

Abstract
Objectives
Loss of tumor‐inherent type I interferon (IFN) signalling has been closely linked to accelerated metastatic progression via decreased immunogenicity and antitumor immunity. Previous studies in murine models of triple‐negative breast cancer (TNBC) demonstrate that systemic IFN inducers are effective antimetastatic agents, via sustained antitumor CD8+ T‐cell responses. Repeated systemic dosing with recombinant IFNs or IFN inducers is associated with significant toxicities; hence, the use of alternate intratumoral agents is an active area of investigation. It is critical to investigate the impact of intratumoral agents on subsequent metastatic spread to predict clinical impact.
Methods
In this study, the local and systemic impact of the intratumoral Toll‐like receptor (TLR) 7/8 agonist 3M‐052 alone or in combination with anti‐PD1 was evaluated in metastatic TNBC models. The IFN‐α receptor (IFNAR1) blocking antibody, MAR1‐5A3, along with immune‐deficient mice and ex vivo assays are utilised to examine the key targets of this agent that are critical for an antimetastatic response.
Results
Single intratumoral administration of 3M‐052 reduced mammary tumor growth, induced a T‐cell‐inflamed tumor microenvironment (TME) and reduced metastatic spread to lung. Metastasis suppression was reliant on IFN signalling and an antitumor immune response, in contrast to primary tumor growth inhibition, which was retained in NSG and CD8+ T‐cell‐depleted mice. 3M‐052 action was demonstrated via dendritic cell activation and production of type I IFN and other pro‐inflammatory cytokines to initiate a T‐cell‐inflamed TME and promote tumor cell antigen presentation.
Conclusion
This work supports neoadjuvant TLR agonist‐based immunotherapeutics as realistic options for immune activation in the TME and long‐term metastatic protection in TNBC.
Keywords: CD8+ T cell, immunotherapy, interferon, metastasis, TLR agonist, triple‐negative breast cancer
Here, we present preclinical findings on the efficacy of direct tumor adminstration of an interferon inducer in triple‐negative breast cancer. The use of an intratumoral Toll‐like receptor agonist (3M‐052), formulated for retention at the primary tumor site, was sufficient to induce a T‐cell‐inflamed tumor microenvironment in mouse models of triple‐negative breast cancer. A single injection of 3M‐052 into the primary tumor dramatically reduced subsequent metastatic spread to lung, and this was dependent on the antitumor immune response.

Introduction
Up to one in five breast cancers are classified triple negative, lacking oestrogen and progesterone receptors along with human epidermal growth factor receptor 2 (HER2). Due to the absence of therapeutically targetable markers, over 20% of patients with triple‐negative breast cancer (TNBC) develop rapid metastatic disease despite standard chemotherapy. 1 The requirement for more targeted therapeutic options, along with the reported link between tumor‐infiltrating lymphocyte (TIL) accumulation and a favorable prognosis in TNBC, 2 has prompted investigations into use of immunotherapy for this breast cancer subtype.
Accumulating data now closely links the immune landscape in TNBC with chemotherapeutic response and the risk of metastatic relapse. Reduced TILs are associated with an enhanced risk of metastasis, 2 , 3 and further interrogation of the nature of TILs has revealed that an increased ratio of immune effector lymphocytes (particularly CD8+ T cells) to immunosuppressive cell types, such as myeloid‐derived suppressor cells (MDSCs) and regulatory T cells (Tregs), predicts a more favorable prognosis. 4 , 5 , 6 In fact, measurement of a subset of CD8+ T cells implicated in tumor immune surveillance, tissue‐resident memory T (TRM) cells, is a superior predictor of improved patient survival in TNBC. 7 The tumor cell‐inherent properties that dictate TIL numbers and composition remain largely unknown. However, our previous studies have implicated tumor cell type I interferon (IFN) signalling in immune activation and metastatic suppression in TNBC and uncovered tumor IFN biomarkers that are associated with elevated TRM signatures and improved patient survival. 8 , 9 In line with this, enhanced tumor‐inherent type I IFN signalling has been implicated in chemotherapy‐driven immune activation and response 8 , 10 Our work in breast and prostate cancer demonstrates that loss of the type I IFN pathway in tumor cells promotes immune escape and metastatic spread. 9 , 11 The mechanism of downregulation of the pathway is largely unknown, yet the expression of the key transcription factors responsible for IFN production and response (IFN regulatory factors) is silenced in aggressive cancers, 8 , 11 , 12 possibly via epigenetic silencing of the pathway, 11 , 13 , 14 or reduced IFN signalling in the surrounding microenvironment. 15
Type I IFN (‐α/β) plays a myriad of roles in the antitumor response, including direct growth‐inhibitory and pro‐apoptotic effects, and modulation of immune cells and signalling pathways to promote immune‐mediated tumor surveillance and destruction. 16 Along with functions in innate immunity, type I IFN provides the initiating signal to enable co‐stimulation for T‐cell priming by dendritic cells (DCs), leading to effector T‐cell proliferation and function. Essential for an effective T‐cell response, IFN signalling also promotes antigen presentation and tumor immunogenicity via the upregulation of antigen processing and presentation machinery, which culminates in the increased expression of self‐peptide‐containing major histocompatibility complex (MHC) molecules on the cell surface. 17 , 18 Given that type I IFN signalling also leads to production of pro‐inflammatory cytokines to attract effector cells, 16 it is conceivable that therapeutic restoration of IFN signals in the tumor microenvironment (TME) may offer a potential route to increase tumor immunogenicity along with T‐cell inflammation to prevent metastasis and/or increase response to immune checkpoint inhibitors. 19
Whilst initially conceived as a revolutionary treatment strategy to combat malignancies through immunomodulation and antiproliferative effects, systemic use of recombinant type I IFN in solid cancers has been restricted because of the dose‐limiting side effects and a lack of benefit against advanced, metastatic disease. 16 , 20 , 21 Alternative strategies have since been employed to stimulate cellular IFN production and signalling. This includes utilising agonists of specific Toll‐like receptors (TLRs) to induce IFN production in the target cell, such as the viral mimetic and TLR3 agonists poly I:C and poly(ICLC); however, these agonists also cause toxicities from systemic delivery but have shown lessened toxicity when administered locally. 22 , 23 , 24 Administration of poly A:U has lessened toxicity profiles in patients with breast cancer; however, it is yet to be investigated using alternate delivery routes. 25 Such systemic approaches have reported antimetastatic activity in preclinical models of TNBC, 26 yet have collectively varied in efficacy in clinical trials. 27 , 28 Use of local rather than systemic administration of TLR or stimulator of IFN gene (STING) agonists has been trialled in preclinical models and demonstrated a degree of antimetastatic activity, yet has similarly failed to completely eliminate systemic and off‐target immune activation, likely because of active drainage from the administration site and induction of a robust pro‐inflammatory cytokine milieu. 29 , 30 , 31 , 32 Recently, an intratumoral TLR7/8 agonist 3M‐052 (also known as MEDI9197) has been described that is formulated for retention at the tumor site. 33 This agent was demonstrated to alter the tumor immune landscape upon multiple intratumoral injections with limited associated systemic immune activation in preclinical subcutaneous tumor models. It remains to be established if such an agent can stimulate a T‐cell‐inflamed response in IFN‐low mammary tumors implanted in the orthotopic site and, importantly, an antimetastatic response to impact overall survival.
Here, we utilise murine TNBC models, including the aggressive 4T1.2 model that rapidly metastasises to lung, to investigate the antitumor and antimetastatic efficacy of a single intratumoral injection of 3M‐052. We compare its use as a single agent or in combination with checkpoint inhibition, and provide mechanistic evidence by which intratumoral 3M‐052 specifically prolongs metastasis‐free survival.
Results
Intratumoral 3M‐052 delivery delays tumor progression and enhances TILs
We first investigated the impact of a single intratumoral dose of 3M‐052 on intramammary fat pad (IMFP) growth of the weakly metastatic E0771 C57BL/6 cell line. Once palpable, E0771 IMFP tumors were directly injected with 3M‐052 or vehicle (sesame oil) control and tumor growth was monitored via calliper measurements (Figure 1a, day 13). Treatment with 3M‐052 decreased primary tumor volume and extended time to ethical primary tumor endpoint. These changes appeared to be independent of any major alterations to the tumor immune infiltrate at endpoint (Supplementary figure 1: flow cytometry gating strategy; Supplementary figure 2a and b). Upon tumor dissociation, we observed, at the macroscopic level, that the oil‐based vehicle was present in primary tumors at least 21 days after i.t injection, suggesting sufficient retention of the drug over this time period, as recently reported in subcutaneous tumors. 33 Given our previous data on the key function of type I IFN in preventing metastasis, 9 we moved forward to assess the impact of 3M‐052 treatment in the aggressive IFN‐low 4T1.2 model.
Figure 1.

Intratumoral 3M‐052 delivery slows primary tumor growth and prolongs survival in immunologically cold tumors. (a) C57BL6/J mice were injected with 1 × 105 E0771 cells into the 4th mammary fat pad, and palpable tumors were i.t. injected with vehicle or 3M‐052 on day 13 post‐inoculation and monitored for size. (b) BALB/c mice were injected with 1 × 105 4T1.2 cells into the 4th mammary fat pad, and palpable tumors were i.t. injected with vehicle or 3M‐052 on day 5 post‐inoculation and primary tumors were measured for size. Primary tumors from a separate group of mice resected on day 12 post‐inoculation were either (c) fixed and stained by IHC, with representative images for Ki67, c‐caspase‐3 and PD1 shown (10× magnification except H&E 4× magnification, scale bars = 100 µm, representative images shown); or (d) analysed by flow cytometry for primary tumor cellularity. (e) A proportion of lungs were taken on day 21 for H&E staining to assess metastatic burden; tumor regions are denoted by dotted line and ‘T’ (4× and 20× magnification, scale bars = 200 µm, representative images shown). (f) At endpoint, relative lung tumor burden (RTB) was assessed by RT‐qPCR on day 21. (g) A proportion of mice were assessed for metastasis‐free survival (Kaplan–Meier). MΦ: macrophage. Metastasis assay: n = 8–10 mice/group. Survival experiments: n = 10 mice/group; IHC: n = 3 mice/group; and flow cytometry: n = 7 mice/group. Experiments were independently repeated at least twice, and representative data are shown. Statistical analysis was performed by (a, b) one‐way RM ANOVA with post hoc Bonferroni's multiple comparison test; (d, f) the Student's two‐tailed t‐test; and (g) the Mantel–Cox log‐rank test. Error bars are SEM. *P < 0.05; **P < 0.01; and ****P < 0.0001.
4T1.2 IMFP tumors were treated with 3M‐052 or vehicle when palpable (Figure 1b, day 5). Single 3M‐052 administration delayed primary tumor growth and reduced tumor volume and weight at endpoint (Figure 1b, Supplementary figure 2c). Analysis of circulating immune cells three days post‐treatment revealed an increase in activation of natural killer (NK) cells, yet no change in CD4+ and CD8+ T‐cell activation (Supplementary figure 2d–f). To mimic a neoadjuvant treatment setting and allow assessment of primary tumor infiltrates at a single time point, we repeated the same treatment regime yet resected primary tumors on day 14, after which differences in tumor morphology and intratumoral immune infiltrates were assessed (Supplementary figure 2g and h). 3M‐052‐treated tumors showed a visible decrease in tumor cell content and enhanced areas of cell death, with an associated increase in cleaved‐caspase‐3 (c‐caspase‐3) expression and decreased Ki67 expression (Figure 1c). 3M‐052 treatment enhanced the infiltration of NK, B and T cells – including CD8+ T cells – whilst changes in Ly6G+ myeloid cell infiltrates were not observed (Figure 1d). As expected from enhanced IFN signalling [given PD‐L1 and PD1 are documented IFN‐stimulated genes (ISGs) 26 , 34 , 35 ], elevated PD1 was observed in the treatment group (Figure 1c, Supplementary figure 2i and j). Importantly, the impact of 3M‐052 intratumoral administration was not limited to the primary tumor. Upon histological assessment of lungs from all mice at the time of first sign of metastasis in the experiment, visible macrometastases were only evident in the vehicle‐treated group and were undetectable by histology in the 3M‐052 treatment group (Figure 1e). This reduction in metastatic burden was confirmed by real‐time (RT)‐qPCR detection of 4T1.2 cells (mCherry+) in lung (Figure 1f), confirming the extent of metastatic suppression from only a single intratumoral dose of this agent. Assessment of metastasis‐free survival post‐treatment also confirmed that 3M‐052 suppressed metastatic progression and burden in an independent experiment (Figure 1g).
3M‐052 is a superior single agent compared with anti‐PD1 in an aggressive model of TNBC
The enhanced T‐cell infiltrate and PD1 expression (Figure 1d, Supplementary figure 2i and j) post‐3M‐052 treatment, along with our previous studies on synergy between systemic TLR agonists and anti‐PD1, 26 prompted combination immunotherapy trials in the 4T1.2 model. 3M‐052 has previously shown enhanced benefit in combination with checkpoint inhibition in a preclinical model of melanoma 33 ; however, this has not been assessed for TNBC. To mimic a neoadjuvant treatment setting where immunotherapies have shown most promise in TNBC mouse models, 26 we treated 4T1.2 tumor‐bearing mice with i.t 3M‐052 and systemic anti‐PD1 therapy prior to primary tumor resection on day 14 (Figure 2a). Treatment with 3M‐052 alone and in combination with anti‐PD1 reduced primary tumor size at the time of resection (Figure 2b), and this was supported by reduced expression of the proliferative marker Ki67 and elevated c‐caspase‐3 expression (Figure 2c). As previously reported in this model, 26 single‐agent anti‐PD1 did not alter primary tumor growth or proliferation (Figure 2b and c). We next evaluated the impact of combination versus single‐agent therapy on metastatic spread post‐primary tumor resection. Upon in vivo bioluminescent imaging to detect luc2‐labelled cells, we observed a clear reduction in signal in the thoracic cavity and a confirmed absence in bioluminescence in lungs ex vivo upon 3M‐052 treatment (Figure 2d). The antimetastatic impact of 3M‐052 as a single agent was confirmed in separate metastasis‐free survival studies, where 3M‐052 treatment significantly extended metastasis‐free survival independent of administration of anti‐PD1 mAb treatment (Figure 2e).
Figure 2.

Combination of 3M‐052 with PD‐1 blockade decreases lung metastasis. (a) Experimental intervention timeline. BALB/c mice were injected with 1 × 105 4T1.2 cells into the 4th mammary fat pad, and palpable tumors were i.t. injected with vehicle or 3M‐052 on day 7 post‐inoculation. Mice were treated with four doses of isotype or anti‐PD1 mAb in a neoadjuvant setting before primary tumor resection on day 14. Primary tumors were assessed for (b) weight and (c) were assessed by IHC for Ki67, c‐caspase‐3 and PD1 expression (40× magnification, scale bars = 50 µm, representative images shown). (d) Day 21 bioluminescence imaging of the thoracic cavity and lungs. (e) Kaplan–Meier survival curve comparing metastasis‐free survival. Survival assay: n = 10 mice/group; and IHC: n = 3 mice/group. Experiments were independently repeated twice, and representative data are shown. Statistical analysis was performed by (b) one‐way ANOVA with post hoc Tukey's multiple comparison test and (e) the Mantel–Cox log‐rank test. Error bars are SEM. **P < 0.01; ***P < 0.001; and ****P < 0.0001.
To better understand the antimetastatic mechanism of intratumorally delivered 3M‐052, we investigated the proportions and phenotype of primary tumor cell types. 3M‐052 treatment completely altered the immune composition of the tumor, inducing an immune active TME. Not only did a single i.t injection of 3M‐052 increase the number of TILs and decrease the proportion of isolated tumor cells (Supplementary figure 3a), the balance of immune suppressor and effector cells was skewed to an immune‐reactive state. 3M‐052 treatment decreased immune‐suppressive M2 macrophage numbers, increased cytotoxic CD8+ T‐cell numbers and decreased the proportion FoxP3+ regulatory CD4+ cells (Tregs) in the CD4+ T‐cell pool, leading to an elevated CD8+ T‐cell:Treg ratio (Figure 3a–c, Supplementary figure 3b and c). No difference was observed in Ly6G+CD11b+ cell numbers (Supplementary figure 3d). DCs and macrophages isolated from 3M‐052‐treated tumors demonstrated increased MHC‐I and MHC‐II expression (Figure 3d–f). Furthermore, tumor cells isolated from 3M‐052‐treated tumors had increased expression of MHC‐I, the peptide‐containing ligand recognised by CD8+ T cells for tumor cell lysis (Figure 3g). Importantly, this impact on both immune activation and MHC‐I expression was only observed upon treatment with 3M‐052, with anti‐PD1 having no additional impact on immune reactivity. Crucially, 3M‐052 treatment led to enhanced activation (CD69+) and functionality of T cells, with more CD8+ T cells producing IFN‐γ and TNF‐α when re‐stimulated with 4T1.2 cells ex vivo (Figure 3h–j, Supplementary figure 3e). The impact of this single dose of 3M‐052 on the TME is highlighted by the decreased ratio of tumor:CD8+ T cells (Figure 3k). This increase in CD8+ T‐cell functionality was further observed systemically in peripheral blood in combination‐treated mice (Figure 3l, Supplementary figure 3f and g).
Figure 3.

Combination 3M‐052/PD‐1 blockade increases primary tumor immunogenicity and adaptive immune responses. BALB/c mice were injected with 1 × 105 4T1.2 cells into the 4th mammary fat pad, and palpable tumors were i.t. injected with vehicle or 3M‐052 on day 7 post‐inoculation. Mice were treated with four doses of isotype or anti‐PD1 mAb in a neoadjuvant setting before primary tumor resection on day 14. Primary tumors were assessed for (a) number of CD206+ F4/80+ cells; (b) number of TCRβ+CD8+ cells; (c) ratio of TCRβ+CD8+: FoxP3+CD25+ TCRβ+CD4+ cells; (d) MHC‐I (H2‐Kd) and (e) MHC‐II expression of F4/80+ cells; MHC‐I expression of (f) CD11b+ DCs and (g) isolated tumor cells (mCherry+); number of (h) CD69+ TCRβ+CD4+ cells, (i) CD69+ TCRβ+CD8+ cells and (j) IFN‐γ+TCRβ+CD8+ cells; and (k) ratio of tumor: TCRβ+CD8+ cells. (l) TCRβ+CD8+ cells from day 18 peripheral blood (PB) was assessed for specificity against 4T1.2 cells denoted by IFN‐γ production. Experiments were independently repeated twice, and representative data are shown. MFI: mean fluorescence intensity. All experiments n = 7 mice/group except (c, k) n = 8 or 9 mice/group, (l) n = 10 mice/group. Statistical analysis was performed by one‐way ANOVA with post hoc Tukey's multiple comparison test. Error bars are SEM. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
CD8+ T‐cell depletion blocks the metastasis‐specific actions of 3M‐052
To determine whether the primary tumor and metastatic impact of 3M‐052 were reliant on an immune response, we sought to investigate the impact of 3M‐052 treatment in 4T1.2 tumor‐bearing, immunodeficient NOD‐scid IL2Rγnull (NSG) mice. Here, mice lacking functional B, T and NK cells were inoculated IMFP with 4T1.2 cells and administered intratumoral 3M‐052 upon tumor palpation followed by measurement of primary tumor weight at resection and metastasis‐free analysis (Figure 4a). Despite lacking an adaptive immune system, 3M‐052 treatment in NSG mice reduced primary tumor weight compared with vehicle treatment (Figure 4b, Supplementary figure 4a), comparable to that observed in immunocompetent BALB/c mice (Figure 2b) indicating the contribution of an immune‐independent mechanism in suppressing primary tumor growth. These results were confirmed in the absence of an immune response in vitro, where higher concentrations of 3M‐052 were capable of decreasing 4T1.2 or E0771 viability (Supplementary figure 4b). When the cellularity of primary tumors was assessed, 3M‐052 treatment suppressed CD206+F4/80+ M2 macrophage infiltration and increased macrophage, DC subset and tumor MHC‐I expression, as seen previously in immune‐competent mice (Figure 4c and d). When overall survival was assessed, despite alterations in primary tumor size, no difference in lung metastatic burden was observed between groups as indicated by metastasis‐free survival time and histology (Figure 4e and f), supporting a critical function of the immune system in 3M‐052‐induced metastasis suppression.
Figure 4.
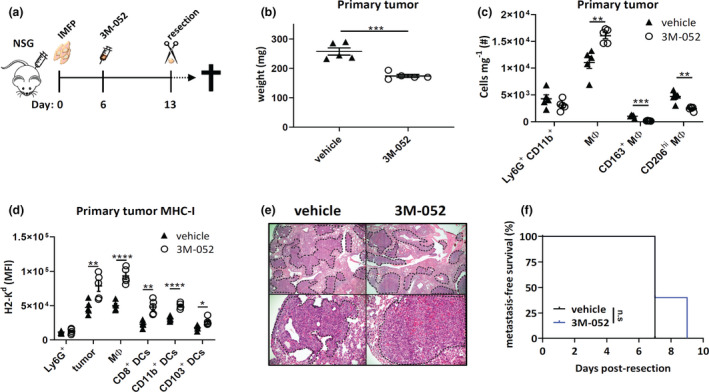
3M‐052 antimetastatic effects require intact immunity. (a) NSG mice were injected with 1 × 105 4T1.2 cells into the 4th mammary fat pad, and palpable tumors were i.t. injected with vehicle or 3M‐052 on day 6 post‐inoculation. Primary tumors were resected on day 13 and were assessed for (b) weight (mg); (c) number of CD11b+Ly6G+ cells and macrophage phenotype; and (d) MHC‐I (H2‐Kd) expression of various cell types by flow cytometry. (e) Lungs were taken at endpoint for H&E staining to assess metastatic burden; tumor regions are denoted by dotted line and ‘T’ (4× and 20× magnification, scale bars = 200 µm, representative images shown). (f) Kaplan–Meier survival curve comparing metastasis‐free survival. MΦ: macrophage. MFI: mean fluorescence intensity. Data representative of one experiment. All data n = 5 mice/group except (e) n = 3 mice/group. Statistical analysis was performed by (b–d) the Student's two‐tailed t‐test and (f) the Mantel–Cox log‐rank test. Error bars are SEM. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
CD8+ T cells are implicated in control of primary tumor progression and metastasis given their ability to persist for extended periods in tissues and their rapid cytolytic response to antigenic recognition. 36 The increase in CD8+ T‐cell number and functional response in tumors treated with 3M‐052 (Figure 3j) prompted study of the functional role of these cells in metastasis suppression. To test this, anti‐CD8 mAb was incorporated in a therapeutic regime (Figure 5a) similar to previous experiments, and CD8+ T‐cell depletion was confirmed in peripheral blood prior to 3M‐052 treatment (Figure 5b). At resection, 3M‐052 treatment decreased primary tumor weight compared with control treatment despite CD8+ T‐cell depletion (Figure 5c). As expected, CD8‐depletion abolished primary tumor IFN‐γ+ CD8+ T cells (Figure 5d), yet did not impact MDSC, Treg and M2 macrophage numbers, as well as MHC‐I and PD‐L1 expression in various DC and macrophage subsets post‐3M‐052 treatment (Figure 5e–h, Supplementary figure 5a–c). In line with NSG experiments, we detected a metastasis‐specific impact of CD8+ T‐cell depletion in response to 3M‐052. Whilst CD8‐depleted 3M‐052‐treated mice had prolonged metastasis‐free survival compared with vehicle‐treated groups, intact CD8+ T cells were required for sustained metastasis‐free survival (Figure 5i) and reduced lung macrometastases at experimental endpoint (Figure 5j), suggesting that CD8+ T cells are important for metastatic control in the periphery. This impact of CD8+ T cells on metastasis suppression was further supported by the presence of enhanced numbers of tumor‐specific CD103+CD8+ TRM present in the lungs of 3M‐052‐treated mice (Figure 5k).
Figure 5.

CD8+ T‐cell depletion does not impact 3M‐052 antiprimary tumor effects but attenuates enhanced long‐term survival. (a) Experimental intervention timeline. BALB/c mice were injected with 1 × 105 4T1.2 cells into the 4th mammary fat pad, and palpable tumors were i.t. injected with vehicle or 3M‐052 on day 7 post‐inoculation. Mice were treated with five doses of isotype or anti‐CD8 mAb, and primary tumors were resected on day 13. (b) Day 5 flow cytometry analysis of peripheral blood (PB) TCRβ+CD8+ cell proportions. Primary tumors were assessed for (c) weight (mg); (d) IFN‐γ+TCRβ+CD8+ T cells; (e) CD206+F4/80+ macrophages; and (f) CD11b+ and (g) CD103+ DC MHC‐I and (h) F4/80+ macrophage MHC‐I (H2‐Kd) expression. (i) Kaplan–Meier survival curve comparing metastasis‐free survival. (j) A proportion of lungs were taken at endpoint for H&E staining to assess metastatic burden; tumor regions are denoted by dotted line and ‘T’ (4× and 20× magnification, scale bars = 200 µm, representative images shown). (k) Lungs were taken at endpoint and assessed for tumor‐specific CD103+CD8+ T cells in an ICS for IFN‐γ by flow cytometry. MΦ: macrophage. MFI: mean fluorescence intensity. Data are representative of one experiment. (b, c, i) n = 8 mice/group; (d–h, k) n = 6 mice/group; and (j) n = 2 mice/group. Statistical analysis was performed by (b) the Student's two‐tailed t‐test; (c–h, k) one‐way ANOVA with post hoc Tukey's multiple comparison test; and (i) the Mantel–Cox log‐rank test. Error bars are SEM. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
3M‐052 antitumor action indirectly through DCs
Next, we sought to investigate the cells that were direct targets of 3M‐052, responsible for both tumor and immune cell alterations. It was possible that 4T1.2 cells could directly uptake the oil‐based compound actively through phagocytosis or diffusion of the TLR7/8 agonist across the cell membrane, leading to stimulation of downstream ISGs, including MHC‐I. Alternatively, another cell type such as well‐known phagocytic IFN‐producing macrophages or DCs could engulf the compound, with the resulting IFN produced acting directly on tumor cells. To investigate the first possibility, 4T1.2 cells were stimulated in vitro with various IFN inducers, including soluble 3M‐052. Interestingly, following 48 h, MHC‐I cell surface expression remained unchanged in response to 3M‐052 and another TLR7/8 agonist, in contrast to direct induction by recombinant IFN‐α and the TLR3 agonist poly I:C (Figure 6a). An explanation for this is the lack of Tlr7 and Tlr 8 transcript in 4T1.2 cells by RT‐qPCR, compared with the high levels of transcript present in bone marrow‐derived DCs (BMDCs) (Supplementary figure 6a and b).
Figure 6.

3M‐052 acts indirectly on tumor cells through cytokine production from DCs. (a) 4T1.2 cells were treated with various IFN agonists for 48 h and assessed for MHC‐I (H2‐Kd) expression. Poly I:C‐transfected cells were compared with Lipofectamine‐treated control cells; (b) BMDCs and (c) BM macrophages were stimulated for 24 h with various IFN inducers and assessed for MHC‐I (H2‐Kd) expression by flow cytometry and production of (d) IFN‐β by enzyme‐linked immunosorbent assay and (e) IL‐6, (f) IL‐1β and (g) TNF‐α by cytometric bead array (CBA). Conditioned medium (s/n) from 3M‐052‐treated (h) 4T1.2, (i) BMDCs and (j) BM macrophages was transferred onto fresh 4T1.2 cells ± IFNAR1 blocking mAb, MAR1‐5A3. Following 48 h, recipient 4T1.2 MHC‐I (H2‐Kd) expression was assessed by flow cytometry. BM MΦ: bone marrow‐derived macrophages. MFI: mean fluorescence intensity. n = 3–6 biological replicates/group. Experiments were independently repeated twice, and representative data are shown. All data n = 3 replicates/group except (d) n = 6 replicates/group. Statistical analysis was performed by one‐way ANOVA with post hoc Tukey's multiple comparison test. Error bars are SEM. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
Given that we had observed an increase in tumor cell, DC and macrophage MHC‐I expression in vivo (Figure 3), we next tested the direct impact of 3M‐052 on BMDCs and bone marrow‐derived macrophages (BM macrophages), both are well‐known surveyors of the extracellular environment for immune monitoring purposes. To explore this, BM macrophages and BMDCs were stimulated in vitro with soluble 3M‐052. Following 48 h of 3M‐052, MHC‐I expression was exclusively enhanced in BMDCs and not in BM macrophages (Figure 6b and c). We next confirmed that 3M‐052 BMDC stimulation resulted in IFN‐β production (Figure 6d) that could impact 4T1.2 cell immunogenicity. We further assessed the cytokine output of stimulated 4T1.2, BMDC and BM macrophages and found that 3M‐052‐stimulated BMDCs produced large amounts of MIP‐1α, IL‐6, IL‐1β, RANTES, MCP‐1 and TNF‐α (Figure 6e–g, Supplementary figure 6c–j). As previously observed, 3M‐052 had no effect on 4T1.2 and BM macrophage cytokine output. To determine whether this cytokine production was sufficient to increase immune visibility in 4T1.2 cells, cultured supernatant from 3M‐052‐stimulated 4T1.2, BMDCs and BM macrophages was transferred onto fresh 4T1.2 cells for a subsequent 48 h. We detected an increase in tumor cell MHC‐I expression from the BMDC‐stimulated supernatant specifically, and this was observed with use of both the soluble and oil‐based forms of 3M‐052 (Figure 6i). Importantly, we were able to deduce this increase occurred via DC type I IFN production as this effect was inhibited with the addition of the IFN‐α receptor (IFNAR1) blocking antibody, MAR1‐5A3 (Figure 6h and i, Supplementary figure 6k).
Type I IFN signalling is critical for 3M‐052‐induced tumor and metastasis suppression
Our results indicate that 3M‐052 treatment results in IFN‐β production by DCs, which is capable of increasing immune visibility in vitro (Figure 6). Furthermore, we observed increased functional CD8+ T‐cell responses and increased MHC‐I on tumor cells and various antigen‐presenting cells ex vivo following 3M‐052 i.t delivery. From this, we hypothesised that 3M‐052 treatment was inducing an IFN‐driven increase in CD8+ T‐cell priming and tumor cell visibility culminating in the stimulation of metastatic immunosurveillance and prolonged metastasis‐free survival, and that blocking type I IFN recognition would ameliorate this effect. To investigate this, we performed the standard 4T1.2 neoadjuvant experimental setup, yet treated mice with the IFNAR1 blocking antibody (MAR1‐5A3) and assessed the primary tumor and metastatic impact of blocking the type I IFN response (Figure 7a). Our results indicated that the antitumor effect of 3M‐052 was IFN‐dependent, with MAR1‐5A3 treatment blocking the reduction in tumor size at the time of resection observed in the control antibody group (Figure 7b). This was also evident by an absence of the antiproliferative and pro‐apoptotic primary tumor impact of 3M‐052 (Figure 7c). Furthermore, MAR1‐5A3 treatment blocked 3M‐052‐induced NK activation and maturation in the TME (Figure 7d and e, Supplementary figure 7a) and increased the number of M2 macrophages (Figure 7f). Tumor cell MHC‐I expression was also reduced upon addition of MAR1‐5A3, as was expression on various DC and macrophage subsets, confirming reliance of IFN on 3M‐052‐induced MHC‐I elevation (Figure 7g–j). MAR1‐5A3 treatment attenuated the 3M‐052‐induced increase in tumor‐specific CD8+ T cells within the primary tumor (Figure 7k). The quality of T cells in the TME was also reduced, with higher proportions of B220+ T cells in 3M‐052‐treated mice that were administered the MAR1‐5A3 (Supplementary figure 7b), a phenotype consistent with dying, dysfunctional T cells. 37 The reduction in tumor immunogenicity and immune‐activated infiltrate by treatment with MAR1‐5A3 correlated with a complete removal of the antimetastatic effect of 3M‐052, as evident by increased bioluminescent detection of thoracic tumors at a single time point (Figure 7l) and a complete lack of metastasis‐free survival benefit compared with mice that received the vehicle control (Figure 7m). Together, this demonstrates that type I IFN is required for the tumor‐suppressive and antimetastatic effects of 3M‐052.
Figure 7.
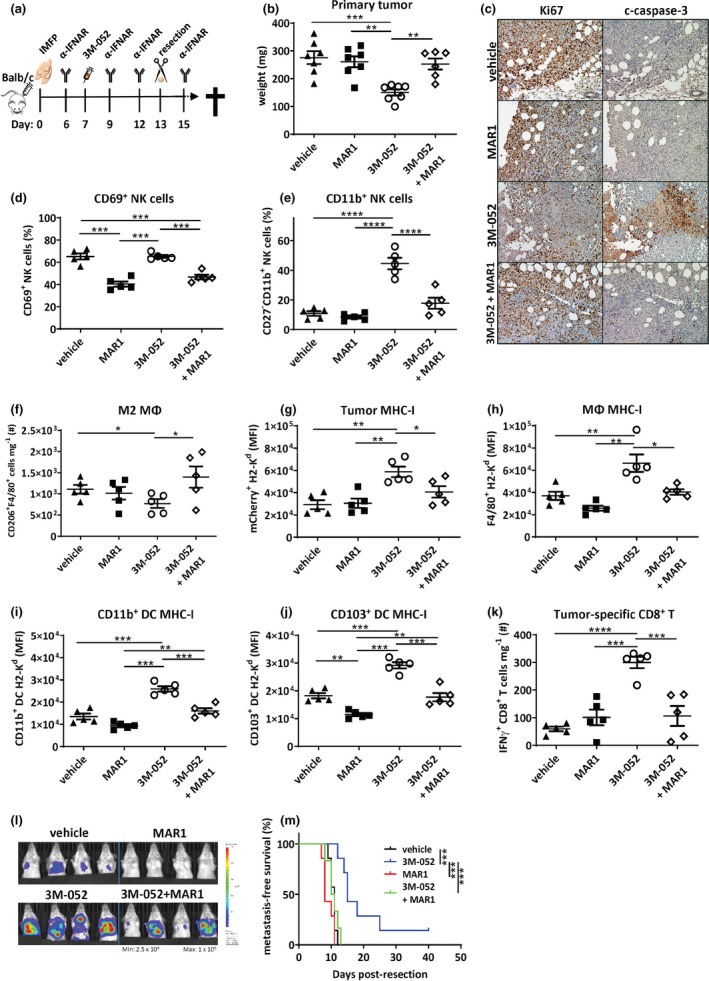
Type I IFN signalling critical for 3M‐052 primary tumor and long‐term survival effects. (a) Experimental intervention timeline. BALB/c mice were injected with 1 × 105 4T1.2 cells into the 4th mammary fat pad, and palpable tumors were i.t. injected with vehicle or 3M‐052 on day 7 post‐inoculation. Mice were treated with four doses of isotype or MAR1‐5A3 mAb and primary tumors resected on day 13. Resected primary tumors were assessed for (b) weight (mg); (c) Ki67 and c‐caspase‐3 staining of formalin‐fixed primary tumors (20× magnification, scale bars = 100 µm, representative images shown); (d) proportion of CD69+TCRβ−NKp46+ cells; (e) proportion of CD11b+TCRβ−NKp46+ cells; (f) number of CD206+F4/80+ macrophages; and (g) tumor cell, (h) F4/80+ macrophage, (i) CD11b+CD11c+ and (j) CD103+CD11c+ DC MHC‐I (H2‐Kd) expression; and (k) number of IFN‐γ+TCRβ+CD8+ T cells. (l) Day 20 bioluminescence imaging of the thoracic cavity and (m) Kaplan–Meier survival curve comparing metastasis‐free survival. MΦ: macrophage. MFI: mean fluorescence intensity. Data are representative of one experiment. (b, l, m) n = 7 mice/group; (c) n = 2 mice/group; and (d–k) n = 5 mice/group. Statistical analysis was performed by one‐way ANOVA with post hoc Tukey's multiple comparison test and (m) the Mantel–Cox log‐rank test. Error bars are SEM. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
Type I IFN enhances immunogenicity of human TNBC cell lines
We next investigated the responsiveness of human breast cancer cell lines to 3M‐052 alone or IFN‐α, to assess whether either agent was capable of increasing immune visibility following treatment, as we had observed in mouse cell lines. Analysis of cell surface expression in ER+ (MCF7), TNBC (CAL‐120, MDA‐MB‐231, MDA‐MB‐468) and Her2+ (MDA‐MB‐453) cell lines post‐3M‐052 or direct IFN‐α treatment revealed that all cell lines were receptive to increases in HLA following IFN‐α treatment and the MDA‐MB‐231 TNBC cell line also had a modest increase following 3M‐052 stimulation (Figure 8a). Our results revealed that the CAL‐120 and MDA‐MB‐231 TNBC cell lines increased surface expression of IFN‐responsive gene, PD‐L1, following IFN‐α treatment (Figure 8b). The direct impact of 3M‐052 on MDA‐MB‐231 cells was explained by analysis of TLR expression in the cell panel, with expression of TLR7 only apparent in these cells (Figure 8c and d). Together, these data suggest that enhanced IFN production in the TME could be sufficient to increase the immunogenicity of human BC lines and that MHC‐I expression in TNBC can be rescued via stimulation‐enhanced IFN in the TME.
Figure 8.

Human TNBC cell lines enhance immunogenicity in response to type I IFN or 3M‐052. MCF7, CAL‐120, MDA‐MB‐231, MDA‐MB‐453 and MDA‐MB‐468 cells were stimulated with either DMSO, 3M‐052 or IFN‐α for 48 h, and expression of (a) HLA‐ABC and (b) PD‐L1 was assessed by flow cytometry. (c) TLR7 and (d) TLR8 expression of various human TNBC cell lines and THP1. MFI: mean fluorescence intensity. Experiments were independently repeated twice, and representative data are shown. (a, b) n = 4 biological replicates/group and (c, d) n = 3 biological replicates/group. Data are one representative of two independent experiments. Statistical analysis was performed by one‐way ANOVA with post hoc Tukey's multiple comparison test. Error bars are SEM. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
Discussion
This study presents evidence that a single intratumoral injection of the TLR7/8 agonist 3M‐052 can both shrink orthotopic tumor size and decrease metastatic burden in models of TNBC. Our results demonstrate that 3M‐052 administration completely alters the immune TME in TNBC, transforming IFN‐low and immune‐suppressed tumors into an immune‐reactive, T‐cell‐inflamed microenvironment. This was demonstrated by widespread alterations in tumor cell immunogenicity, immune cell activation and production of immune‐activating cytokines that culminate into the induction of an adaptive immune response. For the first time, we demonstrate an immune‐dependent impact of this agonist on spontaneous metastasis from the orthotopic site. Further, tumor‐specific TRM cells were identified in lungs of 3M‐052‐treated mice well after primary tumor treatment and were crucial for protection against systemic disease re‐emergence.
Our study represents a valid neoadjuvant therapeutic strategy for TNBC aimed at stimulating intratumoral IFN signalling early to promote an immune‐reactive TME and antitumor memory. Although type I IFNs are now widely implicated in antitumor immunity, the use of recombinant IFNs for treatment of solid malignancies has been somewhat hampered by associated toxicities, highlighting the need for alternate approaches to stimulate IFN signalling at the tumor site in order to limit systemic side effects and enhance the direct impact on the TME. 16 The TLR7/8 agonist 3M‐052 has been formulated for retention at the tumor site, as recently confirmed in subcutaneous models using multiple injections. 33 Our data demonstrate that 3M‐052 delivery stimulates the production of a wide range of pro‐inflammatory cytokines including IL‐6, IL‐1β, TNF‐α and IFN‐β from murine DCs, which were shown to express both TLR7 and TLR8 receptors. IL‐6, IL‐1β, RANTES and TNF‐α aid in the attraction of key effector cell types such as NK and T cells, which can work synergistically with IL‐12/IFN‐β and IL‐1β to increase DC co‐stimulation integral for T‐cell priming of CD8+ and CD4+ T cells, respectively. 38 , 39 , 40 , 41 , 42 These results further expand and give insight into the findings of Mullins et al. 33 where they demonstrated increased type I IFN and effector gene signatures in B16‐F10 tumors treated with 3M‐052 (MEDI9197). Furthermore, type I IFNs have been shown to be required for effective priming of T‐cell populations. 17 , 43 Enhanced T‐cell priming following 3M‐052 treatment likely resulted in the increased T‐cell number advantage over tumor and immune‐suppressive cell types, thus facilitating more effective elimination of metastases. Moreover, our data demonstrate increased numbers of TRM within both 3M‐052 primary tumors and lung that led to increased metastasis‐free survival, correlating with increased TRM presence in TNBC patients who have better outcomes. 7
The current study strengthens previous evidence highlighting the need for increased immunogenicity within the TME for effective therapeutic benefit, with many IFN inducers, oncolytic viruses and STING agonists currently being assessed. 11 Given the altered delivery mechanisms, including systemic versus intratumoral treatment, it will be critical to compare the metastatic versus primary tumor antiproliferative and immune‐activating functions. Our study has revealed that immune activity within the primary tumor was crucial for the antimetastatic action. Assessing these immune cell alterations in primary tumors post‐therapy is likely essential to predicting the metastatic response to agents and supports further investigation comparing systemic and intratumoral TLR agonist approaches for enhancing antimetastatic impact. Previously used therapeutics, such as poly A:U that has shown promise in an adjuvant setting for breast cancer (albeit with mild toxicities), may be of further benefit if given by intratumoral administration. Our previous work using multiple injections of the TLR agonist poly I:C and that of Mullins et al. using 3M‐052 in B16 melanoma reported combined benefit of IFN induction and anti‐PD1 therapy. 26 , 33 In the current study, single‐dose 3M‐052 administration was effective as a single agent and a further survival benefit was not observed with anti‐PD1 checkpoint inhibition therapy. Given that sustained IFN signalling induces secondary immune suppression, including enhanced expression of the IRGs PD1 and PD‐L1, comparisons of dosing and site of delivery need to be investigated in the future to determine the potential of combination therapies.
Our work supports DCs as a relevant target of this TLR7/8 agonist. DCs, particularly plasmacytoid DCs (pDCs), are well‐known producers of type I IFNs. Our DC stimulation studies utilised BMDCs that were generated using GM‐CSF and were highly biased towards a CD11b+ conventional DC cell type and demonstrated production of IFN‐β following activation by 3M‐052. Although not investigated in the current study, human pDCs and myeloid DCs express TLR7 and TLR8, respectively, and, as such, would also have the capacity to further boost local type I IFN production upon exposure to 3M‐052 33 in a similar manner to that described from studies utilising R848. 44 , 45 , 46 The lack of a direct induction of tumor cell MHC‐I expression by 3M‐052 is not surprising as our data demonstrate that 4T1.2 cells do not basally express either TLR7 or TLR8. Although previous reports indicate that murine macrophages express TLR8, the specific class of drug (imidazoquinolines) is only recognised by human TLR8. 47 , 48 As such, administration into patients may further promote pro‐inflammatory cytokine secretion from patient macrophages to promote a TH1 bias, further supporting an immune‐reactive TME. Indeed in our study, 3M‐052 did not promote production of IL‐4 cytokine, which is known to be implicated in M1 → M2 switching, an encouraging finding given its documented immunosuppressive role. 49 , 50 Our work demonstrates that 3M‐052 is a potent immunostimulatory compound in a neoadjuvant setting. TLR7/8 agonists, such as R848, have also been utilised in a post‐resection setting, demonstrating that local delivery of the drug has the capacity to limit Treg accumulation and control metastasis in a similar but slightly less aggressive murine TNBC model. 31
The lynch‐pin of both NK and CD8+ T‐cell function is the recognition of MHC‐I complexes on the cell surface of potential target cells. 51 , 52 Not surprisingly, many cancers mutate in a fashion to find a grey area of MHC‐I expression to avoid being killed by both cell types. 53 Interestingly, many components of the antigen processing and presentation pathway are ISGs. 54 A major consequence of decreased type I IFN signalling is the subsequent decrease in MHC‐I presentation, effectively leaving CD8+ T cells ‘blind’ to unrecognisable tumor cells. 53 The 3M‐052‐induced activation and maturation of NK cells observed in our studies, along with the incomplete suppression of therapeutic effect upon CD8+ T‐cell depletion, suggests future work into the alterations and critical functions of NK cells in the antimetastatic response is important. Our results indicate that 3M‐052 treatment was capable of increasing MHC‐I levels in multiple cell types, including DCs, macrophages and, importantly, tumor cells ex vivo. Our results demonstrating that secreted factors from 3M‐052‐treated DCs upregulate this pathway locally in 4T1.2 cells suggest that this pathway is basally suppressed, not mutated, giving confidence that treatments like the one described in this study have the potential to eliminate breast cancer in the clinic. This theory is further solidified given that all human breast cancer cell lines tested were capable of increasing HLA expression following IFN‐α stimulation, which warrants further exploration in patient‐derived tumors in future studies. Our results using the IFNAR blocking antibody, MAR1‐5A3, also confirm that of our previous studies, that type I IFN signalling is critical in the control of TNBC. 9 These results, along with NSG immune‐deficient mouse survival experiments, highlight the critical interplay between type I IFN signalling and the adaptive immune response in the control of metastasis in TNBC.
A critical component of use of IFN inducers in TNBC is appropriate patient selection, along with individualised combination treatment strategies. This is important given hyperactivation of the type I IFN pathway in a subset of breast cancers is associated with a poor response to conventional therapies, 55 , 56 , 57 contradictory to our findings and those of others in TNBC. 8 , 10 This phenomenon is likely because of unresponsive pathways from chronic IFN stimulation and subsequent initiation of a suppressive TME, and has led to the development of a tumor IFN‐related DNA damage resistance signature (IRDS+) to predict therapeutic resistance. 55 We believe IFN‐based immune‐activating therapies such as 3M‐052 would be most beneficial for patients harbouring tumors with hypoactivation of the type I IFN pathway, those that are immune cold and those that are phenotypically distinct from those described above. Encouragingly, in this study 3M‐052 treatment did not initiate an immunosuppressive TME, nor did it induce increased IL‐10 production, as previously described for IRDS+ tumors. However, such studies highlight that monitoring cytokine alterations during and post‐3M‐052 will be an important consideration in future trials. Timing of treatment will also be of importance. Our previous research, along with that of others, suggests the use of immune‐activating therapies in a neoadjuvant setting, 26 , 58 where the tumor load and hence antigen exposure are sufficient to drive effective immune priming and memory. 26 We recently highlighted an intratumoral IFN marker that predicts long‐term chemotherapeutic benefit in a neoadjuvant trial, the loss of which indicated a cold tumor that was associated with low TRM signatures, poor prognosis 8 and patients who would likely benefit from immune agents that bolster immune reactivity in the tumor. Future trials are required to determine the efficacy of TLR agonists in combination with chemotherapy and other immunotherapies for TNBC.
In summary, 3M‐052 intratumoral administration is capable of induction of tumor‐inherent type I IFN signalling and generation of a long‐lived, adaptive immune response that is capable of suppressing distant metastasis. Therapies of this nature may be equally relevant in re‐igniting an immune response in other cancers with cold TMEs that are associated with a lack of response to conventional and checkpoint‐based therapies and a poor prognosis. The use of neoadjuvant immunotherapeutics that enhance tumor immunogenicity and immune effector cell accumulation may be a viable option for reducing metastatic progression in patients with TNBC, particularly those who do not benefit from neoadjuvant chemotherapy.
Methods
Mice and models
C57BL6/J and BALB/c mice were obtained from the Walter and Eliza Hall Institute of Medical Research (Melbourne, VIC, Australia). NOD‐scid IL2Rγnull (NSG) mice were sourced from Animal Resource Centre (Perth, WA, Australia). Mice were used between the ages of 8 and 12 weeks. All experiments were approved by the La Trobe Animal Experimentation Ethics Committee or the Peter MacCallum Cancer Centre Animal Experimentation Ethics Committee.
The weakly metastatic E0771‐mCherry‐LMB (E0771) and the highly metastatic 4T1.2 subclone of the 4T1 TNBC line were derived in and sourced from Prof. Robin Anderson's laboratory. 59 , 60 The 4T1.2 line was engineered to express mCherry and luc2. 26 For non‐resection experiments, mice were euthanised when primary tumor size reached 1500 mm3. For metastasis‐free survival experiments, mice were euthanised individually upon paralysis or signs of metastatic distress. Mice excluded because of primary tumor regrowth are indicated as censored points on the Kaplan–Meier curves. For metastasis assays, all mice were euthanised at the same time point.
E0771, CAL‐120 and MDA‐MB‐231 cell lines were cultured in DMEM (Sigma‐Aldrich Japan, Tokyo, Japan) supplemented with 10% FBS. THP‐1, MDA‐MB‐453 and MCF7 cell lines were cultured in RPMI 1640 (Sigma‐Aldrich Japan) supplemented with 10% FBS. The MDA‐MB‐468 cell line was cultured in RPMI 1640 supplemented with 10% FBS and 5 μg mL−1 insulin (Sigma‐Aldrich Japan). 4T1.2 cells were cultured in α‐MEM (Sigma‐Aldrich Japan) supplemented with 5% FBS. All cell lines were maintained at 37°C, 5% CO2. All cell lines were verified Mycoplasma‐negative from regular assessment by the Victorian Infectious Disease References Laboratory (Melbourne, VIC, Australia).
Quantification of metastatic burden
Real‐time qPCR was used to quantify metastatic burden by comparing the ratio of mCherry (present in tumor cells) to vimentin (Vim, NC_000068, present in all cells) sequences in genomic DNA preparations from homogenised and proteinase K (100 μg mL−1, Sigma‐Aldrich Japan)‐digested lungs, as previously described. 26 PCR primer sequences are listed in Supplementary table 1. Metastatic burden (arbitrary units, AU) was calculated on the quantification cycle (C q) for mCherry relative to Vim and displayed as relative tumor burden (RTB) using the following equation:
where ΔC q = C q (target gene) − C q (control).
Bone marrow‐derived dendritic cell and bone marrow‐derived macrophage (BM macrophage) culture
Bone marrow‐derived DCs were propagated for 8 days in RPMI 1640 supplemented with 10% FBS and 10% X‐63‐GM‐CSF (containing granulocyte‐macrophage colony‐stimulating factor, GM‐CSF) in methods previously described. 61 , 62 BM macrophages were propagated for 8 days in RPMI 1640 supplemented with 10% FBS and 10% L929 supernatant (containing M‐CSF), in methods previously described. 63 , 64
MTT cell viability assay
The 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyl tetrazolium bromide (MTT; Invitrogen, Carlsbad, CA, USA) assay was used to assess the ability of test agents to inhibit cell growth and/or evoke cytotoxicity and was performed following 72 h of exposure of cells to test agents, as previously described. 65
In vivo treatment and survival analysis
For in vivo experiments, 1 × 105 cells (4T1.2 or E0771) in PBS (20 µL) were injected into the 4th mammary fat pad (IMFP) on day 0. Tumor volume was measured thrice weekly with electronic callipers, using the equation (length × width2/2). Upon tumor palpation (~ day 5 as indicated in figure workflows), 20 µL of vehicle or 3M‐052 (0.8 mg mL−1) was administered intratumorally (i.t) using doses previously described. 66 In vivo metastatic tumor burden (bioluminescent intensity) was assessed by IVIS Lumina XR‐III (Caliper Life Sciences, Waltham, MA, USA) under inhaled isoflurane anaesthesia 10 min post‐intraperitoneal (i.p) injection of 3 mg ᴅ‐Luciferin (Gold Biotechnology, St Louis, MO, USA). At endpoint, lungs were imaged ex vivo. Blocking and depletion antibodies for PD‐1 (250 µg; anti‐PD1: clone RMP1‐14, isotype: clone 2A3), IFNAR (250 µg; anti‐IFNAR1: clone MAR1‐5A3) and CD8 (250 µg; anti‐CD8: clone 53‐6.7, isotype: clone 2A3) were injected via i.p injection in 200 µl on the days indicated in figure workflows. RMP1‐14, 2A3 and 53‐6.7 in vivo antibodies were from Bio X Cell (Lebanon, NH, USA).
For resection experiments, mammary tumors were surgically removed at day 13 (~ 250 mg) post‐tumor cell inoculation and weighed. Lung metastasis was confirmed by histology or RT‐qPCR. The Kaplan–Meier survival curves were generated using GraphPad Prism (San Diego, CA, USA) and compared via log‐rank (Mantel–Cox) test.
RNA expression analysis
RNA was extracted from cells using the TRIzol (Invitrogen) method 67 and reverse‐transcribed into cDNA using the iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA, USA). RT‐qPCR using SYBR technology was used to quantify transcript expression via CFX384 (Bio‐Rad). PCR primer sequences are listed in Supplementary table 1. Murine and human Tlr 7 (mTLR7, hTLR7) and Tlr 8 (mTLR8, hTLR8) were compared with housekeeping genes mHprt and hHprt. Gene expression (AU) was based on the quantification cycle (C q) for gene of interest relative to housekeeping genes (Hprt) and displayed as relative transcript abundance, using the equation as above for quantitating metastatic burden.
Flow cytometry
For analysis of individual cell populations, single‐cell suspensions of primary tumor or lung were prepared through mechanical and enzymatic digestion as previously described. 26 Samples were then stained with antibodies (listed in Supplementary table 2) and assessed by flow cytometry using either a Cytoflex (Beckman Coulter, Brea, CA, USA) or FACSARIA III (BD Biosciences, Franklin Lakes, NJ, USA), and data were analysed using FlowJo software (Tree star, Ashland, OR, USA), as previously described. 68 FSC and SSC gating was used to discriminate between live cells and debris/dead cells. Isolated tumor cells were identified as positive for mCherry expression and negative for lymphoid and myeloid cell markers. For Treg enumeration, FoxP3 expression was assessed using the mouse FoxP3 staining kit and FoxP3‐AF488 mAb as per the manufacturer's instructions (Becton Dickinson, Franklin Lakes, NJ, USA).
Gating strategies are shown in Supplementary figure 1.
Histology and immunohistochemistry
Tissues were fixed in 10% neutral buffered formalin (NBF) and embedded in paraffin. For morphological tissue analysis, haematoxylin and eosin (H&E) staining was performed. Immunohistochemistry (IHC) of specific proteins was performed as previously described. 26 Briefly, slides were dewaxed and antigen retrieval was performed in sodium citrate (pH 6) for 5 min at 110°C. Slides were blocked for 1 h at RT in 3% normal goat serum, with overnight primary antibody incubation at 4°C [Ki67: Ab15580 1 μg mL−1 (Abcam, Cambridge, UK); cleaved‐caspase 3: #9661 0.1038 μg mL−1 (Cell Signaling, Danvers, MA, USA)]. Slides were incubated with secondary antibody goat anti‐rabbit IgG (BA‐1000 1/250; Vector Laboratories, Burlingame, CA, USA) at RT for 1 h and washed, and endogenous peroxidase was quenched in 0.3% H2O2 for 30 min, washed and incubated in ABC reagent for 1 h before washing and DAB development. Primary and secondary antibodies are listed in Supplementary table 2.
Murine PD1 was visualised using the OPAL seven‐colour manual IHC kit (NEL811001KT, PerkinElmer, Waltham, MA, USA) as previously described. 8 Primary antibody (anti‐PD1, 2 μg mL−1; Proteintech, Rosemont, IL, USA) was incubated for 1 h at RT prior to addition of donkey anti‐rabbit HRP (AP182P; 1:2000, Chemicon, Boronia, VIC, Australia) and DAPI nuclear stain. Sections were imaged using the VECTRA 3.0 (PerkinElmer) at 10× magnification and analysed using inForm software (PerkinElmer).
Ex vivo intracellular cytokine staining assays
Single‐cell suspensions of primary tumor and organ samples were generated as described above. Intracellular cytokine staining (ICS) for IFN‐γ and TNF‐α was performed as previously described. 37 To quantitate tumor‐specific CD8+ T cells using a functional assay, ex vivo cell suspensions were incubated with fresh 4T1.2 cells for 5 h at 37°C, 5% CO2, in the presence of 10 µg mL−1 Brefeldin A (Sigma‐Aldrich Japan), and stained for cell surface markers before fixation with 1% paraformaldehyde and permeabilisation with 0.4% saponin (Sigma‐Aldrich Japan) and incubation with intracellular antibodies.
In vitro treatments and co‐culture experiments
Various IFN inducers were used to stimulate cultured cells in vitro: 3M‐052 (10 µm; 3M, Saint Paul, MN, USA); R848 (10 µm; Invivogen, San Diego, CA, USA; doses previously described 66 ); murine or human recombinant IFN‐α used at 1 × 103 IU mL−1; and poly I:C (10 μg mL−1) transfected into cells using Lipofectamine 2000 (10 μg mL−1; Life Technologies, Carlsbad, CA, USA) 24 h prior to flow cytometry.
4T1.2, BMDCs or BM macrophages were stimulated with 10 µm 3M‐052 or equivalent DMSO. Following 48 h, cell culture supernatants were centrifuged and added to fresh 4T1.2 cells and cultured for a further 48 h, and recipient cells were then assessed for MHC‐I expression by flow cytometry as described above.
Enzyme‐linked immunosorbent assay and cytometric bead array
IFN‐α and IFN‐β enzyme‐linked immunosorbent assay was performed using standard molecular biology techniques as described previously 11 ; IFN‐α capture antibody (clone RMMA‐1, 1/500, 0.16 μg mL−1; PBL Assay Science, Piscataway, NJ, USA), IFN‐α detection antibody (1/500 rabbit polyclonal mouse IFN‐α: 32100‐1; 4 μg mL−1; PBL Assay Science), IFN‐β capture antibody (clone RMMB‐1, 1/500, PBL Assay Science), IFN‐β detection antibody (1/500 rabbit polyclonal mouse: 32400‐1; PBL Assay Science). Cytometric bead assays for IL‐1β, IL‐4, IL‐6, IL‐10, IL‐12p70, MIP‐1α, MCP‐1, RANTES and TNF‐α were conducted as per the manufacturer's instructions (Becton Dickinson).
Statistics
All error bars are SEM. Experiments with multiple groups were analysed by one‐way ANOVA with post hoc Tukey's multiple comparison test. Student's two‐tailed t‐tests were used to evaluate significant differences between groups. The Mantel–Cox log‐rank tests were used to evaluate differences in survival time. Primary tumor growth curves were analysed by one‐way RM ANOVA with post hoc Bonferroni's multiple comparison test. GraphPad Prism software was used for all analyses. P < 0.05 was considered statistically significant.
Author Contributions
Damien Zanker: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Writing‐original draft; Writing‐review & editing. Alex Spurling: Investigation; Methodology. Natasha Brockwell: Data curation; Formal analysis; Investigation; Methodology; Writing‐review & editing. Katie Owen: Conceptualization; Methodology; Writing‐review & editing. Jasmine Zakhour: Data curation. Tina Robinson: Data curation; Investigation. Hendrika Duivenvoorden: Data curation; Formal analysis. Paul John Hertzog: Resources; Supervision. Stefanie Mullins: Conceptualization; Methodology; Resources; Writing‐review & editing. Robert Wilkinson: Conceptualization; Methodology; Writing‐review & editing. Belinda Parker: Conceptualization; Funding acquisition; Resources; Supervision; Writing‐review & editing.
Conflict of Interest
Financial support and reagents were provided by AstraZeneca. SRM and RWW are employed by AstraZeneca Ltd.
Supporting information
¶
¶
¶
¶
¶
¶
¶
¶
¶
Acknowledgments
This work was supported by funding from Cancer Council Victoria (Grant‐in‐aid APP1127757; BS Parker), AstraZeneca and the Victorian Cancer Agency (Fellowship to BS Parker).
References
- 1.Available from https://www.cancer.org/cancer/breast‐cancer/understanding‐a‐breast‐cancer‐diagnosis/breast‐cancer‐survival‐rates.html. [e‐pub ahead of print].
- 2. Loi S, Drubay D, Adams S et al Tumor‐infiltrating lymphocytes and prognosis: a pooled individual patient analysis of early‐stage triple‐negative breast cancers. J Clin Oncol 2019; 37: 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Loi S, Michiels S, Salgado R et al Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol 2014; 25: 1544–1550. [DOI] [PubMed] [Google Scholar]
- 4. Gajewski TF, Corrales L, Williams J, Horton B, Sivan A, Spranger S. Cancer immunotherapy targets based on understanding the T cell‐inflamed versus non‐T cell‐inflamed tumor microenvironment. Adv Exp Med Biol 2017; 1036: 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu J, Du W, Yan F et al Myeloid‐derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol 2013; 190: 3783–3797. [DOI] [PubMed] [Google Scholar]
- 6. Miyashita M, Sasano H, Tamaki K et al Prognostic significance of tumor‐infiltrating CD8+ and FOXP3+ lymphocytes in residual tumors and alterations in these parameters after neoadjuvant chemotherapy in triple‐negative breast cancer: a retrospective multicenter study. Breast Cancer Res 2015; 17: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Savas P, Virassamy B, Ye C et al Single‐cell profiling of breast cancer T cells reveals a tissue‐resident memory subset associated with improved prognosis. Nat Med 2018; 24: 986–993. [DOI] [PubMed] [Google Scholar]
- 8. Brockwell NK, Rautela J, Owen KL et al Tumor inherent interferon regulators as biomarkers of long‐term chemotherapeutic response in TNBC. NPJ Precis Oncol 2019;3: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bidwell BN, Slaney CY, Withana NP et al Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med 2012; 18: 1224–1231. [DOI] [PubMed] [Google Scholar]
- 10. Sistigu A, Yamazaki T, Vacchelli E et al Cancer cell‐autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014; 20: 1301–1309. [DOI] [PubMed] [Google Scholar]
- 11. Owen KL, Gearing LJ, Zanker DJ et al Prostate cancer cell‐intrinsic interferon signaling regulates dormancy and metastatic outgrowth in bone. EMBO Rep 2020; 21: e50162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Touati N, Tryfonidis K, Caramia F et al Correlation between severe infection and breast cancer metastases in the EORTC 10994/BIG 1–00 trial: investigating innate immunity as a tumour suppressor in breast cancer. Eur J Cancer 2017; 72: 95–102. [DOI] [PubMed] [Google Scholar]
- 13. Luo N, Nixon MJ, Gonzalez‐Ericsson PI et al DNA methyltransferase inhibition upregulates MHC‐I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun 2018; 9: 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chiappinelli KB, Strissel PL, Desrichard A et al Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015; 162: 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katlinski KV, Gui J, Katlinskaya YV et al Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 2017; 31: 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 2016; 16: 131–144. [DOI] [PubMed] [Google Scholar]
- 17. Casey KA, Fraser KA, Schenkel JM et al Antigen‐independent differentiation and maintenance of effector‐like resident memory T cells in tissues. J Immunol 2012; 188: 4866–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keskinen P, Ronni T, Matikainen S, Lehtonen A, Julkunen I. Regulation of HLA class I and II expression by interferons and influenza A virus in human peripheral blood mononuclear cells. Immunology 1997; 91: 421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bald T, Landsberg J, Lopez‐Ramos D et al Immune cell‐poor melanomas benefit from PD‐1 blockade after targeted type I IFN activation. Cancer Discov 2014; 4: 674–687. [DOI] [PubMed] [Google Scholar]
- 20. Pronzato P, Bertelli G, Amoroso D et al Cisplatin and recombinant alpha interferon in advanced breast cancer. Ann Oncol 1990; 1: 150–151. [DOI] [PubMed] [Google Scholar]
- 21. Quesada JR, Talpaz M, Rios A, Kurzrock R, Gutterman JU. Clinical toxicity of interferons in cancer patients: a review. J Clin Oncol 1986; 4: 234–243. [DOI] [PubMed] [Google Scholar]
- 22. Robinson RA, DeVita VT, Levy HB, Baron S, Hubbard SP, Levine AS. A phase I‐II trial of multiple‐dose polyriboinosic‐polyribocytidylic acid in patieonts with leukemia or solid tumors. J Natl Cancer Inst 1976; 57: 599–602. [DOI] [PubMed] [Google Scholar]
- 23. Levine AS, Sivulich M, Wiernik PH, Levy HB. Initial clinical trials in cancer patients of polyriboinosinic‐polyribocytidylic acid stabilized with poly‐L‐lysine, in carboxymethylcellulose [poly(ICLC)], a highly effective interferon inducer. Cancer Res 1979; 39: 1645–1650. [PubMed] [Google Scholar]
- 24. Caskey M, Lefebvre F, Filali‐Mouhim A et al Synthetic double‐stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med 2011; 208: 2357–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laplanche A, Alzieu L, Delozier T et al Polyadenylic‐polyuridylic acid plus locoregional radiotherapy versus chemotherapy with CMF in operable breast cancer: a 14 year follow‐up analysis of a randomized trial of the Federation Nationale des Centres de Lutte contre le Cancer (FNCLCC). Breast Cancer Res Treat 2000; 64: 189–191. [DOI] [PubMed] [Google Scholar]
- 26. Brockwell NK, Owen KL, Zanker D et al Neoadjuvant interferons: critical for effective PD‐1‐based immunotherapy in TNBC. Cancer Immunol Res 2017; 5: 871–884. [DOI] [PubMed] [Google Scholar]
- 27. Ducret JP, Caille P, Sancho Garnier H et al A phase I clinical tolerance study of polyadenylic‐polyuridylic acid in cancer patients. J Biol Response Mod 1985; 4: 129–133. [PubMed] [Google Scholar]
- 28. Lacour J, Lacour F, Spira A et al Adjuvant treatment with polyadenylic‐polyuridylic acid (Polya.Polyu) in operable breast cancer. Lancet 1980; 2: 161–164. [DOI] [PubMed] [Google Scholar]
- 29. Bourquin C, Hotz C, Noerenberg D et al Systemic cancer therapy with a small molecule agonist of toll‐like receptor 7 can be improved by circumventing TLR tolerance. Cancer Res 2011; 71: 5123–5133. [DOI] [PubMed] [Google Scholar]
- 30. Cheng N, Watkins‐Schulz R, Junkins RD et al A nanoparticle‐incorporated STING activator enhances antitumor immunity in PD‐L1‐insensitive models of triple‐negative breast cancer. JCI Insight 2018; 3(22):e120638–e120658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park CG, Hartl CA, Schmid D, Carmona EM, Kim HJ, Goldberg MS. Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci Transl Med 2018; 10: eaar1916. [DOI] [PubMed] [Google Scholar]
- 32. Corrales L, Glickman LH, McWhirter SM et al Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015; 11: 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mullins SR, Vasilakos JP, Deschler K et al Intratumoral immunotherapy with tlr7/8 agonist medi9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J Immunother Cancer 2019; 7: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Terawaki S, Chikuma S, Shibayama S et al IFN‐α directly promotes programmed cell death‐1 transcription and limits the duration of T cell‐mediated immunity. J Immunol 2011; 186: 2772–2779. [DOI] [PubMed] [Google Scholar]
- 35. Dong H, Strome SE, Salomao DR et al Tumor‐associated B7–H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8: 793–800. [DOI] [PubMed] [Google Scholar]
- 36. Malik BT, Byrne KT, Vella JL et al Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol 2017; 2: eaam6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zanker D, Xiao K, Oveissi S, Guillaume P, Luescher IF, Chen W. An optimized method for establishing high purity murine CD8+ T cell cultures. J Immunol Methods 2013; 387: 173–180. [DOI] [PubMed] [Google Scholar]
- 38. Ben‐Sasson SZ, Hogg A, Hu‐Li J et al IL‐1 enhances expansion, effector function, tissue localization, and memory response of antigen‐specific CD8 T cells. J Exp Med 2013; 210: 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brehm MA, Daniels KA, Welsh RM. Rapid production of TNF‐α following TCR engagement of naive CD8 T cells. J Immunol 2005; 175: 5043–5049. [DOI] [PubMed] [Google Scholar]
- 40. Ben‐Sasson SZ, Hu‐Li J, Quiel J et al IL‐1 acts directly on CD4 T cells to enhance their antigen‐driven expansion and differentiation. Proc Natl Acad Sci USA 2009; 106: 7119–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med 1999; 189: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Montoya M, Schiavoni G, Mattei F et al Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 2002; 99: 3263–3271. [DOI] [PubMed] [Google Scholar]
- 43. Diamond MS, Kinder M, Matsushita H et al Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011; 208: 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kadowaki N, Ho S, Antonenko S et al Subsets of human dendritic cell precursors express different toll‐like receptors and respond to different microbial antigens. J Exp Med 2001; 194: 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur J Immunol 2001; 31: 3388–3393. [DOI] [PubMed] [Google Scholar]
- 46. Gibson SJ, Lindh JM, Riter TR et al Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell Immunol 2002; 218: 74–86. [DOI] [PubMed] [Google Scholar]
- 47. Hemmi H, Kaisho T, Takeuchi O et al Small anti‐viral compounds activate immune cells via the TLR7 MyD88‐dependent signaling pathway. Nat Immunol 2002; 3: 196–200. [DOI] [PubMed] [Google Scholar]
- 48. Jurk M, Heil F, Vollmer J et al Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R‐848. Nat Immunol 2002; 3: 499. [DOI] [PubMed] [Google Scholar]
- 49. Kuroda E, Ho V, Ruschmann J et al SHIP represses the generation of IL‐3‐induced M2 macrophages by inhibiting IL‐4 production from basophils. J Immunol 2009; 183: 3652–3660. [DOI] [PubMed] [Google Scholar]
- 50. La Flamme AC, Kharkrang M, Stone S, Mirmoeini S, Chuluundorj D, Kyle R. Type II‐activated murine macrophages produce IL‐4. PLoS One 2012; 7: e46989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol 2013; 31: 443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol 2016; 37: 724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bubenik J. Tumour MHC class I downregulation and immunotherapy (Review). Oncol Rep 2003; 10: 2005–2008. [PubMed] [Google Scholar]
- 54. Rusinova I, Forster S, Yu S et al Interferome v2.0: an updated database of annotated interferon‐regulated genes. Nucleic Acids Res 2013; 41: D1040–D1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Weichselbaum RR, Ishwaran H, Yoon T et al An interferon‐related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci USA 2008; 105: 18490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boelens MC, Wu TJ, Nabet BY et al Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014; 159: 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Odnokoz O, Yu P, Peck AR et al Malignant cell‐specific pro‐tumorigenic role of type I interferon receptor in breast cancers. Cancer Biol Ther 2020; 21: 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu J, Blake SJ, Yong MC et al Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov 2016; 6: 1382–1399. [DOI] [PubMed] [Google Scholar]
- 59. Casey AE, Laster WR Jr, Ross GL. Sustained enhanced growth of carcinoma EO771 in C57 black mice. Proc Soc Exp Biol Med 1951; 77: 358–362. [DOI] [PubMed] [Google Scholar]
- 60. Lelekakis M, Moseley JM, Martin TJ et al A novel orthotopic model of breast cancer metastasis to bone. Clin Exp Metastasis 1999; 17: 163–170. [DOI] [PubMed] [Google Scholar]
- 61. Zanker D, Waithman J, Yewdell JW, Chen W. Mixed proteasomes function to increase viral peptide diversity and broaden antiviral CD8+ T cell responses. J Immunol 2013; 191: 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lutz MB, Kukutsch N, Ogilvie AL et al An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 1999; 223: 77–92. [DOI] [PubMed] [Google Scholar]
- 63. Weischenfeldt J, Porse B. Bone marrow‐derived macrophages (BMM): isolation and applications. CSH Protoc 2008; 12: pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 64. Fan X, Biskobing DM, Fan D, Hofstetter W, Rubin J. Macrophage colony stimulating factor down‐regulates MCSF‐receptor expression and entry of progenitors into the osteoclast lineage. J Bone Miner Res 1997; 12: 1387–1395. [DOI] [PubMed] [Google Scholar]
- 65. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65: 55–63. [DOI] [PubMed] [Google Scholar]
- 66. Smirnov D, Schmidt JJ, Capecchi JT, Wightman PD. Vaccine adjuvant activity of 3M–052: an imidazoquinoline designed for local activity without systemic cytokine induction. Vaccine 2011; 29: 5434–5442. [DOI] [PubMed] [Google Scholar]
- 67. Rio DC, Ares M Jr, Hannon GJ, Nilsen TW. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc 2010; 2010: pdb.prot5439. [DOI] [PubMed] [Google Scholar]
- 68. Zanker D, Pang K, Oveissi S et al LMP2 immunoproteasome promotes lymphocyte survival by degrading apoptotic BH3‐only proteins. Immunol Cell Biol 2018; 96: 981–993. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
¶
¶
¶
¶
¶
¶
¶
¶
¶