

Abstract
In the present in vitro studies, evidence is provided showing that glutathione (GSH) can undergo spontaneous, nonenzymatic auto-degradation. The initial cleavage of the Glu-Cys bond involves nucleophilic attack of the N-terminal amino group of GSH at the γ-carbonyl side chain, followed by an unusual cleavage of the Cys-Gly amide bond that is complete within 3–5 weeks in the physiological pH range. These observations may be useful for the development of novel biodegradable polymers and releasable prodrugs for sustained and targeted drug delivery applications.
Introduction.
Glutathione (GSH) is the most abundant thiol species in the cytoplasm and the major reducing agent for almost all biochemical processes. GSH is important in the synthesis of proteins and DNA, transport, enzyme activity, metabolism, and protection of cells [1]. Cellular GSH levels must be maintained, since they are constantly fluctuating due to degradation and consumption in various biological processes [2]. The intracellular concentration of GSH (1–10 mm) is substantially higher than the extracellular concentration (e.g., 2–20 μm in plasma) [3]. Extracellular GSH concentrations are relatively low, because GSH is readily oxidized to glutathione disulfide (GSSG) by direct interaction with free radicals or, more often, when GSH acts as a cofactor for antioxidant enzymes such as GSH peroxidases [4]. The efflux of GSSG from cells contributes to a net loss of intracellular GSH. GSH/GSSG is a very important redox couple that plays a crucial role in antioxidant defense, nutrient metabolism, and the regulation of this pathway is essential for whole body homeostasis [5]. GSH Deficiency in the cell contributes to oxidative stress, which plays a key role in aging and the pathogenesis of many diseases including Alzheimer s disease, Parkinson s disease, AIDS, cancer, and myocardial infarction [6]. The chemical structure of GSH is shown in Fig. 1.
Fig. 1. Chemical representation of GSH.
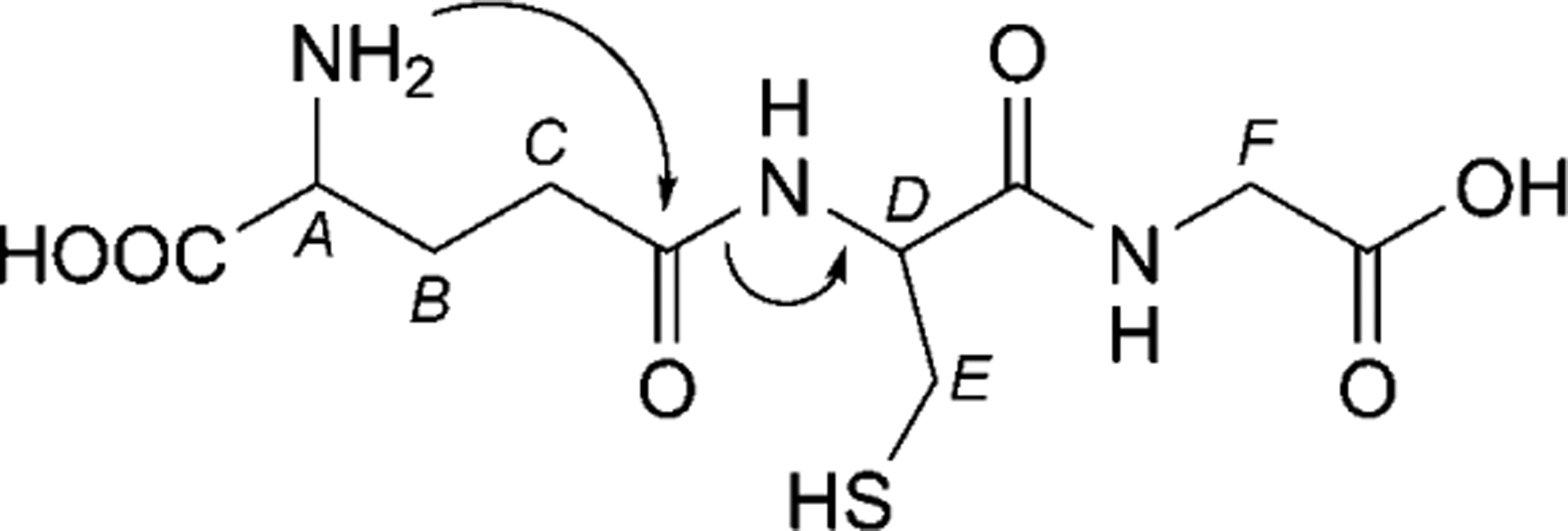
Annotations denote chemical shift assignments in the NMR spectra: A: Glu-α, B: Glu-β, C: Glu-γ, D: Cys-α, E: Cys-β, F: Gly-α. Arrows denote the first step in the proposed degradation mechanism.
The biosynthesis and metabolism of GSH, which proceeds through the γ-glutamyl cycle, has been studied and thoroughly characterized [7][8]. GSH Degradation is initiated by γ-glutamyl transpeptidase, which involves either the cleavage and transfer of the γ-glutamyl moiety from GSH to an acceptor amino acid, or the hydrolysis and release of glutamate accompanied by the release of cysteinyl-glycine. The resulting γ-glutamyl amino acids formed by γ-glutamyl transpeptidase are transported into cells where γ-glutamyl cyclotransferase converts these compounds into the corresponding amino acid and 5-oxo-l-proline. The intracellular enzyme 5-oxo-l-prolinase catalyzes the conversion of 5-oxo-l-proline to l-glutamate. The cysteinyl-glycine formed in the transpeptidase reaction is split by dipeptidase. It is known that γ-glutamylcysteine is formed intracellularly by the action of γ-glutamylcysteine synthetase [9], and intracellular γ-glutamylcysteine is converted into GSH by the action of glutathione synthetase [10].
Although, the γ-glutamyl cycle is well-established, the possibility of an alternative nonenzymatic GSH degradation pathway has not been explored. The oxidation of glutathione into GSSG using oxidizing agents and formation of pyroglutamic acid from GSH at high temperature was suggested in previous studies [11]. In addition to the well-known enzymatic reactions of GSH, there are previously unreported nonenzymatic side reactions occurring, which include oxidation and peptide bond cleavage. Such nonenzymatic reactions may be applicable for designing prodrugs that can be converted to active drugs by an intrinsic mechanism.
Prodrugs that are activated by intramolecular reaction (i.e., prodrugs that are partly or completely activated without the need for enzymatic contribution) are of great interest. A few examples of this type of prodrugs have been reported [12]. One natural example is the biosynthesis of luteinizing hormone releasing hormone (LHRH), in which spontaneous cyclization of the N-terminal glutamine residue results in the formation of pyroglutamyl peptide with release of NH3 [12][13].
We hypothesize that the nonenzymatic degradation of GSH (Scheme 1) occurs by intramolecular rearrangement similar to the LHRH reaction in which nucleophilic attack of the N-terminal amino group breaks the amide bond between Glu and Cys with the release of the N-terminal primary amine of Cys, thereby initiating peptide degradation. To determine the validity of the proposed mechanism, the nonenzymatic degradation of GSH was systematically investigated at three different pH values: 6.2, 6.8 (i.e., representative of a common pH range in cancer cells), and 7.4 (i.e., pH of plasma and the most commonly found pH in the body). In the current study, we demonstrate that nonenzymatic reactions contribute significantly to the degradation of GSH. In addition, the observed pH-dependent degradation mechanism may be exploited as a prodrug strategy for the selective cleavage of GSH and the release of chemotherapeutic agents in the tumor environment.
Scheme 1. Proposed Degradation Mechanism of GSH.
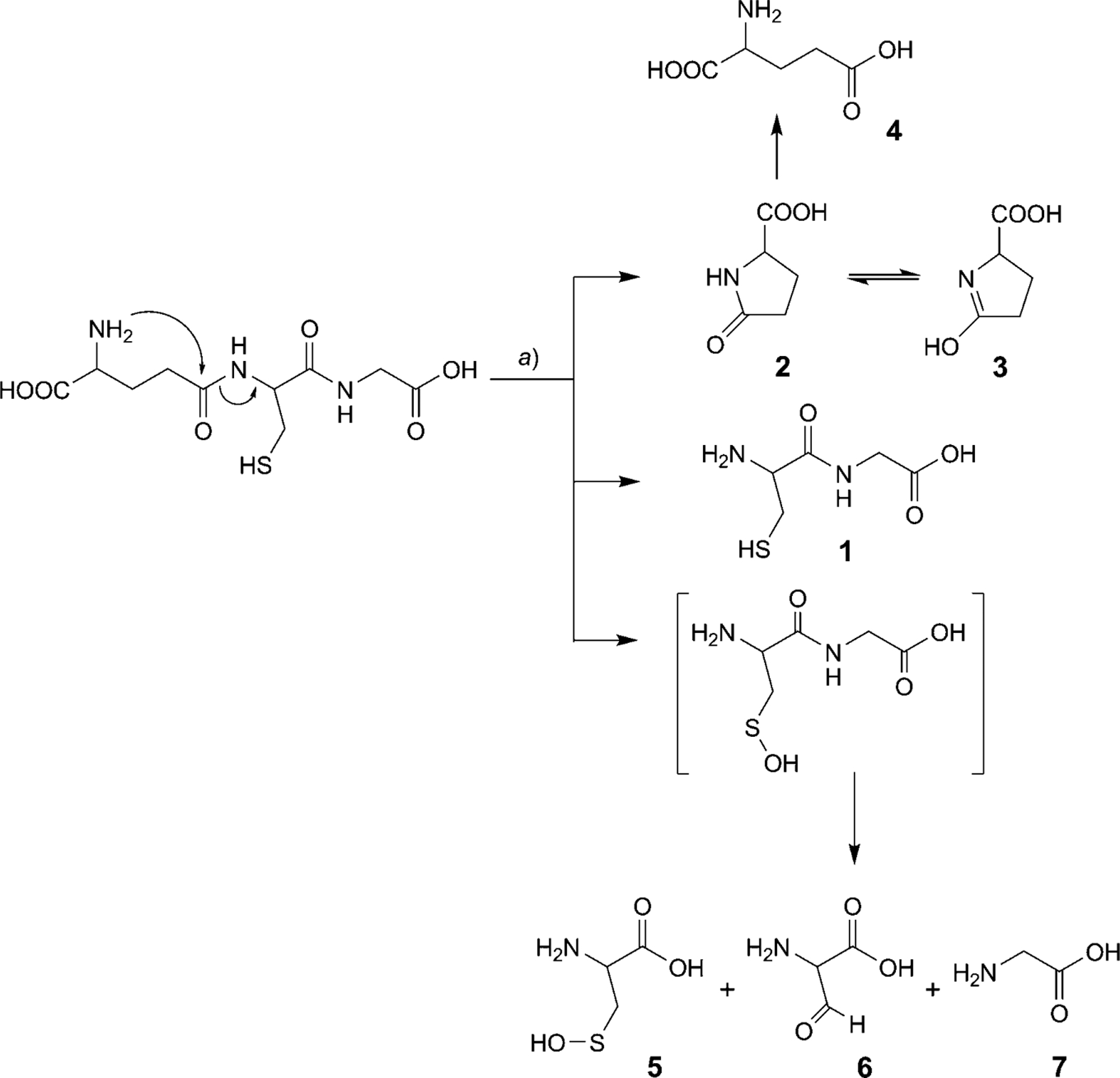
a) 37°, 100 mm sodium phosphate buffer at three different pH values (6.2, 6.8, and 7.4).
Results and Discussion.
GSH (150 mg) was dissolved in 100 mm sodium phosphate buffer (10 ml) at pH 6.2, 6.8, or 7.4, and incubated at 37°. Aliquots (100 μl) were taken at various time points from each of the three reaction mixtures. Sample aliquots were dried using a CentriVap and analyzed by 1H-NMR (for all time points) and LC/MS (for the last time point, when degradation was complete).
The NMR spectrum of GSH before degradation showed a well-resolved pattern of peaks (Exper. Part). The H-atoms (see Fig. 1) at A and D showed a triplet at 3.69 and 4.41 ppm, respectively, whereas the F H-atom showed a singlet at 3.82 ppm. Other signals belonging to the H-atoms B, C, and E appeared at chemical shifts 2.02, 2.41, and 2.8 ppm, respectively. It was observed that the singlet of the F H-atom shifted in all three pH samples. At pH 6.2, it is shifted upfield from 3.82 ppm and appears at 3.74 ppm, whereas, at pH 6.8 and 7.4, it has shifted from 3.82 to 3.61 ppm. The intensity of the Cys-α triplet (at 4.41 ppm, D) decreased with time. The rate of disappearance of the Cys-α peak differs with the change in pH (from 6.2 to 7.4). From the disappearance profile of the Cys-α triplet (D), we can unambiguously confirm that the amide bond between Glu and Cys breaks during the incubation at 37° giving rise to degradation products. NMR Resonance of D was, therefore, chosen for quantification studies (vide infra). In the later time points, signals arising due to degradation products can be seen as well. t-BuOH (0.15 μl per 750 μl of sample) was used as an internal standard for NMR studies, because it does not interact with GSH or with any degradation product as confirmed by comparison of NMR spectra with and without t-BuOH.
Half-lives for the amide bond cleavage between Glu and Cys in GSH at three different pH values, i.e., 7.4, 6.8, and 6.2, were determined by quantitative 1H-NMR recorded with 100 scans and a recycle delay of 15 s. Fig. 2,d, shows the changes in the 1H integral value for peak D derived from quantitative NMR spectra recorded with an interval of 2 d during the studies. Standard kinetics models were applied to the degradation data. Unfortunately, the data did not show a good fit to any standard kinetic model (i.e., zero-, first-, or second-order degradation). This suggests that the degradation mechanism follows a complex order. It was observed that degradation is faster at pH 7.4 (t1/2=12 d) than at pH 6.8 (t1/2=14 d) and pH 6.2 (t1/2=17 d).
Fig. 2. Section of 1D 1H-NMR spectra of GSH degradation studies recorded at pH 6.2 (a), 6.8 (b), and 7.4 (c).
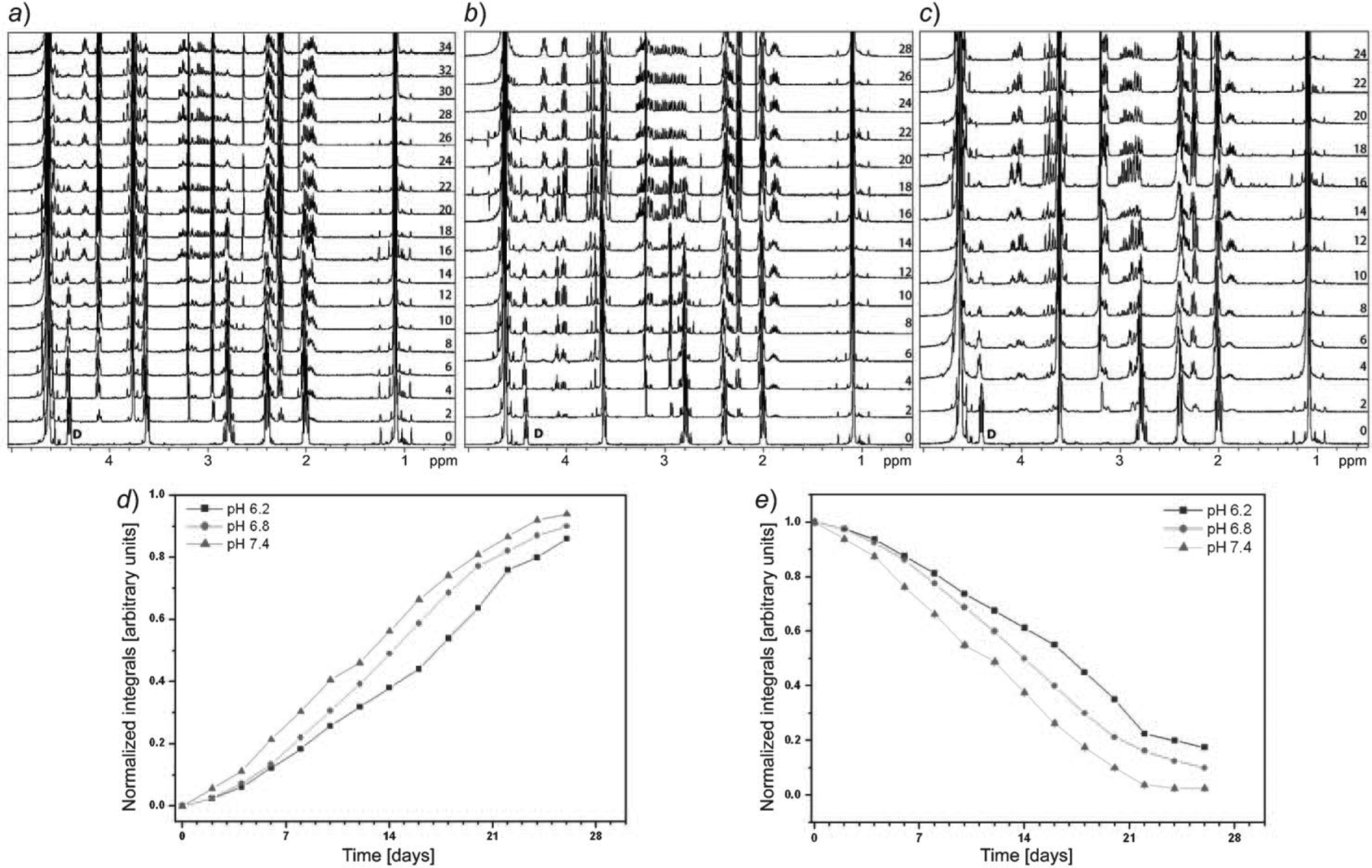
Drop in the intensity of the peak arising from protons D (annotated in 0 day spectrum) indicate cleavage of Glu-Cys bond in GSH which follows proposed degradation pathway. Numbers in panel a, b, and c denote days on which the spectra has been recorded during the studies. d) Half-life for the cleavage of the amide bond in GSH at three pH values: 7.4 (12 d), 6.8 (14 d), and 6.2 (17 d). NMR Integrals are derived for 1H peak arising from D. e) The increase in the concentration of the ‘β-pyroglutamic acid proton’ (at 1.89 ppm) during the course of study which is the first cleavage products. The error in the integral is generated from integrating the noise at a fixed ppm (9.9–10.0 ppm).
In the presence of γ-glutamyl transpeptidase, the amide bond cleavage between Glu and Cys in GSH was reported to occur in ca. 4–6 h as found in tissue extracts [14]. However, total formation of the first product in the absence of the enzyme varies with pH and requires ca. 34 d at pH 6.2, 28 d at pH 6.8, and 24 d at pH 7.4 (Fig. 2,a–c).
NMR Data show that, during the time course, the signal intensities of the α (4.02 ppm) and β (1.89 and 2.5 ppm) H-atoms of pyroglutamic acid (at all three pH values; Fig. 2,a–c) increases with time. For the quantification studies of the first cleavage product, pyroglutamic acid, NMR resonance of the β-pyroglutamic acid H-atom (1.89 ppm) was integrated, and the concentration vs. time curve of pyroglutamic acid is shown in Fig. 2,e.
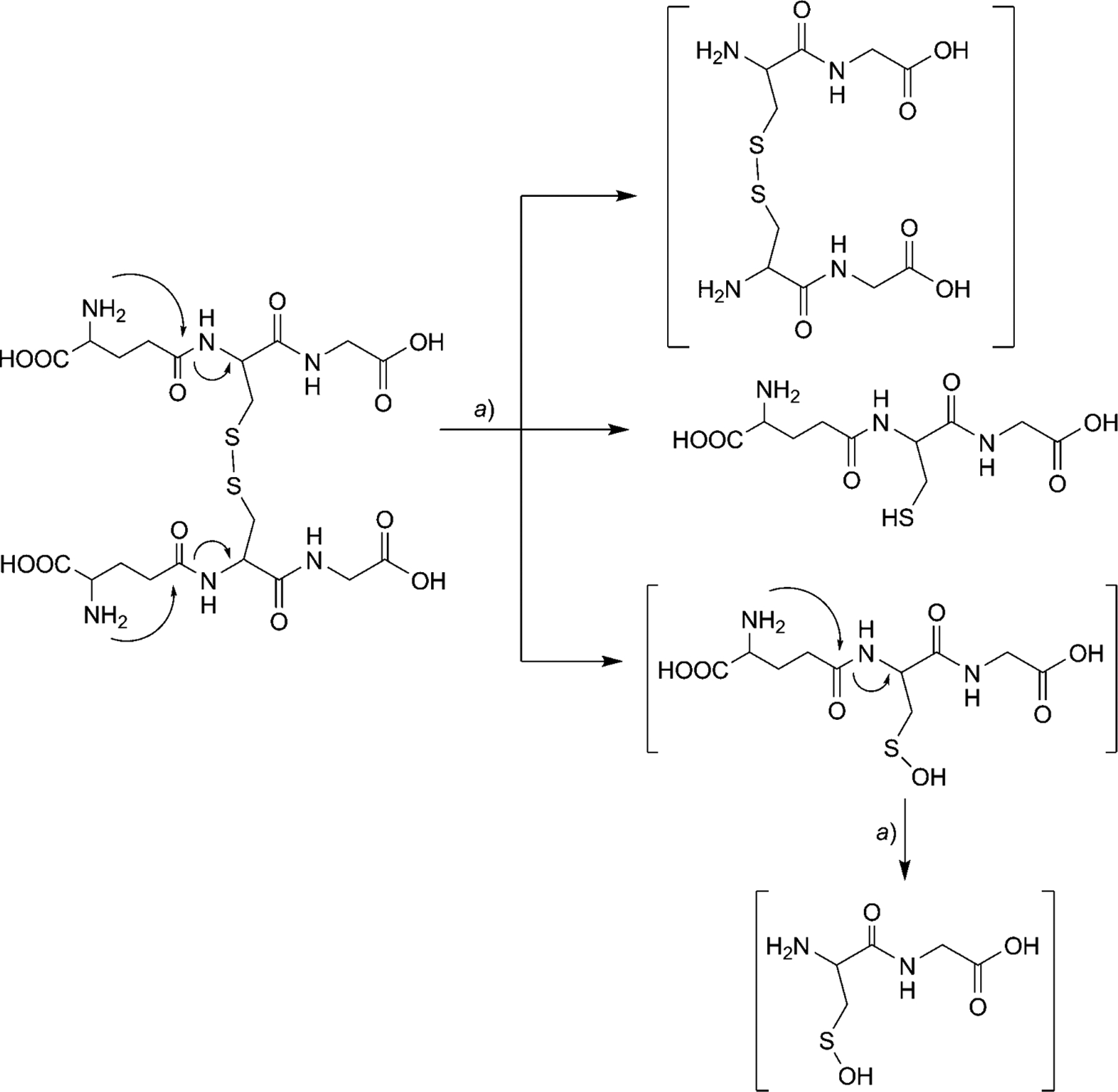
After GSH degradation was essentially complete (99%), the identification of products 5, 6, and 7 strongly suggests the formation of two disulfide intermediates: GS-SG (Scheme 2) and (Cys-Gly)2 (Scheme 3). However, the disulfide intermediates were not observed at any pH studied as confirmed by LC/MS (Fig. 3). In situ disulfide bond-formation between GS-SG or (Cys-Gly)2 is theoretically possible. The disulfide bond should reduce as per the Schoberl mechanism [15][16], in which HO− attacks the disulfide bond producing cysteine and the unstable cysteinesulfenic acid, which is further converted to 2-amino-3-oxopropanoic acid with the release of H2S. The same mechanism was observed in the degradation studies of GSH at all three pH values. The oxidized compound (GS-SG) is reduced, when HO− attacks the disulfide bond, producing GSH and GSH-sulfenic acid. At the same time, the amide bond between Glu and Cys is cleaved via the nucleophilic attack of the N-terminal amino group, and (Cys-Gly)2 and Cys-Gly-sulfenic acid are formed after the release of pyroglutamic acid (2⇌3). The results show that the disulfide bond in (Cys-Gly)2 reduces and gives Cys-Glysulfenic acid and Cys-Gly. This possible degradation mechanism is described in Schemes 2 and 3. Further, Cys-Gly-sulfenic acid breaks the amide bond between Cys and Gly and forms cysteinesulfenic acid (5) and glycine (7). It was observed that cysteinesulfenic acid (5) is not stable, and it hydrolyzes to 2-amino-3-oxopropanoic acid (6). Formation of cysteinesulfenic acid (5), 2-amino-3-oxopropanoic acid (6), and glycine (7) through in situ reduction of (Cys-Gly)2 and GS-SG, is based on the precedence of the Schoberl mechanism.
Scheme 2. The Cleavage Mechanism of the GS-SG Intermediate Involves Disulfide Bond Reduction and Cleavage of Glutamic Acid.
a) 37°, 100 mm sodium phosphate buffer with three different pH values (6.2, 6.8, and 7.4).
Scheme 3. The Cleavage Mechanism of the (Cys-Gly)2 Intermediate Involves Disulfide Bond Reduction.
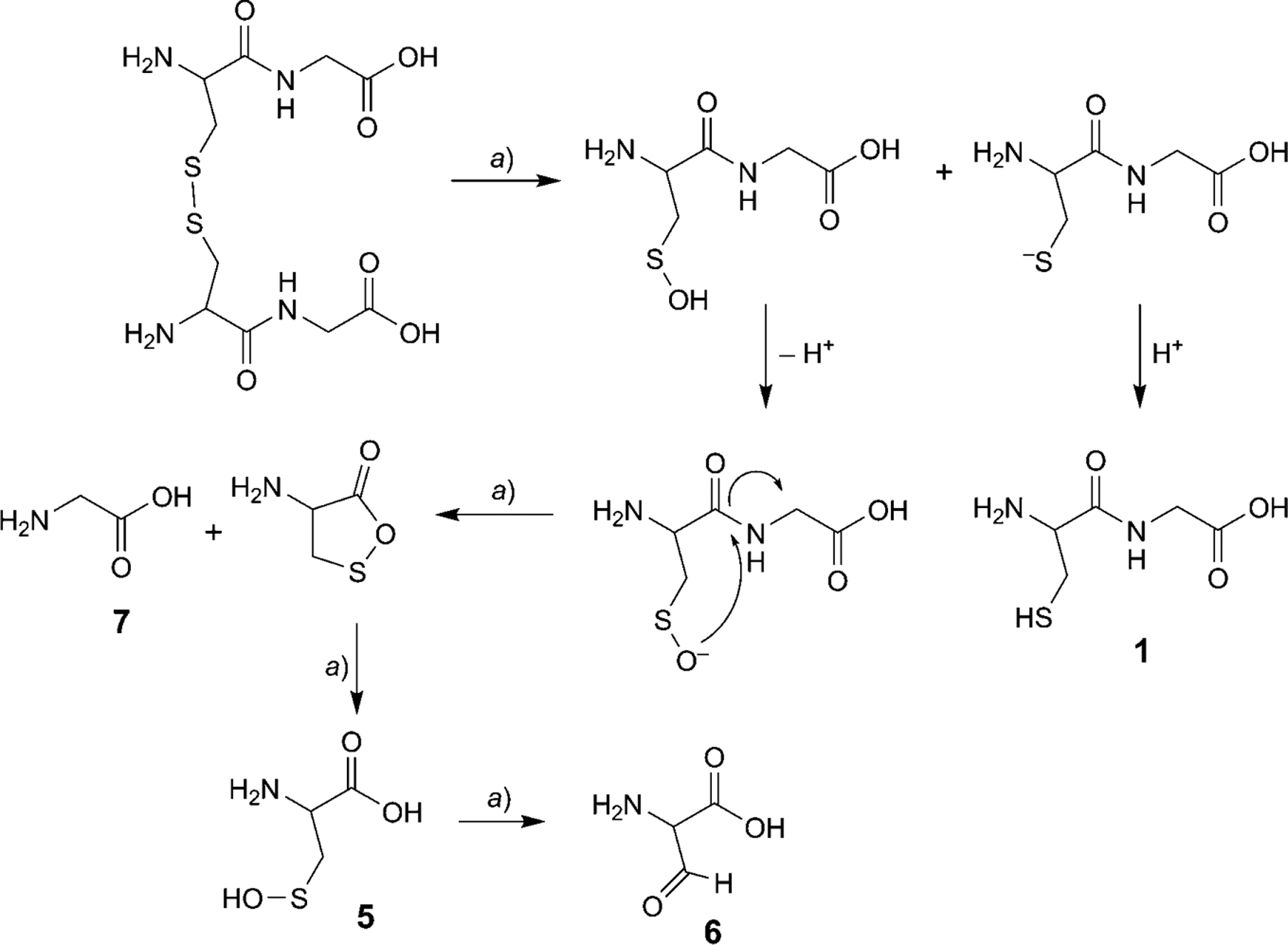
a) 37°, 100 mm sodium phosphate buffer with three different pH values (6.2, 6.8, and 7.4).
Fig. 3. LC/MS of the crude reaction mixture containing seven compounds.
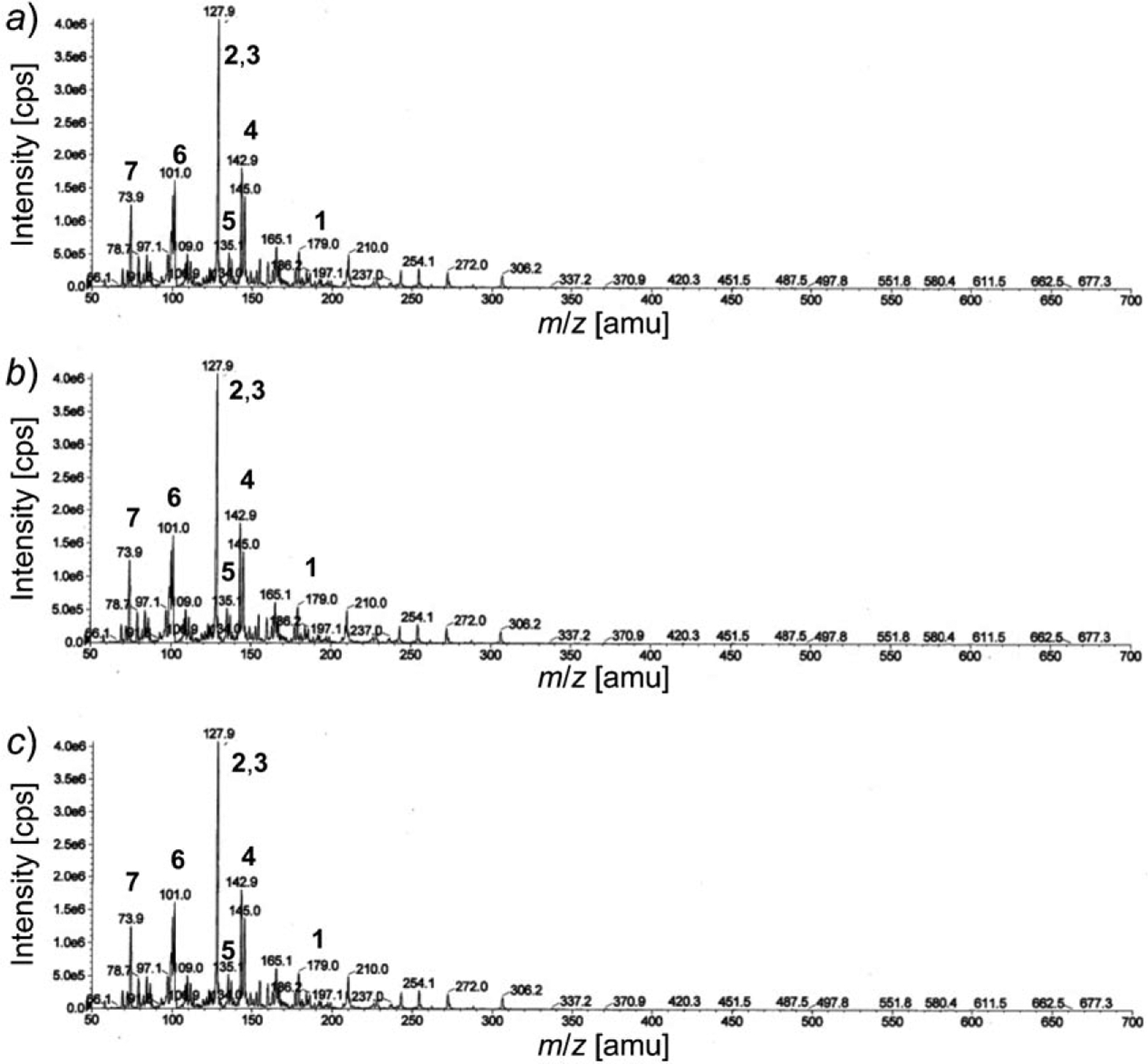
Signals are marked corresponding to peaks for cysteinyl-glycine (1), pyroglutamic acid (2) and 5-oxoproline (3), glutamic acid (4), cysteinesulfenic acid (5), 2-amino-3-oxopropanoic acid (6), and glycine (7). Degradation studies were carried out at 37° and pH 7.4 (a), 6.8 (b), and 6.2 (c).
Thus, both initial amide bond cleavage between Glu and Cys and total GSH degradation (99.9%) is fastest at pH 7.4 (24 d) and decreases with pH (pH 6.8: 28 d and pH 6.2: 34 d). Peak intensities of other protons A, B, C, E, and F (Fig. 1) also decrease, while new peaks for degradation products are observed (Scheme 1).
Based on the LC/MS analysis (Fig. 3), a nonenzymatic degradation pathway was deduced. In this pathway, the GSH ion at m/z 307 decreased with time until it was undetectable. After complete degradation of GSH, intense peaks at m/z 128 for pyroglutamic acid and its tautomer, 5-oxoproline (2), as well as at m/z 145 for glutamic acid (4) were observed. All three compounds were derived from the same glutamic acid moiety. Also seen was the peak at m/z 179 corresponding to Cys-Gly (1), which constitutes the remainder of the peptide after the initial cleavage reaction. There is a peak at m/z 135 evidencing the presence of cysteinesulfenic acid (5), apparently resulting from oxidation of the disulfide bond in (Cys-Gly)2, followed by intramolecular cleavage of amide bond between Cys-Gly. The cysteinesulfenic acid was converted into 2-amino-3-oxopropanoic acid (6), the peak of which was observed at m/z 101. The peak for glycine (7) was observed at m/z 75, completing the assignment of the ions derived from the GSH degradation.
Additional studies were performed with 100 mm sodium phosphate buffer and in the absence of O2 (by flushing with Ar) at all three pH values. In the absence of oxidative environment, no changes were observed to the initial kinetics and degradation (Fig. 4,a–c). Mass spectra of the degradation products show that degradation is greatly hindered after the cleavage of the first amide bond between Glu and Cys, suggesting further degradation requires the presence of oxidative agents (Fig. 5,a). To understand the effect of the presence of a chelating agent, we have repeated degradation studies, in the presence of 1 mm EDTA (containing 100 mm sodium phosphate buffer). We found that the rate of hydrolysis was unaffected in all three cases of pH (Fig. 4,d–f, and Fig. 5,b). To study the effect of the ionic strength on degradation, similar studies were performed using 20 mm sodium phosphate buffer. It was observed that different ionic strength at the same pH does not affect the degradation time (Fig. 4,g–i, and Fig. 5,c). Hence, this supports the conclusion that the auto-degradation of GSH follows the pathway proposed in this article. The composite data indicates that the elimination reaction, initiated by the nucleophilic attack of the N-terminal amino group in GSH (Scheme 1), is the most likely possibility.
Fig. 4. Section of 1H-NMR of the GSH degradation studies performed in the absence of O2.
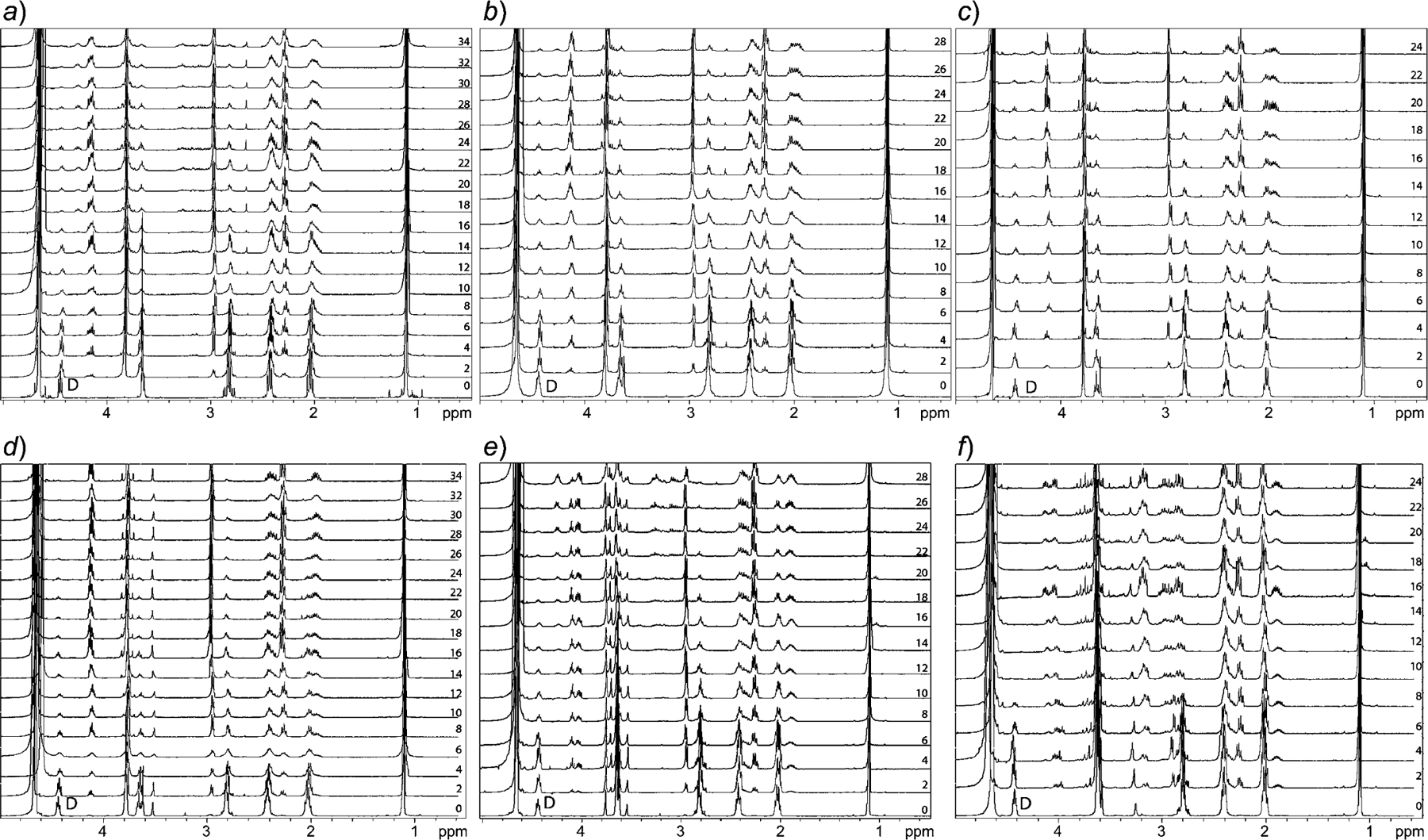
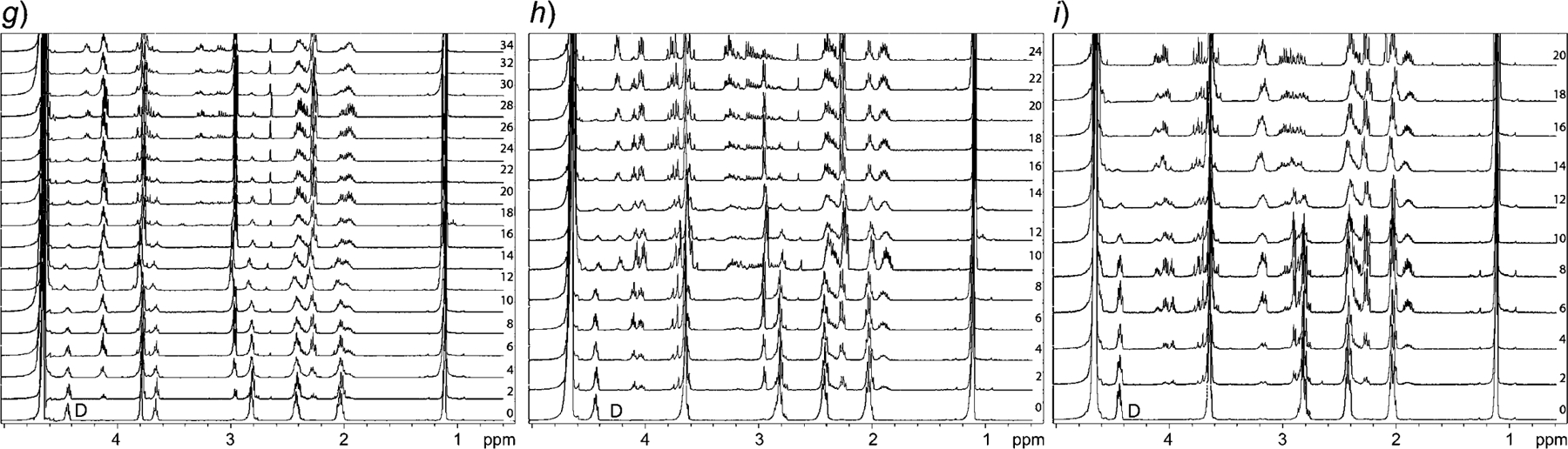
In 100 mm sodium phosphate buffer at pH 6.2 (a), 6.8 (b), 7.4 (c); with 1 mm EDTA in 100 mm sodium phosphate buffer at pH 6.2 (d), 6.8 (e), 7.4 (f); and with 20 mm sodium phosphate buffer at pH 6.2 (h), 6.8 (i), 7.4 (j). Several degradation products are now missing (e.g., peaks in the region of 2.8 to 3.2 ppm) in spectra recorded from a to c. Drop in the intensity of the peak arising from H-atoms D (annotated in 0 day spectrum in all panels) indicates cleavage of Glu-Cys bond in GSH, which follows proposed degradation pathway. Numbers in panels denote days on which the spectra have been recorded during the studies.
Fig. 5. Mass spectra of the crude reaction mixtures.
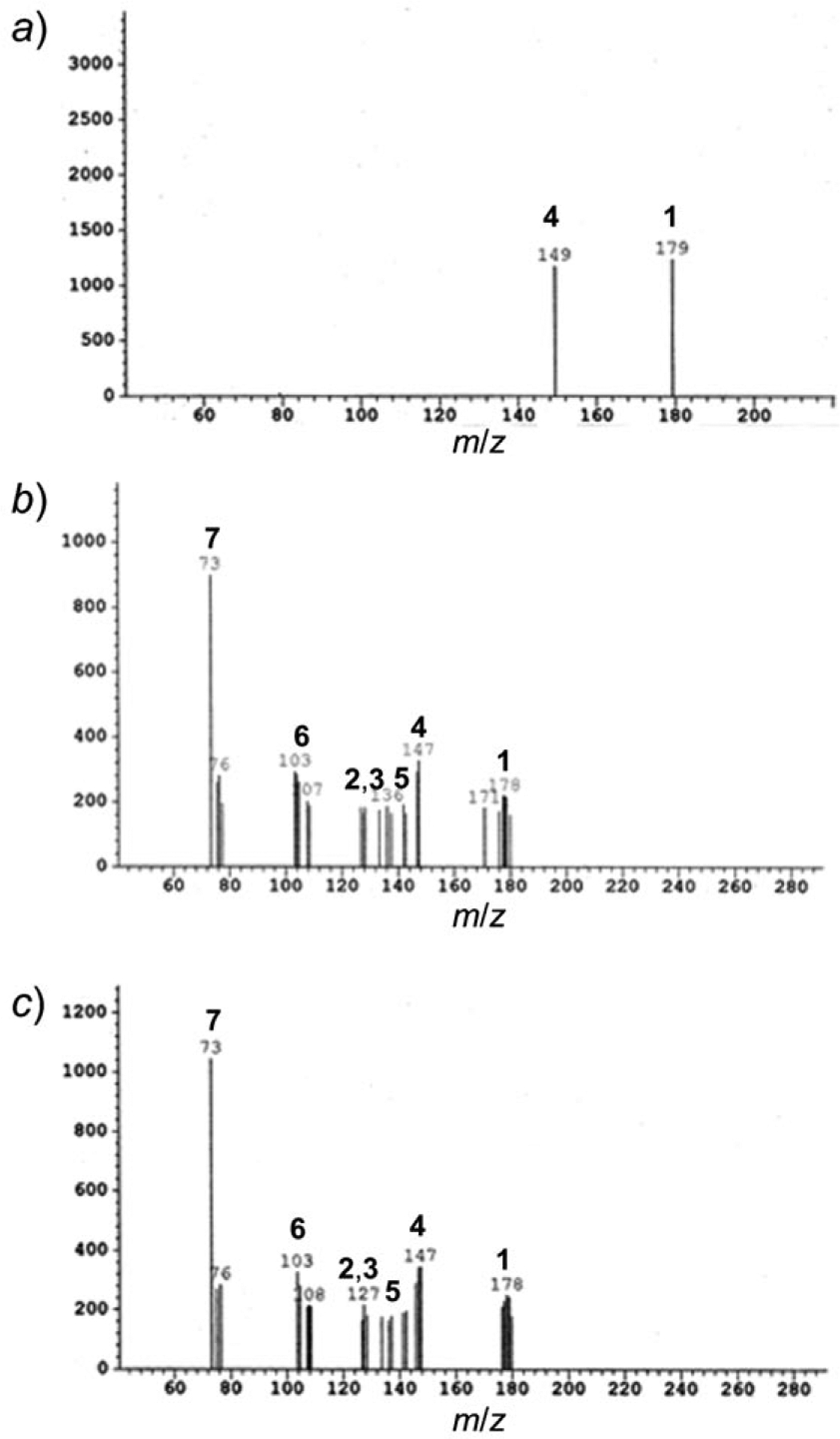
Degradation studies were carried out at pH 7.4, a) in the absence of oxygen, b) in 100 mm sodium phosphate buffer containing 1 mm EDTA, and c) 20 mm sodium phosphate buffer. Similar results were obtained for other two pH values. Signals are marked corresponding to peaks for cysteinyl-glycine (1), pyroglutamic acid (2) and 5-oxoproline (3), glutamic acid (4), cysteinesulfenic acid (5), 2-amino-3-oxopropanoic acid (6), and glycine (7).
Another possibility for the degradation of GSH is via thiolysis of the free −SH group of Cys with the γ-COOH group of Glu followed by thioester bond cleavage. This is not consistent with the data. The products obtained after the degradation, cysteinesulfenic acid (5), followed by 2-amino-3-oxopropanoic acid (6) and Gly (7), would not occur due to thiolysis followed by thioester bond degradation. Thiolysis would cause the shift of D proton from 4.41 to 3.4–3.7 ppm. Instead, it was observed that the intensity of the Cys-α triplet (D) gradually decreased with time, and the resonance disappeared completely within 24 d at pH 7.4. Furthermore, after the disappearance of the D signal (after complete degradation), the thiolysis product was not observed in the LC/MS spectrum. The absence of the thiolysis product (thiolysis of free −SH group of Cys with γ-COOH group of Glu) was confirmed at all three pH values (Fig. 3). Further proof was obtained by analyzing the controls (tert-butoxy)-carbonyl- and trityl-protected GSH, and other peptide sequences (GABA-Arg, εAhx-Arg) containing γ-aminobutyric acid (GABA) and 6-aminohexanoic acid (ε-Ahx) instead of glutamic acid, which did not undergo degradation. These studies prove that other peptides (γ-amino acid or ε-amino acid) do not degrade as per the proposed mechanism and confirm that the initial cleavage of the Glu-Cys bond involves nucleophilic attack of the N-terminal amino group of GSH at the γ-carbonyl side chain, followed by an unusual cleavage of the Cys-Gly amide.
Conclusions.
In the present study, we demonstrated for the first time that GSH degradation occurs in the absence of γ-glutamyl transpeptidase. GSH undergoes spontaneous nonenzymatic auto-degradation at various rates, depending on pH. This pH range covers values typically observed in normal physiological (i.e., plasma pH 7.4) and pathological conditions (i.e., pH 6.2 and 6.8 commonly found in the local tumor environment). The mechanism of the nonenzymatic degradation of GSH involves the formation of two disulfide intermediates, GS-SG and (Cys-Gly)2. The initial cleavage is at the Glu-Cys amide bond by nucleophilic attack of the N-terminal amino group of GSH at the γ-carbonyl side chain, followed by an unusual cleavage of the Cys-Gly amide bond. The proposed nonenzymatic degradation mechanism may have utility in drug targeting especially for diseases where local pH is altered from typical physiological levels. Furthermore, the time scale (t1/2 ca. 2–3 weeks) is useful for sustained release applications.
Data of Glutathione (GSH).
1H-NMR (300 MHz, D2O; Fig. 6): 4.41 (t, 3J(Cys-α,Cys-β) = 6.59, 1 H, Cys-α (D)); 3.82 (br. s, 2 H, Gly-α (F)); 3.69 (t, 3J(Glu-α,Glu-β) =6.32, 1 H, Glu-α (A)); 2.8 (dd, 3J(Cys-β,Cys-β)=6.59, 3J(Cys-β,Cys-α)=3.4, 2 H, Cys-β (E)); 2.41 (ddd, 2J(Glu-γ,Glu-γ) =15.24, 3J(Glu-γ ,Glu-β)=7.41, 3J(Glu-γ,Glu-β)=2.74, 2 H, Glu-γ (C)); 2.02 (q, 3J(Glu-β,Glu-γ) =7.42, 2 H, Glu-β (B)). LC/MS: 307 ([M+1]
Fig. 6.
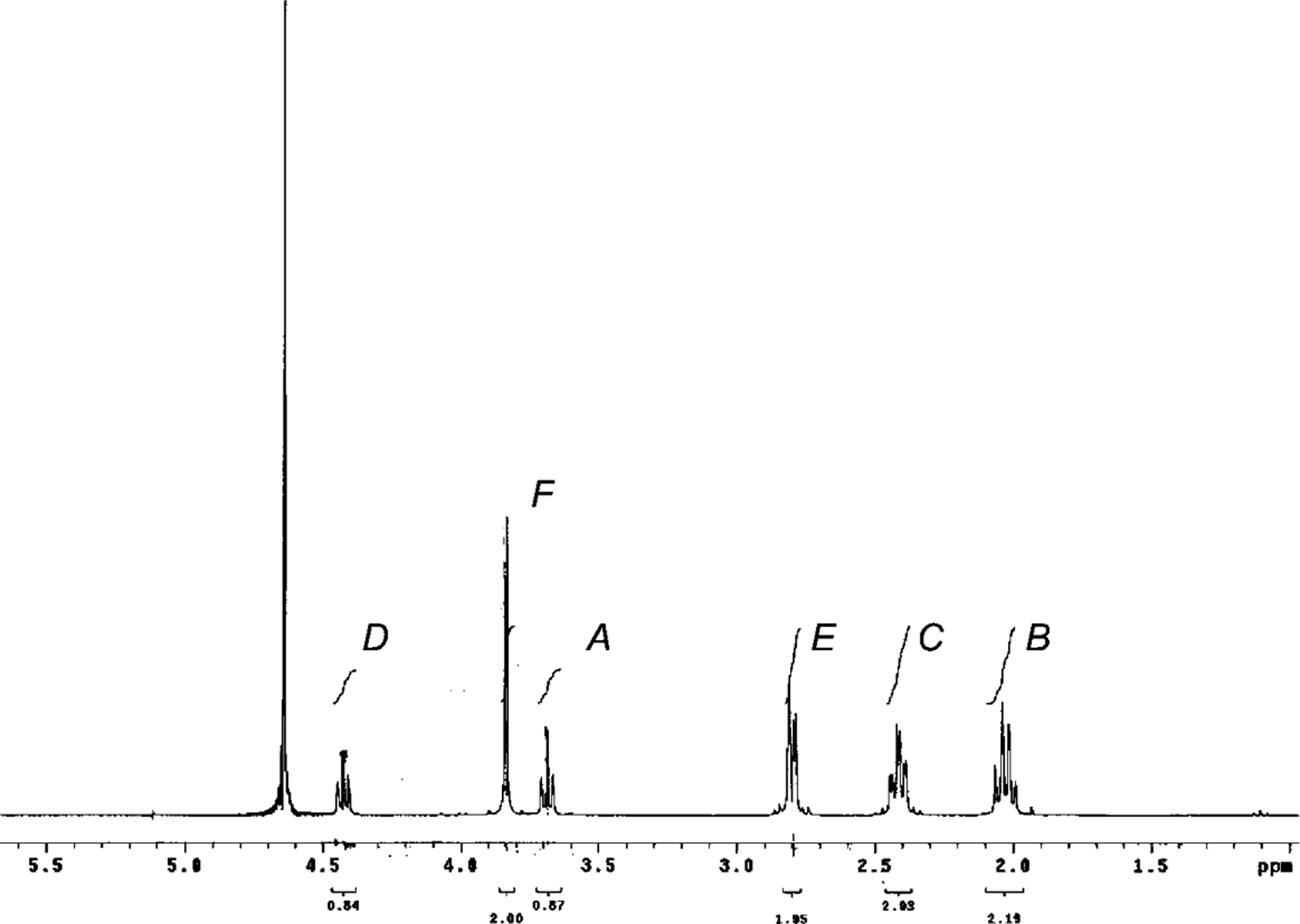
1H-NMR Spectrum of GSH in D2O
Acknowledgments
This research is supported by the Parke-Davis Endowed Chair in Pharmaceutics and Controlled Drug Delivery, Hikma Pharma PLC, and National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Award #U54AR055073). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the federal government. We would like to thank Drs. Simi Gunaseelan, Yashveer Singh, and Dayuan Gao for their assistance and fruitful discussions. Also we would like to thank Scott Pfeil for his help.
General. Abbreviations:
- GSH
glutathione
- Glu
glutamic acid
- Cys
cysteine
- Gly
glycine
- GABA
γ-aminobutyric acid
- ε-Ahx
6-aminohexanoic acid
- LC/MS
Liquid chromatography/tandem mass spectrometry
- Solvents
sodium phosphate buffer. Glutathione was purchased from Sigma-Aldrich
- 1H-and 13C-NMR spectra
400-MHz spectrometer (Varian) with the chemical shift values reported in d units (ppm)
- LC/MS
API-150, Applied Biosciences
REFERENCES
- [1].Meister A, Anderson ME, Annu. Rev. Biochem 1983, 52, 711. [DOI] [PubMed] [Google Scholar]
- [2].Perrone GG, Grant CM, Dawes IW, Mol. Biol. Cell 2005, 16, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hassan SSM, Rechnitz GA, Anal. Chem 1982, 54, 1972; [Google Scholar]; Jones DP, Carlson JL, Samiec PS, Sternberg P Jr., Mody VC Jr., Reed RL, Brown LAS, Clin. Chim. Acta 1998, 275, 175; [DOI] [PubMed] [Google Scholar]; Anderson ME, Chem.-Biol. Interact 1998, 111–112, 1; [DOI] [PubMed] [Google Scholar]; Friesen C, Kiess Y, Debatin K-M, Cell Death Differ. 2004, 11, S73. [DOI] [PubMed] [Google Scholar]
- [4].Balendiran GK, Dabur R, Fraser D, Cell Biochem. Funct 2004, 22, 343. [DOI] [PubMed] [Google Scholar]
- [5].Griffith OW, Free Radical Biol. Med 1999, 27, 922. [DOI] [PubMed] [Google Scholar]
- [6].Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND, Nutr J. 2004, 134, 489. [DOI] [PubMed] [Google Scholar]
- [7].Meister A, Tate SS, Annu. Rev. Biochem 1976, 45, 559; [DOI] [PubMed] [Google Scholar]; Tate SS, Meister A, Mol. Cell. Biochem 1981, 39, 357; [DOI] [PubMed] [Google Scholar]; Griffith OW, Bridges RJ, Meister A, Proc. Natl. Acad. Sci. U.S.A 1979, 76, 6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Meister A, Tate SS, Griffith OW, Methods Enzymol. 1981, 77, 237. [DOI] [PubMed] [Google Scholar]
- [9].Meister A, ‘The Enzymes’, 3rd edn., Academic, New York, 1974, p. 10, p. 671; [Google Scholar]; Griffith OW, Bridges RJ, Meister A, Proc. Natl. Acad. Sci. U.S.A 1981, 78, 2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Griffith OW, Meister A, Proc. Natl. Acad. Sci. U.S.A 1979, 76, 268;34150 [Google Scholar]; Griffith OW, Meister A, Proc. Natl. Acad. Sci. U.S.A 1980, 77, 3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hopkins FG, J. Biol. Chem 1929, 84, 269; [Google Scholar]; Mason HL, J. Biol. Chem 1931, 90, 409; [Google Scholar]; Kendall EC, Mason HL, McKenzie BF, J. Biol. Chem 1930, 88, 409; [Google Scholar]; Mason HL, J. Biol. Chem 1931, 90, 25. [Google Scholar]
- [12].Testa B, Mayer JM, ‘Hydrolysis in Drug and Prodrug Metabolism’, Verlag Helvetica Chimica Acta, Z rich, Wiley-VCH, Weinheim, 2003, p. 235. [Google Scholar]
- [13].Lehninger AL, ‘Biochemistry: The Molecular Basis of Cell Structure and Functions’, Worth Publishers, New York, 1975, p. 583, p. 807; [Google Scholar]; Chen G, Russell JB, J. Bacteriol 1989, 171, 2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Palekar AG, Tate SS, Meister A, Proc. Natl. Acad. Sci. U.S.A 1974, 71, 293; [DOI] [PMC free article] [PubMed] [Google Scholar]; Schroeder EF, Munro MP, Weil L, J. Biol. Chem 1935, 110, 181. [Google Scholar]
- [15].Schöberl A, Justus Liebigs Ann. Chem 1933, 507, 111. [DOI] [PubMed] [Google Scholar]
- [16].Robbins FM, Fioriti JA, Nature (London) 1963, 200, 577. [DOI] [PubMed] [Google Scholar]