

Abstract
Inflammatory cell infiltration contributes to the pathogenesis of acute respiratory distress syndrome (ARDS). Protectin DX (PDX), an endogenous lipid mediator, shows anti‐inflammatory and proresolution bioactions. In vivo, the mice were intraperitoneally injected with PDX (0.1 µg/mouse) after intratracheal (1 mg/kg) or intraperitoneal (10 mg/kg) LPS administration. Flow cytometry was used to measure inflammatory cell numbers. Clodronate liposomes were used to deplete resident macrophages. RT‐PCR, and ELISA was used to measure MIP‐2, MCP‐1, TNF‐α and MMP9 levels. In vitro, sorted neutrophils, resident and recruited macrophages (1 × 106) were cultured with 1 μg/mL LPS and/or 100 nmol/L PDX to assess the chemokine receptor expression. PDX attenuated LPS‐induced lung injury via inhibiting recruited macrophage and neutrophil recruitment through repressing resident macrophage MCP‐1, MIP‐2 expression and release, respectively. Finally, PDX inhibition of neutrophil infiltration and transmembrane was associated with TNF‐α/MIP‐2/MMP9 signalling pathway. These data suggest that PDX attenuates LPS‐stimulated lung injury via reduction of the inflammatory cell recruitment mediated via resident macrophages.
Keywords: acute respiratory distress syndrome, neutrophil, protectin DX, recruited macrophage, resident macrophage
1. INTRODUCTION
Acute respiratory distress syndrome (ARDS) is a foetal disease, 1 the hallmarks of which are massive pro‐inflammatory cytokine secretion and pro‐inflammatory cell infiltration. 2 , 3 , 4 Emerging evidence has revealed that macrophages, containing resident macrophages and circulating monocyte‐derived macrophages, are central in the pathogenesis of ARDS. 5 , 6
Resident macrophages reside in the alveolar space and perform tissue‐specific, homeostatic functions. 7 Growing evidence indicating that resident macrophage depletion protects against lung injury. 1 However, recruited macrophages generate cytokines that are connected with glycolytic and arginine metabolism. 8 A recent study showed that in a liposaccharide (LPS)‐induced ARDS mouse model, depletion of circulating monocytes attenuated lung infiltration by neutrophils and the severity of ARDSI. 9 , 10
Many cytokines, such as IL‐6, TNF‐α and matrix metalloproteinase 9 (MMP9), exhibit elevated expression in ARDS. Moreover, many chemokines are also involved in ARDS, such as macrophage inflammatory protein 2 (MIP‐2), monocyte chemoattractant protein‐1 (MCP‐1). 9 , 11 , 12 MCP‐1 is a widely expressed chemoattractant of monocytes and macrophages. In the lungs, MCP‐1 enhances monocyte trafficking and alveolar macrophage pool expansion, and MIP‐2 promotes neutrophils recruitment through binding to CCR2 and CXCR2, respectively. 12 , 13 , 14 , 15
Protectin DX (10S,17S‐dihydroxydocosa‐4Z,7Z,11E,13Z,15E,19Z‐hexaenoic acid) (PDX), an endogenous lipid mediator, exerts potent anti‐inflammatory and proresolution bioactions, including inhibiting neutrophil infiltration in murine peritonitis. 16 , 17 Previous study showed that PDX reduced LPS‐induced secretion of pro‐inflammatory cytokines, such as TNF‐α and MCP‐1. 18
We previously reported that PDX promoted alveolar fluid clearance and alleviated lung injury. 16 We also found that PDX attenuated bleomycin‐induced lung fibrosis and dysfunction in mice. 19
In the present study, we hypothesize that PDX attenuates LPS‐induced lung injury via inhibiting inflammatory cells recruitment. The secondary hypothesis is that PDX reduces recruited macrophage and neutrophil recruitment via repressing resident macrophage MCP‐1, MIP‐2 expression and release, respectively. Finally, we have been suggested that PDX inhibits neutrophil infiltration and transmembrane was associated with TNF‐α/MIP‐2/MMP9 signalling pathway.
2. MATERIALS AND METHODS
2.1. Materials
PDX was from Cayman Chemical Company (Ann Arbor, MI, USA). LPS (Escherichia coli serotype 055:B5) was from Sigma (St. Louis, MO, USA). TNF‐α, MIP‐2, MCP‐1 and MMP9 ELISA kits were from R&D Systems (Minneapolis, MN, USA). CXCR2 inhibitor, CCR2 inhibitor, TNF‐α inhibitor and MMP9 inhibitor were from MedChem Express (Monmouth Junction, NJ, USA). Anti‐MIP‐2 FITC‐conjugated, anti‐MCP‐1 FITC‐conjugated, anti‐Ly6c FITC‐conjugated, anti‐Ly6g FITC‐conjugated, anti‐F4/80 PE‐Cyanine7‐conjugated, anti‐CD11c PerCP‐Cyanine5.5‐conjugated and anti‐CD11b APC‐conjugated antibodies were from Invitrogen (Carlsbad, CA, USA).
2.2. Animal preparation
C57BL/6 mice (20‐25 g) were obtained from Slac Laboratory Animal (Shanghai, China). Mice were caged with free access to food and fresh water in a temperature‐controlled room on a standard day‐night cycle. The use of animals in the present study was approved by Animal Studies Ethics Committee of the Second Affiliated Hospital of Wenzhou Medical University.
For the experiment groups, mice received PDX (0.1 µg/mouse, intraperitoneal, ip) 10 minutes after atomization inhalation of LPS (ih, 1 mg/kg) or intraperitoneal injection of LPS (ip, 1 mg/kg). For the inhibitor groups, mice received the CXCR2 inhibitor (CXCR2i, 2 mg/kg), CCR2 inhibitor (CCR2i, 30 mg/kg), TNFR inhibitor (TNFRi, 15 mg/kg) or MMP9 inhibitor (MMP9i, 10 mg/kg) with or without PDX after LPS interruption. 24 hours later, the bronchoalveolar lavage fluid (BALF) and lung tissue samples were harvested.
2.3. Pathological studies
Lung lobe was collected and fixed in 4% paraformaldehyde for 24 hours, then embedded in paraffin and stained with haematoxylin and eosin (H&E) for light microscopy. Alveolar congestion, haemorrhage, neutrophil infiltration or aggregation, alveolar wall thickness/hyaline membrane formation were used to assay lung injury. 20 No injury = 0; slight injury = 1 (25%); moderate injury = 2 (50%); severe injury = 3 (75%); and very severe injury = 4 (almost 100%).
2.4. Flow cytometry
Freshly collected and isolated BALF cells were incubated with anti‐F4/80 PE‐Cyanine7‐conjugated, anti‐Ly6c FITC‐conjugated, anti‐Ly6g FITC‐conjugated, anti‐CD11b APC‐conjugated and anti‐CD11c PerCP‐Cyanine5.5‐conjugated antibodies for 30 minutes. Then cells were cultured with fluorescence‐activated cell sorting (FACS) lysis solution for another 10 minutes. After 5 minutes 400 g centrifuge, cells were analysed by CytExpert 2.0 (Beckman Coulter).
Macrophage was identified by high expression of F4/80. Resident macrophage was defined by high expression of CD11b, and recruited macrophage was defined by high expression of CD11c, Ly6c. Neutrophil was identified by high expression of Ly6c and Ly6g.
2.5. Resident macrophages depletion
To deplete resident macrophages, clodronate liposome was given intratracheal in a volume of 50 µl (5 mg/mL) 72 hours before LPS challenge. PBS liposome was used as control. Next, mice were stimulated with LPS (1 mg/kg) with or without PDX (0.1 µg/mouse) for 24 hours. Then BALF was harvested.
2.6. Quantitative real‐time RT‐PCR
Total RNA from lung tissues was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA), according to the manufacturer's protocol. The cDNA was synthesized using a reverse transcription Kit. Gene expression was detected using SYBR green super‐mix PCR kit. Then MIP‐2, MCP‐1 and TNF‐α mRNA level were measured.
The primer pairs for each gene were as follows: MIP‐2:5′‐CCACTCACCTGCTGCTACTCATTC‐3′ and reverse 5′‐CTGCTGCTGGTGATCCTCTTGTAG‐3′; MCP‐1:5‐CCACTCACCTGCTGCTACTCATTC‐3, 5‐CTGCTGCTGGTGATCCTCTTGTAG‐3; and TNF‐α: 5‐GCGACGTGGAACTGGCAGAAG‐3, 5‐GCCACAAGCAGGAATGAGAAGAGG‐3.
Next, Neutrophils, resident and recruited macrophages were sorted and stimulated with 1 μg/mL LPS and/or 100 nmol/L PDX for 24 hours, then CXCR2, CCR2 and TNFR expression were assayed by real‐time PCR.
2.7. ELISA
MIP‐2, MCP‐1, TNF‐α and MMP‐9 concentrations in lung tissue homogenates and BALF were assessed using ELISA kits, according to the manufacturer's protocol.
2.8. Statistical analysis
Data are presented as the mean ± SEM. All data were analysed by one‐way ANOVA, followed by Tukey test for post hoc comparison. Significance was considered at the P < .05. Statistical analyses were performed using Prism 6.0 software (GraphPad Software, San Diego, CA, USA).
3. RESULTS
3.1. PDX protected lung tissue from LPS‐induced lung injury
As shown in Figure 1A, the lung tissues were seriously injured in both LPS (ih) and LPS (ip) groups. Treatment with PDX alleviated LPS‐induced lung injury. Acute lung injury scores were in line with pathophysiological changes (Figure 1B). In addition, the lung tissue homogenate TNF‐α level was significantly higher in both the LPS (ih) and LPS (ip) groups than in the CTR group and was lower in the PDX treatment group (Figure 1C). But there were no significant differences between the CTR group and PDX group (P > .05).
FIGURE 1.

PDX attenuated LPS‐induced lung tissue damage. Mice received LPS by intratracheal atomization (ih) (1 mg/kg) or intraperitoneal injection (ip) (10 mg/kg) and then received PDX (0.1 µg/mouse) by intraperitoneal injection. Lung histological changes were assessed 24 h later by haematoxylin and eosin staining (A) and acute lung injury scoring (B). Aerosol inhalation and intraperitoneal injection of LPS both significantly increased the TNF‐α concentration in lung tissue homogenates, and this effect was markedly attenuated by PDX treatment. Data are presented as the mean ± SEM. n = 6‐8. *P < .05, **P < .01, ***P < .001
3.2. PDX reduced inflammatory cell accumulation in LPS‐induced lung injury
F4/80‐Ly6c+Ly6g+ neutrophil, F4/80+Ly6C‐CD11chiCD11bint resident macrophage and F4/80+Ly6c+CD11cloCD11bhi recruited macrophage (Figure 2A‐D) in the BALF were separated as previously described. 21
FIGURE 2.

PDX reduced recruited macrophage and neutrophil infiltration in LPS‐induced lung injury in vivo. F4/80‐Ly6c+Ly6g+ neutrophils, F4/80+Ly6C‐CD11chiCD11bint resident macrophages and F4/80+Ly6c+CD11cloCD11bhi recruited macrophages in the BALF were separated by flow cytometry (A). The numbers of neutrophils, resident macrophages and recruited macrophages in the BALF were determined by flow cytometry after LPS inhalation (1 mg/kg) (B) or intraperitoneal injection (10 mg/kg) (C). Next, PDX (0.1 µg/mouse) was administered to mice 10 min after LPS (1 mg/kg) inhalation; 24 h later, the three cell types were counted by flow cytometry (D). Data are presented as the mean ± SEM. n = 6‐8. *P < .05, **P < .01, ***P < .001
In the LPS‐ (ih) (Figure 2E) or LPS (ip) (Figure 2F)‐induced lung injury model, neutrophils started infiltrating the lungs at 2 hours after LPS intervention, and the number of neutrophils peaked at 24 hours and then decreased until 48 hours. The number of recruited macrophages was increased at 6 hours, peaked at 24 hours and then gradually decreased until 48 hours, but the number of resident macrophages generally remained constant during the course of LPS‐induced inflammation. Inflammatory cell numbers were much more stable in the LPS (ih) group (Figure 2E) than in the LPS (ip) group (Figure 2F). Therefore, the LPS inhalation (ih)‐induced lung injury model was used in subsequent experiments.
The recruited macrophages and neutrophils were decreased at 24 hours in the LPS + PDX group compared with the LPS group (Figure 2G) (P < .05). But there was no significant difference between the CTR group and PDX group (Figure 2G) (P > .05).
3.3. PDX reduced inflammatory cell infiltration in LPS‐induced lung injury via resident macrophages
Clodronate liposome was used to eliminate resident macrophage (Figure 3A). As shown in Figure 3B, clodronate liposome depleted the vast majority of resident macrophages. Compared with LPS + PBS liposome group, the recruited macrophages and neutrophils were decreased in LPS + PDX+PBS liposome group (P < .05), but not resident macrophages (P > .05) (Figure 3C‐E), suggesting that PDX reduced inflammatory cells infiltration after LPS challenge, but had no effect on resident macrophage numbers. Compared with LPS + PBS liposome group, the resident macrophages, recruited macrophages and neutrophils were decreased in LPS + clodronate liposome group (P < .05), indicating that while clodronate liposome eliminates resident macrophages, recruited macrophage and neutrophils numbers also reduced (P < .05) (Figure 3C‐E). However, PDX had no effect on inflammatory cell numbers after clodronate liposome stimulation (Figure 3D,E), indicating that PDX reduced inflammatory cell infiltration in LPS‐induced lung injury via resident macrophages.
FIGURE 3.

The inhibition of LPS‐induced inflammatory cell infiltration by PDX was dependent on resident macrophages. Resident macrophages were depleted in the lungs 72 h after intratracheal administration of 50 µl clodronate liposomes, and the administration of PDX (0.1 µg/mouse) occurred 10 min after LPS (1 mg/kg) stimulation (A). The numbers of resident macrophages (B, C), recruited macrophages (B, D) and neutrophils (B, E) in the BALF were measured by flow cytometry. CL = clodronate liposome, PB = PBS liposome. The data are presented as the mean ± SEM. n = 6‐8. *P < .05, **P < .01, ***P < .001
3.4. PDX reduced resident macrophage MIP‐2 and MCP‐1 production and release in LPS‐induced lung injury
LPS application not only increased MIP‐2 and MCP‐1 mRNA expression in tissues, but also up‐regulated MIP‐2 and MCP‐1 level in the BALF compared with CTR treatment (P < .05). Treatment with PDX observably weakened the MIP‐2 and MCP‐1 concentration in tissues and BALF compared with LPS group (P < .05) (Figure 4A,B,D,E). However, there was no difference in the CXCR2 expression on neutrophils and CCR2 expression on recruited macrophages among these groups (P > .05) (Figure 4C,F).
FIGURE 4.

PDX down‐regulated LPS‐stimulated resident macrophage MIP‐2 and MCP‐1 expression and release to inhibit inflammatory cell infiltration. Mice received 1 mg/kg LPS by intratracheal atomization and then received PDX (0.1 µg/mouse) by intraperitoneal injection. MIP‐2 and MCP‐1 mRNA expression in lung tissue homogenates (A) and protein levels in the BALF (B) were measured 24 h later. Next, the sorted neutrophils (1 × 106) and recruited macrophages (1 × 106) were incubated with LPS (1 μg/mL) in the presence or absence of PDX (100 nmol/mL) for 24 h, respectively. CXCR2 mRNA expression on neutrophils and CCR2 mRNA level on recruited macrophages were measured by real‐time PCR (C,F). In addition, mice received a CXCR2 inhibitor (2 mg/kg) or CCR2 inhibitor (30 mg/kg) in the presence or absence of PDX via intraperitoneal injection 10 min after LPS administration. The number of neutrophils (G) and recruited macrophages (H) in the BALF was evaluated by flow cytometry. In addition, the mean fluorescence intensity (MFI) of MIP‐2 (I) and MCP‐1 (J) was assessed by flow cytometry. MIP‐2 and MCP‐1 were mainly expressed on resident macrophages, but this expression was strongly down‐regulated by PDX (I, J). CXCR2i = CXCR2 inhibitor, CCR2i = CCR2 inhibitor. Data are presented as the mean ± SEM. n = 6‐8. *P < .05, **P < .01
Next, CXCR2i (MIP‐2 receptor inhibitor) and CCR2i (MCP‐1 receptor inhibitor) were administered via intraperitoneal injection. As shown in Figure 4G,H, with or without PDX, treatment with CXCR2i and CCR2i reduced the number of neutrophils and recruited macrophages, respectively, indicating that the basic function of PDX was disappeared after using CXCR2 and CCR2 inhibitors.
In addition, Figure 4I,J showed that MIP‐2 and MCP‐1 were mostly presented on resident macrophages, and LPS stimulation increased the MIP‐2 and MCP‐1 mean fluorescence intensity (MFI). Furthermore, the up‐regulation of MIP‐2 and MCP‐1 expression induced by LPS was reduced by PDX.
3.5. PDX reduced neutrophil recruitment via recruited macrophage TNF‐α/MIP‐2 signalling pathway
As shown in Figure 5A, TNF‐α was mostly presented on recruited macrophages, and treatment with PDX suppressed the TNF‐α MFI compared with LPS group (P < .05). Up‐regulation of lung tissue TNF‐α mRNA and BALF TNF‐α concentration in the LPS group could be eliminated by PDX treatment (Figure 5B,C). Figure 5D showed that TNFR was mainly presented on macrophages, but there was no significant difference among these groups (P > .05) (Figure 5E).
FIGURE 5.
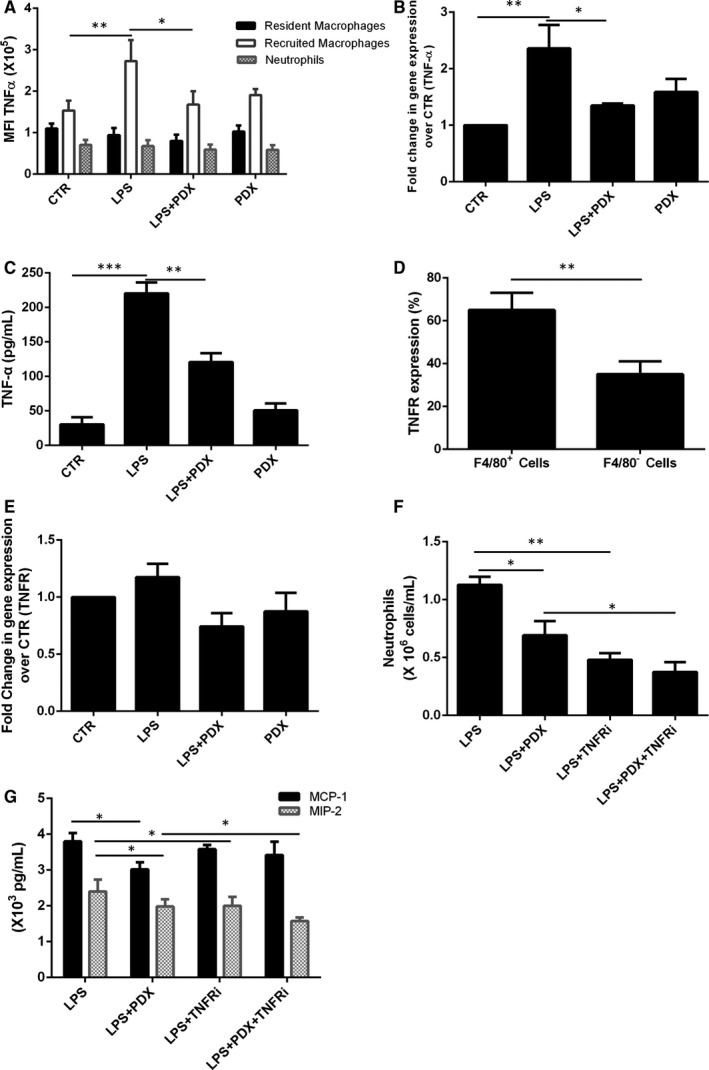
PDX inhibited neutrophil infiltration via recruited macrophage TNF‐α/MIP‐2 signalling pathway. Mice received 1 mg/kg LPS by intratracheal atomization and then received PDX (0.1 µg/mouse) by intraperitoneal injection. Lung homogenates were collected and processed into single‐cell suspensions 24 h later. The mean fluorescence intensity (MFI) of TNF‐α was assessed by flow cytometry (A). TNF‐α was mainly expressed on resident macrophages, but this expression was strongly down‐regulated by PDX (B). TNF‐α mRNA expression in lung tissue homogenates (B) and TNF‐α protein levels in the BALF (C) were measured 24 h later. Then, TNF‐α expression in different kinds of cells was assessed by flow cytometry (D). Next, the sorted resident macrophages (1 × 106) were incubated with LPS (1 μg/mL) in the presence or absence of PDX (100 nmol/mL) for 24 h. TNF‐α mRNA expression on resident macrophages was measured by real‐time PCR (E). In addition, mice received a TNFR inhibitor (15 mg/kg) in the presence or absence of PDX via intraperitoneal injection 10 min after LPS administration. The number of neutrophils (F) in the BALF was evaluated by flow cytometry, and the MIP‐2, MCP‐1 level was measured by ELISA (G). TNFRi = TNFR inhibitor. Data are presented as the mean ± SEM. n = 6‐8. *P < .05, **P < .01
In addition, TNFRi (a TNF‐α receptor inhibitor) was administered via intraperitoneal injection. With or without PDX, treatment with TNFRi reduced neutrophil numbers and MIP‐2 level, but not MCP‐1, indicating that the basic function of PDX was disappears after using TNFR inhibitors (Figure 5F,G).
3.6. PDX inhibited neutrophil transmigration into the alveolar space in connection with TNF‐α/MIP‐2/MMP9 signalling pathway
The MMP‐9 level was reduced in the LPS + PDX group, LPS + CXCR2i group and LPS + TNFRi group compared with the LPS group (P < .05) (Figure 6A). Moreover, MMP‐9 level was decreased in the LPS + PDX+CXCR2i group and LPS + PDX+TNFRi group compared with the LPS + PDX group (P < .05) (Figure 6A). We also found that MMP‐9 was mainly produced by Ly6g+ cells (>90%) (Figure 6B).
FIGURE 6.

PDX regulated neutrophil transmigration into the alveolar space in connection with TNF‐α/MIP‐2/MMP9 signalling pathway. A CXCR2 inhibitor (2 mg/kg), TNFR inhibitor (15 mg/kg) or MMP9 inhibitor (15 mg/kg) in the presence or absence of PDX was administered via intraperitoneal injection after LPS administration. MMP9 expression in the BALF was measured by ELISA (A). MMP9 expression on cells was assessed by flow cytometry (B). Neutrophil numbers in the BALF and lung tissues (C) were measured by flow cytometry. Data are presented as the mean ± SEM. n = 6‐8. *P < .05, **P < .01, ***P < .001
In addition, the neutrophil numbers in the BALF were much lower than those in the lung homogenate in the LPS + PDX+MMP9i group (P < .05) (Figure 6C). Compared with those in the LPS + PDX group, the neutrophil numbers in the LPS + PDX + MMP9i group were decreased in the BALF and increased in the lung homogenate (P < .05) (Figure 6C).
4. DISCUSSION
In the present study, two LPS‐induced lung injury models were created, one established by atomization inhalation of LPS and the other established by intraperitoneal injection of LPS. We found that both approaches could significantly damage the lungs and increase lung TNF‐α expression. Moreover, 2 hours after LPS stimulation, neutrophils started infiltrating into the lungs and recruited macrophages followed at 6 hours after LPS stimulation. PDX effectively alleviated lung injury, down‐regulated TNF‐α concentration and inhibited inflammatory cells recruitment. However, the data were much more stable after LPS inhalation; therefore, the LPS inhalation‐induced lung injury model was used in our experiments.
In the present study, we showed that recruited macrophage and neutrophil numbers decreased when the resident macrophages were depleted, indicating that resident macrophages are associated with inflammatory cell accumulation. Our work was consistent with previous report that resident macrophages recruit helper macrophages into the infected bladder. 22 PDX had no effect on inflammatory cell numbers when the resident macrophages were depleted, suggesting that PDX reducing inflammatory cell accumulation depends on resident macrophages.
MIP‐2 and MCP‐1 play crucial roles in inflammatory cell infiltration, and increased expression of MIP‐2 and MCP‐1 has been reported in various pulmonary diseases, including chronic obstructive pulmonary disease, 23 ARDS 24 and asthma. 25 We have reported that MIP‐2 recruited neutrophils to the damaged lung. 26 A previous report also demonstrated that MCP‐1 recruited inflammatory monocytes to facilitate breast tumour metastasis. 27 Here, we showed that the expression of MIP‐2 and MCP‐1 was rapidly induced in LPS‐induced lung injury and PDX abolished the LPS‐induced up‐regulation of lung tissues MIP‐2, MCP‐1 mRNA and BALF MIP‐2, MCP‐1 protein levels, but PDX had no effect on CXCR2 and CCR2 mRNA expression, indicating that PDX reduces MIP‐2, MCP‐1 production and release, but not CXCR2 and CCR2. Moreover, the repressive effects of PDX on neutrophil numbers were abolished by CXCR2 inhibitor, and recruited macrophage numbers were abolished by CCR2 inhibitor, suggesting that PDX inhibits neutrophil accumulation via MIP‐2, and suppress recruited macrophage infiltration through MCP‐1. Since MIP‐2 and MCP‐1 were important in inflammatory cells infiltration, we asked which cells secrete MIP‐2 and MCP‐1. We found that MIP‐2 and MCP‐1 were mainly present on resident macrophage. PDX down‐regulated MIP‐2 and MCP‐1 level, indicating that PDX could reduce MIP‐2 and MCP‐1 expression on resident macrophage to repress inflammatory cell accumulation.
TNF‐α is a central mediator of inflammation and plays an important role in the host response to injury, but overexpression of TNF‐a can result in severe tissue damage and underlies a number of disease states, such as rheumatoid arthritis, ARDS and malignancy. 28 , 29 In the present study, we found that PDX could inhibit TNF‐α expression to protect lung tissues, which was consistent with a previous study showing that PDX abolished zymosan‐A‐induced TNF‐α production. 30 We also found that TNF‐α was mainly expressed on recruited macrophages and worked by binding with TNFR, which was expressed on resident macrophages. Our work was consistent with previous research, which showed that chemical or genetic depletion of macrophages suggested that early recruited macrophages expressed TNF‐α 31 and indicated that recruited exudative macrophages produced TNF‐α after stimulation with LPS. 32 We also found that PDX inhibiting the MIP‐2 level and neutrophil numbers was associated with TNF‐α.
MMP9 derived from neutrophils during the inflammatory process can change alveolar capillary permeability and mediate neutrophil transmigration into the alveolar space. 33 In the present study, we found that MMP9 was mainly expressed on Ly6g+ cells, which were identified as neutrophils. PDX reduced MMP9 expression, which was consistent with a previous study showing that MMP‐9 expression was reduced in the corneas of HSV‐1‐infected mice by treatment with AT‐PDX. 34 Interestingly, in our study, TNFR inhibitor and CXCR2 inhibitor could inhibit neutrophil infiltration and MMP9 expression with or without PDX in LPS‐induced lung injury, indicating that MMP9 expression on neutrophils could be regulated by TNF‐α and MIP‐2. In vivo studies have shown that TNF‐a can induce the overexpression of MMP9 in cholangiocarcinoma cell lines. 35 Moreover, an MMP9 inhibitor significantly reduced neutrophil transmigration into the alveolar space after treatment with PDX. Collectively, these data indicated that PDX mediated neutrophil transmigration into the alveolar space in connection with TNF‐α/MIP‐2/MMP9 signalling pathway.
In conclusion, we have shown that PDX alleviates lung injury through inhibition of MIP‐2 and MCP‐1 production and release by resident macrophages in LPS‐induced lung injury. PDX inhibited recruited macrophage TNF‐α expression. We also found that PDX inhibited neutrophil infiltration in connection with TNF‐α/MIP‐2/MMP9 signalling pathway (Figure 7). Our findings suggest that PDX may provide a new therapy for the treatment of ARDS.
FIGURE 7.

Graphical summary of the sequence of events. Our findings document the following sequence of events (Figure 7): 1) resident macrophages sensed LPS stimulation and produced the chemokine MIP‐2 and MCP‐1, which recruited neutrophils and recruited macrophages; 2) recruited macrophages produced TNF‐α; 3) TNF‐α and MIP‐2 caused MMP9 expression in neutrophils, which allowed these cells to transmigrate into the alveolar space; and 4) these processes were regulated by PDX
CONFLICT OF INTEREST
The authors confirm that there are no conflicts of interest.
AUTHOR CONTRIBUTION
Yang Ye: Investigation (equal); Methodology (equal). Hua‐Wei Zhang: Investigation (equal); Methodology (equal). Hong‐Xia Mei: Data curation (lead). Hao‐Ran Xu: Data curation (lead). Shu‐Yang Xiang: Data curation (supporting). Qian Yang: Writing‐original draft (lead). Sheng‐Xing Zheng: Writing‐original draft (supporting). Fang Gao Smith: Writing‐original draft (lead). Sheng‐Wei Jin: Conceptualization (equal); Writing‐review & editing (equal). Qian Wang: Conceptualization (equal); Writing‐review & editing (equal).
ACKNOWLEDGEMENTS
This work was sponsored by grants from the National Natural Science Foundation of China (no. 81571862, no. 81270132, and no. 81870065) and by the Natural Science Foundation of Zhejiang Province (LY18H010005, LQ15H010002), and Research Fund for Lin He's Academician Workstation of New Medicine and Clinical Translation (19331102).
Ye Y, Zhang H‐W, Mei H‐X, et al. PDX regulates inflammatory cell infiltration via resident macrophage in LPS‐induced lung injury. J Cell Mol Med. 2020;24:10604–10614. 10.1111/jcmm.15679
Yang Ye and Hua‐Wei Zhang contributed equally to this work:
Contributor Information
Sheng‐Wei Jin, Email: jinshengwei69@163.com.
Qian Wang, Email: wqian84@163.com.
DATA AVAILABILITY STATEMENT
I confirm that my article contains a Data Availability Statement even if no data are available (list of sample statements) unless my article type does not require one (eg Editorials, Corrections, Book Reviews, etc). I confirm that I have included a citation for available data in my references section, unless my article type is exempt.
REFERENCES
- 1. Huang X, Xiu H, Zhang S, et al. The role of macrophages in the pathogenesis is of ALI/ARDS. Mediators Inflamm. 2018;1264913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tu GW, Shi Y, Zheng YJ, et al. Glucocorticoid attenuates acute lung injury through induction of type 2 macrophage. J Transl Med. 2017;15:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hamid U, Krasnodembskaya A, Fitzgerald M, et al. Aspirin reduces lipopolysaccharide‐induced pulmonary inflammation in human models of ARDS. Thorax. 2017;72:971‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334‐1349. [DOI] [PubMed] [Google Scholar]
- 5. Lomas‐Neira J, Chung C‐S, Perl M, et al. Role of alveolar macrophage and migrating neutrophils in hemorrhage‐induced priming for ALI subsequent to septic challenge. Am J Physiol Lung Cell Mol Physiol. 2006;290:51‐58. [DOI] [PubMed] [Google Scholar]
- 6. Johnston LK, Rims CR, Gill SE, et al. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury”. Am J Respir Cell Mol Biol. 2012;47:417‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Srivastava S, Ernst JD, Desvignes L. Beyond macrophages: the diversity of mononuclear cells in tuberculosis. Immunol Rev. 2014;262:179‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zasłona Z, Przybranowski S, Wilke C, et al. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. 2014;193:4245‐4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang Z, Zhou Q, Gu C, et al. Depletion of circulating monocytes suppresses IL‐17 and HMGB1 expression in mice with LPS‐induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2017;312:231‐242. [DOI] [PubMed] [Google Scholar]
- 10. Dhaliwal K, Scholefield E, Ferenbach D, et al. Monocytes control secondphase neutrophil emigration in established lipopolysaccharide‐induced murine lung injury. Am J Respir Crit Care Med. 2012;186:514‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang X, Sun CY, Zhang YB, et al. Kegan Liyan oral liquid ameliorates lipopolysaccharide‐induced acute lung injury through inhibition of TLR4‐mediated NF‐B signaling pathway and MMP‐9 expression. J Ethnopharmacol. 2016;186:91‐102. [DOI] [PubMed] [Google Scholar]
- 12. Belperio JA, Keane MP, Burdick MD, et al. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator‐induced lung injury. J Clin Invest. 2002;110:1703‐1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liang J, Jung Y, Tighe RM, et al. A macrophage subpopulation recruited by CC chemokine ligand‐2 clears apoptotic cells in noninfectious lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;302:933‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miura K, Matsuo J, Rahman MA, et al. Ehrlichia chaffeensis induces monocyte inflammatory responses through MyD88, ERK, and NF‐κB but not through TRIF, interleukin‐1 receptor 1 (IL‐1R1)/IL‐18R1, or toll‐like receptors. Infect Immun. 2011;79:4947‐4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haraguchi K, Kawamoto A, Isami K, et al. TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J Neurosci. 2012;32:3931‐3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhuo XJ, Hao Y, Cao F, et al. Protectin DX increases alveolar fluid clearance in rats with lipopolysaccharide‐induced acute lung injury. Exp Mol Med. 2018;50:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Serhan CN, Gotlinger K, Hong S, et al. Anti‐inflammatory actions of neuroprotectin D1/protectin D1 and its naturalstereoisomers: assignments of dihydroxy‐containing docosatrienes. J Immunol. 2006;176:1848‐1859. [DOI] [PubMed] [Google Scholar]
- 18. Jung TW, Chung YH, Kim HC, et al. Protectin DX attenuates LPS‐induced inflammation and insulin resistance in adipocytes via AMPK‐mediated suppression of the NF‐κB pathway. Am J Physiol Endocrinol Metab. 2018;315:E543‐E551. [DOI] [PubMed] [Google Scholar]
- 19. Li H, Hao Y, Zhang H, et al. Posttreatment with Protectin DX ameliorates bleomycin‐induced pulmonary fibrosis and lung dysfunction in mice. Sci Rep. 2017;7:46754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu D, Zeng BX, Zhang SH, et al. Rosiglitazone, a peroxisome proliferator‐activated receptor‐gamma agonist, reduces acute lung injury in endotoxemic rats. Crit Care Med. 2005;33:2309‐2316. [DOI] [PubMed] [Google Scholar]
- 21. Ueno M, Maeno T, Nishimura S, et al. Alendronate inhalation ameliorates elastase‐induced pulmonary emphysema in mice by induction of apoptosis of alveolar macrophage. Nat Commun. 2015;6:6332. [DOI] [PubMed] [Google Scholar]
- 22. Schiwon M, Weisheit C, Franken L, et al. Crosstalk between sentinel and helper macrophages permits neutrophil migration into infected uroepithelium. Cell. 2014;156:456‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Boer WI, Sont JK, van Schadewijk A, et al. Monocyte chemoattractant protein 1, interleukin 8, and chronic airways inflammation in COPD. J Pathol. 2000;190:619‐626. [DOI] [PubMed] [Google Scholar]
- 24. Parsons PE, Fowler AA, Hyers TM, et al. Chemotactic activity in bronchoalveolar lavage fluid from patients with adult respiratory distress syndrome. Am Rev Respir Dis. 1985;132:490‐493. [DOI] [PubMed] [Google Scholar]
- 25. Tillie‐leblond I, Hammad H, Desurmont S, et al. CC chemokines and interleukin‐5 in bronchial lavage fluid from patients with status asthmaticus. Potential implication in eosinophil recruitment. Am J Respir Crit Care Med. 2000;162:586‐592. [DOI] [PubMed] [Google Scholar]
- 26. Zhang HW, Wang Q, Mei HX, et al. RvD1 ameliorates LPS‐induced acute lung injury via the suppression of neutrophil infiltration by reducing CXCL2 expression and release from resident alveolar macrophages. Int Immunopharmacol. 2019;76:105877. [DOI] [PubMed] [Google Scholar]
- 27. Qian BZ, Li J, Zhang H, et al. MCP‐1 recruits inflammatory monocytes to facilitate breast‐tumour metastasis. Nature. 2011;475:222‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pfeiffer JR, Brooks SA. Cullin 4B is recruited to tristetraprolin‐containing messenger ribonucleoproteins and regulates TNF‐α mRNA polysome loading. J Immunol. 2012;188:1828‐1839. [DOI] [PubMed] [Google Scholar]
- 29. Pugin J, Ricou B, Steinberg KP, et al. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin‐1. Am J Respir Crit Care Med. 1996;153:1850‐1856. [DOI] [PubMed] [Google Scholar]
- 30. Tan W, Chen L, Wang YX, et al. Protectin DX exhibits protective effects in mouse model of lipopolysaccharide‐induced acute lung injury. Chin Med J. 2018;131:1167‐1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nguyen‐Chi M, Laplace‐Builhé B, Travnickova J, et al. TNF signaling and macrophages govern fin regeneration in zebrafish larvae. Cell Death Dis. 2017;8:e2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tighe RM, Liang J, Liu N, et al. Recruited exudative macrophages selectively produce CXCL10 after noninfectious lung injury. Am J Respir Cell Mol Biol. 2011;45:781‐788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang F, Hu L, Wu YX, et al. Doxycycline alleviates paraquat‐induced acute lung injury by inhibiting neutrophil‐derived matrix metalloproteinase 9. Int Immunopharmacol. 2019;72:243‐251. [DOI] [PubMed] [Google Scholar]
- 34. Rajasagi NK, Bhela S, Varanasi SK, et al. Frontline Science: Aspirin‐triggered resolvin D1 controls herpes simplex virus‐induced corneal immunopathology. J Leukoc Biol. 2017;102:1159‐1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Onodera M, Zen Y, Harada K, et al. Fascin is involved in tumor necrosis factor‐alpha‐dependent production of MMP9 in cholangiocarcinoma. Lab Invest. 2009;89:1261‐1274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
I confirm that my article contains a Data Availability Statement even if no data are available (list of sample statements) unless my article type does not require one (eg Editorials, Corrections, Book Reviews, etc). I confirm that I have included a citation for available data in my references section, unless my article type is exempt.