

Abstract
Background and Purpose
IL‐1β produced by macrophages via the NOD‐, LRR‐ and pyrin domain‐containing 3 (NLRP3) inflammasome, mediates the inflammatory crosstalk between macrophages and adipocytes. In our previous study, (16S,20S,24R)‐12β‐acetoxy‐16,23‐epoxy‐24,25‐dihydroxy‐3β‐(β‐D‐xylopyranosyloxy)‐9,19‐cyclolanost‐22(23)‐ene (AEDC), a cycloartane triterpenoid isolated from Actaea vaginata (Ranunculaceae), was found to possess anti‐inflammatory effect on LPS‐treated RAW264.7 macrophages. This study was designed to investigate whether AEDC modulates macrophage–adipocyte crosstalk to alleviate adipose tissue inflammation.
Experimental Approach
The anti‐inflammatory effect of AEDC was evaluated on LPS plus ATP‐induced THP‐1 macrophages and C57BL/6J mice. The expression of autophagy‐related and NLRP3 inflammasome complex proteins was analysed by western blots, immunofluorescence staining and co‐immunoprecipitation. The pro‐inflammatory cytokines levels were determined by ELISA kits. The adipose tissue inflammation was evaluated by histological analysis and immunohistochemical staining.
Key Results
AEDC (5 and 10 μM) activated autophagy, which in turn suppressed the NLRP3 inflammasome activation and IL‐1β secretion in THP‐1 macrophages. AEDC increased the expression of SIRT3 deacetylase and enhanced its deacetylating activity to reverse mitochondrial dysfunction and activate AMP‐activated protein kinase, which together induced autophagy. Moreover, AEDC (10 μM) attenuated macrophage conditioned medium‐induced inflammatory responses in adipocytes and blocked THP‐1 macrophages migration towards 3T3‐L1 adipocytes. In inflammation mice, AEDC (5 and 20 mg·kg−1) treatment reduced the levels of pro‐inflammatory cytokines in serum and epididymal adipose tissue and reduced macrophage infiltration to alleviate adipose tissue inflammation.
Conclusion and Implications
AEDC attenuated the inflammatory crosstalk between macrophages and adipocytes through SIRT3‐autophagy‐mediated NLRP3 inflammasome inhibition, which might used for the treatment of adipose tissue inflammation‐related metabolic disorders.
Keywords: adipocytes; adipose tissue inflammation; autophagy; cycloartane triterpenoid; IL‐1β, macrophages; NLRP3 inflammasome
Abbreviations
- AEDC
(16S,20S,24R)‐12β‐acetoxy‐16,23‐epoxy‐24,25‐dihydroxy‐3β‐(β‐D‐xylopyranosyloxy)‐9,19‐cyclolanost‐22(23)‐ene
- AMPK
AMP‐activated kinase
- Arg1
arginase 1
- ASC
apoptosis‐associated speck‐like protein containing a carboxy‐terminal caspase‐recruitment domain
- CC
compound C dorsomorphin
- CD206
cluster of differentiation 206
- CETSA
cellular thermal shift assay
- CM
conditioned media
- Cxcl10
C‐X‐C motif chemokine 10
- Cx3cl1
C‐X3‐C motif ligand 1
- DEX
dexamethasone
- eWAT
epididymal white adipose tissue
- JC‐1
5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazolcarbocyanine iodide
- MCP‐1
monocyte chemoattractant protein‐1
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- NLRP3
NOD‐ like receptor family pyrin domain‐containing 3
- PMA
phorbol 12‐myristate 13‐acetate
- SIRT3
NAD+‐dependent deacetylase sirtuin‐3
What is already known
AEDC possesses anti‐inflammatory effect on LPS‐treated RAW264.7 macrophages.
IL‐1β from macrophages via the NLRP3 inflammasome mediates the inflammatory crosstalk between macrophages and adipocytes.
What this study adds
AEDC prevents IL‐1β secretion from macrophages and macrophage–adipocyte crosstalk through SIRT3‐autophagy‐mediated NLRP3 inflammasome inhibition.
AEDC reduces macrophage infiltration to alleviate adipose tissue inflammation in LPS‐induced acute inflammation mice.
What is the clinical significance
AEDC could be developed as a candidate for treatment of adipose tissue inflammation‐related metabolic disorders.
1. INTRODUCTION
Growing evidence suggests that a state of low‐grade chronic inflammation links excess fat to metabolic disorders (Lumeng & Saltiel, 2011). Adipose tissue inflammation is characterized by augmented filtration and pro‐inflammatory polarization of macrophages (Weisberg et al., 2003), which leads to an abnormal production and secretion of pro‐inflammatory cytokines and chemokines, including IL‐1β, IL‐6, TNF‐α, monocyte chemoattractant protein‐1 (MCP‐1 or CCL2 ), C‐X‐C motif chemokine 10 (CXCL10), CXCL11 and C‐X3‐C motif ligand 1 (CX3C1 or fractalkine) (Chacón et al., 2007; Deng & Scherer, 2010). The crosstalk between macrophages and adipocytes has been demonstrated to modulate inflammatory responses in adipose tissue. Hence, suppressing the inflammatory state and infiltration of macrophages in adipose tissue is critical to attenuate adipose tissue inflammation.
Inflammasomes are multi‐protein oligomer platforms for initiating and sustaining inflammation (Latz, 2010). Inflammasome comprises an intracellular sensor, such as NOD‐ like receptor family pyrin domain‐containing 3 (NLRP3), which is coupled with pro‐caspase 1 and the adaptor ASC (apoptosis‐associated speck‐like protein containing a carboxy‐terminal caspase‐recruitment domain) (Schroder & Tschopp, 2010). Activation of inflammasome complex triggers the maturation of caspase 1 (Martinon, Burns, & Tschopp, 2002). IL‐1β, a crucial pro‐inflammatory cytokine produced mainly by monocytes and macrophages, is activated through caspase‐1 via the NLRP3 inflammasome complex (Agostini et al., 2004). IL‐1β is a key inflammatory cytokine and is involved in causing insulin resistance and thus type 2 diabetes. Macrophages‐derived IL‐1β impairs the insulin signalling pathway and induces expression of pro‐inflammatory factors in human adipocytes (Gao et al., 2014). Blocking IL‐1β activity reduces hyperglycaemia and tissue inflammation in obese mice and diabetic rats (Ehses et al., 2009). Hence, inhibition of NLRP3–caspase‐1‐mediated IL‐1β production in macrophages is vital to ameliorate adipose tissue inflammation.
Autophagy is an evolutionarily conserved cellular process, which facilitates the turnover of damaged proteins and organelles (Oh & Lee, 2014). Accumulating evidence indicates that autophagy acts as a negative regulator of NLRP3 inflammasome activation at different levels (Deretic, 2005). Autophagosomes sequester and degrade pro‐IL‐1β, damaged mitochondria and inflammasome components, thus limiting IL‐1β secretion (Levine & Deretic, 2007). ATG16LI deleted mice produced exaggerated amounts of IL‐1β in response to LPS (Saitoh et al., 2008). Autophagy deficiency in macrophages increased inflammasome activation in response to metabolic or extrinsic stress (Lee et al., 2016). Consistently, autophagy induction by rapamycin (sirolimus) suppressed the production of IL‐1β and caspase 1 activation (Ko, Yoon, Lee, & Oh, 2017). Thus, activation of autophagy in macrophages is a potential way to alleviate adipose tissue inflammation.
The rhizomes of Actaea vaginata (Ranunculaceae) are commonly used as a traditional Tibetan medicine for the treatment of conjunctivitis, stomatitis, pharyngitis and enteritis (Lee et al., 2018). Our previous study found that (16S,20S,24R)‐12β‐acetoxy‐16,23‐epoxy‐24,25‐dihydroxy‐3β‐(β‐D‐xylopyranosyloxy)‐9,19‐cyclolanost‐22(23)‐ene (AEDC), a cycloartane triterpenoid isolated from the whole plant of A. vaginata, possesses anti‐inflammatory effect on LPS‐treated RAW264.7 macrophages (Fang et al., 2019). Herein, we investigated the regulatory role of AEDC on crosstalk between macrophages and adipocytes, both in vitro and in vivo, and uncovered the underlying mechanisms.
2. METHODS
2.1. Cell culture and differentiation
Human THP‐1 cells (RCB Cat# RCB3686, RRID:CVCL_0006) were obtained from ATCC (Manassas, VA, USA) and maintained in RPMI 1640 medium supplemented with 10% heat‐inactivated FBS. THP‐1 cells were stimulated by phorbol 12‐myristate 13‐acetate (PMA) (100 ng·ml−1) for 12 h to differentiate into macrophages. Primary peritoneal macrophages were isolated from 3‐month‐old mice and incubated in RPMI 1640 supplemented with 10% FBS and penicillin–streptomycin (P/S) at 37°C. After 72 h, nonadherent cells were removed by repeated washing with culture medium. The adherent cells were maintained for further experiments. 3T3‐L1 preadipocytes (JCRB Cat# IFO50416, RRID:CVCL_0123) were obtained from ATCC and maintained in DMEM containing 10% calf serum and 1% P/S. Cells were differentiated into adipocytes as reported previously (Li et al., 2018). Briefly, 2 days post‐confluent, 3T3‐L1 preadipocytes were stimulated with DMEM supplemented with 10% FBS, 1 μM dexamethasone (DEX), 0.5 mM IBMX and 5 μg·ml−1 insulin for 2 days. Cells were subsequently cultured in maintaining medium (DMEM supplemented with 10% FBS and 5 μg·ml−1 insulin) for 6 days. The medium was changed every other day. The fully differentiated 3T3‐L1 cells were checked by microscopic observation and Oil Red O staining. Cells were cultured in a humidified incubator with 5% (v·v−1) CO2 at 37°C.
2.2. Macrophage–adipocyte co‐culture
Macrophage–adipocyte co‐culture was performed as described previously (Li et al., 2018). THP‐1 macrophages or primary peritoneal macrophages were treated with or without 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h. Subsequently, the cells were stimulated with LPS (1 μg·ml−1) for 4 h and then 1 mM ATP for 1 h. After removing the medium, THP‐1 macrophages or primary peritoneal macrophages were rinsed twice with RPMI 1640 medium and then incubated in RPMI 1640 medium (serum free, 0.2% endotoxin and fatty acid‐free BSA) for 24 h. The medium was harvested and centrifuged at 400 g for 10 min and the supernatant was collected as macrophage conditioned media (CM). The fully differentiated 3T3‐L1 adipocytes were incubated with migration medium (serum free, 0.2% endotoxin and fatty acid‐free BSA in DMEM) for 24 h. Subsequently, the adipocytes were incubated with macrophage conditioned media for 24 h and then harvested for further studies.
For macrophage migration assay, Transwell inserts with an 8 μm membrane pore size (Millipore, Bedford, MA, USA) were used. The fully differentiated 3T3‐L1 adipocytes were incubated with migration medium for 24 h. The medium was harvested and centrifuged at 400 g for 10 min and the supernatant was collected as adipocyte conditioned media. THP‐1 macrophages or primary peritoneal macrophages were seeded onto the inserts at a density of 5 × 104 cells per well and treated with or without 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h. Then, the macrophages were co‐cultured with adipocyte conditioned media for 4 h at 37°C. The macrophages in the lower compartment were fixed with 4% formaldehyde for 20 min, stained with DAPI and counted as described previously (Marcotorchino et al., 2012).
2.3. Cell viability
Cell viability was determined by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay as described previously (Feng et al., 2018). Human THP‐1 cells were seeded in 96‐well plates at a density of 1 × 104 cells per well. The PMA‐treated THP‐1 macrophages were treated with the indicated concentrations of AEDC for 24 h. Then, cell viability was determined by incubation with DMEM containing MTT (1 mg·ml−1) for 4 h, followed by dissolving the formazan crystals with DMSO. The absorbance at 570 nm was measured by a SpectraMax M5 microplate reader (Molecular Devices, CA, USA). The calculation equation for relative cell viability was as following: cell viability (%) = (As − A0)·(Ac − A0)−1·100%, where As, A0 and Ac were the absorptions of test sample, blank control and negative control (DMSO).
2.4. Protein harvest from cell culture medium
THP‐1 cell culture media were harvested and centrifuged at 15,000 g for 10 min at 4°C. Then, the supernatant was transferred into a new tube and mixed well with 700‐μl methanol and 175‐μl chloroform. After sitting for 5 min at room temperature, the mixture was centrifuged at 21,000 g for 10 min at 4°C. The white intermediate layer was transferred to a new tube, washed by 700‐μl methanol and then dissolved in SDS sample buffer for western blotting analysis.
2.5. Determination of cytokines
Cell medium, blood serum and tissue lysates from mice were centrifuged at 1,500 g for 10 min at 4°C, and the supernatant was collected. The cytokines in cell medium, mice serum and tissue lysates were determined by using commercial ELISA kits (Neobioscience Technology Co., Ltd., Shenzhen, China), following the manufacturer's instruction. The cytokine levels in tissues were further normalized by protein content.
2.6. Western blotting analysis
Western blotting was performed as described previously (Lin et al., 2012). 3T3‐L1 adipocytes, THP‐1 macrophages and peritoneal macrophages and epidydymal adipose tissue from mice were lysed with RIPA lysis buffer. Protein concentration was determined using a BCA Protein Assay Kit. Equal amount of proteins (20–30 μg) were separated by SDS‐PAGE, transferred to PVDF membranes, blocked with 5% non‐fat milk in TBST buffer (Tris‐Buffered Saline and Tween 20, 100 mM NaCl, 10 mM Tris‐HCl, pH 7.5 and 0.1% Tween‐20) for 1 h at room temperature and incubated with specific primary antibodies (Table S1) overnight at 4°C. After washing with TBST three times, an HRP‐conjugated secondary antibody was added and incubated for 1 h at room temperature. Signals were developed by using a SuperSignal West Femto Maximum Sensitivity Substrate kit. Then, specific protein bands were visualized using the ChemiDoc MP Imaging System (Bio‐Rad) and quantitated using Image Lab 5.1 (RRID:SCR_014210, Bio‐Rad) as described previously (Zhang, Liu, Tong, & Lin, 2020). Western blotting procedures and analysis comply and adhere with BJP guidelines (Alexander et al., 2018).
2.7. Cellular thermal shift assay (CETSA)
THP‐1 cells were pretreated with or without 10 μM AEDC for 12 h. The cells were lysed by RIPA lysis buffer. The cell lysates were incubated in ice for 10 min and then centrifuged at 12,000 g for 10 min at 4°C. The protein concentration was determined using a BCA Protein Assay Kit and adjusted to 2 μg·μl−1 using RIPA lysis buffer; 50‐μL cell lysates was transferred to new tubes and heated for 5 min at different temperature (50–90°C) using a thermal cycler. After incubation in ice for 10 min, soluble proteins were separated by centrifugation at 12,000 g for 20 min at 4°C and analysed by western blotting analysis (Jafari et al., 2014; Martinez et al., 2013).
2.8. SIRT3 deacetylating activity
To measure cellular SIRT3 deacetylating activity, THP‐1 macrophages treated with or without AEDC (10 μM) and 3‐TYP (50 μM) were lysed with RIPA lysis buffer. SIRT3 deacetylating activity in THP‐1 cell lysates was determined with a SIRT3 Fluorometric Assay Kit (Sigma‐Aldrich) according to the manufacturer's instruction. The fluorescence intensities were measured with a microplate fluorimeter (excitation wavelength = 360 nm; emission wavelength = 460 nm). All values were represented as percentage of the control group.
2.9. Determination of superoxide dismutase 2 (SOD2) activity
To measure cellular SOD2 activity, THP‐1 macrophages were treated with or without 10 μM AEDC for 6 h. Subsequently, the cells were stimulated with LPS (1 μg·ml−1) for 4 h and then 1 mM ATP for 1 h. Then, cells were lysed with RIPA lysis buffer. The SOD2 activity in THP‐1 cells was determined by using the commercial assay kit (Nanjing Jiancheng, Nanjing, Jiangsu, China) in accordance with the manufacturer's protocol. Protein concentration was quantitated by the BCA Protein Assay Kit. SOD2 activity was then normalized by protein concentration.
2.10. Co‐immunoprecipitation
Cell lysates were mixed with the indicated antibody and subsequently 20‐μl protein A/G‐agarose beads (Santa Cruz Biotechnology) and incubated on a rotator for 4 h at 4°C. The beads were washed twice with PBS and then twice with lysis buffer supplemented with complete mini protease inhibitor cocktail. Bound proteins were boiled in sample preparation buffer for 5 min and then used for western blotting analysis.
2.11. Immunofluorescence staining
Immunofluorescence staining was performed as described previously (Liu et al., 2017). THP‐1 cells were grown on collagen‐precoated glass coverslips. After various treatments, cells were fixed with 4% paraformaldehyde. The slides were then incubated with a primary antibody (1:100 dilution) at 4°C for 30 min. The slides were washed and incubated with corresponding secondary antibody (1:1000 dilution) for 30 min at room temperature. The autophagic flux alterations and NLRP3 components were detected using a confocal microscopy (Olympus, Tokyo, Japan). Paraffin sections from the same adipose tissues used for morphological analysis were processed for conventional immunofluorescence labelling using anti‐F4/80 antibody (RRID:AB_1122717) for macrophages (1:100 dilution, Santa Cruz, CA, USA) and anti‐perilipin antibody (RRID:AB_10829911) for adipocytes (1:100 dilution, Cell Signaling Technologies, MA, USA). The corresponding secondary antibodies (Thermo Fisher Scientific, NY, USA) were used for fluorescence detection. The images were captured by confocal laser scanning microscope.
2.12. Real‐time RT‐PCR
Total RNA was isolated from cells and epididymal adipose tissue using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instruction. The cDNA was synthesized from 1 μg RNA using the SuperScript III First Strand Synthesis System for RT‐PCR. The quantitative PCR experiments were conducted on StepOnePlus Real‐Time PCR System using SYBR Green PCR Master Mix with gene specific primers (Table S2). The 18S RNA was used as an internal control.
2.13. MitoROS determination
The superoxide levels in THP‐1 macrophages were measured with MitoSOX Red (Yeasen, Shanghai, China). The cells were treated with or without AEDC and incubated with MitoSOX Red for 30 min. After washed with PBS twice, the cellular fluorescence intensity was measured by FACSCalibur flow cytometry (RRID:SCR_000401, Becton Dickinson, San Jose, CA, USA).
2.14. Mitochondrial membrane potential (ΔΨm) assay
THP‐1 macrophages were grown on collagen‐precoated glass coverslips. After treatments, the cells were incubated with 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazolcarbocyanine iodide (JC‐1, 1 μg·ml−1) in culture medium for 20 min at 37°C. The cells were rinsed with ice‐cold PBS twice. The fluorescence images were obtained by confocal laser scanning microscope. JC‐1 monomers positive cells intensity was analysed by the ImageJ software (RRID:SCR_003070).
2.15. Ethics
All animal experiments were approved by the Animal Ethical and Welfare Committee of University of Macau (no. ICMS‐AEC‐2015‐07). All procedures involved in the animal experiments were carried out in accordance with the approved guidelines and regulations. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and the editorial on reporting animal studies (McGrath & Lilley, 2015), and with the recommendations made by the British Journal of Pharmacology. Experimental protocols and design are reported in compliance with the guidelines (Curtis et al., 2018).
Male C57BL/6J mice (8‐ to 10‐week‐olds, RRID:IMSR_JAX:000664) were obtained from the Faculty of Health Science, University of Macau (Macau, China). The mice were housed at 22 ± 1°C with 12 h light–dark cycles and fed with a regular chow diet (Guangdong Medical Lab Animal Center, Guangzhou, Guangdong, China) and water ad libitum under standard conditions (specific pathogen‐free) with air filtration. The authors declare that the data supporting the findings of this study are available with the article.
2.16. LPS‐induced acute inflammation mice
Thirty‐five mice were randomly divided into seven groups according to body weight (n = 5). The vehicle and LPS + ATP groups of mice were i.p. injected with 10 ml·kg−1 PEG 400 solution (PEG 400:0.9% saline, 6:4 v·v−1). The other five groups of mice were i.p. injected with 4 mg·kg−1 dexamethasone (0.4 mg·ml−1 dissolved in PEG 400 solution), 5 mg·kg−1 AEDC (AEDC‐L, 0.5 mg·ml−1 dissolved in PEG 400 solution), 20 mg·kg−1 AEDC (AEDC‐H, 2 mg·ml−1 dissolved in PEG 400 solution), 4 mg·kg−1 3‐TYP (3‐TYP, 0.4 mg·ml−1 dissolved in PEG 400 solution) and the combination of 4 mg·kg−1 3‐TYP and 20 mg·kg−1 AEDC (0.4 mg·ml−1 3‐TYP and 2 mg·ml−1 AEDC dissolved in PEG 400 solution), respectively. The mice were administrated once a day for 5 days. On the sixth day, the vehicle group of mice was i.p. injected with 10 ml·kg−1 PBS and the other six groups of mice were i.p. injected with 4 mg·kg−1 LPS (0.4 mg·ml−1, serotype O111:B4, dissolved in PBS) for 4 h followed by 30 mg·kg−1 ATP (3 mg·ml−1, dissolved in PBS, pH 6.2). Half hour after ATP injection, the blood samples were collected from mice under anaesthesia (inhalation of 3% isoflurane gas at 0.5 L·min−1). Then, the mice were killed by carbon dioxide inhalation and the peritoneal macrophages and epididymal adipose tissues were collected and stored at −80°C.
2.16. Histological analysis
For histological analysis, a part of the adipose tissue was fixed in 10% buffered formalin and embedded in paraffin. Sections were stained with haematoxylin and eosin (H&E) according to standard protocols. Histological scoring was performed by a pathologist. Histological evaluation of H&E‐stained adipose sections was graded as follows:‐ 0, no signs of inflammation; 1, low macrophage infiltration; 2, moderate macrophage infiltration; 3, high macrophage infiltration and 4, transmural infiltrations, massive accumulation of macrophage and dead adipocytes emerging.
2.17. Immunohistochemistry
For immunohistochemistry, 10‐μm dewaxed sections of epididymal adipose tissue were treated with 3% hydrogen peroxide to inactivate endogenous peroxidase, followed by normal goat serum to reduce non‐specific staining and then incubated with anti‐CD11c (RRID:AB_626859, 1:100; Santa Cruz) and anti‐CD206 (RRID:AB_2800175, 1:100, Cell Signaling Technologies) antibodies overnight at 4°C. The slides were incubated with biotinylated HRP‐conjugated secondary antibodies (Cell Signaling Technologies) for 2 h at room temperature. Histochemical reactions were performed using DAb as a substrate. Nucleus were counterstained with haematoxylin. The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
2.18. Data and analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Studies were designed to generate groups of equal size and no data points were excluded from the analysis in any experiment. The n value for each experiment was shown in the figure legend. This is adopted based on our previous results (Shen, Liao, Liu, et al., 2019; Shen, Liao, Zhang, et al., 2019; Zhang, Liu, Shen, et al., 2020) and also for the purpose of carrying out statistical analysis according to the guidelines of British Journal of Pharmacology (Curtis et al., 2018). Cells and mice were randomly assigned to each treatment group and experiment. All the quantifications and data analysis were blinded and the analyst did not know the origin of the data during statistical analysis. The outliers were included in data analysis and presentation. Data normalization was undertaken to control for sources of variation of baseline parameters and to allow comparison of the magnitude of drug effects in different conditions. The units of a variable were determined as fold mean of the controls. Data were expressed as mean ± SEM based on at least three independent experiments and analysed on GraphPad Prism 7 (RRID:SCR_002798, GraphPad Software, San Diego, CA, USA). Statistical analysis was undertaken only for studies where each group size was at least n = 5. The significance of differences between groups was assessed by one‐way ANOVA followed by Tukey's post hoc test using SPSS software 16.0 (RRID:SCR_002865, Chicago, IL, USA). Post hoc tests were run only if F achieved P < .05 and there was no significant variance inhomogeneity. P < .05 was considered statistically significant.
2.19. Materials and reagents
DMEM, FBS, PBS, P/S, RPMI 1640 medium and 0.25% (w/v) trypsin‐EDTA were obtained from Gibco (Gaithersburg, MD, USA). Calf serum was provided by HyClone (Logan, UT, USA). 3‐Methyladenine (3‐MA), PMA, LPS, ATP, IBMX, dexamethasone, insulin and compound C (CC or dorsomorphin) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). BCA protein assay kit and SuperSignal West Femto Maximum Sensitivity Substrate kit were obtained from Thermo Fisher Scientific (Grand Island, NY, USA). mRFP‐GFP‐LC3 plasmid and RIPA lysis buffer were purchased from Beyotime (Shanghai, China). Triton X‐100 and PVDF membranes were purchased from Bio‐Rad (Hercules, CA, USA).
2.20. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. AEDC alleviated LPS plus ATP‐induced IL‐1β secretion in THP‐1 macrophages by restoring impaired autophagy
IL‐1β has been considered as a culprit in macrophage‐induced adipocyte malfunction in obesity (Gao et al., 2014). AEDC (Figure 1a) possesses anti‐inflammatory property on LPS‐stimulated RAW264.7 macrophages. Herein, PMA‐primed THP‐1 macrophages were treated with or without AEDC and then stimulated with LPS plus ATP to induce IL‐1β secretion. AEDC did not show obvious cytotoxicity on THP‐1 macrophages up to 10 μM (Figure S1a). Intriguingly, AEDC dose‐dependently alleviated LPS plus ATP‐induced increase of IL‐1β production and release, assessing by immunofluorescence staining (Figure 1b) and ELISA (Figure 1c), respectively. The NLRP3 inflammasome in macrophages modulates the maturation and release of IL‐1β (Chen & Nunez, 2010). As AEDC inhibited the production of IL‐1β, the monitoring of AEDC on NLRP3 inflammasome activation was taken into consideration. As expected, AEDC dose‐dependently inhibited NLRP3 expression and caspase‐1 activation in LPS plus ATP‐induced THP‐1 macrophages (Figure 1d).
FIGURE 1.

AEDC alleviated LPS plus ATP‐induced IL‐1β secretion in THP‐1 macrophages by restoring impaired autophagy. (a) The chemical structure of compound AEDC (16S,20S,24R)‐12β‐acetoxy‐16,23‐epoxy‐24,25‐dihydroxy‐3β‐(β‐D‐xylopyranosyloxy)‐9,19‐cyclolanost‐22(23)‐ene). THP‐1 cells were treated with or without different concentrations of AEDC for 12 h, followed by stimulation with LPS for 18 h and then ATP for 1 h. (b) Immunofluorescence staining of IL‐1β was performed (n = 5). Scale bar = 10 μm. (c) The level of IL‐1β in the culture medium from THP‐1 cells were determined by ELISA (n = 6). THP‐1 cells were treated with or without different concentrations of AEDC for 12 h, followed by stimulation with LPS for 4 h and then ATP for 1 h. (d) NLRP3 and Caspase‐1 in the lysates of THP‐1 cells were detected by Western blotting (n = 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (e) Expression of autophagy‐related proteins were detected by Western blotting (n = 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (f) THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 5 mM 3‐MA for 6 h, followed by stimulation of LPS for 4 h and then ATP for 1 h. Expression of autophagy‐related proteins were detected by Western blotting (n = 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (g) THP‐1 cells were transiently infected with the mRFP‐GFP‐LC3 lentivirus for 24 h. Then, the cells were treated with or without 10 μM AEDC for 12 h and 5 mM 3‐MA for 6 h, followed by stimulation of LPS for 4 h and ATP for 1 h. mRFP‐GFP‐LC3 puncta were measured using a confocal microscope (n = 5). Scale bar = 5 μm. (h) THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 5 mM 3‐MA for 6 h, followed by stimulation of LPS for 18 h and then ATP for 1 h. The levels of IL‐1β in the culture medium were determined by ELISA (n = 6). Data are expressed as means ± SEM. # P < 0.05 LPS + ATP vs. ctrl; *P < 0.05 LPS + ATP + AEDC (3‐MA) vs. LPS + ATP; & P < 0.05 LPS + ATP + AEDC vs. LPS + ATP + AEDC + 3‐MA
Defective autophagy in macrophages results in NLRP3 inflammasome activation and inflammatory responses (Levine, Mizushima, & Virgin, 2011). As shown in Figure 1e, LPS plus ATP treatment significantly decreased the protein levels of Atg5, Atg7, Beclin1 and the ratio of LC3‐II to LC3‐I to approximately 42–65% and increased the level of p62 to 378%, compared with those of the control cells, suggesting impaired autophagy in THP‐1 macrophages. Interestingly, AEDC treatment increased the levels of Atg5, Atg7, Beclin1 and the ratio of LC3‐II to LC3‐I and decreased the level of p62 in dose‐dependent manners (Figure 1e). In the LC3 turnover assay, the difference of LC3‐II level in the presence or absence of the autophagy inhibitor 3‐MA was greater in AEDC‐treated cells (Figure 1f), indicating the autophagic flux was enhanced in AEDC‐treated macrophages. Interestingly, AEDC also activated autophagy in unstimulated THP‐1 cells, indicating by the increased levels of Atg5, Atg7, Beclin1 and the ratio of LC3‐II to LC3‐I and decreased level of p62, which were reversed by the co‐treatment of 3‐MA (Figure S1b). To further verify whether AEDC enhanced autophagic flux, THP‐1 cells were infected with mRFP‐GFP‐LC3 adenovirus that serves as a sensor of autolysosome formation. Both GFP and mRFP are expressed in autophagosomes; when autophagosomes merge with lysosomes, GFP is quenched easily in acidic environments and loses its fluorescence in autolysosomes, whereas mRFP is stable under acidic conditions (Yoshii & Mizushima, 2017). As shown in Figure 1g, treatment with LPS and ATP decreased both red and green puncta, suggesting impaired autophagy. As expected, more mRFP‐LC3 puncta were observed in AEDC‐treated cells, indicating autophagic flux was enhanced without disrupting the lysosomal function and/or autophagosome–lysosome fusion, which was reversed by co‐treatment with 3‐MA (Figure 1g). Similarly, AEDC also enhanced autophagic flux in unstimulated THP‐1 cells, in accordance with the autophagy inducer rapamycin (Figure S1c). AEDC obviously reversed LPS plus ATP‐induced increase of IL‐1β secretion in the cell medium, which was totally abolished by co‐treatment of 3‐MA (Figure 1h). These results indicated that AEDC suppressed IL‐1β level in LPS plus ATP‐induced THP‐1 cells through activating autophagy.
3.2. AEDC inhibited the activation of NLRP3 inflammasome by enhancing autophagy
NLRP3 inflammasome can be degraded by autophagy (Latz, Xiao, & Stutz, 2013). Therefore, we questioned whether AEDC inactivated NLRP3 inflammasome through inducing autophagy. As shown in Figure 2a, AEDC suppressed NLRP3‐mediated caspase‐1 activation and mature IL‐1β secretion, which was partially reversed in combination with 3‐MA. Consistently, the immunofluorescence staining results showed that NLRP3 level was largely decreased by AEDC treatment in LPS plus ATP‐treated THP‐1 cells and this trend was blocked by co‐treatment of 3‐methyladenine (3‐MA; Figure 2b). To further verify the role of autophagy in NLRP3 inflammasome inactivation, the co‐localization of NLRP3 and autophagy components was detected by immunofluorescence staining. The distributions of p62 and NLRP3 in LPS and ATP‐treated THP‐1 cells were superposed, indicating the co‐localization of the autophagosomes and NLRP3 inflammasomes, which was attenuated by AEDC treatment (Figure 2c). In addition, the co‐immunoprecipitation showed greater level of p62 was pulled‐down by NLRP3 antibody after LPS and ATP administration, compared with the control cells, which was attenuated by AEDC treatment (Figure 2d). NLRP3 inflammasome is a caspase‐1‐ASC‐assembled multicomponent complex. As shown in Figure 2e, stimulation with LPS plus ATP induced NLRP3 expression and ASC–caspase 1 complex formation, while AEDC disrupted these trends. Taken together, these results indicated that AEDC suppressed the activation of NLRP3 inflammasomes by enhancing p62‐mediated autophagy in LPS and ATP‐induced THP‐1 macrophages.
FIGURE 2.
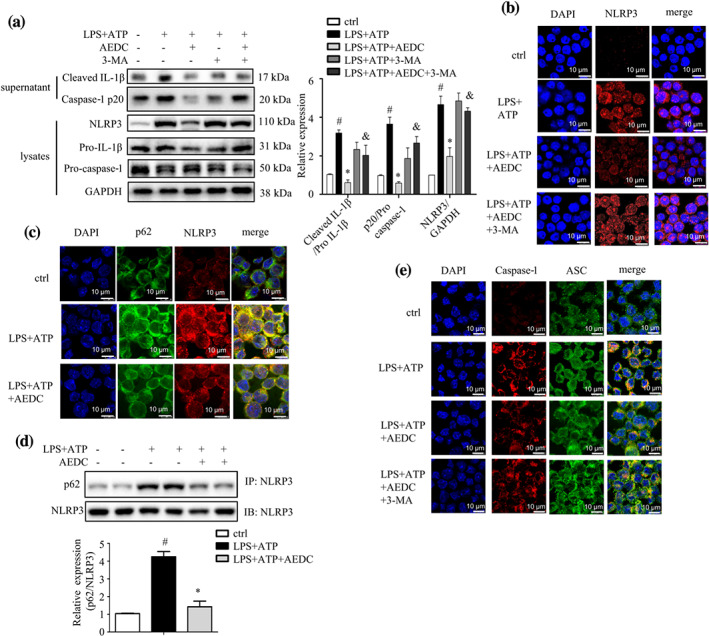
AEDC inhibited the activation of NLRP3 inflammasome by enhancing autophagy. THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 5 mM 3‐MA for 6 h, followed by stimulation with LPS for 4 h and then ATP for 1 h. (a) NLRP3, pro‐caspase 1 and pro‐IL‐1β in the lysates and cleaved caspase‐1 and IL‐1β in the supernatant were analysed by Western blotting (n = 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (b) Immunofluorescence staining of NLRP3 was performed (n = 5). Scale bar = 10 μm. (c) Immunofluorescence stainings of endogenous NLRP3 and p62 were performed (n = 5). Scale bar = 10 μm. (d) The level of co‐precipitated p62 with NLRP3 in THP‐1 cells (n = 5). (d) Immunofluorescence stainings of caspase‐1 and ASC were performed (n = 5). Scale bar = 10 μm. Data are expressed as means ± SEM. # P < 0.05 LPS + ATP vs. ctrl; *P < 0.05 LPS + ATP + AEDC vs. LPS + ATP; & P < 0.05 LPS + ATP + AEDC vs. LPS + ATP + AEDC + 3‐MA
3.3. AEDC suppressed IL‐1β production and NLRP3 inflammasome activation by SIRT3‐AMPK‐mediated autophagy
SIRT3 overexpression induces macrophage autophagy and attenuates NLRP3 inflammasome activation (Liu et al., 2018). Interestingly, AEDC treatment dose‐dependently increased SIRT3 expression in LPS and ATP‐induced THP‐1 cells (Figure 3a). To experimentally confirm that AEDC interacts with SIRT3 deacetylase, CETSA was performed on THP‐1 cells treated with or without AEDC. AEDC strongly induced the thermal stability of SIRT3 at a variety of temperatures, compared with the control cells (Figure 3b). Moreover, AEDC enhanced the SIRT3 deacetylating activity, which was totally abolished by co‐treatment with 3‐TYP (3‐(1H‐1,2,3‐triazol‐4‐yl)pyridine), a confirmed selective SIRT3 inhibitor (Figure 3c). 3‐TYP treatment almost reversed AEDC‐driven increases of Beclin1 protein and ratio of LC3‐II to LC3‐I and decrease of p62 protein (Figure 3d). Consistently, the mRFP‐GFP‐LC3 fluorescence images indicated that the co‐treatment of 3‐TYP and AEDC reduced the red puncta, indicating impaired autophagic flux (Figure 3e). Moreover, the inhibitory effect of AEDC of NLRP3 inflammasome activation was almost abolished by the co‐treatment of 3‐TYP in LPS plus ATP‐treated THP‐1 cells (Figure 3f). As expected, AEDC suppressed the production of IL‐1β, which was reversed by the co‐treatment of 3‐TYP (Figure 3g).
FIGURE 3.
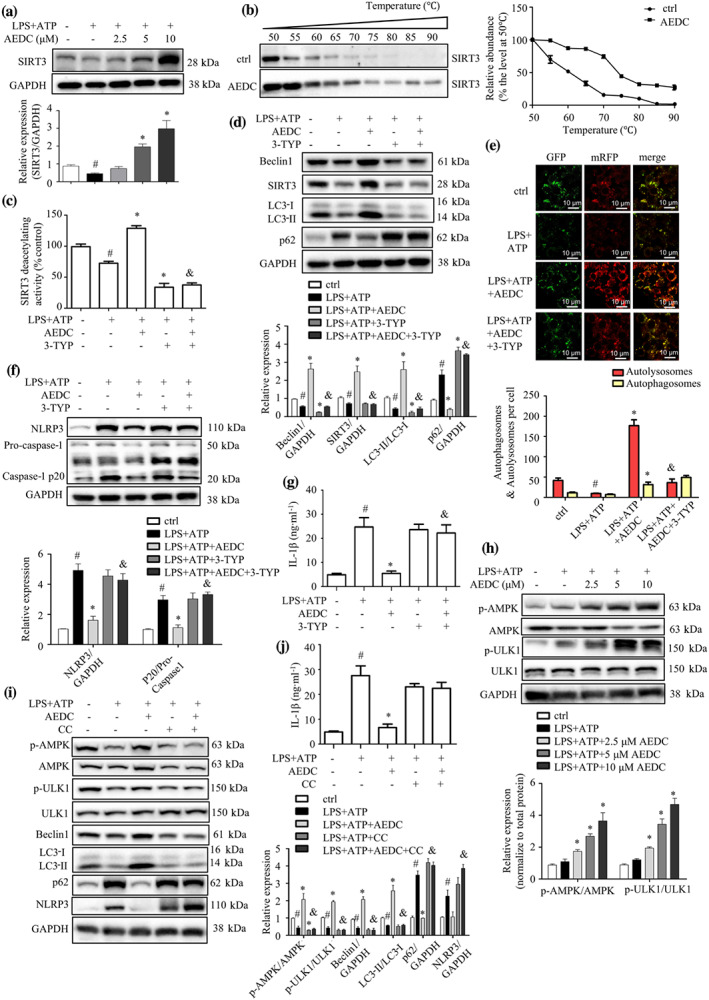
AEDC suppressed IL‐1β production and NLRP3 inflammasome activation by SIRT3‐AMPK‐mediated autophagy. (a) THP‐1 cells were treated with different concentrations of AEDC for 12 h, followed by stimulation with LPS for 4 h and then ATP for 1 h. Expression of SIRT3 was detected by Western blotting (n = 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (b) CETSA was performed on THP‐1 cells treated with or without 10 μM AEDC for 12 h. Data were normalized to the mean value of the respective group at 50°C (n = 5). THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h, followed by stimulation with LPS for 4 h and then ATP for 1 h. (c) The SIRT3 deacetylating activity was evaluated. Data were normalized to the mean value of the control group (n = 6). (d) Expression of autophagy‐related proteins was detected by Western blotting (n = 5). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (e) THP‐1 cells were transiently infected with the mRFP‐GFP‐LC3 lentivirus for 24 h. mRFP‐GFP‐LC3 puncta were measured using a confocal microscope (n = 5). Scale bar = 10 μm. (f) NLRP3 and caspase‐1 in the cell lysates were detected by Western blotting (n = 5). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (g) The level of IL‐1β in the cell culture medium was determined by ELISA, in THP‐1 cells treated with or without 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h, followed by stimulation of LPS for 18 h and ATP for 1 h (n = 6). Data are expressed as means ± SEM. # P < 0.05 LPS + ATP vs. ctrl; *P < 0.05 LPS + ATP + AEDC (3‐TYP) vs. LPS + ATP; & P < 0.05 LPS + ATP + AEDC vs. LPS + ATP + AEDC + 3‐TYP. (h) THP‐1 cells were treated with or without different concentrations of AEDC for 12 h, followed by stimulation with LPS for 4 h and then ATP for 1 h. p‐AMPK, AMPK, p‐ULK1 and ULK1 were detected by Western blotting (n = 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (i) THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 4 μM compound C (CC) for 6 h, followed by stimulation with LPS for 4 h and then ATP for 1 h. The expression of autophagy‐related proteins was examined by Western blotting (n = 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (j) The level of IL‐1β in the culture medium was determined by ELISA, in THP‐1 cells treated with or without 10 μM AEDC for 12 h and 4 μM CC for 6 h, followed by stimulation of LPS for 18 h and ATP for 1 h (n = 6). Data are expressed as means ± SEM. # P < 0.05 LPS + ATP vs. ctrl; *P < 0.05 LPS + ATP + AEDC (CC) vs. LPS + ATP; & P < 0.05 LPS + ATP + AEDC vs. LPS + ATP + AEDC + CC
SIRT3 activates autophagy through AMP‐activated kinase (AMPK)–unc‐51 like autophagy activating kinase 1 (ULK1) pathway in palmitate‐stressed hepatocytes (Zhang, Liu, Shen, et al., 2020). To decipher the mechanism of AEDC on SIRT3‐mediated autophagy, the phosphorylation of AMPK and ULK1 was evaluated in AEDC‐treated THP‐1 macrophages. As expected, the AEDC dose‐dependently increased the phosphorylated AMPK and ULK1 in unstimulated THP‐1 cells (Figure 3h). Compound C, an AMPK inhibitor, remarkably abolished the effect of AEDC on autophagy activation and NLRP3 inactivation (Figure 3i). As shown in Figure 3j, treatment of AMPK inhibitor reversed the inhibitory effect of AEDC on IL‐1β secretion, supporting that AEDC suppressed IL‐1β production in THP‐1 cell via activating AMPK signalling pathway. These results indicated that the inhibitory effect of AEDC on IL‐1β production and NLRP3 activation was mediated through SIRT3‐AMPK‐induced autophagy.
3.4. AEDC prevented LPS plus ATP‐induced mitochondrial perturbation by activating SIRT3
Mitochondrial damage caused by excessive ROS contributes to NLRP3 inflammasome activation (Zhou, Yazdi, Menu, & Tschopp, 2011). To determine whether AEDC inhibited NLRP3 inflammasome activation by preventing mitochondrial damage, the mitochondrial ROS (mtROS) and mitochondrial membrane potential were detected. AEDC treatment reduced the mtROS levels (Figure 4a) and restored the mitochondria membrane potential (Figure 4b) in LPS plus ATP‐treated THP‐1 cells, which were blocked by the co‐treatment of 3‐TYP. SOD2 is a substrate of SIRT3 deacetylase, to scavenge ROS in mitochondria (Liu et al., 2017). As expected, the co‐immunoprecipitation results showed greater level of acetylated SOD2 in LPS plus ATP‐stimulated THP‐1 cells, compared with that of the control cells; AEDC treatment markedly reduced the acetylation level of SOD2, which was reversed by 3‐TYP (Figure 4c). Consistently, SOD2 activity was reduced in LPS plus ATP‐treated THP‐1 cells and AEDC treatment greatly restored SOD2 activity, which was blocked by co‐treatment of 3‐TYP (Figure 4c). These results indicated that AEDC reduced ROS accumulation in LPS plus ATP‐treated THP‐1 cells through activating SIRT3–SOD2.
FIGURE 4.

AEDC prevented LPS plus ATP‐induced mitochondrial perturbation by activating SIRT3. THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h, followed by stimulation with LPS for 4 h and then ATP for 1 h. (a) The mtROS level was determined by MitoSOX (n = 6). (b) Mitochondrial membrane potential was evaluated by JC‐1 staining. The percentage of JC‐1 monomers positive cells was quantified (n = 6). Scale bar = 10 μm. (c) The acetylated and total SOD2 protein levels and SOD2 activity were determined (n = 6). Data were normalized to the mean value of the control group. (d) Immunofluorescence stainings of Tubulin Tracker and MitoTracker were performed (n = 5). Scale bar = 2 μm. (e) Immunofluorescence stainings of LC3‐II and MitoTracker were performed (n = 5). Scale bar = 5 μm. (f) Expression of autophagy‐related proteins and NLRP3 in mitochondria and cytosol was detected by Western blotting (n = 5). Data were normalized to the mean value of the control group. Data are expressed as means ± SEM. # P < 0.05 LPS + ATP vs. ctrl; *P < 0.05 LPS + ATP + AEDC (3‐TYP) vs. LPS + ATP; & P < 0.05 LPS + ATP + AEDC vs. LPS + ATP + AEDC + 3‐TYP
Mitochondria are transported along microtubules in membrane nanotubes to rescue distressed cells (Shen et al., 2018). ATP treatment caused detyrosinated microtubules breakdown (Infante, Stein, Zhai, Borisy, & Gundersen, 2000). AEDC increased the mitochondrial content, protected the microtubule network and promoted mitochondria dispersion on impaired microtubules (Figure 4d). In addition, more mitochondria were overlapped with lipidated LC3 (LC3‐II) in AEDC‐treated cells (Figure 4e), which indicated that AEDC activated autophagy mainly on mitochondria. The AEDC‐driven increases of Beclin1 level and ratio of LC3‐II to LC3‐I and decrease of NLRP3 level mainly occurred in mitochondrial fraction but not in cytosol, which were partially reversed in combination with 3‐TYP (Figure 4f). The above results suggested that AEDC prevented LPS plus ATP‐induced mitochondrial perturbation by activating SIRT3.
3.5. AEDC attenuated macrophage conditioned media or TNF‐α induced inflammatory responses in adipocytes
To assess the effect of macrophage‐derived cytokines on adipocyte inflammation, the fully differentiated 3T3‐L1 adipocytes were incubated with conditioned media from THP‐1 macrophages treated with or without AEDC (Figure S2a). The treatment of macrophage conditioned media significantly increased NO production, TNF‐α, IL‐6 and MCP‐1 levels in adipocytes, compared with those of RPMI 1640 medium‐treated cells, whereas pretreatment of AEDC in macrophages reversed the increases of NO production and cytokine levels (Figure 5a–d). The effects of AEDC were partially eliminated by the co‐treatment of 3‐TYP (Figure 5a–d). Next, the migration capacity of THP‐1 macrophages towards 3T3‐L1 adipocytes conditioned media was evaluated using a transwell chemotaxis assay (Figure S2b). Pretreatment of AEDC to the macrophages prevented the migration of macrophages towards adipocyte conditioned media (Figure 5e). The above results were further confirmed by the co‐culture of primary peritoneal macrophages and 3T3‐L1 adipocytes (Figure S3a–e). Taken together, these results indicated that AEDC suppressed macrophage conditioned media‐induced inflammatory responses in adipocytes and macrophage migration towards adipocytes.
FIGURE 5.
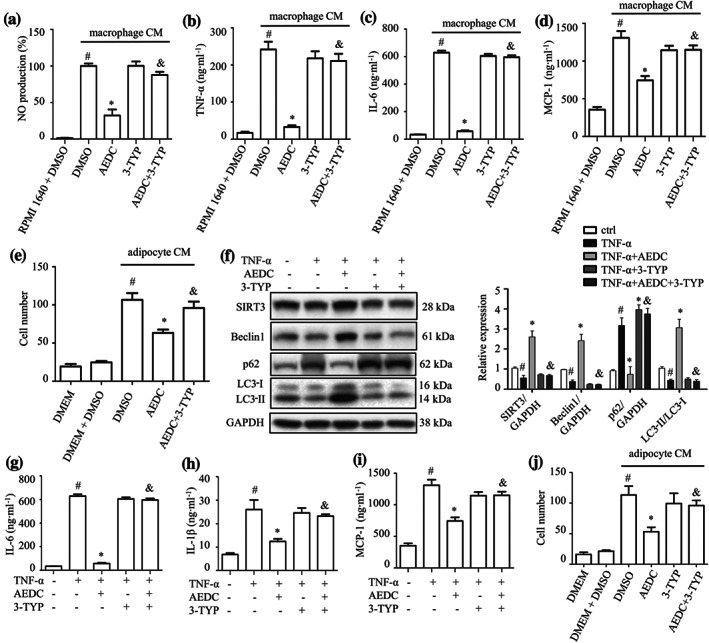
AEDC attenuates macrophage conditioned media (CM) or TNF‐α‐induced inflammatory responses in adipocytes. THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h. Subsequently, the cells were stimulated with LPS for 4 h and then 1 mM ATP for 1 h. Then, the cells were changed to fresh medium. After 24 h, the medium supernatants were collected as macrophage conditioned media (CM). The fully differentiated 3T3‐L1 adipocytes were incubated in macrophage for 24 h. (a) NO production and the levels of (b) TNF‐α, (c) IL‐6 and (d) MCP‐1 in 3T3‐L1 adipocytes were determined. Data are expressed as means ± SEM (n = 6). # P < 0.05 RPMI 1640 + DMSO vs. macrophage CM + DMSO; *P < 0.05 macrophage CM + AEDC vs. macrophage CM + DMSO; & P < 0.05 macrophage CM + AEDC vs. macrophage CM + AEDC + 3‐TYP. (e) THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h, followed by co‐culture with adipocyte CM for 4 h. Migrated THP‐1 macrophages were visualized by DAPI staining and quantified. Data are expressed as means ± SEM (n = 6). # P < 0.05 DMEM + DMSO vs. adipocyte CM + DMSO; *P < 0.05 adipocyte CM + AEDC vs. adipocyte CM + DMSO; & P < 0.05 adipocyte CM + AEDC vs. adipocyte CM + AEDC + 3‐TYP. Fully differentiated 3T3‐L1 adipocytes were treated with 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h, followed by stimulation with TNF‐α (15 ng·ml−1) for 24 h. (f) The expression of autophagy‐related proteins were examined by Western blotting (n = 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. The levels of (g) IL‐6, (h) IL‐1β and (i) MCP‐1 in the culture medium from 3T3‐L1 adipocytes were determined by ELISA (n = 6). Data are expressed as means ± SEM. # P < 0.05 TNF‐α vs. ctrl; *P < 0.05 TNF‐α + AEDC vs. TNF‐α; & P < 0.05 TNF‐α + AEDC vs. TNF‐α + AEDC + 3‐TYP. (j) Fully differentiated 3T3‐L1 adipocytes were treated with 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h. Then, the cells were changed to fresh medium. After 24 h, the medium supernatants were collected as adipocyte CM. THP‐1 macrophages were cultured in adipocyte CM for 4 h and the migrated THP‐1 macrophages were visualized by DAPI staining and quantified (n = 5). Data are expressed as means ± SEM. # P < 0.05 DMEM + DMSO vs. adipocyte CM + DMSO; *P < 0.05 adipocyte CM + AEDC vs. adipocyte CM + DMSO; & P < 0.05 adipocyte CM + AEDC vs. adipocyte CM + AEDC + 3‐TYP
Macrophages‐derived TNF‐α induces MCP‐1 and inflammatory cytokines expression in adipocytes, which is crucial for macrophage infiltration into adipose tissue (Gao et al., 2014). The anti‐inflammatory effect of AEDC was determined in TNF‐α‐stimulated adipocytes. TNF‐α treatment significantly decreased the protein levels of SIRT3 and Beclin1 and the ratio of LC3‐II to LC3‐I to approximately 52–68% and increased the level of p62 to 315%, compared with those of the control cells (Figure 5f), suggesting impaired autophagy in adipocytes. AEDC totally reversed these changes, which was partially blocked by the co‐treatment of 3‐TYP (Figure 5f). Additionally, TNF‐α significantly increased the levels of IL‐6, IL‐1β and MCP‐1 in adipocytes, compared with those of the control cells, whereas pretreatment of AEDC reversed the increases of cytokines (Figure 5g–i). In the THP‐1 macrophages migration assay, pretreatment of AEDC to the adipocytes prevented the migration of macrophages towards adipocyte conditioned media (Figure 5j). The effects of AEDC were partially eliminated by the co‐treatment of 3‐TYP (Figure 5g–j). Thus, AEDC alleviates TNF‐α‐mediated inflammation in adipocytes and blocks macrophage migration towards the adipocytes conditioned media, presumably through activating SIRT3‐mediated autophagy.
3.6. AEDC treatment alleviated LPS plus ATP‐mediated inflammatory responses in mice
To examine the anti‐inflammatory effect of AEDC in vivo, an LPS plus ATP‐induced acute inflammation mouse model was implemented (Figure 6a). AEDC treatment did not change the body weight obviously, indicating AEDC had no toxicity on mice (Figure S4). LPS plus ATP treatment significantly increased IL‐1β, TNF‐α, IL‐6 and MCP‐1 levels in serum, whereas pretreatment of AEDC reversed the increases of these cytokines, which was in coincident with the positive control group (dexamethasone) (Figure 6b–e). These trends were partially eliminated by the co‐treatment of 3‐TYP (Figure 6b–e). Thus, AEDC attenuates LPS plus ATP‐mediated inflammatory responses in mice, which might be through activating SIRT3.
FIGURE 6.

AEDC ameliorated LPS and ATP‐mediated inflammatory responses in mice. (a) The experimental procedure of LPS plus ATP‐induced acute inflammation. The male C57BL/6J mice were i.p. administrated with or without compound (AEDC, dexamethasone (DEX), or 3‐TYP) once a day for 5 days. On the sixth day, the control group of mice were i.p. injected with 10 ml·kg−1 PBS and the other six groups of mice were i.p. injected with 4 mg·kg−1 LPS (0.4 mg·ml−1 in PBS) for 4 h followed by 30 mg·kg−1 ATP (3 mg·ml−1 in PBS). Half hour later, the blood samples were collected and the mice were dissected. DEX: 4 mg·kg−1 DEX; AEDC‐L: 5 mg·kg−1 AEDC; AEDC‐H: 20 mg·kg−1 AEDC; 3‐TYP: 4 mg·kg−1 3‐TYP; AEDC + 3‐TYP: 20 mg·kg−1 AEDC and 4 mg·kg−1 3‐TYP. The serum levels of (b) IL‐1β, (c) TNF‐α, (d) MCP‐1 and (e) IL‐6 were determined by ELISA. Data are expressed as means ± SEM (n = 5). # P < 0.05 LPS + ATP vs. vehicle; *P < 0.05 AEDC‐L, AEDC‐H, or DEX vs. LPS + ATP; & P < 0.05 AEDC‐H vs. AEDC + 3‐TYP
3.7. AEDC suppressed inflammation in the peritoneal macrophages from LPS plus ATP‐induced mice
In the peritoneal macrophages, AEDC ameliorated LPS and ATP‐induced increase of IL‐1β, assessed by both RT‐PCR analysis (Figure 7a) and ELISA kit (Figure 7b). The ELISA results also indicated that AEDC reduced the levels of other pro‐inflammatory cytokines, including TNF‐α, IL‐6 and MCP‐1, in the peritoneal macrophages from LPS and ATP‐induced mice (Figure 7c–e). These trends were totally abolished by the co‐treatment of 3‐TYP (figure 7a–e). AEDC treatment increased the levels of Beclin1, Atg5, Atg7, SIRT3 and the ratio of LC3‐II to LC3‐I and reduced the level of p62, which were reversed in combination with 3‐TYP (Figure 7f). Consistently, AEDC inhibited NLRP3 inflammasome activation in the peritoneal macrophages from LPS and ATP‐induced mice, which was reversed by the co‐treatment of 3‐TYP (Figure 7g). These results suggested that AEDC suppressed the inflammatory responses in peritoneal macrophages through SIRT3‐mediated autophagy and NLRP3 inflammasome activation.
FIGURE 7.
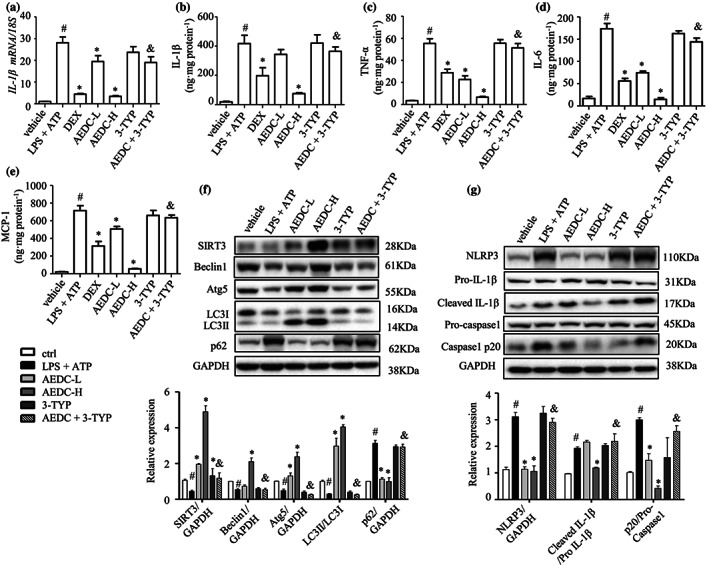
AEDC suppressed inflammation in the peritoneal macrophages from LPS plus ATP‐induced mice. The level of IL‐1β was determined by (a) quantitative RT‐PCR and (b) ELISA. The levels of (c) TNF‐α, (d) IL‐6 and (e) MCP‐1 were determined by ELISA. (f) Expression of autophagy‐related proteins was detected by Western blotting. GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (g) NLRP3, pro‐IL‐1β, cleaved IL‐1β, pro‐caspase 1 and caspase 1 p20 in peritoneal macrophages were analysed by Western blotting. GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. dexamethasone (DEX): 4 mg·kg−1 DEX; AEDC‐L: 5 mg·kg−1 AEDC; AEDC‐H: 20 mg·kg−1 AEDC; 3‐TYP: 4 mg·kg−1 3‐TYP; AEDC + 3‐TYP: 20 mg·kg−1 AEDC and 4 mg·kg−1 3‐TYP. Data are expressed as means ± SEM (n = 5). # P < 0.05 LPS + ATP vs. vehicle; *P < 0.05 AEDC‐L, AEDC‐H, or DEX vs. LPS + ATP; & P < 0.05 AEDC‐H vs. AEDC + 3‐TYP
3.8. AEDC treatment attenuated adipose tissue inflammation
The adipose tissue inflammation is characterized by increased pro‐inflammatory cytokines and augmented macrophages filtration. The H&E staining of epididymal white adipose tissue (eWAT) from LPS and ATP‐treated mice exhibited more infiltration of macrophages when compared with the vehicle control mice, whereas AEDC treatment significantly reduced the amount of macrophage infiltration in epididymal white adipose tissue, which was comparable with the dexamethasone mice (Figure 8a). The immunofluorescent staining results further supported the above observation (Figure 8b). The ELISA results showed that the cytokine levels in epididymal white adipose tissue from LPS and ATP‐induced mice, including IL‐1β, TNF‐α, IL‐ and MCP‐1, were increased, which were significantly reduced in epididymal white adipose tissue from AEDC‐treated mice (Figure 8c–f), indicating that AEDC treatment attenuated adipose tissue inflammation. Interestingly, these trends were reversed by the co‐treatment of 3‐TYP (Figure 8a–f).
FIGURE 8.
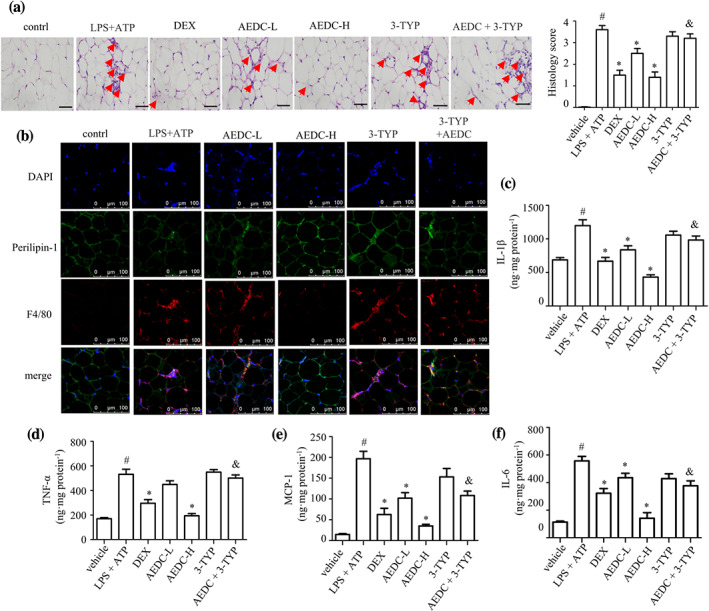
AEDC reduced macrophage content in epididymal adipose tissue from LPS plus ATP‐induced mice. (a) H&E staining and histopathological score of epididymal white adipose tissue (eWAT). The infiltrated macrophages in eWAT were indicated by the red arrows. Scale bar = 200 μm. (b) Immunofluorescence staining of perilipin‐1 (green) and F4/80 (red) in eWAT. Scale bar = 100 μm. The levels of (c) IL‐1β, (d) TNF‐α, (e) MCP‐1 and (f) IL‐6 in eWAT were determined by ELISA. Dexamethasone (DEX): 4 mg·kg−1 DEX; AEDC‐L: 5 mg·kg−1 AEDC; AEDC‐H: 20 mg·kg−1 AEDC; 3‐TYP: 4 mg·kg−1 3‐TYP; AEDC + 3‐TYP: 20 mg·kg−1 AEDC and 4 mg·kg−1 3‐TYP. Data are expressed as means ± SEM (n = 5). # P < 0.05 LPS + ATP vs. vehicle; *P < 0.05 AEDC‐L, AEDC‐H, or DEX vs. LPS + ATP; & P < 0.05 AEDC‐H vs. AEDC + 3‐TYP
Next, the expression of macrophage markers was examined. The protein levels of macrophage markers, F4/80 and CD68, in epididymal white adipose tissue were increased in LPS and ATP‐induced mice and significantly reduced in AEDC‐treated mice (Figure 9a). The levels of M1 macrophage marker, CD11c and M2 macrophage marker, CD206, were both increased in epididymal white adipose tissue from LPS and ATP‐treated mice; remarkably, AEDC down‐regulated the expression of CD11c and CD206 (Figure 9a). The immunohistochemistry results further supported the above observation (Figure 9b). Furthermore, the quantitative RT‐PCR data indicated that AEDC treatment suppressed the levels of macrophage markers, F4/80 and CD68, and M1 macrophage marker, CD11c, but not M2 macrophage marker, CD206, in epididymal white adipose tissue from LPS and ATP‐treated mice (Figure 9c–f). Interestingly, the effects of AEDC were almost abolished by the co‐treatment of 3‐TYP (Figure 9a–f). The mRNA levels of chemokines, including MCP‐1, MIP‐1α, Cxcl10, Ccl5 and Ccl11, were significantly elevated in epididymal white adipose tissue from LPS and ATP‐treated mice when compared with those of the vehicle control mice and AEDC treatment greatly suppressed the expressions of these chemokines, which were blocked when co‐treated with 3‐TYP (Figure 9g–k), suggesting that AEDC prevents LPS and ATP‐induced macrophage infiltration into adipose tissue. These data suggested that AEDC reduces macrophages infiltration to alleviate adipose tissue inflammation in LPS and ATP‐induced mice.
FIGURE 9.

AEDC suppressed the expression of chemokines and macrophage markers in epididymal white adipose tissue (eWAT) from LPS plus ATP‐induced mice. (a) Expression of F4/80, CD68, CD11c and CD206 was detected by Western blotting. GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (b) Immunohistochemical stainings of CD206 and CD11c in eWAT. The mRNA levels of macrophage markers in eWAT, including (c) F4/80, (d) CD68, (e) CD11c and (f) CD206. The mRNA levels of chemokines, including (g) MCP‐1, (h) MIP‐1α, (i) Cxcl10, (j) Ccl11 and (k) Ccl5. 18S was used as an internal control. DEX: 4 mg·kg−1 DEX; AEDC‐L: 5 mg·kg−1 AEDC; AEDC‐H: 20 mg·kg−1 AEDC; 3‐TYP: 4 mg·kg−1 3‐TYP; AEDC + 3‐TYP: 20 mg·kg−1 AEDC and 4 mg·kg−1 3‐TYP. Data are expressed as means ± SEM (n = 5). # P < 0.05 LPS + ATP vs. vehicle; *P < 0.05 AEDC‐L, AEDC‐H vs. LPS + ATP; & P < 0.05 AEDC‐H vs. AEDC + 3‐TYP
4. DISCUSSION
Adipose tissue inflammation is associated with many metabolic diseases such as type‐2 diabetes, insulin resistance and obesity (Harte et al., 2013; Weisberg et al., 2003). Recruitment and pro‐inflammatory polarization of macrophages in the expanding adipose tissues promote low‐grade inflammation in adipose tissue (Suganami, Nishida, & Ogawa, 2005). The majority of adipose tissue‐derived cytokines originate from infiltrating macrophages, which ultimately increase circulating pro‐inflammatory cytokines to develop low‐grade chronic inflammatory state (Lumeng, Bodzin, & Saltiel, 2007). Many molecules have been reported to alleviate adipose tissue inflammation (Li, Zhang, Lu, Peng, & Lin, 2020). Herein, AEDC was found to ameliorate LPS plus ATP‐induced adipose tissue inflammation in mice, mainly due to reduced macrophage infiltration but not pro‐inflammatory macrophage polarization. Due to technical ease and high reproducibility, LPS has been widely used to induce acute inflammation in different organs, with earlier and greater cytokine responses (Seemann, Zohles, & Lupp, 2017). LPS model does not exactly reproduce the characteristic features of adipose tissue inflammation that is chronic low‐grade inflammation. LPS always causes systematic inflammation, including lung, spleen and nervous system. In the current study, we found LPS plus ATP‐induced dramatically inflammatory responses in spleen (data not shown). To fully understand the role of AEDC in alleviating adipose tissue inflammation, diet‐induced obese mice model and/or genetically obese mice model should be recruited in the future.
Chemokines and cytokines are involved in a variety of physiologic and pathologic processes. MCP‐1 and MIP‐1α (CCL3) are the key chemokines for macrophage recruitment into adipose tissue (Surmi & Hasty, 2008). Currently, AEDC reversed LPS plus ATP‐induced increase of chemokines in THP‐1 macrophages, 3T3‐L1 adipocytes, peritoneal macrophages and epididymal white adipose tissue. Pretreatment of AEDC in either macrophages or adipocytes impaired the migration capacity of macrophages towards adipocyte conditioned media. Thus, AEDC impairs chemokine‐induced migration of macrophages into adipose tissue, resulting in improved inflammatory responses in adipose tissue.
IL‐1β, a major pro‐inflammatory cytokine, is produced mainly by macrophages; biologically active IL‐1β is formed through cleavage of pro‐IL‐1β by NLRP3 inflammasome‐activated caspase 1 (Sims & Smith, 2010). IL‐1β has been considered as the culprit of the inflammatory crosstalk between macrophages and adipocytes in obese objects (Gao et al., 2014). Exogenous stimuli, such as LPS and excessive lipids, promote the production and secretion of IL‐1β in macrophages, which in turn induces local inflammation in adipocytes, resulting in systematic inflammation. AEDC was identified to suppress the production and release of IL‐1β in macrophages, resulting in improved inflammatory responses in adipocytes and adipose tissue from acute inflammation mice. AEDC‐induced inhibition of IL‐1β production and secretion might put a brake on the vicious cycle of macrophage infiltration and escalate inflammatory response in adipose tissue, thereby improving adipose tissue inflammation.
Accumulating evidence has indicated that NLRP3 inflammasome inactivation acts as a protective mechanism against inflammation and metabolic disorders (Kastner, Aksentijevich, & Goldbach‐Mansky, 2010). Ablation of NLRP3 in mice prevented obesity‐induced inflammasome activation in fat depots (Vandanmagsar et al., 2011). Herein, AEDC suppressed NLRP3 inflammasome activation in THP‐1 macrophages in vitro and in visceral adipose tissue and peritoneal macrophages from LPS plus ATP‐stimulated mice in vivo. On the other hand, excessive mtROS and mitochondrial dysfunction are essential for inflammasome activation, which acts as a common event upstream of the NLRP3 inflammasome machinery. MtROS triggers the NLRP3 inflammasome, which in turn leads to IL‐1β secretion (Zhou et al., 2011). Currently, LPS plus ATP treatment enhanced the level of mtROS, induced mitochondrial membrane potential collapse and prevented mitochondria relocates on damaged microtubules. AEDC suppressed LPS‐induced inflammation by rescuing mitochondrial dysfunction. Thus, targeting mitochondrial function could be an efficient strategy to cure NLRP3‐inflammasome‐IL‐1β‐related diseases.
Autophagy clears damaged mitochondria and inhibits NLRP3 inflammasome activation and the autophagic adaptors, p62 and LC3, participate in this process (Nakahira et al., 2011; Shi et al., 2012). Autophagy deficiency in macrophages contributes to the mitochondrial dysfunction, inflammasome activation and metabolic deterioration in mice (Lee et al., 2016; Razani et al., 2012). AEDC alleviated LPS plus ATP‐induced inflammation and inhibited the activation of NLRP3 inflammasome by restoring impaired autophagy. Mitophagy, or autophagy of the mitochondria, is important for mitochondrial quality control, thereby essential for providing cellular energy, calcium homoeostasis and redox signalling; p62 recognizes ubiquitinated proteins and brings mitochondria to autophagosomes by binding to LC3 (Park, Choi, Yoo, Son, & Jung, 2014). AEDC repressed LPS and ATP‐promoted p62 to bond with NLRP3 and improved the translocation of lipidated LC3 to mitochondria. These evidences suggested that AEDC might recruit autophagosomal membranes and associated machinery to damaged mitochondria, to activate mitophagy and maintain mitochondrial homoeostasis. Consistently, depletion of LC3‐II and Beclin1 enhanced NLRP3 inflammasome activation and promoted accumulation of dysfunctional mitochondria to LPS and ATP in macrophages (Nakahira et al., 2011).
Sirtuins are a family of NAD+‐dependent deacetylases and there are seven sirtuin isoforms (SIRT1 to 7) in mammalian cells. SIRT3 is primarily located in mitochondria and plays an anti‐inflammatory role in macrophages (Xu, Hertzel, Steen, & Bernlohr, 2016). Similarly, prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly through SIRT3‐mediated SOD2 activation (Traba et al., 2017). Moreover, SIRT3‐induced autophagy inhibited NLRP3 inflammasome activation in THP‐1 macrophages and SIRT3 expression is negatively correlated with NLRP3 inflammasome activation (Liu et al., 2018). Our studies demonstrated that SIRT3 level was decreased in inflammatory macrophages and in peritoneal macrophages from acute inflammatory mice. AEDC increased SIRT3 expression in these models and enhanced SIRT3 deacetylating activity. Moreover, AEDC suppressed NLRP3 inflammasome activation and mitochondrial dysfunction by SIRT3‐mediated autophagy and ROS scavenging in THP‐1 macrophages. AEDC inhibited NLRP3 inflammasome activation in the peritoneal macrophages and epididymal white adipose tissue from LPS plus ATP‐induced mice, through SIRT3‐mediated autophagy. Thus, SIRT3 activators might be potential therapeutic agents for the treatment of adipose tissue inflammation‐related diseases.
Cycloartane triterpenoids have been reported with a wide range of bioactivities (Molnar et al., 2006). Herein, AEDC was identified as a naturally occurring autophagy activator, mediating the inactivation of NLRP3 inflammasome and anti‐inflammatory effect in macrophages and adipocytes. Moreover, SIRT3 is involved in AEDC‐driven autophagy. Drug target engagement is a critical factor for the pharmacological effects of drugs and therapeutic target validation at the cellular level. CETSA takes advantage of drug‐induced changes in the thermal stability of a target protein. The greatest advantage of this method is that native small molecules without chemical modification, such as biotin or fluorescent tags, or photoaffinity labels, can be used. Herein, the CETSA results indicated that AEDC interacts with the SIRT3 deacetylase. To further decipher the binding domain, a virtual docking was performed on AutoDock. Resveratrol was used as a positive control. The docking score of the complex of resveratrol and SIRT3 is −5.447 and π–π stacking interaction between them exists. Unfortunately, the interaction between AEDC and SIRT3 was hard to detect and the docking score of this complex was different to estimate. AEDC might bind with SIRT3 deacetylase at an undefined domain. Further studies are needed to fully elucidate it.
In summary, AEDC, a newly isolated cycloartane triterpenoid, suppresses IL‐1β production and secretion in LPS plus ATP‐treated macrophages and mice, possibly through SIRT3‐autophagy‐mediated NLRP3 inflammasome inactivation and SIRT3–SOD2‐mediated ROS scavenging (Figure 10). AEDC attenuates the inflammatory crosstalk between macrophages and adipocytes and blocks the migration of macrophages towards adipocytes (Figure 10). Moreover, AEDC alleviates adipose tissue inflammation through preventing macrophage accumulation (Figure 10). AEDC might be further developed as a candidate for treating adipose tissue inflammation and its related metabolic disorders.
FIGURE 10.
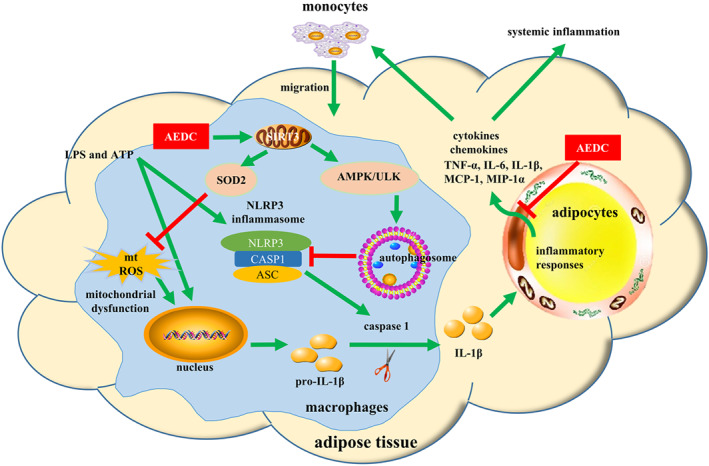
Schematic models of molecular targets of AEDC in attenuating visceral adipose tissue inflammation signalling pathways
AUTHOR CONTRIBUTIONS
T.Z. conducted experiments, analysed the data and wrote the manuscript; Z.F. supplied the compound and proofread the paper; K‐G.L. and J.L. conducted experiments; L.G. and L.L. designed the experiments and wrote the paper; and L.L. conceived the study.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Natural Product Research, Design and Analysis, Immunoblotting and Immunochemistry and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1. Antibodies for Western blotting analysis
Table S2. List of oligonucleotide primer pairs used in qRT‐PCR.
Figure S1. Cytotoxicity and autophagy inducing activity of AEDC on THP‐1 cells. (A) THP‐1 cells were treated with different concentrations of AEDC for 24 h. Cell viability was detected by MTT assay (n= 9).(B) THP‐1 cells were treated with or without AEDC(10 μM) for 12 h and 3‐MA (5 mM) for 6h. Expression of autophagy related proteins was detected by Western blotting (n= 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (E) THP‐1 cells were transiently infected with the mRFP‐GFP‐LC3 lentivirus. mRFP‐GFP‐LC3 puncta were measured using a confocal microscope (n= 5). Scale bar = 10 μm. Data are expressed as means ± SEM. *P< 0.05 AEDC vs. ctrl; #P< 0.05 AEDC vs. AECD + 3‐MA.
Figure S3. AEDC attenuates the primary peritoneal macrophages‐CM induced inflammatory responses in adipocytes. THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h. Subsequently, the cells were stimulated with LPS (1 μg·mL‐1) for 4 h and then 1 mM ATP for 1 h. Then, the cells were changed to fresh medium. After 24 h, the medium supernatants were collected as peritoneal macrophage CM. The fully differentiated 3T3‐L1 adipocytes were incubated in peritoneal macrophage CM for 24 h. NO production (A) and the levels of IL‐β (B), TNF‐α (C), IL‐6 (D) and MCP‐1 (E) in 3T3‐L1 adipocytes were determined by ELISA kits. Data are expressed as means ± SEM (n= 6). #P <0.05, RPMI‐1640+ DMSO vs. peritoneal macrophage CM+ DMSO; *P <0.05, peritoneal macrophage CM + AEDC vs. peritoneal macrophage CM + DMSO; &P <0.05, peritoneal macrophage CM + AEDC vs. peritoneal macrophage CM + AEDC + 3‐TYP.(F) The primary peritoneal macrophages were treated with or without 10 μM AEDC for 12 hand50 μM 3‐TYP for 6 h, followed by co‐culture with adipocyte CM for 4 h. Migrated primary peritoneal macrophages were visualized by DAPI staining and quantified. Data are expressed as means ± SEM (n= 6). #P <0.05, DMEM + DMSO vs. adipocyte CM + DMSO; *P <0.05 adipocyte CM + AEDC vs. adipocyte CM + DMSO; &P <0.05 adipocyte CM + AEDC vs. adipocyte CM + AEDC + 3‐TYP.7
Figure S4. The body weight of different group of LPS plus ATP‐induced mice model. DEX: 4 mg·Kg‐1 dexamethasone; AEDC‐L: 5 mg·Kg‐1 AEDC; AEDC‐H: 20 mg·Kg‐1 AEDC; 3‐TYP: 4 mg·Kg‐1 3‐TYP; AEDC + 3‐TYP: 20 mg·Kg‐1 AEDCand4 mg·Kg‐1 3‐TYP.
ACKNOWLEDGEMENTS
Financial support by the National Natural Science Foundation of China (81872754 and 81872756), the Science and Technology Development Fund, Macau SAR (file no. FDCT 0031/2019/A1), the University of Macau (file nos MYRG2017‐00109‐ICMS and MYRG2018‐00037‐ICMS) and the Zhejiang Provincial Natural Science Foundation of China (LR17H300001) is gratefully acknowledged.
Zhang T, Fang Z, Linghu K‐G, Liu J, Gan L, Lin L. Small molecule‐driven SIRT3‐autophagy‐mediated NLRP3 inflammasome inhibition ameliorates inflammatory crosstalk between macrophages and adipocytes. Br J Pharmacol. 2020;177:4645–4665. 10.1111/bph.15215
Tian Zhang and Zhujun Fang contributed equally to this work.
Contributor Information
Lishe Gan, Email: ganlishe@163.com.
Ligen Lin, Email: ligenl@um.edu.mo.
REFERENCES
- Agostini, L. , Martinon, F. , Burns, K. , McDermott, M. F. , Hawkins, P. N. , & Tschopp, J. (2004). NALP3 forms an IL‐1beta‐processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity, 20, 319–325. 10.1016/s1074-7613(04)00046-9 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … Collaborators, C. (2019). The concise guide to pharmacology 2019/20: Introduction and other protein targets. British Journal of Pharmacology, 176(Suppl 1), S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology . British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacón, M. R. , Fernandez‐Real, J. , Richart, C. , Megía, A. , Gómez, J. , Miranda, M. , … Vendrell, J. (2007). Monocyte chemoattractant protein‐1 in obesity and type 2 diabetes. Relationship with insulin‐resistance and proinflammatory cytokines. Obesity, 15, 664–672. 10.1038/oby.2007.578 [DOI] [PubMed] [Google Scholar]
- Chen, G. Y. , & Nunez, G. (2010). Sterile inflammation: Sensing and reacting to damage. Nature Reviews Immunology, 10, 826–837. 10.1038/nri2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Y. F. , & Scherer, P. E. (2010). Adipokines as novel biomarkers and regulators of the metabolic syndrome. Annals of the New Youk Academy of Sciences, 1212, E1–E19. 10.1111/j.1749-6632.2010.05875.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic, V. (2005). Autophagy in innate and adaptive immunity. Trends in Immunology, 26, 523–528. 10.1016/j.it.2005.08.003 [DOI] [PubMed] [Google Scholar]
- Ehses, J. A. , Lacraz, G. , Giroix, M. H. , Schmidlin, F. , Coulaud, J. , Kassis, N. , … Donath, M. Y. (2009). IL‐1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proceedings of the National Academy of Sciences of the United States of America, 106, 13998–14003. 10.1073/pnas.0810087106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Z. J. , Zhang, T. , Chen, S. X. , Wang, Y. L. , Zhou, C. X. , Mo, J. X. , … Gan, L. S. (2019). Cycloartane triterpenoids from Actaea vaginata with anti‐inflammatory effects in LPS‐stimulated RAW264.7 macrophages. Phytochemistry, 160, 1–10. 10.1016/j.phytochem.2019.01.003 [DOI] [PubMed] [Google Scholar]
- Feng, Z. L. , Zhang, T. , Liu, J. X. , Chen, X. P. , Gan, L. S. , Ye, Y. , & Lin, L. G. (2018). New podolactones from the seeds of Podocarpus nagi and their anti‐inflammatory effect. Journal of Natural Medicines, 72, 882–889. 10.1007/s11418-018-1219-5 [DOI] [PubMed] [Google Scholar]
- Gao, D. , Madi, M. , Ding, C. , Fok, M. , Steele, T. , Ford, C. , … Bing, C. (2014). Interleukin‐1β mediates macrophage‐induced impairment of insulin signaling in human primary adipocytes. American Journal of Physiology‐Endocrinology and Metabolism, 307, E289–E304. 10.1152/ajpendo.00430.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte, A. L. , Tripathi, G. , Piya, M. K. , Barber, T. M. , Clapham, J. C. , Al‐Daghri, N. , … McTernan, P. G. (2013). NFκB as a potent regulator of inflammation in human adipose tissue, influenced by depot, adiposity, T2DM status, and TNFα. Obesity, 21, 2322–2330. 10.1002/oby.20336 [DOI] [PubMed] [Google Scholar]
- Infante, A. S. , Stein, M. S. , Zhai, Y. , Borisy, G. G. , & Gundersen, G. G. (2000). Detyrosinated (Glu) microtubules are stabilized by an ATP‐sensitive plus‐end cap. Journal of Cell Science, 113, 3907–3919. [DOI] [PubMed] [Google Scholar]
- Jafari, R. , Almqvist, H. , Axelsson, H. , Ignatushchenko, M. , Lundback, T. , Nordlund, P. , & Martinez Molina, D. (2014). The cellular thermal shift assay for evaluating drug target interactions in cells. Nature Protocol, 9, 2100–2122. 10.1038/nprot.2014.138 [DOI] [PubMed] [Google Scholar]
- Kastner, D. L. , Aksentijevich, I. , & Goldbach‐Mansky, R. (2010). Autoinflammatory disease reloaded: A clinical perspective. Cell, 140, 784–790. 10.1016/j.cell.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko, J. H. , Yoon, S. O. , Lee, H. J. , & Oh, J. Y. (2017). Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome‐p38 MAPK‐NFκB pathways in autophagy‐ and p62‐dependent manners. Oncotarget, 8, 40817–40831. 10.18632/oncotarget.17256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz, E. (2010). The inflammasomes: Mechanisms of activation and function. Current Opinion in Immunology, 22, 28–33. 10.1016/j.coi.2009.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz, E. , Xiao, T. S. , & Stutz, A. (2013). Activation and regulation of the inflammasomes. Nature Reviews Immunology, 13, 397–411. 10.1038/nri3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D. H. , Shin, J. S. , Kang, S. Y. , Lee, S. B. , Lee, J. S. , Ryu, S. M. , … Jang, D. S. (2018). Iridoids from the roots of Patrinia scabra and their inhibitory potential on LPS‐induced nitric oxide production. Journal of Natural Products, 81, 1468–1473. 10.1021/acs.jnatprod.8b00229 [DOI] [PubMed] [Google Scholar]
- Lee, H. Y. , Kim, J. , Quan, W. , Lee, J. C. , Kim, M. S. , Kim, S. H. , … Lee, M. S. (2016). Autophagy deficiency in myeloid cells increases susceptibility to obesity‐induced diabetes and experimental colitis. Autophagy, 12, 1390–1403. 10.1080/15548627.2016.1184799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, B. , & Deretic, V. (2007). Unveiling the roles of autophagy in innate and adaptive immunity. Nature Reviews Immunology, 7, 767–777. 10.1038/nri2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, B. , Mizushima, N. , & Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature, 469, 323–335. 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Liu, Q. , Sun, W. , Chen, X. , Wang, Y. , Sun, Y. , & Lin, L. (2018). 1,3,6,7‐Tetrahydroxy‐8‐prenylxanthone ameliorates inflammatory responses resulting from the paracrine interaction of adipocytes and macrophages. British Journal of Pharmacology, 175, 1590–1606. 10.1111/bph.14162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Zhang, T. , Lu, J. , Peng, C. , & Lin, L. (2020). Natural constituents from food sources as therapeutic agents for obesity and metabolic diseases targeting adipose tissue inflammation. Critical Reviews in Food Science and Nutrition., 1–19. 10.1080/10408398.2020.1768044 [DOI] [PubMed] [Google Scholar]
- Lin, L. G. , Pang, W. J. , Chen, K. Y. , Wang, F. , Gengler, J. , Sun, Y. X. , & Tong, Q. (2012). Adipocyte expression of PU.1 transcription factor causes insulin resistance through upregulation of inflammatory cytokine gene expression and ROS production. American Journal of Physiology‐Endocrinology and Metabolism, 302, E1550–E1559. 10.1152/ajpendo.00462.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Li, D. , Zhang, T. , Tong, Q. , Ye, R. D. , & Lin, L. (2017). SIRT3 protects hepatocytes from oxidative injury by enhancing ROS scavenging and mitochondrial integrity. Cell Death & Disease, 8, e3158 10.1038/cddis.2017.564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P. H. , Huang, G. J. , Wei, T. , Gao, J. , Huang, C. L. , Sun, M. W. , … Shen, W. L. (2018). Sirtuin 3‐induced macrophage autophagy in regulating NLRP3 inflammasome activation. Biochimica et Biophysica Acta‐Molecular Basis of Disease, 1864, 764–777. 10.1016/j.bbadis.2017.12.027 [DOI] [PubMed] [Google Scholar]
- Lumeng, C. N. , Bodzin, J. L. , & Saltiel, A. R. (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. Journal of Clinical Investigation, 117, 175–184. 10.1172/JCI29881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng, C. N. , & Saltiel, A. R. (2011). Inflammatory links between obesity and metabolic disease. Journal of Clinical Investigation, 121, 2111–2117. 10.1172/JCI57132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotorchino, J. , Romier, B. , Gouranton, E. , Riollet, C. , Gleize, B. , Malezet‐Desmoulins, C. , & Landrier, J. F. (2012). Lycopene attenuates LPS‐induced TNF‐α secretion in macrophages and inflammatory markers in adipocytes exposed to macrophage‐conditioned media. Molecular Nutrition & Food Research, 56, 725–732. 10.1002/mnfr.201100623 [DOI] [PubMed] [Google Scholar]
- Martinez, M. D. , Jafari, R. , Ignatushchenko, M. , Seki, T. , Larsson, E. A. , Dan, C. , … Nordlund, P. (2013). Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science, 341, 84–87. 10.1126/science.1233606 [DOI] [PubMed] [Google Scholar]
- Martinon, F. , Burns, K. , & Tschopp, J. (2002). The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Molecular Cell, 10, 417–426. 10.1016/s1097-2765(02)00599-3 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP . British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar, J. , Gyemant, N. , Tanaka, M. , Hohmann, J. , Bergmann‐Leitner, E. , Molnar, P. , … Ferreira, M. J. U. (2006). Inhibition of multidrug resistance of cancer cells by natural diterpenes, triterpenes and carotenoids. Current Pharmaceutical Design, 12, 287–311. 10.2174/138161206775201893 [DOI] [PubMed] [Google Scholar]
- Nakahira, K. , Haspel, J. A. , Rathinam, V. A. K. , Lee, S. J. , Dolinay, T. , Lam, H. C. , … Choi, A. M. K. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology, 12, 222–230. 10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, J. E. , & Lee, H. K. (2014). Pattern recognition receptors and autophagy. Frontiers in Immunology, 5, 300 10.3389/fimmu.2014.00300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. , Choi, S. G. , Yoo, S. M. , Son, J. H. , & Jung, Y. K. (2014). Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy, 10, 1906–1920. 10.4161/auto.32177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani, B. , Feng, C. , Coleman, T. , Emanuel, R. , Wen, H. T. , Hwang, S. , … Semenkovich, C. F. (2012). Autophagy links inflammasomes to atherosclerotic progression. Cell Metabolism, 15, 534–544. 10.1016/j.cmet.2012.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh, T. , Fujita, N. , Jang, M. H. , Uematsu, S. , Yang, B. G. , Satoh, T. , … Akira, S. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1 beta production. Nature, 456, 264–268. 10.1038/nature07383 [DOI] [PubMed] [Google Scholar]
- Schroder, K. , & Tschopp, J. (2010). The inflammasomes. Cell, 140, 821–832. 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- Seemann, S. , Zohles, F. , & Lupp, A. (2017). Comprehensive comparison of three different animal models for systemic inflammation. Journal of Biomedical Science, 24, 60 10.1186/s12929-017-0370-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Zhang, J. H. , Xiao, H. , Wu, J. M. , He, K. M. , Lv, Z. Z. , … Zhang, Y. Y. (2018). Mitochondria are transported along microtubules in membrane nanotubes to rescue distressed cardiomyocytes from apoptosis. Cell Death & Disease, 9, 81 10.1038/s41419-017-0145-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, S. , Liao, Q. , Liu, J. , Pan, R. , Lee, S. M. , & Lin, L. (2019). Myricanol rescues dexamethasone‐induced muscle dysfunction via a sirtuin 1‐dependent mechanism. Journal of Cachexia Sarcopenia and Muscle, 10, 429–444. 10.1002/jcsm.12393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, S. , Liao, Q. , Zhang, T. , Pan, R. , & Lin, L. (2019). Myricanol modulates skeletal muscle‐adipose tissue crosstalk to alleviate high‐fat diet‐induced obesity and insulin resistance. British Journal of Pharmacology, 176, 3983–4001. 10.1111/bph.14802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, C. S. , Shenderov, K. , Huang, N. N. , Kabat, J. , Abu‐Asab, M. , Fitzgerald, K. A. , … Kehrl, J. H. (2012). Activation of autophagy by inflammatory signals limits IL‐1β production by targeting ubiquitinated inflammasomes for destruction. Nature Immunology, 13, 255–263. 10.1038/ni.2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims, J. E. , & Smith, D. E. (2010). The IL‐1 family: Regulators of immunity. Nature Reviews Immunology, 10, 89–102. 10.1038/nri2691 [DOI] [PubMed] [Google Scholar]
- Suganami, T. , Nishida, J. , & Ogawa, Y. (2005). A paracrine loop between adipocytes and macrophages aggravates inflammatory changes—Role of free fatty acids and tumor necrosis factor α. Arteriosclerosis Thrombosis and Vascular Biology, 25, 2062–2068. 10.1161/01.ATV.0000183883.72263.13 [DOI] [PubMed] [Google Scholar]
- Surmi, B. K. , & Hasty, A. H. (2008). Macrophage infiltration into adipose tissue: Initiation, propagation and remodeling. Future Lipidol, 3, 545–556. 10.2217/17460875.3.5.545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traba, J. , Geiger, S. S. , Kwarteng‐Siaw, M. , Han, K. , Ra, O. H. , Siegel, R. M. , … Sack, M. N. (2017). Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3‐mediated activation of superoxide dismutase 2. Journal of Biological Chemistry, 292, 12153–12164. 10.1074/jbc.M117.791715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar, B. , Youm, Y. H. , Ravussin, A. , Galgani, J. E. , Stadler, K. , Mynatt, R. L. , … Dixit, V. D. (2011). The NLRP3 inflammasome instigates obesity‐induced inflammation and insulin resistance. Nature Medicine, 17, 179–188. 10.1038/nm.2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg, S. P. , McCann, D. , Desai, M. , Rosenbaum, M. , Leibel, R. L. , & Ferrante, A. W. (2003). Obesity is associated with macrophage accumulation in adipose tissue. Journal of Clinical Investigation, 112, 1796–1808. 10.1172/JCI19246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H. L. , Hertzel, A. V. , Steen, K. A. , & Bernlohr, D. A. (2016). Loss of fatty acid binding protein 4/aP2 reduces macrophage inflammation through activation of SIRT3. Molecular Endocrinology, 30, 325–334. 10.1210/me.2015-1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii, S. R. , & Mizushima, N. (2017). Monitoring and measuring autophagy. International Journal of Molecular Sciences, 18, 1865 10.3390/ijms18091865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Liu, J. , Shen, S. , Tong, Q. , Ma, X. , & Lin, L. (2020). SIRT3 promotes lipophagy and chaperon‐mediated autophagy to protect hepatocytes against lipotoxicity. Cell Death & Differetiation, 27, 329–344. 10.1038/s41418-019-0356-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Liu, J. , Tong, Q. , & Lin, L. (2020). SIRT3 acts as a positive autophagy regulator to promote lipid mobilization in adipocytes via activating AMPK. International Journal of Molecular Sciences, 21, 372 10.3390/ijms21020372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, R. B. , Yazdi, A. S. , Menu, P. , & Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature, 469, 221–225. 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Antibodies for Western blotting analysis
Table S2. List of oligonucleotide primer pairs used in qRT‐PCR.
Figure S1. Cytotoxicity and autophagy inducing activity of AEDC on THP‐1 cells. (A) THP‐1 cells were treated with different concentrations of AEDC for 24 h. Cell viability was detected by MTT assay (n= 9).(B) THP‐1 cells were treated with or without AEDC(10 μM) for 12 h and 3‐MA (5 mM) for 6h. Expression of autophagy related proteins was detected by Western blotting (n= 6). GAPDH was used as an internal loading control. Data were normalized to the mean value of the control group. (E) THP‐1 cells were transiently infected with the mRFP‐GFP‐LC3 lentivirus. mRFP‐GFP‐LC3 puncta were measured using a confocal microscope (n= 5). Scale bar = 10 μm. Data are expressed as means ± SEM. *P< 0.05 AEDC vs. ctrl; #P< 0.05 AEDC vs. AECD + 3‐MA.
Figure S3. AEDC attenuates the primary peritoneal macrophages‐CM induced inflammatory responses in adipocytes. THP‐1 cells were treated with or without 10 μM AEDC for 12 h and 50 μM 3‐TYP for 6 h. Subsequently, the cells were stimulated with LPS (1 μg·mL‐1) for 4 h and then 1 mM ATP for 1 h. Then, the cells were changed to fresh medium. After 24 h, the medium supernatants were collected as peritoneal macrophage CM. The fully differentiated 3T3‐L1 adipocytes were incubated in peritoneal macrophage CM for 24 h. NO production (A) and the levels of IL‐β (B), TNF‐α (C), IL‐6 (D) and MCP‐1 (E) in 3T3‐L1 adipocytes were determined by ELISA kits. Data are expressed as means ± SEM (n= 6). #P <0.05, RPMI‐1640+ DMSO vs. peritoneal macrophage CM+ DMSO; *P <0.05, peritoneal macrophage CM + AEDC vs. peritoneal macrophage CM + DMSO; &P <0.05, peritoneal macrophage CM + AEDC vs. peritoneal macrophage CM + AEDC + 3‐TYP.(F) The primary peritoneal macrophages were treated with or without 10 μM AEDC for 12 hand50 μM 3‐TYP for 6 h, followed by co‐culture with adipocyte CM for 4 h. Migrated primary peritoneal macrophages were visualized by DAPI staining and quantified. Data are expressed as means ± SEM (n= 6). #P <0.05, DMEM + DMSO vs. adipocyte CM + DMSO; *P <0.05 adipocyte CM + AEDC vs. adipocyte CM + DMSO; &P <0.05 adipocyte CM + AEDC vs. adipocyte CM + AEDC + 3‐TYP.7
Figure S4. The body weight of different group of LPS plus ATP‐induced mice model. DEX: 4 mg·Kg‐1 dexamethasone; AEDC‐L: 5 mg·Kg‐1 AEDC; AEDC‐H: 20 mg·Kg‐1 AEDC; 3‐TYP: 4 mg·Kg‐1 3‐TYP; AEDC + 3‐TYP: 20 mg·Kg‐1 AEDCand4 mg·Kg‐1 3‐TYP.