

Abstract
The use of biologics (peptide and protein based drugs) has increased significantly over the past few decades. However, their development has been limited by their short half-life, immunogenicity and low membrane permeability, restricting most therapies to extracellular targets and administration by injection. Lipidation is a clinically-proven post-translational modification that has shown great promise to address these issues: improving half-life, reducing immunogenicity and enabling intracellular uptake and delivery across epithelia. Despite its great potential, lipidation remains an underutilized strategy in the clinical translation of lead biologics. We review how lipidation can overcome common challenges in biologics development as well as highlight gaps in our understanding of the effect of lipidation on therapeutic efficacy, where increased research and development efforts may lead to next-generation drugs.
1. Introduction
The use of biologics in the treatment of numerous diseases has increased steadily over the past several years. The biologics market grew at an annual rate of 8% in 2014, doubling the annual growth rate of conventional pharma [1]. In fact, pharmaceutical companies are increasingly shifting their focus towards biologics: biologics-based revenue is expected to account for over 32% of total revenue by 2023, from 22% in 2013 [1,2].
Biologics have grown as a class of therapies due to the high specificities afforded by their complex structures, often leading to reduced toxicity and side-effects [3]. This specificity has allowed biologics to be approved at consistently higher rates than small molecules in recent years [4]. For example, from 2012 to 2014, biologics had twice the approval rate (18%) of small molecules [4]. Biologics also have had a lower number of withdrawals due to safety concerns at only 2% (0% if antibodies are excluded) from 1982 to 2013, compared to 3% for small molecules [5].
Despite the success and future promise of biologics, peptide and protein-based drugs in general continue to face many challenges not shared with small molecules, as illustrated in Fig. 1. Because peptides and proteins generally cross epithelia less readily than small molecules and are more prone to host metabolism, most biologic drugs are administered using either subcutaneous or intravenous injections. Compared to non-invasive and self-administered modes of delivery, this leads to greater costs and lower patient compliance (Fig. 1a) [6,7]. Without special modifications to increase their half-lives, most peptides and proteins are cleared from the serum in the range of minutes to hours by intra- and extracellular host metabolism and renal elimination (Fig. 1b) [8]. This short serum half-life, coupled with slow uptake into target tissues, demands increased dose size and frequency for therapeutic effect and therefore tends to increase cost, reduce compliance and narrow the therapeutic window. An additional major limitation on the therapeutic window and efficacy of biologics is their potential for immunogenicity (Fig. 1c). Protein and peptide drugs elicit anti-drug antibodies (ADAs) much more effectively than small molecules, both because their larger and more complex structures are more effective at stimulating B cells and because they are uniquely able to stimulate T cells. In turn, these ADAs and other anti-drug immune responses can reduce drug efficacy and may cause severe adverse health effects [9]. Finally, biologic drugs tend to be poorly permeable across cell membranes, and routes of cellular uptake that are available often lead to drug degradation in lysosomes (Fig. 1d) [10]. Consequently, the vast region of potential drug target space that lies inside cells is virtually inaccessible to biologics at present.
Fig. 1.

Current challenges and solutions facing biologic therapeutics (a) (i) The majority of biologics are administered through injections while alternative routes including pulmonary, oral, and topical administration remain underused. (ii) Injections can lower therapy effectiveness through increased patient non-compliance (Data from [7]). (b) Half-life of biologic therapeutics are shortened by (i) by proteolysis and (ii & iii) renal clearance from the bloodstream. (c) Biologics can also induce immune reactions. (i) Antigen presenting cells (APC) can present peptide fragments to T-cells inducing an immune response through cytokine release that leads to rapid clearance. (ii) B-cell receptors recognize protein antigens and secrete antigen specific immunoglobulin into the serum which can neutralize the therapeutic activity and mark therapeutics for clearance. (d) Biologics mainly enter cells through endocytosis. Endocytosis requires endosomal escape for the therapeutic to gain access to the cytosol without which the biologic is degraded in the lysosome. (e) Challenges and current solutions for biologic therapeutics. Approaches listed in overlapping regions address multiple challenges. *Denotes controversial data on PEGylation that supports both reductions and increases in immunogenicity of biologic therapeutics.
One means of addressing these key challenges has been to covalently modify the pharmacologically-active peptide or protein with other groups that confer new properties. Examples can be found of approved or investigational drugs covalently modified to overcome each of the aforementioned challenges, and several general strategies have emerged as depicted in Fig. 1e. Success in non-invasive administration has mostly come from recombinant fusions with groups that enable epithelial transport by receptor-mediated trafficking [11,12], or with groups that facilitate endocytosis and translocation, such as cell-penetrating peptides [13–15]. Increased half-life has been achieved with both chemical and recombinant modifications and has relied on three primary mechanisms: (i) increased hydrodynamic radius to reduce renal elimination, (ii) steric shielding from proteases, and (iii) receptor-mediated endosomal escape and recycling. All three mechanisms may be employed by fusions to Fc [16], albumin [17], or groups that bind to albumin [18–21]. Modification with the polymer polyethylene glycol (PEG), by far the most widely used covalent modification for biologics to date [22–24], relies on the first two of these mechanisms. PEGylation is also a potential means of reducing immunogenicity, though anti-drug antibodies have remained a significant concern even for some PEGylated therapeutics [25,26]. Other approaches such as fusion to tolerogenic Fcs have been proposed as potential platform technologies in this area [27]. Finally, cytosolic uptake of peptide and protein drugs has been improved by fusions that enable specific receptor-mediated endocytosis (e.g. by antibodies against human insulin receptor [28] or the transferrin receptor [29]) or cell-penetrating peptides enabling non-specific endocytosis or translocation [30].
Though there has been much success with covalent modifications of biologics to date, there are still significant problems that are not well addressed by any of the above well-explored strategies. For example, the function of protein and peptide drugs is exquisitely sensitive to their tertiary structure, which is easily disturbed by fusions and poorly-placed modifications. With respect to cytosolic uptake, most current strategies rely on endocytosis and thus face the challenge of subsequent escape from the endosome, as opposed to direct translocation or membrane permeabilization. Finally, because many biologic drug candidates face several of the major challenges outlined above, it is desirable to find modification strategies that can simultaneously improve performance in as many of these areas as possible. Lipidation, or acylation, is a clinically-proven strategy with the potential to address each of the key challenges in protein and peptide drug development while overcoming many of the challenges of current alternative methods.
Lipidation involves the transfer of a lipid group to a protein [31]. In nature, lipidation is used by the cell to control the function and intracellular location of several proteins [31,32]. When used to modify therapeutics, lipidation can greatly increase their half-life and cell membrane permeability [33,34]. Moreover, lipidation has been shown, in some cases, to have superior performance to PEGylation, the current standard post-translational modification [35]. Currently, there are several FDA-approved lipidated therapeutics with more in development, as summarized in Table 1. Potential advantages of lipidation can primarily be attributed to several key effects, including improving half-life, enhanced delivery, enabling alternative routes of administration, improving pharmacologic potency and reducing immunogenicity.
Table 1.
Current acylated therapeutic peptide and protein based drugs.
| Drug | Parent molecule | Indication | Mode of delivery | Stage | Half-life (h) |
|---|---|---|---|---|---|
| Daptomycin | N/A | Bacterial infections | Injection | Approved | 8.6 [36] |
| Polymixin B | N/A | Bacterial infections | Topical/Injection | Approved | 4–6 [37] |
| Tesamorelin | Growth hormone releasing factor | HIV-associated lipodystrophy | Injection | Approved | 0.4–0.6 [38] |
| Detemir | Insulin | Diabetes | Injection | Approved | 5–7 [39] |
| Degludec | Insulin | Diabetes | Injection | Approved | 25 [40] |
| Liraglutide | GLP-1 | Diabetes | Injection | Approved | 13 [41] |
| Obesity | Injection | Approved | |||
| Semaglutide | GLP-1 | Diabetes | Injection | Approved | 168 [42] |
| Oral | Phase 3 | ||||
| Obesity | Injection | Phase 3 | |||
| Non-alcoholic fatty liver disease | Injection | Phase 2 | |||
| Somapacitan | Human growth hormone | Growth disorders | Injection | Phase 3 | 34–45 [43] |
| MEDI0382 | Glucagon/GLP-1 | Diabetes and obesity | Injection | Phase 2 | 9.5–12 [44] |
| LY3298176 | GLP-1/Gastric inhibitory polypeptide (GIP) | Diabetes | Injection | Phase 2 | 116.7 [45] |
| Tri-agonist 1706 | Glucagon/GLP-1/GIP | Obesity | Phase 1 [46] |
Despite the several potential advantages of lipidation, this post-translational modification remains underutilized for developing therapeutics. We will discuss the mechanisms by which lipidation alters the properties of peptide and protein therapeutics as well as highlight important variables that may impact therapeutic development and efficacy.
1.1. Improvements in drug half-life
Lipidated molecules have demonstrated significant improvements in half-life ranging from ~10-fold increase for liraglutide, a lipidated GLP-1 analogue indicated for diabetes and obesity (13 h half-life vs. 1.5 h for native GLP-1), to an over 100-fold improvement for semaglutide (168 h half-life), a next-generation lipidated GLP-1 analogue [41,42]. Another recent advance is the development of a lipidated human growth hormone analogue [47], which can be administered once weekly thanks to its ~40 h half-life [48].
Lipidation improves drug half-life primarily by enabling albumin binding, as shown for the drugs in Table 1 and other leads in development [49–52]. Albumin, a 66.5 kDa protein and the most abundant protein in blood at 40 mg/ml [53], has a very long half-life (20 days) because it can bind to neonatal Fc receptors (FcRn) in growing endosomes which recycle albumin by sorting it away from lysosomes [54]. Albumin’s main role is to transport hydrophobic molecules in the bloodstream, including fatty acids [55]. When incubated in vitro with plasma at clinically relevant concentrations, a large fraction (> 95%) of lipidated biologics bind to albumin [52,56]. Albumin binding improves half-life [57,58] by limiting the access of proteases to the drug [59], greatly reducing renal clearance [49,51,60], and leveraging FcRn-mediated recycling (Fig. 2a). Because albumin binding is reversible (Fig. 2), lipidated therapeutics are free to elicit their effect once unbound [52]. Importantly, under normal physiological conditions only 2 out of 7 binding sites on albumin are occupied with fatty acids, eliminating the threat of lipidated therapeutics competing for binding sites with albumin’s natural cargo [58,61,62].
Fig. 2.

Half-life extension of lipidated therapeutics through albumin binding. (a) non-lipidated molecules have increased rates of (i) proteolysis and (ii) renal clearance. (iii) Lipidation enables reversible albumin binding (iv) reducing renal clearance and degradation by proteases, such as dipeptidyl peptidase 4 (relevant for GLP-1 analogues; PDB ID: 4APD). (b) Seven sites are available on albumin for medium and long-chain fatty acids. Interactions between the lipid carboxylate and polar amino acids at some of these sites (top close-up, dark blue) along with increased nonpolar interactions resulting from longer lipids (bottom close-up) can be leveraged for tighter albumin binding (PDB ID: 1E7H).
Affinity to albumin’s fatty acid binding sites results from both electrostatic interactions of the lipid carboxylate group and polar side-chains as well as key binding of the nonpolar fatty acid chain as shown in Fig. 2b [63,64]. Increasing the length of the attached fatty acid on lipidated drugs generally leads to tighter albumin binding and increased therapeutic half-life [41,50,58,65]. Recent efforts to increase lipidated therapeutic binding to albumin have resulted in the use of longer lipids (18 or 20 carbons) and the transition from monocarboxylic acids to dicarboxylic acids [33,47]. The improved half-life associated with the use of these lipids has so far been attributed to tighter albumin binding [33,47]. Linear fatty acids also tend to outperform bulkier albumin binders since the latter category results in either decreased therapeutic potency or half-life [66]. Also, unsaturated bonds do not appear to significantly affect albumin binding [66]. Importantly, the lipid attachment site within the lipidated therapeutic does not affect albumin binding [41].
1.2. Enhanced drug delivery
Another key advantage of lipidation is that it enables enhanced drug delivery. First, lipidation enables a depot offering delayed drug absorption after subcutaneous injection. Second, lipidation enables improved cellular permeabilization and cellular drug uptake.
Lipidated therapeutics are routinely delivered subcutaneously and they have been shown to reversibly form multimers at the injection site as illustrated in Fig. 3. These multimers act like a depot that delays absorption from the injection site to the bloodstream (Fig. 3a) [65]. Although lipidated therapeutics are still able to bind albumin at the interstitium and this contributes to some absorption delay, the formation of multimers has a greater contribution [56,59,65,67,68]. For example, the formation of a subcutaneous depot almost doubles the half-life of liraglutide as compared to intravenous injections of the same molecule (Fig. 3b) [69]. Once the therapeutic has entered the bloodstream, albumin binding takes over as the primary mechanism for extending half-life.
Fig. 3.

Enhanced drug delivery due to lipidation. (a) Lipidated therapeutics are routinely injected subcutaneously where they form a drug depot through reversible self-association causing (b) a delay of drug absorption into the body, which accounts, for example, for almost doubling the half-life of liraglutide, a GLP-1 analogue used to treat diabetes and obesity (data taken adapted from [69]). (c) Lipidation also increases cell uptake through (i) a flip-flop mechanism or (ii) endocytosis. (d) Increases in lipid length results in increased cell uptake (data taken adapted from [72 to 74]).
Another major consequence of lipidation is an increase in cell membrane permeability of peptide drugs. The lipid groups increase affinity towards the cell membrane and can enable insertion into the membrane [70,71]. Increased affinity towards the cell membrane also promotes higher cellular uptake with potential implications for intracellular delivery [50]. Generally, increasing the lipid length leads to increased cell uptake (Fig. 3d) [72–74]. However the peptide portion can also affect the magnitude of cell internalization and can sometimes shift the optimal lipid length for maximal cell uptake to shorter lipids [75,76]. For example, lipidation of nona-arginine (9 arginines) with laurate (12 carbons) resulted in a 1.5- to 3-fold increase in uptake when compared to lipidation with myristate (14 carbons) or palmitate (16 carbons), respectively [75]. It has been hypothesized that the formation of stable multimers, a common occurrence for lipidated molecules, can lead to decreased cell membrane interactions [34]. In these cases, elongating the lipid chain may lead to more stable multimers due to increased non-polar interactions. The competing equilibria between multimer formation and cell membrane interaction may alter the specific impact of lipid length on cellular uptake.
Studies on the uptake of lipidated peptides have been performed and point to both endocytosis and direct translocation as pertinent mechanisms (Fig. 3c) [75–78]. Intracellular delivery is a highly sought-after goal, as only one third of the proteome is accessible from outside cells [30,79]. The direct translocation of lipidated peptides would eliminate the need for endosomal escape in intracellular delivery. However, uptake through direct translocation has been highly disputed, in part due to studies using fixed cells in confocal microscopy experiments [80]. Cell fixation causes an artifact where peptides are observed in the cytoplasm after uptake (suggesting direct translocation or endosomal escape), contrary to what is observed in non-fixed cells [77,80]. Still, experiments looking at quenching kinetics of fluorescently labeled lipidated peptides loaded into artificial vesicles indicate lipidated peptides can flip-flop between the inner and outer membrane of the vesicles [81]. Moreover, using FRET probes, it was observed that a family of lipidated therapeutics, pepducins, can enter cells and localize in the inner leaflet of the cell membrane [82].
Direct translocation is further supported by the observation that lipidated peptides accumulate in the cytoplasm after incubation at 4 °C, at which temperature endocytosis is stopped [72,75,76,83]. While these experiments are well-established and easy to perform, they erroneously assume that direct translocation, which can be an energy-dependent process, is not significantly affected at 4 °C; thus, they likely underestimate the magnitude of direct translocation at elevated temperatures [76]. In addition, membrane fluidity is also reduced at temperatures below 20 °C [76,77]. The latter concern is especially relevant to lipidated peptides since they cross cell membranes in fluid-phase domains after perturbing membrane packing [34,78]. In fact, the use of unsaturated lipids as opposed to saturated ones can increase relative rates of direct translocation because they can interfere with membrane packing to a higher extent [78].
1.3. Routes of administration
The increased cell membrane permeability of lipidated therapeutics also has implications for alternative routes of administration, including oral, topical and pulmonary delivery [49]. Current research is ongoing to utilize lipidation to enhance the oral delivery of peptide-based drugs. Studies using Caco-2 cell monolayers, a model for drug intestinal absorption, have shown that lipidation increases translocation of the lipidated therapeutic through the monolayer [34,74]. Notably, the optimal lipid length for monolayer translocation was shorter (12 carbons) than the optimal length for cellular uptake (16 carbons), probably due to higher retention of longer chain lipidated biologics in cell membranes [34,72,74]. Lipidated therapeutics can also act as permeation enhancers on Caco-2 monolayers in a detergent-like manner [34]. Higher concentrations and lower pH favor increased permeability [34]. In addition to lipid length, lipid attachment site also influences changes in permeability, with lipid length trends even reversing for different attachment positions [34]. It was hypothesized that a dependence on lipidation site may be due to the formation of different multimers where more stable multimers would lead to decreased interactions with the cell monolayer and thus decreased permeability [34]. This is in agreement with previous observations that only monomers, as opposed to multimers, are internalized by cells [77]. Thus, dissociation of single lipidated peptides from multimers seems to be a necessary step prior to uptake.
An important recent development that highlights the potential of lipidation to improve biologic-based therapies has been a novel oral formulation for semaglutide, as depicted in Fig. 4. Semaglutide is currently approved for once-weekly administration through subcutaneous injections, and its new daily oral formulation is currently undergoing Phase 3 clinical trials [84]. The oral semaglutide formulation needs at least 20 times more lipidated GLP-1 analogue (20 mg) than an injection of the same molecule (1 mg) to achieve the same efficacy [84,85]. Absorption into the bloodstream happens in the gastric epithelium requiring patients to be fasting before ingesting the pill (Fig. 4a) [86]. The permeability of semaglutide alone through the gastric epithelium does not offer sufficient bioavailability, so semaglutide is supplemented with the hydrophobic permeation enhancer sodium N-8(-[2-hydroxylbenzoyl]amino) caprylate (SNAC) (Fig. 4b, c) [87]. SNAC has also been used to increase the efficacy of oral insulin and peptide YY administration [88]. In addition to enhancing the permeability of semaglutide, SNAC also increases the local pH around the tablet in the stomach, protecting the therapeutic against degradation [86].
Fig. 4.
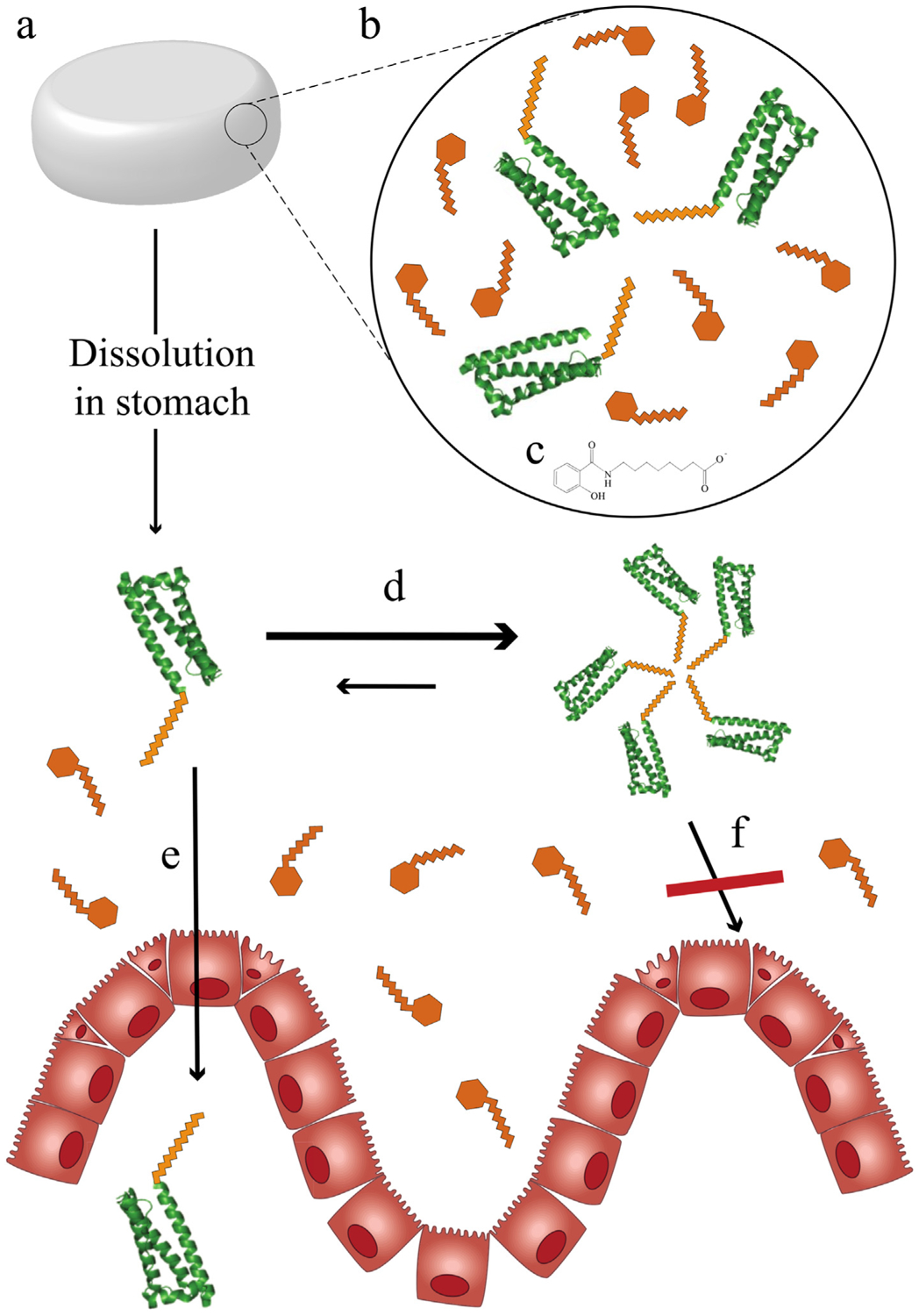
Oral delivery of peptides enabled by lipidation. (a) Tablet formulations are now possible for lipidated therapeutics, such as semaglutide, a GLP-1 analogue used to treat diabetes. (b) Besides the active ingredient (eg semaglutide), permeation enhancers are needed to increase therapeutic bioavailability such as (c) N-[8-(2-hydroxybenzoyl) amino] caprylate (SNAC) for semaglutide’s oral formulation. (d) Self-association of lipidated therapeutics may negatively affect permeation of lipidated therapeutics through the gastric endothelium as (e) monomers may be able pass, (f) but not multimers.
Semaglutide maintains a very long half-life after oral delivery (~153–160 h compared to 168 h after injection) [89], but as discussed its bioavailability is drastically reduced, requiring higher daily doses. Future lipidated therapeutics intended for oral delivery may benefit from lipidation with shorter lipids (12 carbons) than used in semaglutide (18 carbons) which may increase bioavailability. Lipidated therapeutics that do not form stable multimers in the stomach may also have higher permeability (and bioavailability) due to increased interactions with the membrane (Fig. 4d–f). Generally, more efficacious lipidated therapeutics intended for oral delivery have the potential to be designed specifically for that route of administration where higher bioavailability is balanced with improvements in half-life.
Lipidation has also been widely used for topical delivery. Specifically, lipidation of small peptides increases skin permeability [90,91], where increasing the fatty acid chain length increases permeability, but also retention of the peptides in the first skin layer, the stratum corneum [92]. However, lipidated peptides still have poor half-lives when applied on the skin [90] and further studies looking to optimize dermal delivery using lipidation have not been extensively performed.
Among non-invasive routes of administration for peptides and proteins, the lungs have an exceptional potential for rapid and complete systemic uptake of lipidated molecules. The alveolar epithelium in particular is very thin, extensive, covered only by a small volume of fluid, and generally contains far lower levels of drug-metabolizing enzymes than either the skin or the gastrointestinal tract [93]. However, approvals for inhaled biologics have been rare despite many years of extensive efforts [94]. The use of lipidation in pulmonary delivery efforts is unexplored, but promising. In particular, drug stabilization and transport across the alveolar epithelium might be supported by binding of lipidated drugs to albumin either in situ or in the formulated drug product, because albumin is present in the alveolar epithelial lining fluid and is actively transported into the capillaries at significant rates [95].
Despite the route of administration, lipidated therapeutics have a unique biodistribution. Lipidation primarily results in increased accumulation in the liver when compared to non-lipidated counterparts [35,49,51,52], though the mechanisms behind this accumulation are unclear. Liver targeting may be an advantage for certain therapies, but accumulation in the liver may also be undesirable if other tissues are targeted, due to drug unavailability or liver toxicity. Although, to date, lipidated therapeutics have not been observed to cause liver damage and importantly, dosage modification has not been required on patients with hepatic impairment [137,138], future potential therapies such as cytotoxic cancer drugs may face a different reality. Targeting moieties that have been previously shown to lessen hepatic accumulation of lipidated agents, may be of future use to alter biodistribution [106].
1.4. Changes in pharmacologic potency
Lipidation, like other drug modifications, can have a profound effect on the potency and pharmacologic activity of the target therapeutic. The impact can be a function of the length of the lipid, as well as the site of lipidation, the linker used, and any structural interactions between the lipid or linker and the therapeutic or its target. As a result, the impact of lipidation on potency can be variable, unpredictable and drug-specific, as illustrated in Fig. 5.
Fig. 5.

Changes in drug potency due to lipidation. (a) (i) Variations in lipid length, (ii) site of lipid attachment and (iii) type of linker used to attach the lipid influence resulting drug potency. (b) In some cases, increasing lipid length leads to decreased potency where lipidation at a particular site may result in a series of lipidated analogues with higher potency than at a different site (eg for GLP-1 analogues, lipidation at K36 results in analogues with higher potency than lipidation at K26) (Data adapted from [41]). (c) Longer, hydrophilic linkers allow the attachment of longer lipids at lower potency loss such as the use of a γ-glutamyl linker elongated with amino-3,6-dioxaoctanoic acid (OEG) which results in more potent GLP-1 analogues when compared to the use of only the γ-glutamyl linker or no linker (Data adapted from [33]). (d) In some other cases, because lipidation increases affinity towards the cell membrane, it can lower the energy barrier for the formation of therapeutic-target complex (e) which results in increased drug potency with increasing lipid length (Data adapted from [70,104,105]). (f) In some other cases, increasing the lipid length results in oscillating potencies with maxima and minima (Data adapted from [103]).
Lipid length can be a powerful modifier of drug potency. Lipidation has been shown to either (i) decrease potency with increasing lipid length (Fig. 5b–c), (ii) increase potency with increasing lipid length (Fig. 5e) or (iii) result in optimal potency at intermediate lipid lengths (Fig. 5f). Furthermore, altering the lipidation site can result in molecules with varying potencies [41,65], and the use of different lipids at one site may result in a series of analogues with higher potency than at a different site (Fig. 5b). As such, it is important to evaluate different lipid attachment sites and lipid lengths to optimize therapeutic efficacy [41,47,65].
The linker used to attach the lipid moiety to the target therapeutic can also impact potency [33,41,50]. For molecules whose potency decreases with increasing lipid length, the use of a long hydrophilic linker for attachment of longer fatty acids can minimize the potency cost of lipidation (Fig. 5c) [33,41]. Conformational changes resulting from lipidation can modify the affinity of the therapeutic with its target and modify potency, and increasing the linker length may minimize the extent of these changes by increasing the distance between the attached lipid and the drug. In fact, screening different linkers can completely compensate for the potency loss of attaching long fatty acids [33,47]. In general, however, it is unpredictable how linker choice may affect potency [33], as linker length has also been associated with affecting half-life, as exemplified by a 2-fold reduction in half-life when shorter linkers are used in lipidated human growth hormone analogues [47].
Recently, better-performing linkers have been designed. The use of γ-Glu has been most successful, complemented with further linker elongation using repeats of ethylene groups (PEG) (eg. semaglutide, a GLP-1 analogue used for diabetes) or amino-3,6-dioxaoctanoic acid (OEG) (eg. somapacitan, a growth hormone analogue used for growth disorders) [33,47,96–99]. The hydrophilicity of these linkers may aid in the synthesis and formulation since solubility issues are common for lipidated therapeutics [100]. Importantly, initial clinical studies have shown no significant safety concerns for these PEG-based and OEG-based linkers [48,101], though their long-term effects are yet to be evaluated [102]. It is unclear what specific features of these new linkers (besides length) favor higher drug potencies. More in-depth structure-activity relationship studies may be required on a case-by-case basis, including crystallographic studies of different linkers and lipids, although these have proven difficult in the past [47].
If the therapeutic target of the lipidated biologic is membrane-bound, insertion into the cell membrane aided by the lipid group can reduce the overall energy requirements for the formation of ligand-receptor complexes (Fig. 5d) [103]. For these cases, increasing the lipid length elevates the affinity for the cell membrane and thus increases potency (Fig. 5e) [70,103–105]. Interestingly, lipidated therapeutics have also been shown to preferentially bind to membranes of cell types expressing their target receptors, which may lower concerns related to the retention of lipidated therapeutics at non-target tissues [70,106].
However, as discussed above, the relationship between lipid length and drug-target affinity can be complex, as shown in Fig. 5f, and even small changes to the overall structure can greatly impact potency. The large impact of these small changes in lipid length can be exploited to improve drug specificity, such as in targeting specific melanocortin receptors [103]. For example, Fig. 5f illustrates a maximum local potency against Melanocortin Receptor 1 for a drug with a lipid length of 17 carbons, which also corresponds to a local affinity minimum for an alternative receptor: Melanocortin Receptor 4 [103]. These results again highlight the need for experimentation, and perhaps better structural characterization, to identify lipid and linker design principles that may very well be drug and target specific.
A better understanding of design principles may allow for the use of very long chain fatty acids (20 carbons or more), a virtually untapped therapeutic space with few examples [47]. Studies have shown that while very long fatty acid chains are able to bind to albumin, they preferentially partition to a greater extent with lipoproteins [107,108]. The effect of lipoprotein binding on half-life has not been investigated, but selective binding to lipoproteins as opposed to albumin could be leveraged to use them as drug carriers or to treat lipoprotein-related conditions [109–111].
1.5. Reduced immunogenicity
Lipidation has also been shown to decrease the immunogenicity of potential biologics, which has been a major hurdle in the clinical translation of protein-based therapeutics [112,113]. As of 2015, 89% of all approved biologics have reported some immunogenic response and 55% of these reactions have had an impact on therapy efficacy [114]. These responses include life-threatening autoimmunity, allergic reactions and accelerated blood clearance due to formation of anti-drug antibodies [26,112]. Furthermore, some post-translational modifications intended to increase therapeutic half-life are themselves immunogenic, limiting efficacy [26,115].
Patients treated with lipidated therapeutics have also been found to develop low levels of anti-drug antibodies, although to date the presence of these antibodies has not been shown to reduce therapy efficacy [116,117]. On the other hand, lipidated peptides have been shown in some cases to be less immunogenic than their non-lipidated counterparts, as depicted in Fig. 6 [118,119]. Of course, this excludes highly immunogenic bacteria-inspired lipid epitopes (eg. Pam2/3Cys), which are known to activate immune responses and are used for vaccine development [120,121].
Fig. 6.

Reduced immunogenicity due to lipidation. (a) Peptide and protein-based drugs can become immunogenic through the MHC II antigen presentation pathway. (b) Lipidated therapeutics can potentially be presented through both MHC I and MHC II antigen presentation pathways but have been shown to have lower immunogenic potential than non-lipidated counterparts. (c) Comparison of non-lipidated with lipidated therapeutics at different steps in the antigen presentation pathway. (i) Lipidation causes increased antigen uptake by antigen presenting cells (APC) (Data adapted from [124]). (ii) Lipidation have been shown to be resistant to proteolytic degradation, though this has not been shown in APCs specifically (Data adapted from [90,126,127]). Lipidation causes an overall decrease in binding to MHC II molecules (Data adapted from [118]). (iv) Lipidation can greatly decrease the magnitude of T-cell responses (Data adapted from [118]).
One key determinant of the immunogenicity of lipidated drugs is the stability of the bond between the lipid and the peptide. An early report showed that use of a thioester bond (which is labile in the intracellular environment), resulted in significantly higher immunogenicity compared to the use of an amide bond, which is more stable [122]. Another study showed that lipidating a peptide using an amide linker could convert an immunogenic peptide into a non-immunogenic peptide whereas the use of a thioester linker would do the opposite [119].
To explain this phenomenon, it was suggested that lipidation increased the uptake of the peptide by antigen presenting cells (APCs), increasing immunogenicity, but also that the attached lipid could interfere with antigen processing or T cell activation, decreasing immunogenicity [122,123]. Lipidation does increase cellular uptake regardless of the nature of the bond [124], however if the bond is stable, it is hypothesized that the lipid remains attached inside APCs and interference with immune activation occurs, outweighing increased APC uptake and lowering overall immunogenicity [119]. A recent study by Schultz et al. determined that lipidation can interfere with T cell activation because it can decrease binding to MHC II molecules and significantly reduce T cell responses (Fig. 6) [118,125]. The exact mechanisms for decreased MHC II binding and T-cell responses are still under investigation, but lipidated peptides are known to be protease-resistant [90,126,127], and impaired antigen proteolysis may prevent downstream processes from happening [128]. It has been observed that increasing the lipid chain length decreases immunogenicity [118]. The lipid attachment site also affects any decrease in immunogenicity, with several cases showing complete immune response evasion [118]. These changes are not necessarily additive, as lipidation at multiple sites does not further reduce immunogenicity and, in some cases, can increase it [118,129].
Lipidated peptides should also be capable of being presented to the immune system through the MHC I pathway since they can access the cytoplasm after cellular uptake. MHC I molecules that bind lipidated peptides have been described and shown to bind shorter peptides (5-mer lipidated peptides) than regular MHC I molecules (9-mer peptides) [130]. The shorter peptide length is a result of the lipid portion also interacting with the antigen binding groove of the MHC I molecule [130]. The impact of these shorter peptides on MHC I presentation and immune activation is still unknown, and the effects of lipidation on MHC I pathway presentation are still under investigation. Studying how lipidation interferes with proper antigen processing may also help explain why some long (> 20 amino acids) amide-bond lipidated peptides derived from pathogens, used for vaccine development, become immunogenic upon lipidation [131]. Lipidation in these cases may increase APC uptake but may not confer sufficient proteolysis resistance, leading to proper antigen processing, presentation and immune stimulation. The sequence-specific degradation of lipidated peptides may also explain why lipidation at multiple sites is not beneficial, since multiple lipid groups do not provide additional protection against proteolysis [126].
1.6. Discussion and future outlook
While the use of lipidation as a post-translational modification in biologics has seen significant success and shows great promise in future drug development, there are some applications were its potential benefits may be limited. For example, in short-term anticoagulation therapies, used to prevent post-operative complications [132], rapid drug clearance may be desirable as opposed to half-life extension. In addition, lipidation of monoclonal antibodies, the largest group of biologic therapeutics to date, may provide limited advantages. Antibodies already possess a long half-life on the order of days, due to natural FcRn-mediated recycling [133] and absorption of subcutaneously injected antibodies into the bloodstream is already a slow process [134]. While lipidation of antibodies may increase therapy potency [106] and has been used to enable intracellular targeting [135], in general antibody lipidation remains understudied, with perhaps limited advantages.
In some applications, the use mixtures of lipidated drugs with their non-lipidated counterparts, may present some advantages. These mixtures could be used to balance potency and extensions in half-life. For example, in wound healing applications [136] a high initial therapeutic effect could be provided by a potent, short-lived non-lipidated therapeutic, and followed by a lower sustained effect from longer-lived, less potent, lipidated therapeutics.
Furthermore, even in applications where lipidation may offer a significant advantage, many challenges remain to the generalized use of lipidation. These challenges include: (i) a lack of mechanistic understanding of the effect of lipidation on cell uptake and epi/endothelium permeability, (ii) a lack of mechanistic understanding of the effect of lipidation on APC processing and immunogenicity and (iii) a lack of methodologies for the rapid synthesis of lead compounds.
A key understudied aspect of lipidated peptides and proteins is the effect of structure on cell uptake and permeability. There remains a need for studies that better quantify cellular uptake of lipidated therapeutics and elucidate the underlying uptake mechanisms at biologically relevant concentrations. Methodologies developed to study cell penetrating peptides can readily be translated to study lipidated drugs. These methods would allow for the precise quantification of uptake by direct translocation as well as endocytosis and the kinetics of such processes in both artificial vesicles (lacking endocytosis machinery) and live cells [139,140]. Future studies need to determine the properties that affect uptake through direct translocation (vs. endocytosis) such as degree of unsaturation, peptide hydrophobicity, charge and polarity, amphiphilicity, and concentration. Although this discussion has focused on the cellular uptake of peptides, it would also be interesting to determine any molecular weight cut-offs for direct translocation that may impact the development of larger protein based lipidated drugs.
Importantly, the specific role of multimers in permeability and uptake also needs further study. Lipidated therapeutics need to be in monomeric form to enter cells [77]. Thus, formation of stable structures may decrease, rather than increase, cell uptake [34]. However, it has also been observed that increasing the lipid length increases cell uptake despite the formation of structures with longer lipids [72]. The key to understanding this relationship may be the stability of the multimers formed and the nature of the multiple equilibria involved, as opposed to whether or not structures are formed. Future studies on cell uptake and membrane permeability need to be coupled with robust structural characterization studies. Formation of multimers also protects against proteases [126], but it is unknown whether this can occur in vivo inside APCs at biologically relevant concentrations.
Further investigations on the effects of lipidation on immunogenicity are also needed. Thus far, clinically successful lipidated therapeutics have been based on human proteins (e.g., GLP-1) with low intrinsic immunogenicity, as seen in Table 1, where all therapeutics, besides a couple of antibiotics, are human protein-based. However, expanding the repertoire of attainable lipidated therapeutics may include the use of intrinsically more immunogenic non-human-based proteins (e.g., peptide agonists to therapeutic targets). While there is an indication that lipidation can lower immunogenicity, the exact mechanisms remain unclear. Future immunological studies should evaluate the presentation through MHC I (non-endocytic) pathways and subsequent T-cell activation. The effect of lipidation on antigen processing and MHC antigen presentation (not just binding) are also unknown.
As discussed above, one challenge with all post-translational modifications, including lipidation, is that the effect of the modification on potency and/or immunogenicity may be unpredictable [113,141,142]. These unpredictable effects are often dealt with on a case-by-case basis and overcome by screening many compounds for optimized properties [33,47]. This often requires changing (i) the attached lipid moeity, (ii) the lipid attachment site, and (iii) the linker used for the attachment in order to optimize the potency as well as the pharmacokinetic and pharmacodynamic properties of the lead molecule. This non-trivial case-by-case optimization requires the synthesis of a panel of compounds, which is generally first screened for improved half-life while minimizing potency losses. To date, the synthesis of these panels has relied on purely chemical methods that have been limited in the number and nature of compounds that can be generated.
Site-selectivity is a major restriction in current chemical synthesis approaches, where only a few types of amino acids can be used for conjugation. Small lipidated peptides can generally be synthesized using solid phase synthesis wherein a unique amino acid can then lipidated, most commonly lysine [41] or where lipidated amino acid building blocks are premade and incorporated during synthesis [143]. For example, this strategy has been used to introduce a free cysteine into human growth hormone which, after production in E coli, allows site-specific lipidation [47]. Site-selectivity becomes a more significant challenge when lipidating proteins because this can require mutations to either introduce or reduce the number of sites available for conjugation, which may carry a risk of immunogenicity. Another strategy is to produce a truncated protein that can later be ligated with a chemically synthesized lipidated peptide. These ligation reactions include native chemical ligation, MIC ligation and sortease-mediated ligation, but all of these have sequence restrictions on the lipid attachment site and on the peptides/proteins that can be ligated. These approaches have been recently reviewed by Mejuch and Waldmann [143]. Microbial transglutaminase has also been used to site-specifically lipidate proteins, but also has substrate specificity restrictions [47,144]. Purely biosynthetic routes are attractive and have the potential to enable the synthesis of a large diversity of non-natural lipidated peptides or proteins. But, current biological methodologies rely on media supplementation with free fatty acids which are then used for lipidation of natural protein substrates by existing post-translational lipidation machinery [145]. To broaden the applicability of this approach, significant improvements in enzymes are needed because the substrate specificity of the lipidation machinery is restricted. Altogether, the production of next-generation lipidated biologics will require the development of new synthesis technologies that expand the array of readily obtained compounds.
In summary, new lipidation methodologies coupled with a better understanding of the mechanisms underlying the efficacy of lipidated therapeutics will lead to next-generation lipidated drugs with more diverse and complex properties, tailored to specific applications, accelerating the development of new therapies for a large variety of diseases.
Acknowledgements
We would like to acknowledge the following support: the North Carolina Biotechnology Center grant # 2018-BIG-6503, ONR YIP # 12043956, and DOE EERE grant #EE0007563, as well as the NIH CBTE Training Grant #T32GM008555.
Footnotes
Conflicts of interest
R. Menacho-Melgar and M.D. Lynch have prepared and filed patent application on methods for the biosynthesis of lipidated peptides and proteins.
References
- [1].Otto R, Santagostino A, Schrader U, Rapid Growth in Biopharma: Challenges and Opportunities, https://www.mckinsey.com/industries/pharmaceuticals-and-medical-products/our-insights/rapid-growth-in-biopharma, (2018) , Accessed date: 7 October 2018.
- [2].Economist The, Large Going, https://www.economist.com/business/2014/12/30/going-large, (2018) , Accessed date: 7 October 2018.
- [3].Fosgerau K, Hoffmann T, Peptide therapeutics: current status and future directions, Drug Discov. Today 20 (1) (2015) 122–128. [DOI] [PubMed] [Google Scholar]
- [4].Smietana K, Siatkowski M, Moller M, Trends in clinical success rates. Nature reviews, Drug Des. Discov 15 (6) (2016) 379–380. [DOI] [PubMed] [Google Scholar]
- [5].Kinch MS, An overview of FDA-approved biologics medicines, Drug Discov. Today 20 (4) (2015) 393–398. [DOI] [PubMed] [Google Scholar]
- [6].Hooven MD, Opportunities and Challenges in Biologic Drug Delivery, https://www.americanpharmaceuticalreview.com/Featured-Articles/345540-Opportunities-and-Challenges-in-Biologic-Drug-Delivery, (2018) , Accessed date: 29 October 2018.
- [7].Cutfield WS, Derraik JG, Gunn AJ, Reid K, Delany T, Robinson E, Hofman PL, Non-compliance with growth hormone treatment in children is common and impairs linear growth, PLoS One 6 (1) (2011) e16223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kontermann RE, Strategies for extended serum half-life of protein therapeutics, Curr. Opin. Biotechnol 22 (6) (2011) 868–876. [DOI] [PubMed] [Google Scholar]
- [9].Yin L, Chen X, Vicini P, Rup B, Hickling TP, Therapeutic outcomes, assessments, risk factors and mitigation efforts of immunogenicity of therapeutic protein products, Cell. Immunol 295 (2) (2015) 118–126. [DOI] [PubMed] [Google Scholar]
- [10].Varkouhi AK, Scholte M, Storm G, Haisma HJ, Endosomal escape pathways for delivery of biologicals, J. Control. Release 151 (3) (2011) 220–228. [DOI] [PubMed] [Google Scholar]
- [11].Dumont JA, Bitonti AJ, Clark D, Evans S, Pickford M, Newman SP, Delivery of an erythropoietin-Fc fusion protein by inhalation in humans through an immunoglobulin transport pathway, J. Aerosol Med 18 (3) (2005) 294–303. [DOI] [PubMed] [Google Scholar]
- [12].Vallee S, Rakhe S, Reidy T, Walker S, Lu Q, Sakorafas P, Low S, Bitonti A, Pulmonary administration of interferon Beta-1a-fc fusion protein in non-human primates using an immunoglobulin transport pathway, J. Interferon Cytokine Res 32 (4) (2012) 178–184. [DOI] [PubMed] [Google Scholar]
- [13].Patel LN, Wang J, Kim KJ, Borok Z, Crandall ED, Shen WC, Conjugation with cationic cell-penetrating peptide increases pulmonary absorption of insulin, Mol. Pharm 6 (2) (2009) 492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gautam A, Nanda JS, Samuel JS, Kumari M, Priyanka P, Bedi G, Nath SK, Mittal G, Khatri N, Raghava GPS, Topical delivery of protein and peptide using novel cell penetrating peptide IMTeP8, Sci. Rep 6 (2016) 26,278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kauffman WB, Fuselier T, He J, Wimley WC, Mechanism matters: a taxonomy of cell penetrating peptides, Trends Biochem. Sci 40 (12) (2015) 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ducore JM, Miguelino MG, Powell JS, Alprolix (recombinant Factor IX Fc fusion protein): extended half-life product for the prophylaxis and treatment of hemophilia B, Expert. Rev. Hematol 7 (5) (2014) 559–571. [DOI] [PubMed] [Google Scholar]
- [17].Golor G, Bensen-Kennedy D, Haffner S, Easton R, Jung K, Moises T, Lawo JP, Joch C, Veldman A, Safety and pharmacokinetics of a recombinant fusion protein linking coagulation factor VIIa with albumin in healthy volunteers, J. Thromb. Haemost 11 (11) (2013) 1977–1985. [DOI] [PubMed] [Google Scholar]
- [18].Adabi E, Saebi F, Moradi Hasan-Abad A, Teimoori-Toolabi L, Kardar GA, Evaluation of an albumin-binding domain protein fused to recombinant human IL-2 and its effects on the bioactivity and serum half-life of the cytokine, Iran. Biomed. J 21 (2) (2017) 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Miyakawa N, Nishikawa M, Takahashi Y, Ando M, Misaka M, Watanabe Y, Takakura Y, Prolonged circulation half-life of interferon gamma activity by gene delivery of interferon gamma-serum albumin fusion protein in mice, J. Pharm. Sci 100 (6) (2011) 2350–2357. [DOI] [PubMed] [Google Scholar]
- [20].Langenheim JF, Chen WY, Improving the pharmacokinetics/pharmacodynamics of prolactin, GH, and their antagonists by fusion to a synthetic albumin-binding peptide, J. Endocrinol 203 (3) (2009) 375–387. [DOI] [PubMed] [Google Scholar]
- [21].Kim D, Jeon H, Ahn S, Choi WI, Kim S, Jon S, An approach for half-life extension and activity preservation of an anti-diabetic peptide drug based on genetic fusion with an albumin-binding aptide, J. Control. Release 256 (2017) 114–120. [DOI] [PubMed] [Google Scholar]
- [22].Swierczewska M, Lee KC, Lee S, What is the future of PEGylated therapies? Expert Opin. Emerg. Drugs 20 (4) (2015) 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Harris JM, Martin NE, Modi M, Pegylation: a novel process for modifying pharmacokinetics, Clin. Pharmacokinet 40 (7) (2001) 539–551. [DOI] [PubMed] [Google Scholar]
- [24].Zhang C, Desai R, Perez-Luna V, Karuri N, PEGylation of lysine residues improves the proteolytic stability of fibronectin while retaining biological activity, Biotechnol. J 9 (8) (2014) 1033–1043. [DOI] [PubMed] [Google Scholar]
- [25].Gefen T, Vaya J, Khatib S, Harkevich N, Artoul F, Heller ED, Pitcovski J, Aizenshtein E, The impact of PEGylation on protein immunogenicity, Int. Immunopharmacol 15 (2) (2013) 254–259. [DOI] [PubMed] [Google Scholar]
- [26].Mima Y, Hashimoto Y, Shimizu T, Kiwada H, Ishida T, Anti-PEG IgM is a major contributor to the accelerated blood clearance of polyethylene glycol-conjugated protein, Mol. Pharm 12 (7) (2015) 2429–2435. [DOI] [PubMed] [Google Scholar]
- [27].Levin D, Golding B, Strome SE, Sauna ZE, Fc fusion as a platform technology: potential for modulating immunogenicity, Trends Biotechnol. 33 (1) (2015) 27–34. [DOI] [PubMed] [Google Scholar]
- [28].Giugliani R, Giugliani L, de Oliveira Poswar F, Donis KC, Corte AD, Schmidt M, Boado RJ, Nestrasil I, Nguyen C, Chen S, Pardridge WM, Neurocognitive and somatic stabilization in pediatric patients with severe Mucopolysaccharidosis Type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): an open label phase 1–2 trial, Orphanet J. Rare Dis 13 (1) (2018) 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sonoda H, Morimoto H, Yoden E, Koshimura Y, Kinoshita M, Golovina G, Takagi H, Yamamoto R, Minami K, Mizoguchi A, Tachibana K, Hirato T, Takahashi K, A blood-brain-barrier-penetrating anti-human transferrin receptor antibody fusion protein for neuronopathic mucopolysaccharidosis II, Mol. Ther 26 (5) (2018) 1366–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Guidotti G, Brambilla L, Rossi D, Cell-penetrating peptides: from basic research to clinics, Trends Pharmacol. Sci 38 (4) (2017) 406–424. [DOI] [PubMed] [Google Scholar]
- [31].Lanyon-Hogg T, Faronato M, Serwa RA, Tate EW, Dynamic protein acylation: new substrates, mechanisms, and drug targets, Trends Biochem. Sci 42 (7) (2017) 566–581. [DOI] [PubMed] [Google Scholar]
- [32].Guan X, Fierke CA, Understanding protein palmitoylation: biological significance and enzymology, Sci. China Chem 54 (12) (2011) 1888–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lau J, Bloch P, Schaffer L, Pettersson I, Spetzler J, Kofoed J, Madsen K, Knudsen LB, McGuire J, Steensgaard DB, Strauss HM, Gram DX, Knudsen SM, Nielsen FS, Thygesen P, Reedtz-Runge S, Kruse T, Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide, J. Med. Chem 58 (18) (2015) 7370–7380. [DOI] [PubMed] [Google Scholar]
- [34].Trier S, Linderoth L, Bjerregaard S, Strauss HM, Rahbek UL, Andresen TL, Acylation of salmon calcitonin modulates in vitro intestinal peptide flux through membrane permeability enhancement, Eur. J. Pharm. Biopharm 96 (2015) 329–337. [DOI] [PubMed] [Google Scholar]
- [35].Bellmann-Sickert K, Elling CE, Madsen AN, Little PB, Lundgren K, Gerlach LO, Bergmann R, Holst B, Schwartz TW, Beck-Sickinger AG, Long-acting lipidated analogue of human pancreatic polypeptide is slowly released into circulation, J. Med. Chem 54 (8) (2011) 2658–2667. [DOI] [PubMed] [Google Scholar]
- [36].Kullar R, Chin JN, Edwards DJ, Parker D, Coplin WM, Rybak MJ, Pharmacokinetics of single-dose daptomycin in patients with suspected or con-firmed neurological infections, Antimicrob. Agents Chemother 55 (7) (2011) 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Thomas TA, Broun EC, Abildskov KM, Kubin CJ, Horan J, Yin MT, Cremers S, High performance liquid chromatography-mass spectrometry assay for polymyxin B1 and B2 in human plasma, Ther. Drug Monit 34 (4) (2012) 398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Stanley TL, Chen CY, Branch KL, Makimura H, Grinspoon SK, Effects of a growth hormone-releasing hormone analog on endogenous GH pulsatility and insulin sensitivity in healthy men, J. Clin. Endocrinol. Metab 96 (1) (2011) 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Danne T, Lupke K, Walte K, Von Schuetz W, Gall MA, Insulin detemir is characterized by a consistent pharmacokinetic profile across age-groups in children, adolescents, and adults with type 1 diabetes, Diabetes Care 26 (11) (2003) 3087–3092. [DOI] [PubMed] [Google Scholar]
- [40].Haahr H, Heise T, A review of the pharmacological properties of insulin degludec and their clinical relevance, Clin. Pharmacokinet 53 (9) (2014) 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thogersen H, Wilken M, Agerso H, Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration, J. Med. Chem 43 (9) (2000) 1664–1669. [DOI] [PubMed] [Google Scholar]
- [42].Jensen L, Helleberg H, Roffel A, van Lier JJ, Bjornsdottir I, Pedersen PJ, Rowe E, Derving Karsbol J, Pedersen ML, Absorption, metabolism and excretion of the GLP-1 analogue semaglutide in humans and nonclinical species, Eur. J. Pharm. Sci 104 (2017) 31–41. [DOI] [PubMed] [Google Scholar]
- [43].Battelino T, Rasmussen MH, De Schepper J, Zuckerman-Levin N, Gucev Z, Savendahl L, Somapacitan, a once-weekly reversible albumin-binding GH derivative, in children with GH deficiency: a randomized dose-escalation trial, Clin. Endocrinol 87 (4) (2017) 350–358. [DOI] [PubMed] [Google Scholar]
- [44].Ambery PD, Klammt S, Posch MG, Petrone M, Pu W, Rondinone C, Jermutus L, Hirshberg B, MEDI0382, a GLP-1/glucagon receptor dual agonist, meets safety and tolerability endpoints in a single-dose, healthy-subject, randomized, Phase 1 study, Br. J. Clin. Pharmacol 84 (10) (2018) 2325–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, Cui X, Briere DA, Cabrera O, Roell WC, Kuchibhotla U, Moyers JS, Benson CT, Gimeno RE, D’Alessio DA, Haupt A, LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept, Mol. Metab 18 (December 2018) 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Novo Nordisk A/S, Research Study of a New Medicine (NNC9204–1706) in People With Overweight or Obesity (ClinicalTrials.gov Identifier: NCT03661879), https://clinicaltrials.gov/ct2/show/NCT03661879, (2018) , Accessed date: 6 November 2018.
- [47].Ramírez-Andersen HS, Behrens C, Buchardt J, Fels JJ, Folkesson CG, Jianhe C, Nørskov-Lauritsen L, Nielsen PF, Reslow M, Rischel C, Su J, Thygesen P, Wiberg C, Zhao X, Wenjuan X, Johansen NL, Long-acting human growth hormone analogue by noncovalent albumin binding, Bioconjug. Chem 29 (9) (2018) 3129–3143. [DOI] [PubMed] [Google Scholar]
- [48].Johannsson G, Feldt-Rasmussen U, Håkonsson IH, Biering H, Rodien P, Tahara S, Toogood A, Rasmussen MH, Karges W, Mann A, Christiansen JS, Hansen TK, Andersen M, Borresen S, Borson-Chazot F, Kerlan V, Cariou B, Verges B, Matsuno A, Takano K, Tagami T, Takahashi Y, Takahashi T, Yamamoto M, Höybye C, Erfurth EM, Drake W, Higham C, Murray R, Brooke A, Safety and convenience of once-weekly somapacitan in adult GH deficiency: a 26-week randomized, controlled trial, Eur. J. Endocrinol 178 (2018) 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang J, Chow D, Heiati H, Shen WC, Reversible lipidization for the oral delivery of salmon calcitonin, J. Control. Release 88 (3) (2003) 369–380. [DOI] [PubMed] [Google Scholar]
- [50].Wang J, Wu D, Shen WC, Structure-activity relationship of reversibly lipidized peptides: studies of fatty acid-desmopressin conjugates, Pharm. Res 19 (5) (2002) 609–614. [DOI] [PubMed] [Google Scholar]
- [51].Yuan L, Wang J, Shen WC, Reversible lipidization prolongs the pharmacological effect, plasma duration, and liver retention of octreotide, Pharm. Res 22 (2) (2005) 220–227. [DOI] [PubMed] [Google Scholar]
- [52].Hamilton-Wessler M, Ader M, Dea M, Moore D, Jorgensen PN, Markussen J, Bergman RN, Mechanism of protracted metabolic effects of fatty acid acylated insulin, NN304, in dogs: retention of NN304 by albumin, Diabetologia 42 (10) (1999) 1254–1263. [DOI] [PubMed] [Google Scholar]
- [53].Andersen JT, Dalhus B, Viuff D, Ravn BT, Gunnarsen KS, Plumridge A, Bunting K, Antunes F, Williamson R, Athwal S, Allan E, Evans L, Bjoras M, Kjaerulff S, Sleep D, Sandlie I, Cameron J, Extending serum half-life of albumin by engineering neonatal Fc receptor (FcRn) binding, J. Biol. Chem 289 (19) (2014) 13,492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, Anderson CL, The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan, J. Exp. Med 197 (3) (2003) 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Quinlan GJ, Martin GS, Evans TW, Albumin: biochemical properties and therapeutic potential, Hepatology (Baltimore, Md.) 41 (6) (2005) 1211–1219. [DOI] [PubMed] [Google Scholar]
- [56].Plum A, Jensen LB, Kristensen JB, In vitro protein binding of liraglutide in human plasma determined by reiterated stepwise equilibrium dialysis, J. Pharm. Sci 102 (8) (2013) 2882–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Matthews JE, Stewart MW, De Boever EH, Dobbins RL, Hodge RJ, Walker SE, Holland MC, Bush MA, Pharmacodynamics, pharmacokinetics, safety, and tolerability of albiglutide, a long-acting glucagon-like peptide-1 mimetic, in patients with type 2 diabetes, J. Clin. Endocrinol. Metab 93 (12) (2008) 4810–4817. [DOI] [PubMed] [Google Scholar]
- [58].Markussen J, Havelund S, Kurtzhals P, Andersen AS, Halstrom J, Hasselager E, Larsen UD, Ribel U, Schaffer L, Vad K, Jonassen I, Soluble, fatty acid acylated insulins bind to albumin and show protracted action in pigs, Diabetologia 39 (3) (1996) 281–288. [DOI] [PubMed] [Google Scholar]
- [59].Jacobsen LV, Flint A, Olsen AK, Ingwersen SH, Liraglutide in type 2 diabetes mellitus: clinical pharmacokinetics and pharmacodynamics, Clin. Pharmacokinet 55 (2016) 657–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Malm-Erjefalt M, Bjornsdottir I, Vanggaard J, Helleberg H, Larsen U, Oosterhuis B, van Lier JJ, Zdravkovic M, Olsen AK, Metabolism and excretion of the once-daily human glucagon-like peptide-1 analog liraglutide in healthy male subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase, Drug Metab. Dispos 38 (11) (2010) 1944–1953. [DOI] [PubMed] [Google Scholar]
- [61].Kurtzhals P, Havelund S, Jonassen I, Kiehr B, Larsen UD, Ribel U, Markussen J, Albumin binding of insulins acylated with fatty acids: characterization of the ligand-protein interaction and correlation between binding affinity and timing of the insulin effect in vivo, Biochem. J 312 (Pt 3) (1995) 725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fujiwara S, Amisaki T, Fatty acid binding to serum albumin: molecular simulation approaches, Biochim. Biophys. Acta 1830 (12) (2013) 5427–5434. [DOI] [PubMed] [Google Scholar]
- [63].Spector AA, Fatty acid binding to plasma albumin, J. Lipid Res 16 (3) (1975) 165–179. [PubMed] [Google Scholar]
- [64].Bhattacharya AA, Grune T, Curry S, Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin, J. Mol. Biol 303 (5) (2000) 721–732. [DOI] [PubMed] [Google Scholar]
- [65].Wang Y, Lomakin A, Kanai S, Alex R, Belli S, Donzelli M, Benedek GB, The molecular basis for the prolonged blood circulation of lipidated incretin peptides: Peptide oligomerization or binding to serum albumin? J. Control. Release 241 (2016) 25–33. [DOI] [PubMed] [Google Scholar]
- [66].Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thogersen H, Wilken M, Johansen NL, Structure-activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness, J. Med. Chem 50 (24) (2007) 6126–6132. [DOI] [PubMed] [Google Scholar]
- [67].Havelund S, Plum A, Ribel U, Jonassen I, Volund A, Markussen J, Kurtzhals P, The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin, Pharm. Res 21 (8) (2004) 1498–1504. [DOI] [PubMed] [Google Scholar]
- [68].Dea MK, Hamilton-Wessler M, Ader M, Moore D, Schaffer L, Loftager M, Volund A, Bergman RN, Albumin binding of acylated insulin (NN304) does not deter action to stimulate glucose uptake, Diabetes 51 (3) (2002) 762–769. [DOI] [PubMed] [Google Scholar]
- [69].Elbrond B, Jakobsen G, Larsen S, Agerso H, Jensen LB, Rolan P, Sturis J, Hatorp V, Zdravkovic M, Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects, Diabetes Care 25 (8) (2002) 1398–1404. [DOI] [PubMed] [Google Scholar]
- [70].Wexler-Cohen Y, Shai Y, Membrane-anchored HIV-1 N-heptad repeat peptides are highly potent cell fusion inhibitors via an altered mode of action, PLoS Pathog. 5 (7) (2009) e1000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].D’Errico G, D’Ursi AM, Marsh D, Interaction of a peptide derived from glyco-protein gp36 of feline immunodeficiency virus and its lipoylated analogue with phospholipid membranes, Biochemistry 47 (19) (2008) 5317–5327. [DOI] [PubMed] [Google Scholar]
- [72].Bode SA, Thévenin M, Bechara C, Sagan S, Bregant S, Lavielle S, Chassaing G, Burlina F, Self-assembling mini cell-penetrating peptides enter by both direct translocation and glycosaminoglycan-dependent endocytosis, Chem. Commun 57 (2012). [DOI] [PubMed] [Google Scholar]
- [73].Oh D, Nasrolahi Shirazi A, Northup K, Sullivan B, Tiwari RK, Bisoffi M, Parang K, Enhanced cellular uptake of short polyarginine peptides through fatty acylation and cyclization, Mol. Pharm 11 (8) (2014) 2845–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Trier S, Linderoth L, Bjerregaard S, Andresen TL, Rahbek UL, Acylation of glucagon-like peptide-2: interaction with lipid membranes and in vitro intestinal permeability, PLoS One 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lee JS, Tung CH, Lipo-oligoarginines as effective delivery vectors to promote cellular uptake, Mol. BioSyst 6 (10) (2010) 2049–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Nelson AR, Borland L, Allbritton NL, Sims CE, Myristoyl-based transport of peptides into living cells, Biochemistry 46 (51) (2007) 14,771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Missirlis D, Khant H, Tirrell M, Mechanisms of peptide amphiphile internalization by SJSA-1 cells in vitro, Biochemistry 48 (15) (2009) 3304–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Swiecicki JM, Di Pisa M, Lippi F, Chwetzoff S, Mansuy C, Trugnan G, Chassaing G, Lavielle S, Burlina F, Unsaturated acyl chains dramatically enhanced cellular uptake by direct translocation of a minimalist oligo-arginine lipopeptide, Chem. Commun. (Camb., Engl.) 51 (78) (2015) 14656–14659. [DOI] [PubMed] [Google Scholar]
- [79].Miersch S, Sidhu SS, Intracellular targeting with engineered proteins, F1000Research 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B, Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake, J. Biol. Chem 278 (1) (2003) 585–590. [DOI] [PubMed] [Google Scholar]
- [81].Eisele F, Kuhlmann J, Waldmann H, Synthesis and membrane binding properties of a lipopeptide fragment from influenza virus a hemagglutinin, Chemistry (Weinheim an der Bergstrasse, Germany) 8 (15) (2002) 3362–3376. [DOI] [PubMed] [Google Scholar]
- [82].Tsuji M, Ueda S, Hirayama T, Okuda K, Sakaguchi Y, Isono A, Nagasawa H, FRET-based imaging of transbilayer movement of pepducin in living cells by novel intracellular bioreductively activatable fluorescent probes, Org. Biomol. Chem 11 (18) (2013) 3030–3037. [DOI] [PubMed] [Google Scholar]
- [83].Katayama S, Hirose H, Takayama K, Nakase I, Futaki S, Acylation of octaarginine: Implication to the use of intracellular delivery vectors, J. Control. Release 149 (1) (2011) 29–35. [DOI] [PubMed] [Google Scholar]
- [84].Aroda VR, Rosenstock J, Terauchi Y, Jeppesen OLE, Christiansen E, Hertz CL, Haluzik M, Effect and safety of oral semaglutide monotherapy in type 2 diabetes—PIONEER 1 trial, Diabetes 67 (Supplement 1) (2018). [Google Scholar]
- [85].Davies M, Pieber TR, Hartoft-Nielsen ML, Hansen OKH, Jabbour S, Rosenstock J, Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes: a randomized clinical trial, JAMA 318 (15) (2017) 1460–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Connor A, Borregaard J, Buckley ST, Donsmark M, Hartoft-Nielsen ML, Maarbjerg SJ, Sondergaard FL, Vegge A, Knudsen LB, Baekdal TA, Site of absorption of an oral formulation of semaglutide, Diabetes 66 (Supplement 1) (2017). [Google Scholar]
- [87].Bjerregaard S, Nielsen FS, Sauerberg P, Solid Compositions Comprising a GLP-1 Agonist and a Salt of N-(8-(2-Hydroxybenzoyl)Amino)Caprylic Acid. WO2012080471A1, (2012).
- [88].Karsdal MA, Riis BJ, Mehta N, Stern W, Arbit E, Christiansen C, Henriksen K, Lessons learned from the clinical development of oral peptides, Br. J. Clin. Pharmacol 79 (5) (2015) 720–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Granhall C, Donsmark M, Golor G, Sondergaard FL, Thomsen M, Safety, tolerability, and pharmacokinetics of multiple once-daily dosing of oral semaglutide in healthy males and in males with T2D, Diabetes 66 (Supplement 1) (2017). [Google Scholar]
- [90].Choi YL, Park EJ, Kim E, Na DH, Shin YH, Dermal stability and in vitro skin permeation of collagen pentapeptides (KTTKS and palmitoyl-KTTKS), Biomol. Ther. (Seoul) 22 (2014) 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Rocco D, Ross J, Murray PE, Caccetta R, Acyl lipidation of a peptide: effects on activity and epidermal permeability in vitro, Drug Des. Dev. Ther 10 (2016) 2203–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yamamoto A, Setoh K, Murakami M, Shironoshita M, Kobayashi T, Fujimoto K, Okada N, Fujita T, Muranishi S, Enhanced transdermal delivery of phenylalanylglycine by chemical modification with various fatty acids, Int. J. Pharm 250 (1) (2003) 119–128. [DOI] [PubMed] [Google Scholar]
- [93].Patton JS, Fishburn CS, Weers JG, The lungs as a portal of entry for systemic drug delivery, Proc. Am. Thorac. Soc 1 (4) (2004) 338–344. [DOI] [PubMed] [Google Scholar]
- [94].Morales JO, Fathe KR, Brunaugh A, Ferrati S, Li S, Montenegro-Nicolini M, Mousavikhamene Z, McConville JT, Prausnitz MR, Smyth HDC, Challenges and future prospects for the delivery of biologics: oral mucosal, pulmonary, and transdermal routes, AAPS J. 19 (3) (2017) 652–668. [DOI] [PubMed] [Google Scholar]
- [95].Buchackert Y, Rummel S, Vohwinkel CU, Gabrielli NM, Grzesik BA, Mayer K, Herold S, Morty RE, Seeger W, Vadasz I, Megalin mediates transepithelial albumin clearance from the alveolar space of intact rabbit lungs, J. Physiol 590 (20) (2012) 5167–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M, Jansson-Löfmark R, Naylor J, Rossi A, Bednarek MA, Bhagroo N, Salari H, Will S, Oldham S, Hansen G, Feigh M, Klein T, Grimsby J, Maguire S, Jermutus L, Rondinone CM, Coghlan MP, Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates, Diabetes Obes. Metab 18 (2016) 1176–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Dahl K, Schaffer L, Kruse T, Human Amylin Analogues. WO2013156594A1, (2013).
- [98].Ostergaard S, Wulff BS, Frieboes KWC, Wieczorek B, Selective pyy Compounds and Uses Thereof, (2016).
- [99].Wieczorek B, Tagmose TM, Sassorum K, Andersen B, Olsen J, Fgf21 Derivatives and Uses Thereof. WO2016102562A1, (2016).
- [100].van Witteloostuijn SB, Mannerstedt K, Wismann P, Bech EM, Thygesen MB, Vrang N, Jelsing J, Jensen KJ, Pedersen SL, Neoglycolipids for prolonging the effects of peptides: self-assembling glucagon-like peptide 1 analogues with albumin binding properties and potent in vivo efficacy, Mol. Pharm 14 (1) (2017) 193–205. [DOI] [PubMed] [Google Scholar]
- [101].Ikushima I, Jensen L, Flint A, Nishida T, Zacho J, Irie S, A randomized trial investigating the pharmacokinetics, pharmacodynamics, and safety of subcutaneous semaglutide once-weekly in healthy male japanese and caucasian subjects, Adv. Ther 35 (2018) 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Bech EM, Pedersen SL, Jensen KJ, Chemical strategies for half-life extension of biopharmaceuticals: lipidation and its alternatives, ACS Med. Chem. Lett 9 (7) (2018) 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Todorovic A, Holder JR, Bauzo RM, Scott JW, Kavanagh R, Abdel-Malek Z, Haskell-Luevano C, N-terminal fatty acylated His-dPhe-Arg-Trp-NH(2) tetrapep-tides: influence of fatty acid chain length on potency and selectivity at the mouse melanocortin receptors and human melanocytes, J. Med. Chem 48 (9) (2005) 3328–3336. [DOI] [PubMed] [Google Scholar]
- [104].Doyle JR, Krishnaji ST, Zhu G, Xu ZZ, Heller D, Ji RR, Levy BD, Kumar K, Kopin AS, Development of a membrane-anchored chemerin receptor agonist as a novel modulator of allergic airway inflammation and neuropathic pain, J. Biol. Chem 289 (19) (2014) 13,385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Skovbakke SL, Heegaard PM, Larsen CJ, Franzyk H, Forsman H, Dahlgren C, The proteolytically stable peptidomimetic Pam-(Lys-betaNSpe)6-NH2 selectively inhibits human neutrophil activation via formyl peptide receptor 2, Biochem. Pharmacol 93 (2) (2015) 182–195. [DOI] [PubMed] [Google Scholar]
- [106].Chekhonin VP, Kabanov AV, Zhirkov YA, Morozov GV, Fatty acid acylated Fab-fragments of antibodies to neurospecific proteins as carriers for neuroleptic targeted delivery in brain, FEBS Lett. 287 (1–2) (1991) 149–152. [DOI] [PubMed] [Google Scholar]
- [107].Choi JK, Ho J, Curry S, Qin D, Bittman R, Hamilton JA, Interactions of very long-chain saturated fatty acids with serum albumin, J. Lipid Res 43 (7) (2002) 1000–1010. [DOI] [PubMed] [Google Scholar]
- [108].Shafrir E, Gatt S, Khasis S, Partition of fatty acids of 20–24 carbon atoms between serum albumin and lipoproteins, Biochim. Biophys. Acta 98 (1965) 365–371. [DOI] [PubMed] [Google Scholar]
- [109].Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, Zimmermann T, Koteliansky V, Manoharan M, Stoffel M, Mechanisms and optimization of in vivo delivery of lipophilic siRNAs, Nat. Biotechnol 25 (10) (2007) 1149–1157. [DOI] [PubMed] [Google Scholar]
- [110].Thaxton CS, Rink JS, Naha PC, Cormode DP, Lipoproteins and lipoprotein mimetics for imaging and drug delivery, Adv. Drug Deliv. Rev 106 (Pt A) (2016) 116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Stoekenbroek RM, Stroes ES, Hovingh GK, ApoA-I mimetics, Handb. Exp. Pharmacol 224 (2015) 631–648. [DOI] [PubMed] [Google Scholar]
- [112].Baker MP, Reynolds HM, Lumicisi B, Bryson CJ, Immunogenicity of protein therapeutics: the key causes, consequences and challenges, Self 1 (4) (2010) 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ, Garratty G, Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients, Cancer 110 (1) (2007) 103–111. [DOI] [PubMed] [Google Scholar]
- [114].Wang YM, Wang J, Hon YY, Zhou L, Fang L, Ahn HY, Evaluating and reporting the immunogenicity impacts for biological products–a clinical pharmacology perspective, AAPS J. 18 (2) (2016) 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Zhang P, Sun F, Liu S, Jiang S, Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation, J. Control. Release 244 (Pt B) (28 December 2016) 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Buse JB, Garber A, Rosenstock J, Schmidt WE, Brett JH, Videbaek N, Holst J, Nauck M, Liraglutide treatment is associated with a low frequency and magnitude of antibody formation with no apparent impact on glycemic response or increased frequency of adverse events: results from the Liraglutide Effect and Action in Diabetes (LEAD) trials, J. Clin. Endocrinol. Metab 96 (6) (2011) 1695–1702. [DOI] [PubMed] [Google Scholar]
- [117].Bartley PC, Bogoev M, Larsen J, Philotheou A, Long-term efficacy and safety of insulin detemir compared to Neutral Protamine Hagedorn insulin in patients with Type 1 diabetes using a treat-to-target basal-bolus regimen with insulin aspart at meals: a 2-year, randomized, controlled trial, Diabet. Med 25 (4) (2008) 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Schultz HS, Ostergaard S, Sidney J, Lamberth K, Sette A, The effect of acylation with fatty acids and other modifications on HLA class II:peptide binding and T cell stimulation for three model peptides, PLoS One 13 (5) (2018) e0197407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Cloake NC, Beaino W, Trifilieff E, Greer JM, Thiopalmitoylation of altered peptide ligands enhances their protective effects in an animal model of multiple sclerosis, J. Immunol. (Baltim., Md.) 192 (5) (2014) 2244–2251. [DOI] [PubMed] [Google Scholar]
- [120].Jackson DC, Lau YF, Le T, Suhrbier A, Deliyannis G, Cheers C, Smith C, Zeng W, Brown LE, A totally synthetic vaccine of generic structure that targets Toll-like receptor 2 on dendritic cells and promotes antibody or cytotoxic T cell responses, Proc. Natl. Acad. Sci. U. S. A 101 (43) (2004) 15,440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zeng W, Eriksson EM, Lew A, Jackson DC, Lipidation of intact proteins produces highly immunogenic vaccine candidates, Mol. Immunol 48 (4) (2011) 490–496. [DOI] [PubMed] [Google Scholar]
- [122].Beekman NJ, Schaaper WM, Tesser GI, Dalsgaard K, Kamstrup S, Langeveld JP, Boshuizen RS, Meloen RH, Synthetic peptide vaccines: palmitoylation of peptide antigens by a thioester bond increases immunogenicity, J. Pept. Res 50 (5) (1997) 357–364. [DOI] [PubMed] [Google Scholar]
- [123].Pfender NA, Grosch S, Roussel G, Koch M, Trifilieff E, Greer JM, Route of uptake of palmitoylated encephalitogenic peptides of myelin proteolipid protein by antigen-presenting cells: importance of the type of bond between lipid chain and peptide and relevance to autoimmunity, J. Immunol. (Baltim., Md.) 180 (3) (2008) 1398–1404. [DOI] [PubMed] [Google Scholar]
- [124].Pfender N, Guéin E, Greer JM, Trifilieff E, Solid-phase synthesis of a biotin-labeled thiopalmitoylated myelin proteolipid protein epitope and application in the study of uptake of antigen by macrophages, Lett. Pept. Sci 10 (5) (2003) 581–588. [Google Scholar]
- [125].Bueno C, Lee KK, Chau LA, Lee-Chan E, Singh B, Strejan GH, Madrenas J, Mechanism of modulation of T cell responses by N-palmitoylated peptides, Eur. J. Immunol 34 (12) (2004) 3497–3507. [DOI] [PubMed] [Google Scholar]
- [126].Asada H, Douen T, Mizokoshi Y, Fujita T, Murakami M, Yamamoto A, Muranishi S, Stability of acyl derivatives of insulin in the small intestine: relative importance of insulin association characteristics in aqueous solution, Pharm. Res 11 (8) (1994) 1115–1120. [DOI] [PubMed] [Google Scholar]
- [127].Yuan L, Wang J, Shen WC, Reversible lipidization of somatostatin analogues for the liver targeting, Eur. J. Pharm. Biopharm 70 (2) (2008) 615–620. [DOI] [PubMed] [Google Scholar]
- [128].Antoniou AN, Blackwood SL, Mazzeo D, Watts C, Control of antigen presentation by a single protease cleavage site, Immunity 12 (4) (2000) 391–398. [DOI] [PubMed] [Google Scholar]
- [129].Deliyannis G, Jackson DC, Ede NJ, Zeng W, Hourdakis I, Sakabetis E, Brown LE, Induction of long-term memory CD8(+) T cells for recall of viral clearing responses against influenza virus, J. Virol 76 (9) (2002) 4212–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Morita D, Yamamoto Y, Mizutani T, Ishikawa T, Suzuki J, Igarashi T, Mori N, Shiina T, Inoko H, Fujita H, Iwai K, Tanaka Y, Mikami B, Sugita M, Crystal structure of the N-myristoylated lipopeptide-bound MHC class I complex, Nat. Commun 7 (2016) 10,356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].BenMohamed L, Thomas A, Druilhe P, Long-term multiepitopic cytotoxic-T-lymphocyte responses induced in chimpanzees by combinations of Plasmodium falciparum liver-stage peptides and lipopeptides, Infect. Immun 72 (2004) 4376–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Gunaratne R, Kumar S, Frederiksen JW, Stayrook S, Lohrmann JL, Perry K, Bompiani KM, Chabata CV, Thalji NK, Ho MD, Arepally G, Camire RM, Krishnaswamy S, Sullenger BA, Combination of aptamer and drug for reversible anticoagulation in cardiopulmonary bypass, Nat. Biotechnol 36 (7) (2018) 606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Keizer RJ, Huitema AD, Schellens JH, Beijnen JH, Clinical pharmacokinetics of therapeutic monoclonal antibodies, Clin. Pharmacokinet 49 (8) (2010) 493–507. [DOI] [PubMed] [Google Scholar]
- [134].Zhao L, Ji P, Li Z, Roy P, Sahajwalla CG, The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model, J. Clin. Pharmacol 53 (3) (2013) 314–325. [DOI] [PubMed] [Google Scholar]
- [135].Cruikshank WW, Doctrow SR, Falvo MS, Huffman K, Maciaszek J, Viglianti G, Raina J, Kornfeld H, Malfroy B, A lipidated anti-Tat antibody enters living cells and blocks HIV-1 viral replication, J. Acquir. Immune Defic. Syndr. Hum. Retrovirol 14 (3) (1997) 193–203. [DOI] [PubMed] [Google Scholar]
- [136].Huang X, Brazel CS, On the importance and mechanisms of burst release in matrix-controlled drug delivery systems, J. Control. Release 73 (2–3) (2001) 121–136. [DOI] [PubMed] [Google Scholar]
- [137].Jensen L, Kupcova V, Arold G, Pettersson J, Hjerpsted JB, Pharmacokinetics and tolerability of semaglutide in people with hepatic impairment, Diabetes Obes. Metab 20 (4) (2018) 998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Flint A, Nazzal K, Jagielski P, Hindsberger C, Zdravkovic M, Influence of hepatic impairment on pharmacokinetics of the human GLP-1 analogue, liraglutide, Br. J. Clin. Pharmacol 70 (6) (2010) 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Henriques ST, Melo MN, Castanho MA, How to address CPP and AMP trans-location? Methods to detect and quantify peptide internalization in vitro and in vivo (Review), Mol. Membr. Biol 24 (3) (2007) 173–184. [DOI] [PubMed] [Google Scholar]
- [140].Bechara C, Sagan S, Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 587 (12) (2013) 1693–1702. [DOI] [PubMed] [Google Scholar]
- [141].Sundy JS, Baraf HS, Yood RA, Edwards NL, Gutierrez-Urena SR, Treadwell EL, Vazquez-Mellado J, White WB, Lipsky PE, Horowitz Z, Huang W, Maroli AN, Waltrip RW 2nd, Hamburger SA, Becker MA, Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: two randomized controlled trials, JAMA 306 (7) (2011) 711–720. [DOI] [PubMed] [Google Scholar]
- [142].Ehrlich GK, Michel H, Truitt T, Riboulet W, Pop-Damkov P, Goelzer P, Hainzl D, Qureshi F, Lueckel B, Danho W, Conde-Knape K, Konkar A, Preparation and characterization of albumin conjugates of a truncated peptide YY analogue for half-life extension, Bioconjug. Chem 24 (12) (2013) 2015–2024. [DOI] [PubMed] [Google Scholar]
- [143].Mejuch T, Waldmann H, Synthesis of Lipidated Proteins, (2016). [DOI] [PubMed]
- [144].Abe H, Goto M, Kamiya N, Protein lipidation catalyzed by microbial transglutaminase, Chem. Eur. J 17 (50) (2011) 14,004–14,008. [DOI] [PubMed] [Google Scholar]
- [145].Gluck JM, Hoffmann S, Koenig BW, Willbold D, Single vector system for efficient N-myristoylation of recombinant proteins in E. coli, PLoS One 5 (4) (2010) e10081. [DOI] [PMC free article] [PubMed] [Google Scholar]