cholesterol efflux in macrophages
P12 more strongly induced cholesterol efflux in THP‐1 cells than in RAW264.7 cells (Figure 4a). The results of cholesterol efflux determination using flow cytometry are shown in Figure 4b. Compared with the findings with the peptide PP (apoA‐I221–240), both P12 and L‐4F significantly promoted the efflux of intracellular cholesterol at 10 μM in both RAW264.7 (efflux rates of 3.2% and 4.56%, respectively) and THP‐1 cells (efflux rates of 5.5% and 9.89%, respectively). ApoA‐I221–240 did not display distinctive cholesterol efflux activity in either cell line (efflux rates of 0.18% and 0.59%, respectively, at 10 μM).
FIGURE 4.
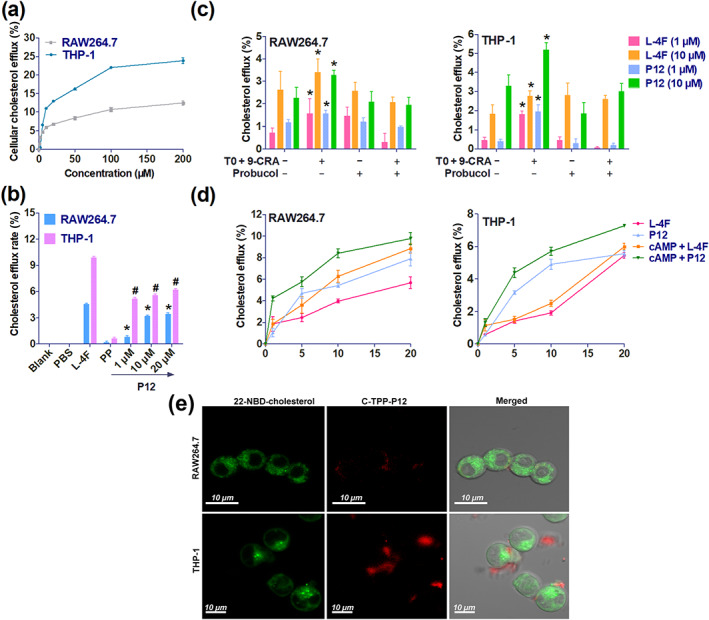
Comparison of cholesterol efflux activity of P12 in RAW264.7 and THP‐1 cells, ATP‐binding cassette transporter A1 (ABCA1)‐dependent cellular cholesterol efflux, and the action site of 5‐(4‐carboxyphenyl)‐10,15,20‐triphenylporphyrin (C‐TPP)‐labelled P12 in macrophages. (a) Cholesterol efflux rates induced by P12 in RAW264.7 and THP‐1 cells were measured using a multimode reader. (b) Cholesterol efflux induced by P12 (1, 10, and 20 μM) in 22‐NBD‐cholesterol‐labelled RAW264.7 and THP‐1 was measured using a flow cytometer. Blank represents unlabelled cells using 22‐NBD‐cholesterol. All peptides were dissolved in PBS. * P < 0.05, significantly different from PP (apoA‐I221–240) in RAW264.7 cells; # P < 0.05 significantly different from PP (apoA‐I221–240) in THP‐1 cells; one‐way ANOVA followed by Dunnett's post hoc test. (c) ABCA1‐dependent cellular cholesterol efflux induced by 5 μM of 9‐cis‐retinoic acid (9‐CRA) and TO‐901317 (TO) for 10 h. To suppress ABCA1, cells were treated with 20‐μM probucol for 2 h. * P < 0.05, significantly different from untreated cells by TO and 9‐CRA; Student's t test. (d) ABCA1‐dependent cellular cholesterol efflux induced by 0.3‐M cAMP 12 h. (e) Laser scanning confocal microscope images of C‐TPP‐P12 and 22‐NBD‐cholesterol in RAW264.7 and THP‐1 cells. C‐TPP‐P12 staining was observed outside the cell and on the cell membrane. Membrane C‐TPP‐P12 staining was stronger in THP‐1 cells than in RAW264.7 cells. Each data point represents the mean ± SEM. n = 5. Each experiment was repeated independently five times
2.11.2. P12 induced cholesterol efflux through ABCA1
After treatment with 9‐cis‐retinoic acid and TO‐901317, P12 increased cholesterol efflux in RAW264.7 cells by 0.40% at 1 μM and 1.02% at 10 μM. Meanwhile, probucol suppressed the cholesterol efflux induced by P12. Similar results were obtained for L‐4F. ABCA1‐dependent cellular cholesterol efflux was more strongly induced by 9‐cis‐retinoic acid and TO‐901317 in THP‐1 cells than in RAW264.7 cells. P12 increased cholesterol efflux by 1.53% at 1 μM and 1.92% at 10 μM in THP‐1 cells. Meanwhile, in the presence of probucol, P12‐induced cellular cholesterol efflux was reduced by 1.75% at 1 μM and 2.18% at 10 μM (Figure 4c).
In both RAW264.7 and THP‐1 cells, P12‐induced cholesterol efflux was obviously enhanced by treatment with cAMP. Unexpectedly, L‐4F was cytotoxic in THP‐1 cells, leading to cholesterol release at concentrations exceeding 10 μM (Figure 4d). Our data show that P12 had higher ABCA1‐dependent cholesterol efflux activity than L‐4F.
2.11.3. P12 promoted cholesterol efflux by acting on the cell membrane
To further confirm the site of action of P12, the fluorescence of 22‐NBD‐cholesterol and P12 labelled with C‐TPP was examined in RAW264.7 and THP‐1 cells. As shown in Figure 4e, 22‐NBD‐cholesterol had a strong fluorescent signal throughout RAW264.7 and THP‐1 cells excluding the nucleus at 24 h, but C‐TPP‐P12 fluorescence was only observed outside the cell or on the cell membrane. Moreover, THP‐1 cells exhibited much greater membrane fluorescence for C‐TPP‐P12 than RAW264.7 cells. This result was consistent with the results in Figure 4a,b.
3.4. Animal experiments
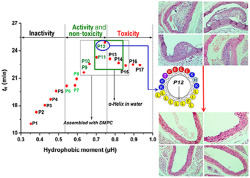
3.4.1. P12 suppressed the abnormal thickening of the arterial wall
The typical pathological characteristics of atherosclerosis were confirmed in apoE−/− mice (Figure S9). Data on the therapeutic effect of P12 in apoE−/− mice is presented in Figure 5a,b. The relative intimal thickening and plaque size of each sample were significantly decreased by P12. The relative thickness of the arterial wall in male and female apoE−/− mice was markedly reduced by L‐4F, their parent peptide (PP), and low and high doses of P12.
FIGURE 5.
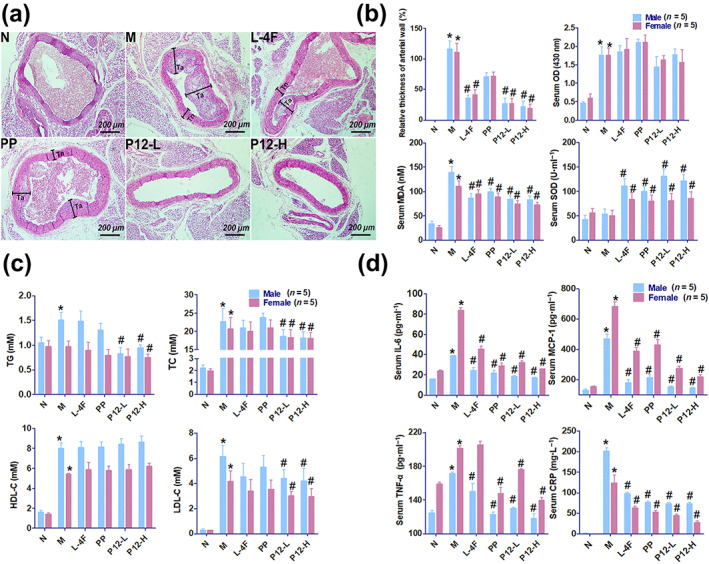
The arterial plaque‐reducing effect of P12 in apoE−/− mice and relative thickness of arterial wall (%), serum malondialdehyde (MDA), SOD, lipid (triacylglycerol [TG], total cholesterol [TC], HDL‐C, and LDL‐C), and inflammatory factor levels (IL‐6, CCL2, TNF‐α, and C‐reactive protein [CRP]), and serum turbidity (OD, OD430 nm) in mice. (a) The effects of P12 on atherosclerosis in apoE−/− mice (cross section of arteries, staining with haematoxylin and eosin, normal thickness of the arterial wall is indicated by black lines and Tn, and abnormal thickness of arterial wall is indicated by black lines and Ta). (b) Effects of P12 on the relative thickness of arterial wall and serum turbidity, MDA, and SOD levels. Relative thickness of the arterial wall (%) = (Ta − Tn)/Tn × 100%. Five mice were taken from each group, and three samples were taken from each mouse. Serum turbidity was presented as OD430 nm. The relative thickness of arterial wall, serum turbidity, and serum MDA levels were decreased by P12 treatment, whereas serum SOD level was increased. (c) Effects of P12 on serum lipid levels (TG, TC, HDL‐C, and LDL‐C). Serum TG, TC, and LDL‐C levels were decreased by P12. (d) Effects of P12 on serum inflammatory factor levels (IL‐6, CCL2, TNF‐α, and CRP). Serum IL‐6, CCL2, TNF‐α, and CRP levels were decreased by P12 treatment in apoE−/− mice. Each data point represents the mean ± SEM. * P < 0.05, significantly different from normal control. # P < 0.05, significantly different from model control; oOne‐way ANOVA followed by Dunnett's post hoc test. n = 10, including five male and five female mice. P12‐L and P12‐H indicate treatment with 10 and 20 mg·kg−1 P12 respectively. L‐4F, L‐4F (10 mg·kg−1) treatment group; M, model control group; N, normal control group; PP, parent peptide apoA‐I221–240 (10 mg·kg−1) treatment group
2.11.4. P12 has lipid‐lowering, anti‐inflammatory, and antioxidative effects in apoE−/− mice
Serum turbidity and MDA, TG, TC, HDL‐C, and LDL‐C levels were significantly higher in apoE−/− mice than in wild‐type mice after 4 months of feeding. P12 significantly prevented lipid accumulation and peroxidation in blood vessels. SOD levels were increased by P12 in apoE−/− mice. Serum HDL‐C levels were higher in untreated apoE−/− mice than in wild‐type mice after 4 months. HDL‐C levels were increased by high doses of P12 in apoE−/− mice. TC, TG, and LDL‐C levels were dramatically reduced by P12 in apoE−/− mice. After 4 months of feeding, the gender differences in serum lipids (TG, TC, HDL‐C, and LDL‐C) were obvious. Serum lipids levels were lower in female mice than in male mice. Serum TG and TC levels were more strongly lowered by P12 in male mice than in female mice. P12 had no significant effect on serum HDL‐C levels in female or male mice, whereas it reduced LDL‐C levels in female and male mice by the same extent (Figure 5b,c).
After high‐fat‐diet feeding, serum CRP, IL‐6, CCL2 and TNF‐α levels were significantly higher in apoE−/− mice than in wild‐type mice. Meanwhile, P12 significantly suppressed these inflammatory indices. The apoA‐I221–240 (PP) had no cholesterol efflux activity in vitro and no satisfactory lipid‐lowering activity in vivo, although it exhibited favourable anti‐inflammatory effects. CRP levels were close to 0 mg·L−1 in wild‐type mice compared with more than 150 mg·L−1 in apoE−/− mice. Serum CRP levels in apoE−/− mice were reduced by approximately 70% by P12 treatment compared with those in untreated apoE−/− mice. After 4 months of feeding, the gender differences in serum levels of inflammatory factors were very clear. Excluding CRP, serum inflammatory factor levels (IL‐6, CCL2 and TNF‐α) were lower in all of male mice than in all of female mice (Figure 5d).
3.5.3. P12 has a short half‐life in wild‐type C57BL/6J and apoE−/− mice
The plasma clearance curve and fitted half‐life of P12 are presented in Figure 6a. No significant difference was observed in the plasma clearance rate of P12 between apoE−/− and C57BL/6J mice. However, the t 1/2 of P12 was longer in apoE−/− mice (3.09 h) than in C57BL/6J mice (2.40 h). After intravenous injection, P12 remained in the plasma for approximately 25 h.
FIGURE 6.
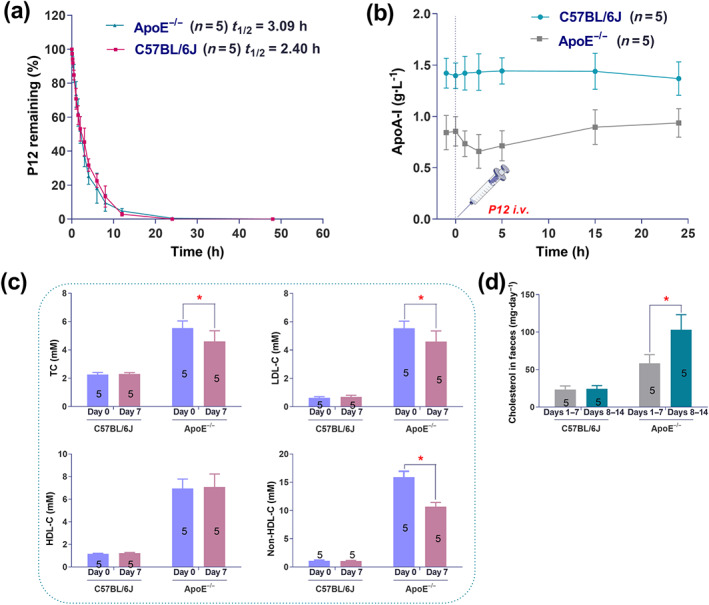
Half‐life of P12 and effects of P12 on plasm apoA‐I levels, blood lipid levels, and cholesterol excretion. (a) Plasma clearance and half‐life (t 1/2) of P12 (10 mg·kg−1 i.v.). The plasma t 1/2 of P12 was 3.09 h in apoE−/− and 2.40 h in C57BL/6J mice. P12 was cleared with first‐order kinetics in both mouse strains. (b) Effect of P12 (10 mg·kg−1 i.v.) on plasma apoA‐I levels. P12 slowly increased plasma apoA‐I levels in apoE−/− mice within 24 h, whereas it had no effect on plasma apoA‐I levels in C57BL/6J mice. (c) Effects of 1 week of P12 treatment on serum total cholesterol (TC), LDL‐C, HDL‐C, and non‐HDL‐C levels in apoE−/− and normal C57BL/6J mice. Day 0, the measurement was conducted 1 day before the experiment began. Day 7, the measurement was conducted on the seventh day. * P < 0.05, significantly different from blood lipid levels in apoE−/− mice at Day 0. Student's t test. (d) Effect of 2 weeks of P12 treatment on cholesterol excretion. * P < 0.05, significantly different from cholesterol excretion of apoE−/− mice at Days 1–7; Student's t test. Data are presented as the mean ± SEM; n = 5. Each experiment was repeated independently five times
3.5.4. P12 can slowly increase plasma apoA‐I levels in apoE−/− mice within 24 h
As shown in Figure 6b, plasma apoA‐I levels were lower in apoE−/− mice than in C57BL/6J mice. P12 had no effect on plasma apoA‐I in C57BL/6J mice. After the intravenous injection of P12, plasma apoA‐I in apoE−/− mice displayed a downward trend from 0 to 2.5 h and an upward trend from 2.5 to 24 h. However, after the intravenous injection of P12 for 24 h, plasma apoA‐I levels were increased in apoE−/− mice by 0.094 g·L−1.
3.5.5. P12 decreased blood lipid levels and increased cholesterol excretion in apoE−/− mice
P12 did not change serum lipid levels in wild‐type C57BL/6J mice and HDL‐C levels in apoE−/− mice after 7 days of treatment, whereas this AAMP significantly reduced serum TC, LDL‐C, and non‐HDL‐C levels in apoE−/− mice (Figure 6c). Similarly, P12 had no effect on cholesterol excretion in wild‐type mice after 7 days, whereas its treatment clearly increased the cholesterol excretion in apoE−/− mice (Figure 6d).
3.6. P12 can bind to plasma HDL and transform α‐HDL into preβ‐HDL
The results in Figure 7 show that the peak HDL area was smaller in plasma treated with P12 than in plasma incubated with saline. The peak HDL area was significantly increased by incubation with C‐TPP‐P12 and plasma. The increased peak fluctuation of the red line was the absorbance of C‐TPP‐P12 (Figure 7a). As shown in Figure 7b, most of the cholesterol in saline‐treated plasma was concentrated in HDL, whereas the cholesterol content of HDL was significantly reduced in P12‐treated plasma. As shown in Figure 7b,c, P12 reduced TC content. C‐TPP‐P12 was concentrated in the peak period of HDL, which fully indicated that C‐TPP‐P12 can bind to mature α‐HDL.
FIGURE 7.
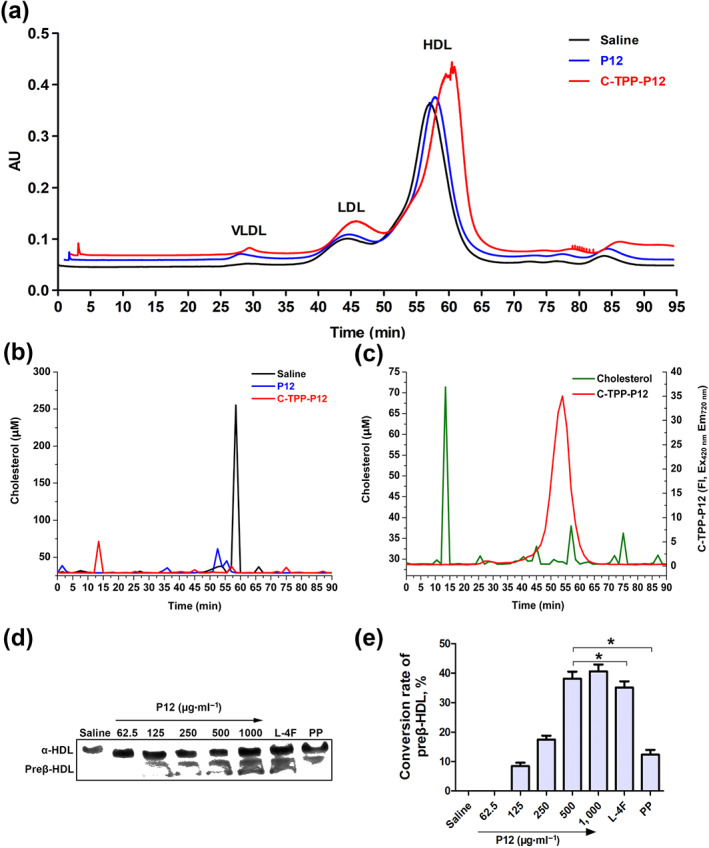
The separation of lipoproteins in the plasma of mice after co‐incubation with P12. (a) The separation chromatogram of lipoproteins obtained using size exclusion chromatography (Superose 6 column, 10 × 1,000 mm) after incubation with P12 (0.25 mg·ml−1), 5‐(4‐carboxyphenyl)‐10,15,20‐triphenylporphyrin (C‐TPP)‐P12 (0.25 mg·ml−1), and saline (blank control) at 280 nm. (b) Size exclusion chromatography fractions (1.5 min per fraction). The relative cholesterol content of each fraction measured using a cholesterol kit. (c) C‐TPP‐P12 and cholesterol were measured in each size exclusion chromatography fraction after co‐incubation of C‐TPP‐P12 with plasma. (d) The native polyacrylamide gel‐separated plasma lipoprotein after co‐incubation with P12 (62.5–1,000 μg·ml−1), L‐4F (500 μg·ml−1), and parent peptide apoA‐I221–240 (PP, 500 μg·ml−1). (e) Western blotting band after analysing the grey value using Photoshop and calculating the conversion rates of preβ‐HDL induced by the peptides. Each experiment was repeated independently five times. Each data point represents the mean ± SEM; n = 5. * P < 0.05, significantly different from P12 (500 μg·ml−1); one‐way ANOVA followed by Dunnett's post hoc test
As shown in Figure 7d,e, P12 converted α‐HDL to preβ‐HDL, and the conversion rates increased with increasing P12 concentrations, from 62.5 ‐1 000 μg ml‐1. At 500 μg·ml−1, the preβ‐HDL conversion rate in the presence of L‐4F was slightly, but significantly, less than that of P12, whereas the conversion rate with the parent peptide apoA‐I221–240 was markedly less than that of P12.
4. DISCUSSION
This study aimed to design new short apoA‐I mimetics exhibiting the biological functions of apoA‐I and called AAMPs. Considering that these AAMPs exert anti‐atherosclerotic effects through RCT, the effect of hydrophobicity on their lipid affinity and ability to transport cholesterol is a primary factor of consideration (Datta et al., 2001; Van Lenten et al., 2009). In this study, fragment 221–240 of apoA‐I was used as the parent peptide (PP) because it includes the amino acid sequence essential for cholesterol efflux and also contains the ABCA1‐binding domain (Chroni, Koukos, Duka, & Zannis, 2007). In order to maintain the original sequence of apoA‐I221–240 as much as possible, the novel AAMPs were designed using a strict amphiphilic construct. The hydrophobic surface is beneficial for affinity with the fatty chain of phospholipids. The positively charged amino acids at the amphiphilic junction facilitate them binding to the negatively charged phospholipid head, and the negatively charged amino acids in the middle of the hydrophilic surface can balance the positive charge and stabilize ABCA1.
This study found that the new AAMPs induced cholesterol efflux when the hydrophobic value was ≥0.260 (calculated using HeliQuest), whereas their cytotoxicity was observed at higher values, ≥0.548. Furthermore, when their hydrophobic value was ≥0.699, AAMPs induced slight haemolysis at high concentration. According to the in vitro mechanism studies, the possible explanations of these results are as follows: (i) Hydrophobicity affects the secondary structure of new AAMPs. As hydrophobicity increased, the secondary structure of new AAMPs changed from a disordered structure (P1–P10) to an α‐helical (P11–P14) or β‐sheet (P15–P17) structure. In line with this finding, prior research uncovered that AAMPs must have an amphipathic α‐helical structure to induce cholesterol efflux (Beaufils et al., 2007). In this study, P11–P14 always maintained a stable α‐helical structure whether in an aqueous or the simulated biofilm environment, leading to their stronger activity and weaker cytotoxicity compared with the other AAMPs. (ii) Hydrophobicity affects lipid affinity and amphipathicity (represented by the hydrophobic moment) of AAMPs. With an increasing hydrophobicity, AAMPs exhibit increasing ability to disperse DMPC vesicles. We found that P9–P11 had good lipid affinity and appropriate amphipathicity (in line with prior findings that the moment balance between hydrophobic and hydrophilic face is important for lipid interactions; Natarajan et al., 2004), thereby explaining their ability to rapidly bind to micro‐DMPC vesicles in water and then disperse them into nanovesicles. Although lipid affinity is the basis of their cholesterol efflux activity, P12 more strongly induced cholesterol efflux than P9–P11 despite its weaker lipid affinity. This finding also suggested that other factors, such as ABCA1 dependence, affect the cholesterol efflux activity of AAMPs in addition to lipid affinity and amphipathicity (Yao, Gordon, Barochia, Remaley, & Levine, 2016).
Several studies reported that apoA‐I can promote the dephosphorylation of the PEST sequence of ABCA1, reduce the calpain‐mediated degradation of ABCA1, and allow permit greater ABCA1 expression on the cell membrane surface, leading to the enhancement of cholesterol efflux from cells (Arakawa et al., 2004; Martinez, Agerholm‐Larsen, Wang, Chen, & Tall, 2003). ApoA‐I promotes cholesterol efflux by binding to ABCA1 rather than impairing cell membranes. ABCA1 overexpression can enhance the cellular binding of apoA‐I (Jin et al., 2016; Xie, Zhao, & Li, 2010). The process of cholesterol efflux through ABCA1 is mild, and it does not impair structure or function of cell membranes, leading to a smooth concentration–effect curve. Similarly, our results indicated that P12 had a nearly smooth concentration–effect curve and functioned together with ABCA1. LSCM also confirmed that P12 acted only on the cell membrane surface without impairing cellular integrity.
Numerous studies have confirmed that the function of HDL is more important to anti‐atherosclerotic effects than its quantity (Chapman, Orsoni, Robillard, Therond, & Giral, 2018; Tsompanidi, Brinkmeier, Fotiadou, Giakoumi, & Kypreos, 2010). In this study, P12 clearly regressed the atherosclerotic plaques in apoE−/− mice, although serum HDL‐C levels did not change after the 12 weeks of P12 treatment. A possible mechanism for P12‐induced plaque suppression might be the promotion of HDL function. Moreover, P12 can slowly increase the concentration of apoA‐I within 24 h. These findings illustrate that P12 can increase the ability of HDL to block the accumulation of harmful lipids in the body. Unexpectedly, the hypolipidaemic effect of P12 was better at a dose levels of 10 mg·kg−1 than at 20 mg·kg−1 of P12, suggesting that excessive P12 affected some functions of HDL. In addition to RCT, HDL also prevents atherosclerosis through anti‐inflammatory effects. Interestingly, apoA‐I221–240 had excellent anti‐inflammatory effects in atherosclerotic mice, suggesting that P12 retained the anti‐inflammatory properties of its parent peptide. The findings indicate that P12 can potentially treat atherosclerosis by lowering blood lipid levels, promoting cholesterol excretion, suppressing inflammation, and reducing lipid peroxidation. In addition, gender differences leading to differences in blood lipids, lipid peroxidation, and inflammatory factors were observed.
As mentioned previously, HDL consists of nascent HDL particles (pre β‐HDL, size approximately 7.2 nm) and mature HDL particles (α‐HDL, size approximately 12.2 nm) according to its electrophoretic mobility (Tsompanidi et al., 2010). Compared with α‐HDL, preβ‐HDL is a cholesterol‐unsaturated moiety containing less cholesterol and cholesterol ester content. When preβ‐HDL loads cholesterol to saturation, it becomes α‐HDL. Thus, preβ‐HDL has a stronger ability to carry and load cholesterol than α‐HDL, making it a key molecule for the HDL function (Hafiane & Genest, 2015). Our results showed that P12 promoted the function of HDL by remodelling α‐HDL into preβ‐HDL. Further analysis indicated that P12 can bind with the phospholipids of HDL to promote RCT, thereby improving its efficiency.
Overall, as a novel developed AAMP with an amphipathic α‐helical structure, the peptide P12 had a stronger ABCA1‐dependent cholesterol efflux activity and lower cytotoxicity than L‐4F, and it significantly suppressed the formation of atherosclerotic plaque by promoting cholesterol efflux from the plaques through ABCA1 and combined with cholesterol‐saturated α‐HDL particles in the plasma to jointly carry cholesterol to the liver. Furthermore, P12 can also convert α‐HDL into nascent preβ‐HDL particles, which can more efficiently transport cholesterol in the atherosclerotic plaques to the liver and remove excess cholesterol from the arteries. Moreover, P12 can alleviate atherosclerosis through anti‐inflammatory and antioxidant effects in apoE−/− mice.
Therefore, P12 is a promising candidate peptide for development as a new generation atheroprotective agent for cardiovascular disease. P12 represents a new technical innovation that allows us to respond to the “resistance to statins”, without contributing to the associated side effects and mortality epidemics or losing the required mild and effective anti‐atherosclerotic effect.
AUTHOR CONTRIBUTIONS
S.G. performed the experiments and data processing. L.W., G.B., and B.L. did the peptide identification and modification. C.Z. participated in the peptide purification. X.C. and X.O. participated in the data statistical analysis. X.O. and S.G. participated in the animal studies. X.C., Y.Z., and H.L. did the manuscript reading and correction. J.N. and S.G. conceived, supervised, and funded the work. J.N. and S.G. wrote the paper. All authors reviewed the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Figure S1 Synthesis reaction equation of C‐TPP.
Figure S2 Basic properties of the fluorescent probe C‐TPP‐P12. C‐TPP‐P12 was synthesized by Fmoc‐based on solid‐phase peptide synthesis. It is green in acidic solution and red in alkaline or neutral solution. The molecular weight is 3091.7314. Maximum excitation wavelengths are 420–450, 530, 560 and 580 nm, and Maximum emission wavelengths are 650 and 720 nm.
Figure S3 CD spectra of L‐4F and C‐TPP‐P12 in the water or 50% trifluoroethanol (TFE).
Figure S4 Net cholesterol efflux rates from 0 to 20 μM of apoA‐I221–240, P1 − P17 and L‐4F in RAW264.7 for 12 h.
Figure S5 Effects of different active peptides on cholesterol release in RAW264.7 cells. (a) RAW264.7 treated with the non‐toxic or toxic concentration of peptides observed on the optical microscope. At toxic concentrations of peptide, the cells morphologies were changed, and even some cells were broken or differentiation. (b) Different dose–response curves of peptides with different cholesterol efflux activity. The dose‐effect curve of P12 was an ideal. From 2 μM to 200 μM, P17 damaged the cell membrane lead to lots of cholesterol leak from RAW264.7, and its dose‐effect curve was over the dose‐effect curve of P12.
Figure S6 Hemolytic toxicity of the parent peptides apoA‐I221–240, P1 − P17 and L‐4F in C57BL/6J mice erythrocytes for 1 h.
Figure S7 Dose‐climbing experiment result of P12 in two mice. n = 6.
Figure S8 Body weight of the experimental mice in 16 weeks.
Figure S9 Four arterial typical pathological phenotypes of apoE−/− atherosclerotic mice. (1‐foam cells, 2‐cholesterol crystal, 3‐abnormally proliferating VSMCs and endothelium cells and 4‐calcified plaque) apoE−/− mice were fed high‐fat diets for 4 months.
ACKNOWLEDGEMENT
This work was financially supported by the funding from the National Natural Science Foundation of China (81273440 and 81773564).
Gou S, Wang L, Zhong C, et al. A novel apoA‐I mimetic peptide suppresses atherosclerosis by promoting physiological HDL function in apoE−/− mice. Br J Pharmacol. 2020;177:4627–4644. 10.1111/bph.15213
REFERENCES
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators . (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Nuclear hormone receptors. British Journal of Pharmacology, 176, S229–S246. 10.1111/bph.14750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … CGTP Collaborators . (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology, 176, S397–S493. 10.1111/bph.14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar, M. J. , D'Souza, W. , Turner, S. , Demosky, S. , Sviridov, D. , Stonik, J. , … Remaley, A. T. (2010). 5A apolipoprotein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. The Journal of Pharmacology and Experimental Therapeutics, 334, 634–641. 10.1124/jpet.110.167890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa, R. , Hayashi, M. , Remaley, A. T. , Brewer, B. H. , Yamauchi, Y. , & Yokoyama, S. (2004). Phosphorylation and stabilization of ATP binding cassette transporter A1 by synthetic amphiphilic helical peptides. The Journal of Biological Chemistry, 279, 6217–6220. 10.1074/jbc.C300553200 [DOI] [PubMed] [Google Scholar]
- Avdulov, N. A. , Chochina, S. V. , Igbavboa, U. , & Wood, W. G. (2000). Cholesterol efflux to high‐density lipoproteins and apolipoprotein A‐I phosphatidylcholine complexes is inhibited by ethanol: Role of apolipoprotein structure and cooperative interaction of phosphatidylcholine and cholesterol. Biochemistry, 39, 10599–10606. 10.1021/bi0008534 [DOI] [PubMed] [Google Scholar]
- Beaufils, C. , Alexopoulos, C. , Petraki, M. P. , Tselepis, A. D. , Coudevylle, N. , Sakarellos‐Daitsiotis, M. , … Cung, M. T. (2007). Conformational study of new amphipathic α‐helical peptide models of apoA‐I as potential atheroprotective agents. Biopolymers, 88, 362–372. 10.1002/bip.20651 [DOI] [PubMed] [Google Scholar]
- Carballo‐Jane, E. , Chen, Z. , O'Neill, E. , Wang, J. , Burton, C. , Chang, C. H. , … Bianchi, E. (2010). ApoA‐I mimetic peptides promote pre‐β HDL formation in vivo causing remodeling of HDL and triglyceride accumulation at higher dose. Bioorganic & Medicinal Chemistry, 18, 8669–8678. 10.1016/j.bmc.2010.09.074 [DOI] [PubMed] [Google Scholar]
- Chapman, M. J. , Orsoni, A. , Robillard, P. , Therond, P. , & Giral, P. (2018). Duality of statin action on lipoprotein subpopulations in the mixed dyslipidemia of metabolic syndrome: Quantity vs quality over time and implication of CETP. Journal of Clinical Lipidology, 12, 784–800. 10.1016/j.jacl.2018.02.001 [DOI] [PubMed] [Google Scholar]
- Chroni, A. , Koukos, G. , Duka, A. , & Zannis, V. I. (2007). The carboxy‐terminal region of apoA‐I is required for the ABCA1‐dependent formation of α‐HDL but not preβ‐HDL particles in vivo. Biochemistry, 46, 5697–5708. 10.1021/bi602354t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chroni, A. , Liu, T. , Gorshkova, I. , Kan, H. Y. , Uehara, Y. , Von Eckardstein, A. , & Zannis, V. I. (2003). The central helices of ApoA‐I can promote ATP‐binding cassette transporter A1 (ABCA1)‐mediated lipid efflux—Amino acid residues 220–231 of the wild‐type ApoA‐I are required for lipid efflux in vitro and high density lipoprotein formation in vivo. Journal of Biological Chemistry, 278, 6719–6730. 10.1074/jbc.M205232200 [DOI] [PubMed] [Google Scholar]
- Datta, G. , Chaddha, M. , Hama, S. , Navab, M. , Fogelman, A. M. , Garber, D. W. , … Anantharamaiah, G. M. (2001). Effects of increasing hydrophobicity on the physical–chemical and biological properties of a class A amphipathic helical peptide. Journal of Lipid Research, 42, 1096–1104. [PubMed] [Google Scholar]
- D'Souza, W. , Stonik, J. A. , Murphy, A. , Demosky, S. J. , Sethi, A. A. , Moore, X. L. , … Sviridov, D. (2010). Structure/function relationships of apolipoprotein A‐I mimetic peptides: Implications for antiatherogenic activities of high‐density lipoprotein. Circulation Research, 107, 217–227. 10.1161/CIRCRESAHA.110.216507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, E. A. , Feig, J. E. , Hewing, B. , Hazen, S. L. , & Smith, J. D. (2012). High‐density lipoprotein function, dysfunction, and reverse cholesterol transport. Arteriosclerosis, Thrombosis, and Vascular Biology, 32, 2813–2820. 10.1161/ATVBAHA.112.300133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz, G. S. , & Reardon, C. A. (2011). Apolipoprotein A‐I and A‐I mimetic peptides: A role in atherosclerosis. Journal of Inflammation Research, 4, 83–92. 10.2147/JIR.S12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafiane, A. , & Genest, J. (2015). High density lipoproteins: Measurement techniques and potential biomarkers of cardiovascular risk. BBA Clinical, 3, 175–188. 10.1016/j.bbacli.2015.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingenito, R. , Burton, C. , Langella, A. , Chen, X. , Zytko, K. , Pessi, A. , … Bianchi, E. (2010). Novel potent apoA‐I peptide mimetics that stimulate cholesterol efflux and pre‐β particle formation in vitro. Bioorganic & Medicinal Chemistry Letters, 20, 236–239. 10.1016/j.bmcl.2009.10.128 [DOI] [PubMed] [Google Scholar]
- Islam, R. M. , Pourmousa, M. , Sviridov, D. , Gordon, S. M. , Neufeld, E. B. , Freeman, L. A. , … Remaley, A. T. (2018). Structural properties of apolipoprotein A‐I mimetic peptides that promote ABCA1‐dependent cholesterol efflux. Scientific Reports, 8, 2956 10.1038/s41598-018-20965-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, X. T. , Sviridov, D. , Liu, Y. , Vaisman, B. , Addadi, L. , Remaley, A. T. , & Kruth, H. S. (2016). ABCA1 (ATP‐binding cassette transporter A1) mediates ApoA‐I (apolipoprotein A‐I) and ApoA‐I mimetic peptide mobilization of extracellular cholesterol microdomains deposited by macrophages. Arteriosclerosis Thrombosis and Vascular Biology, 36, 2283–2291. 10.1161/ATVBAHA.116.308334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera, A. V. , & Rader, D. J. (2010). Future therapeutic directions in reverse cholesterol transport. Current Atherosclerosis Reports, 12, 73–81. 10.1007/s11883-009-0080-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman, L. J. , Maryanoff, B. E. , & Ghadiri, M. R. (2014). Molecules that mimic apolipoprotein A‐I: Potential agents for treating atherosclerosis. Journal of Medicinal Chemistry, 57, 2169–2196. 10.1021/jm4005847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, G. F. , & Rader, D. J. (2005). New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circulation Research, 96, 1221–1232. 10.1161/01.RES.0000170946.56981.5c [DOI] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. , Cirino, G. , Docherty, J. R. , … Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology. https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, L. O. , Agerholm‐Larsen, B. , Wang, N. , Chen, W. , & Tall, A. R. (2003). Phosphorylation of a pest sequence in ABCA1 promotes calpain degradation and is reversed by ApoA‐I. The Journal of Biological Chemistry, 278, 37368–37374. 10.1074/jbc.M307161200 [DOI] [PubMed] [Google Scholar]
- Mei, X. H. , & Atkinson, D. (2015). Lipid‐free apolipoprotein A‐I structure: Insights into HDL formation and atherosclerosis development. Archives of Medical Research, 46, 351–360. 10.1016/j.arcmed.2015.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan, P. , Forte, T. M. , Chu, B. , Phillips, M. C. , Oram, J. F. , & Bielicki, J. K. (2004). Identification of an apolipoprotein A‐I structural element that mediates cellular cholesterol efflux and stabilizes ATP binding cassette transporter A1. The Journal of Biological Chemistry, 279, 24044–24052. 10.1074/jbc.M400561200 [DOI] [PubMed] [Google Scholar]
- Navab, M. , Anantharamaiah, G. M. , Hama, S. , Garber, D. W. , Chaddha, M. , Hough, G. , … Fogelman, A. M. (2002). Oral administration of an ApoA‐I mimetic peptide synthesized from d‐amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation, 105, 290–292. 10.1161/hc0302.103711 [DOI] [PubMed] [Google Scholar]
- Navab, M. , Reddy, S. T. , Anantharamaiah, G. M. , Imaizumi, S. , Hough, G. , Hama, S. , & Fogelman, A. M. (2011). Intestine may be a major site of action for the apoA‐I mimetic peptide 4F whether administered subcutaneously or orally. Journal of Lipid Research, 52, 1200–1210. 10.1194/jlr.M013144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi, R. , Mu, H. , Wang, X. , Yao, Q. , & Chen, C. (2005). Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM: An International Journal of Medicine, 98, 845–856. 10.1093/qjmed/hci136 [DOI] [PubMed] [Google Scholar]
- Osei‐Hwedieh, D. O. , Amar, M. , Sviridov, D. , & Remaley, A. T. (2011). Apolipoprotein mimetic peptides: Mechanisms of action as anti‐atherogenic agents. Pharmacology & Therapeutics, 130, 83–91. 10.1016/j.pharmthera.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palgunachari, M. N. , Mishra, V. K. , LundKatz, S. , Phillips, M. C. , Adeyeye, S. O. , Alluri, S. , … Segrest, J. P. (1996). Only the two end helixes of eight tandem amphipathic helical domains of human apo A‐I have significant lipid affinity: Implications for HDL assembly. Arteriosclerosis Thrombosis and Vascular Biology, 16, 328–338. 10.1161/01.ATV.16.2.328 [DOI] [PubMed] [Google Scholar]
- Panagotopulos, S. E. , Witting, S. R. , Horace, E. M. , Hui, D. Y. , Maiorano, J. N. , & Davidson, W. S. (2002). The role of apolipoprotein A‐I helix 10 in apolipoprotein‐mediated cholesterol efflux via the ATP‐binding cassette transporter ABCA1. Journal of Biological Chemistry, 277, 39477–39484. 10.1074/jbc.M207005200 [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, S. T. , Navab, M. , Anantharamaiah, G. M. , & Fogelman, A. M. (2014). Searching for a successful HDL‐based treatment strategy. Biochimica et Biophysica Acta, 1841, 162–167. 10.1016/j.bbalip.2013.10.012 [DOI] [PubMed] [Google Scholar]
- Shimizu, T. , Tanigawa, H. , Miura, S. , Kuwano, T. , Takata, K. , Suematsu, Y. , … Saku, K. (2015). Newly developed apolipoprotein A‐I mimetic peptide promotes macrophage reverse cholesterol transport in vivo. International Journal of Cardiology, 192, 82–88. 10.1016/j.ijcard.2015.05.012 [DOI] [PubMed] [Google Scholar]
- Stoekenbroek, R. M. , Stroes, E. S. , & Hovingh, G. K. (2015). ApoA‐I mimetics. Handbook of Experimental Pharmacology, 224, 631–648. 10.1007/978-3-319-09665-0_21 [DOI] [PubMed] [Google Scholar]
- Sviridov, D. O. , Andrianov, A. M. , Anishchenko, I. V. , Stonik, J. A. , Amar, M. J. , Turner, S. , & Remaley, A. T. (2013). Hydrophobic amino acids in the hinge region of the 5A apolipoprotein mimetic peptide are essential for promoting cholesterol efflux by the ABCA1 transporter. The Journal of Pharmacology and Experimental Therapeutics, 344, 50–58. 10.1124/jpet.112.198143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsompanidi, E. M. , Brinkmeier, M. S. , Fotiadou, E. H. , Giakoumi, S. M. , & Kypreos, K. E. (2010). HDL biogenesis and functions: Role of HDL quality and quantity in atherosclerosis. Atherosclerosis, 208, 3–9. 10.1016/j.atherosclerosis.2009.05.034 [DOI] [PubMed] [Google Scholar]
- Uehara, Y. , Ando, S. , Yahiro, E. , Oniki, K. , Ayaori, M. , Abe, S. , … Saku, K. (2013). FAMP, a novel apoA‐I mimetic peptide, suppresses aortic plaque formation through promotion of biological HDL function in ApoE‐deficient mice. Journal of the American Heart Association, 2, e000048 10.1161/JAHA.113.000048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushasri, C. , Jaipal, K. , Srinivas, G. , Avinash Raj, T. , Mahesh, K. J. , Giribabu, L. , … Pande, G. (2014). Synthesis and functional characterization of a fluorescent peptide probe for noninvasive imaging of collagen in live tissues. Experimental Cell Research, 327, 91–101. 10.1016/j.yexcr.2014.05.005 [DOI] [PubMed] [Google Scholar]
- Van Lenten, B. J. , Wagner, A. C. , Anantharamaiah, G. M. , Navab, M. , Reddy, S. T. , Buga, G. M. , & Fogelman, A. M. (2009). Apolipoprotein A‐I mimetic peptides. Current Atherosclerosis Reports, 11, 52–57. 10.1007/s11883-009-0008-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wool, G. D. , Reardon, C. A. , & Getz, G. S. (2014). Mimetic peptides of human apoA‐I helix 10 get together to lower lipids and ameliorate atherosclerosis: Is the action in the gut? Journal of Lipid Research, 55, 1983–1985. 10.1194/jlr.E053538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Q. , Zhao, S. P. , & Li, F. (2010). D‐4F, an apolipoprotein A‐I mimetic peptide, promotes cholesterol efflux from macrophages via ATP‐binding cassette transporter A1. The Tohoku Journal of Experimental Medicine, 220, 223–228. 10.1620/tjem.220.223 [DOI] [PubMed] [Google Scholar]
- Yao, X. , Gordon, E. M. , Barochia, A. V. , Remaley, A. T. , & Levine, S. J. (2016). The A's have it: Developing apolipoprotein A‐I mimetic peptides into a novel treatment for asthma. Chest, 150, 283–288. 10.1016/j.chest.2016.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , He, J. , Jiang, C. , Zhang, W. , Yang, Y. , Wang, Z. , & Liu, J. (2017). Plaque‐hyaluronidase‐responsive high‐density‐lipoprotein‐mimetic nanoparticles for multistage intimal‐macrophage‐targeted drug delivery and enhanced anti‐atherosclerotic therapy. International Journal of Nanomedicine, 12, 533–558. 10.2147/IJN.S124252 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Synthesis reaction equation of C‐TPP.
Figure S2 Basic properties of the fluorescent probe C‐TPP‐P12. C‐TPP‐P12 was synthesized by Fmoc‐based on solid‐phase peptide synthesis. It is green in acidic solution and red in alkaline or neutral solution. The molecular weight is 3091.7314. Maximum excitation wavelengths are 420–450, 530, 560 and 580 nm, and Maximum emission wavelengths are 650 and 720 nm.
Figure S3 CD spectra of L‐4F and C‐TPP‐P12 in the water or 50% trifluoroethanol (TFE).
Figure S4 Net cholesterol efflux rates from 0 to 20 μM of apoA‐I221–240, P1 − P17 and L‐4F in RAW264.7 for 12 h.
Figure S5 Effects of different active peptides on cholesterol release in RAW264.7 cells. (a) RAW264.7 treated with the non‐toxic or toxic concentration of peptides observed on the optical microscope. At toxic concentrations of peptide, the cells morphologies were changed, and even some cells were broken or differentiation. (b) Different dose–response curves of peptides with different cholesterol efflux activity. The dose‐effect curve of P12 was an ideal. From 2 μM to 200 μM, P17 damaged the cell membrane lead to lots of cholesterol leak from RAW264.7, and its dose‐effect curve was over the dose‐effect curve of P12.
Figure S6 Hemolytic toxicity of the parent peptides apoA‐I221–240, P1 − P17 and L‐4F in C57BL/6J mice erythrocytes for 1 h.
Figure S7 Dose‐climbing experiment result of P12 in two mice. n = 6.
Figure S8 Body weight of the experimental mice in 16 weeks.
Figure S9 Four arterial typical pathological phenotypes of apoE−/− atherosclerotic mice. (1‐foam cells, 2‐cholesterol crystal, 3‐abnormally proliferating VSMCs and endothelium cells and 4‐calcified plaque) apoE−/− mice were fed high‐fat diets for 4 months.