

Abstract
Background and Purpose
Prenatal caffeine exposure (PCE) can cause developmental toxicity of long bones in offspring, but the long‐term effects and the underlying mechanism have not been fully clarified. Here, we investigated the effects of PCE peak bone mass accumulation and osteoporosis susceptibility in offspring and its intrauterine programming mechanism.
Experimental Approach
Pregnant Wistar rats were administrated intragastrically with saline or caffeine (120 mg·kg−1·day−1) on gestational days 9–20. The serum and bone samples were collected from the fetal and postnatal offspring for bone mass, genes expression and corticosterone analysis. Then, rat bone marrow mesenchymal stem cells (BMSCs) were treated with corticosterone in vitro to confirm the molecular mechanism.
Key Results
PCE caused fetal bone dysplasia in male and female offspring. In adulthood, PCE reduced peak bone mass and increased osteoporosis susceptibility in male offspring but not in females. Meanwhile, PCE only decreased the H3K9ac and expression levels of 11β‐hydroxysteroid dehydrogenase 2 (11β‐HSD2) before and after birth in the male offspring but not in the females. Moreover, the high level of corticosterone induced by PCE down‐regulated the H3K9ac and expression levels of 11β‐HSD2 through promoting glucocorticoid receptor (GR; NR3C1) into the nucleus of bone marrow mesenchymal stem cells (BMSCs) and recruiting histone deacetylase 11 (HDAC11) binding to 11β‐HSD2 promoter region, which further enhanced the effect of corticosterone on suppressing osteogenic function of BMSCs.
Conclusion and Implications
PCE caused osteoporosis susceptibility in male adult offspring, which attributed to the low‐functional programming of 11β‐HSD2 induced by corticosterone via GR/HDAC11 signalling.
Keywords: 11β‐hydroxysteroid dehydrogenase 2, glucocorticoid, histone acetylation, intrauterine programming, peak bone mass, prenatal caffeine exposure
Abbreviations
- 11β‐HSD2
11β‐hydroxysteroid dehydrogenase 2
- BMSCs
bone marrow‐derived mesenchymal stem cells
- GR
glucocorticoid receptor/NR3C1
- H3K9ac
histone 3 lysine 9 acetylation
- HDAC11
histone deacetylase 11
- Md. Ar
mineralized area
- Tb. N
trabecular number
- Tb. Sp
trabecular separation
- Tb. Th
trabecular thickness
What is already known
Prenatal caffeine exposure can cause developmental toxicity of long bones in offspring.
What this study adds
Low‐expression of 11β‐HSD2 mediated osteoporosis susceptibility induced by prenatal caffeine exposure in male adult offspring.
What is the clinical significance
11β‐HSD2 could be an easly therapeutic target forp caffeine exposure‐induced osteoporosis in male adult offspring.
1. INTRODUCTION
Caffeine is widely present in coffee, tea, soft drinks, some drugs and caffeine consumption during pregnancy is common in women (Frary, Johnson, & Wang, 2005). Also, in clinical settings, caffeine is commonly used to treat premature infants with pulmonary dysplasia (Dekker et al., 2017). Whereas, caffeine exposure during pregnancy can do a variety of short‐term and long‐term harm to offspring. In our previous studies, we found that prenatal caffeine exposure caused developmental toxicities of multiple organs in fetal offspring and susceptibility to several kinds of diseases in adulthood (He et al., 2019; Shangguan et al., 2017; Tan et al., 2012; Xu et al., 2018). However, the effects of prenatal caffeine exposure on peak bone mass accumulation and osteoporosis susceptibility in offspring and the underlying mechanism have not been fully clarified.
Peak bone mass is the maximum bone mass in humans or animals under physiological conditions and it is a determinant factor for osteoporosis occurrence. Epidemiological studies have found that low birth weight infants have a lower bone mineral density in adulthood than healthy infants (Balasuriya et al., 2017). Moreover, the adverse environments during pregnancy, such as maternal dexamethasone administration, as well as smoking and undernutrition during pregnancy contributed to the low peak bone mass and increased occurrence risk of osteoporosis in adult offspring (Heppe, Medina‐Gomez, Hofman, Rivadeneira, & Jaddoe, 2015; Xiao et al., 2018; Yin, Dwyer, Riley, Cochrane, & Jones, 2010). These studies suggested that low peak bone mass was closely related to bone development in utero, which further contributes to the susceptibility to osteoporosis.
In the physiological condition, maternal glucocorticoids, including cortisol in humans and corticosterone in rodents, are mainly responsible for promoting fetal bone development. We previously found that prenatal caffeine exposure up‐regulated the maternal serum corticosterone and then opened the placenta barrier for glucocorticoids, which caused a large amount of maternal corticosterone to enter the fetal serum through the placenta, thereby leading to an increased corticosterone in fetal serum (Xu et al., 2012). Furthermore, the increased corticosterone in fetal serum due to prenatal caffeine exposure was involved in multiple organ dysplasia in the offspring (He et al., 2019; Hu et al., 2019; Shangguan et al., 2017). The effects of glucocorticoids are known to be regulated by 11β‐hydroxysteroid dehydrogenase 2 (11β‐HSD2; Chapman, Holmes, & Seckl, 2013), which is widely expressed in multi‐organ tissues, including bone tissue. 11β‐HSD2 can inactivate glucocorticoids and prevent glucocorticoids from playing their role (O'Brien et al., 2004; Zallocchi, Matkovic, Calvo, & Damasco, 2004; Zuo et al., 2017). This suggests that the stable expression of 11β‐HSD2 can regulate the level of active glucocorticoids in bone tissue and ensures normal bone growth and development.
In this research, by measuring the bone mass index and functional gene expression in offspring rats, we aimed to investigate whether prenatal caffeine exposure could lead to low peak bone mass and the susceptibility to osteoporosis in adult offspring. Furthermore, we explored whether the enhanced effect of corticosterone on suppressing osteogenic function induced by low expression of 11β‐HSD2 contributed to the increased susceptibility to osteoporosis and its epigenetic mechanism. This study would be help to uncover any the long‐term harmful effects on bone development in response to prenatal caffeine exposure and to clarify its intrauterine actions. Meanwhile, this study provides experimental evidence to support the theory of “Developmental Origins of Health and Disease” (Baird et al., 2017).
2. METHODS
2.1. Materials
Caffeine (CAS #58‐08‐2, >99% purity) was purchased from Sigma‐Aldrich Co., Ltd. (St Louis, MO, USA). The histone deacetylase 11 (HDAC11) siRNA oligo and 11β‐HSD2 overexpression plasmid were purchased from Gene Pharma Co. (Jiangsu, China). Mifepristone (RU486) the glucocorticoid receptor antagonist was purchased from Sigma‐Aldrich (St. Louis, MO, USA). The antibodies for 11β‐HSD2 (ab80317, RRID: AB_1658782) were purchased from Abcam plc. (Cambridge, Cambridge shire, UK). The antibodies for glucocorticoid receptor (sc‐376426, RRID: AB_11150486) and HDAC11 (sc‐390737, RRID: AB_2715508) were purchased from Santa Cruz Biotech Co. (Santa Cruz, CA, USA). The antibodies for histone 3 lysine 9 acetylation (H3K9ac; A17560, RRID: AB_2768208), H3K14ac (A7254, RRID: AB_2737401) and H3K27ac (A7253, RRID: AB_2767797) were purchased from ABclonal Biotech Co., Ltd. (Wuhan, China). Rat corticosterone ELISA kits were all obtained from Assaypro (Saint Charles Missouri, USA). The α‐minimum essential medium (MEM) was purchased from HyClone Co. (Logan, UT, United States) and FBS was purchased from Gibco Co. (Detroit, MI, United States). TRIZOL was purchased from Invitrogen Co. (Carlsbad, CA, USA). Reverse transcription and real‐time quantitative PCR (RT‐qPCR) kits were purchased from Takara Biotech Co., Ltd. (Dalian, China). The SYBR Green dye was purchased from Applied Biosystems through Thermo Fisher Scientific (ABI; Foster City, CA, USA). All primers were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
2.2. Animals and treatment
Specific pathogen‐free adult Wistar rats (RGD_10044, No. 2012‐2014, certification number: 42000600002258, license number: SCXK [Hubei]) weighing 209 ± 12 g (females) and 258 ± 17 g (males) were purchased from the Experimental Center of the Hubei Medical Scientific Academy (Wuhan, China). The rats were housed in a temperature‐controlled room (temperature: 18–22°C; humidity: 40%–60%; light cycle: 12 h light–dark cycle) and allowed free access to food and water. Animal experiments were performed in the Center for Animal Experiment of Wuhan University (Wuhan, China), which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International). All animal experimental procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the Chinese Animal Welfare Committee and the Committee on the Ethics of Animal Experiments of the Wuhan University School of Medicine approved the animal experimental protocol (Permit No. 201709). Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology 2020. The above rats were acclimated for 1 week before starting the experiment. Next, two female rats were placed together with one male rat overnight in a cage for mating. Gestational 0 was determined upon confirmation of mating by the appearance of sperm in a vaginal smear. Pregnant rats were transferred to individual cages and then randomly divided into the control and prenatal caffeine exposure groups (n = 16 per group). To reduce bias in animal experiments, rats were housed and treated by a technician, while different co‐authors performed samples harvesting and data analysis, respectively. From gestational day 9 to 20, the prenatal caffeine exposure group was established, comprising rats administered intragastrically 120 mg·kg−1 caffeine daily (8:00–9:00 a.m.) and the control group was administered the same volume of distilled water.
To obtain fetal bone and serum samples, some of the pregnant rats from the control and prenatal caffeine exposure group were killed after anaesthesia with isoflurane on gestational day 20 (n = 8 per group). The foetuses from each littermate were decapitated immediately to collect blood and the serum samples from each littermate were pooled and immediately frozen at −80°C for subsequent analyses. The left femurs and tibias were collected from the foetuses that were randomly selected from each litter and then fixed with 4% paraformaldehyde overnight and embedded in paraffin for histological or immunohistochemistry analysis, while the right femurs and tibias were frozen immediately in liquid nitrogen and then conserved in a refrigerator at −80°C for further analysis. The rest of the pregnant rats, including the control and prenatal caffeine exposure groups, went into spontaneous labour to produce offspring (n = 8 per group). The pregnant rats with litter size (8–14) were normalized to 10; the male/female ratio was approximately 1:1. There were eight pregnant rats in each group. Then, the male and female pups were born and raised normally until postnatal week or 28. The corresponding rats were anaesthetized with isoflurane or ether and decapitated to collect blood and spongy bone tissue samples. The left femurs were dissected and fixed in 70% ethanol for micro‐CT analysis. The left tibias were fixed with 4% paraformaldehyde and then embedded in paraffin for histological or immunohistochemistry analysis, while the right femurs and tibias were frozen immediately in liquid nitrogen and then conserved at −80°C in a refrigerator for further analysis.
2.3. Serum and bone corticosterone concentration analysis
The content of serum corticosterone was measured by an ELISA kit, following the manufacturers protocols. To test the content of corticosterone in bone tissue, 1 ml of PBS for homogenate was added to equal weights of bone tissue from each group and centrifuged to collect the supernatant. Then, the content of bone corticosterone was measured by the ELISA kit following the manufacturer's protocols (n = 8 per group). The intra‐assay and inter‐assay coefficients of variation for corticosterone determination were 5.0% and 7.2%, respectively.
2.4. Histological and immunohistochemical analysis
To explore morphology and calcification of hindlimb long bones in the fetal rats, serial longitudinal sections (5‐μm thick) were cut and one out of six sections were stained with 5% AgNO3 until they become dark brown for von Kossa staining to quantify the bone mass in the primary ossification centre (n = 8 per group). The primary indices, including the length of fetal bone and primary ossification centre, bone trabecula perimeter and mineralized area (Md. Ar), were measured on von Kossa‐stained sections at ×100 magnification using Image‐Pro Plus 6.0 (RRID:SCR_007369) as described previously (Dempster et al., 2013).
For immunohistochemistry analysis, the bone tissue slices were cut mentioned above at each time point (n = 5 per group). Then these slices were deparaffinized and hydrated through a graded series of ethanol (100%–70%). Antigen retrieval was achieved by boiling the sections in 0.01 M sodium citrate buffer (pH 6.0) at approximately 95°C for 10–15 min. After antigen retrieval, the hydrated sections were then incubated in 3% H2O2 for 15 min to quench the endogenous peroxidase activity. Sections were then blocked in 3% BSA (Servicebio, Wuhan, China) at room temperature for 1 h and incubated with a primary antibody for 11β‐HSD2 (1:100 dilution) and glucocorticoid receptor (1:200) at 4°C overnight. After washing with PBS, sections were incubated with a biotinylated secondary antibody (1:100 dilution) and then with an avidin‐biotinylated HRP complex solution according to the manufacturer's directions. Finally, peroxidase activity was revealed by immersion in diaminobenzidine (DAB). For negative controls in immunohistochemistry, immunostaining was performed on parallel sections in which the primary antibody was replaced with non‐immune rabbit IgG. The intensity of immunostaining was determined by measuring the mean optical density in six sections from different samples and using the Photo Imaging System (Nikon H550S, Japan). The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
2.5. Micro‐CT analysis
Femurs were collected and the attached soft tissue was removed thoroughly and fixed in 70% ethanol (n = 8 per group). The bone mass was scanned and analysed with the micro‐CT system (VivaCT 40; Scanco, Basserdorf, Switzerland) as described previously (Bouxsein et al., 2010). To determine bone volume/total volume (BV/TV), trabecular number (Tb. N), trabecular thickness (Tb. Th) and trabecular separation, 0.5–5.5 mm below the lowest point of growth plate was selected as the region of interest and cross‐sectional images were scanned at 21‐μm resolution. These parameters of trabecular bone microarchitecture described above were computed using 3D model‐independent algorithms.
2.6. Cell culture and treatment
Bone marrow‐derived mesenchymal stem cells (BMSCs) were extracted from 1‐month‐old male rats as described previously (Zhang et al., 2012). These rats were killed by overdose of anaesthetic and soaked in 75% ethanol for 10 min. Then, their femurs and tibias were separated under aseptic conditions. Both ends of the epiphyses were sterile cut off and the bone marrow cavity was rinsed with a syringe with 5 ml of α‐minimum essential medium (α‐MEM). The evenly dispersed rinse was then filtered through a 200‐mesh nylon filter and the supernatant was centrifuged at room temperature at 111 g for 10 min. Next, the cells were collected and plated at a density of 4 × 105 cells per well in six‐well plates with growth medium (α‐MEM with 10% FBS, 100 mg·ml−1 streptomycin and 100 U·ml−1 penicillin). When the cells reached 80% confluence, growth medium was switched to osteogenic differentiation medium (α‐MEM with 10% FBS, 100 mg·ml−1 streptomycin, 100 U·ml−1 penicillin, 10‐mM β‐glycerophosphate, 50 μg·ml−1 ascorbic acid and 10‐nM dexamethasone). Then, the cells were treated with various concentrations of corticosterone or co‐treated with mifepristone (RU486) for further analysis.
2.7. siRNA knockdown of HDAC11 in BMSCs
RNA interference technology was used to knockdown HDAC11 expression. The HDAC11 siRNA sequences were GCAGACAUCACACUGGCUATT and UAGCCAGUGUGA‐UGUCUGCTT, while the negative control sequences were UUCUCCGAACGUGUCACGUTT and ACGUGACACGUUCGGAGAATT. Prior to transfection, BMSCs were seeded in six‐well plates at a density of 4 × 105 cells per well. Twenty‐four hours later, the cells were then transfected with 30 nM of HDAC11 siRNA using Lipofectamine 3000 according to the manufacturer's protocol. Eight hours later, the medium was exchanged for a fresh medium, and the cells were treated with corticosterone. The cells were harvested after 3 days for further analysis (n = 5 per group).
2.8. Overexpression of 11β‐HSD2 in BMSCs
11β‐HSD2 was ligated into pcDNA3.1 at the NheI and KpnI sites to construct pcDNA3.1.11β‐HSD2 vector. Prior to transfection, BMSCs were seeded in six‐well plates at a density of 4 × 105 cells per well. Twenty‐four hours later, the cells were then transfected respectively with pcDNA3.1.11β‐HSD2 vector, empty pcDNA3.1 vector or empty control vector using Lipofectamine 3000 according to the manufacturer's protocol. Eight hours later, the medium was exchanged for a fresh medium, and the cells were treated with 500‐nM corticosterone. The cells were harvested after 3 days for further analysis (n = 5 per group).
2.9. Cell immunofluorescence analysis
For immunofluorescence staining, the BMSCs were treated with corticosterone for 3 days (n = 5 per group), and then the BMSCs were rehydrated in PBS and permeabilized with 0.3% Triton X‐100 for 15 min at room temperature. After washing with PBS, blocking solution (5% BSA + 0.1% Triton X‐100) was applied for 20 min at room temperature. After another PBS wash, BMSCs were treated with the primary antibody for glucocorticoid receptor (1:200; sc‐376426) at 4°C overnight. The next day, BMSCs were washed four times with PBS and incubated with secondary antibodies for 1.5 h at room temperature. The nuclei were stained with DAPI at 1:500 dilution for 5 min. After three wash steps with PBS, the BMSCs were observed in the fluorescence microscope. All of the images were captured and then analysed using a Nikon NIS Elements BR light microscope (Nikon, Tokyo, Japan). The staining intensity was determined by measuring the integrated optical density (IOD) in six different fields for each sample.
2.10. Luciferase constructs and reporter assay
The cDNA of 1 kb region directly upstream of 11β‐HSD2 was synthesized by GenePharma (Shanghai, China). The amplified DNA sequences were inserted into the GV238‐basic at the KpnI and XhoI sites (GenePharma Co. Ltd, Shanghai, China) to generate GV238‐11β‐HSD2‐WT. Similarly, we produced a vector that contains mutated 11β‐HSD2 promoter, generating GV238‐11β‐HSD2‐MUT. For the dual luciferase assay, 1.2 × 104 BMSCs in a 96‐well plate were transfected with 50‐nM GV238‐11β‐HSD2‐WT or GV238‐11β‐HSD2‐MUT. The BMSCs were then co‐transfected with corticosterone or glucocorticoid receptor antagonist mifepristone (RU486). Forty‐eight hours post‐transfection, luciferase activity was measured using the Dual‐Luciferase® Reporter Assay System (Promega, Madison, WI, USA). The firefly luciferase activity value was normalized to the renilla activity value. Promoter transcription activity was presented as the fold induction of relative luciferase unit (RLU) compared with basic GV238 vector control (the RLU was the value of the firefly luciferase unit divided by the value of the renilla luciferase unit; n = 5 per group).
2.11. Total RNA extract and RT‐qPCR
Total RNA was isolated from bone tissue (n = 8 per group) and BMSCs (n = 5 per group) using TRIzol Reagent following the manufacturer's protocol. The total RNA was reverse transcribed using a first strand cDNA synthesis kit. Then, RT‐qPCR was performed using a SYBR Green qPCR Master Mix Kit and ABI StepOnePlus cycler (Applied Biosystems, Foster City, CA, USA) with 40 cycles. Relative gene expression was calculated for each gene by the 2−ΔΔCT method with glyceraldehyde 3‐phosphatedehydrogenase (GAPDH) for normalization. The data were conducted data normalization, which means the correction of test values to control group values. Results were shown as fold mean of the controls and labelled it in Y axis. The rat primer sequences for the genes used in this study are shown in Table S1.
2.12. Western blotting
Western blotting was conducted the experimental detail provided conforms with BJP guidelines (Alexander et al., 2018). Cells were washed twice with ice‐cold PBS and lysed in 200‐μl RIPA Lysis Buffer and 1 mM of phenylmethylsulfonyl fluoride (PMSF) for 5 min on ice to extract total protein. Cytoplasmic protein and nucleic protein were, respectively, extracted by the nuclear and cytoplasmic protein extraction kit following the manufacturer's protocols. Equal amounts of protein lysates (30 μg per lane) were resolved by SDS‐PAGE on 10% polyacrylamide gels, transferred to polyvinylidene difluoride membranes, and blotted with the primary antibodies for 11β‐HSD2 (1:1000, ab80317, RRID: AB_1658782, Abcam plc, Cambridge, UK), glucocorticoid receptor (1:500, sc‐376426, RRID: AB_11150486, Santa Cruz, CA, USA) or HDAC11 (1:1000, sc‐390737, RRID: AB_2715508, Santa Cruz, CA, USA) at 4°C overnight. Band intensity was quantified using Quantity One (Bio‐Rad, Shanghai, China; n = 5 per group). The data were conducted data normalization. The results were shown as fold mean of the controls and labelled it in Y axis.
2.13. Co‐immunoprecipitation (CO‐IP) assay
Co‐IP assay was performed to detect the interaction of glucocorticoid receptor with HDAC11. After washing with ice‐cold PBS, cells were lysed in 1.2 ml of lysis buffer at −80°C for 30 min. The samples were centrifuged with 21,756 g at 4°C for 15 min, and the supernatant was transferred to a new column. Then, the samples were divided into three parts: the first was used for input protein, and the other two were performed using 1 μg of either a mock antibody (IgG) as a control or glucocorticoid receptor antibody, respectively, followed by incubation overnight at 4°C with gentle shaking. The samples were incubated with Protein G magnetic beads (Millipore, 16‐157) at 4°C for 6 h. After immunoprecipitation, the samples were washed with lysis buffer. After three washings, retained proteins were mixed with 30 μl of loading buffer at 100°C for 10 min. Protein complexes and input protein were then detected by Western blotting (n = 5 per group).
2.14. Chromatin immunoprecipitation (ChIP) assay
Cell suspensions from bone tissue (n = 8 per group) and BMSCs (n = 5 per group) were collected and fixed in 1% formaldehyde for chromatin cross‐linking, and 125 mM of glycine was added to stop the reaction. The samples were then centrifuged and resuspended in 0.5 ml of lysis buffer containing protease inhibitors. Cell lysates were sonicated to shear DNA to lengths of approximately 200 base pairs and transferred to a new tube with ChIP dilution buffer. Chromatin was incubated overnight at 4°C on a nutator/rocker with specific antibodies for H3K9ac (1:50, A17560, RRID: AB_2768208, ABclonal Biotech, Wuhan, China), H3K14ac (1:100, A7254, RRID: AB_2737401, ABclonal Biotech, Wuhan, China), or H3K27ac (1:50, A7253, RRID: AB_2767797, ABclonal Biotech, Wuhan, China), and BSA‐treated Protein G beads to reduce nonspecific background binding. The immunoprecipitated DNA‐protein complex with beads was collected by centrifugation and washed sequentially with low‐salt, high‐salt, LiCl immune complex, and Tris‐EDTA washing buffer solutions. Freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3) was used to elute the DNA protein complex. The samples were then placed in 65°C water baths overnight to reverse formaldehyde cross‐linking and subsequently purified using DNA purification kits. The isolated DNA was then assayed using RT‐qPCR. The rat primer sequences of 11β‐HSD2 promoter region in this study were as follows: forward, GAGTGACTCTGAGTGGGGAC; reverse, AAGAGTTCACTGGCCCTTC. The predicted binding sites for glucocorticoid receptor in the 11β‐HSD2 promoter region were performed using the JASPAR database (http://jaspar.genereg.net/; RRID:SCR_003030). Numbers are given relative to the first base of the transcription start site (TSS). The rat primer sequences used for testing the binding of glucocorticoid receptor and 11β‐HSD2 promoter region were as follows: primer1: (nt‐1500/‐1351) forward, GAGTGACTCTGAGTGGGGAC; reverse, TGAAGGGCCAGTGAACTCTT, primer2: (nt‐900/‐751): forward, AGGACCTTCATACACACAGGG, reverse, CTTCCCCAAGAACCCAGCTA. The rat primer sequences used for distant region (nt‐4200/‐4001) in 11β‐HSD2 were as follows: forward, CCTCCACAACCAGACACTCA, reverse, TGAGTCTTTTGCCCTCCCTT. Gene enrichment was quantified relative to input controls by RT‐qPCR using primers specific for the promoter regions of 11β‐HSD2. Results were shown as fold enrichment of RT‐qPCR value over IgG and labelled fold enrichment in Y axis.
2.15. Data and analysis
This study complied with the recommendations of the British Journal of Pharmacology on experimental design and analysis (Curtis et al., 2018). The group size selection for each protocol was based on our previous studies (Shangguan et al., 2017; Wen et al., 2019), which is verifiable. SPSS 20.0 (RRID:SCR_002865, SPSS Science Inc., Chicago, Illinois, USA) and Prism 7.0 (RRID:SCR_002798, GraphPad Software, La Jolla, CA, USA) were used to analyse experimental data. The group sizes of all figures in this study were at least five (n ≥ 5), and the statistical analysis was undertaken for these figures. The declared group size was the number of independent values, and statistical analysis was done using these independent values. All data were expressed as mean ± SEM. The outliers were included in data analysis and presentation. To reduce unwanted sources of variation, some data were conducted data normalization, which means the correction of test values to baseline or control group values. Student's two‐tailed t test was performed on one factor of prenatal caffeine treatment (control or prenatal caffeine exposure). For the data from multigroup studies, a post hoc Dunnett's t test or a post hoc Bonferroni t test was conducted if F in one‐way ANOVA (or equivalent) achieved the chosen necessary level of statistical significance and there was no significant variance inhomogeneity. The Pearson correlation analysis was used to analyse the correlation between two indicators. A value of P < 0.05 was considered statistically significant. The level of P value in the present study was deemed to constitute the threshold for statistical significance for determining whether groups differ, and this P value was not varied later in Section 3.
2.16. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Fabbro, et al., 2019; Alexander, Kelly, et al., 2019).
3. RESULTS
3.1. Effects of prenatal caffeine exposure on bone development and osteogenic differentiation in fetal rat offspring
First, we observed the effects of prenatal caffeine exposure on fetal bone development. Vonkossa staining and quantitative analysis results showed that the indexes of bone development, including length of bone and primary ossification centre, bone trabecula perimeter and mineralized area, were significantly reduced by prenatal caffeine exposure in both male and female offspring Figure 1a,b). These results indicated that prenatal caffeine exposure caused bone dysplasia in male and female foetuses. Osteoblasts are known to be differentiated from BMSCs, which play an important osteogenesis role in bone development (Li et al., 2019). Thus, we detected the osteogenic differentiation marker genes, such as Runx2, osterix, bone sialoprotein (BSP) and osteocalcin (OCN) expressed in primary ossification centres and found that prenatal caffeine exposure significantly down‐regulated the expression of these marker genes in male and female offspring (Figure 1c). The above results suggested that prenatal caffeine exposure could cause inhibition of osteogenic differentiation, which participated in fetal bone dysplasia in the both male and female offspring.
FIGURE 1.
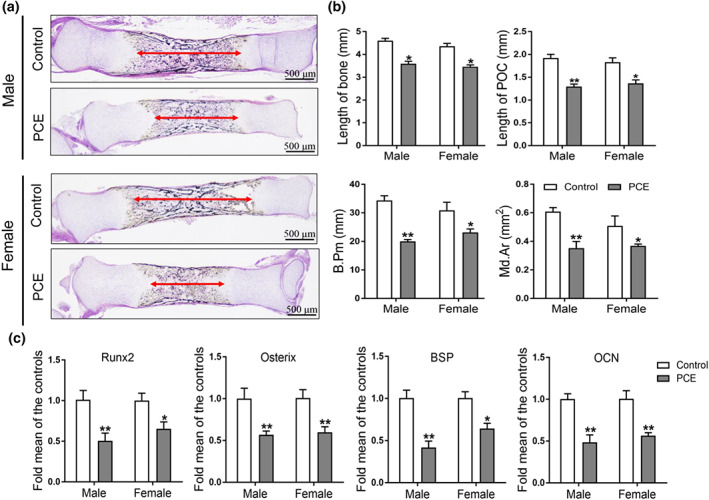
Effects of prenatal caffeine exposure (PCE) on bone development and osteogenic function in male and female fetal offspring rats. (a) Representative von Kossa staining images of bones from the control and PCE groups on gestation day (GD) 20; (b) quantification of the length of fetal bone and primary ossification centre (POC), bone trabecula perimeter (B. Pm) and mineralized area (Md. Ar; n = 8 per group); (c) RT‐qPCR analysis of gene expression of osteogenic differentiation markers, including Runx2, osterix, bone sialoprotein (BSP) and osteocalcin (OCN) in bone tissue on GD20 (n = 8 per group). Mean ± SEM, *P < 0.05 compared with the control. Scale bar = 500 μm
3.2. Effects of prenatal caffeine exposure on serum/bone corticosterone concentration and bone 11β‐HSD2 expression in fetal rat offspring
Next, we detected the corticosterone concentration in fetal serum and bone tissue, and the results showed that prenatal caffeine exposure significantly increased the serum and bone corticosterone concentration in male and female offspring (Figure 2a,b). We further found that prenatal caffeine exposure significantly reduced the mRNA expression of 11β‐HSD2 in fetal bone tissue (Figure 2c). Moreover, the immunohistochemical results indicated that the 11β‐HSD2 protein level in fetal bone tissue was also reduced by prenatal caffeine exposure (Figure 2d,e). Meanwhile, the correlation analysis results in male and female offspring indicated that there was a significant negative correlation between serum corticosterone concentration and bone 11β‐HSD2 mRNA and protein expression (Figure 2f) and the same change also existed in the correlation between bone 11β‐HSD2 mRNA and protein expression and bone corticosterone concentration (Figure 2g). This suggested that prenatal caffeine exposure increased the fetal serum and bone corticosterone concentration, which was related to the down‐regulated 11β‐HSD2 expression in bone tissue.
FIGURE 2.
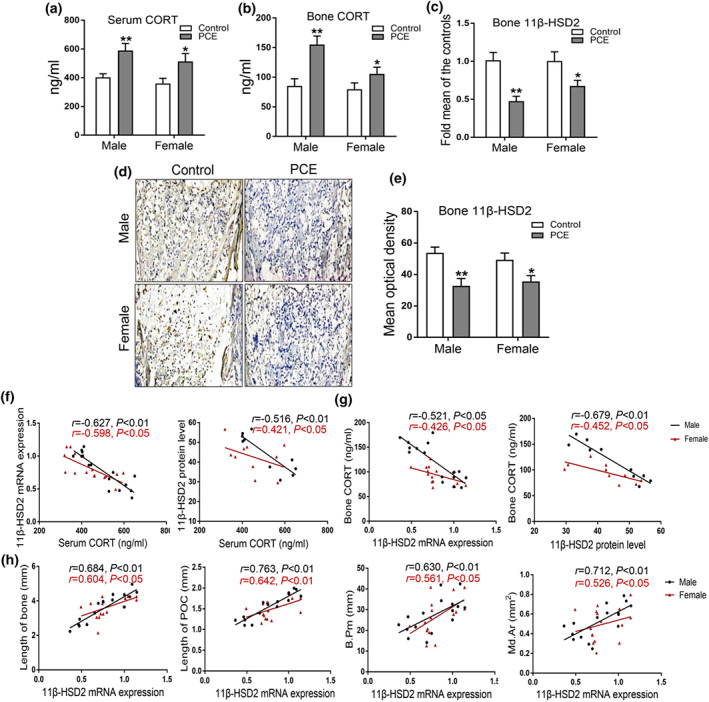
Effects of prenatal caffeine exposure (PCE) on corticosterone (CORT) concentration and 11β‐hydroxysteroid dehydrogenase 2 (11β‐HSD2) expression and in male and female fetal offspring. (a) CORT concentration of fetal serum (n = 8 per group); (b) CORT concentration of fetal bone tissue (n = 8 per group); (c) RT‐qPCR analysis of 11β‐HSD2 mRNA expression (n = 8 per group); (d) representative immunohistochemical images of 11β‐HSD2 in fetal bone tissue; (e) quantitative immunohistochemical analysis of the mean optical density of 11β‐HSD2 protein (n = 5 per group); (f) the correlation analysis between fetal serum CORT concentration and bone 11β‐HSD2 mRNA and protein expression; (g) the correlation analysis between bone 11β‐HSD2 mRNA and protein and bone CORT concentration; (h) the correlation analysis between 11β‐HSD2 mRNA expression and bone length, primary ossification centre (POC) length, bone trabecula perimeter (B. Pm, and mineralized area (Md. Ar). Mean ± SEM, *P < 0.05 compared with the control
To clarify the effect of 11β‐HSD2 expression on fetal bone development, we analysed the correlation between bone 11β‐HSD2 expression and bone development index in male and female offspring. The results showed that there were positive correlations between bone 11β‐HSD2 expression and the length of bone and primary ossification centre, bone trabecula perimeter, and mineralized area (Figure 2h). Collectively, this indicated that bone development dysplasia was associated with the down‐regulated bone 11β‐HSD2 expression induced by prenatal caffeine exposure in both male and female foetuses.
3.3. Effects of prenatal caffeine exposure on peak bone mass accumulation and osteogenic differentiation in adult rat offspring
To investigate the effect of prenatal caffeine exposure on peak bone mass accumulation in adult male and female offspring, we measured the bone mass by micro‐CT at postnatal week 12, the high speed of peak bone mass accumulation around this time point (Heaney et al., 2000). It was found that prenatal caffeine exposure reduced bone mass in male offspring at postnatal week 12, which was manifested as the significant decreased bone volume/total volume, trabecular number, and trabecular thickness and increased trabecular separation (Figure 3a,b). We further found that the expression levels of osteogenic markers such as Runx2, osterix, BSP, and OCN were all down‐regulated by prenatal caffeine exposure in male offspring at postnatal week 12 (Figure 3c). However, there were no significant changes in peak bone mass accumulation and osteogenic function in the prenatal caffeine exposure female adult offspring (Figure 3a–c). We also measured the bone mass by micro‐CT at postnatal week 28, when the peak bone mass is reached (Heaney et al., 2000). The results showed that prenatal caffeine exposure significantly reduced the peak bone mass in male offspring at P postnatal week 8 (Figure 3d,e), but the peak bone mass in female offspring was not affected by prenatal caffeine exposure (Figure 3d,e). These results demonstrated that prenatal caffeine exposure induced the low peak bone mass and increased the susceptibility to osteoporosis in the male adult offspring, but not in female adult offspring.
FIGURE 3.
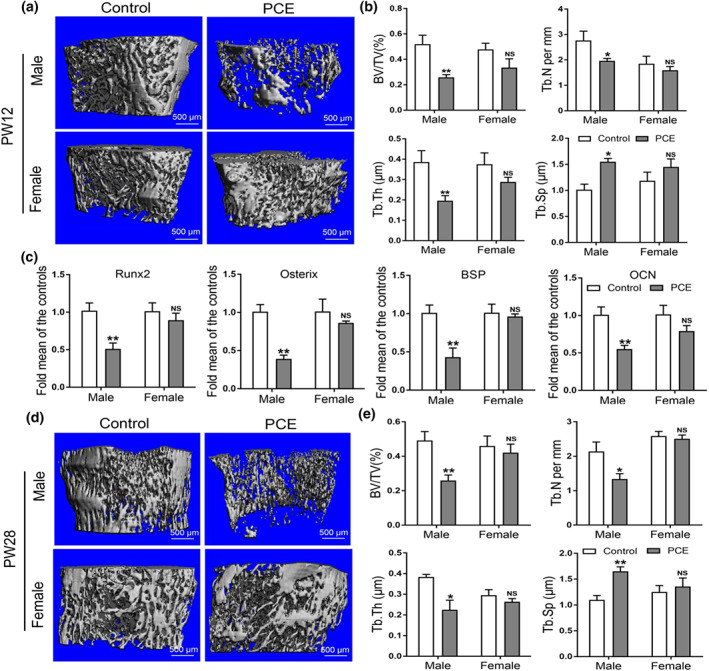
Effects of prenatal caffeine exposure (PCE) on peak bone mass accumulation and osteogenic function in male and female adult offspring. (a) Representative micro‐CT images of femur from control and PCE offspring at 12 weeks old; (b) quantitative micro‐CT analysis of trabecular bone microarchitecture at 12 weeks old (n = 8 per group); (c) RT‐qPCR analysis of osteogenic marker genes expression, including Runx2, osterix, bone sialoprotein (BSP) and osteocalcin (OCN) in bone tissue at 12 weeks old (n = 8 per group). (d) Representative micro‐CT images of femur from control and PCE offspring at 28 weeks old; (e) quantitative micro‐CT analysis of trabecular bone microarchitecture at 28 weeks old (n = 8 per group); mean ± SEM, *P < 0.05 compared with the control; NS, no significance. Scale bar = 500 μm. BV/TV, bone volume/trabecula volume; Tb. N, trabecula number; Tb. Th, trabecula thickness; Tb. Sp, trabecula space
3.4. Effects of prenatal caffeine exposure on serum/bone corticosterone concentration and bone 11β‐HSD2 expression in adult rat offspring
We further detected the corticosterone concentration in serum and bone tissue in male and female adult offspring. The results showed that even though the serum corticosterone concentration was not affected by prenatal caffeine exposure in male offspring (Figure 4a), while prenatal caffeine exposure significantly increased the bone corticosterone concentration in male offspring (Figure 4b). However, prenatal caffeine exposure did not affect both the serum and bone corticosterone concentration in female adult offspring (Figure 4a,b). When analysing the expression of 11β‐HSD2 in bone tissue, we found that prenatal caffeine exposure consistently significantly reduced 11β‐HSD2 mRNA expression in male offspring (Figure 4c). Also, the immunohistochemical results indicated that the 11β‐HSD2 protein level in male adult bone tissue was significantly down‐regulated by prenatal caffeine exposure (Figure 4d,e). Nevertheless, prenatal caffeine exposure did not cause significant changes in the mRNA and protein expression of 11β‐HSD2 in female adult offspring (Figure 4c–e). The correlation analysis results in male offspring showed that there was no apparent correlation between serum corticosterone concentration and bone 11β‐HSD2 expression (Figure 4f), but there was a significantly negative correlation between bone 11β‐HSD2 expression and bone corticosterone concentration (Figure 4g,h). In female adult offspring, the correlations between serum corticosterone concentration and bone 11β‐HSD2 expression, bone 11β‐HSD2 expression and bone corticosterone showed no obvious changes (Figure 4f–h).
FIGURE 4.
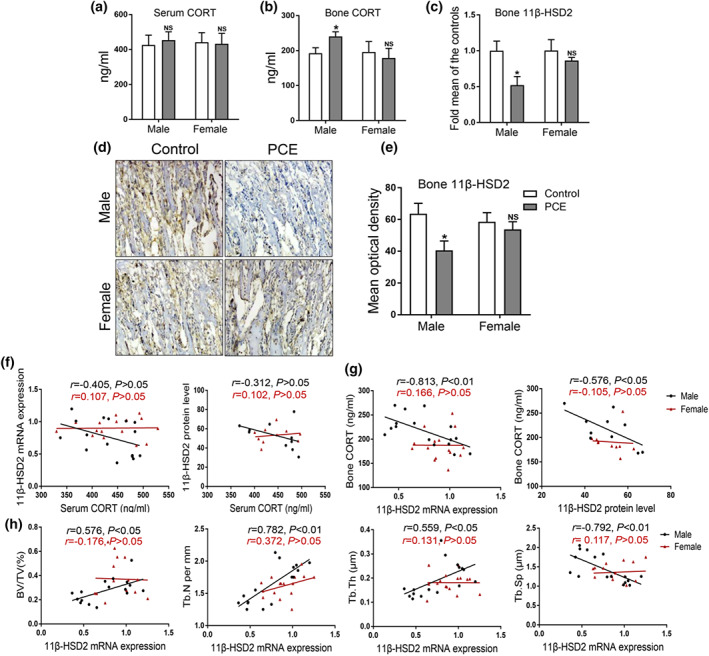
Effects of prenatal caffeine exposure (PCE) on corticosterone concentration and 11β‐hydroxysteroid dehydrogenase 2 (11β‐HSD2) expression in 12‐week‐old male and female offspring. (a) Serum corticosterone (CORT) concentration (n = 8 per group); (b) bone tissue CORT concentration (n = 8 per group); (c) RT‐qPCR analysis of bone 11β‐HSD2 mRNA expression (n = 8 per group); (d) representative immunohistochemical images of 11β‐HSD2 in adult bone tissue; (e) quantitative immunohistochemical analysis of the mean optical density of 11β‐HSD2 protein (n = 5 per group); (f) the correlation analysis between serum CORT concentration and bone 11β‐HSD2 mRNA and protein expression; (g) the correlation analysis of bone 11β‐HSD2 mRNA and protein expression and bone CORT; (h) the correlation analysis of bone 11β‐HSD2 mRNA expression and BV/TV, Tb. N, Tb. Th and Tb. Sp. Mean ± SEM, *P < 0.05 compared with the control. BV/TV, bone volume/trabecula volume; Tb. N, trabecula number; Tb. Th, trabecula thickness; Tb. Sp, trabecula space
To determine the effect of 11β‐HSD2 expression on bone mass formation, we analysed the correlation between bone 11β‐HSD2 expression and bone mass index. The results demonstrated that there were positive correlations between bone 11β‐HSD2 expression and bone volume/total volume, trabecular number and trabecular thickness in male adult offspring (Figure 4i). However, there were no evident changes in the correlation between bone 11β‐HSD2 expression and bone mass indicators in female adult offspring (Figure 4i). Collectively, the low peak bone mass was closely related to the reduced bone 11β‐HSD2 expression in prenatal caffeine exposure male adult offspring, but not in females.
3.5. The intrauterine programming mechanism of low‐expressional 11β‐HSD2 induced by prenatal caffeine exposure in male rat offspring
Then, we investigated whether the epigenetic modification alteration participated in the consistently decreased 11β‐HSD2 expression induced by prenatal caffeine exposure in male offspring before and after birth. A ChIP assay was used to detect the histone acetylation level in the 11β‐HSD2 promoter region. We found that prenatal caffeine exposure significantly decreased the H3K9ac levels in 11β‐HSD2 promoter region from gestational day 20 to postnatal week 12 in male offspring (Figure 5a,b). These results indicated that prenatal caffeine exposure continuously down‐regulated the H3K9ac level of 11β‐HSD2, which mediated the decreased 11β‐HSD2 expression in male offspring.
FIGURE 5.
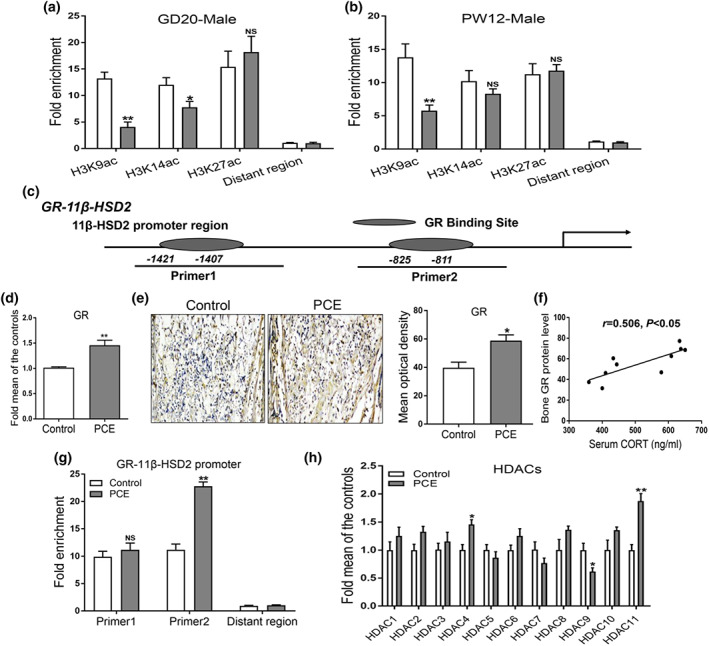
Effects of prenatal caffeine exposure (PCE) on histone acetylation level of 11β‐hydroxysteroid dehydrogenase 2 (11β‐HSD2) and its intrauterine programming mechanism in male offspring. (a) ChIP assay of the histone acetylation level in 11β‐HSD2 promoter region on gestational day 20 (n = 8 per group); (b) ChIP assay of the histone acetylation level in 11β‐HSD2 promoter region at postnatal week (PW) 12 (n = 8 per group); (c) analysis of 11β‐HSD2 promoter region; (d) RT‐qPCR analysis of glucocorticoid receptor (GR) mRNA expression in fetal bone tissue (n = 8 per group); (e) representative immunohistochemical images and quantitative analysis of GR protein in fetal bone tissue (n = 5 per group); (f) the correlation analysis between serum CORT concentration and bone GR protein level on GD20; (g) detection of GR binding to 11β‐HSD2 promoter region through ChIP‐PCR (n = 8 per group); (h) RT‐qPCR analysis of gene expression of HDACs in fetal bone tissue (n = 8 per group). Mean ± SEM, *P < 0.05 compared with the control
We further explored the potential mechanism of the decreased H3K9ac level of 11β‐HSD2 in response to prenatal caffeine exposure in utero. When analysing the promoter region of 11β‐HSD2, we found that there were glucocorticoid receptor binding sites in the 11β‐HSD2 promoter region (Figure 5c). Moreover, the mRNA and protein expression of glucocorticoid receptor were significantly increased by prenatal caffeine exposure in fetal bone tissue (Figure 5d,e). Meanwhile, there was a positive correlation between serum corticosterone concentration and bone glucocorticoid receptor protein level (Figure 5f). The ChIP assay results also suggested that prenatal caffeine exposure could prompt glucocorticoid receptor binding to 11β‐HSD2 promoter region (Figure 5g). It is known that glucocorticoids usually regulate the histone deacetylation modification of target genes through activating glucocorticoid receptor and recruiting histone deacetylases (HDACs; Ratman et al., 2013). Thus, we detected the expression of HDACs in male fetal bone tissue between the control and prenatal caffeine exposure groups and found that HDAC11 expression was significantly up‐regulated by prenatal caffeine exposure (Figure 5h). These above findings suggested that the increased fetal serum corticosterone concentration caused by prenatal caffeine exposure might promote glucocorticoid receptor activation and recruit HDAC11, which co‐operatively decreased the H3K9ac level of 11β‐HSD2.
3.6. Corticosterone inhibits osteogenic differentiation of BMSCs by down‐regulating 11β‐HSD2 expression in vitro
To investigate whether corticosterone could inhibit osteogenic differentiation through decreasing 11β‐HSD2 expression in vitro, BMSCs were treated with different concentrations of corticosterone (250, 500 and 1,000 nM) in the process of osteogenic differentiation culture. We found that the expression of osteogenic differentiation markers (Runx2, osterix, BSP and OCN) were all significantly down‐regulated by corticosterone (Figure 6a). Moreover, a high concentration of corticosterone significantly down‐regulated 11β‐HSD2 mRNA expression (Figure 6b). The western blotting assay showed that the protein level of 11β‐HSD2 was also reduced by 500‐nM corticosterone (Figure 6c). We further determined whether up‐regulating 11β‐HSD2 expression by using 11β‐HSD2 plasmid could alleviate the inhibited osteogenic differentiation attributed to corticosterone. It was shown that the 11β‐HSD2 plasmid remarkably up‐regulated the protein level of 11β‐HSD2 but had no damage effects on the cell activity, osteoblastic differentiation and mineralization in the osteogenic differentiation culture (Figure S1). When co‐treating BMSCs with corticosterone and 11β‐HSD2 plasmid, we found that the 11β‐HSD2 plasmid significanlty alleviated the inhibitory effect of osteogenic differentiation caused by corticosterone (Figure 6d). These findings indicated that corticosterone suppressed BMSCs osteogenic differentiation by decreasing the expression of 11β‐HSD2.
FIGURE 6.
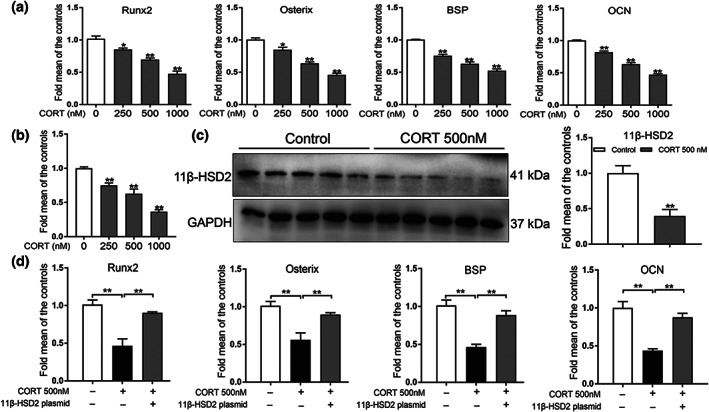
Effects of corticosterone (CORT) on osteogenic differentiation and 11β‐hydroxysteroid dehydrogenase 2 (11β‐HSD2) expression in bone marrow mesenchymal stem cells (BMSCs). (a) RT‐qPCR analysis of gene expression of Runx2, osterix, bone sialoprotein (BSP) and osteocalcin (OCN) in BMSCs cultured in the osteogenic medium and treated with different concentrations of CORT for 3 days (n = 5 per group); (b) RT‐qPCR analysis of gene expression of 11β‐HSD2 in BMSCs cultured in the osteogenic medium and treated with different concentrations of CORT for 3 days (n = 5 per group); (c) western blotting assay of 11β‐HSD2 in BMSCs with 500 nM CORT for 3 days (n = 5 per group). (d) RT‐qPCR analysis of gene expression of Runx2, osterix, BSP and OCN in BMSCs cultured in the osteogenic medium and treated with 500‐nM CORT and 11β‐HSD2 plasmid for 3 days (n = 5 per group). Mean ± SEM, *P < 0.05 compared with the untreated cells; NS, no significance
3.7. The molecular mechanism of the down‐regulated 11β‐HSD2 expression level in response to corticosterone
We then explored the molecular mechanism of the down‐regulated 11β‐HSD2 expression induced by corticosterone in vitro. When treating BMSCs with corticosterone in the process of osteogenic differentiation culture, we found that the glucocorticoid receptor protein level was decreased in the cytoplasm but increased in the nucleus (Figure 7a). Moreover, the immunofluorescence results indicated that a high concentration of corticosterone led to an increase of glucocorticoid receptor in the BMSCs nucleus (Figure 7b). This demonstrated that corticosterone could activate glucocorticoid receptor by promoting the transfer of glucocorticoid receptor into the nucleus. Meanwhile, the protein level of HDAC11 was increased in the BMSC nucleus in response to corticosterone, and mifepristone (RU486), the glucocorticoid receptor antagonist partially reversed the above performance (Figure 7c). The ChIP assay results indicated that corticosterone decreased the H3K9ac level in the 11β‐HSD2 promoter region, whereas the mifepristone (RU486) and HDAC11 siRNA both attenuated the decrease in H3K9ac and the expression levels of 11β‐HSD2 induced by corticosterone (Figure 7d). Meanwhile, the mifepristone (RU486) and HDAC11 siRNA could partly reverse the decreased expression levels of 11β‐HSD2 caused by corticosterone (Figure 7e). Taken together, this indicated that corticosterone promoted HDAC11 into the nucleus via activating glucocorticoid receptor, which further cooperatively decreased H3K9ac and expression levels of 11β‐HSD2 in the process of osteogenic differentiation.
FIGURE 7.
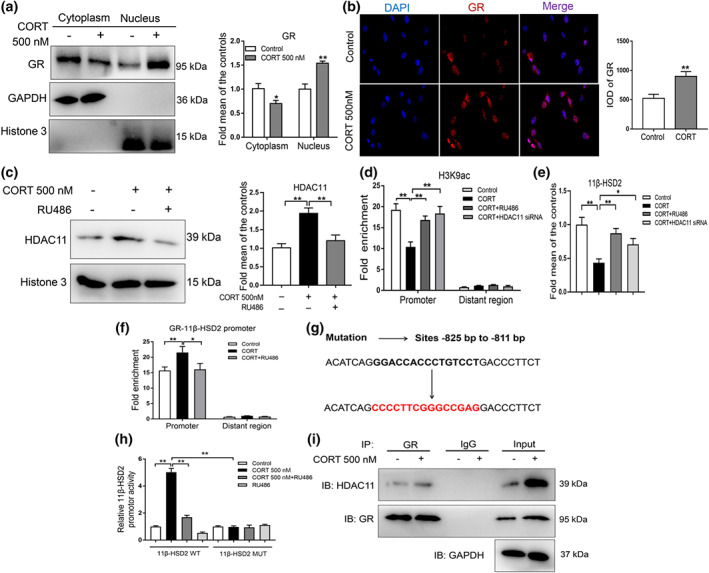
The molecular mechanism of the decreased histone acetylation level of 11β‐hydroxysteroid dehydrogenase 2 (11β‐HSD2) induced by corticosterone (CORT) in bone marrow mesenchymal stem cells (BMSCs). (a) Western blotting assay of glucocorticoid receptor (GR) protein level in cytoplasm and nucleus after treating BMSCs with 500‐nM CORT for 3 days (n = 5 per group); (b) immunofluorescence analysis of GR after treating BMSCs with 500‐nM CORT for 3 days (n = 5 per group); (c) western blotting assay of histone deacetylase 11 (HDAC11) protein level in nucleus after treating BMSCs with 500‐nM CORT or co‐treating BMSCs with 500‐nM CORT and 30‐μM mifepristone (RU486) for 3 days (n = 5 per group); (d) ChIP assay of the histone 3 lysine 9 acetylation (H3K9ac) in 11β‐HSD2 promoter region in BMSCs treated with 500‐nM CORT or co‐treated with CORT and RU486 or HDAC11 siRNA for 3 days (n = 5 per group). (e) RT‐qPCR analysis of gene expression of 11β‐HSD2 in BMSCs treated with 500‐nM CORT or co‐treated with CORT and RU486 or HDAC11 siRNA for 3 days (n = 5 per group). (f) Detection of GR binding to 11β‐HSD2 promoter region through ChIP‐PCR after treating BMSCs with 500‐nM CORT or co‐treating BMSCs with CORT and RU486 for 3 days (n = 5 per group); (g) the sequence of the mutant site in 11β‐HSD2 promoter region; (h) luciferase activities assay for the binding site of GR with 11β‐HSD2 promoter region (n = 5 per group). (i) Co‐IP assay of GR and HDAC11 (n = 5 per group). Mean ± SEM, *P < 0.05, compared with the untreated cells
Next, we clarified how these factors, including glucocorticoid receptor and HDAC11, co‐operatively decreased the H3K9ac level of 11β‐HSD2 in vitro. It was found that corticosterone could promote glucocorticoid receptor binding to the promoter region of 11β‐HSD2 (−825 bp to −811 bp), and the above changes can be reversed by glucocorticoid receptor antagonist mifepristone (RU486) (Figure 7f). We next constructed the luciferase reporter vectors containing a wild‐type or a mutant binding site to confirm the binding site of glucocorticoid receptor with the 11β‐HSD2 promoter region (Figure 7g). We observed that under treatment with corticosterone, the luciferase activities of GV238‐11β‐HSD2‐WT remarkably increased, which was significantly reversed by glucocorticoid receptor antagonist mifepristone (RU486) (Figure 7h). When GV238‐11β‐HSD2‐MUT was transferred into the BMSCs, corticosterone failed to affect the luciferase activities (Figure 7h). These results indicated that a high concentration of corticosterone could promote glucocorticoid receptor binding to the 11β‐HSD2 promoter region, and the binding site was located from −825 bp to −811 bp. We further found that corticosterone promoted the glucocorticoid receptor interaction with HDAC11 through CO‐IP assay (Figure 7i). Collectively, a high concentration of corticosterone activated glucocorticoid receptor into the BMSCs nucleus and then directly bind to the promoter region of 11β‐HSD2 and recruit HDAC11, resulting in the decreased H3K9ac level in 11β‐HSD2 promoter region.
4. DISCUSSION
4.1. Prenatal caffeine exposure led to low peak bone mass and increased osteoporosis susceptibility in male offspring but not in females
An adverse environment during pregnancy can result in fetal growth restriction, characterized by low birth weight and functional disorder of multiple organs (Rossner et al., 2011). As a typically adverse environment in utero, prenatal caffeine exposure also can cause multi‐organ developmental toxicity in foetuses and susceptibility to multiple diseases in adults such as non‐alcoholic fatty liver, osteoarthritis and metabolic syndrome (Hu et al., 2019; Pei et al., 2017; Tan et al., 2018). Epidemiologic evidence indicated that a caffeine intake of 300 mg per day in pregnant women is associated with an increased risk of intrauterine growth retardation in foetuses (Guilbert, 2003). Based on the dose conversion between human and rat (1:6.17; Reagan‐Shaw, Nihal, & Ahmad, 2008), 300 mg per day caffeine intake for a 60 kg pregnant woman approximately corresponds to 30 mg·kg−1 per day caffeine intake for a pregnant rat. Therefore, in our previous study, the pregnant rats were exposed to caffeine at these doses, including 30, 60 and 120 mg·kg−1·day−1 to induce intrauterine growth retardation offspring rats, which showed a good dose‐dependent effect, and we observed a remarkable phenomenon of bone development retardation in the intrauterine growth retardation foetuses with a caffeine exposure dose of 120 mg·kg−1 daily (Shangguan et al., 2017). Hence, in the present study, the pregnant rats were administrated with the dose caffeine of 120 mg·kg−1·day−1 to investigate the long‐term effects of bone development toxicity induced by prenatal caffeine exposure.
Previous data showed that prenatal caffeine exposure could lead to bone dysplasia in female foetuses, which was related to the inhibited apoptosis of hypertrophic chondrocytes in growth plate (Shangguan et al., 2017). In this study, we compared the effects of prenatal caffeine exposure on bone development and peak bone mass accumulation in male and female offspring to reveal the long‐term harmful phenomenon about bone development and its gender differences induced by prenatal caffeine exposure. Our present findings indicated that prenatal caffeine exposure caused bone dysplasia in both male and female fetal rats, and these changes were related to the inhibition of osteogenic differentiation. We further found prenatal caffeine exposure only induced the low peak bone mass in male adult offspring, accompanied by the continuous inhibition of osteogenic differentiation, but prenatal caffeine exposure did not affect the osteogenic function and peak bone mass accumulation in female adult offspring. This demonstrated that there were gender differences in the effect of prenatal caffeine exposure on the peak bone mass and osteoporosis susceptibility in adult offspring, showing as a low peak bone mass and increased osteoporosis susceptibility in the males, but not in females.
4.2. The low‐expressional programming of 11β‐HSD2 contributed to osteoporosis susceptibility induced by prenatal caffeine exposure in male adult offspring
It was indicated that the high level of glucocorticoids in the intrauterine period induced by adverse environment during pregnancy is the main factor for programming multiple‐organ dysplasia (Drake, Tang, & Nyirenda, 2007; Moisiadis & Matthews, 2014). In this study, we found that prenatal caffeine exposure significantly increased the serum corticosterone in male foetuses, however the serum corticosterone level was not affected by prenatal caffeine exposure in adulthood. We previously shown that prenatal caffeine exposure could elevate the serum corticosterone in the maternal body and opened the placenta barrier of glucocorticoids, which caused a large amount of maternal corticosterone to enter the fetus serum through the placenta, thereafter leading to an increased corticosterone in fetal serum (Xu et al., 2012). Hence, the increased serum corticosterone in foetuses was due to overexposure to excessive maternal corticosterone following prenatal caffeine exposure. After birth, the prenatal caffeine exposure offspring has been out of the environment of excessive maternal corticosterone, which caused the serum corticosterone returning to the normal level in adulthood.
Furthermore, we found that the corticosterone concentration in bone tissue was continuously increased by prenatal caffeine exposure in male offspring from fetal period to adulthood. Meanwhile, prenatal caffeine exposure reduced the bone 11β‐HSD2 expression in male offspring before and after birth. Previous studies reported that the stable expression of 11β‐HSD2 in bone tissue can inactivate serum corticosterone and effectively regulate the bone corticosterone level to ensure the normal bone development (Henneicke et al., 2017; McNeil et al., 2007). So, these findings suggested that the continuously increased bone corticosterone was attributed to the reduced bone 11β‐HSD2 expression in prenatal caffeine exposure male offspring. The correlation analysis further indicated that the reduced peak bone mass accumulation was related to the consistently decreased bone 11β‐HSD2 expression in male adult offspring. In vitro, we also found that high levels of corticosterone could down‐regulate the expression of 11β‐HSD2 in the osteogenic differentiation process of BMSCs, while 11β‐HSD2 overexpression plasmid partly reversed the osteogenic function suppressed by corticosterone. Collectively, these findings indicated that the low expression of bone 11β‐HSD2 consistently led to an increase in bone corticosterone, thereafter, inhibiting osteogenic differentiation and causing osteoporosis susceptibility in prenatal caffeine exposure male offspring.
4.3. The glucocorticoid receptor/HDAC11 cooperatively decreased the H3K9ac level in the 11β‐HSD2 promoter region in response to glucocorticoids
Epigenetics refers to situations in which the DNA sequence does not change but the heritability gene expression changes, including histone acetylation modification, which exists in the normal development of mammals (Igarashi, Ideta‐Otsuka, & Narita, 2017). However, histone acetylation abnormalities participated in the occurrence and development of various environment‐genetic interaction‐related diseases (Tzika, Dreker, & Imhof, 2018). Histone acetylation is known to have a variety of modification forms, such as H2A, H2B, H3, and H4 acetylation, which can regulate the transcriptional activity of target genes (Kameda, Awazu, & Togashi, 2019). Our recent study indicated that prenatal caffeine exposure reduced the H3K14 acetylation level in IGF1 promoter region, which caused a decrease in IGF1 expression and testicular dysplasia (Pei et al., 2019). It suggested that the abnormal H3 acetylation modification on the functional gene participated in organ dysplasia in the prenatal caffeine exposure rat model. Hence, in this research, we studied the role of H3 acetylation modification on the consistently low‐expressional 11β‐HSD2 induced by prenatal caffeine exposure in male offspring rats. Our present results show that prenatal caffeine exposure only decreased the H3K9ac levels in the 11β‐HSD2 promoter region before and after birth in male offspring, which then induced the low expression of 11β‐HSD2.
Glucocorticoids mainly participate in regulating cell proliferation and differentiation through interacting with multiple transcription factors via glucocorticoid receptor (Kadmiel & Cidlowski, 2013). In this research, we found that there were glucocorticoid receptor binding sites in the 11β‐HSD2 promoter region. Moreover, corticosterone could promote the transfer of glucocorticoid receptor into the nucleus of BMSCs and binding to the promoter region of 11β‐HSD2, and the binding site was located from ‐825 bp to ‐811 bp. It is known that glucocorticoids usually regulate the histone deacetylation modification of target genes through activating glucocorticoid receptor and recruiting HDACs (Ratman et al., 2013). Thus, we detected the expression of HDACs in male fetal bone tissue and found that the expression levels of HDAC4 and HDAC11 were up‐regulated in bone tissue. In vitro, we found that knockdown HDAC4 expression by HDAC4 siRNA could not reversed the reduced H3K9ac level of 11β‐HSD2 due to corticosterone, but the HDAC11 siRNA alleviated the decreased H3K9ac and expression levels of 11β‐HSD2 induced by corticosterone. These finding indicated that HDAC11 mediated the decreased H3K9ac level in the 11β‐HSD2 promoter region caused by corticosterone. Moreover, corticosterone could promote the interaction of glucocorticoid receptor with HDAC11 in the 11β‐HSD2 promoter region. This indicated that corticosterone promoted HDAC11 into the nucleus via activating glucocorticoid receptor, which further cooperatively decreased H3K9ac levels of 11β‐HSD2 in the process of osteogenic differentiation.
4.4. The possible mechanism for gender differences of susceptibility to osteoporosis in adult offspring due to prenatal caffeine exposure
In the present study, we found that the male offspring continued to have low bone mass due to prenatal caffeine exposure, while there was an increase in bone mass in the postnatal female offspring. Meanwhile, there were gender differences in the bone 11β‐HSD2 expression in adult offspring following prenatal caffeine exposure, shown as a continuous decrease in 11β‐HSD2 expression in males, but an increasing expreesion in females after birth. Intriguingly, the correlation analysis between bone 11β‐HSD2 expression and bone mass indicators demonstrated that the gender difference of bone mass was closely related to the bone 11β‐HSD2 expression in adult offspring. Previous research revealed that antenatal betamethasone exposure induced a gender difference in placental 11β‐HSD2 expression, which contributed to the increased incidence of poor outcomes observed in preterm male foetuses but not in females (Stark, Wright, & Clifton, 2009). Also, studies indicated that the gonadal differences between male and female lead to variation in sex hormones produced (testosterone in men and estrogen in women), which are responsible for gender differences in gene expression and causing different phenotype (Quinn, Ramamoorthy, & Cidlowski, 2014). Collectively, it suggested that the gender difference in bone 11β‐HSD2 expression may contribute to the gender differences in bone mas changes caused by prenatal caffeine exposure, which was considered to be due to their sex hormones level in adult offspring. However, its in‐depth mechanism of this remains to be elucidated in our future research.
5. CONCLUSIONS
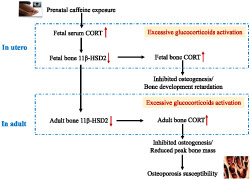
This study revealed that prenatal caffeine exposure caused gender differences of the peak bone mass accumulation in adult offspring rats. Moreover, it indicated that prenatal caffeine exposure only led to low peak bone mass and increased the susceptibility to osteoporosis in male adult offspring, which was mediated by the enhanced effects of corticosterone on suppressing osteogenic differentiation in response to low‐expressional programming of 11β‐HSD2 via glucocorticoid receptor/HDAC11 epigenetic mechanism (as shown in Figure 8). This research helps to uncover the long‐term harm phenomenon of bone development induced by prenatal caffeine exposure, to investigate the early prevention and treatment target and to provide an experimental basis for clarifying the theory of “Developmental Origins of Health and Disease”.
FIGURE 8.
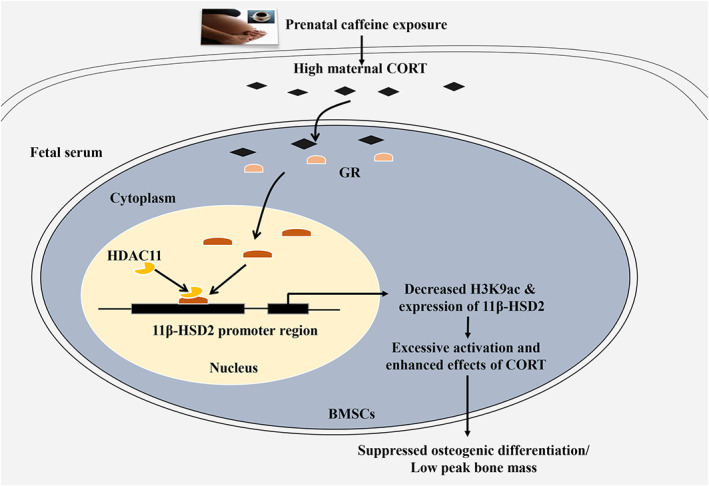
The low‐expression programming of 11β‐HSD2 mediates osteoporosis susceptibility induced by prenatal caffeine exposure in male offspring rats. Corticosterone (CORT); 11β‐HSD2, 11 β‐hydroxysteroid dehydrogenases; GR, glucocorticoid receptor; HDAC11, histone deacetylation 11; H3K9ac, histone 3 lysine 9 acetylation; BMSCs, bone marrow mesenchymal stem cells
AUTHOR CONTRIBUTIONS
H. W., L. B. C. and H. X. designed the research study; H. X., Z. X. W. and Y. F. S. performed the research; H. X., Z. X. W., B. L., L. B. C. and H. W. analysed the data; H. X., Z. X. W., B. L., L. B. C., H. W., J. M. and J. F. S. wrote and revised the paper; all authors approved the final manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Figure S1. The effects of manipulation of 11β‐HSD2 expression by 11β‐HSD2 2 plasmid on preosteoblast cells. A. MTS assay for cell activity; B. Alizarin red staining for mineralization detection; C. RT‐qPCR analysis of gene expression of Runx2, osterix, bone sialoprotein (BSP), osteocalcin (OCN). n = 5 per group, mean ± S.E.M., NS, no significance.
Figure S2. Western blotting assay of HDAC11 or 11β‐HSD2 protein level after treating bone marrow mesenchymal stem cells with HDAC11 siRNA or 11β‐HSD2 plasmid. n = 5 per group, mean ± S.E.M., **P < 0.01 compared with the control group.
Figure S3. ChIP assay of the H3K9ac level in 11β‐HSD2 promoter region after treating bone marrow mesenchymal stem cells with 500 nM CORT or combined with HDAC4 siRNA. n = 5 per group, mean ± S.E.M., **P < 0.01 compared with the control; NS, no significance.
Table S1. Primers used in quantitative real‐time PCR
Table S2. The offspring's body weight in control and prenatal caffeine exposure
Data S1 Supporting information
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (81673490, 81673524, 81972036 and 81430089), the National Key Research and Development Program of China (2017YFC 1001300) and the Medical Science Advancement Program (Basic Medical Sciences) of Wuhan University (TFJC2018001).
Xiao H, Wu Z, Li B, et al. The low‐expression programming of 11β‐HSD2 mediates osteoporosis susceptibility induced by prenatal caffeine exposure in male offspring rats. Br J Pharmacol. 2020;177:4683–4700. 10.1111/bph.15225
Hao Xiao and Zhixin Wu contributed equally to this work.
Contributor Information
Liaobin Chen, Email: lbchen@whu.edu.cn.
Hui Wang, Email: wanghui19@whu.edu.cn.
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Watts, V. (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … Wong, S. S. (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Introduction and Other Protein Targets. British Journal of Pharmacology, 176, S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Roberts, R. E. , Broughton, B. R. , Christopher, G. S. , George, C. H. , Stanford, S. C. , … Insel, P. A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology . British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird, J. , Jacob, C. , Barker, M. , Fall, C. H. , Hanson, M. , Harvey, N. C. , … Cooper, C. (2017). Developmental origins of health and disease: A lifecourse approach to the approach of non‐communicable diseases. Healthcare, 5, 14–26. 10.3390/healthcare5010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasuriya, C. N. D. , Evensen, K. A. I. , Mosti, M. P. , Brubakk, A. M. , Jacobsen, G. W. , Indredavik, M. S. , … Syversen, U. (2017). Peak bone mass and bone microarchitecture in adults born with low birth weight preterm or at term: A cohort study. The Journal of Clinical Endocrinology and Metabolism, 102, 2491–2500. 10.1210/jc.2016-3827 [DOI] [PubMed] [Google Scholar]
- Bouxsein, M. L. , Boyd, S. K. , Christiansen, B. A. , Guldberg, R. E. , Jepsen, K. J. , & Muller, R. (2010). Guidelines for assessment of bone microstructure in rodents using micro‐computed tomography. Journal of Bone and Mineral Research, 25, 1468–1486. 10.1002/jbmr.141 [DOI] [PubMed] [Google Scholar]
- Chapman, K. , Holmes, M. , & Seckl, J. (2013). 11β‐hydroxysteroid dehydrogenases: Intracellular gate‐keepers of tissue glucocorticoid action. Physiological Reviews, 93, 1139–1206. 10.1152/physrev.00020.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker, J. , Hooper, S. B. , van Vonderen, J. J. , Witlox, R. , Lopriore, E. , & te Pas, A. B. (2017). Caffeine to improve breathing effort of preterm infants at birth: A randomized controlled trial. Pediatric Research, 82, 290–296. 10.1038/pr.2017.45 [DOI] [PubMed] [Google Scholar]
- Dempster, D. W. , Compston, J. E. , Drezner, M. K. , Glorieux, F. H. , Kanis, J. A. , Malluche, H. , … Parfitt, A. M. (2013). Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. Journal of Bone and Mineral Research, 28, 2–17. 10.1002/jbmr.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake, A. J. , Tang, J. I. , & Nyirenda, M. J. (2007). Mechanisms underlying the role of glucocorticoids in the early life programming of adult disease. Clinical Science (London, England), 113, 219–232. 10.1042/CS20070107 [DOI] [PubMed] [Google Scholar]
- Frary, C. D. , Johnson, R. K. , & Wang, M. Q. (2005). Food sources and intakes of caffeine in the diets of persons in the United States. Journal of the American Dietetic Association, 105, 110–113. 10.1016/j.jada.2004.10.027 [DOI] [PubMed] [Google Scholar]
- Guilbert, J. J. (2003). The world health report 2002—Reducing risks, promoting healthy life. Education for Health (Abingdon, England), 16, 230. [DOI] [PubMed] [Google Scholar]
- He, Z. , Zhang, J. , Huang, H. , Yuan, C. , Zhu, C. , Magdalou, J. , & Wang, H. (2019). Glucocorticoid‐activation system mediated glucocorticoid‐insulin‐like growth factor 1 (GC‐IGF1) axis programming alteration of adrenal dysfunction induced by prenatal caffeine exposure. Toxicology Letters, 302, 7–17. 10.1016/j.toxlet.2018.12.001 [DOI] [PubMed] [Google Scholar]
- Heaney, R. P. , Abrams, S. , Dawson‐Hughes, B. , Looker, A. , Marcus, R. , Matkovic, V. , & Weaver, C. (2000). Peak bone mass. Osteoporosis International, 11, 985–1009. 10.1007/s001980070020 [DOI] [PubMed] [Google Scholar]
- Henneicke, H. , Li, J. , Kim, S. , Gasparini, S. J. , Seibel, M. J. , & Zhou, H. (2017). Chronic mild stress causes bone loss via an osteoblast‐specific glucocorticoid‐dependent mechanism. Endocrinology, 158, 1939–1950. 10.1210/en.2016-1658 [DOI] [PubMed] [Google Scholar]
- Heppe, D. H. , Medina‐Gomez, C. , Hofman, A. , Rivadeneira, F. , & Jaddoe, V. W. (2015). Does fetal smoke exposure affect childhood bone mass? The Generation R Study. Osteoporosis International, 26, 1319–1329. 10.1007/s00198-014-3011-z [DOI] [PubMed] [Google Scholar]
- Hu, S. , Xia, L. , Luo, H. , Xu, Y. , Yu, H. , Xu, D. , & Wang, H. (2019). Prenatal caffeine exposure increases the susceptibility to non‐alcoholic fatty liver disease in female offspring rats via activation of GR‐C/EBPα‐SIRT1 pathway. Toxicology, 417, 23–34. 10.1016/j.tox.2019.02.008 [DOI] [PubMed] [Google Scholar]
- Igarashi, K. , Ideta‐Otsuka, M. , & Narita, M. (2017). The current state and future development of epigenetic toxicology. Yakugaku Zasshi, 137, 265–271. 10.1248/yakushi.16-00230-3 [DOI] [PubMed] [Google Scholar]
- Kadmiel, M. , & Cidlowski, J. A. (2013). Glucocorticoid receptor signaling in health and disease. Trends in Pharmacological Sciences, 34, 518–530. 10.1016/j.tips.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameda, T. , Awazu, A. , & Togashi, Y. (2019). Histone tail dynamics in partially disassembled nucleosomes during chromatin remodeling. Frontiers in Molecular Biosciences, 6, 133 10.3389/fmolb.2019.00133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P. , Deng, Q. , Liu, J. , Yan, J. , Wei, Z. , Zhang, Z. , … Li, B. (2019). Roles for HB‐EGF in mesenchymal stromal cell proliferation and differentiation during skeletal growth. Journal of Bone and Mineral Research, 34, 295–309. 10.1002/jbmr.3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. , Cirino, G. , Docherty, J. R. , … Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177, 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil, C. J. , Nwagwu, M. O. , Finch, A. M. , Page, K. R. , Thain, A. , McArdle, H. J. , & Ashworth, C. J. (2007). Glucocorticoid exposure and tissue gene expression of 11β HSD‐1, 11β HSD‐2, and glucocorticoid receptor in a porcine model of differential fetal growth. Reproduction, 133, 653–661. 10.1530/rep.1.01198 [DOI] [PubMed] [Google Scholar]
- Moisiadis, V. G. , & Matthews, S. G. (2014). Glucocorticoids and fetal programming part 1: Outcomes. Nature Reviews. Endocrinology, 10, 391–402. 10.1038/nrendo.2014.73 [DOI] [PubMed] [Google Scholar]
- O'Brien, C. A. , Jia, D. , Plotkin, L. I. , Bellido, T. , Powers, C. C. , Stewart, S. A. , … Weinstein, R. S. (2004). Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology, 145, 1835–1841. 10.1210/en.2003-0990 [DOI] [PubMed] [Google Scholar]
- Pei, L. G. , Yuan, C. , Guo, Y. T. , Kou, H. , Xia, L. P. , Zhang, L. , … Wang, H. (2017). Prenatal caffeine exposure induced high susceptibility to metabolic syndrome in adult female offspring rats and its underlying mechanisms. Reproductive Toxicology, 71, 150–158. 10.1016/j.reprotox.2017.06.045 [DOI] [PubMed] [Google Scholar]
- Pei, L. G. , Zhang, Q. , Yuan, C. , Liu, M. , Zou, Y. F. , Lv, F. , … Wang, H. (2019). The GC‐IGF1 axis‐mediated testicular dysplasia caused by prenatal caffeine exposure. The Journal of Endocrinology, 242, M17–M32. 10.1530/JOE-18-0684 [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn, M. , Ramamoorthy, S. , & Cidlowski, J. A. (2014). Sexually dimorphic actions of glucocorticoids: Beyond chromosomes and sex hormones. Annals of the new York Academy of Sciences, 1317, 1–6. 10.1111/nyas.12425 [DOI] [PubMed] [Google Scholar]
- Ratman, D. , Vanden Berghe, W. , Dejager, L. , Libert, C. , Tavernier, J. , Beck, I. M. , & de Bosscher, K. (2013). How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Molecular and Cellular Endocrinology, 380, 41–54. 10.1016/j.mce.2012.12.014 [DOI] [PubMed] [Google Scholar]
- Reagan‐Shaw, S. , Nihal, M. , & Ahmad, N. (2008). Dose translation from animal to human studies revisited. FASEB, 22, 659–661. 10.1096/fj.07-9574LSF [DOI] [PubMed] [Google Scholar]
- Rossner, P. Jr. , Tabashidze, N. , Dostal, M. , Novakova, Z. , Chvatalova, I. , Spatova, M. , & Sram, R. J. (2011). Genetic, biochemical, and environmental factors associated with pregnancy outcomes in newborns from the Czech Republic. Environmental Health Perspectives, 119, 265–271. 10.1289/ehp.1002470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangguan, Y. , Jiang, H. , Pan, Z. , Xiao, H. , Tan, Y. , Tie, K. , … Wang, H. (2017). Glucocorticoid mediates prenatal caffeine exposure‐induced endochondral ossification retardation and its molecular mechanism in female fetal rats. Cell Death & Disease, 8, e3157 10.1038/cddis.2017.546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark, M. J. , Wright, I. M. , & Clifton, V. L. (2009). Sex‐specific alterations in placental 11β‐hydroxysteroid dehydrogenase 2 activity and early postnatal clinical course following antenatal betamethasone. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 297, R510–R514. 10.1152/ajpregu.00175.2009 [DOI] [PubMed] [Google Scholar]
- Tan, Y. , Liu, J. , Deng, Y. , Cao, H. , Xu, D. , Cu, F. , … Wang, H. (2012). Caffeine‐induced fetal rat over‐exposure to maternal glucocorticoid and histone methylation of liver IGF‐1 might cause skeletal growth retardation. Toxicology Letters, 214, 279–287. 10.1016/j.toxlet.2012.09.007 [DOI] [PubMed] [Google Scholar]
- Tan, Y. , Lu, K. , Li, J. , Ni, Q. , Zhao, Z. , Magdalou, J. , … Wang, H. (2018). Prenatal caffeine exprosure increases adult female offspring rat's susceptibility to osteoarthritis via low‐functional programming of cartilage IGF‐1 with histone acetylation. Toxicology Letters, 295, 229–236. 10.1016/j.toxlet.2018.06.1221 [DOI] [PubMed] [Google Scholar]
- Tzika, E. , Dreker, T. , & Imhof, A. (2018). Epigenetics and metabolism in health and disease. Frontiers in Genetics, 9, 361 10.3389/fgene.2018.00361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, Y. X. , Shangguan, Y. F. , Pan, Z. Q. , Hu, H. , Magdalou, J. , Chen, L. B. , & Wang, H. (2019). Activation of local bone RAS by maternal excessive glucocorticoid participated in the fetal programing of adult osteopenia induced by prenatal caffeine exposure. Toxicology and Applied Pharmacology, 363, 1–10. 10.1016/j.taap.2018.11.003 [DOI] [PubMed] [Google Scholar]
- Xiao, H. , Wen, Y. , Pan, Z. , Shangguan, Y. , Qin, J. , Tan, Y. , … Wang, H. (2018). Increased H3K27ac level of ACE mediates the intergenerational effect of low peak bone mass induced by prenatal dexamethasone exposure in male offspring rats. Cell Death & Disease, 9, 638 10.1038/s41419-018-0701-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, D. , Luo, H. W. , Hu, W. , Hu, S. W. , Yuan, C. , Wang, G. H. , … Wang, H. (2018). Intrauterine programming mechanism for hypercholesterolemia in prenatal caffeine‐exposed female adult rat offspring. The FASEB Journal, 32, 5563–5576. 10.1096/fj.201701557R [DOI] [PubMed] [Google Scholar]
- Xu, D. , Zhang, B. , Liang, G. , Ping, J. , Kou, H. , Li, X. , … Wang, H. (2012). Caffeine‐induced activated glucocorticoid metabolism in the hippocampus causes hypothalamic‐pituitary‐adrenal axis inhibition in fetal rats. PLoS ONE, 7, e44497 10.1371/journal.pone.0044497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, J. , Dwyer, T. , Riley, M. , Cochrane, J. , & Jones, G. (2010). The association between maternal diet during pregnancy and bone mass of the children at age 16. European Journal of Clinical Nutrition, 64, 131–137. 10.1038/ejcn.2009.117 [DOI] [PubMed] [Google Scholar]
- Zallocchi, M. L. , Matkovic, L. , Calvo, J. C. , & Damasco, M. C. (2004). Adrenal gland involvement in the regulation of renal 11β‐hydroxysteroid dehydrogenase 2. Journal of Cellular Biochemistry, 92, 591–602. 10.1002/jcb.20078 [DOI] [PubMed] [Google Scholar]
- Zhang, P. , Men, J. , Fu, Y. , Shan, T. , Ye, J. , Wu, Y. , … Jiang, H. (2012). Contribution of SATB2 to the stronger osteogenic potential of bone marrow stromal cells from craniofacial bones. Cell and Tissue Research, 350, 425–437. 10.1007/s00441-012-1487-4 [DOI] [PubMed] [Google Scholar]
- Zuo, R. , Liu, X. , Wang, W. , Li, W. , Ying, H. , & Sun, K. (2017). A repressive role of enhancer of zeste homolog 2 in 11β‐hydroxysteroid dehydrogenase type 2 expression in the human placenta. The Journal of Biological Chemistry, 292, 7578–7587. 10.1074/jbc.M116.765800 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The effects of manipulation of 11β‐HSD2 expression by 11β‐HSD2 2 plasmid on preosteoblast cells. A. MTS assay for cell activity; B. Alizarin red staining for mineralization detection; C. RT‐qPCR analysis of gene expression of Runx2, osterix, bone sialoprotein (BSP), osteocalcin (OCN). n = 5 per group, mean ± S.E.M., NS, no significance.
Figure S2. Western blotting assay of HDAC11 or 11β‐HSD2 protein level after treating bone marrow mesenchymal stem cells with HDAC11 siRNA or 11β‐HSD2 plasmid. n = 5 per group, mean ± S.E.M., **P < 0.01 compared with the control group.
Figure S3. ChIP assay of the H3K9ac level in 11β‐HSD2 promoter region after treating bone marrow mesenchymal stem cells with 500 nM CORT or combined with HDAC4 siRNA. n = 5 per group, mean ± S.E.M., **P < 0.01 compared with the control; NS, no significance.
Table S1. Primers used in quantitative real‐time PCR
Table S2. The offspring's body weight in control and prenatal caffeine exposure
Data S1 Supporting information