

Abstract
Background
Nager syndrome is a rare genetic syndrome characterized by craniofacial and preaxial limb anomalies. Haploinsufficiency of the SF3B4 gene has been identified as a significant reason for Nager syndrome. Treacher Collins syndrome (TCS) has similar facial features; however, the TCOF1, POLR1D, and POLR1C genes have been reported as the critical disease‐causing genes. Similar phenotypes make it easy to misdiagnose.
Case report
In this report, we have presented a case of one newborn with acrofacial dysostosis, who was first diagnosed with TCS. Expanded next‐generation sequencing eventually detected a (c.1A>G) heterozygous mutation in the SF3B4 gene at chr1:149899651 that was confirmed by Sanger sequencing. Combined with his preaxial limb anomalies discovered after his death, a diagnosis of Nager syndrome was made.
Conclusions
This report presents one patient with Nager syndrome who was initially misdiagnosed with TCS. Correct genetic testing will be beneficial to future prenatal diagnosis.
Keywords: acrofacial dysostosis, Nager syndrome, next‐generation sequencing, SF3B4 mutation, Treacher collins syndrome
1. INTRODUCTION
Craniofacial anomalies account for approximately one‐third of all congenital disabilities. Treacher Collins syndrome (TCS), Miller syndrome, Goldenhar syndrome, and Nager syndrome (NS) all manifest as craniofacial anomalies. 1
Treacher Collins syndrome is a rare genetic disorder of craniofacial development. Patients with TCS suffer life‐threatening airway complications and functional difficulties involving sight, hearing, speech, and feeding. Deformation of facial structures produces a characteristic appearance that includes malar hypoplasia, periorbital soft tissue anomalies, and maxillomandibular hypoplasia. Mutations in TCOF1 (78%‐93%) and POLR1C or POLR1D (8%) cause the disease. 2 , 3 For Miller syndrome, patients have limb deformities besides mandibular dysostosis. However, limb deformities of Miller syndrome are behind of shaft, usually accompanied by all the legs of the fifth fingers dysplasia. The mutation of dihydroorotate dihydrogenase (DHODH) contributes to it. 4 Goldenhar syndrome is usually characterized by asymmetric facial atrophy, ears, mouth, and mandible developmental disorders. The etiology of this disease is still unclear. Multiple factors including chromosomal abnormalities, maternal diabetes mellitus or drug use, and influence of environment during pregnancy may involve it. 5 , 6
Nager syndrome was first described by Slingenberg in 1908, but the eponym was attributed to Felix R. Nager in 1948. Nager syndrome is a group of malformations characterized by acrofacial dysostosis and preaxial limb deformities, resulting from problems in the development of the first and second branchial arches and limb buds. 7 , 8 Serious acrofacial dysostosis can result in feeding difficulties and/or severe breathing difficulties during infancy. If it were not taken seriously, Nager syndrome could lead to neonatal death. 9 Although the exact pathogenesis remains uncertain, the SF3B4 gene has been confirmed as the significant disease‐causing gene in dominant forms of Nager syndrome. 10
Previous reports estimated an incidence of 3 per 1 000 000 for Nager syndrome. To date, less than 100 cases have been reported in the medical literature. 11 The similar phenotypes, along with the small number of reported cases, make it difficult to diagnose this rare disease. Next‐generation sequencing (NGS) is widely used to diagnose genetic diseases. 12 Here, we report a case which was initially diagnosed as TCS. However, gene detection by next‐generation sequencing helped us to arrive at a final diagnosis of Nager syndrome.
2. METHODS AND MATERIALS
2.1. Case presentation
A newborn (male) was born through vaginal delivery at 41 weeks’ gestation with a birth weight of 2995 g, length of 48 cm, frontal occipital circumference (OFC) of 33 cm, and an Apgar score of 10. The baby manifested an unusual facial manifestation including bilaterally symmetric malar hypoplasia, severe mandibular hypoplasia with retrognathia, maxillary protrusion, downward slanted palpebral fissures, stenosis (pin size) of the external auditory canals, a broad and high nasal bridge (rare among Chinese individuals), macrostomia, a contracted tongue, and dysplastic ears that leaned back (Figure 1A). A diagnosis of TCS was made according to these characteristic bird‐like facial abnormalities. The baby's parents and grandparents had no physical appearance abnormalities. There was no obvious abnormality during his mother's pregnancy except a relatively high amniotic fluid index (AFI) and slow growth rate of the fetal biparietal diameter (BPD) since 32 weeks’ gestation (Figure 2).
FIGURE 1.
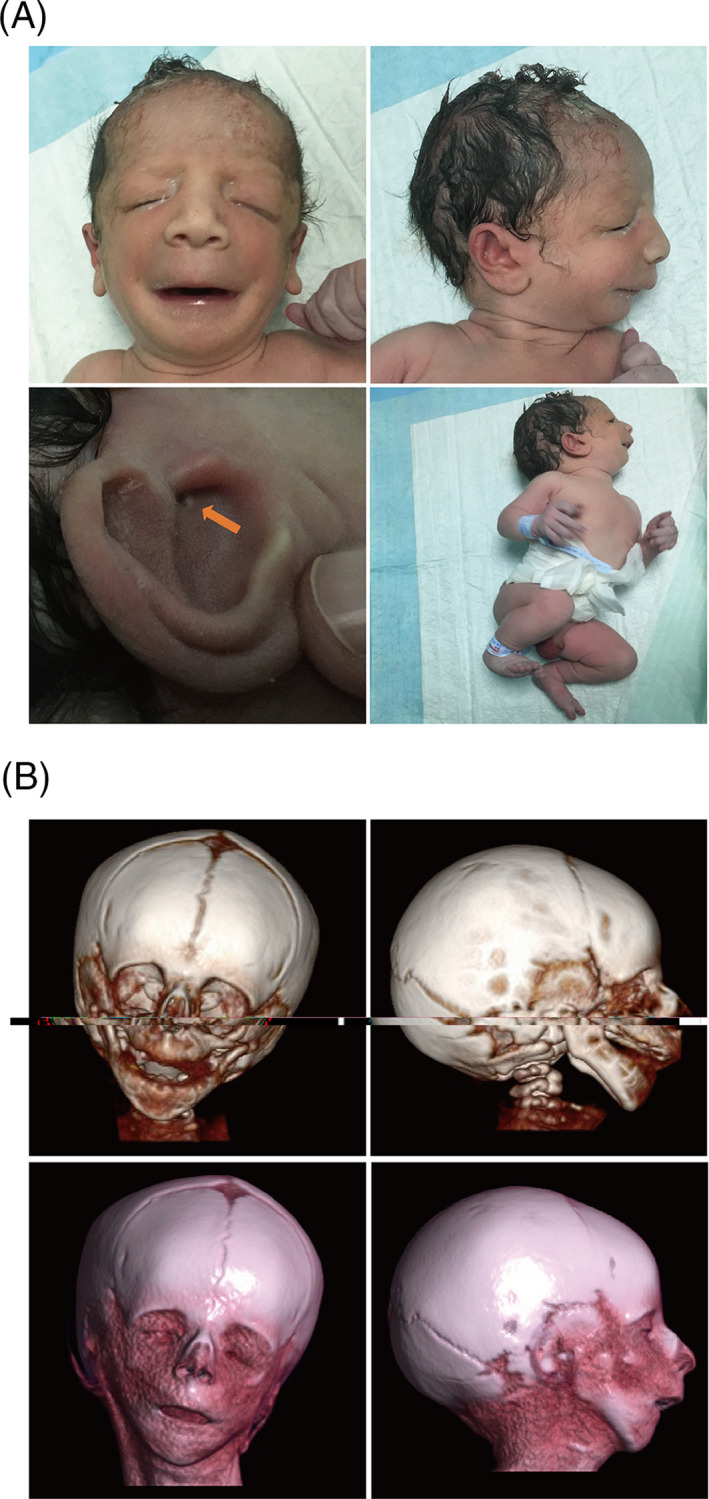
Clinical manifestation of the newborn. A, The craniofacial anomalies of the newborn. B, Complex facial malformations with mandibular hypoplasia and malar hypoplasia were constructed using three‐dimensional CT reconstruction images
FIGURE 2.
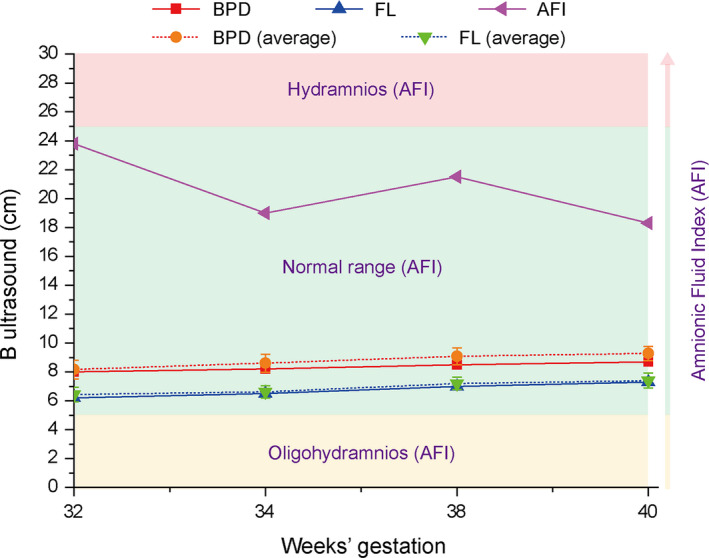
The ultrasonography data from 32 to 40 wk’ gestation
The newborn was immediately transferred to the department of pediatrics for further evaluation and treatment. On the first day of birth, hemoglobin was 226.0 g/L, hematocrit was 0.65, total serum bilirubin was 104.3 µmol/L, and uncorrected bilirubin was 94.1 µmol/L. The baby was treated with phototherapy (blue light) and 10 mL of 0.9% normal saline to dilute the blood. The following day, during feeding, nurses noticed that glucose water was draining from the nasal cavity, and the baby started vomiting. Oral feedings were not well established. The initial treatment consisted of gastrointestinal decompression and total parenteral nutrition therapy. A chest X‐ray and an abdominal ultrasound were normal, except for an unapparent gastric bubble. The baby's parents decided to forego treatment and took the baby home. A small blood sample was preserved for further examination. Follow‐up revealed that the baby died one month later. A postmortem three‐dimensional computed tomography (CT) reconstruction was performed and revealed complex facial malformations, with mandibular hypoplasia and malar hypoplasia (Figure 1B).
Cytogenetic analysis revealed a normal karyotype. NGS was applied to sequence the exons and exon‐intron boundaries of the TCOF1, POLR1D, and POLR1C genes (key disease‐causing genes in TCS) that revealed no mutations. Considering that the genetic testing result was not consistent with our original clinical diagnosis, we expanded the screening to include all known disease‐causing genes for single‐gene diseases associated with craniofacial anomalies. We reanalyzed the NGS data to check other related genes (ERCC1, ERCC2, ERCC6, OFD1, EVC, B3GAT3, TMCO1, EFNB1, ALX4, DHODH, SF3B4, and EFTUD2). To our surprise, a (c.1A>G) heterozygous mutation in the SF3B4 gene at chr1:149899651 was detected and confirmed using Sanger sequencing. The SF3B4 gene has been identified as the major disease‐causing gene in Nager syndrome, suggesting that we misdiagnosed the baby with TCS. Nager syndrome is an autosomal dominant disorder, so we re‐examined the proband's parents. Neither had acrofacial dysostosis, the limb abnormalities, or the same mutation as the proband (Figure 3A). The parents’ karyotypes were normal. We further focused on whether the patient had limb abnormalities. Then, we checked with his parents and found that movement of the baby's right thumb was restricted. We managed to review photographs taken when the newborn was alive and performed an X‐ray on the corpse. We noticed contracture of the first web and dysplasia of the right thumb and forefinger (Figure 3B). However, because of poor cryopreservation of the corpse, X‐ray films were of relatively low resolution.
FIGURE 3.
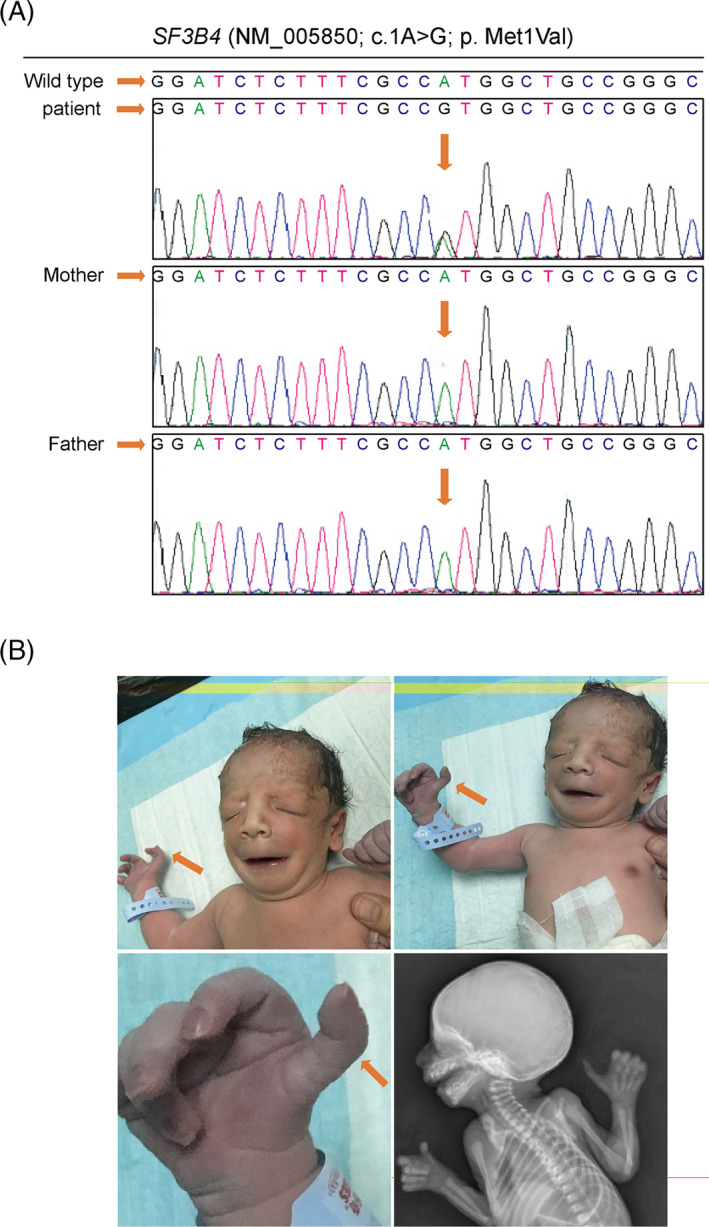
The SF3B4 gene mutation analysis and dysplasia of the right thumb and forefinger. A, Analysis of the SF3B4 gene mutation in the male newborn and his parents. B, Dysplasia of the right thumb and forefinger and contracture of the first web was apparent on photographs
Based on the combination of the neonate's clinical features and the genetic report, a diagnosis of Nager syndrome was made. Two years later, the mother gave birth to a healthy baby boy with a normal physical appearance.
3. DISCUSSION
Given how rare Nager syndrome is, its diagnosis relies on clinical features, patient history, and genetic testing. CT scans and X‐ray methods can reveal detailed craniofacial and preaxial limb bone anatomy in Nager syndrome and, thus, are more beneficial for diagnosis. Although ultrasonography is the most convenient way for prenatal examination, it has low accuracy in detecting bony abnormalities. So far, only four cases have been reported using prenatal ultrasonography that were confirmed by genetics or pathology. 13 Therefore, prenatal diagnosis of Nager syndrome is challenging. In this case report, ultrasound examinations did not reveal any craniofacial or preaxial limb bone anomalies. Although a relatively high AFI and poor growth rate of the BPD were detected using prenatal ultrasound, the evidence was not enough to suggest a Nager syndrome diagnosis. Nager syndrome comprises some malformations similar to those of other syndromes, including TCS, Miller syndrome, Edwards syndrome, and Goldenhar syndrome. The diagnostic criteria to differentiate Nager syndrome from other branchial arches syndromes are its unique preaxial limb deformities. In this case, the facial phenotype comprised malar and mandibular hypoplasia, down‐slanting palpebral fissures, and external ear dysplasia, overlapping the gestalt of TCS. However, the inconspicuous dysplasia of the right thumb and forefinger was overlooked. This is related to our limited experience with this syndrome, which should be taken seriously by fellow clinicians.
Within the past six years, haploinsufficiency of the SF3B4 gene has been confirmed as the major disease‐causing gene in dominant forms of Nager syndrome. Bernier et al 9 identified 18 heterogeneous mutations in the SF3B4 gene in 61% of 41 patients with Nager syndrome. The SF3B4 gene encodes SAP49, a component of the pre‐mRNA spliceosomal complex, on chromosome 1q12‐q21. Petit et al also confirmed mutations in SF3B4 in 9/14 families (64%) diagnosed with Nager syndrome. 14 However, more than one‐third of tested patients with Nager syndrome do not have an SF3B4 mutation. In this case, the newborn was, at first, considered to have TCS based on his severe acrofacial dysostosis, but the NGS test for TCS showed no mutations on the TCOF1, POLR1D, or POLR1C genes. Eventually, the diagnosis of TCS was withdrawn after a more in‐depth analysis of the NGS data. The initiation code variant, SF3B4 c.1A>G, was first classified as a PVS1 null variant (PVS1_Moderate). The variant has been reported as a de novo mutation in several unrelated individuals with Nager syndrome and is apparent in the patient's clinical phenotype (PS4 + PM6 + PP1). 10 , 14 The variant has not been recorded in the 1,000 human genome database or several population databases (PM2). Mutations in the SF3B4 gene have been associated with the autosomal dominant Nager syndrome, and mutation‐related disease is highly consistent with the patient's clinical phenotype (PP4). Based on the available evidence (PVS1 + PS4 + PM2 + PM6 + PP1 + PP4), this variant has been classified as pathogenic.
Therefore, we suggest performing broader spectrum tests such as exome sequencing, especially for those patients with normal results on the NGS panel.
Treatment should be tailored to the specific needs of each individual. Nager syndrome is a malformation resulting from problems in the development of the first and second branchial arches and limb buds. 1 The cause of the abnormal development of the pharyngeal arches is unknown. The first arch develops into muscles and nerves for chewing, as well as the mandible, two middle ear bones, and part of the auricles. The second arch develops into muscles and nerves for facial expression, as well as one middle ear bone, the majority of the external ear, and a portion of the palate. 15 Functional impairments mainly include difficulties in breathing and swallowing, craniofacial deformity, and hearing impairment, with subsequently delayed speech development. Dysphagia in Nager syndrome is often attributed to mandibular hypoplasia, temporomandibular joint problems, poor jaw opening, cleft palate, and an absent epiglottis. 16 Besides focusing on the corrective surgeries and early airway intervention, swallowing rehabilitation should be started early. However, some patients still do not have a normal swallowing function after corrective surgeries. Combined anatomical and sensory deficits in the pharynx may preclude normal swallowing. 16 Whether patients with swallowing impairment can experience improvement with aggressive swallowing rehabilitation remains to be determined. In this case, the parent of the newborn withdrew from treatment because of uncertainty regarding efficacy.
In conclusion, there are still difficulties in clinical diagnosis and treatments for Nager syndrome. Haploinsufficiency of the SF3B4 gene has been identified as the major disease‐causing gene in Nager syndrome. Genetic testing is crucial in diagnosing Nager syndrome to exclude other diseases with similar clinical features.
AUTHOR CONTRIBUTIONS
Jue Zhao is responsible for the intellectual content, literature research, clinical studies, data acquisition and analysis, statistical analysis, manuscript preparation, and editing; Liwei Yang is responsible for guaranteeing the integrity of the entire report, report concepts and design, and manuscript review. All authors read and approved the final manuscript.
ETHICAL APPROVAL
The report was approved by the Ethics Committee of Zhejiang Provincial Hospital. Informed consent was obtained.
ACKNOWLEDGEMENTS
We are grateful to Dr. Jiong Gao (BGI Genomics, BGI‐Shenzhen, Shenzhen 518083, China) for analyzing the genetic data.
Zhao J, Yang L. Broad‐spectrum next‐generation sequencing‐based diagnosis of a case of Nager syndrome. J Clin Lab Anal. 2020;34:e23426 10.1002/jcla.23426
REFERENCES
- 1. Terrazas K, Dixon J, Trainor PA, Dixon MJ. Rare syndromes of the head and face: mandibulofacial and acrofacial dysostoses. Wiley Interdiscip Rev Dev Biol. 2017;6:e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aljerian A, Gilardino MS. Treacher collins syndrome. Clin Plast Surg. 2019;46:197‐205. [DOI] [PubMed] [Google Scholar]
- 3. Wang P, Fan X, Fan Y. The research progress of Treacher Collins syndrome. J Clin Otorhinolaryngol Head Neck Surg. 2016;30:333‐338. [PubMed] [Google Scholar]
- 4. Kinoshita F, Kondoh T, Komori K, Matsui T, Harada N, Yanai A. Miller syndrome with novel dihydroorotate dehydrogenase gene mutations. Pediatr Int. 2011;53:587‐591. [DOI] [PubMed] [Google Scholar]
- 5. Ng YY, Hu JM, Su PH, Chen JY, Yang MS, Chen SJ. Goldenhar syndrome (oculoauriculovertebral dysplasia): report of one case. Acta Paediatr Taiwan. 2006;47:142‐145. [PubMed] [Google Scholar]
- 6. Bogusiak K, Puch A, Arkuszewski P. Goldenhar syndrome: current perspectives. World J Pediatr. 2017;13:405‐415. [DOI] [PubMed] [Google Scholar]
- 7. Sulik KK, Smiley SJ, Turvey TA, Speight HS, Johnston MC. Pathogenesis of cleft palate in Treacher Collins, Nager, and Miller syndromes. Cleft Palate J. 1989;26:209‐216; discussion 216. [PubMed] [Google Scholar]
- 8. Czeschik JC, Voigt C, Alanay Y, et al. Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum Genet. 2013;132:885‐898. [DOI] [PubMed] [Google Scholar]
- 9. Herrmann BW, Karzon R, Molter DW. Otologic and audiologic features of Nager acrofacial dysostosis. Int J Pediatr Otorhinolaryngol. 2005;69:1053‐1059. [DOI] [PubMed] [Google Scholar]
- 10. Bernier FP, Caluseriu O, Ng S, et al. Haploinsufficiency of SF3B4, a component of the pre‐mRNA spliceosomal complex, causes Nager syndrome. Am J Hum Genet. 2012;90:925‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wieczorek D. Human facial dysostoses. Clin Genet. 2013;83:499‐510. [DOI] [PubMed] [Google Scholar]
- 12. Izevbaye I, Liang LY, Mather C, El‐Hallani S, Maglantay R Jr, Saini L. Clinical validation of a myeloid next‐generations sequencing panel for single nucleotide variants, indels, and fusion genes. J Mol Diagn. 2020;22(2):208‐219. [DOI] [PubMed] [Google Scholar]
- 13. Lund IC, Vestergaard EM, Christensen R, Uldbjerg N, Becher N. Prenatal diagnosis of Nager syndrome in a 12‐week‐old fetus with a whole gene deletion of SF3B4 by chromosomal microarray. Eur J Med Genet. 2016;59:48‐51. [DOI] [PubMed] [Google Scholar]
- 14. Petit F, Escande F, Jourdain AS, et al. Nager syndrome: confirmation of SF3B4 haploinsufficiency as the major cause. Clin Genet. 2014;86:246‐251. [DOI] [PubMed] [Google Scholar]
- 15. Abdollahi Fakhim S, Shahidi N, Mousaviagdas M. A case report: nager acrofacial dysostosis. Iran J Otorhinolaryngol. 2012;24:45‐50. [PMC free article] [PubMed] [Google Scholar]
- 16. Tay SY, Loh WS, Lim TC. A case report of absent epiglottis in children with nager syndrome: its impact on swallowing. Cleft Palate Craniofac J. 2017;54:754‐757. [DOI] [PubMed] [Google Scholar]