

Abstract
Background and Purpose
Klotho is a membrane‐bound or soluble protein, originally identified as an age‐suppressing factor and regulator of mineral metabolism. Klotho deficiency is associated with the development of renal disease, but its role in cardiac function in the context of uraemic cardiomyopathy is unknown.
Experimental Approach
We explored the effects of Klotho on cardiac Ca2+ cycling. We analysed Ca2+ handling in adult cardiomyocytes from Klotho‐deficient (kl/kl) mice and from a murine model of 5/6 nephrectomy (Nfx). We also studied the effect of exogenous Klotho supplementation, by chronic recombinant Klotho treatment, or endogenous Klotho overexpression, using transgenic mice overexpressing Klotho (Tg‐Kl), on uraemic cardiomyopathy. Hearts from Nfx mice were used to study Ca2+ sensitivity of ryanodine receptors and their phosphorylation state.
Key Results
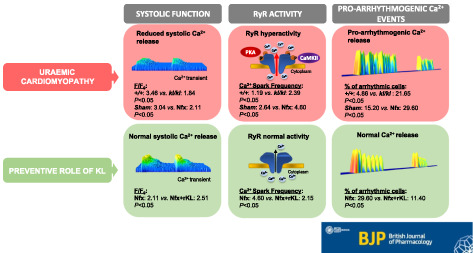
Cardiomyocytes from kl/kl mice showed decreased amplitude of intracellular Ca2+ transients and cellular shortening together with an increase in pro‐arrhythmic Ca2+ events compared with cells from wild‐type mice. Cardiomyocytes from Nfx mice exhibited the same impairment in Ca2+ cycling as kl/kl mice. Changes in Nfx cardiomyocytes were explained by higher sensitivity of ryanodine receptors to Ca2+ and their increased phosphorylation at the calmodulin kinase type II and protein kinase A sites. Ca2+ mishandling in Nfx‐treated mice was fully prevented by chronic recombinant Klotho administration or transgenic Klotho overexpression.
Conclusions and Implications
Klotho emerges as an attractive therapeutic tool to improve cardiac Ca2+ mishandling observed in uraemic cardiomyopathy. Strategies that improve Klotho availability are good candidates to protect the heart from functional cardiac alterations in renal disease.
Keywords: Ca2+ mishandling, chronic kidney disease, Klotho, ryanodine receptors, uraemic cardiomyopathy
Abbreviations
- BUN
blood urea nitrogen
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- CKD
chronic kidney disease
- EF
ejection fraction
- FGF‐23
fibroblast growth factor‐23
- FGFR
fibroblast growth factor receptor
- Nfx
5/6 nephrectomy
- PKA
protein kinase A
- rKL
recombinant Klotho
- sKL
soluble Klotho
- SR
sarcoplasmic reticulum
- SERCA2a
sarcoplasmic reticulum Ca2+‐ATPase 2a
- RyR
ryanodine receptor
What is already known
CKD patients present alterations in mineral metabolism components including a decline in Klotho protein expression.
Recent studies point to Klotho as a new anti‐aging factor with cardioprotective properties.
What this study adds
Klotho deficiency alters systolic Ca2+ release and contractile function and increases pro‐arrhythmogenic diastolic Ca2+ leak.
Recombinant Klotho treatment prevents cardiac Ca2+ mishandling and RyR hypersensitivity in CKD.
What is the clinical significance
Recombinant Klotho is a promising new prophylactic strategy to reduce cardiac functional alterations in CKD.
1. INTRODUCTION
The risk of cardiovascular disease increases as renal function declines (Go, Chertow, Fan, McCulloch, & Hsu, 2004; Herzog et al., 2011), and cardiovascular disease is the major cause of mortality in patients with chronic kidney disease (CKD). Among the components of bone mineral metabolism, alterations in Klotho protein and the fibroblast growth factor (FGF)‐23 axis seem to be important contributors to the high cardiovascular risk in this patient group (Gutiérrez et al., 2008; Isakova et al., 2011; Isakova et al., 2018). FGF‐23 is a circulatory hormone that mainly targets the kidney, where it controls systemic phosphate homeostasis. The phosphaturic action of FGF‐23 is dependent on renal expression of membrane‐bound Klotho, a powerful regulator of aging and lifespan (Kuro‐o et al., 1997). Klotho is predominantly synthesised in the kidney where it binds to FGF receptors (FGFRs), increasing their affinity for FGF‐23 and promoting urinary phosphate excretion (Urakawa et al., 2006). Klotho expression diminishes as renal function deteriorates, and this reduction might be responsible for the resistance to FGF‐23 observed in the course of CKD (Pavik et al., 2013). Membrane‐bound Klotho can be shed by the action of secretases and released into circulation as a soluble form. Circulating soluble Klotho (sKL) has several functions that are poorly understood, including an apparently cardioprotective action that is independent of FGF‐23 and phosphate (Hu et al., 2017; Xie et al., 2012; Xie, Yoon, An, Kuro‐o, & Huang, 2015). Accordingly, high levels of sKL are associated with a lower risk of developing cardiovascular disease, and low‐sKL levels are related to cardiac dysfunction, at least when advanced age is considered (Semba et al., 2011). Several recent clinical studies have shown that low circulating sKL levels are associated with an elevated risk of cardiovascular mortality or hospitalisation due to cardiovascular events (including heart failure development, myocardial infarction, or stroke) in patients with stable ischaemic heart disease (Bergmark et al., 2019) or on haemodialysis (Memmos et al., 2019). Thus, there is a need to determine whether Klotho can directly regulate cardiomyocyte function and cardiac rhythm in the setting of renal disease and to elucidate the underlying mechanisms involved in the potential cardioprotective actions of Klotho.
The increased cardiovascular morbidity and mortality linked to renal dysfunction are due, at least in part, to the high prevalence of heart failure and arrhythmias in patients with CKD, mainly those undergoing dialysis (Charytan et al., 2016; Verde et al., 2016; Wanner, Amann, & Shoji, 2016). Indeed, cardiovascular deaths account for ~50% of all deaths in CKD, especially in patients on dialysis, which is chiefly due to fatal arrhythmias (Coll et al., 2018). Alterations in intracellular Ca2+ cycling, such as changes in Ca2+ release from the sarcoplasmic reticulum (SR) mediated by ryanodine receptor (RyR) channels in cardiomyocytes, are well‐documented mechanisms involved in cardiac dysfunction and arrhythmias (Bers, 2014; Nattel, Maguy, Le Bouter, & Yeh, 2007). Cardiac muscle contraction is triggered by an action potential that induces a small Ca2+ influx via sarcolemmal L‐type Ca2+ (I CaL) channels, triggering a greater release of Ca2+ from the SR by RyR channels. This elevation in cardiomyocyte Ca2+ transients stimulates cardiomyocyte contraction (Bers, 2002). Relaxation is initiated when Ca2+ is pumped out from the cytosol by two chief mechanisms: (i) Ca2+ re‐uptake to the SR by the Ca2+‐ATPase 2a pump (SERCA2a) (~92%) and (ii) Ca2+ extrusion to the extracellular medium by the Na+/Ca2+ exchanger (NCX) (~7%) (Bers, 2002). An increase in RyR channel sensitivity, evoking higher spontaneous diastolic SR‐Ca2+ leak, is known to increase the risk of malignant arrhythmias. Whether Ca2+ handling alterations also occur in the context of CKD and whether Klotho could induce cardioprotection in this context by modulating intracellular Ca2+ handling remain unknown.
Here, we used Klotho‐deficient (kl/kl) mice to determine whether Klotho is necessary for correct cardiomyocyte function. Furthermore, we established a murine model of CKD by 5/6 nephrectomy to test the hypothesis that strategies directed to improve Klotho availability—supplied exogenously (recombinant) or endogenously (by enhancing its expression) could prevent cardiac dysfunction related to intracellular Ca2+ mishandling in uraemic cardiomyopathy.
2. METHODS
2.1. Animals
All animal care and experimental procedures complied with the guidelines for ethical NIH Guide for the Care and Use of Laboratory Animals and Guidelines for Ethical Care and Welfare (2013/175) of Experimental Animals of the European Union (2010/63/EU) and were approved by the Bioethical Committee of the Universidad Autónoma de Madrid, and by the General Direction of Agriculture and the Environment at the Environment Council of Madrid (PROEX 053/16). Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020).
Male C57BL/6 J mice (20–23 g, 6 weeks of age; IMSR_JAX:000664) were purchased from Charles River Laboratories International Inc. (Wilmington, MA, USA; SCR_003792). Mice were bred and housed under specific pathogen‐free conditions in the Experimental Animal Centre of Hospital Universitario 12 de Octubre, Madrid, Spain. Animals were maintained at controlled temperature (23–25 °C) on a 12‐hr light/dark cycle with ad libitum access to water and a standard diet (ROD14, Altromin Spezialfutter GmbH & Co. KG, Lage, Germany). Animals were housed in groups of four per cage of 553 cm2 by 20.8‐cm depth (polysulfone cage type II L, SODISPAN, Madrid, Spain) with standard wood chip (ECO‐PURE 7 Chips, Tapvei®, Estonia). All efforts were made to reduce the number of animals required to obtain reliable results and minimise the suffering of the animals based on the 3Rs rule of replacement, refinement, and reduction. The viability of the ventricular cardiomyocyte isolation process was established when the percentage of relaxed and rod‐shaped cardiomyocytes in total cell count was equal or greater than 70% (Shioya, 2007). No difference was observed in the viability of the cardiomyocyte isolation process between all experimental groups.
2.2. Experimental model of chronic kidney disease and treatments
Mice underwent Sham or 5/6 nephrectomy (Nfx) surgery to induce CKD. Nfx was performed in a two‐step procedure under isoflurane anaesthesia (1.5% v/v, isoflurane/oxygen). In the first step, an abdominal incision was made, and the upper and lower poles of the left kidney were removed, leaving an intact segment around the hilum (experimental week 0). One week later, a dorsal incision was made, and the right kidney was completely removed (Gagnon & Gallimore, 1988). Sham‐operated mice followed the same protocol without the removal of the kidneys. Meloxicam (0.06 ml·kg−1, s.c. single dose) was used as analgaesia after surgeries. After that, Sham or Nfx surgery mice were blindly randomised to receive either vehicle solution (0.9% sodium chloride) or murine recombinant Klotho (rKL, 0.01 mg·kg−1 per day). Mice were treated at the same hour in the morning every day for 6 weeks by i.p. injection as described before (Hu et al., 2017). Macroscopic parameters were analysed in 16 Sham, 20 Nfx, 17 Sham+rKL and 22 Nfx+rKL mice. Ten mice in each group were used for biochemical parameters, five to seven mouse hearts per group were used for ryanodine binding analysis, and five mice per group were used for echocardiogram analysis. Cardiomyocytes obtained from five Sham, six Nfx, five Sham+rKL, and six Nfx+rKL mice were used for Ca2+ handling analysis.
2.3. Echocardiography
Mouse hearts were visualised by echocardiography, using the Vivid Q ultrasound system (GE Healthcare), with a coupled linear sonar of 14 MHz. A single investigator, blinded to the experimental groups, performed the analysis. Measurements were carried out with mice under isoflurane anaesthesia (1.5% v/v, isoflurane/oxygen) in order to keep a heart rate of about 400 beats min‐1. Body hair was shaved, and parasternal images of the short axis of the heart were obtained in a bidimensional mode (B), for anatomical visualisation. Taking as a reference the papillary muscles, we carried out the functional analysis of cardiac contractility by an ultrasound M mode, to determine ejection fraction (EF) using cardiac analysis software.
2.4. Klotho‐hypomorphic and Klotho‐overexpressing mice
Transgenic (Tg‐Kl) and Klotho‐hypomorphic (kl/kl) mice were kindly provided by Dr. Kuro‐o and bred in our animal facilities.
Male kl/kl mice (8 g of weight) and their +/+ littermates (20 g of weight) were used at 6–8 weeks of age. Macroscopic parameters were analysed in 22 +/+ and 19 kl/kl mice. Ten mice in each group were used for biochemical parameter analysis, and five mice per group were used for Ca2+ handling analysis.
Tg‐Kl 6‐week‐old mice with a weight between 20 and 22 g underwent Sham (Sham‐Tg‐Kl) or Nfx (Nfx‐Tg‐Kl) surgery as described before. Biochemical parameters were analysed in five Sham‐Tg‐Kl and seven Nfx‐Tg‐Kl mice, and five Sham‐Tg‐Kl mice and Nfx‐Tg‐Kl six mice were used for Ca2+ handling analysis.
2.5. Serum and urine biochemistry
Plasma levels of phosphorus (Abcam, Cambridge, UK), urea and blood urea nitrogen (BUN) (BioAssays System, Hayword, CA), and FGF‐23 (Immunotopics, Inc., San Clemente, CA) were measured by ELISA following the manufacturers' instructions. Mouse soluble α‐Klotho assay kit (IBL International, Hamburg, Germany) was used to measure sKL in urine samples.
2.6. Cardiomyocyte isolation
Adult male mice (6–8‐week‐old +/+ or kl/kl, 14‐week‐old WT‐Sham, Nfx, Sham+rKL or Nfx+rKL, and Tg‐Kl‐Sham or Tg‐Kl‐Nfx, blindly selected) were anaesthetised with sodium pentobarbital‐heparin (100 mg/kg, i.p.). Mice were sacrificed, and hearts were rapidly removed and cannulated via the ascending aorta on a Langendorff perfusion system (Navarro‐García et al., 2019; Ruiz‐Hurtado et al., 2015). Hearts were retrogradely perfused with calcium‐free Tyrode's solution supplemented with EGTA (0.2 mmol·L−1) for 3 min followed by Tyrode's solution containing 0.1 mmol·L−1 of CaCl2, 1 mg·mL−1 of type II collagenase (Worthington, Lakewood, NY), and 1 mg·mL−1 of BSA for 3–4 min at room temperature. Digested hearts were filtered through a nylon mesh (250 μm) and centrifuged at 280 x g for 3 min at room temperature. The pellet was re‐suspended in Tyrode's solution containing 0.5 mmol·L−1 of CaCl2 and centrifuged again. Finally, the pellet was re‐suspended in Tyrode's solution containing CaCl2 (1 mmol·L−1). Tyrode's solution contained (in mmol·L−1): 130 NaCl, 5.4 KCl, 0.4 NaH2PO4, 0.5 MgCl2, 25 HEPES, and 22 glucose. The pH was adjusted to 7.4 with LiOH.
2.7. Confocal Ca2+ image analysis
Isolated ventricular cardiomyocytes were loaded with the membrane‐permeant fluorescent Ca2+ indicator Fluo‐3 AM (5 μmol·L−1; Invitrogen, Carlsbad, CA) for 30 min at room temperature. Line‐scan Ca2+ images were obtained using confocal microscopy (Meta Zeiss LSM 510, objective w.i. 40×, n.a. 1.2) at the speed of 1.5 ms/line (1,000 lines per image) with a pixel size of 0.07 μm for recording Ca2+ transients, Ca2+ sparks, and Ca2+ wave images, while for caffeine and arrhythmia protocols, confocal images were obtained at a speed of 3 ms/line (10,000 lines per image) for a total duration of 30 s. All Ca2+ images were corrected for the background fluorescence. In a set of experiments, Fluo‐3‐loaded cells were placed in a field stimulation chamber with two parallel platinum electrodes and filled with Tyrode's solution. Cells were allowed to settle at the bottom of the chamber and paced at 2 Hz with a pulse generator (CS‐420, Cibertec) during 1–2 min to allow steady‐state intracellular Ca2+ transients. Then, a line was selected always in parallel to the longitudinal cell axis to be able to measure the associated cell shortening; and Ca2+ imaging recordings were manually initiated with the time‐lapse plug‐in included in Zen 2009 Imaging Software (12.09, ZEISS).
The amplitude of Ca2+ transients was calculated by averaging the fluorescence values in a 1.4‐μm frame over time. The amplitude of Ca2+ transients was estimated as F/F 0, where F was the maximum value of fluorescence signal during electrical stimulation and F 0 was the basal fluorescence at rest at each position determined as the average of the 50 lowest values on the fluorescence transient. Moreover, ΔF/F 0 was calculated using the following formula ΔF/F 0 = (F − F 0)/F 0. SERCA2a function was indirectly estimated by the decay time constant of intracellular Ca2+ transients, termed Tau, which was obtained by fitting the descending phase of the fluorescence trace to a single exponential. K SERCA2a refers to the SR‐dependent fraction of the rate constant of decay of the systolic Ca2+ transient and was measured by subtracting the rate constant of decay of the caffeine‐evoked transients from that of the systolic Ca2+ transients (Bode et al., 2011). Cell shortening was measured as the difference of cardiomyocytes length between electrical stimulation and resting. Cell shortening was expressed as the percentage of cell's shortening length. Cell shortening profiles were obtained offline by measuring the cell length from each line of Ca2+ transient images. SR Ca2+ load was estimated by perfusing cardiomyocytes with 10 mmol·L−1 of caffeine immediately after field stimulation. Caffeine depletes the SR of Ca2+ stores. Caffeine‐evoked Ca2+ transients' amplitude was measured as peak F/F 0 and was used to rate the global SR Ca2+ load.
In a set of experiments, spontaneous Ca2+ sparks were recorded in quiescent Fluo‐3‐loaded cardiomyocytes when electrical stimulation was stopped. Ca2+ sparks were considered as brief, located, and rapid increments in Ca2+ fluorescence. Ca2+ spark regions are determined as the sites where fluorescence signal increased by at least four times the standard deviation of the image fluorescence. These inclusion criteria limited false events while detecting most sparks (Cheng et al., 1999). Spontaneous Ca2+ transients and Ca2+ waves in quiescent cardiomyocytes were detected as substantial spontaneous Ca2+ synchronic or non‐synchronic release, respectively. The arrhythmic activity was measured as abnormal spontaneous Ca2+ transients with automatic contractions in cardiomyocytes during a specific protocol, consisting of three cycles of seven electric pulses followed by a recovery period.
All Ca2+ images were processed and analysed offline using IDL (RSI) software and homemade routines.
2.8. Ryanodine binding analysis
[3H]‐Ryanodine binding experiments were performed with homogenates of left ventricles from individual hearts, as described earlier (de Alba‐Aguayo et al., 2017). Specific binding was defined as the difference between the binding in the absence (total binding) and presence (non‐specific binding) of 20 μmol·L−1 of unlabelled ryanodine. The incubation medium contained (in mmol·L−1 ) the following: KCl 200, HEPES 20, and EGTA‐K salt 1, pH 7.2 with KOH. CaCl2 was added as necessary to establish free Ca2+ at 100 nmol·L−1 (equivalent to diastolic Ca2+ concentration) and 10 μmol·L−1 (equivalent to systolic Ca2+ concentration). Ca2+‐EGTA ratios were calculated using the WEBMAXCLITE program v1.15 (https://web.stanford.edu/~cpatton/webmaxc/webmaxclite115.htm). Data analysis was performed using Origin (v8.1; OriginLab Corporation, Northampton, MA, USA).
2.9. Western blotting
Immunoblotting analysis complies with the recommendations on immunoblotting and immunohistochemistry in pharmacology (Alexander et al., 2018). Hearts were homogenised in 400 μl of homogenisation buffer (0.05 mol·L−1 of Tris, 0.32 mol·L−1 of sucrose, 0.5% CHAPS, 0.5 μmol·L−1 of okadaic acid, and the protease inhibitors PMSF (0.1 mmol·L−1), leupeptin (12 μmol·L−1), aprotinin (0.2 μmol·L−1) and benzamidine (0.5 mmol·L−1), and the pH was adjusted to 7.0. Homogenates were centrifuged at 2800 xg for 10 min at 4 °C. Supernatants were collected in microtubes, and 20 μg of total protein was used for western blotting. Samples were re‐suspended in SDS‐PAGE loading buffer (4× Laemmli Sample Buffer; Bio‐Rad, Hercules, CA, USA) with 10% β‐mercaptoethanol and were heated for 5 min at 90 °C. Samples were loaded on 4–20% SDS‐PAGE gradient gels (Criterion™ TGX™ Precast Gels, Bio‐Rad). Proteins were electrophoretically transferred onto PVDF membranes (Trans‐Blot® Turbo™ Midi Format, 0.2 μmol·L−1 PVDF, Bio‐Rad) on a semi‐dry transfer system (Trans‐Blot SD, Bio‐Rad) at 2.5 A, 25 V for 10 min. Membranes were blocked with 5% BSA and TBS‐T20 buffer (50 mmol·L−1 of Tris–HCl, 150 mmol·L−1 of NaCl, and 0.1% Tween‐20). Fresh antibodies solutions were used for immunoblotting. Antibodies used were as follows: rabbit polyclonal IgG anti‐phosphoRyR Ser2814 (AB_2617055) at 1:2,000 dilution and rabbit polyclonal IgG anti‐phosphoRyR Ser2030 at 1:2,000 dilution (A010‐31 and A010‐32, respectively, both from Badrilla, Leeds, UK), mouse monoclonal IgG1 anti‐RyR (AB_2183054) at 1:2,500 dilution, and mouse monoclonal IgG1 anti‐GAPDH (AB_2536381) at 1:400,000 dilution (MA3‐916 and AM4300, respectively, both from Thermo Fisher Scientific, Waltham, MA, USA). HRP‐conjugated secondary antibodies used were as follows: anti‐mouse IgG kappa (AB_2687626) at 1:5,000 dilution and mouse anti‐rabbit (AB_628497) at 1:5,000 dilution (sc‐516102 and sc‐2357, respectively, both from Santa Cruz Biotechnology, Dallas, TX, USA).
2.10. RNA isolation and quantitative real‐time PCR
Total RNA was isolated from mouse hearts using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Quality and quantity of RNA were assessed with NanoDrop One Microvolume UV–Vis Spectrophotometer (Thermo Fischer Scientific, Waltham, MA), and 2 μg of RNA was reverse‐transcribed to cDNA using High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative RT‐PCR was performed using FastStart Essential DNA Green Master (Roche, Basel, Switzerland) in 10‐μl total reaction volume on a LightCycler® 480 II (Roche, Basel, Switzerland) at optimised thermocycling settings. Relative gene expression was normalised to the ribosomal housekeeping gene 36b4 (RPLP0) and evaluated using the 2−ΔΔCt method. The primer sequences (5′–3′) for SERCA2a (Atp2a2), 36b4, collagen I (col1a1), collagen III (col3a1), and atrial natriuretic peptide (Nppa) were as follows: Atp2a2‐Forward TAAATGCCCGCTGTTTTGCT; Atp2a2‐Reverse TTGTCATCTGCCAGGACCAT; 36b4‐Forward AGATGCAGCAGATCCGCAT; 36b4‐Reverse GTTCTTGCCCATCAGCACC; col1a1‐Forward AATGGCACGGCTGTCTGCGA; col1a1‐Reverse AGCACTCGCCCTCCCGTCTT; col3a1‐Forward CTGTAACATGGAAACTGGGGAAA; col3a1‐Reverse CCATAGCTGAACTGAAAACCACC; Nppa‐Forward CTGGGACCCCTCCGATAGAT; and Nppa‐Reverse CACTCTGGGCTCCAATCCTG.
2.11. Data and statistical analysis
Group size estimation was designed to be equal, yielding 90% power. Inferential statistics were used to summarise the data from ≥5 animals per group. Any inequalities between experimental groups were exclusively owing to the assumed animal losses related to the specific surgery (5/6 nephrectomy) used in the study. The number of animals used in this study was based on power calculation and was a total of 122. The mouse was considered as the experimental unit for echocardiography, western blotting, and ryanodine binding analyses; and the whole heart was used for measurement and referred to as “N.” For all Ca2+ handling studies, the cardiomyocyte was considered as the experimental unit and referred to as “n” after assessing the absence of false positives and independence of the obtained data and the absence of clustering estimated by the intra‐cluster correlation coefficient (ICC) tests for all cellular data sets and experimental models used. For Ca2+ handling studies, both cell number (n) and animal number (N) are shown, and cardiomyocytes were isolated from at least five mice in each experiment. Data are presented as mean ± SEM. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). The data that support the results obtained in this study are available upon reasonable request from the corresponding author. Statistical significance was evaluated using paired Student's test, χ 2 test, or ANOVA with Newman–Keuls multiple‐comparison tests when appropriate (only with homogeneity and if F was significant). The Kolmogorov–Smirnov test was used to determine the Gaussian distribution of the values. All P‐values are two‐tailed, and P‐values < 0.05 were considered statistically significant. All analyses were performed using GraphPad Prism v6.0 (GraphPad Software Inc., San Diego, CA; SCR_002798) or Origin Pro v9.0 (OriginLab Corp., Northampton, MA; SCR_014212).
2.12. Materials
EGTA, sodium chloride, potassium chloride, monosodium phosphate, magnesium chloride, HEPES, glucose, lithium hydroxide, potassium hydroxide, leupeptin, aprotinin, benzamidine, β‐mercaptoethanol, Tween 20, and caffeine were purchased from Sigma‐Aldrich (Milan, Italy). Isoflurane was purchased from Abbie Spain (Madrid, Spain). CHAPS was purchased from GE Healthcare (Chicago, IL, USA). Okadaic acid‐K salt was purchased from Santa Cruz Biotechnology (Dallas, TX, USA); 4× Laemmli Sample Buffer and TBS were purchased from Bio‐Rad (Hercules, CA, USA). Pentobarbital was purchased from Vetoquinol (Madrid, Spain). Heparin was purchased from ROVI (Madrid, Spain). All the reagents have been dissolved in distilled H2O, when necessary.
2.13. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY (http://www.guidetopharmacology.org) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Fabbro et al., 2019a, b; Alexander, Kelly et al., 2019; Alexander, Mathie et al., 2019).
3. RESULTS
3.1. Klotho deficiency alters intracellular Ca2+ handling and cardiomyocyte shortening and increases pro‐arrhythmogenic Ca2+ events
To examine whether Klotho is needed for a correct cardiac function related to cardiomyocyte Ca2+ handling, we used an experimental model of uraemia characterised by deficiency in Klotho, the Klotho‐hypomorphic (kl/kl) mouse. Macroscopic analysis showed that kl/kl mice were significantly smaller than their littermates +/+ (Figure 1a, Table 1). Similarly, heart and kidney weights were significantly lower in kl/kl mice than in +/+ mice (Table 1). Heart weight to body weight ratio (HW/BW) was significantly higher in kl/kl mice than in +/+ mice (Table 1). However, this increase in HW/BW ratio was not accompanied by signs of cellular hypertrophy in kl/kl mice, because cardiomyocyte area was significantly smaller in kl/kl mice than in +/+ mice (1,471 ± 39.7 vs. 2,428 ± 67.7 μm2). We also measured mRNA expression levels of the atrial natriuretic peptide (ANP; Nppa) by quantitative RT‐PCR in hearts from kl/kl and +/+ mice as a classic marker of cellular hypertrophy, finding Nppa gene expression not different between groups (1.037 ± 0.13 for +/+ vs. 0.974 ± 0.07 for kl/kl, arbitrary units). Moreover, to assess whether the absence of Klotho expression could be accompanied by myocardial fibrosis development, collagen I and III levels were also analysed through col1a1 or col3a1 gene expression levels. The kl/kl mice showed reduced levels of collagen I and III, compared with those in +/+ mice (1.008 ± 0.06 vs. 0.267 ± 0.03 arbitrary units, for col1a1; 1.162 ± 0.03 vs. 1.014 ± 0.08, for col3a1). Therefore, the increment in HW/BW ratio in kl/kl mice was not due to cardiomyocyte hypertrophy or cardiac fibrosis development. On the other hand, as expected, kidney function was impaired in kl/kl mice, as reflected by the significant increase in the levels of urea, BUN, and FGF‐23 relative to +/+ littermates (Table 1). However, no differences in serum levels of phosphate were observed between both groups (Table 1), possibly as consequence of the phosphaturic action induced by the very high FGF‐23 levels.
FIGURE 1.
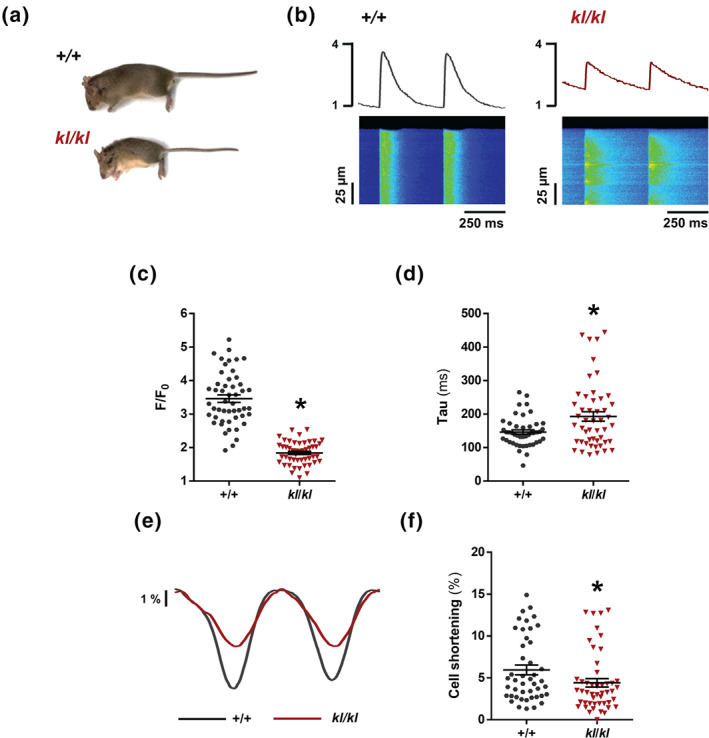
Klotho deficiency impairs systolic Ca2+ release and induces contractile dysfunction. (A) Appearance of 7‐week‐old wild‐type (+/+) mouse and its Klotho‐hypomorphic (kl/kl) littermate. (B) Line‐scan images and fluorescence profiles of cardiomyocytes under 2‐Hz field stimulation. (C and D) Mean values of peak (F/F 0) (C) and time of decay (Tau) (D) of electrically evoked Ca2+ transients in +/+ (n = 50 cells/N = 5 mice) and kl/kl samples(n = 49 cells/N = 5 mice). (E) Cell shortening profiles of cardiomyocytes. (F) Percentage of cell contraction of +/+ (n = 50 cells/N = 5 mice) and kl/kl (n = 49 cells/N = 5 mice). Data are shown as mean ± SEM. * P < 0.05, significantly different from +/+ mice
TABLE 1.
Macroscopic and biochemical parameters in Klotho‐hypomorphic mice
| +/+ | kl/kl | |
|---|---|---|
| Macroscopic parameters | ||
| Body weight (BW, g) | 23.7 ± 0.3 (N = 22) | 8.4 ± 0.3* (N = 19) |
| Heart weight (HW, mg) | 169.0 ± 6.2 (N = 22) | 102.7 ± 5.3* (N = 19) |
| HW/BW ratio | 7.10 ± 0.20 (N = 22) | 12.37 ± 0.67* (N = 19) |
| Kidney weight (KW, mg) | 155.9 ± 4.4 (N = 22) | 58.1 ± 2.3* (N = 19) |
| Biochemical parameters | ||
| Urea (mg·L−1) | 357 ± 25 (N = 10) | 646 ± 49* (N = 10) |
| BUN (mg·L−1) | 167 ± 12 (N = 10) | 302 ± 23* (N = 10) |
| Phosphorus (mg·L−1) | 133. ± 7 (N = 10) | 1215 ± 8 (N = 10) |
| FGF‐23 (pg·ml−1) | 268.3 ± 36.7 (N = 10) | 457,318 ± 57,566* (N = 10) |
Note. Data for macroscopic and biochemical parameters per experimental group are reported as mean ± SEM. BUN, blood urea nitrogen; BW, body weight; FGF‐23, fibroblast growth factor 23; HW, heart weight; KW, kidney weight.
P < 0.05, significantly different from +/+ mice.
We next analysed intracellular Ca2+ handling in cardiomyocytes isolated from both experimental groups. We studied systolic Ca2+ release through electrically evoked intracellular Ca2+ transients. Figure 1b shows representative line‐scan Ca2+ images (bottom panels) and fluorescence profiles (upper panels) obtained during electrical field stimulation at 2 Hz, corresponding to +/+ (left panel) and kl/kl (right panel) cardiomyocytes. Similar differences in the amplitude of Ca2+ transients (F/F 0) were found between +/+ and kl/kl mice analysed by cells or by hearts (3.46 ± 0.11 vs. 1.84 ± 0.05 or 3.46 ± 0.18 vs. 1.84 ± 0 ± 0.08, respectively). Intracellular Ca2+ transients amplitude (F/F 0) was significantly lower in kl/kl cells than in +/+ cells (Figure 1c). In addition, cardiomyocytes from kl/kl mice showed a decrease in ΔF/F 0, indicating a significant decrease in the global intracellular Ca2+ transient in kl/kl cells (Figure S1). The time constant of Ca2+ transients decay, Tau, was significantly longer in kl/kl cells than in +/+ cells (Figure 1d). This is also illustrated in the representative fluorescence profiles normalised by the peak of Ca2+ transients from +/+ and kl/kl cells (Figure S2), indicating that the SERCA2a pump was working less efficiently in kl/kl cardiomyocytes. Because this slower time decay of Ca2+ transients could indicate a worse SR‐Ca2+ re‐uptake by SERCA2a, we analysed the cardiac mRNA levels of SERCA2a in hearts from +/+ and kl/kl mice. As shown in Figure S3, SERCA2a expression was significantly reduced in the hearts of kl/kl mice compared with +/+ mice. Moreover, K SERCA2a, the SR‐dependent contribution of the rate constant of decay of the systolic Ca2+ transient, was also determined by subtracting the rate constant of decay of the caffeine‐evoked transients from that of the systolic Ca2+ transients. K SERCA2a was significantly lower in kl/kl mice than in +/+ mice (Figure S4), supporting a diminished SERCA2a‐mediated Ca2+ re‐uptake. We next addressed whether the decrease in systolic Ca2+ release impaired contractile function. Representative cell shortening profiles of cardiomyocytes are shown in Figure 1e. Cell shortening was significantly lower in kl/kl cells than in +/+ cells (Figure 1f). Changes in systolic Ca2+ release can be related to alterations in SR‐Ca2+ load. To study this, we applied caffeine to isolated Fluo‐3‐loaded cardiomyocytes to empty the SR of Ca2+. Representative line‐scan images of caffeine‐evoked Ca2+ transients are shown in Figure 2a. Results showed that the amplitude (F/F 0) of caffeine‐evoked Ca2+ transients was significantly lower in kl/kl cardiomyocytes than in cells from their littermates (Figure 2b). Caffeine‐evoked Ca2+ transient decay in cardiomyocytes from kl/kl mice was not different from that in +/+ mice, suggesting no alterations in Ca2+ extrusion through the NCX (Figure S5).
FIGURE 2.
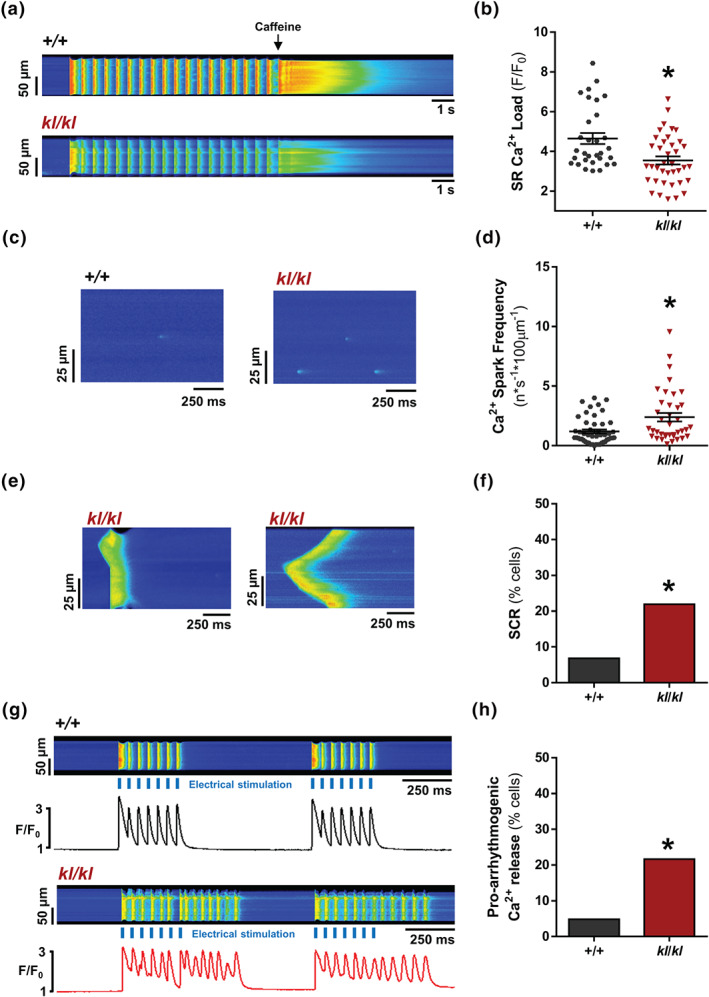
Klotho deficiency reduces SR‐Ca2+ load and increases diastolic Ca2+ leak and pro‐arrhythmogenic events. (A) Line‐scan images of cardiomyocytes under 2‐Hz field stimulation perfused with caffeine. (B) Mean values of caffeine‐evoked Ca2+ transients amplitude expressed as peak (F/F 0) in +/+ (n = 31 cells/N = 5 mice) and kl/kl (n = 39 cells/N = 5 mice). (C) Line‐scan images of spark recordings in quiescent cardiomyocytes. (D) Mean values of Ca2+ spark frequency in +/+ (664 sparks, n = 48 cells/N = 5 mice) and kl/kl (630 sparks, n = 39 cells/N = 5 mice). (E) Line‐scan images of spontaneous Ca2+ release (SCR) as spontaneous Ca2+ transients (left panel) or Ca2+ waves (right panel) in quiescent kl/kl cardiomyocytes. (F) Occurrence of SCR in +/+ (n = 48 cells/N = 5 mice) and kl/kl (n = 39 cells/N = 5 mice). (G) Line‐scan images and fluorescence profiles of cardiomyocytes paced at 2‐Hz field stimulation. (H) Occurrence of pro‐arrhythmogenic events related to automatic Ca2+ transients and automatic contractions in +/+ (n = 26 cells/N = 5 mice) and kl/kl (n = 38 cells/N = 5 mice) mice. Data are shown as mean ± SEM. * P < 0.05, significantly different from +/+ mice
We next analysed whether the change in SR‐Ca2+ load was related to alterations in diastolic Ca2+ leak. Representative line‐scan images of Ca2+ spark recordings are shown in Figure 2c. We observed a significantly higher frequency of Ca2+ sparks in kl/kl cardiomyocytes than in +/+ cells (Figure 2d). Spontaneous calcium release (SCR) was also studied during diastolic recordings; spontaneous intracellular Ca2+ transients and Ca2+ waves were considered forms of SCR (Figure 2e, left and right panels, respectively). kl/kl cardiomyocytes had a significantly higher prevalence of SCR than +/+ cells (Figure 2f). Finally, we measured pro‐arrhythmogenic Ca2+ events in cardiomyocytes isolated from both groups of mice. Figure 2g shows representative line‐scan images of the pacing protocol. Pro‐arrhythmogenic Ca2+ release was significantly higher in kl/kl cardiomyocytes than in +/+ cells (Figure 2h).
3.2. Chronic recombinant Klotho administration prevents the cardiac dysfunction and impaired systolic Ca2+ release induced by experimental chronic kidney disease
We next employed a classical model of CKD (5/6 nephrectomy) to conduct a detailed analysis of the functional cardiac consequences in a uraemic context. Moreover, based on previous evidence demonstrating the cardioprotective action of exogenous Klotho supplementation (Hu et al., 2017; Nowak et al., 2014) and after confirming that Nfx mice have significantly decreased Klotho levels compared with Sham mice (Figure S6), we analysed the effect of recombinant Klotho (rKL) administration on cardiac function and Ca2+ cycling. The experimental design is shown in Figure 3a. Macroscopic analysis revealed that Nfx‐induced overall body weight loss independently of subsequent treatment (Table 2). Kidney weight was also significantly lower in Nfx mice than in Sham mice, both in vehicle‐treated and rKL‐treated mice. No differences were observed in heart weight or HW/BW ratio among groups (Table 2). No evidence of cellular hypertrophy was observed in CKD mice as demonstrated by similar cardiomyocyte area (3,192 ± 93.2 vs. 3,187 ± 92.7) and ANP expression levels (1.170 ± 0.51 vs. 0.300 ± 0.05) between Sham and Nfx mice. To assess whether uremic cardiomyopathy induces myocardial fibrosis development, collagen I and III levels were analysed through col1a1 or col3a1 expression levels by quantitative PCR in hearts from Sham and Nfx mice. No differences were detected between Sham and Nfx mice (1.031 ± 0.13 vs. 1.136 ± 0.11 arbitrary units, for col1a1; 1.058 ± 0.18 vs. 1.29 ± 0.11, for col3a1). Therefore, no signs of cardiac hypertrophy or fibrosis were detected in Nfx mice. Kidney function was impaired in Nfx mice, as demonstrated by significantly higher levels of plasma urea and BUN in the mice that underwent Nfx, compared with those in Sham mice (Table 2). These parameters remained elevated in rKL‐treated Nfx mice, indicating that chronic rKL treatment did not restore renal function. FGF‐23 levels were significantly elevated in Nfx mice, and also in Nfx animals chronically treated with rKL. By contrast, phosphorus levels were similar in all groups, likely due to the compensatory and rapid phosphaturic action evoked by the high FGF‐23 levels. EF was measured by echocardiography as indicator of in vivo left ventricle global systolic function. EF was significantly reduced in Nfx mice, compared with Sham mice, whereas Nfx treated with rKL for 6 weeks exhibited significantly improved EF compared with that in Nfx treated with vehicle, which were not different from Sham+rKL mice (Figure 3b). Electrically evoked Ca2+ transients of cardiomyocytes were studied under field stimulation of 2 Hz. Regarding contractile function, representative cell shortening profiles are shown for all groups in Figure 3c, and we observed a significant decrease in cardiomyocyte shortening in Nfx compared with Sham cells (Figure 3d). Whereas no effect of rKL was found in Sham cells for shortening, Nfx+rKL cells showed a significant increase in cell contraction, compared with vehicle‐treated Nfx cardiomyocytes (Figure 3c, d). Figure 3e shows representative line‐scan images (bottom panels) and fluorescence profiles (upper panels) corresponding to intracellular Ca2+ transients in all experimental groups. Similar differences in the amplitude of Ca2+ transients (F/F 0) were found between Sham and Nfx mice, analysed in cells or in hearts (3.04 ± 0.12 vs. 2.11 ± 0.06 or 3.18 ± 0.32, 2.12 ± 0.14, respectively). Ca2+ transients amplitude (F/F 0) was significantly lower in Nfx cells than in Sham cells and was significantly higher in rKL‐treated Nfx cells than in vehicle‐treated Nfx cells (Figure 3f). Moreover, longer Tau values observed in Nfx cells (Figure 3g) indicate an impairment in SERCA2a function, which was also evident in the fluorescence profiles normalised to the peak of Ca2+ transients obtained from Sham, compared with Nfx cells (Figure S7). Similarly, K SERCA2a was significantly lower in Nfx mice than in Sham mice (Figure S8). rKL treatment prevented the increase in Tau (Figure 3g). No differences were found for Tau in Sham+rKL and Nfx+rKL cardiomyocytes (Figure 3g). Thus, chronic rKL treatment prevented the cardiac dysfunction and impairment in cell shortening observed in Nfx cardiomyocytes.
FIGURE 3.
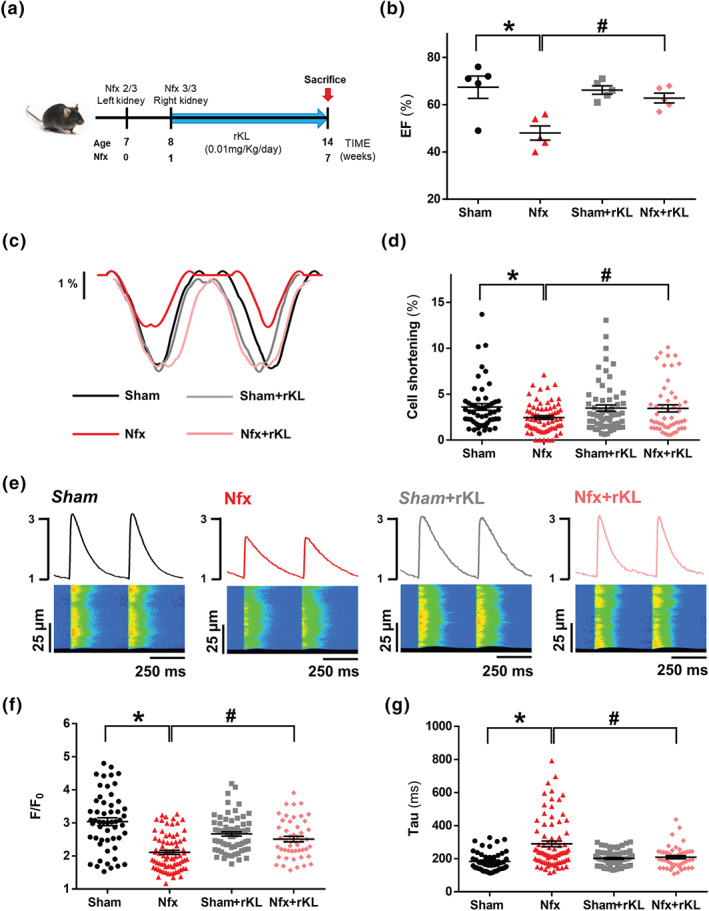
Recombinant Klotho prevents contractile cellular dysfunction and reduction in systolic Ca2+. (A) Schematic design of chronic kidney disease (CKD) animal model (5/6 nephrectomy) and treatments. (B) Mean values of ejection fraction (EF) from Sham (N = 5 mice), Nfx (N = 5 mice), Sham+rKL (N = 5 mice), and Nfx+rKL (N = 5 mice) mouse hearts. (C) Cell shortening profiles of cardiomyocytes. (D) Percentage of cell contraction of Sham (n = 52 cells/N = 5 mice), Nfx (n = 73 cells/N = 6 mice), Sham+rKL (n = 62 cells/N = 5 mice), and Nfx+rKL (n = 48 cells/N = 6 mice). (E) Line‐scan images and fluorescence profiles of cardiomyocytes under 2‐Hz field stimulation. (F and G) Mean values of peak (F/F 0) (F) and time of decay (Tau) (G) of electrically evoked Ca2+ transients in Sham (n = 54 cells/N = 5 mice), Nfx (n = 77 cells/N = 6 mice), Sham+rKL (n = 67 cells/N = 5 mice), and Nfx+rKL (n = 49 cells/N = 6 mice). Data shown mean ± SEM. * P < 0.05, significantly different from Sham; # P < 0.05, significantly different from Nfx
TABLE 2.
Macroscopic and biochemical parameters in an experimental CKD model
| Sham | Nfx | Sham+rKL | Nfx+rKL | |
|---|---|---|---|---|
| Macroscopic parameters | ||||
| Body weight (BW, g) | 27.5 ± 0.5 (N = 16) | 25.4 ± 0.6* (N = 20) | 27.2 ± 0.4 (N = 17) | 25.2 ± 0.6 † (N = 22) |
| Heart weight (HW, mg) | 200.9 ± 6.2 (N = 16) | 187.7 ± 8.1 (N = 20) | 186.1 ± 3.9 (N = 17) | 185.6 ± 4.6 (N = 22) |
| HW/BW ratio | 7.31 ± 0.17 (N = 16) | 7.45 ± 0.21 (N = 20) | 6.80 ± 0.13 (N = 17) | 7.40 ± 0.17 (N = 22) |
| Kidney weight (KW, mg) | 165.9 ± 6.5 (N = 16) | 146.1 ± 6.2* (N = 20) | 165.9 ± 4.5 (N = 17) | 134.8 ± 4.5 † (N = 22) |
| Biochemical parameters | ||||
| Urea (mg·L−1) | 516.1 ± 26.2 (N = 10) | 1103.7 ± 93.0* (N = 10) | 453.9 ± 47.6 (N = 10) | 1070.1 ± 91 † (N = 10) |
| BUN (mg·L−1) | 241.2 ± 12.2 (N = 10) | 515.4 ± 43.5* (N = 10) | 212.1 ± 22.2 (N = 10) | 500.0 ± 43 † (N = 10) |
| Phosphorus (mg·L−1) | 80.1 ± 9 (N = 10) | 72.2 ± 7 (N = 10) | 67.6 ± 7 (N = 10) | 79.0 ± 7 (N = 10) |
| FGF‐23 (pg·ml−1) | 144.6 ± 14.4 (N = 10) | 352.7 ± 41.7* (N = 10) | 138.6 ± 18.8 (N = 10) | 368.5 ± 39.5 † (N = 10) |
Note. Data for macroscopic and biochemical parameters per experimental group are reported as mean ± SEM. BUN, blood urea nitrogen; BW, body weight; FGF‐23, fibroblast growth factor 23; HW, heart weight; KW, kidney weight.
P < 0.05, significantly different from Sham;
P < 0.05, significantly different from Sham+rKL.
3.3. Increased diastolic Ca2+ leak induced by experimental chronic kidney disease is prevented by recombinant Klotho administration
We evaluated SR‐Ca2+ load in experimental CKD and the effect of rKL treatment. Figure 4a shows representative line‐scan images of caffeine‐induced Ca2+ release in each experimental group. CKD development led to a significant decrease in caffeine‐evoked Ca2+ transient amplitude (F/F 0) (Figure 4b), and this was partly prevented in rKL‐treated Nfx mice (Figure 4b). We tested whether the diastolic Ca2+ leak could be involved in the decreased SR‐Ca2+ content evident in CKD. We next analysed the diastolic Ca2+ leak by measuring Ca2+ spark frequency and SCR. Figure 4c shows line‐scan examples of Ca2+ spark recordings in the different groups of cardiomyocytes. Ca2+ spark frequency was significantly higher in Nfx cardiomyocytes than in Sham cardiomyocytes (Figure 4d). This increase was not observed in cardiomyocytes from Nfx mice treated with rKL (Figure 4d). We also analysed diastolic SCR in the form of Ca2+ waves (Figure 4e, left panel) and spontaneous Ca2+ transients (Figure 4e, right panel). The occurrence of SCR was 1.8‐fold higher in Nfx cardiomyocytes (as Ca2+ spark frequency shown in Figure 4d) than in Sham cardiomyocytes (Figure 4f). However, chronic rKL treatment in Nfx mice prevented the increase of cardiomyocytes with SCR: 4% in rKL‐treated Nfx mice and 20% in vehicle‐treated Nfx mice (Figure 4f), which was even lower than that from Sham or rKL‐treated Sham mice (12%, Figure 4f).
FIGURE 4.
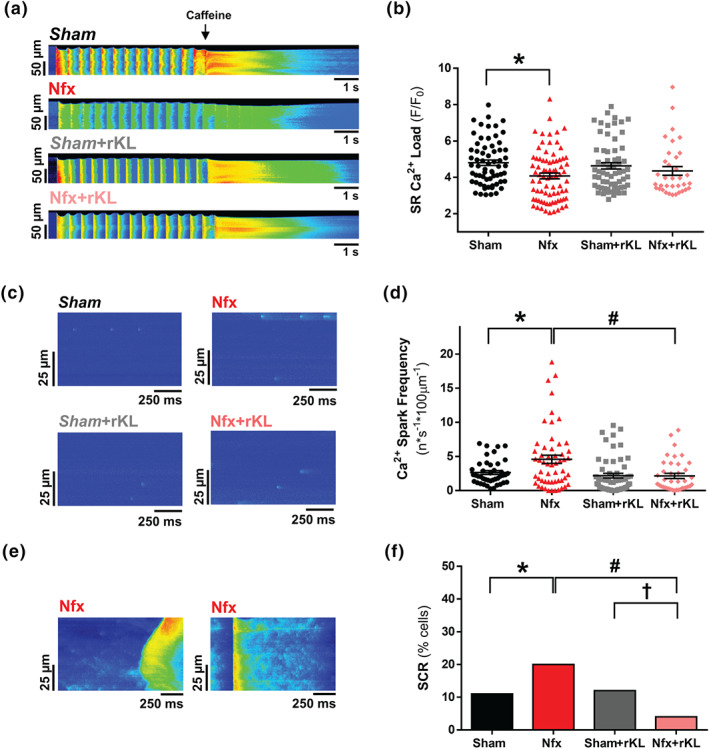
Recombinant Klotho prevents SR‐Ca2+ load and diastolic SR‐Ca2+ leak in experimental chronic kidney disease (CKD). (A) Line‐scan images of cardiomyocytes under 2‐Hz field stimulation perfused with caffeine. (B) Mean values of caffeine‐evoked Ca2+ transients amplitude expressed as peak (F/F 0) in Sham (n = 66 cells/N = 6 mice), Nfx (n = 79 cells/N = 6 mice), Sham+rKL (n = 67 cells/N = 6 mice), and Nfx+rKL (n = 49 cells/N = 6 mice) cardiomyocytes. (C) Line‐scan images of spark recordings in quiescent cardiomyocytes. (D) Mean values of Ca2+ spark frequency in Sham (1,270 sparks, n = 42 cells/N = 5 mice), Nfx (2,985 sparks, n = 55 cells/N = 6 mice), Sham+rKL (1,188 sparks, n = 56 cells/N = 5 mice), and Nfx+rKL (1,075 sparks, n = 39 cells/N = 6 mice). (E) Line‐scan images of spontaneous Ca2+ release (SCR) as Ca2+ waves (left panel) or spontaneous Ca2+ transients (right panel) in quiescent Nfx cardiomyocytes. (F) Occurrence of SCR in Sham (n = 42 cells/N = 5 mice), Nfx (n = 55 cells/N = 6 mice), Sham+rKL (n = 56 cells/N = 5 mice), and Nfx+rKL (n = 39 cells/N = 6 mice) cardiomyocytes. Data are shown as mean ± SEM. * P < 0.05, significantly different from Sham; # P < 0.05, significantly different from Nfx; and † P < 0.05, significantly different from Sham+rKL
3.4. Chronic recombinant Klotho treatment prevents pro‐arrhythmogenic Ca2+ events, RyR hyperactivity, and phosphorylation promoted by experimental chronic kidney disease
We examined for the presence of arrhythmic behaviour dependent on Ca2+ cycling alterations during electrical pacing in the experimental CKD model. Figure 5a shows representative line‐scan Ca2+ images of ventricular cardiomyocytes from Sham or Nfx mice, with or without rKL treatment and exposed to trains of electrical stimulation. When compared with Sham, Nfx cardiomyocytes presented a significant increase of automatic Ca2+ transients triggering automatic contractions between electrical pulses and after electrical pacing, and this was prevented by rKL treatment (Figure 5b). Given these findings, we next assessed whether alterations in RyR sensitivity could explain the arrhythmic behaviour in experimental CKD. Considering that ryanodine binds only to open RyRs, we determined the levels of functionally active RyRs in equivalent systolic and diastolic Ca2+ conditions in vitro. During the systolic Ca2+ recordings, we found a significant 2.9‐fold increase in [3H]‐ryanodine binding in Nfx hearts (Figure 6a), and this activity was effectively decreased by rKL, although it did not reach the levels in Sham animals, suggesting that additional mechanisms (i.e., SR‐Ca2+ load) participate in decreasing the activity of RyRs. During diastolic Ca2+ recordings, Nfx hearts showed a significant 3.2‐fold increase in the binding of [3H]‐ryanodine at 100 nmol L‐1 of free [Ca2+] (Figure 6b), and again, rKL treatment reduced the Ca2+ sensitivity of RyRs in Nfx hearts (Figure 6b). As Nfx did not alter total RyR protein expression levels (Figure 6c and d, upper panels), the results indicate that RyRs from Nfx hearts are more sensitive to Ca2+. To determine the mechanism involved in RyR hyperactivity, we examined the phosphorylation status of cardiac RyR at two serine residues: Ser2030, key phosphorylation site for protein kinase A (PKA), and Ser2814, the specific phosphorylation site for Ca2+/calmodulin‐dependent protein kinase II (CaMKII). RyR phosphorylation at Ser2030 and Ser2814 was significantly higher in extracts from Nfx mice than from Sham mice (Figure 6c and d). This increased phosphorylation for Ser2030 and Ser2814 was not observed in Nfx mice treated with rKL, where basal levels of phosphorylation were similar to those of Sham mice.
FIGURE 5.
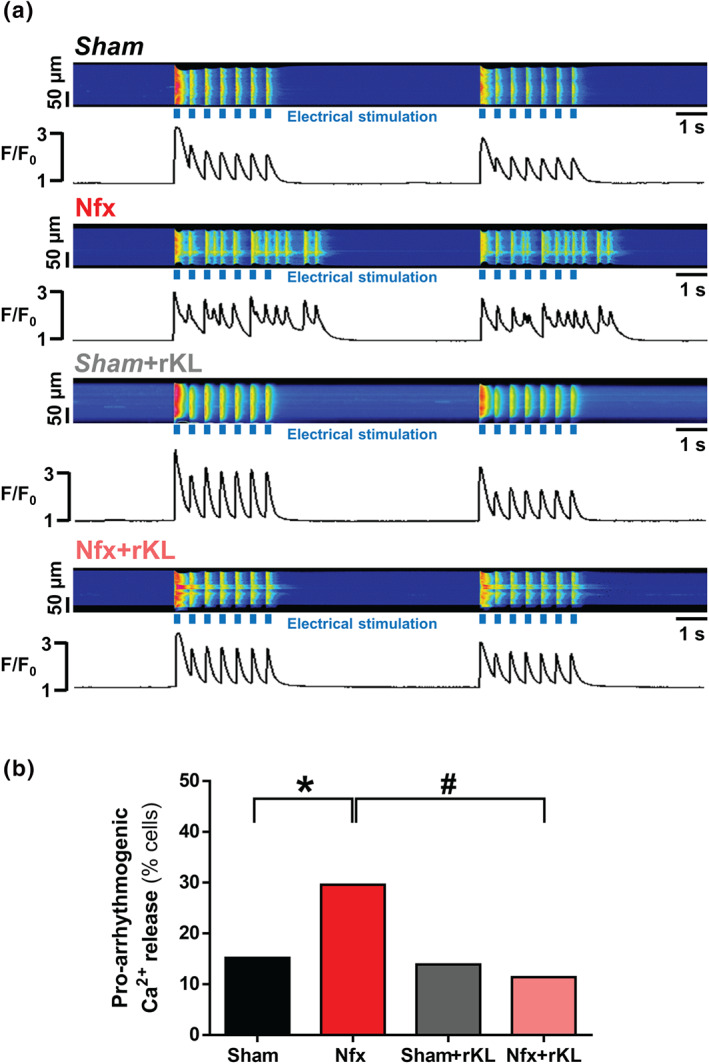
Recombinant Klotho treatment impedes pro‐arrhythmogenic events induced by Nfx. (A) Line‐scan images and fluorescence profiles of cardiomyocytes paced at 2‐Hz field stimulation. (B) Occurrence of pro‐arrhythmogenic events related to automatic Ca2+ I transients and automatic contractions in Sham (n = 92 cells/N = 5 mice), Nfx (n = 142 cells/N = 6 mice), Sham+rKL (n = 72 cells/N = 5 mice), and Nfx+rKL (n = 149 cells/N = 7 mice) mice. Data are shown as mean ± SEM. * P < 0.05, significantly different from Sham; # P < 0.05, significantly different from Nfx
FIGURE 6.
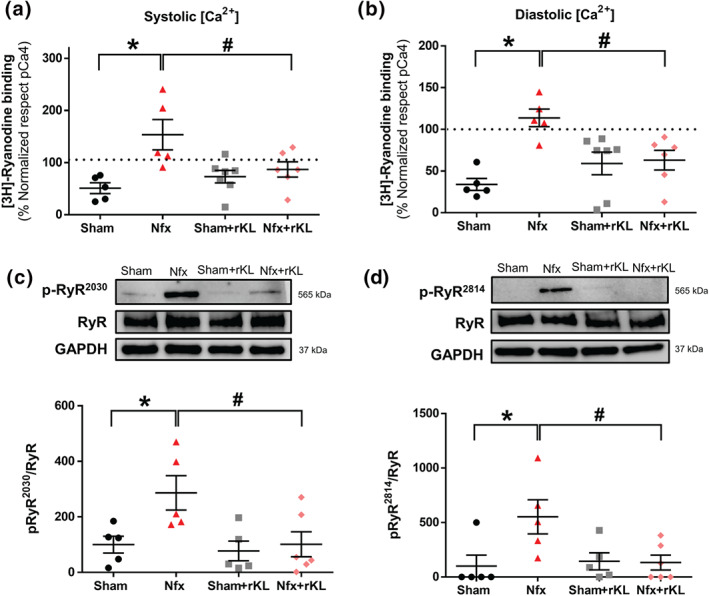
Recombinant Klotho protects against RyR hyperactivation in experimental chronic kidney disease (CKD). (A and B) Specific [3H]‐ryanodine binding at (A) 10 μM (equivalent to the intracellular Ca2+ rise in systole) and (B) 100 nM (equivalent to basal levels of intracellular Ca2+ in quiescent conditions or diastole) and free Ca2+ concentrations of heart homogenates from Sham (N = 5 mice), Nfx (N = 5 mice), Sham+rKL (N = 7 mice), and Nfx+rKL (N = 6 mice). [3H]‐Ryanodine binding values were normalised to the specific [3H]‐ryanodine binding at 100 μmol L‐1 of free [Ca2+]. (C and D) Western blots (upper panels) and quantification (bottom panels) of RyR phosphorylation at Ser2030 (p‐RyR2030) or at Ser2814 (p‐RyR2814) normalised to total RyR from Sham (N = 5 mice), Nfx (N = 7 mice), Sham+rKL (N = 5 mice), and Nfx+rKL (N = 7 mice) hearts. Data are shown as mean ± SEM. * P < 0.05 significantly different from Sham; # P < 0.05, significantly different from Nfx
3.5. Klotho overexpression protects against cardiomyocyte contractile dysfunction and pro‐arrhythmic Ca2+ events in chronic kidney disease but does not eliminate renal damage
We used Tg‐Kl mice to examine the cardioprotective role of endogenous Klotho overexpression in a uraemic context. Serum levels of BUN were significantly higher in Tg‐Kl mice subjected to Nfx than Sham‐Tg‐Kl mice (Figure 7a). No differences were observed in serum phosphorus levels between both groups (Figure 7b), but serum FGF‐23 levels were significantly higher in Nfx‐Tg‐Kl mice than in Sham‐Tg‐Kl mice (Figure 7c). Analysis of Ca2+ cycling in isolated cardiomyocytes from both groups revealed no differences in Ca2+ spark frequency (Figure 7d), SCR (Figure 7e), SR Ca2+ load (Figure 7f), intracellular Ca2+ transients (Figure 7g), or cell shortening (Figure 7h) between them. Overall, these results show that endogenous Klotho overexpression protects against changes in Ca2+ cycling, preventing contractile dysfunction and diastolic pro‐arrhythmogenic Ca2+ release in Nfx mice.
FIGURE 7.
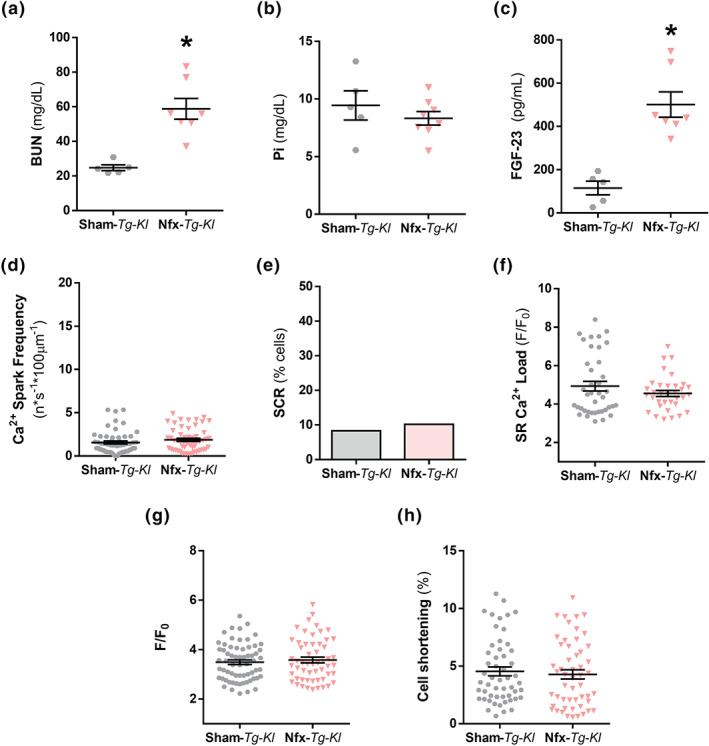
Klotho overexpression protects from electrical cardiac and Ca2+ cycling alterations induced by chronic kidney disease (CKD). (A–C) Mean serum levels of (A) blood urea nitrogen (BUN), (B) phosphates (Pi), and (C) fibroblast growth factor‐23 (FGF‐23) in Sham‐Tg‐Kl (N = 5 mice) and Nfx‐Tg‐Kl (N = 7 mice). (D) Mean values of Ca2+ spark frequency in Sham‐Tg‐Kl (995 sparks, n = 54 cells/N = 5 mice) and Nfx‐Tg‐Kl (893 sparks, n = 55 cells/N = 6 mice). (E) Occurrence of SCR in Sham‐Tg‐Kl (n = 54 cells/N = 5 mice) and Nfx‐Tg‐Kl (n = 55 cells/N = 6 mice). (F) Mean values of caffeine‐evoked Ca2+ transients amplitude expressed as peak (F/F 0) in Sham‐Tg‐Kl (n = 40 cells/N = 5 mice) and Nfx‐Tg‐Kl (n = 34 cells/N = 6 mice). (G) Mean values of peak (F/F 0) of electrically evoked Ca2+ transients in Sham‐Tg‐Kl (n = 64 cells/N = 5 mice) and Nfx‐Tg‐Kl (n = 55 cells/N = 6 mice). (H) Percentage of cell contraction of Sham‐Tg‐Kl (n = 56 cells/N = 5 mice) and Nfx‐Tg‐Kl (n = 52 cells/N = 6 mice). Data are shown as mean ± SEM. * P < 0.05, significantly different from Sham‐Tg‐Kl
4. DISCUSSION
Klotho was initially discovered as an age‐suppressing gene in mice that extends lifespan when overexpressed (Kuro‐o et al., 1997). Klotho is expressed mainly in the kidney and is composed of a very short intracellular domain, a transmembrane domain, and a large extracellular domain that is susceptible to ectodomain shedding, resulting in two forms, membrane and secreted or sKL (Kuro‐o, 2011a). In the kidney, membrane Klotho acts as a co‐receptor for FGF‐23, increasing the affinity of FGFRs for FGF‐23‐mediated phosphaturic actions (Urakawa et al., 2006). sKL is a humoral factor with pleiotropic functions, including a cardioprotective action through the regulation of ion channels (Xie et al., 2012) that is independent of FGF‐23 and phosphate (Xie et al., 2015). CKD is considered a Klotho‐deficient state (Asai et al., 2012; Kuro‐O, 2011b). CKD is accompanied by premature aging and is considered a global health concern, because of its high prevalence of around 10% in the general population (Coresh et al., 2007). At the experimental level, models of premature aging, such as Klotho‐hypomorphic mice, develop early‐stage CKD. In addition, other authors have recently described that uremic environment increases with aging in the experimental CKD model (Heveran et al., 2019). However, no studies have described overlapping effects of CKD and aging on cardiac function. Cardiac dysfunction and fatal arrhythmias, in many cases asymptomatic ventricular arrhythmias, are the leading causes of mortality in patients with CKD, especially in those with end‐stage renal disease (Banerjee, 2016; Verde et al., 2016). However, the underlying mechanisms involved in this cardiorenal interaction remain to be defined and clinicians remain uncertain about how to reduce the burden of cardiovascular events in CKD.
Our study provides the first demonstration, to our knowledge, that experimental uraemia induced by Klotho deficiency or nephrectomy with reduced Klotho levels is linked to a strong defect in Ca2+ handling, characterised by decreased systolic Ca2+ release and contractile dysfunction and increased diastolic pro‐arrhythmogenic Ca2+ release and leak, accompanied by RyR hyperactivity. Our data also support the concept that preserving Klotho availability, exogenously using prophylactic chronic treatment with rKL or endogenously enhancing Klotho expression, could be potential therapeutic strategies to protect the heart from the uraemic environment, including alterations in intracellular Ca2+ cycling that trigger contractile dysfunction and pro‐arrhythmic mechanisms such as the hyperactivation of RyRs observed in CKD (see Graphical Abstract). Our results show an increase in RyR activity in Nfx mice with higher spontaneous diastolic Ca2+ release and a diminution in SR‐Ca2+ load. Both mechanisms are known to be associated with cardiac dysfunction and the predisposition to arrhythmias. The decrease in systolic Ca2+ release accompanied by a worsened contractile response, concomitant with extra‐contractions, and Ca2+ leak in the form of Ca2+ sparks and waves during diastole were pro‐arrhythmic Ca2+ events in cardiomyocytes from kl/kl and CKD mice. Furthermore, we identified a CKD‐related increase in RyR phosphorylation as an underlying molecular explanation for the high RyR activity. Accordingly, RyR was robustly phosphorylated at its PKA (3‐fold higher) and CaMKII (5‐fold higher) sites in CKD mice. Similar results of PKA‐mediated and CaMKII‐mediated RyR phosphorylation and its relationship with the predisposition to trigger fatal ventricular arrhythmias have also been found in experimental models of heart failure (Grimm et al., 2015; Xiao et al., 2005). This post‐translational RyR modification of increased phosphorylation can be explained by the higher Ca2+ sensitivity of RyR at systolic and diastolic Ca2+ concentrations in CKD mice, which was prevented by rKL administration. Dhindwal et al. proposed that phosphorylation induces RyR to adopt a protein conformation that requires less energy for the transition to the open state (Dhindwal et al., 2017). Therefore, a more flexible RyR conformation might exist when the phosphorylation state is significantly increased, as in CKD mice, in both phosphorylation sites (Ser2030 and Ser2814). Moreover, a similar increase in Ca2+ sensitivity has been observed in ventricular arrhythmia in experimental catecholaminergic polymorphic ventricular tachycardia (Fernández‐Velasco et al., 2009). Gender‐related differential activation of PKA and CaMKII pathways exists in rodent hearts also under pathological conditions (Bell et al., 2015; Parks, Ray, Bienvenu, Rose, & Howlett, 2014). For example, in diabetic cardiomyopathy, the contribution of PKA‐dependent and CaMKII‐dependent pathways is less relevant in female mice than in male mice, with a significant diminution of PKA‐induced and CaMKII‐induced RyR phosphorylation sites correlating with decreased spark‐mediated calcium leak in female mice (Delgado, Gomez, Samia El Hayek, Ruiz‐Hurtado, & Pereira, 2019; Pereira et al., 2014). Thus, it would be very likely that these pathways could be also less active in female mice than in male mice, in the setting of uremic cardiomyopathy. However, in our study, female mice were not studied, to avoid the potential cardioprotective effects mediated by oestrogens, which could be overlapping the Klotho‐mediated cardioprotective actions in experimental conditions of CKD. Further experimental studies are needed to assess the contribution of oestrogens to the Ca2+ mishandling observed under uremic cardiomyopathy conditions.
This study has several potential clinical and therapeutic applications for renal patients. First, our results show that decreased Klotho availability, together with the presence of pathologically elevated FGF‐23 levels, might be a warning sign for increased risk of developing arrhythmic events in patients with CKD. Supporting this idea, several studies have previously shown that FGF‐23 exposure alters cardiac contractility and Ca2+ handling in isolated adult ventricular cardiomyocytes (Navarro‐García et al., 2019; Touchberry et al., 2013) and in HL‐1 atrial cells (Kao et al., 2014). These findings support the notion that systemic elevation of FGF‐23 is likely to have deleterious and direct effects on heart function in the complex pathological setting of uraemic cardiomyopathy (Faul, 2018). However, in the clinical setting, further studies are still needed to establish whether FGF‐23 itself is an independent uremic factor able to play a relevant role in the cardiac events accompanying the decline of renal function. Accordingly, a logical experimental approach would be the use of FGF‐23 blocking antibodies as the most direct strategy to inhibit FGF‐23 actions in the uraemic context. Yet in a preclinical model of CKD, rats treated with a neutralising FGF‐23 antibody presented elevated systemic serum phosphate and developed severe cardiovascular complications (Shalhoub et al., 2012). Thus, the key point would be not to fully suppress FGF‐23 actions, such as the physiological phosphaturic action, but only to block its damaging actions on the heart. The second application would involve exogenous Klotho administration as a therapeutic option to block the deleterious FGF‐23 actions on the heart. Klotho is emerging as a potential cardioprotective factor for uraemic cardiomyopathy. Indeed, Klotho protects against vascular calcification in human (Lim et al., 2012) and experimental models (Hu et al., 2011) of CKD. Moreover, sKL deficiency in CKD seems to render the myocardium more susceptible to stress‐induced injury (Xie et al., 2012). Similarly, some authors have proposed that FGF‐23 induces cardiac toxicity only when serum levels of sKL are reduced in CKD (Hu et al., 2015), whereas other authors posit that Klotho prevents uraemic cardiomyopathy by a direct effect on cardiomyocytes independently of FGF‐23 and phosphates (Xie et al., 2015). Nevertheless, clinical studies on the predictive value of Klotho have produced conflicting results, with some reporting that plasma sKL levels did not predict cardiovascular events or death in a large CKD cohort (Seiler et al., 2013), while others demonstrated a clear association between preserved sKL levels and reduced cardiovascular morbidity and mortality in dialysis‐dependent CKD (Hu et al., 2011). The idea that sKL could function as a decoy of FGF‐23 actions in the heart is in agreement with our results. Thus, we observed a cardioprotective effect of rKL administration on preventing contractile dysfunction and diastolic Ca2+ leak related to pro‐arrhythmic events in cardiomyocytes from Nfx mice. In this respect, Klotho has been shown to produce beneficial cardiac actions on ionic channels, including the downregulation of TRPC6 channels in conditions of cardiac hypertrophy (Xie et al., 2012; Xie et al., 2015). However, in physiological conditions, the contribution of TRPC6 channels to the total Ca2+ entry in cardiomyocytes is small and is higher in conditions of cardiac hypertrophy. In the present study, we found no evidence of cardiac morphology changes such as hypertrophy or fibrosis in Klotho‐hypomorphic mice and mice after 6 weeks of 5/6 nephrectomy. The kl/kl mice showed an increase in HW/BW ratio that was not accompanied by an increase in cardiomyocyte area, or ANP and collagen expression as specific markers of cellular hypertrophy or fibrosis, respectively. A possible explanation for these results is that Klotho‐hypomorphic mice showed reduced BW compared with +/+ mice and they also present an important growth retardation affecting all organs, which could explain the reduced cardiomyocyte areas or collagen levels, compared with their +/+ littermates. Besides, the absence of Klotho expression also induces important ectopic calcification in arterial walls and cardiac muscle (Kuro‐o et al., 1997; Yoshida, Fujimori, & Nabeshima, 2002). All these facts might favour a higher HW/BW ratio than that expected for its body weight and animal size. Therefore, these results support the conclusion that Ca2+ mishandling observed in kl/kl mice is not due to cardiomyocyte hypertrophy or deleterious remodelling development, although the influence of systemic calcaemia and alterations in vitamin D homeostasis in this Klotho mutant mice (Yoshida et al., 2002) could not be discarded, especially on the elevated diastolic Ca2+ release and pro‐arrhythmic events of kl/kl cardiomyocytes.
In general, the experimental CKD model shows a compromised EF and cardiac dysfunction in mice (Bro, Bollano, Brüel, Olgaard, & Nielsen, 2008; Chen et al., 2017). However, there are apparently opposite results related to hypertrophy development that might appear together with this cardiac dysfunction. Thus, some authors have described Nfx‐induced cardiac dysfunction with a significant increment of the heart weight (Chen et al., 2017), while other authors using the same experimental CKD model did not observe any increase in the heart weight (Bro et al., 2008) as we have also observed in the present study. These apparently opposing results could be explained by the age of the animals used and also the time employed from 5/6 Nfx surgery to the cardiac analysis. In our hands, this experimental model of CKD developed a dysfunctional cardiac phenotype without hypertrophy or fibrosis development. This is an ideal in vivo model to study functional cardiac events such as heart failure and the predisposition to arrhythmia related to Ca2+ mishandling in the context of uraemic cardiomyopathy without any overlap due to structural myocardial alterations. Using this model, we have demonstrated a cardioprotective role of Klotho through the normalisation of cardiomyocyte Ca2+ cycling. Mechanistically, we propose that RyRs are a novel target of Klotho, and Klotho could act as a “brake” for the pro‐arrhythmogenic Ca2+ leak from the SR by impeding phosphorylation and hyperactivation of RyRs previously observed in CKD. By halting Ca2+ leakage, Klotho helps to maintain an adequate cellular shortening due to the recovery of systolic Ca2+ release in the form of Ca2+ transients and, at the same time, impedes the increase in diastolic Ca2+ release in the form of Ca2+ sparks or waves that might favour reaching a intracellular Ca2+ threshold able to evoke automatic Ca2+ transients and extra‐contractions. Finally, one of the most relevant findings of our study is that we show for the first time that Klotho can protect the heart from Ca2+ mishandling associated with CKD, without any associated improvement in renal dysfunction. Thus, Nfx mice overexpressing Klotho or chronically treated with rKL present the same high systemic levels of FGF‐23 and BUN as those Nfx mice treated with vehicle. We believe these findings are clinically relevant because strategies directed to maintain adequate Klotho levels could be a new therapeutic goal to directly protect the heart from a uraemic environment and guaranteeing FGF‐23 phosphaturic action.
One limitation of the present study is the lack of data in larger animal models that exhibit closer physiological characteristics to humans, such as rabbits or pigs. Moreover, further investigation will be required to determine, in detail, whether the Klotho deficiency, a very common aspect in CKD patients, might influence diastolic Ca2+ levels and therefore compromise cardiac contractility. We envision the need to corroborate the cardioprotective role of Klotho throughout the development of renal disease from initial stages, including the use of larger animal models in further studies.
In conclusion, this study has provided new insights into how Klotho availability determines cardiac function in uraemic cardiomyopathy via regulation of Ca2+ handling. The present investigation revealed the dysregulated cardiac physiological mechanisms in uraemia modelled by Klotho deficiency or nephrectomy, including defects in systolic Ca2+ release and contractile dysfunction together with RyR hyperactivation, which can explain the elevated presence of pro‐arrhythmogenic Ca2+ events found in a uraemic setting. All of these alterations in Ca2+ handling observed in CKD mice were prevented when Klotho was present, indicating that therapeutic strategies directed to improve Klotho availability might be an effective approach to protecting the heart from cardiac dysfunction and arrhythmogenic potential events in CKD.
AUTHOR CONTRIBUTIONS
G.R.‐H. and J.A.N.‐G. designed and conceptualised the experiments. J.A.N.‐G., T.R.‐G., and G.R.‐H. performed the experiments. J.A.N.‐G., A.R., T.R.‐G., E.R.‐S., L.G.‐L., and G.R.‐H. analysed the data. C.Z. carried out, analysed, and interpreted the echocardiograms. G.R.‐H., M.F.‐V., L.M.R., and M.K. provided reagents and materials. J.A.N.‐G., M.F.‐V., L.M.R., and M.K. helped with data interpretation. J.A.N.‐G. and G.R.‐H. wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry and Animal Experimentation, and as recommended by funding agencies, publishers, and other organisations engaged with supporting research.
Supporting information
Figure S1. Change in peak amplitude as ΔF/F0=(F‐F0/F0) in cardiomyocytes from +/+ (n=50 cells/N=5 mice) and kl/kl (n=49 cells/N=5 mice) mice. Data are shown as mean±SEM. *P<0.05 vs. +/+ mice.
Figure S2. Representative normalisation of Ca2+ transients to the peak of fluorescence obtained in cardiomyocytes from a +/+ (dark grey line) and kl/kl (dark red line) mice.
Figure S3. mRNA expression of SERCA2a (Atp2a2) in +/+ (N=6 mice) and kl/kl (N=6 mice) mice. Data are shown as mean±SEM. *P<0.05 vs. +/+ mice.
Figure S4: K SERCA in cardiomyocytes from +/+ (n=18 cells/N=5 mice) and kl/kl (n=27 cells/N=5 mice) mice. Data are shown as mean±SEM. *P<0.05 vs. +/+ mice.
Figure S5. Time of decay (Tau) of caffeine‐evoked intracellular Ca2+ transients in +/+ (n=37 cells/N=5 mice) and kl/kl (n=46 cells/N=5 mice) mice.
Figure S6. Urine levels of sKL in Sham (N=7 mice) compared to Nfx mice (N=10 mice). *P<0.05 vs. Sham.
Figure S7. Representative normalisation of Ca2+ transients to the same peak of fluorescence obtained in cardiomyocytes from a Sham (black line) and Nfx (red line) mice.
Figure S8: K SERCA in cardiomyocytes from Sham (n=35 cells/N=5 mice) and Nfx (n=48 cells/N=6 mice) mice. Data are shown as mean±SEM. *P<0.05 vs. Sham mice.
ACKNOWLEDGEMENTS
We thank Dr. Monserrat Grau Sanz for her excellent assistance with experimental animal facilities, Dr. Inés García‐Consuegra Galiana (from the Proteomic facility of Hospital 12 de Octubre (i+12)) for her technical assistance, Dr. Agustín Guerrero‐Hernández (Department of Biochemistry, Cinvestav‐IPN) for helpful recommendations on Ca2+ measurement analysis, and Dr. Blanca Miranda for her continuous support. This work was supported by several projects from the Instituto de Salud Carlos III, Ministry of Economy, Industry and Competitiveness (PI14/01841, CP15/00129, PI17/01093, PI17/01193, PI14/01078, PI17/01344, and MSII16/00047), Fundación SENEFRO, Sociedad Española de Cardiología (SEC), and Fundación Renal Íñigo Álvarez de Toledo (FRIAT), co‐funded by the European Regional Development Fund (Fondos FEDER), and approved by the ethics committees of our institution.
Navarro‐García JA, Rueda A, Romero‐García T, et al. Enhanced Klotho availability protects against cardiac dysfunction induced by uraemic cardiomyopathy by regulating Ca2+ handling. Br J Pharmacol. 2020;177:4701–4719. 10.1111/bph.15235
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators . (2019a). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Catalytic receptors. British Journal of Pharmacology, 176, S247–S296. 10.1111/bph.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators . (2019b). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … CGTP Collaborators . (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology, 176, S397–S493. 10.1111/bph.14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Mathie, A. , Peters, J. A. , Veale, E. L. , Striessnig, J. , Kelly, E. , … CGTP Collaborators . (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology, 176, S142–S228. 10.1111/bph.14749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai, O. , Nakatani, K. , Tanaka, T. , Sakan, H. , Imura, A. , Yoshimoto, S. , … Saito, Y. (2012). Decreased renal α‐Klotho expression in early diabetic nephropathy in humans and mice and its possible role in urinary calcium excretion. Kidney International, 81, 539–547. 10.1038/ki.2011.423 [DOI] [PubMed] [Google Scholar]
- Banerjee, D. (2016). Sudden cardiac death in haemodialysis: Clinical epidemiology and mechanisms. Journal of Electrocardiology, 49, 843–847. 10.1016/j.jelectrocard.2016.07.016 [DOI] [PubMed] [Google Scholar]
- Bell, J. R. , Raaijmakers, A. J. , Curl, C. L. , Reichelt, M. E. , Harding, T. W. , Bei, A. , … Delbridge, L. M. (2015). Cardiac CaMKIIδ splice variants exhibit target signaling specificity and confer sex‐selective arrhythmogenic actions in the ischemic‐reperfused heart. International Journal of Cardiology, 181, 288–296. 10.1016/j.ijcard.2014.11.159 [DOI] [PubMed] [Google Scholar]
- Bergmark, B. A. , Udell, J. A. , Morrow, D. A. , Jarolim, P. , Kuder, J. F. , Solomon, S. D. , … Sabatine, M. S. (2019). Klotho, fibroblast growth factor‐23, and the renin‐angiotensin system—an analysis from the PEACE trial. European Journal of Heart Failure, 21, 462–470. 10.1002/ejhf.1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers, D. M. (2002). Cardiac excitation–contraction coupling. Nature, 415, 198–205. 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- Bers, D. M. (2014). Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annual Review of Physiology, 76, 107–127. 10.1146/annurev-physiol-020911-153308 [DOI] [PubMed] [Google Scholar]
- Bode, E. F. , Briston, S. J. , Overend, C. L. , O'Neill, S. C. , Trafford, A. W. , & Eisner, D. A. (2011). Changes of SERCA activity have only modest effects on sarcoplasmic reticulum Ca2+ content in rat ventricular myocytes. The Journal of Physiology, 589, 4723–4729. 10.1113/jphysiol.2011.211052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bro, S. , Bollano, E. , Brüel, A. , Olgaard, K. , & Nielsen, L. B. (2008). Cardiac structure and function in a mouse model of uraemia without hypertension. Scandinavian Journal of Clinical and Laboratory Investigation, 68, 660–666. 10.1080/00365510802037272 [DOI] [PubMed] [Google Scholar]
- Charytan, D. M. , Foley, R. , McCullough, P. A. , Rogers, J. D. , Zimetbaum, P. , Herzog, C. A. , … MiD Investigators and Committees . (2016). Arrhythmia and sudden death in hemodialysis patients: protocol and baseline characteristics of the monitoring in dialysis study. Clinical Journal of the American Society of Nephrology, 11, 721–734. 10.2215/CJN.09350915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Kieswich, J. E. , Chiazza, F. , Moyes, A. J. , Gobbetti, T. , Purvis, G. S. , … Collino, M. (2017). IκB kinase inhibitor attenuates sepsis‐induced cardiac dysfunction in CKD. Journal of the American Society of Nephrology, 28, 94–105. 10.1681/ASN.2015060670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, H. , Song, L. S. , Shirokova, N. , González, A. , Lakatta, E. G. , Ríos, E. , … Stern, M. D. (1999). Amplitude distribution of calcium sparks in confocal images: Theory and studies with an automatic detection method. Biophysical Journal, 76, 606–617. 10.1016/S0006-3495(99)77229-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll, M. , Ferrer‐Costa, C. , Pich, S. , Allegue, C. , Rodrigo, E. , Fernández‐Fresnedo, G. , … Brugada, R. (2018). Role of genetic and electrolyte abnormalities in prolonged QTc interval and sudden cardiac death in end‐stage renal disease patients. PLoS ONE, 13, e0200756 10.1371/journal.pone.0200756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coresh, J. , Selvin, E. , Stevens, L. A. , Manzi, J. , Kusek, J. W. , Eggers, P. , … Levey, A. S. (2007). Prevalence of chronic kidney disease in the United States. JAMA, 298, 2038–2047. 10.1001/jama.298.17.2038 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. P. H. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alba‐Aguayo, D. R. , Pavón, N. , Mercado‐Morales, M. , Miranda‐Saturnino, M. , López‐Casamichana, M. , Guerrero‐Hernández, A. , … Rueda, A. (2017). Increased calcium leak associated with reduced calsequestrin expression in hyperthyroid cardiomyocytes. Cell Calcium, 62, 29–40. 10.1016/j.ceca.2017.01.009 [DOI] [PubMed] [Google Scholar]
- Delgado, C. , Gomez, A. M. , Samia El Hayek, M. , Ruiz‐Hurtado, G. , & Pereira, L. (2019). Gender‐dependent alteration of Ca. Frontiers in Physiology, 10, 40 10.3389/fphys.2019.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhindwal, S. , Lobo, J. , Cabra, V. , Santiago, D. J. , Nayak, A. R. , Dryden, K. , & Samsó, M. (2017). A cryo‐EM‐based model of phosphorylation‐ and FKBP12.6‐mediated allosterism of the cardiac ryanodine receptor. Science Signaling, 10(480), eaai8842 10.1126/scisignal.aai8842 [DOI] [PubMed] [Google Scholar]
- Faul, C. (2018). FGF23 effects on the heart‐levels, time, source, and context matter. Kidney International, 94, 7–11. 10.1016/j.kint.2018.03.024 [DOI] [PubMed] [Google Scholar]
- Fernández‐Velasco, M. , Rueda, A. , Rizzi, N. , Benitah, J. P. , Colombi, B. , Napolitano, C. , … Gómez, A. M. (2009). Increased Ca2+ sensitivity of the ryanodine receptor mutant RyR2R4496C underlies catecholaminergic polymorphic ventricular tachycardia. Circulation Research, 104, 201–209212p following 209. 10.1161/CIRCRESAHA.108.177493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon, R. F. , & Gallimore, B. (1988). Characterization of a mouse model of chronic uremia. Urological Research, 16, 119–126. 10.1007/BF00261969 [DOI] [PubMed] [Google Scholar]
- Go, A. S. , Chertow, G. M. , Fan, D. , McCulloch, C. E. , & Hsu, C. Y. (2004). Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. The New England Journal of Medicine, 351, 1296–1305. 10.1056/NEJMoa041031 [DOI] [PubMed] [Google Scholar]
- Grimm, M. , Ling, H. , Willeford, A. , Pereira, L. , Gray, C. B. , Erickson, J. R. , … Brown, J. H. (2015). CaMKIIδ mediates β‐adrenergic effects on RyR2 phosphorylation and SR Ca(2+) leak and the pathophysiological response to chronic β‐adrenergic stimulation. Journal of Molecular and Cellular Cardiology, 85, 282–291. 10.1016/j.yjmcc.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez, O. M. , Mannstadt, M. , Isakova, T. , Rauh‐Hain, J. A. , Tamez, H. , Shah, A. , … Wolf, M. (2008). Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. The New England Journal of Medicine, 359, 584–592. 10.1056/NEJMoa0706130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog, C. A. , Asinger, R. W. , Berger, A. K. , Charytan, D. M. , Díez, J. , Hart, R. G. , … Ritz, E. (2011). Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney International, 80, 572–586. 10.1038/ki.2011.223 [DOI] [PubMed] [Google Scholar]
- Heveran, C. M. , Schurman, C. A. , Acevedo, C. , Livingston, E. W. , Howe, D. , Schaible, E. G. , … Ferguson, V. L. (2019). Chronic kidney disease and aging differentially diminish bone material and microarchitecture in C57Bl/6 mice. Bone, 127, 91–103. 10.1016/j.bone.2019.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, M. C. , Shi, M. , Cho, H. J. , Adams‐Huet, B. , Paek, J. , Hill, K. , … Moe, O. W. (2015). Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. Journal of the American Society of Nephrology, 26, 1290–1302. 10.1681/ASN.2014050465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, M. C. , Shi, M. , Gillings, N. , Flores, B. , Takahashi, M. , Kuro‐O, M. , & Moe, O. W. (2017). Recombinant α‐Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney International, 91, 1104–1114. 10.1016/j.kint.2016.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, M. C. , Shi, M. , Zhang, J. , Quiñones, H. , Griffith, C. , Kuro‐o, M. , … Moe, O. W. (2011). Klotho deficiency causes vascular calcification in chronic kidney disease. Journal of the American Society of Nephrology, 22, 124–136. 10.1681/ASN.2009121311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakova, T. , Cai, X. , Lee, J. , Xie, D. , Wang, X. , Mehta, R. , … Chronic Renal Insufficiency Cohort (CRIC) Study Investigators . (2018). Longitudinal FGF23 trajectories and mortality in patients with CKD. Journal of the American Society of Nephrology, 29, 579–590. 10.1681/ASN.2017070772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakova, T. , Xie, H. , Yang, W. , Xie, D. , Anderson, A. H. , Scialla, J. , … Chronic Renal Insufficiency Cohort (CRIC) Study Group . (2011). Fibroblast growth factor 23 and risks of mortality and end‐stage renal disease in patients with chronic kidney disease. JAMA, 305, 2432–2439. 10.1001/jama.2011.826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, Y. H. , Chen, Y. C. , Lin, Y. K. , Shiu, R. J. , Chao, T. F. , Chen, S. A. , … Chen, Y. J. (2014). FGF‐23 dysregulates calcium homeostasis and electrophysiological properties in HL‐1 atrial cells. European Journal of Clinical Investigation, 44, 795–801. 10.1111/eci.12296 [DOI] [PubMed] [Google Scholar]
- Kuro‐o, M. (2011a). Klotho and the aging process. The Korean Journal of Internal Medicine, 26, 113–122. 10.3904/kjim.2011.26.2.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro‐O, M. (2011b). Phosphate and Klotho. Kidney International, 121, S20–S23. 10.1038/ki.2011.26 [DOI] [PubMed] [Google Scholar]
- Kuro‐o, M. , Matsumura, Y. , Aizawa, H. , Kawaguchi, H. , Suga, T. , Utsugi, T. , … Nabeshima, Y. I. (1997). Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature, 390, 45–51. 10.1038/36285 [DOI] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. H. , Cirino, G. , Docherty, J. R. , … Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology. https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K. , Lu, T. S. , Molostvov, G. , Lee, C. , Lam, F. T. , Zehnder, D. , … Hsiao, L. L. (2012). Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation, 125, 2243–2255. 10.1161/CIRCULATIONAHA.111.053405 [DOI] [PubMed] [Google Scholar]
- Memmos, E. , Sarafidis, P. , Pateinakis, P. , Tsiantoulas, A. , Faitatzidou, D. , Giamalis, P. , … Papagianni, A. (2019). Soluble Klotho is associated with mortality and cardiovascular events in hemodialysis. BMC Nephrology, 20, 217 10.1186/s12882-019-1391-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattel, S. , Maguy, A. , Le Bouter, S. , & Yeh, Y. H. (2007). Arrhythmogenic ion‐channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiological Reviews, 87, 425–456. 10.1152/physrev.00014.2006 [DOI] [PubMed] [Google Scholar]
- Navarro‐García, J. A. , Delgado, C. , Fernández‐Velasco, M. , Val‐Blasco, A. , Rodríguez‐Sánchez, E. , Aceves‐Ripoll, J. , … Ruiz‐Hurtado, G. (2019). Fibroblast growth factor‐23 promotes rhythm alterations and contractile dysfunction in adult ventricular cardiomyocytes. Nephrology, Dialysis, Transplantation, 34(11), 1864–1875. 10.1093/ndt/gfy392 [DOI] [PubMed] [Google Scholar]
- Nowak, A. , Friedrich, B. , Artunc, F. , Serra, A. L. , Breidthardt, T. , Twerenbold, R. , … Mueller, C. (2014). Prognostic value and link to atrial fibrillation of soluble Klotho and FGF23 in hemodialysis patients. PLoS ONE, 9(7), e100688 10.1371/journal.pone.0100688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, R. J. , Ray, G. , Bienvenu, L. A. , Rose, R. A. , & Howlett, S. E. (2014). Sex differences in SR Ca(2+) release in murine ventricular myocytes are regulated by the cAMP/PKA pathway. Journal of Molecular and Cellular Cardiology, 75, 162–173. 10.1016/j.yjmcc.2014.07.006 [DOI] [PubMed] [Google Scholar]
- Pavik, I. , Jaeger, P. , Ebner, L. , Wagner, C. A. , Petzold, K. , Spichtig, D. , … Serra, A. L. (2013). Secreted Klotho and FGF23 in chronic kidney disease stage 1 to 5: A sequence suggested from a cross‐sectional study. Nephrology, Dialysis, Transplantation, 28, 352–359. 10.1093/ndt/gfs460 [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol, 18, e3000410 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, L. , Ruiz‐Hurtado, G. , Rueda, A. , Mercadier, J. J. , Benitah, J. P. , & Gómez, A. M. (2014). Calcium signaling in diabetic cardiomyocytes. Cell Calcium, 56, 372–380. 10.1016/j.ceca.2014.08.004 [DOI] [PubMed] [Google Scholar]
- Ruiz‐Hurtado, G. , Li, L. , Fernández‐Velasco, M. , Rueda, A. , Lefebvre, F. , Wang, Y. , … Gómez, A. M. (2015). Reconciling depressed Ca2+ sparks occurrence with enhanced RyR2 activity in failing mice cardiomyocytes. The Journal of General Physiology, 146, 295–306. 10.1085/jgp.201511366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler, S. , Wen, M. , Roth, H. J. , Fehrenz, M. , Flügge, F. , Herath, E. , … Heine, G. H. (2013). Plasma Klotho is not related to kidney function and does not predict adverse outcome in patients with chronic kidney disease. Kidney International, 83, 121–128. 10.1038/ki.2012.288 [DOI] [PubMed] [Google Scholar]
- Semba, R. D. , Cappola, A. R. , Sun, K. , Bandinelli, S. , Dalal, M. , Crasto, C. , … Ferrucci, L. (2011). Plasma klotho and cardiovascular disease in adults. Journal of the American Geriatrics Society, 59, 1596–1601. 10.1111/j.1532-5415.2011.03558.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalhoub, V. , Shatzen, E. M. , Ward, S. C. , Davis, J. , Stevens, J. , Bi, V. , … Richards, W. G. (2012). FGF23 neutralization improves chronic kidney disease‐associated hyperparathyroidism yet increases mortality. The Journal of Clinical Investigation, 122, 2543–2553. 10.1172/JCI61405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioya, T. (2007). A simple technique for isolating healthy heart cells from mouse models. The Journal of Physiological Sciences, 57, 327–335. 10.2170/physiolsci.RP010107 [DOI] [PubMed] [Google Scholar]
- Touchberry, C. D. , Green, T. M. , Tchikrizov, V. , Mannix, J. E. , Mao, T. F. , Carney, B. W. , … Wacker, M. J. (2013). FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. American Journal of Physiology. Endocrinology and Metabolism, 304, E863–E873. 10.1152/ajpendo.00596.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakawa, I. , Yamazaki, Y. , Shimada, T. , Iijima, K. , Hasegawa, H. , Okawa, K. , … Yamashita, T. (2006). Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature, 444, 770–774. 10.1038/nature05315 [DOI] [PubMed] [Google Scholar]
- Verde, E. , Pérez de Prado, A. , López‐Gómez, J. M. , Quiroga, B. , Goicoechea, M. , García‐Prieto, A. , … Luño, J. (2016). Asymptomatic intradialytic supraventricular arrhythmias and adverse outcomes in patients on hemodialysis. Clinical Journal of the American Society of Nephrology, 11, 2210–2217. 10.2215/CJN.04310416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner, C. , Amann, K. , & Shoji, T. (2016). The heart and vascular system in dialysis. Lancet, 388, 276–284. 10.1016/S0140-6736(16)30508-6 [DOI] [PubMed] [Google Scholar]
- Xiao, B. , Jiang, M. T. , Zhao, M. , Yang, D. , Sutherland, C. , Lai, F. A. , … Chen, S. R. W. (2005). Characterization of a novel PKA phosphorylation site, serine‐2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circulation Research, 96, 847–855. 10.1161/01.RES.0000163276.26083.e8 [DOI] [PubMed] [Google Scholar]
- Xie, J. , Cha, S. K. , An, S. W. , Kuro‐O, M. , Birnbaumer, L. , & Huang, C. L. (2012). Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nature Communications, 3, 1238 10.1038/ncomms2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, J. , Yoon, J. , An, S. W. , Kuro‐o, M. , & Huang, C. L. (2015). Soluble Klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. Journal of the American Society of Nephrology, 26, 1150–1160. 10.1681/ASN.2014040325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, T. , Fujimori, T. , & Nabeshima, Y. (2002). Mediation of unusually high concentrations of 1,25‐dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1alpha‐hydroxylase gene. Endocrinology, 143, 683–689. 10.1210/endo.143.2.8657 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Change in peak amplitude as ΔF/F0=(F‐F0/F0) in cardiomyocytes from +/+ (n=50 cells/N=5 mice) and kl/kl (n=49 cells/N=5 mice) mice. Data are shown as mean±SEM. *P<0.05 vs. +/+ mice.
Figure S2. Representative normalisation of Ca2+ transients to the peak of fluorescence obtained in cardiomyocytes from a +/+ (dark grey line) and kl/kl (dark red line) mice.
Figure S3. mRNA expression of SERCA2a (Atp2a2) in +/+ (N=6 mice) and kl/kl (N=6 mice) mice. Data are shown as mean±SEM. *P<0.05 vs. +/+ mice.
Figure S4: K SERCA in cardiomyocytes from +/+ (n=18 cells/N=5 mice) and kl/kl (n=27 cells/N=5 mice) mice. Data are shown as mean±SEM. *P<0.05 vs. +/+ mice.
Figure S5. Time of decay (Tau) of caffeine‐evoked intracellular Ca2+ transients in +/+ (n=37 cells/N=5 mice) and kl/kl (n=46 cells/N=5 mice) mice.
Figure S6. Urine levels of sKL in Sham (N=7 mice) compared to Nfx mice (N=10 mice). *P<0.05 vs. Sham.
Figure S7. Representative normalisation of Ca2+ transients to the same peak of fluorescence obtained in cardiomyocytes from a Sham (black line) and Nfx (red line) mice.
Figure S8: K SERCA in cardiomyocytes from Sham (n=35 cells/N=5 mice) and Nfx (n=48 cells/N=6 mice) mice. Data are shown as mean±SEM. *P<0.05 vs. Sham mice.