

Abstract
Background
Exogenous gene introduction by transfection is one of the most important approaches for understanding the function of specific genes at the cellular level. Electroporation has a long-standing history as a versatile gene delivery technique in vitro and in vivo. However, it has been under-utilized in vitro because of technical difficulty and insufficient transfection efficiency.
New Method
We have developed an electroporation technique that combines the use of large glass electrodes, tetrodotoxin-containing artificial cerebrospinal fluid and mild electrical pulses. Here, we describe the technique and compare it with existing methods.
Results
Our method achieves a high transfection efficiency (~80 %) in both excitatory and inhibitory neurons with no detectable side effects on their function. We demonstrate this method is capable of transferring at least three different genes into a single neuron. In addition, we demonstrate the ability to transfect different genes into neighboring cells.
Comparison with Existing Methods
The majority of existing methods use fine-tipped glass electrodes (i.e., > 10 MΩ) and apply high voltage (10 V) pulses with high frequency (100 Hz) for 1 second. These parameters contribute to practical difficulties thus lowering the transfection efficiency. Our unique method minimizes electrode clogging and therefore procedure duration, increasing transfection efficiency and cellular viability.
Conclusions
Our modifications, relative to current methods, optimize electroporation efficiency and cell survival. Our approach offers distinct research strategies not only in elucidating cell-autonomous functions of genes but also for assessing genes contributing to intercellular functions, such as trans-synaptic interactions.
Keywords: Single-cell electroporation, neuron, gene delivery, electrophysiology, organotypic slice culture, mouse, hippocampus
Graphical abstract
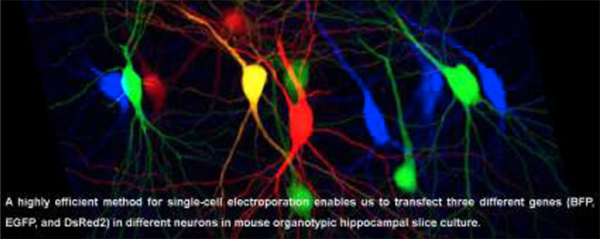
1. Introduction
Gene transfer is an essential approach in molecular biology, in vivo and in vitro, to elucidate the function of a gene of interest. Various in vitro gene transfer methods have been developed based on three different approaches: biological (e.g. viral vectors), chemical (e.g. lipid and calcium phosphate) and physical (e.g. biolistic gene gun and electroporation) (Kim and Eberwine, 2010; Washbourne and McAllister, 2002). Viral vectors are highly efficient and capable of delivering transgenes in a specific cell population by integrating genetic tools (e.g. Cre-LoxP systems). However, specificity is limited to the development of genetic tools (e.g. cell type-specific lines expressing Cre recombinase) and it is not possible to transfect transgenes to individual cells. In addition, the packaging limitation of transgenes, and the toxicity and potential hazard of viral vectors remain an issue. Chemical gene transfection is a powerful approach in vitro, but the types of cells transfected are highly random, and application is mainly restricted to primary cells. The biolistic gene gun is a simple physical gene transfer tool, but offers minimal control over cell-specific transfection and has low transfection efficiency.
Electroporation has been considered one of the best and most versatile transfection methods. The first in vitro and in vivo electroporations were demonstrated in mouse lyoma cells (Neumann et al., 1982) and skin cells (Titomirov et al., 1991), respectively. Since the first in vitro electroporation method was established in post-mitotic neurons using a field microporator (Teruel et al., 1999), multiple in vitro protocols have been developed to provide higher specificity (Rathenberg et al., 2003; Tanaka et al., 2009; Wiegert et al., 2017). These protocols demonstrate the feasibility of performing single-cell gene transfection in a specified cell type. While Rathenberg et al. (2003) describe their method as highly efficient (up to 80 % in cortical neurons), thorough details regarding electroporation condition optimization, besides improving cell membrane and pipette tip visualization under high magnification, was not reported.
We identified technical challenges in performing these methods, thus lowering successful gene transfection. The majority of published protocols use fine-tipped glass electrodes (i.e. > 10 MΩ) and apply high voltage pulses with high frequency and long duration (i.e. 10 V amplitude at 200 Hz for 1 second) (Rathenberg et al., 2003; Tanaka et al., 2009; Wiegert et al., 2017). We identified two problems when attempting electroporation with these experimental settings. First, we were not able to achieve a high transfection efficiency, possibly due to cell death from application of strong electrical pulses. Second, we observed pipette tip clogging with the plasmid-containing internal solution during repeated electroporations. We found that a modified protocol, using larger glass electrodes and milder electroporation parameters together with an innovative pressure cycling step, enables transfection of genes of interest into both excitatory and inhibitory hippocampal neurons with higher efficiency.
2. Materials and Methods
2.1. Animal and organotypic slice culture preparation:
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Massachusetts Medical School. Organotypic hippocampal slice cultures were prepared from postnatal 6- to 7-day-old mice of either sex, as described previously (Stoppini et al., 1991). Mice were wild-type (C57BL/6J, Jax # 000664) or were expressing interneuron-specific TdTomato (Sst-TdTomato), generated by crossing Sst-Cre (Jax #013044) and a TdTomato reporter line (Jax #007905).
2.2. DNA constructs:
Enhanced green fluorescent protein (EGFP, Clontech), Tagblue fluorescent protein (Tag-BFP, Evrogen) and Discosoma sp. red fluorescent protein variant 2 (DsRed2, Clontech) genes were subcloned into a pCAG vector. Plasmids (0.1 μg / μl) were purified by endotoxin-free purification kits (Qiagen Endofree kit) and were dissolved in internal solution (see below) at 55 °C for 10–15 min, followed by storage at −20 °C.
2.3. Single-cell electroporation:
A standard whole-cell electrophysiology rig equipped with a mounted upright microscope (Olympus BX61WI) with a 60X objective lens was used. The head stage of the electroporator (Axoporator, Molecular Devices) was installed in a regular micromanipulator (Sutter) and speakers were connected to the electroporator. The borosilicate glass capillaries with filament (2.0 mm, Warner Instruments) were pulled (P-1000, Sutter) to prepare glass electrodes at a resistance of 3.5 – 6.0 MΩ. The glass pipettes were baked at 200 °C overnight for sterilization. Prior to the experiments, the perfusion lines were cleaned by perfusing with bleach for 5 min followed by sterile deionized water for at least 30 min. The slice cultures were perfused with aCSF consisting of (in mM): 119 NaCl, 2.5 KCl, 0.5 CaCl2, 5 MgCl2, 26 NaHCO3, 1 NaH2PO4, and 11 glucose, gassed with 5% CO2/95% O2, pH 7.4, unless otherwise noted. In some experiments, 0.001 mM tetrodotoxin (TTX, Hello Bio Inc) was added to aCSF. The intracellular solution was filter sterilized (0.22 μm, GP Millipore Express PLUS membrane). Patch pipettes were filled with plasmid (0.1 μg / μl). The internal solution was made with diethyl pyrocarbonate-treated water and consisted of (in mM): 140 K-methanesulfonate, 0.2 EGTA, 2 MgCl2, and 10 HEPES, pH adjusted to 7.3 with KOH.
The slice cultures were stored in a 5% CO2/95% O2 incubator at 35 °C and removed for electroporation for no longer than 30 min to prevent unforeseen side effects, such as homeostatic changes in neuronal function (Ibata et al., 2008). The method for approaching cells with the electrode for electroporation is similar to the approach used for whole-cell recording. Light positive pressure was applied during the approach to the target cell. After observing the dimple on the cell surface (Figure 1A, top), mild negative pressure was applied by mouth to form a loose seal between the glass tip and plasma membrane, thereby increasing pipette resistance up to 2.5-fold above the initial resistance (Figure 1A, middle). The plasma membrane was then expelled by applying mild positive pressure (Figure 1A, top). This procedure, named “pressure cycling,” was repeated 3 – 4 times at a frequency of approximately 1 cycle per second before the electrical pulse (amplitude: −5 V, square pulse, train: 500 ms, frequency: 50 Hz, pulse width: 500 μs) was applied during the final application of negative pressure to the cell. The timing of the electroporation was determined by listening for the tone in the speakers that reached a stable apex in pitch, indicating peak electrical resistance. The pulse was sent after the peak resistance was held for at least 1 second. After electroporation, the electrode was gently retracted approximately 100 μm from the cell without applying pressure to the electrode (Figure 1A, bottom). The next electroporation attempt was then made by re-adding positive pressure to the electrode as another cell was approached. Typically, 10 to 20 electroporations were performed per electrode. Repeated electroporations sometimes caused the pipette resistance to increase even when not in contact with a cell; pipettes whose resistance increased by more than 15% of their initial resistance were discarded. High pipette resistance could be caused by clogging, also indicated through an abnormal, consistent increase in tone between electroporations; in most cases, positive pressure application relieved minor clogging. After electroporation, the slice was transferred on a culture insert (Millipore) with slice culture media (1 ml) (Stoppini et al., 1991) in a 3.5 cm petri dish and incubated in a 5% CO2 incubator at 35 °C for up to 2–3 days.
Figure 1. Electroporation procedure.

(A) The experimental flow of the electroporation procedure. Immediately after confirming the dimple on the cell body (top), mild negative pressure was applied by mouth (middle). This cycle was rapidly repeated three to four times after which electroporation was applied. Afterward, the glass electrode was gently removed from the cell body without applying pressure (bottom). (B) Summary bar graph of the transfection efficiency in CA1 pyramidal neurons using two different methods: applying electroporation either immediately after the first negative pressure (Single, left bar) or after 3–4 cycles of pressure (Pressure Cycling, right bar). Each symbol represents the transfection efficiency obtained from one organotypic slice culture (5 slice cultures from 2 mice for each condition). Student’s t-test: total number of transfected and electroporated neurons: single suction, 56 transfected neurons/ 158 electroporated neurons; pressure cycling, 112/ 143. ***p<0.001. Data shown are means ± SEM. (C) Confocal image of EGFP-transfected hippocampal CA1 pyramidal neurons.
2.4. Electrophysiology:
The extracellular solution for recording consisted of (in mM): 119 NaCl, 2.5 KCl, 4 CaCl2, 4 MgCl2, 26 NaHCO3, 1 NaH2PO4, 11 glucose, and 0.01 2-chloroadenosine (Sigma), gassed with 5% CO2 and 95% O2, pH 7.4. Thick-walled borosilicate glass pipettes were pulled to a resistance of 2.5 – 4.5 MΩ. Whole-cell voltage clamp recordings were performed with internal solution containing (in mM): 115 cesium methanesulfonate, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 ATP disodium salt, 0.4 guanosine triphosphate trisodium salt, 10 sodium phosphocreatine, and 0.6 EGTA, adjusted to pH 7.25 with CsOH. For current clamp recordings, cesium in the internal solution was substituted with potassium. GABAA receptor-mediated inhibitory postsynaptic currents (IPSCs) were measured at Vhold ± 0 mV. Stimulus strength was set to produce an IPSC amplitude of 500 – 1000 pA in untransfected pyramidal neurons. α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor- (AMPAR-) mediated excitatory postsynaptic currents (EPSCs) were evoked at Vhold −70 mV in the presence of picrotoxin (0.1 mM, Sigma). The stimulus strength was set to evoke an AMPAR-EPSC amplitude of 50 – 200 pA. NMDAR-EPSCs were measured at Vhold +40 mV with picrotoxin and NBQX (0.003 mM, Ascent Scientific). 40 to 50 consecutive stable postsynaptic currents were evoked at 0.2 Hz with a stimulating electrode placed in the stratum radiatum of the hippocampal CA1 region. Recordings were performed using a MultiClamp 700B amplifier and Digidata 1440, digitized at 10 kHz and filtered at 4 kHz with a low-pass filter. Data were acquired and analyzed using pClamp (Molecular Devices).
2.5. Confocal imaging:
Images were acquired using a confocal microscope (LSM 700, Zeiss). Organotypic hippocampal slice cultures were fixed in 4% paraformaldehyde (PFA) in 0.01 M phosphate-buffered saline (PBS) for at least 30 min, then washed with PBS and plated onto glass microscope slides with mounting media (Vectashield). The slices were then imaged with 10X, 25X, or 40X objective lenses at a size of 1024 × 1024 pixels. Each image collected was a Z-series projection of x-y images taken at a depth interval of 1 μm. Image analysis was performed using ImageJ software.
2.6. Statistical analyses:
Results are reported as mean ± SEM. The statistical significance was evaluated by two-way ANOVA with post hoc Tukey for multiple comparison, Mann-Whitney U-test and Student’s t-test for two-group comparison. Statistical significance was set at p < 0.05.
3. Results
3.1. Optimization of electroporation protocol:
The majority of published protocols for electroporation use sharp glass electrodes, typically with a resistance of > 10 MΩ, with high frequency stimulation (e.g. 10 V at 100 Hz for 1 s) (Rathenberg et al., 2003; Tanaka et al., 2009; Wiegert et al., 2017). However, we found that using these parameters resulted in poor transfection efficiency. We performed electroporations to 42 CA1 pyramidal neurons in five slice cultures following the protocol published by Rathenberg et al. (2003) (pipette resistance: 10–15 MΩ, amplitude: −10 V, square pulse, train: 1 sec, frequency: 25 Hz, pulse width: 1 ms), and obtained only 11 neurons transfected with EGFP (transfection efficiency: 26 %). Moreover, more than 80 % of glass electrodes (12 out of 14) had clogging issues after 2 to 3 consecutive electroporations. Therefore, we refined the classical electroporation protocol, testing whether larger glass electrodes (3.5 – 6.0 MΩ) could be used with the same voltage pulse of −10 V used previously (Rathenberg et al., 2003). With this modification to pipette resistance, we found that applying the electrical pulse immediately after applying the first negative pressure was feasible and led to successful electroporation, although the transfection efficiency did not reach 40 % (Figure 1B). We noticed that electroporation following a single suction often caused strong adhesion of the plasma membrane to the inside glass wall, which may result in significant physical damage to the cell and thus lead to poor cell viability. During the course of protocol optimization, we found that cycling between positive and negative pressure at least 3–4 times (Figure 1A) was critical for efficient electroporation. Following this pressure cycling, it was easier to retract the glass electrodes from cell bodies with minimal damage. This method significantly increased our transfection efficiency (~80 %) in hippocampal CA1 pyramidal neurons even with weaker voltage pulses (−5 V) (Figure 1B-C). We also found that larger glass electrodes greatly reduced the incidence of clogging during repeated electroporations. Most of the glass electrodes (33 out of 41 glass electrodes) could be used to perform electroporations to more than 10 neurons without significant clogging.
Next, we tested our protocol in hippocampal CA1 interneurons. Interneurons located in the hippocampal CA1 oriens region were morphologically identified by their shape and relative distance between neurons, as distinct from the pyramidal cell layer. To our surprise, the transfection efficiency of our electroporation method in interneurons was much lower (~20 %) than that of pyramidal neurons (Figure 2A). We speculated that electroporation may overexcite interneurons, leading to cytotoxicity and cell death. To test this, we used tetrodotoxin (TTX), a sodium channel blocker, to minimize the propagation of excitation induced by the voltage pulses during electroporation. Importantly, we found that the addition of TTX to the perfusion media during electroporation significantly improved transfection efficiency in hippocampal CA1 inhibitory interneurons (Figure 2A). We did not see a significant improvement in the transfection efficiency by TTX in CA1 pyramidal neurons. With selective addition of TTX, our newly developed electroporation technique yields high transfection efficiency in both excitatory and inhibitory neurons.
Figure 2. Validation of the electroporation method in both excitatory and inhibitory neurons.

(A) TTX improves the transfection efficiency in inhibitory but not excitatory hippocampal neurons. (Left) Confocal image of EGFP-transfected interneurons in the hippocampal CA1 oriens region. Interneurons were identified by morphology and anatomy. (Right) Summary bar graph of the transfection efficiency of excitatory and inhibitory neurons with or without TTX. Each symbol represents the transfection efficiency obtained from one organotypic slice culture. Numbers in each bar represent the numbers of transfected (numerator) and electroporated (denominator) neurons. Student’s t-test: n = 5 – 7 slice cultures from 2 mice for each condition. Data shown are means ± SEM. ***p<0.001, n.s.: not significant. (B) Confocal images of Tag-BFP, EGFP, and DsRed2 triple gene transfection by electroporation in hippocampal CA1 pyramidal neurons. Internal solution contained 33 ng / μl of each plasmid. Note that all transfected pyramidal neurons expressed all three co-transfected genes. (C) Confocal images of three non-overlapping gene transfections using Tag-BFP, EGFP and DsRed2 electroporated in hippocampal CA1 pyramidal neurons. Three different gene plasmids, Tag-BFP, EGFP, and DsRed2, were loaded into glass pipettes one at a time, and three sets of electroporations were done sequentially in different neurons. The middle left neuron that showed up as yellow was electroporated with both EGFP and DsRed2, leading to their co-expression.
We next evaluated the versatility of gene delivery with this method. We first confirmed that our technique could express at least three different transgenes simultaneously in the same neuron (Figure 2B). This multi-gene expression capability will allow us to perform higher-level studies of target gene functions, such as rescue approach to co-express shRNA- and shRNA-resistant transgene-containing plasmids in neurons. Next, electroporations were performed using Tag-BFP-, EGFP- or DsRed2-containing internal solutions. We confirmed that our method transmits different genes to different CA1 pyramidal neurons (Figure 2C). Off-target labelling has been reported as a complication of single-cell electroporation (Dempsey et al., 2015). To test whether our protocol causes non-targeted gene transfection, we performed 140 electroporation trials without forming a loose seal between the glass tip and plasma membrane (tested in seven slice cultures). We detected no transfected neurons in the slice cultures indicating that correct cell-targeting by a loose seal is essential in our method.
3.2. Basal inhibitory and excitatory synaptic transmission in electroporated CA1 pyramidal neurons:
Compared with other in vitro electroporation methods, our method uses a larger glass pipette with repeated pressure cycling, which may cause unforeseen side effects. To examine the physiological effects of our electroporation method on neuronal function, simultaneous whole-cell recordings of EGFP-transfected and untransfected CA1 pyramidal neurons were performed. GABAAR-mediated IPSCs and AMPAR- and NMDAR-mediated EPSCs were measured by stimulating inhibitory and Shaffer collateral inputs (Figure 3A, B, C). Importantly, transfection of EGFP by electroporation produced no detectable differences in the amplitude of GABAAR-IPSCs and AMPAR- and NMDAR-EPSCs. This preservation of electrophysiological function is similar to what we have observed in EGFP-transfected cells using a biolistic gene gun (Chen et al., 2014; Futai et al., 2013; Futai et al., 2007; Hasegawa et al., 2017; Hoogenraad et al., 2010; Mao et al., 2018). Paired-pulse ratio (PPR) of AMPAR-EPSC or GABAAR-IPSC responses, obtained by double stimulation of Shaffer collateral or inhibitory inputs with a 50 ms interval, indicated comparable levels of facilitation between transfected and untransfected (control) neurons, indicating that our electroporation method does not affect presynaptic release probability (Figure 3D).
Figure 3. Comparable levels of basal synaptic transmission between EGFP-transfected and untransfected CA1 pyramidal neurons.

Electroporation of pCAG-EGFP plasmid by our modified transfection method did not alter inhibitory and excitatory synaptic transmission in hippocampal CA1 pyramidal cells. Recordings were carried out 2 – 4 days following transfection. (A-C) (Top) Sample postsynaptic current (PSC) traces mediated by GABAARs (A), AMPARs (B) and NMDARs (C) from pairs of transfected neurons (Trans, gray traces) and neighboring untransfected neurons (Untrans, black traces). Stimulus artifacts were truncated. PPF: paired-pulse facilitation, PPR: paired-pulse depression. (Bottom) Scatter plots of GABAAR-, AMPAR-, NMDAR- mediated PSC amplitude. Each pair of transfected and neighboring untransfected cells are presented as open symbols. Filled symbols indicate the mean. (D) Paired-pulse ratio (PPR) of AMPAR-EPSCs and GABAAR-IPSCs recorded from transfected and untransfected neurons, as indicated. Left, sample traces, with superimposed EPSCs and IPSCs. Middle, summary graph of AMPAR-PPR. The PPR was calculated by dividing the average amplitude of the second EPSC by that of the first EPSC. Right, summary graph of GABAAR PPR. The PPR was calculated by subtracting the averaged traces of single stimulation. Mann-Whitney U-test: number of cell pairs tested: GABAAR-IPSCs, 14 pairs/ 2 mice; AMPAR-EPSCs, 19/ 2; NMDAR-EPSCs, 11/ 2. Number of slice cultures is equal to that of pairs. n.s.: not significant. Data shown are means ± SEM.
3.3. Membrane excitability is not altered in electroporated CA1 pyramidal neurons:
Neurons transfected with EGFP were compared to untransfected neurons to examine whether electroporation affected action potentials (APs) and basic membrane properties (Figure 4 and Table 1). CA1 pyramidal neurons transfected with EGFP exhibited no significant difference in APs compared to untransfected control neurons (Figure 4A) and showed no detectable abnormality in basic membrane properties (Table 1), indicating that our electroporation method does not change neuronal excitability or membrane properties in hippocampal CA1 pyramidal neurons.
Figure 4. Membrane excitability is not altered by electroporation of CA1 pyramidal neurons.

A pCAG-EGFP plasmid was transfected using our protocol and cells were studied to assess neuronal excitability. (Top) Sample traces from untransfected and transfected CA1 pyramidal neurons in organotypic hippocampal slice cultures. The superimposed traces were elicited by current injections of −100, 0, 200, 500 and 900 pA for 200 ms. (Bottom) Summary graph of the frequency of action potentials in untransfected and transfected neurons. The input–output relationship (number of spikes elicited vs. amount of current injection over a 200 ms duration) was plotted for untransfected and transfected neurons. Neurons were held at resting membrane potentials. Two-way ANOVA with post hoc Tukey: number of cells tested: untrans, 9 cells from 2 mice (9/ 2); trans, (10/ 2). n.s.: not significant.
Table 1. The basic membrane properties of untransfected and EGFP-transfected hippocampal CA1 pyramidal and Sst neurons.
These parameters were not statistically different between electroporation conditions for each cell type (Student’s t-test).
| Table 1 | Untransfected excitatory neurons | GFP-transfected excitatory neurons | Untransfected Sst neurons | GFP-transfected Sst neurons |
|---|---|---|---|---|
| Series Resistance | 9.7 ± 0.4 MΩ (N = 34 neurons) | 10.4 ± 0.5 MΩ (N = 34) | 15.1 ± 2.6 MΩ (N = 14) | 12.8 ± 1.3 MΩ (N = 14) |
| Input Resistance | 136.8 ± 16.4 MΩ (N = 34) | 129.6 ± 13.5 MΩ (N = 34) | 63.9 ± 5.8 MΩ (N = 14) | 72.4 ± 4.5 MΩ (N = 14) |
| Resting Membrane Potential | −62.5 ± 1.5 mV (N = 11) | −63.0 ± 1.2 mV (N = 11) | −56.5 ± 0.9 mV (N = 11) | −57.1 ± 0.7 mV (N = 11) |
3.4. Excitatory synaptic transmission and membrane excitability are not altered in electroporated CA1 Sst-positive interneurons:
Our electroporation method enables us to transfect interneurons with high efficiency (Figure 2A). However, our results strongly suggest that interneurons are more susceptible to damage from electroporation than pyramidal neurons, and it is possible that electroporation-induced stress leads to altered interneuronal function. We therefore addressed whether our electroporation approach causes any side effects on the physiological properties of interneurons. Inhibitory interneurons are highly diverse and have different morphological and electrophysiological properties in the hippocampus (Klausberger and Somogyi, 2008; Somogyi and Klausberger, 2005), making it difficult to evaluate the impact of electroporation. To overcome this issue, we generated mice with TdTomato labeling the Sst interneuron population (Sst-TdTomato). Organotypic slice cultures were prepared from Sst-TdTomato mice and EGFP was transfected into TdTomato-positive Sst-neurons in the hippocampal CA1 oriens region (Figure 5A, left). Electrophysiological recordings from EGFP-transfected and neighboring untransfected TdTomato-positive interneurons were performed (Figure 5A). Importantly, AMPAR-EPSCs, PPR, membrane excitability and basal membrane properties were not significantly different between EGFP-transfected and untransfected Sst neurons (Figure 5B and C, Table 1), suggesting that our electroporation approach does not cause any physiological impairment in Sst-expressing interneurons. It has been reported that short-term application of TTX induces homeostatic synaptic scaling, which may cause undesired changes in neuronal function (Ibata et al., 2008). To address this concern, we compared the membrane excitability of Sst neurons in slices treated with or without TTX for 30 min (Figure 5D, Table 2). Sst neuronal excitability was unchanged regardless of treatment (Figure 5D) and showed no detectable abnormality in basic membrane properties (Table 2), indicating that electroporation under TTX-containing aCSF does not significantly affect the membrane properties of these interneurons.
Figure 5. Comparable levels of excitatory synaptic transmission and membrane excitability in electroporated CA1 Sst-positive inhibitory interneurons.

(A) Configuration of dual whole-cell recording, superimposed TdTomato (left) or EGFP (right) fluorescent and Nomarski images, respectively. (B) (Top) Sample traces mediated by AMPARs obtained from pairs of transfected (Trans, gray traces) and neighboring untransfected Sst-expressing interneurons (Untrans, black). Stimulus artifacts were truncated. (Bottom left) Scatter plot of AMPAR-EPSC amplitude. Each pair of transfected and neighboring untransfected Sst cells are presented as open symbols. Filled symbols indicate the mean ± SEM. (Bottom right) Summary graph of AMPAR-PPR. PPR of AMPAR-EPSCs recorded from trans- and untransfected Sst neurons, as indicated. Number of cell pairs tested: AMPAR-EPSCs, 14 cells/ 2 mice. n.s.: not significant. (C, D) Effect of GFP electroporation (C) and TTX-treatment (D) on neuronal excitability in Sst neurons. (Top) Sample traces from untransfected and transfected (C), and mock- and TTX-treated (D) CA1 Sst cells. The superimposed traces were elicited by current injections of −100, 0, 200, 500 and 900 pA for 200 ms. (Bottom) Summary graph of the frequency of action potentials in GFP transfected (C) and TTX-treated (D) Sst neurons. The input–output relationship was plotted for untransfected and transfected neurons. Neurons were held at resting membrane potentials. Two-way ANOVA with post hoc Tukey: number of cells tested: untrans, 8 cells from 2 mice (8/ 2); trans, (11/ 2); mock, (10/2); TTX (10/2).
Table 2. The basic membrane properties of hippocampal CA1 Sst neurons with or without TTX treatment.
These parameters were not statistically different between treatment conditions (Student’s t-test).
| Table 2 | Mock-treated Sst neurons | TTX-treated Sst neurons |
|---|---|---|
| Series Resistance | 18.4 ± 1.4 MΩ (N = 10) | 18.6 ± 1.5 MΩ (N = 10) |
| Input Resistance | 76.0 ± 5.5 MΩ (N = 10) | 68.4 ± 6.8 MΩ (N = 10) |
| Resting Membrane Potential | −56.8 ± 0.6 mV (N = 10) | −55.8 ± 0.7 mV (N = 10) |
4. Discussion
Electroporation is a versatile method for in vitro gene transfection but remains underutilized due to procedural challenges and concerns regarding transfection efficiency. Our electroporation method demonstrates high transfection efficiency in both excitatory and inhibitory neurons and multi-gene transfection efficacy, and produces no noticeable side effects on basal synaptic transmission, membrane excitability and basic membrane properties.
The resistance of glass electrodes we used in this protocol (3.5 – 6 MΩ) is standard for whole-cell recordings. Therefore, laboratories that already perform whole-cell in vitro electrophysiology studies should be able to perform this method with minimal difficulty or extra cost. Some recent studies have made significant advancements in their incorporation of robotics into electroporation protocols, leading to significant improvements in temporal efficiency of single-cell electroporation of excitatory neurons in vitro and in vivo (Li et al., 2017; Steinmeyer and Yanik, 2012). However, the cost of these instruments can serve as a barrier for implementation, and it remains to be seen if the method is effective in inhibitory neurons. Our pressure cycling step minimizes damage to cells after electroporation and drastically increases transfection efficiency to about 80%, although the possibility of yet undiscovered side effects cannot be ruled out.
We postulate that the usage of low resistance glass electrodes provides two benefits. First, the combination of low resistance electrodes (5 MΩ) with lower voltage pulses (−5 V) gives milder electrical pulses to the neurons compared with the protocol used in Rathenberg et al. (2003) (i.e. 10 MΩ and 10 V). The theoretical voltage applied to the cells (VO) is equal to VIN*(RC/(RE+RC)) where VIN is the command voltage, RC is the resistance of the cleft between the glass electrode and plasma membrane, and RE is the resistance of the electrode (Rae and Levis, 2002). By incorporating the typical range of RC (1 – 3 MΩ), this formula determines that the lower limit of the range of VO in our protocol (using VIN = 5 and RE = 5, VO = −0.71 to −1.88 V) compared with Rathenberg’s parameters (using VIN = 10 and RE = 10, VO = −0.91 to −2.31 V) yields a milder pulse condition (Bae and Butler, 2006; Rae and Levis, 2002). Second, the use of larger glass pipettes diminishes the incidence of pipette clogging during repeated electroporations and allows for upwards of 20 consecutive electroporations with a single pipette, greatly reducing the difficulty and duration of the procedure.
It is unclear why interneurons are especially vulnerable to damage by electroporation (Figure 2A). It has been suggested that immature neurons are more vulnerable to stress factors compared to mature neurons (Pfisterer and Khodosevich, 2017), and the postnatal maturation of excitatory neurons in the hippocampus precedes that of inhibitory interneurons (Danglot et al., 2006). Therefore, it is possible that we have tested excitatory and inhibitory neurons at different maturation stages in early development, which may explain the latter’s distinct vulnerability to electroporation. Notably, inclusion of TTX led to much higher transfection rates (Figure 2), suggesting that electroporation-related action potential propagation contributes to toxicity to interneurons. Indeed, we achieved high rates of transfection of EGFP into TdTomato-expressing Sst interneurons (Figure 5A). Most studies elucidating gene function by utilizing overexpression and knockdown approaches have focused on excitatory neurons, which occupy more than 80 % of the total neuronal population in both the hippocampus and cortex (Bezaire and Soltesz, 2013; Sahara et al., 2012). Our technique, combining single-cell electroporation and fluorescent mouse models holds significant potential for assessing the role of specific genes of interest in virtually any neuronal subtype. In addition, this method enables us to perform transfection of different genes in near-adjacent cells (Figure 2C), a complex task that is much more difficult to accomplish using other gene transfer methods. This new protocol greatly increases the feasibility of studying protein-protein interactions between cells, such as trans-synaptic protein interactions, as we have previously performed using less efficient methods (Futai et al., 2013).
Conclusion
Single-cell electroporation is a powerful tool to express genes in specific neuronal cell types and populations. However, suboptimal transfection efficiency and labor-intensive procedures are major drawbacks to existing methods. We utilized microelectrodes with a larger opening and gentler electrical pulse settings together with a pressure cycling step to improve this method. We demonstrated that these strategies dramatically increased transfection efficiency and reduced procedural difficulties such as pipette clogging. For investigators who find electroporation unreliable or impractical due to its low efficiency, this new protocol will enable more ambitious and complex studies of gene function, including studies that focus on specific neuronal subpopulations.
Highlights.
Current electroporation methods have low efficiency, limiting their usefulness
Our approach achieves 80% transfection efficiency in hippocampal organotypic slices
The method is effective in both excitatory and inhibitory neurons
This novel technique will greatly benefit in vitro studies of neuronal function
Acknowledgements
This work was supported by the grants from the National Institutes of Health Grants (R01NS085215 to K.F., T32 GM107000 to A.C.). The authors thank Ms. Naoe Watanabe for skillful technical assistance. We thank Dr. David Weaver for comments on an earlier draft of the manuscript.
Footnotes
Declaration of interests: The authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bae C, Butler PJ. Automated single-cell electroporation. BioTechniques, 2006; 41: 399–400, 2. 10.2144/000112261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezaire MJ, Soltesz I. Quantitative assessment of CA1 local circuits: knowledge base for interneuronpyramidal cell connectivity. Hippocampus, 2013; 23: 751–85. 10.1002/hipo.22141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang Y, Erturk A, Kallop D, Jiang Z, Weimer RM, Kaminker J, Sheng M. Activity-induced Nr4a1 regulates spine density and distribution pattern of excitatory synapses in pyramidal neurons. Neuron, 2014; 83: 431–43. 10.1016/j.neuron.2014.05.027 [DOI] [PubMed] [Google Scholar]
- Danglot L, Triller A, Marty S. The development of hippocampal interneurons in rodents. Hippocampus, 2006; 16: 1032–60. 10.1002/hipo.20225 [DOI] [PubMed] [Google Scholar]
- Dempsey B, Turner AJ, Le S, Sun QJ, Bou Farah L, Allen AM, Goodchild AK, McMullan S. Recording, labeling, and transfection of single neurons in deep brain structures. Physiological reports, 2015; 3 10.14814/phy2.12246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futai K, Doty CD, Baek B, Ryu J, Sheng M. Specific trans-synaptic interaction with inhibitory interneuronal neurexin underlies differential ability of neuroligins to induce functional inhibitory synapses. J Neurosci, 2013; 33: 3612–23.33/8/3612 [pii] 10.1523/JNEUROSCI.1811-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat Neurosci, 2007; 10: 186–95. 10.1038/nn1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y, Mao W, Saha S, Gunner G, Kolpakova J, Martin GE, Futai K. Luciferase shRNA Presents off-Target Effects on Voltage-Gated Ion Channels in Mouse Hippocampal Pyramidal Neurons. eNeuro, 2017; 4 10.1523/ENEURO.0186-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Popa I, Futai K, Sanchez-Martinez E, Wulf PS, van Vlijmen T, Dortland BR, Oorschot V, Govers R, Monti M, Heck AJ, Sheng M, Klumperman J, Rehmann H, Jaarsma D, Kapitein LC, van der Sluijs P. Neuron specific Rab4 effector GRASP-1 coordinates membrane specialization and maturation of recycling endosomes. PLoS Biol, 2010; 8: e1000283. 10.1371/journal.pbio.1000283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron, 2008; 57: 819–26. 10.1016/j.neuron.2008.02.031 [DOI] [PubMed] [Google Scholar]
- Kim TK, Eberwine JH. Mammalian cell transfection: the present and the future. Analytical and bioanalytical chemistry, 2010; 397: 3173–8. 10.1007/s00216-010-3821-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science, 2008; 321: 53–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Ouellette B, Stoy WA, Garren EJ, Daigle TL, Forest CR, Koch C, Zeng H. A robot for high yield electrophysiology and morphology of single neurons in vivo. Nature communications, 2017; 8: 15604. 10.1038/ncomms15604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao W, Salzberg AC, Uchigashima M, Hasegawa Y, Hock H, Watanabe M, Akbarian S, Kawasawa YI, Futai K. Activity-Induced Regulation of Synaptic Strength through the Chromatin Reader L3mbtl1. Cell Rep, 2018; 23: 3209–22. 10.1016/j.celrep.2018.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann E, Schaefer-Ridder M, Wang Y, Hofschneider PH. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J, 1982; 1: 841–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfisterer U, Khodosevich K. Neuronal survival in the brain: neuron type-specific mechanisms. Cell death & disease, 2017; 8: e2643. 10.1038/cddis.2017.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae JL, Levis RA. Single-cell electroporation. Pflugers Archiv : European journal of physiology, 2002; 443: 664–70. 10.1007/s00424-001-0753-1 [DOI] [PubMed] [Google Scholar]
- Rathenberg J, Nevian T, Witzemann V. High-efficiency transfection of individual neurons using modified electrophysiology techniques. J Neurosci Methods, 2003; 126: 91–8. 10.1016/s0165-0270(03)00069-4 [DOI] [PubMed] [Google Scholar]
- Sahara S, Yanagawa Y, O’Leary DD, Stevens CF. The fraction of cortical GABAergic neurons is constant from near the start of cortical neurogenesis to adulthood. J Neurosci, 2012; 32: 4755–61. 10.1523/JNEUROSCI.6412-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol, 2005; 562: 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeyer JD, Yanik MF. High-throughput single-cell manipulation in brain tissue. PLoS One, 2012; 7: e35603. 10.1371/journal.pone.0035603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods, 1991; 37: 173–82 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Yanagawa Y, Hirashima N. Transfer of small interfering RNA by single-cell electroporation in cerebellar cell cultures. J Neurosci Methods, 2009; 178: 80–6. 10.1016/j.jneumeth.2008.11.025 [DOI] [PubMed] [Google Scholar]
- Teruel MN, Blanpied TA, Shen K, Augustine GJ, Meyer T. A versatile microporation technique for the transfection of cultured CNS neurons. J Neurosci Methods, 1999; 93: 37–48. 10.1016/s01650270(99)00112-0 [DOI] [PubMed] [Google Scholar]
- Titomirov AV, Sukharev S, Kistanova E. In vivo electroporation and stable transformation of skin cells of newborn mice by plasmid DNA. Biochimica et biophysica acta, 1991; 1088: 131–4. 10.1016/0167-4781(91)90162-f [DOI] [PubMed] [Google Scholar]
- Washbourne P, McAllister AK. Techniques for gene transfer into neurons. Curr Opin Neurobiol, 2002; 12: 566–73. 10.1016/s0959-4388(02)00365-3 [DOI] [PubMed] [Google Scholar]
- Wiegert JS, Gee CE, Oertner TG. Single-Cell Electroporation of Neurons. Cold Spring Harb Protoc, 2017; 2017. 10.1101/pdb.prot094904 [DOI] [PubMed] [Google Scholar]