

Abstract
In responses to activation of receptor tyrosine kinases (RTKs), crucial cell fate decisions depend on the duration and dynamics of ERK signaling. In PC12 cells, epidermal growth factor (EGF) induces transient ERK activation that leads to cell proliferation, whereas nerve growth factor (NGF) promotes sustained ERK activation and cell differentiation. These differences have typically been assumed to reflect distinct feedback mechanisms in the Raf-MEK-ERK signaling network, with the receptors themselves acting as simple upstream inputs. We failed to confirm the expected differences in feedback type when investigating transient versus sustained signaling downstream of the EGF receptor (EGFR) and NGF receptor (TrkA). Instead, we found that ERK signaling faithfully followed RTK dynamics when receptor signaling was modulated in different ways. EGFR activation was switched from transient to sustained when receptor internalization was inhibited with drugs or mutations, or in a chimeric receptor likely to have impaired dimerization, and ERK activation kinetics followed. EGFR signaling also became more sustained upon addition of sub-stoichiometric levels of the EGFR inhibitor erlotinib to reduce kinase activation lifetime. Again, ERK signaling dynamics followed receptor activation kinetics. Our results argue that RTK activation kinetics play a crucial role in determining MAP kinase cascade signaling dynamics and cell fate decisions and that signaling outcome can be selected by activating a given RTK in different ways.
Introduction
How different receptor tyrosine kinases (RTKs) can promote distinct cellular responses – despite all appearing to share largely the same set of downstream pathways – has been a much debated question for almost 30 years (1, 2). Now-classic studies in the 1990s using rat pheochromocytoma (PC12) cells provided a major step towards understanding this puzzle (3–6), revealing that signaling specificity is encoded, at least in part, in the dynamics of downstream network responses (7). In comparing responses of PC12 cells to epidermal growth factor (EGF) and nerve growth factor (NGF), transient activation of ERK was associated with proliferation, whereas sustained ERK activation was linked to cell differentiation (4). Substantial advances have since been made in understanding how different durations of ERK signaling are interpreted by a network of immediate early gene (IEG) products (8–11) and later regulatory loops (12). How distinct dynamic modes of ERK activation are dictated by different upstream RTKs, however, remains less clear. Several reports have argued that differential engagement of feedback mechanisms at various ‘downstream’ levels in the ERK pathway determine how ERK activation dynamics are specified by individual RTKs (13–17). Although such feedback is undoubtedly important in defining ERK activation dynamics, several studies have suggested that receptor-level mechanisms (and the kinetics of RTK activation) also play a key role (18). Indeed, it has been argued that the Raf-MEK-ERK protein kinase cascade functions to transmit RTK signals to the level of ERK with high temporal fidelity (19, 20).
We reported that the EGFR is activated with different kinetics by its various ligands (21), in turn leading to altered downstream ERK signaling dynamics. These findings support the developing view that receptor-level mechanisms define key characteristics of ERK activation. They also suggest that, rather than being simple binary ‘on/off switches’, RTKs that dimerize upon ligand binding can display functional selectivity or biased agonism – discriminating between different ligands by eliciting distinct biological responses through a single receptor (22–26). Studies with engineered forms of stem cell factor (SCF) similarly suggested ways of exploiting biased agonism of the RTK that it activates, Kit (27). The idea of functional selectivity in signaling by dimerizing receptors with a single transmembrane domain has also been extended to cytokine receptors such as the erythropoietin and type I interferon receptors (28, 29).
Although the dynamics of ERK responses define cellular outcomes in RTK signaling (7, 30), their origin and relation to the molecular characteristics of receptor activation itself remain unclear. To evaluate the relative contributions of network topology and the intrinsic characteristics of RTKs to signaling network dynamics, we undertook a series of experiments in PC12 and MCF7 cells. We first investigated the consequences of disrupting key positive and negative feedback loops previously proposed to define sustained-versus-transient ERK signaling in response to NGF and EGF respectively (13, 17, 31). None of these manipulations substantially altered ERK signaling dynamics, which always maintained its close temporal relationship with RTK activation kinetics. Our findings extend conclusions drawn from optogenetic studies that the Raf-MEK-ERK cascade functions to transmit dynamic signals from cell surface receptors to the level of ERK (19, 20). We also showed that impairing EGFR internalization efficiency and/or trafficking led to similar alterations in the dynamics of both RTK activation and ERK signaling. Using a chimeric RTK and kinase inhibitors, we further showed that the same receptor can elicit either transient or sustained signaling in the same cells, providing support for our earlier hypothesis (21) that cell signaling characteristics are defined by the kinetics of RTK activation.
Results
Impairing feedback loops thought to define NGF/EGF differences does not affect ERK signaling dynamics
EGF-induced ERK activation in PC12 cells peaked at 5 min and fell back to near-background levels within 15 min (Fig. 1A), whereas NGF-induced ERK activation persisted for >60 mins (Fig. 1B). As a first approach to understanding these well-established differences in ERK signaling dynamics, we began by asking whether they could be explained by differences in network topology downstream of EGFR and TrkA (the NGF receptor). Key differences in network organization that have been proposed to explain these effects include: i) involvement of the small GTPase Rap1 rather than Ras in linking TrkA activation to the Raf-MEK-ERK cascade (17, 31); and ii) differences in the nature of feedback within the Raf-MEK-ERK cascade itself (13).
Fig. 1. Time courses of ERK activation are not defined by Rap1, FRS2 or PKCδ-based feedback.

(A and B) Serum-starved PC12 cells were either left unstimulated or were stimulated with 100 ng/ml EGF (A) or 50 ng/ml NGF (B) for the indicated times. Total and phosphorylated (pThr202/pTyr204) ERK1/2 levels were detected by immunoblotting of total cell lysates, with Grb2 as a protein loading control as described (69). Representative blots of independent experiments are shown (n = 27 for EGF, n = 32 for NGF).
(C) PC12 cells were transiently transfected with either 100 nM siRNA specific for Rap1A/Rap1B (+) or 100 nM non-targeting negative control siRNA(–). 72 hours following transfection, serum-starved cells were stimulated with 50 ng/ml NGF for the indicated times, and levels of total and phosphorylated (pThr202/pTyr204) ERK1/2, Rap1, and Grb2 (as loading control) were determined by immunoblotting of total cell lysates. Blots representative of independent experiments (n = 3) are shown.
(D) Quantitation of pERK levels (normalized to Grb2 and the peak value at 5 min) from (C), with mean ± SD (n = 3 experiments), with no statistically significant differences between plots using the Bonferroni-Dunn method (alpha = 0.05). Datasets for each point were analyzed individually, without assuming consistent SD.
(E) PC12 cells were transiently transfected with either 100 nM siRNA specific for FRS2 (+) or were mock transfected (–). 72 hours after transfection, serum-starved cells were stimulated with 50 ng/ml NGF for the indicated times, and levels of total and phosphorylated (pThr202/pTyr204) ERK1/2, FRS2, and Grb2 (as loading control) were detected by immunoblotting of total cell lysates. Blots representative of independent experiments (n = 3) are shown.
(F) Quantitation of pERK levels (normalized to Grb2 and the peak value at 5 min) from (E), with mean ± SD (n = 3 experiments), with no statistically significant differences between plots using the Wilcoxon rank sum test (for 60 min time point).
(G) Serum-starved PC12 cells were either left untreated (–) or were pretreated (+) with Gö7874 (10 μM, 60 min) before stimulation with 50 ng/ml NGF for the indicated times. Immunoblotting of total cell lysates was used to detect levels of total and phosphorylated (pThr202/pTyr204) ERK1/2 and TrkA (pTyr674/675, pTyr490, pTyr785: a cocktail of the three antibodies, each at 1/1000-fold dilution was used). Grb2 levels were used as protein loading control. Blots representative of independent experiments (n = 3) are shown.
(H) Quantitation of pERK levels (normalized to Grb2 and the peak value at 5 min) from (G), with mean ± SD (n = 3 experiments), with no statistically significant differences between plots using the Bonferroni-Dunn method (alpha = 0.05). Datasets for each point were analyzed individually, without assuming consistent SD.
By activating ERK through Rap1 rather than Ras, TrkA signaling has been proposed to lack a key negative feedback loop responsible for making the EGFR response transient. This key negative feedback loop involves inhibitory phosphorylation of the Ras guanine nucleotide exchange factor Sos by ERK (17, 31, 32) to create transient activation (Fig. 1A). Because C3G, the GDP/GTP exchanger for Rap1, is not subject to this negative feedback regulation, utilization of Rap1 (rather than Ras) in NGF signaling has been proposed (17) to explain the sustained ERK response to NGF (Fig. 1B). To test this hypothesis, we knocked down Rap1A/1B in PC12 cells to levels that were almost undetectable by Western blot (Fig. 1C) and assessed ERK activation dynamics having ensured linearity of the assay (fig. S1). The NGF response remained sustained (Fig. 1C,D), arguing that differential small GTPase utilization is not the primary determinant of ERK signaling dynamics, although it may make some contribution. Further, knocking down (by ~80%) FRS2, which links TrkA to Rap1 (through the adaptor Crk and C3G), also allowed NGF-induced ERK activation to remain sustained, with a robust signal persisting 60 mins after stimulation (Fig. 1E,F).
Studies employing modular response analysis (33) also suggested that a differential network topology for NGF and EGF signaling at the level of the Raf-MEK-ERK cascade defines the sustained versus transient nature of their signaling. As an alternative to the well documented negative feedback from ERK to Raf in the case of EGFR signaling (which yields a transient response), positive feedback in the Raf-MEK-ERK cascade was proposed (13) to explain the observed sustained response to NGF. The positive feedback loop operating in NGF signaling was further reported to depend on protein kinase C-δ (PKCδ) activity (13). In our hands, however, PKCδ inhibition with Gö7874 had little influence on the sustained nature of NGF signaling in PC12 cells (Fig. 1G,H and fig. S2, A and B). Together, these data argue against the idea that distinct network topologies in the Raf-MEK-ERK cascade alone (or in the identity of the GTPase that activates the cascade) define the difference in dynamics of ERK activation by EGF and NGF in PC12 cells.
ERK activation dynamics follow those of ligand-induced RTK phosphorylation
Our failure to confirm downstream network-specific explanations for sustained versus transient ERK activation by NGF and EGF, together with other reports that receptor-level mechanisms encode ERK dynamics (18–20), prompted us to compare RTK and ERK signaling kinetics. When EGFRs and NGF receptors in PC12 cells were saturated by their respective ligands, the characteristic time courses of RTK autophosphorylation and ERK phosphorylation (Fig. 2A) were the same across multiple experiments (transient for EGF, sustained for NGF). This result supports the idea that the Raf-MEK-ERK cascade transmits dynamic signals from the RTKs to ERK with high fidelity (20). The number of binding sites for EGF and NGF in PC12 cells are within ~2-fold of one another (34, 35), suggesting that these different kinetics do not simply reflect RTK levels. Moreover TrkA (36) and EGFR (5) both faithfully retain these kinetic characteristics when overexpressed in PC12 cells. Our earlier work with EGFR has also shown that different growth factor ligands can elicit signaling with distinct dynamic characteristics through the same receptor (21). Whereas EGF at saturating levels induced transient activation of EGFR and ERK in MCF7 breast cancer cells (Fig. 2A), saturating levels of the low-affinity ligands epigen (EPGN) and epiregulin (EREG) instead induced sustained activation of EGFR and ERK (21) – resembling the NGF/TrkA response (Fig. 2B). As a result, EREG and EPGN both promote cytoplasmic lipid accumulation (a response associated with differentiation) through EGFR in MCF7 cells whereas EGF promotes proliferation through the same receptor (21). These data refocused our attention on signaling by the receptors themselves (the input) rather than the nature of the network downstream of these RTKs.
Fig. 2. ERK signaling dynamics track RTK activation kinetics for EGFR and TrkA.

(A and B) Serum-starved PC12 cells were either left unstimulated or were stimulated with 100 ng/ml EGF (A) or 50 ng/ml NGF (B) for the indicated times. Levels of total and phosphorylated (pThr202/pTyr204) ERK1/2 were determined by immunoblotting of total cell lysates (see gel inserts for representative blots) and are plotted in blue. In parallel, growth factor-induced phosphorylation of (A) EGFR (pTyr1173) and (B) TrkA (cocktail of pTyr674/675, pTyr490, pTyr785 antibodies as in Fig. 1E) was monitored by immunoblotting of total cell lysates with phospho-specific antibodies – plotted in red for pEGFR (A) or orange for pTrkA (B). Total EGFR and TrkA levels are also shown. Grb2 immunoblots were used as protein loading controls. Time courses for phosphorylation of ERK (blue) and the RTK were overlaid, with signal intensities divided by the corresponding signal intensities of Grb2 at each time point and then normalized by peak value at 5 minutes. Mean values ± SD are plotted for n = 8 experiments (EGF/pEGFR) and n = 7 experiments (NGF/pTrkA).
Studies with ErbB4/HER4 argue that RTK signaling dynamics is receptor-intrinsic
In our previous studies of EGFR signaling kinetics in MCF7 cells (21), the EGFR ligands that induced sustained signaling (EPGN and EREG) also bind the receptor with lower affinity (>100-fold) than EGF (37). We therefore wondered whether ligand affinity might be a key determinant of signaling kinetics, and thus whether differences in signaling dynamics might also be seen for the high- and low-affinity ligands of EGFR’s close relative, ErbB4/HER4. ErbB4 is stimulated by the neuregulins or NRGs (22, 38). The ErbB4-binding EGF-like domain of NRG1 has two different splice-forms, NRG1α (low affinity) and NRG1β (high affinity), with affinities similar to those of EREG/EPGN and EGF respectively (39). We analyzed endogenous ErbB4 activation by saturating NRG1α and NRG1β in MCF7 cells (Fig. 3A); we switched cells because ErbB4 is undetectable in PC12 cells (40). Sustained ErbB4 autophosphorylation was seen with both ligands, with substantial signal remaining after 1 or 2 hours (Fig. 3B,C). There were no statistically significant differences in the dynamics of receptor phosphorylation or ERK phosphorylation for the two ligands. ERK activation dynamics again followed ErbB4 signaling for NRG1α and NRG1β in these experiments – supporting the argument that RTK dynamics define the kinetic properties of downstream events. ERK phosphorylation did fall off slightly more rapidly with NRG1α than with NRG1β, however, suggesting that there may be some difference in engagement of downstream feedback loops between these two ligands.
Fig. 3. ErbB4 activation with high-affinity or low-affinity NRGs gives sustained signals.
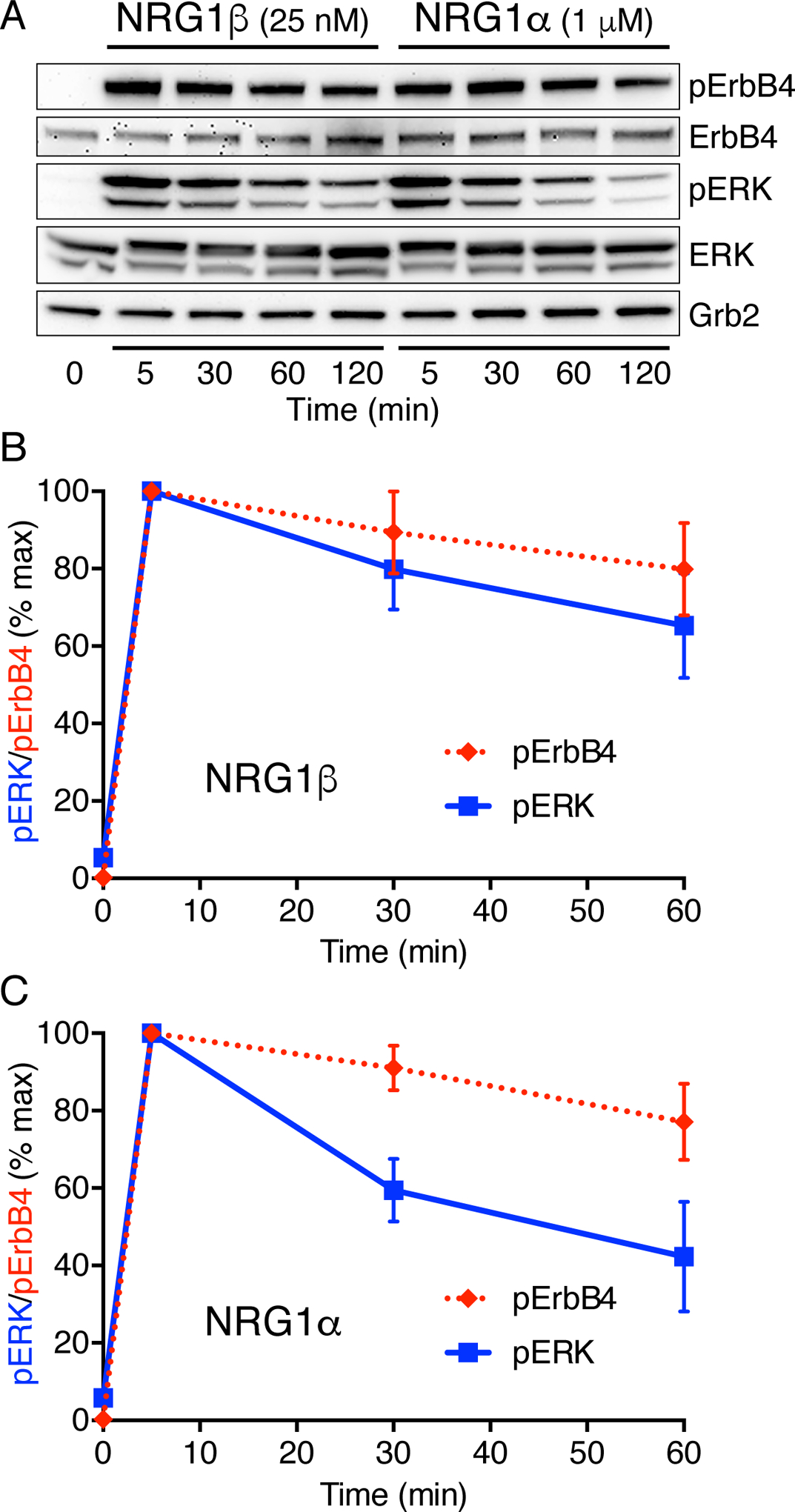
(A) Endogenous ErbB4 in MCF7 cells was stimulated using saturating levels of NRG1β (25 nM/ 200 ng/ml) or NRG1α (1 μM/ 7 μg/ml). Activating phosphorylation of ErbB4 and ERK1/2, as well as total ErbB4 and ERK levels, were monitored by immunoblotting total cell lysates. Grb2 was used as a loading control. Blots are representative of at least three independent experiments. Statistical significance of differences was assessed using the Bonferroni-Dunn method (alpha = 0.05), with datasets for each point analyzed individually without assuming consistent SD.
(B) Quantitation of pERK and pErbB4 levels (normalized to Grb2 and the peak values at 5 min) for NRG1β-activation.
(C) Quantitation of pERK and pErbB4 levels (normalized to Grb2 and the peak values at 5 min) for NRG1α-activation.
These results showed that EGFR and ErbB4 signal with different kinetic characteristics (transient and sustained) when activated by their respective high-affinity ligands (EGF and NRG1β) in MCF7 cells. For EGFR, sustained signaling was associated with reduced strength of the ligand-induced dimer (21). Although the strengths of EGF- and NRG1β-induced dimers of EGFR and ErbB4 have never been compared in cells to our knowledge, studies of extracellular regions suggest similar affinities (41). Previous analyses of NRG1β-induced ErbB4 internalization have suggested that ErbB4 is ‘internalization impaired’ compared with EGF-activated EGFR (42, 43), implying that ErbB4 lacks an important internalization signal which could partially explain its sustained signaling.
Impairing EGFR internalization promotes sustained signaling at both receptor and ERK levels
If downstream signaling dynamics are defined by RTK activation kinetics, at least some of the negative feedback event(s) responsible for transient responses to EGF must directly influence its receptor. The most obvious candidates for such RTK-level negative feedback are endocytosis and protein tyrosine phosphatases (or both). Endocytosis is further implicated by the sustained signaling of NRG1β-activated ErbB4, which is endocytosis-impaired (42). To investigate how internalization influences EGFR signaling kinetics, we inhibited dynamin activity using the dynasore analog Dyngo®−4a (44). Adding 30 μM Dyngo®−4a converted the normally transient autophosphorylation response of EGFR to EGF stimulation to one that was sustained (Fig. 4A). Dyngo®−4a also induced a similar switch in ERK signaling from transient to sustained (Fig. 4A,B). Mutations that impair internalization of EGFR also made EGFR signaling more sustained (Fig. 4C,D). We used a form of human EGFR (21KR/ΔAP2 hEGFR) that has a reduced internalization rate (by ~4–5-fold) because of mutations at ubiquitinylation, acetylation and clathrin-adaptor AP‐2‐binding sites (45). When stably expressed in rat PC12 cells, 21KR/ΔAP2 hEGFR showed substantially more sustained autophosphorylation after EGF stimulation than parental cells (Fig. 4C). Again, this shift from transient to sustained signaling was also reflected at the level of ERK (Fig. 4C,D). Moreover, expression of 21KR/ΔAP2 hEGFR in PC12 cells allowed EGF to promote neurite outgrowth (Fig. 4E). To confirm that the altered signaling dynamics seen for 21KR/ΔAP2 hEGFR (Fig. 4D) did not simply reflect elevated EGFR levels, we confirmed that wild-type hEGFR expressed at the same level in rat PC12 cells signals transiently at both receptor and ERK levels (fig. S3, A, B, and C). These results are consistent with the findings of the Sorkin lab (45), who studied the 21KR/ΔAP2 hEGFR variant in porcine aortic endothelial (PAE) cells and similarly reported more sustained signaling. Moreover, Yamada et al. described a PC12 cell clone (PC12h-R) that differentiates (rather than proliferating) in response to EGF, apparently as a result of reduced EGFR downregulation and more sustained EGFR phosphorylation (46).
Fig. 4. Impaired EGFR internalization leads to sustained signaling.
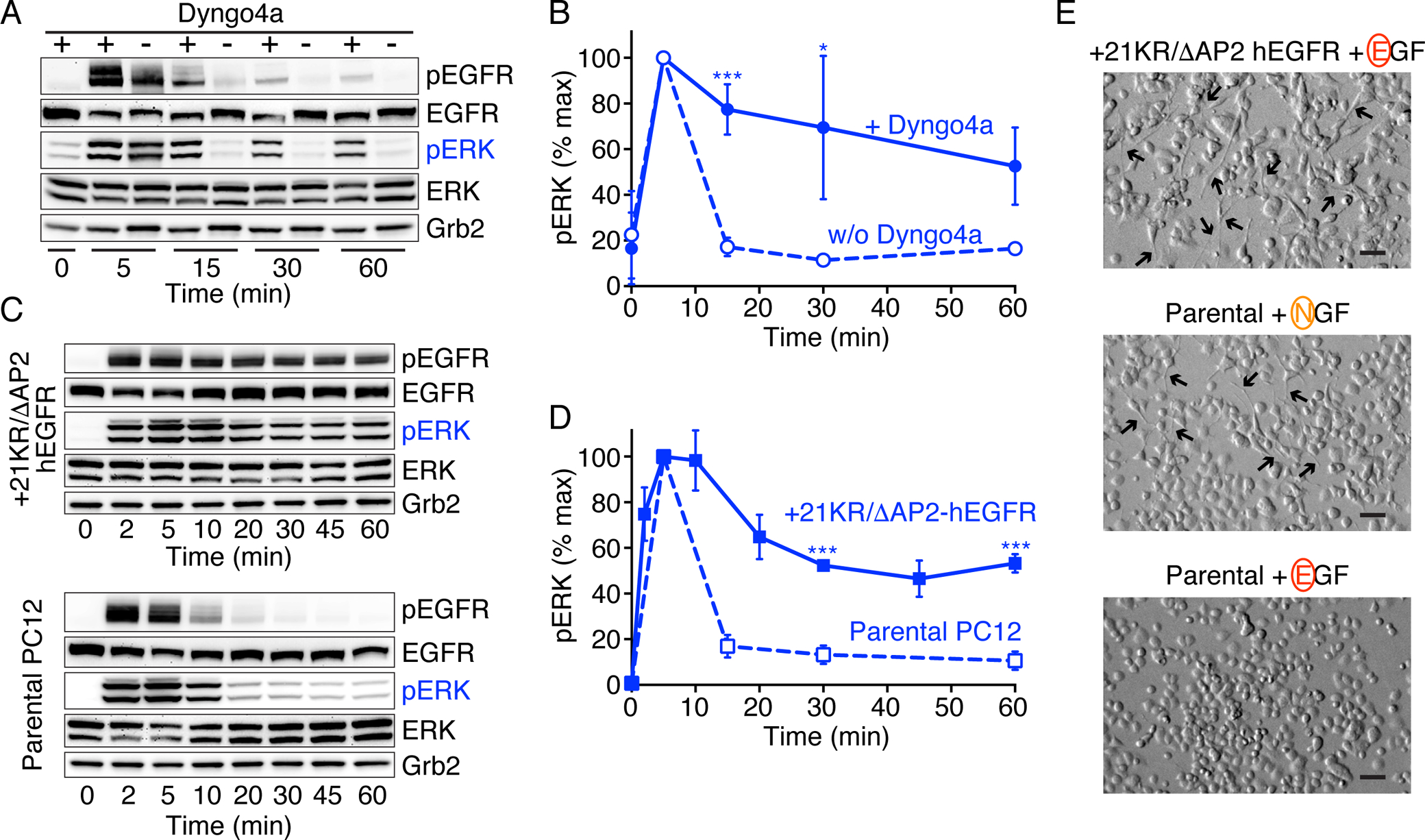
(A) Serum-starved PC12 cells were either pretreated (+) with Dyngo 4a (30 μM, 60 min) or left untreated (–) prior to stimulation with 100 ng/ml EGF for the indicated times. Levels of phosphorylated EGFR (pTyr1173) and ERK1/2 (pThr202/pTyr204), as well as corresponding total protein levels, were then detected by immunoblotting of total cell lysates – with Grb2 levels serving as protein loading controls. Blots are representative of at least three independent experiments.
(B) Quantitation of pERK levels (normalized to Grb2 and the peak value at 5 min) from (A), plotting mean ± SD (n = 3 experiments). Statistical significance was determined using the Bonferroni-Dunn method (alpha = 0.05), and repeats for each data point were analyzed individually, without assuming a consistent SD. Adjusted p values (after Bonferroni correction) were *p < 0.05, **p < 0.01, and ***p < 0.005.
(C) Serum-starved PC12 cells stably transfected with human 21KR/ΔAP2-hEGFR (upper set of blots) or parental PC12 cells expressing only wild-type EGFR (lower set of blots) were either left unstimulated or were stimulated with 100 ng/ml EGF for the indicated times. Levels of phosphorylated EGFR (pTyr1173) and ERK1/2 (pThr202/pTyr204), plus corresponding total protein levels, were then detected by immunoblotting of total cell lysates. Grb2 was used as the loading control. Blots are representative of at least three independent experiments.
(D) Quantitation of pERK levels (normalized to Grb2 and the peak value at 5 min), plotting mean ± SD (n = 3 experiments for 21KR/ΔAP2-hEGFR transfected cells and n = 7 experiments for parental PC12 cells). Statistical significance was determined using the Bonferroni-Dunn method (alpha = 0.05), and repeats for each data point were analyzed individually, without assuming a consistent SD. Adjusted p values (after Bonferroni correction) were *p < 0.05, **p < 0.01, and ***p < 0.005.
(E) Neurite outgrowth inspected by phase contrast microscopy in PC12 cells stably expressing 21KR/ΔAP2-hEGFR stimulated with 100 ng/ml EGF for 47 h or in parental PC12 cells stimulated with either 50 ng/ml NGF or 100 ng/ml EGF for 47 h. Figures are representative fields (n = 2 experiments for 21KR/ΔAP2-hEGFR, n = 4 experiments for parental PC12 cells with EGF or NGF). Scale bar, 70 μm.
Although impairing ligand-induced EGFR internalization converted signaling from being transient to sustained, we do not think that internalization alone defines signaling dynamics. NGF induces efficient endocytosis of TrkA in PC12 cells (47, 48) and the efficiency of NGF internalization in PC12 cells appears comparable with that seen for EGF (49–51) – arguing that lack of internalization does not cause sustained RTK signaling. Indeed, TrkA activation retains its sustained character following NGF stimulation when internalization is impaired, with an associated slight reduction in receptor dephosphorylation (52). Moreover, EREG and EGF promote sustained and transient signaling respectively through EGFR (21) despite triggering similar rates of receptor internalization (53). Thus, rather than differences in internalization per se being the key, it seems likely that differences in the rates (or itinerary) of post-endocytic trafficking and/or the degree of recycling following EGFR activation by different ligands determine RTK signaling dynamics. Indeed, different post-endocytic trafficking itineraries have been proposed to control access of activated receptors to key protein tyrosine phosphatases that reside in different intracellular locations (54, 55). Such controlled access to these phosphatases could determine signaling dynamics and specificity. Inhibiting EGFR endocytosis presumably blocks access of EGF-activated receptor to these intracellular phosphatases, but it is possible for EGFR activated by other ligands (or TrkA) to be endocytosed without subsequently being trafficked to phosphatases that cause signaling to appear transient.
A chimeric RTK displays sustained signaling driven by the EGFR intracellular region
To ask whether the transient signaling characteristics of EGFR track with the origin of the receptor’s intracellular region, we used a commonly used tool in studies of RTKs (56–62) and generated a chimeric receptor. We fused the TrkA extracellular region to the EGFR transmembrane (TM) and intracellular regions (TEE) (Fig. 5A). We stably expressed the TEE chimera in MCF7EGFR-KO cells, from which EGFR had been knocked out using CRISPR/Cas9 (to avoid complications from endogenous EGFR). When these cells were stimulated with 50 ng/ml NGF, sustained phosphorylation of the EGFR intracellular region was seen (Fig. 5B), which contrasted with the transient signal expected for a chimera harboring the EGFR intracellular region. Once again, dynamics of ERK activation also followed those of the receptor and were sustained (Fig. 5B,C). The complementary chimera with EGFR extracellular and TrkA intracellular regions (ETT) showed constitutive activation in most experiments, suggesting that it was misfolded, which prevented us from reliably assessing its signaling kinetics.
Fig. 5. TrkA/EGFR chimera gives sustained signals.

(A) Cartoon of a FLAG tagged chimera containing the TrkA extracellular region fused to the transmembrane and cytoplasmic regions of EGFR (TEE chimera).
(B) MCF7EGFR-KO cells stably expressing the TEE chimera (MCF7EGFR-KO-TEE) were serum starved overnight and stimulated with 50 ng/ml NGF for the indicated times. Levels of phosphorylated EGFR (pTyr1068) and ERK1/2 (pThr202/pTyr204), and of total EGFR and ERK, were detected by immunoblotting of total cell lysates. Grb2 was used as a loading control. Blots are representative of three independent experiments.
(C) Quantitation of pERK (blue) and pEGFR (red) from (B), normalized to Grb2 and the peak value at 5 min, plotting mean ± SD (n = 3 experiments), with no statistically significant differences between plots using the Bonferroni-Dunn method (alpha = 0.05). Repeats for each data point were analyzed individually, without assuming a consistent SD.
Our experiments with the TEE chimera demonstrated that the same RTK intracellular region (from EGFR) can signal with quite different dynamics depending on how it is activated. Similarly, activating the EGFR intracellular region in a PDGFR/EGFR chimera can allow PDGF to promote differentiation of PC12 cells and sustained ERK signaling (61). One possible explanation for these results is suggested by our earlier studies of EGFR activated by different growth factors (21). The different EGFR ligands stabilize dimers with different structural arrangements and strengths. Weaker – or more short-lived – EGFR dimers (as induced by EPGN and EREG) promote sustained signaling by EGFR. EGF induces stronger (and longer-lived) EGFR dimers, which promote transient signaling and cell proliferation. Based on these observations, we suggest that placing the EGFR intracellular region in the context of the TEE and PDGFR/EGFR chimeras may weaken dimerization compared with that seen for EGF/EGFR complexes – likely because cooperation between the extra- and intracellular dimer contacts is impaired in the artificial chimeras. The weakened dimers then induce sustained signaling that mimics EGFR activated by EPGN or EREG (21). Modulation of signaling outcomes arising from different dimer stability in this way may be a general phenomenon in the RTK family. Indeed, other studies with Kit (27) and FGFR (63) have also linked altered dimer strength to signaling specificity and outcome.
Reducing the duration of EGFR activation with inhibitor leads to more sustained signaling
If sustained EGFR signaling is promoted by weakened or shorter-lived receptor dimerization – as for EREG/EPGN-activated EGFR (21) or possibly the TEE chimera (Fig. 5B,C) – we reasoned that the same effect might also be achieved by adding sub-stoichiometric levels of EGFR inhibitors. Low levels of a reversible ATP-competitive inhibitor like erlotinib that binds to and dissociates from the EGFR kinase domain rapidly (64, 65) should reduce the average length of time for which the kinase is active in an EGF-induced receptor dimer. This effect should mimic reduced lifetime (and thus strength) of the activated receptor dimer. To test the hypothesis that reducing the duration of kinase activation leads to sustained signaling at the receptor level, we monitored EGFR autophosphorylation dynamics in EGF-treated MCF7 cells in the presence of increasing erlotinib concentrations (Fig. 6A and fig. S4, A and B). The characteristic transient pEGFR (and ERK) activation responses to EGF were seen in the absence of erlotinib, and these responses remained largely unchanged as erlotinib concentration was increased to 7.5–15 nM. Between 30 nM and 50 nM erlotinib, however, the pEGFR signals at 20 and 60 min appeared greater than those in the absence of erlotinib (Fig. 6B) in the same blots. Adding 40 nM erlotinib actually caused a statistically significant increase in the observed level of EGFR phosphorylation at 60 min following EGF addition (Fig. 6C), despite reducing it by ~35% at the 5 minute time-point. As a result, the EGFR autophosphorylation response to EGF was markedly more sustained at 30–50 nM erlotinib than without inhibitor. Beyond 50 nM erlotinib, the overall signal was progressively further inhibited as expected, decaying almost completely by 1,000 nM. These data support the idea that reducing the lifetime of the activated EGFR kinase causes signaling to become more prolonged or sustained – whether this is achieved by weakening dimerization per se (as in an EREG-induced dimer) or by transiently occupying the activated kinase with a rapidly dissociating inhibitor. As with all of the other manipulations outlined above, more sustained EGFR autophosphorylation in the presence of 30–50 nM erlotinib also led to more sustained ERK activation (Fig. 6A,B; fig. S4, A and B).
Fig. 6. Sub-stoichiometric levels of erlotinib promote sustained EGFR signaling.

(A and B) Serum-starved MCF7 cells were either left untreated (no erlotinib) or were pretreated with increasing doses of erlotinib for 60 min prior to stimulation with 200 ng/ml EGF for the indicated times. Levels of phosphorylated pEGFR (pTyr1068) and ERK1/2 (pThr202/pTyr204) were detected by immunoblotting of total cell lysates, with Grb2 levels as a protein loading control. Total EGFR and ERK levels are shown in fig. S4, A and B.
(B) Comparison of pEGFR levels at 20 min and 60 min in untreated cells and those treated with 30 nM or 40 nM erlotinib. Blots are representative of 3 independent experiments (30 nM) or 7 independent experiments (40 nM erlotinib). Total ERK and EGFR levels are shown in fig. S4, A and B.
(C) Quantitation of pEGFR levels for untreated cells and cells treated with 40 nM erlotinib, normalized to Grb2 (loading control) and the peak level at 5 min for untreated samples. Data represent mean ± SD for 7 independent measurements. Statistical significance was determined using the Bonferroni-Dunn method (alpha = 0.05), with repeats for each data point analyzed individually, without assuming a consistent SD. Adjusted p values (after Bonferroni correction) were *p < 0.05, **p < 0.01, and ***p < 0.005. After Bonferroni correction, p = 0.085 for simple differences between erlotinib treated and untreated groups at 20 min, and p = 0.038 at 60 min. However, if we use the null hypothesis that erlotinib reduces EGFR phosphorylation at later time points by the same percentage (by 35%) as at 5 mins, Bonferroni corrected p values for these differences would be 0.0013 (20 min) and 0.0004 (60 min) – or 0.00045 and 0.00013 without the Bonferroni multiple-comparison correction.
At first view, the increase in the 60 min pEGFR (and pERK) signal with 40 nM erlotinib seems paradoxical. Such effects of reducing duration of EGFR activation would be predicted in the context of kinetic proofreading, however (21, 66). If a progressive multi-site phosphorylation ‘program’ must be completed (or nearly completed) to elicit a negative feedback event (such as endocytosis, phosphatase recruitment, or delivery to phosphatases), reducing duration of the active state could prevent its engagement. Thus, low levels of inhibitor would be expected to blunt the negative feedback regulation that normally makes the response transient. The inhibitor could achieve this through effects on receptor internalization or trafficking – although we cannot exclude other influences. Rashkov et al. (67) have described an analysis of dual phosphorylation/dephosphorylation cycles that predicts similar enhancement of phosphorylation at low inhibitor concentrations, which they term hormesis.
Discussion
Signaling networks downstream of RTKs are highly complex, and full understanding of how RTKs specify cell-fate decisions requires sophisticated computational models. Quantitative models of cellular responses to growth factors such as NGF and EGF (68, 69) typically assume two receptor states, in which the receptor is either active (when dimeric) or inactive (when monomeric and unphosphorylated) – and RTK dimers are considered as a single molecular class (68). The resulting models have been valuable in identifying emergent properties of the signaling networks downstream of RTKs (70, 71), but may underestimate the extent to which non-linear processes at the level of the RTK itself define network properties (18, 55, 72).
Focusing on the classic question of how NGF and EGF promote orthogonal cellular responses, we set out to ask how TrkA and EGFR differentially engage feedback loops in the Raf-MEK-ERK signaling network. Our results argue that the sustained ERK signaling elicited by NGF in PC12 cells cannot be explained by its proposed selective use of Rap1 rather than Ras (17, 31). PKCδ inhibition studies also contradict the proposal that a TrkA-specific positive feedback from ERK to Raf in NGF signaling gives it sustained switch-like properties (13). It is important to note that suggestions of positive ERK-to-Raf feedback emerged from studies using modular response analysis (MRA) in which a module within the larger network is conceptually isolated and subjected to perturbations by siRNA – in this case directed against Raf, MEK, and ERK (13, 33). An important limitation of the MRA approach when comparing NGF/TrkA and EGF/EGFR signaling is that events at the receptor level are assumed equivalent. Defining the module in this way will conflate feedback to the receptor with feedback to Raf, which is at the head of the isolated module. We therefore suggest that the apparent differences seen in Raf-MEK-ERK network topology with the MRA approach may actually reflect feedback to the receptor level. This notion is consistent with other findings that support the importance of receptor-level mechanisms in determining signaling dynamics downstream of RTKs (18–20).
Although we could not switch ERK signaling dynamics by manipulating proposed downstream feedback loops, we could convert the normally transient response of EGFR to EGF to a sustained response through several different receptor-level manipulations. EGF-induced EGFR autophosphorylation was sustained when internalization was impaired by dynamin inhibition or through EGFR mutations. A TrkA/EGFR chimera (TEE) also gave sustained responses to NGF activation. Further, sub-stoichiometric levels of a reversible EGFR kinase inhibitor made EGF-induced EGFR activation more sustained. In each case, ERK activation dynamics mirrored the altered EGFR signaling kinetics. Indeed, the activation kinetics of ERK followed those of RTK in all experiments described here, arguing for a focus on receptor-level mechanisms to more completely understand MAPK network dynamics.
Although our findings cannot precisely explain the different kinetic characteristics of TrkA and EGFR signaling, the fact that the EGFR intracellular region can elicit either sustained or transient signaling (in the same cells) shows that kinetics are not simply defined by ‘RTK identity’. Otherwise, differences might be explainable by recruitment of distinct downstream signaling components. Rather, our results (and previous data (21)) argue that the lifetime and/or strength of the activated RTK dimer may define whether receptor phosphorylation (and thus downstream signaling) is sustained or transient. For RTKs that undergo multi-site autophosphorylation (which is most of them), a kinetic proofreading model (73) can be envisioned in which only the longest-lived dimers can complete the autophosphorylation ‘program’ and fully engage negative feedbacks that require recruitment to late-phosphorylated tyrosines. Shorter-lived dimers would accumulate less autophosphorylation (and thus fail to complete the program before disassociation), but might remain partially phosphorylated for longer if they fail to engage negative feedbacks that promote dephosphorylation. Intracellular trafficking must also play a role, because impairing EGFR internalization switches it from transient to sustained (Fig. 4B,D). Work from the Olsen and Bastiaens labs (54, 55) has argued that, once EGFR is activated at the plasma membrane, its phosphorylation is dynamically shaped by post-endocytic trafficking that controls access of the receptor to 4–5 key protein tyrosine phosphatases in different subcellular locations. By influencing this trafficking ‘itinerary’, changes in vesicular dynamics (or trafficking pathways) of the receptor with different ligands and/or with different dimer lifetimes/strengths could thus define whether receptor phosphorylation (and signaling) is sustained or transient. Differences in degree of internalization itself cannot explain the distinction between EGFR and TrkA signaling or between EGF- and EREG-induced EGFR signaling. Rapid receptor internalization occurs in each case (47, 48, 53). There are known differences in post-endocytic vesicular dynamics, however. NGF-activated TrkA is primarily (~70%) recycled to the plasma membrane after endocytosis in PC12 cells (74), whereas most EGF-activated EGFR (~80%) is targeted for lysosomal degradation in parallel studies. Similarly, EREG-activated EGFR shows much more recycling than EGF-activated receptor (53).
Together with previous results, the work described here suggests that RTK signaling specificity and cellular outcome are defined by receptor activation kinetics – determined at least in part by the lifetime/strength of ligand-induced receptor dimers (21, 27, 63) and/or their consequent intracellular trafficking itineraries (54, 55). This in turn suggests that it might be possible to switch EGFR signaling from transient to sustained in pathological settings such as cancer. Indeed, we show that sub-stoichiometric concentrations of erlotinib can make pEGFR and ERK responses to EGF more sustained, presumably by reducing the lifetime of the activated kinase. If modulating the time for which the kinase is active can alter signaling dynamics for EGFR, it should also be possible to do this with extracellularly targeted antibodies. Early reports described EGFR antibodies selective for particular receptor conformations (75) or that selectively stabilize conformations preferred by one or other of its ligands (76). Rather than developing approaches designed to shut off signaling by the receptors (or other signal transducers), redirecting signal dynamics with such agents may be more effective. Indeed, Hoffmann and colleagues have suggested a more general strategy, based on studies of NF-κB signaling, in which signaling dynamics per se could be treated as a pharmacological target (77).
Materials and Methods
Ligands, antibodies and inhibitors.
Recombinant rat EGF was obtained from PeproTech (Cat. #400–25) and recombinant rat beta-NGF from R&D Systems (#556-NG-100). Recombinant human NRG1α (Cat. #296-HR) and NRG1β (Cat. #396-HB/CF) were obtained from R&D Systems. All ligands were reconstituted in sterile PBS containing 1 mg/ml bovine serum albumin (BSA) (Roche #03116956001). The following antibodies were used: phospho-p44/42 MAPK (ERK1/2) (pThr202/pTyr204) (E10) (Cell Signaling Technology (CST) #9106), p44/42 MAPK (ERK1/2) (L34F12) (CST #4696), Grb2 (C-23) (Santa Cruz #sc-255) and Grb2 (CST #3972), phospho-EGFR (pTyr1173) (53A5) (CST #4407), phospho-EGFR (pTyr845) (CST #2231), phospho-EGFR (pTyr1086) (CST #2220), phospho-EGFR (pTyr845) (CST #2234), hEGFR (Lab Vision Ab-15: ThermoFisher Sci MS-665-P0), EGFR (CST #2232), phospho-TrkA (pTyr674/675) (C50F3) (CST #4621), phospho-TrkA (pTyr490) (CST #9141), phospho-TrkA (pTyr785) (C67C8) (CST #4168), phospho-TrkA (pTyr785) (R&D #AF5479), TrkA (R&D #AF1056), Rap1A/Rap1B (26B4) (CST #2399), FRS2 (Fisher Sci #PA124685), phospho-ErbB4 (pTyr1242) (4C6) (Santa Cruz #sc81491), phospho-ErbB4 (pTyr1284) (21A9) (CST #4757) and anti-FLAG (CST #2044). The following kinase inhibitors were used: Gö7874 (Calbiochem #365252), erlotinib (CST #5083), and the dynamin GTPase inhibitor Dyngo 4a (Abcam #ab120689).
Cell lines and culture conditions
PC12 cells (ATCC #CRL-1721) were cultured in T-75 flasks (Denville # T1225 (1004016)) at 37˚C, humidified 5% CO2 in complete RPMI-1640 medium containing L-glutamine and HEPES (ThermoFisher Sci #22400089), supplemented with 1 mM sodium pyruvate (ThermoFisher Sci #11360070), 5% fetal bovine serum (FBS) (Atlanta Biologicals #S11550), 10% heat inactivated horse serum (ThermoFisher Sci #26050088) as well as 100 U/ml penicillin and 100 μg/ml streptomycin (ThermoFisher Sci #15140122). To subculture or renew medium, cells were harvested, centrifuged at 200 × g for 10 min at room temperature (RT), and supernatant was removed. The cell pellet was resuspended in 5 ml of fresh medium and cells were aspirated 4 times with a 20 ml syringe outfitted with a 22 gauge (1½ in.) needle to break up cell clusters. Appropriate aliquots of the cell suspension were added to new T-75 flasks containing 10–15 ml fresh medium and cells were cultured up to cell density 2–4 × 106 cells/ml. All cell lines were routinely tested for contaminants.
Cell stimulation
Two days before stimulation with ligands, cells were harvested from T-75 flasks, centrifuged and subjected to the needle aspiration procedure described above. Cells were plated on 100 × 20 mm Corning BioCoat collagen I-coated dishes (Fisher Sci #356450) in complete growth medium at a density of 1 × 106 cells/ml (10 ml per dish) and grown for one day at 37˚C in 5% CO2. Cells were starved overnight in RPMI-1640 medium with L-glutamine and HEPES, supplemented with sodium pyruvate and penicillin-streptomycin solution. Where applicable, cells were preincubated with inhibitors, left unstimulated or were stimulated with ligand(s) for different periods at 37˚C in humidified 5% CO2. Inhibitors and ligands were diluted to final concentrations in starvation media. 15 seconds before the end of a given stimulation period, the medium was removed by vacuum aspiration, dishes with cells were placed on ice and 0.6 ml of ice-cold Cell Lysis Buffer (CST #9803) supplemented with Phosphatase Inhibitor Cocktail Tablets PhosSTOP (Roche #04906837001) and Protease Inhibitor Cocktail Tablets Complete (Roche #11697498001) was added. Cells were scraped from the surface using a cell scraper (Fisher Sci #08100241) and harvested into an Eppendorf tube. Total cell lysates were vortexed, incubated for 30 min on ice, then centrifuged at 10,000 × g for 10 min at 4˚C. Equal amounts of solubilized proteins in supernatant were dissolved in NuPAGE LDS sample buffer (ThermoFisher Sci #NP0007) supplemented with 50 mM DTT and heated for 7 min at 75˚C.
Quantitative immunoblotting
Proteins in whole cell extracts were separated by gel electrophoresis using Xcell Surelock Electrophoresis system (ThermoFisher Scientific) and NuPAGE™ Novex™ 4–12% Bis-Tris Protein Gels (ThermoFisher Sci #NP0321BOX). Resolved proteins were transferred onto nitrocellulose membranes (Bio-Rad #162–0112) using the Multistrip Western blotting procedure as described (78, 79). Membranes were blocked in 4% BSA solution (Roche #03116956001) for 1 h at RT, incubated with primary antibodies for 1 h at RT, extensively washed with TBS-T washing buffer (10 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.5% (w/v) Triton X-100), and incubated with appropriate secondary antibodies, either Horse Anti-Mouse IgG, HRP-linked Antibody (CST #7076) or Goat Anti-Rabbit IgG (H+L) Cross Adsorbed Secondary Antibody, HRP conjugate (ThermoFisher Sci #31462) at dilutions 1:10,000 and 1:40,000 respectively, for 1 h at RT. After a final wash with TBS-T, net intensities of chemiluminescent signals from protein bands were detected by ECL using SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher Sci #34080). Bands were visualized and quantified with densitometry analysis using a KODAK Image Station 440CF (Kodak Scientific Imaging Systems, New Haven, CT). The signal intensities for a given protein were normalized by signal intensities of the loading control (Grb2 protein from the same gel) at each time point and were expressed either in arbitrary units or as fold changes over basal levels (in unstimulated cells). As in previous studies (21), we used Grb2 as a loading control for all quantitation because it is less abundant than tubulin or actin (for example), so can more easily be detected with an unsaturated signal – yielding more reliable normalization. Our previous studies have shown that Grb2 levels remain constant for the time frames of the experiments performed here and across ERK signaling conditions (69, 79). We also used Grb2 levels for normalization of pEGFR/pTrkA/pERK levels as in previous work (21, 69, 78, 79), having established that this yields the same results as normalization to total EGFR/TrkA/ERK respectively, and using the Grb2 signal avoids potential problems with effects of phosphorylation on antibody recognition. We demonstrated that Western blots were in a linear range across the experiments described here, by quantitating a 2-fold dilution series of a sample with maximally phosphorylated ERK (fig. S1). To compare time courses of protein phosphorylation measured in different experiments, data points were additionally normalized by the corresponding peak value at 5 min stimulation and expressed in percent. Results were presented by plotting mean ± SD plotted using GraphPad Prism 8. p values were determined using a two-tailed Wilcoxon rank sum test when assessing individual comparisons. For multiple comparisons, the Bonferroni-Dunn method (alpha = 0.05) was used in GraphPad Prism 8, with repeats for each data point analyzed individually, without assuming a consistent SD.
Transient cell transfection with siRNA
PC12 cells were harvested and resuspended in antibiotic-free complete media 30 min before transfection. Cells were aspirated 4 times with a 20 ml syringe outfitted with a 22 gauge (1½ in.) needle to break up cell clusters. 2 × 106 cells per sample were aliquoted into Eppendorf tubes and centrifuged at 100 × g for 10 min at RT. Supernatant was removed and the cell pellet was resuspended in 100 μl of Ingenio transfection solution (Mirus #MIR50111) containing siRNA. The following rat specific siRNA reagents were used: Rap1A/Rap1B siRNA duplexes (Integrated DNA Technologies # RNC.RNAI.N001005765.12.5, # RNC.RNAI.N001005765.12.10, # RNC.RNAI.N134346.12.1, # RNC.RNAI.N134346.12.3), SMARTpool siGENOME p75NTR siRNA (Dharmacon # M-080041-01-0005), FRS2 siRNA (Integrated DNA Technologies #RNC.RNAI.N001108097.12.1), FAK siRNA (Santa Cruz #sc-156037), SMARTpool siGENOME Src siRNA (Dharmacon #M-080144-02-0005). The responses of cells transfected with 100 nM of specific siRNA were compared to those transfected with 100 nM AllStars Negative Control siRNA (Qiagen #1027280). Cell suspensions containing siRNA were electroporated using the U-029 program on Nucleofector 2b device (Lonza, Basel, Switzerland). Immediately after electroporation, 0.5 ml of the pre-equilibrated antibiotic-free complete medium was added to the cuvette and the cell suspension was gently transferred into 60 × 15 mm Corning BioCoat collagen I-coated dishes (Fisher Sci #356401) to a final volume of 2 ml. Cells were allowed to attach for 6 h before addition of penicillin-streptomycin solution. Cell culture medium was aspirated next day and replaced with 5 ml of fresh complete medium. Cell signaling experiments were performed 72 h post-transfection.
Generation of stable cell lines
A DNA construct with wild-type human EGFR cloned into pcDNA3.1(+) was generated earlier (80). The internalization-defective variant 21KR/ΔAP2-hEGFR in pEGFP-N1 vector was kindly provided by Alexander Sorkin (University of Pittsburgh). Transfections of both DNA constructs into PC12 cells were performed using electroporation with Nucleofector 2b device as described above for siRNA procedure. Transfected cells were selected in medium containing 0.5 mg/ml G418. Stable clones were maintained in the presence of 250 mg/ml G418. EGFR expression levels were confirmed by Western blotting using anti-EGFR antibody specific to the human protein (CST #4267).
Cloning of chimeric receptors
A chimeric receptor (TEE) receptor consisting of the extracellular NTRK1 isoform I region (residues 1–385 from UniProt P04629–2, RefSeq NM_001012331.1, using mature protein numbering) linked to the EGFR transmembrane and cytoplasmic regions (residues 622–1186 from UniProt P00533–1, RefSeq NM_005228.3, using mature protein numbers) followed by a FLAG epitope was cloned into pcDNA3.1(+) using Gibson assembly of three overlapping dsDNA fragments (81, 82). The fragments included: (i) pcDNA3.1(+) linearized vector with additional Kozak (GCCACC) and FLAG sequences, (ii) the extracellular region of TrkA, and (iii) the transmembrane and cytoplasmic regions of EGFR. All three DNA fragments were generated by PCR using Q5 Hot Start High-Fidelity DNA polymerase (NEB # M0494S). The primers for each fragment have gene-specific 3’- sequences and 15–20 additional bases at the 5’ end that overlap with the ends of the adjacent fragment. PCR products were purified by QIAquick Gel Extraction Kit (Qiagen # 28704) and used in equimolar amounts in a Gibson assembly reaction consisting of 5 μl DNA, 5 μl H2O, and 10 μl enzyme-reagent NEBuilder HiFi DNA Assembly Master mix (NEB # E2621S). The reaction was incubated at 50˚C for 60 min, and the mixture was directly transformed into chemically competent NEB 10-beta E. coli cells and plated onto LB agar plates containing 100 μg/ml ampicillin overnight. Several colonies were tested by PCR to identify insert-containing constructs using OneTaq DNA polymerase (NEB # M0484S) and a pair of vector and gene-specific primers. The correct plasmid was finally selected by Sanger sequencing of the full-length chimeric receptor using a set of 9 vector and gene-specific primers.
Generation of MCF7EGFR-KO-TEE stable cell line
Transfections of chimeric TEE DNA construct into MCF7EGFR-KO cells were performed using electroporation with the Nucleofector 2b device as described above for siRNA introduction procedure. Transfected cells were selected in medium containing 0.5 mg/ml G418. Stable clones were maintained in the presence of 250 mg/ml G418. Expression levels of the TEE chimera were confirmed by Western blotting using anti-FLAG antibody (CST #2044).
Supplementary Material
fig. S1. Chemiluminescence signal intensity of maximally activated ERK as a function of sample dilution.
fig. S2. Effects of kinase inhibitors on NGF-induced activation of the MAPK cascade.
fig. S3. Levels of mutated EGFR expression in PC12 cells.
fig. S4. Total EGFR and total ERK for pEGFR and pERK blots shown in Fig. 6.
Acknowledgments:
We thank members of the Lemmon and Ferguson laboratories for discussions and comments on the manuscript, and Dr. Wei Wei for advice on statistical analysis.
Funding: This work was supported by the NIH, NCI grants R01CA198164, U54CA209992 and U54CA193417 (to M.A.L.), and K22CA215821 (to D.E.K).
Footnotes
This manuscript has been accepted for publication in Science Signaling. This version has not undergone final editing. Please refer to the complete version of record at: https://stke.sciencemag.org/
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data required to evaluate the conclusions of the paper are present in the main text or the Supplementary Materials of the paper.
References and Notes:
- 1.Chao MV, Growth factor signaling: where is the specificity? Cell 68, 995–997 (1992). [DOI] [PubMed] [Google Scholar]
- 2.Vasudevan HN, Soriano P, A thousand and one receptor tyrosine kinases: Wherein the specificity? Curr. Top. Dev. Biol 117, 393–404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dikic I, Schlessinger J, Lax I, PC12 cells overexpressing the insulin receptor undergo insulin-dependent neuronal differentiation. Curr. Biol 4, 702–708 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Marshall CJ, Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179–185 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Traverse S, Seedorf K, Paterson H, Marshall CJ, Cohen P, Ullrich A, EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr. Biol 4, 694–701 (1994). [DOI] [PubMed] [Google Scholar]
- 6.Traverse S, Gomez N, Paterson H, Marshall C, Cohen P, Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem. J 288, 351–355 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purvis JE, Lahav G, Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–956 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albeck JG, Mills GB, Brugge JS, Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell 49, 249–261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakakuki T, Birtwistle MR, Saeki Y, Yumoto N, Ide K, Nagashima T, Brusch L, Ogunnaike BA, Okada-Hatakeyama M, Kholodenko BN, Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell 141, 884–896 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy LO, MacKeigan JP, Blenis J, A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol. Cell. Biol 24, 144–153 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J, Molecular interpretation of ERK signal duration by immediate early gene products. Nature Cell Biol 4, 556–564 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Avraham R, Yarden Y, Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nature Rev. Mol. Cell Biol 12, 104–117 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Santos SD, Verveer PJ, Bastiaens PI, Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nature Cell Biol 9, 324–330 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Brightman FA, Fell DA, Differential feedback regulation of the MAPK cascade underlies the quantitative differences in EGF and NGF signalling in PC12 cells. FEBS Lett. 482, 169–174 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Cirit M, Wang CC, Haugh JM, Systematic quantification of negative feedback mechanisms in the extracellular signal-regulated kinase (ERK) signaling network. J. Biol. Chem 285, 36736–36744 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kao S, Jaiswal RK, Kolch W, Landreth GE, Identification of the mechanisms regulating the differential activation of the MAPK cascade by epidermal growth factor and nerve growth factor in PC12 cells. J. Biol. Chem 276, 18169–18177 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Sasagawa S, Ozaki Y, Fujita K, Kuroda S, Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nature Cell Biol 7, 365–373 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Sparta B, Pargett M, Minguet M, Distor K, Bell G, Albeck JG, Receptor Level Mechanisms Are Required for Epidermal Growth Factor (EGF)-stimulated Extracellular Signal-regulated Kinase (ERK) Activity Pulses. J. Biol. Chem 290, 24784–24792 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toettcher JE, Weiner OD, Lim WA, Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155, 1422–1434 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bugaj LJ, Sabnis AJ, Mitchell A, Garbarino JE, Toettcher JE, Bivona TG, Lim WA, Cancer mutations and targeted drugs can disrupt dynamic signal encoding by the Ras-Erk pathway. Science 361, eaao3048 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freed DM, Bessman NJ, Kiyatkin A, Salazar-Cavazos E, Byrne PO, Moore JO, Valley CC, Ferguson KM, Leahy DJ, Lidke DS, Lemmon MA, EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics. Cell 171, 683–695 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson KJ, Gilmore JL, Foley J, Lemmon MA, Riese DJ 2nd., Functional selectivity of EGF family peptide growth factors: implications for cancer. Pharmacol. Ther 122, 1–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sweeney C, Carraway KL 3rd., Ligand discrimination by ErbB receptors: Differential signaling through differential phosphorylation site usage. Oncogene 19, 5568–5573 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Sweeney C, Lai C, Riese DJ 2nd., Diamonti AJ, Cantley LC, Carraway KL 3rd., Ligand discrimination in signaling through an ErbB4 receptor homodimer. J. Biol. Chem 275, 19803–19807 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Wilson KJ, Mill C, Lambert S, Buchman J, Wilson TR, Hernandez-Gordillo V, Gallo RM, Ades LM, Settleman J, Riese DJ 2nd., EGFR ligands exhibit functional differences in models of paracrine and autocrine signaling. Growth Factors 30, 107–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macdonald-Obermann JL, Pike LJ, Different epidermal growth factor (EGF) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation. J. Biol. Chem 289, 26178–26188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho CCM, Chhabra A, Starkl P, Schnorr PJ, Wilmes S, Moraga I, Kwon HS, Gaudenzio N, Sibilano R, Wehrman TS, Gakovic M, Sockolosky JT, Tiffany MR, Ring AM, Piehler J, Weissman IL, Galli SJ, Shizuru JA, Garcia KC, Decoupling the functional pleiotropy of stem cell factor by tuning c-Kit signaling. Cell 168, 1041–1052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim AR, Ulirsch JC, Wilmes S, Unal E, Moraga I, Karakukcu M, Yuan D, Kazerounian S, Abdulhay NJ, King DS, Gupta N, Gabriel SB, Lander ES, Patiroglu T, Ozcan A, Ozdemir MA, Garcia KC, Piehler J, Gazda HT, Klein DE, Sankaran VG, Functional selectivity in cytokine signaling revealed through a pathogenic EPO mutation. Cell 168, 1053–1064 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, Nolan GP, Piehler J, Schreiber G, Garcia KC, Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 146, 621–632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selimkhanov J, Taylor B, Yao J, Pilko A, Albeck J, Hoffmann A, Tsimring L, Wollman R, Systems biology. Accurate information transmission through dynamic biochemical signaling networks. Science 346, 1370–1373 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJ, Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392, 622–626 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Langlois WJ, Sasaoka T, Saltiel AR, Olefsky JM, Negative feedback regulation and desensitization of insulin- and epidermal growth factor stimulated p21ras activation. J. Biol. Chem 270, 25320–25323 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Kholodenko BN, Kiyatkin A, Bruggeman FJ, Sontag E, Westerhoff HV, Hoek JB, Untangling the wires: a strategy to trace functional interactions in signaling and gene networks. Proc. Natl. Acad. Sci. U. S. A 99, 12841–12846 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huff K, End D, Guroff G, Nerve growth factor-induced alteration in the response of PC12 pheochromocytoma cells to epidermal growth factor. J. Cell Biol 88, 189–198 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein R, Jing SQ, Nanduri V, O’Rourke E, Barbacid M, The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 65, 189–197 (1991). [DOI] [PubMed] [Google Scholar]
- 36.Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR, Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron 9, 883–896 (1992). [DOI] [PubMed] [Google Scholar]
- 37.Singh B, Carpenter G, Coffey RJ, EGF receptor ligands: recent advances. F1000Res. 5, F1000 Faculty Rev-2270 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Falls DL, Neuregulins: functions, forms, and signaling strategies. Exp. Cell Res 284, 14–30 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Jones JT, Akita RW, Sliwkowski MX, Binding specificities and affinities of egf domains for ErbB receptors. FEBS Lett. 447, 227–231 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Vaskovsky A, Lupowitz Z, Erlich S, Pinkas-Kramarski R, ErbB-4 activation promotes neurite outgrowth in PC12 cells. J. Neurochem 74, 979–987 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Ferguson KM, Darling PJ, Mohan MJ, Macatee TL, Lemmon MA, Extracellular domains drive homo- but not hetero-dimerization of erbB receptors. EMBO J. 19, 4632–4643 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G, All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J. Biol. Chem 271, 5251–5257 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Sorkin A, Goh LK, Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res 314, 3093–3106 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harper CB, Martin S, Nguyen TH, Daniels SJ, Lavidis NA, Popoff MR, Hadzic G, Mariana A, Chau N, McCluskey A, Robinson PJ, Meunier FA, Dynamin inhibition blocks botulinum neurotoxin type A endocytosis in neurons and delays botulism. J. Biol. Chem 286, 35966–35976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goh LK, Huang F, Kim W, Gygi S, Sorkin A, Multiple mechanisms collectively regulate clathrin-mediated endocytosis of the epidermal growth factor receptor. J. Cell Biol 189, 871–883 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamada M, Ikeuchi T, Aimoto S, Hatanaka H, EGF-induced sustained tyrosine phosphorylation and decreased rate of down-regulation of EGF receptor in PC12h-R cells which show neuronal differentiation in response to EGF. Neurochem. Res 21, 815–822 (1996). [DOI] [PubMed] [Google Scholar]
- 47.Jullien J, Guili V, Reichardt LF, Rudkin BB, Molecular kinetics of nerve growth factor receptor trafficking and activation. J. Biol. Chem 277, 38700–38708 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC, Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J. Neurosci 16, 7950–7964 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Opresko LK, Chang CP, Will BH, Burke PM, Gill GN, Wiley HS, Endocytosis and lysosomal targeting of epidermal growth factor receptors are mediated by distinct sequences independent of the tyrosine kinase domain. J. Biol. Chem 270, 4325–4533 (1995). [DOI] [PubMed] [Google Scholar]
- 50.Jing S, Tapley P, Barbacid M, Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron 9, 1067–1079. (1996). [DOI] [PubMed] [Google Scholar]
- 51.Green SH, Rydel RE, Connolly JL, Greene LA, PC12 cell mutants that possess low- but not high-affinity nerve growth factor receptors neither respond to nor internalize nerve growth factor. J. Cell Biol 102, 830–843 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Moheban DB, Conway BR, Bhattacharyya A, Segal RA, Cell surface Trk receptors mediate NGF-induced survival while internalized receptors regulate NGF-induced differentiation. J. Neurosci 20, 5671–5678 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roepstorff K, Grandal MV, Henriksen L, Knudsen SL, Lerdrup M, Grøvdal LM, Willumsen BM, van Deurs B, Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 10, 1115–1127 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Francavilla C, Papetti M, Rigbolt KT, Pedersen AK, Sigurdsson JO, Cazzamali G, Karemore G, Blagoev B, Olsen JV, Multilayered proteomics reveals molecular switches dictating ligand-dependent EGFR trafficking. Nature Struct. Mol. Biol 23, 608–618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stanoev A, Mhamane A, Schuermann KC, Grecco HE, Stallaert W, Baumdick M, Brüggemann Y, Joshi MS, Roda-Navarro P, Fengler S, Stockert R, Roßmannek L, Luig J, Koseska A, Bastiaens PIH, Interdependence between EGFR and phosphatases spatially established by vesicular dynamics generates a growth factor sensing and responding network. Cell Syst. 7, 295–309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berger MB, Mendrola JM, Lemmon MA, ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS Lett. 569, 332–336 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Lev S, Yarden Y, Givol D, Receptor functions and ligand-dependent transforming potential of a chimeric kit proto-oncogene. Mol. Cell. Biol 10, 6064–6068 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piccinini G, Bacchiocchi R, Serresi M, Vivani C, Rossetti S, Gennaretti C, Carbonari D, Fazioli F, A ligand-inducible epidermal growth factor receptor/anaplastic lymphoma kinase chimera promotes mitogenesis and transforming properties in 3T3 cells. J. Biol. Chem 277, 22231–22239 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Riedel H, Dull TJ, Schlessinger J, Ullrich A, A chimaeric receptor allows insulin to stimulate tyrosine kinase activity of epidermal growth factor receptor. Nature 324, 68–70 (1986). [DOI] [PubMed] [Google Scholar]
- 60.Seedorf K, Felder S, Millauer B, Schlessinger J, Ullrich A, Analysis of platelet-derived growth factor receptor domain function using a novel chimeric receptor approach. J. Biol. Chem 266, 12424–12431 (1991). [PubMed] [Google Scholar]
- 61.Tyson DR, Larkin S, Hamai Y, Bradshaw RA, PC12 cell activation by epidermal growth factor receptor: role of autophosphorylation sites. Int. J. Dev. Neurosci 21, 63–74 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Ward MD, Leahy DJ, Kinase activator-receiver preference in ErbB heterodimers is determined by intracellular regions and is not coupled to extracellular asymmetry. J. Biol. Chem 290, 1570–1579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang Z, Tan Y, Gu J, Liu Y, Song L, Niu J, Zhao L, Srinivasan L, Lin Q, Deng J, Li Y, Conklin DJ, Neubert TA, Cai L, Li X , Mohammadi M, Uncoupling the mitogenic and metabolic functions of FGF1 by tuning FGF1-FGF receptor dimer stability. Cell Rep. 20, 1717–1728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barkovich KJ, Hariono S, Garske AL, Zhang J, Blair JA, Fan QW, Shokat KM, Nicolaides T, Weiss WA, Kinetics of inhibitor cycling underlie therapeutic disparities between EGFR-driven lung and brain cancers. Cancer Disc 2, 450–457 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, Alligood KJ, Rusnak DW, Gilmer TM, Shewchuk L, A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 64, 6652–6659 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Swain PS, Siggia ED, The role of proofreading in signal transduction specificity. Biophys. J 82, 2928–2933 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rashkov P, Barrett IP, Beardmore RE, Bendtsen C, Gudelj I, Kinase inhibition leads to hormesis in a dual phosphorylation-dephosphorylation cycle. PLoS Comput. Biol 12, e1005216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kholodenko BN, Demin OV, Moehren G, Hoek JB, Quantification of short term signaling by the epidermal growth factor receptor. J. Biol. Chem 274, 30169–30181 (1999). [DOI] [PubMed] [Google Scholar]
- 69.Fey D, Aksamitiene E, Kiyatkin A, Kholodenko BN, Modeling of receptor tyrosine kinase signaling: Computational and experimental protocols. Methods Mol. Biol 1636, 417–453 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Volinsky N, Kholodenko BN, Complexity of receptor tyrosine kinase signal processing. Cold Spring Harb. Perspect. Biol 5, a009043 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sampattavanich S, Steiert B, Kramer BA, Gyori BM, Albeck JG, Sorger PK, Encoding growth factor identity in the temporal dynamics of FOXO3 under the combinatorial control of ERK and AKT kinases. Cell Syst. 6, 664–678 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sigismund S, Algisi V, Nappo G, Conte A, Pascolutti R, Cuomo A, Bonaldi T, Argenzio E, Verhoef LG, Maspero E, Bianchi F, Capuani F, Ciliberto A, Polo S, Di Fiore PP, Threshold-controlled ubiquitination of the EGFR directs receptor fate. EMBO J. 32, 2140–2157 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McKeithan TW, Kinetic proofreading in T-cell receptor signal transduction. Proc. Natl. Acad. Sci. U. S. A 92, 5042–5046 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen ZY, Ieraci A, Tanowitz M, Lee FS, A novel endocytic recycling signal distinguishes biological responses of Trk neurotrophin receptors. Mol. Biol. Cell 16, 5761–5772 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Defize LH, Boonstra J, Meisenhelder J, Kruijer W, Tertoolen LG, Tilly BC, Hunter T, van PM Bergen en Henegouwen, W. H. Moolenaar, S. W. de Laat, Signal transduction by epidermal growth factor occurs through the subclass of high affinity receptors. J. Cell Biol 109, 2495–2507 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Winkler ME, O’Connor L, Winget M, Fendly B, Epidermal growth factor and transforming growth factor alpha bind differently to the epidermal growth factor receptor. Biochemistry 28, 6373–6378 (1989). [DOI] [PubMed] [Google Scholar]
- 77.Behar M, Barken D, Werner SL, Hoffmann A, The dynamics of signaling as a pharmacological target. Cell 155, 448–461 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aksamitiene E, Hoek JB, Kholodenko B, Kiyatkin A, Multistrip Western blotting to increase quantitative data output. Electrophoresis 28, 3163–3173 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aksamitiene E, Hoek JB, Kiyatkin A, Multistrip Western blotting: a tool for comparative quantitative analysis of multiple proteins. Methods Mol. Biol 1312, 197–226 (2015). [DOI] [PubMed] [Google Scholar]
- 80.Choi SH, Mendrola JM, Lemmon MA, EGF-independent activation of cell-surface EGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene 26, 1567–1576 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Gibson DG, Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 498, 349–361 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO, Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
fig. S1. Chemiluminescence signal intensity of maximally activated ERK as a function of sample dilution.
fig. S2. Effects of kinase inhibitors on NGF-induced activation of the MAPK cascade.
fig. S3. Levels of mutated EGFR expression in PC12 cells.
fig. S4. Total EGFR and total ERK for pEGFR and pERK blots shown in Fig. 6.