

Abstract
A mysterious feature of Crohn’s disease (CD) is the extra-intestinal manifestation of “creeping fat” (CrF), defined as expansion of mesenteric adipose tissue around the inflamed and fibrotic intestine. In the current study, we explore whether microbial translocation in CD serves as a central cue for CrF development. We discovered a subset of mucosal-associated gut bacteria that consistently translocated and remained viable in CrF in CD ileal surgical resections, and identified Clostridium innocuum as a signature of this consortium with strain variation between mucosal and adipose isolates, suggesting preference for lipid-rich environments. Single-cell RNA sequencing characterized CrF as both pro-fibrotic and pro-adipogenic with a rich milieu of activated immune cells responding to microbial stimuli, which we confirm in gnotobiotic mice colonized with C. innocuum. Ex vivo validation of expression patterns suggests C. innocuum stimulates tissue remodeling via M2 macrophages, leading to an adipose tissue barrier that serves to prevent systemic dissemination of bacteria.
Keywords: human microbiome, creeping fat, Crohn’s disease, inflammatory bowel diseases, mesenteric adipose, fibrosis, translocation, macrophages, adipogenesis, ileum
Graphical Abstract
Highlights
-
•
Gut bacterial translocation to mesenteric adipose tissue (MAT) naturally occurs
-
•
MAT from Crohn’s disease (CD) harbors a bacterial consortium defined by C. innocuum
-
•
These bacteria in CD promote restructuring of MAT and formation of “creeping fat”
-
•
Creeping fat expansion and fibrosis prevent systemic translocation of gut bacteria
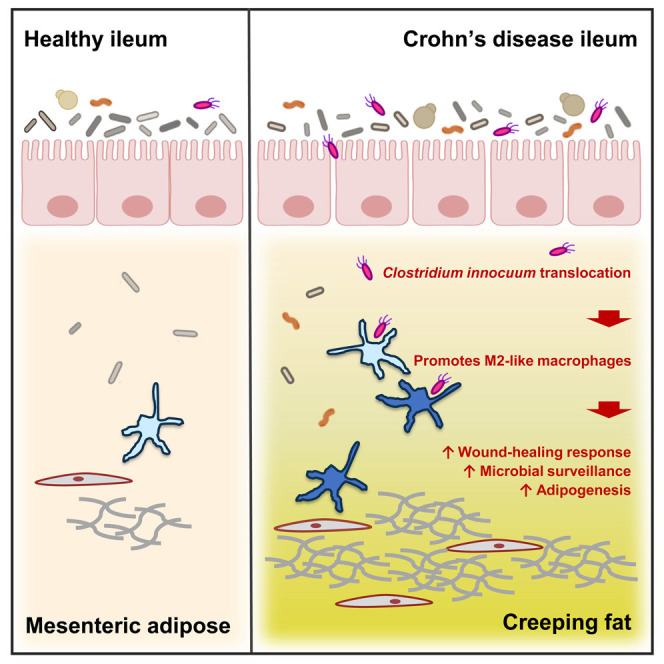
Ha et al. provide evidence that, in humans with inflammatory bowel disease, the phenomenon known as “creeping fat” is a protective response where mesenteric adipose tissue migrates (or “creeps”) to sites of gut barrier dysfunction to prevent systemic dissemination of potentially harmful bacterial antigens that have translocated across the barrier from the gut lumen.
Introduction
The remarkable plasticity of adipose tissue was thought to exist for excess energy storage or for use in times of famine. It is now clear that adipose is not a passive tissue, but rather a complex milieu of multiple cell types that respond to and influence many physiological processes (Cawthorn et al., 2012; Huh et al., 2014; Kershaw and Flier, 2004). One of the clearest examples of this is migration of mesenteric adipose tissue (MAT) to intestinal inflammatory lesions in Crohn’s disease (CD), resulting in so-called “creeping fat.”
Creeping fat (CrF) is an extra-intestinal manifestation of CD often found in patients with fibrotic, stricturing complications and presents as hyperplastic MAT, which expands and wraps specifically around sites of intestinal inflammation, primarily in the small bowel, and most often the ileum. CrF is not seen in ulcerative colitis (UC), the other form of inflammatory bowel diseases (IBDs) (Crohn et al., 1932; Kredel and Siegmund, 2014), and has been a mystery since the first published observation in 1932 (Crohn et al., 1932). CrF is also visually striking because inflammation in the CD intestine is patchy rather than continuous. As a result, CrF is also observed in patches, extending like fingerlike projections gripping the inflamed (involved) segments of intestine (Figure 1 A, top), adjacent to normal (uninvolved) MAT, which is pliable and attached longitudinally to a single border (Figure 1A, bottom). CrF itself and the underlying intestinal tissue tend to be severely fibrotic (Figure 1B); therefore, CrF and intestinal strictures often co-occur (Sheehan et al., 1992). Of note is the adipose encroachment into the intestinal muscularis (Figure 1B, top), which we observe to be a consistent histological feature in these patients. However, the relationship between CrF fibrosis as a driving or mitigating factor in intestinal fibrosis has not yet been explored despite their intimate spatial relationship and consistent presentation across this patient population.
Figure 1.

Characterizing the Defining Features of CrF in CD
(A) Representative adjacent paired involved and uninvolved intestinal segments from a CD patient.
(B) H&E stained CrF at the intestinal interface (top) and mucosa (bottom) from the same CD specimen. Arrows point to fibrotic adipose interleaved with ileal muscularis. Scale bar, 500 μm.
(C) Schematic of experimental design.
Given that inflammation-induced barrier dysfunction is a prominent feature of CD lesions, we hypothesized that the phenomenon of CrF at these lesions is a response to microbial translocation from the inflamed ileum to the surrounding MAT. Increasing evidence suggests that intestinal permeability is an integral component of chronic intestinal inflammatory diseases (Chang et al., 2017; Mankertz and Schulzke, 2007) and that impaired barrier function is among the constellation of accepted pathologies in IBDs (Jäger et al., 2013). This, in turn, can expose other body sites to intestinal microbiota and/or their metabolites (Amar et al., 2011; Zulian et al., 2013), triggering immunological responses and physiological changes in the peri-intestinal adipose.
In the following study, we identify a cultivable, gut-derived bacterial community in human MAT collected from surgical resections, with a microbial signature that distinguishes CD, and a specific translocated bacterium, Clostridium innocuum, that distinguishes CrF. Transcriptomic profiling of MAT across disease and healthy tissue controls, combined with ex vivo validation in patient-derived primary cells, led us to characterize CrF as a primarily fibrotic and immunogenic tissue with cellular phenotypes significantly upregulated for microbial surveillance. C. innocuum’s ability to translocate to the MAT was confirmed prospectively in gnotobiotic mice gavaged with a CrF-derived strain of C. innocuum, resulting in MAT expansion and recovery of C. innocuum from this tissue. Furthermore, the fibrotic and adipogenic phenotypes we identify in both humans and mice are similar to the visceral adipose phenotypes described in obesity (Crewe et al., 2017). This suggests that the microbial-driven MAT expansion we observe in CD may be relevant to the etiopathogenesis of fat expansion more broadly.
Results
Metagenomic Sequencing Reveals Bacterial Translocation, Which Occurs in Both CD and Healthy MAT, But Profile and Function Differ
Paired involved and adjacent uninvolved ileal segments (CD iMUC and uMUC, respectively) with attached CrF and adjacent uninvolved mesenteric adipose (CD MAT), and blood, for a total of five regional sites per patient (Figure 1C), were obtained from 11 patients undergoing surgical resections due to complications from CD. In addition, we collected the analogous regions, involved/uninvolved colon (UC iMUC and uMUC) and UC MAT from 13 UC patients as controls who exhibit intestinal inflammation in the absence of CrF. We also obtained healthy tissue controls from ileal mucosa (H Muc) and attached MAT (H MAT) and blood from four subjects undergoing ileostomy removal after recovery from non-IBD colon surgery. To ensure that luminal content contamination of MAT resulting from surgery was not a confounder, we vetted a detailed standard operating procedure in the operating room for sample collection, which entails carefully suturing each end of the resected specimen to eliminate leakage of luminal content. In the event a leakage occurred or abscesses were identified, these samples were eliminated from analysis. Environmental exposure of the sample was also limited, as specimens were aseptically transported directly to a sterile biosafety cabinet for processing less than 20 min from time of resection, and MAT was always dissected first before removing the intestinal sutures. These samples were placed through a systematic workflow of sample processing and analysis (Figure 1C). Patient metadata including clinical characteristics, medication use, family history, social history, and demographic information of this study cohort are detailed in Table S1.
We performed deep shotgun metagenomic sequencing on a subset of patients to first assess whether bacterial DNA could be detected in mesenteric adipose, and if so, whether this was unique to CD patients or was in fact a natural occurrence. From the 24 paired adipose and mucosal samples from CD (n = 4 patients, 4 tissue sites) and H (n = 4 patients, 2 tissue sites) (Figure 1C), one sample from H MAT had zero bacterial reads after host filtering and decontamination (Figure S1 A), with 2,803 taxa identified in total across the tissues. Bacterial reads were identified in three of four H MAT samples (Figure S1A; Table S2), demonstrating that bacterial translocation from the gut to MAT may not be unusual. To determine if the bacterial footprints in MAT were environmental contaminants, we used SourceTracker2 (Knights et al., 2011) to compare our identified taxa with the Human Microbiome Project (HMP) samples sourced from eight different body sites—largely of skin, oral, nasal, and stool origin (Huttenhower et al., 2012). This analysis determined that our mucosal and adipose sequences aligned most closely with stool samples (Figure 2 A).
Figure S1.

Diversity Analyses of the Metagenomes in Mesenteric Adipose Tissue and Ileal Mucosa in CrF and Healthy MAT Controls, Related to Figure 2
Relative abundance of bacterial phyla across tissue sites. H MAT has one less sample than the other groups because no bacterial DNA could be recovered from this sample.
Figure 2.

MAT Harbors a Diverse Microbiome of Gut Origin
(A) SourceTracker2 prediction of MAT bacterial origin compared against samples from the HMP.
(B) Alpha diversity from metagenomic sequencing comparing CD and H (left); the following paired tissues: CrF and iMUC (middle-left), CD MAT and CD uMUC (middle-right), and H MAT and H Muc (far right).
(C) Pathway analysis using Songbird multinomial regression of the total CD versus H microbiome. Positive values indicate association with CD.
(D) 16S rRNA-based amplicon sequence variants (ASVs) of the MAT and MUC microbiota in CD and UC.
(E) 16S rRNA-based phylogenetic tree of bacterial taxa observed in CD and UC resections. Vertical alignment of a dot represents co-occurrence of taxon across multiple sampling sites.
(F) ITS-based ASVs of the MAT and MUC mycobiota in CD and UC.
(G) Relative abundances of dominant fungal signals in CD and UC MAT and MUC.
Error bars ± SEM. Kruskal-Wallis test (A). Two-sided Mann-Whitney U test (B, far left). Paired Wilcoxon signed rank test (B, middle-left to far right). One-way ANOVA with Tukey’s test for multiple comparisons (D, F, and G). Statistical significance (p < 0.05) is represented by different letters on each bar. See also Figures S1 and S2 and Tables S2 and S3.
Alpha diversity was a key feature that separated CD and H samples. Despite using the same amount of adipose tissue for library construction, bacterial read counts were consistently lower in H MAT versus CD MAT (Table S2), yet H MAT retained greater diversity than CD MAT post-normalization (Figure 2B, far left). This is consistent with studies showing decreased microbial diversity in CD iMUC compared to control tissues (Lloyd-Price et al., 2019; Seksik et al., 2003), and we discover here that decreased mucosal diversity is correlated with a greater bioburden in MAT. Within-CD alpha diversity showed no significant difference between MAT and MUC from involved or uninvolved sites (Figure 2B, middle left and middle right). This was similarly observed in the tissue controls (Figure 2B, far right). Adonis PERMANOVA analysis of weighted and unweighted UniFrac data shows that the greatest distinguishing factor in our metagenomic dataset is inter-individual differences, which has also been shown in much larger IBD cohorts (Lloyd-Price et al., 2019); however, the next most distinguishing factor is sample type (Table S3). Beta diversity analysis demonstrated that although there is high inter-individual variability within our dataset, the community structure of samples can be largely separated by disease and tissue status (Figure S1B).
We interrogated the metabolic potential of the metagenomes from CD and H using MetaCyc pathways and ranked them using Songbird multinomial regression (Morton et al., 2019). Putative pathway analysis reveals that CD- and H-associated microbiomes have distinct preferences for carbon and nitrogen sources. In particular, pathways for sucrose utilization were significantly enriched in CD, as were pathways related to sulfur metabolism, whereas processes related to gut health such as fiber fermentation and vitamin B6 synthesis were enriched in microbiomes from H controls (Figure 2C).
Bacterial and Fungal Patterns Distinguish Tissue Compartments and Disease Status
While microbial differences exist between CD and H, this might be indicative of chronic intestinal inflammation in general, rather than specific signatures of CD. Therefore, we collected samples from UC as another form of chronic intestinal inflammation that does not develop CrF, and performed 16S rRNA sequencing. Irrespective of CD or UC status, we could isolate bacterial DNA from all adipose specimens; however, CD CrF and MAT alpha diversity compared to paired MUC was not significantly different, as was shown in the metagenomic data, whereas UC had significantly greater diversity in MUC compared to MAT and compared to all CD samples (Figure 2D). This likely reflects the bacterial richness of the colon compared to the small bowel. To further determine if the bacterial footprint in MAT is in fact gut derived, we compared the taxa identified in MAT from CD and UC to their respective MUC and looked for overlapping taxa and outliers. Principal coordinate analysis (PCoA) using Bray-Curtis distance showed no unique clustering between MUC and MAT in either CD or UC resections (Figure S2 A), suggesting no significant distinction between the microbiota at these sites. At the individual taxa level, MAT-derived bacteria phylogenetically aligned to members of the MUC microbiota in both CD and UC (Figure 2E), suggesting MAT does not represent a novel microbial niche, but rather translocation from the gut to neighboring MAT. CrF specimens were distinctly characterized by an expansion of Erysipelotrichaceae relative abundance compared to both adjacent CD MAT and underlying CD MUC (Figure S2B).
Figure S2.

16S rRNA and ITS Taxonomic Profiles of Mesenteric Adipose Tissue and Intestinal Mucosa in CD and UC, Related to Figure 2
(A) PCoA of Bray-Curtis distance of MAT- and MUC-associated microbiota in different subtypes of IBD. Specimens from involved (i) and uninvolved (u) resections are separated for analysis.
(B) Relative abundance of bacterial phyla (Left) and families within the Firmicutes phylum (Right) in involved and uninvolved specimens.
(C) Principal coordinate analysis of Bray-Curtis distance of MAT- and MUC-associated mycobiota from CD and UC resections.
In addition to bacterial sequences, to our surprise, fungal DNA was also identified in all MAT specimens from CD and UC, but there was no significant difference in the number of observed internal transcribed spacer (ITS) sequence variants between tissue compartments (Figure 2F). PCoA revealed that fungal communities were largely separated by specimen location (MAT versus MUC) (Figure S2C), suggesting that while there was no disease or tissue specificity in overall fungal diversity, the community structure reflects tissue site difference. Relative abundance of Saccharomyces cerevisiae and Candida metapsilosis, for example, was significantly higher in MAT specimens irrespective of disease (Figure 2G), whereas Malassezia restricta showed significant specificity for CD MUC, which is consistent with the recent discovery of M. restricta as a signature of CD MUC (Limon et al., 2019).
Ileal CD MUC and CrF Is Distinguished by a Distinct Cultivable Microbiota
While sequencing-based profiling is useful for providing a snapshot of microbial DNA present in MAT, whether the associated organisms are passive bystanders dead upon arrival or viable inhabitants of a given niche cannot be reliably determined. We aimed, therefore, to determine if the identified sequences in the MAT came from viable organisms via cultivation, and if so, we hypothesized that the genomic and functional characteristics of these organisms could provide insights into the micro-environment of CrF.
Live bacterial isolates were recovered from the MAT of 9/11 CD patients and 9/13 UC patients despite identifying microbial sequences in all of the patients. We were also able to recover viable bacteria from 4/4 H MAT controls (Figure 3 A). In total, we recovered 229 isolates from CD and UC MAT, which binned into 84 species after full-length 16S rRNA sequencing of each isolate (Table S4). When overlaying the cultivable bacteria with those identified by amplicon sequencing, 41 species were detected by both methods, while others were exclusively detected by sequencing or cultivation (Figure 3B). For example, Akkermansia muciniphilia and Faecalibacterium prausnitzii were frequently detected by sequencing but not by cultivation, yet we validated that the culture media we developed for this study could support the growth of their type strains. This suggests that many intestinal organisms can translocate to MAT but only a subset remain viable, or these bacteria under the given patient-derived conditions may have altered metabolic needs that differ from their type strain. We recovered two live fungal isolates from MAT, Candida albicans and Pseudozyma aphidis, and only in two CD patients. However, isolating specific fungi is notoriously challenging, and it is quite possible that our growth media was not optimal for supporting all fungal growth.
Figure 3.

CD MAT Has a Distinct Cultivable Microbiota Dominated by Clostridium innocuum
(A) Key cultivable organisms recovered from CD, UC, and H MAT. Bacteria found in more than one specimen are shown. Each column represents the cultivable community for an individual patient. Organisms recovered solely from CD, UC, or H are shaded black.
(B) Venn diagram denoting number of unique bacterial species identified by 16S rRNA sequencing and by cultivation methods.
(C) Compositionally coherent log-ratio t tests of metagenomic sequences from the five bacteria exclusively cultivated in CD samples (C. innocuum, E. ramosum, P. distasonis, C. symbiosum, and B. pseudolongum; Figure 3A) to an H-MAT-exclusive bacteria, P. merdae, identified by Songbird multinomial regression.
(D) Whole genome sequencing comparison of C. innocuum isolates recovered from CD CrF (n = 11), CD MAT (n = 6), CD mucosa (n = 8), and UC mucosa (n = 5), as well as a reference genome, C. innocuum 2959 and type strain DSM1286. Disease and tissue distribution of samples are coded on the right.
(E) Differentially abundant KEGG pathways across C. innocuum isolates. R = reference strains.
(F) Functional phenotyping of C. innocuum isolates by Biolog in vitro substrate utilization assay. Growth of each isolate was screened against 95 different substrates as the sole nutrient source. R = DSM1286 type strain.
Student’s t tests were performed for (C) given a priori knowledge of CD and H-MAT-associated bacteria. Kruskal-Wallis test was performed for (E). See also Figure S3 and Tables S4 and S5.
When cultivable adipose bacterial species were stratified by host status, we found a subset of five bacteria exclusive to CD MAT: Clostridium innocuum, Erysipeloclostridium ramosum, Parabacteroides distasonis, Clostridium symbiosum, and Bifidobacterium pseudolongum (Figure 3A). Both C. innocuum and E. ramosum are classified as Erysipelotrichaceae, which was identified as the most significantly expanded family in the 16S rRNA data (Figure S2B). To determine if this reflects a cultivation bias, we asked whether this signature can distinguish CD from healthy tissue controls, and specifically CrF from H MAT in our metagenomic dataset. To compare these microbial abundances in a compositionally coherent way, we calculated a log ratio with abundances of these five CD-specific bacteria in the numerator and P. merdae abundances in the denominator, which Songbird multinomial regression identified as the organism most associated with healthy MAT controls from metagenomic sequencing. This analysis shows that our putative cultivable CD signature significantly discriminates between CD and H tissues (Figure 3C).
Among this CD consortium, C. innocuum was the most frequently isolated. It is characterized as a gram-positive, vancomycin-resistant, spore-forming member of the commensal microbiota and the second most common species to cause extra-intestinal Clostridial infection, second to Clostridium perfringens (Chia et al., 2017). Interestingly, we could isolate live C. innocuum from MUC in UC patients, but never from their MAT, even though other viable bacteria were recovered (Table S5.1), demonstrating that C. innocuum colonizes ileum and colon but only appears to translocate in the small bowel. Moreover, we were unable to recover live C. innocuum from H MAT or MUC.
Comparative Genomic and Functional Analysis of C. innocuum Isolates across Tissue Sites
We performed whole genome sequencing (WGS) on a total of 30 patient-derived C. innocuum isolates from MUC and MAT, including type strain C. innocuum DSM 1286 and C. innocuum 2959 from the HMP as reference to determine degree of strain variation across tissue sites. Comparative genomics revealed that there are 1,627 operational gene units (OGUs) that comprise the conserved core of the C. innocuum genome, present in all isolates, and 3,788 were variably detected. The conserved genes fall under pathways that would suggest a competitive advantage for translocation to more toxic lipid- and oxygen-rich environments such as adipose. Among these are genes for protecting against oxidative damage (OGUc20831 superoxide reductase, OGUc25899 nitrate reductase, OGUc6810 thioredoxin reductase, and OGUc18816 peroxiredoxin Q), genes regulating adhesion and immune evasion (OGUc6982 capsule biosynthesis and OGUc6745 tryptophan synthase), lipid utilization genes (OGUc9722 and c9750 lysophospholipase/monoglyceride lipase and OGUc2822 myosin cross reactive antigen, a fatty acid hydratase that plays a role in oleic acid detoxification), and cell motility (OGUc8054 twitching motility protein) (Table S5.3).
The twitching motility feature stood out because C. innocuum has been described as a non-motile bacterium (Smith and King, 1962), yet our WGS data and the reference genomes of C. innocuum suggest otherwise. We tested bacterial motility using an agar-based motility assay, and indeed, all forms of patient-derived C. innocuum demonstrated notable motility in vitro (representative strains shown in Figure S3 A). The isolates also possess the gene for type IV pili (OGUc6526). Previous studies have shown that the type IV pili required for twitching motility is necessary for some pathogens, such as Pseudomonas aeruginosa, to migrate, adhere to, and invade epithelial cells and promote intracellular division (Mattick, 2002; Nieto et al., 2019). Twitching motility may therefore be an intrinsic feature for facilitating C. innocuum translocation across the intestinal epithelium.
Figure S3.

C. innocuum Functional Motility Assay and Intestinal Barrier Gene Expression, Related to Figure 3 and 4
(A) Motility test of C. innocuum in pre-reduced brain-heart infusion media with 0.3% agar. Motility is designated by growth deviated from the center stab line after 48 h. Non-motile bacteria, Staphylococcus aureus, is included here as negative control.
(B) Biolog assay from Figure 3F with CD CrF and CD MAT isolates grouped by patient source. Number indicates patient ID. For patients with multiple C. innocuum isolates, left-handed column refers to the CrF-derived isolate and the remaining columns for MAT-derived isolates.
(C) qRT-PCR of gut barrier genes comparing involved CD and UC specimens relative to their paired uninvolved specimen. Data below the dotted line represents downregulation of target genes in the involved segment compared to paired uninvolved segment. Data below the dotted line represents reduced expression of target genes in the involved UC tissues when compared to involved CD tissues (Right). (n = 10 for CD; n = 8 for UC).
Error bars ± SEM. Multiple t tests with FDR correction; ∗∗p < 0.01.
These analyses also revealed that the clearest phylogenetic distinction among the C. innocuum isolates was between MUC versus MAT-derived strains (Figure 3D). KEGG pathways differentially present in MUC and MAT isolates and between CD CrF and CD MAT isolates related to infectious diseases, folding, sorting and degradation, carbohydrate metabolism, and nucleotide metabolism (Figure 3E). The infectious disease pathways were particularly interesting as there were only two key genes contributing to this KEGG pathway, serpin B (OGUc5301 and c5312) and arginase (OGUc11046, c11199 and c10216) (Table S5.2). Eukaryotic serpin B is a serine protease inhibitor that protects against neutrophil elastase and has been shown to behave in a similar fashion in bacterial pathogens (Bao et al., 2018). Bacterial arginase is a well described defense mechanism that inhibits macrophage nitric oxide production, thereby promoting bacterial survival and proliferation in host tissue (Das et al., 2010). These features suggest that the CD adipose environment exerts a selection pressure for C. innocuum strains most capable of modulating host defenses.
Anaerobic substrate utilization assays were then performed on the Biolog platform to determine whether phylogenetic similarity of C. innocuum isolates was reflected in functional similarity. Similar to WGS data, this assay revealed core functional features as well as variable features distinguishing tissue source. Among conserved functions, no C. innocuum isolates could effectively utilize amino acids or their derivatives, except for L-phenylalanine, whereas all isolates robustly metabolized sugar and sugar derivatives, as well as nucleotides and their derivatives (Figure 3F). The two most highly metabolized substrates were 3-methyl-D-glucose and β-hydroxybutyrate. The latter is particularly intriguing as it is an abundant ketone byproduct of fatty acid oxidation. However, it was recently described that β-hydroxybutyrate is secreted by mature adipocytes to suppress the fibrogenic potential of surrounding precursor cells (Wang et al., 2019). Therefore, if C. innocuum can robustly metabolize β-hydroxybutyrate secreted by adipocytes, then adipose fibrosis may be allowed to proceed. However, we can only speculate on this at present time.
Despite our finding that only 2/5 CD patients had genetically divergent C. innocuum in their paired CrF and MAT, functionally, each CrF and MAT isolate recovered from the same patient had different metabolic preferences (Figure S3B, grouped by patient). This was true for all five patients. We also observed clear differences in substrate preferences between CD MUC and MAT isolates. Specifically, MUC isolates can metabolize pyruvic acid more effectively than MAT isolates, which may reflect oxygen tension in those tissues. These data demonstrate clear strain divergence of C. innocuum at the genomic level between MUC and MAT isolates; however, functional analyses reveal nuanced substrate preferences that likely reflect overall adaptations to changes taking place in CrF tissue.
Systemic Circulation of Bacterial Products Is Attenuated in CD Despite Impaired Barrier Function
We next turned to examine the host environment that may be facilitating bacterial translocation and the adipose expansion in CrF. We first speculated whether variations in bacterial translocation could be attributed to differences in intestinal permeability between healthy ileal controls and IBD subtypes. We measured intestinal tight junction gene expression for junctional adhesion molecule-A (JAM-A), E-cadherin, Claudins 3, 4, and 7, Mucin 1 (MUC1), tricellulin, and zonulin-1 (ZO-1) from full thickness tissue collected from CD iMUC and uMUC, UC iMUC and uMUC, and H MUC (Figure 4 A). CD and UC cases generally had lower expression for all markers tested compared to H, except for MUC1, which is consistent with MUC1 overexpression reported in human IBDs (Campbell et al., 2001; Rhodes, 1996). Comparison of the paired specimens within each IBD patient showed that CD iMUC had lower expression of all measured barrier genes compared to the CD uMUC, except for MUC1 and ZO-1 (Figure S3C). Despite the consistent impairment of barrier gene expression in both CD and UC-involved tissues, measurements of plasma lipopolysaccharide-binding protein (LBP) and soluble CD14, surrogate markers of intestinal permeability that are proportionately related to the amount of circulating bacterial products in the blood, were significantly lower in CD patients compared to UC, and not significantly different from H MUC (Figure 4B). This could reflect inflammatory status of the host, regional differences in CD and UC disease, or possibly, that the presence of CrF in CD serves to contain the systemic spread of bacteria at the site of inflammatory lesions.
Figure 4.

C. innocuum Translocation Promotes MAT Expansion and Attenuated Systemic Dissemination of Bacterial LPS
(A) Gut barrier gene expression measured by qRT-PCR in CD, UC, and H MUC. Data below the dotted line represent downregulation of target genes compared to H MUC (H MUC, n = 4; CD, n = 10; UC, n = 8).
(B) Plasma LBP and soluble CD14 from the same CD and UC as in (A). Healthy samples are a combination of H patients in (A) (open symbols) and ten additional healthy blood donors (H, n = 14; CD, n = 14; UC, n = 11).
(C) Representative images of ileal-mesenteric region in ASF gnotobiotic mice gavaged with the following: PBS (left), live C. innocuum (middle), and live C. innocuum + DSS (right). Black arrow points to the MAT.
(D) Gnotobiotic mice body weight change compared to baseline. Untreated, n = 3; C. innocuum alone, n = 2; C. innocuum + DSS, n = 2.
(E) Colon lengths.
(F) Translocated bacteria recovered from MAT of mice from (C). Arrows indicate distinct bacterial species (representative isolates; yellow, ASF; blue, C. innocuum).
(G) qRT-PCR of adipogenesis and ECM markers in gnotobiotic MAT.
(H) Endpoint plasma LBP in gnotobiotic mice.
Error bars ± SEM. One-way ANOVA with Tukey’s multiple comparison test. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; #p < 0.05 compared to untreated and C. innocuum + DSS; +p < 0.05 compared to untreated. See also Figure S3.
C. innocuum Translocates to MAT in Gnotobiotic Mice and Promotes Adipose Expansion
Currently, it is impossible to predict which newly diagnosed CD patients will develop fibrotic complications, and access to surgical resections only occurs in advanced cases; therefore, we cannot prospectively observe bacterial translocation and development of CrF in these patients. Furthermore, no animal model of CrF exists that reliably recapitulates the phenomenon seen in humans. Nonetheless, we utilized gnotobiotic mice to determine whether we could prospectively observe C. innocuum translocation to the MAT, and if so, whether it would result in any alterations to the MAT.
We utilized age- and weight-matched gnotobiotic mice raised with altered Schaedler flora (ASF), a well-defined consortium of eight anaerobic bacterial species often used by commercial vendors to promote healthy development of the gut. Dams were colonized with ASF and the consortium was therefore propagated in pups and verified by PCR-based routine monitoring. We chose these mice to help ensure normal development of the intestines—critical when permeability and translocation are readouts. ASF mice were administered a one-time oral gavage of human CrF-derived C. innocuum and confirmed colonization by day 4 post-gavage. Mice were terminated at day 14 post-gavage, along with PBS gavaged control mice. A subset of C. innocuum-gavaged mice were given dextran sulfate sodium (DSS) in the drinking water at day 4 to determine if intestinal injury and impaired barrier function are required for translocation.
Upon termination, we observed dramatic mesenteric adiposity in the C. innocuum-gavaged mice while control mice had trace amounts of MAT similar to typical gnotobiotic wild-type mice (Figure 4C). The observed MAT expansion did not appear to be a consequence of overall weight gain, as oral gavage with C. innocuum did not lead to weight change compared to controls (Figure 4D). While significant body weight loss was observed in the C. innocuum + DSS group, as is expected with DSS treatment, these animals still exhibited noticeable MAT adiposity compared to the control group. Colon length shortening, an indicator of intestinal inflammation, was observed significantly in both C. innocuum groups, but to a greater degree in C. innocuum + DSS (Figure 4E).
Cultivation of MAT tissues across these mice resulted in successful isolation of the gavaged C. innocuum, confirming that it can translocate from gut to MAT, and occurred in both DSS-treated and untreated groups (Figure 4F, blue arrows, pointing to a representative C. innocuum isolate). This suggests that overt inflammation is not a prerequisite for its translocation. We also demonstrate, similarly to the cultivation data from healthy human tissue controls, that bacterial translocation to MAT is a natural occurrence to a degree. We were able to recover two of the eight ASF species from the control group (Figure 4F, left, yellow arrows), which suggests that not all members of the community have the capacity to translocate or survive in the extra-intestinal space, which is in line with the patient data in Figures 3A and 3B. We have experience routinely cultivating all eight organisms from our ASF colony, so we believe this observation is reflective of the true translocation state rather than an artifact of cultivation conditions. Interestingly, the colonization of C. innocuum appears to promote the translocation of four additional members within the ASF consortium (Figure 4F, middle and right, yellow arrows). Similar results were also observed in the DSS treatment group. It has been shown that non-motile microorganisms can attach to motile neighbors, effectively hitchhiking and promoting dispersal and invasion within a host (Samad et al., 2017). This is an intriguing yet unexplored concept in human tissues.
Tissue gene expression of adipogenesis and fibrosis-related genes showed that mice gavaged with C. innocuum alone exhibited upregulation of genes involved in adipogenesis, such as Fabp4, Fasn, Pparg, and Cebpa (Figure 4G). This was not observed in the C. innocuum + DSS and control groups. However, collagens IV and VI, extracellular matrix (ECM) components often implicated in adipose fibrosis, were highly expressed in C. innocuum-gavaged mice ± DSS (Figure 4G), suggestive of a role for C. innocuum in promoting both adipogenesis and ECM production.
To address our hypothesis that MAT expansion attenuates systemic dissemination of bacterial products, we measured plasma LBP across treatment groups. Here, we found that the C. innocuum-alone group had similar, if not lower, LBP levels compared to the untreated ASF controls (Figure 4H), which mirrors the results from our human cohort (Figure 4B). DSS-treated mice, on the other hand, showed significant increase in LBP levels despite their C. innocuum translocation and MAT expansion. Given that acute DSS induces injury throughout the small bowel and colon, leakage or dissemination of microbial products could occur outside the region of adipose expansion. We acknowledge low n’s in this experiment (please see Limitations of the Study); however, these data represent a conceptual demonstration that human CrF-derived C. innocuum, when gavaged into a gnotobiotic mouse with a simplified microbiota, can translocate to the MAT and promote adipose expansion characterized by both adipogenesis and fibrosis.
The Cellular Composition of CrF Is Distinguished by Markers of Tissue Remodeling and Distinct Immune Cell Populations
Given the degree of bacterial translocation to the mesenteric depot across our patients as well as tissue controls, we characterized the cellular milieu of the MAT in CD, UC, and H-MAT, by single-cell RNA sequencing (scRNA-seq) and bulk RNA sequencing and looked for distinguishing cellular phenotypes across tissues and disease.
Bulk RNA-sequencing transcriptional activity of CrF and H MAT revealed that, of the 42,606 genes detected, 1,118 were identified as significantly differentially expressed genes (DEGs), with 834 upregulated and 284 downregulated in the CrF specimens (Figure 5 A; Table S6). As expected, a subset of adipogenesis-related genes was overexpressed in CrF and the expression of negative regulators of adipogenesis was reduced (Figure 5B). However, pathway level analysis indicated that the greatest transcriptional changes were not functions related to lipid metabolism. Genes with the highest fold enrichment scores in CrF related to processes such as cellular response to bacterial products, phagocytosis, B and T cell differentiation and activation, and ECM production and organization (Figure S4 ). This supports that CrF is primarily characterized by an immune response to bacterial translocation and fibrosis. To further refine these analyses, we performed scRNA-seq on the adipose stromal vascular fraction (SVF), which contains the immune, endothelial, and progenitor cell types.
Figure 5.

Single-Cell and Bulk RNA Sequencing of MAT from CD, UC, and H Reveals a Distinct Cellular Profile in CrF
(A) Bulk-RNA-sequencing-generated heatmap of differentially expressed genes (DEGs) in CrF versus H MAT. Genes with adjusted p value < 0.05 are shown.
(B) Fold change in expression level for a given gene in CrF versus H MAT, with adipose differentiation genes highlighted. Significant DEGs with adjusted p value < 0.05 are colored red.
(C) scRNA-seq-generated UMAP plots of CrF and H MAT distinguishing individual cell clusters (left) and tissue source (right). Frequency of cell types indicated by colored bars.
(D) scRNA-seq-generated UMAP plots of CrF and paired CD MAT distinguishing individual cell clusters (left) and tissue source (right). Frequency of cell types indicated by colored bars.
See also Figures S4 and Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure S1, Figure S2, Figure S3, Figure S4, Figure S5and Tables S6 and S7.
Figure S4.

Enriched Functions Determined by Bulk RNA Sequencing in Creeping Fat Compared to Healthy Mesenteric Adipose Tissue, Related to Figure 5
Significant functions that were upregulated in CrF compared to H:
(A) KEGG pathways
(B) Cellular components
(C) Molecular functions
(D) Biological processes
Categories with adjusted p value < 0.05 are shown. Size of the symbol represents the number of differentially expressed genes in creeping fat binned into a specific function. CD: n = 4; Healthy ileal controls: n = 4.
Analysis of combined CrF and H MAT revealed 14 different cell clusters (Figure 5C, left) consisting primarily of progenitor cells (P1–P5), immune cells, and endothelial cells. When the 14 clusters were distinguished by tissue source (Figure 5C, right), the clusters belonging to P3 (FABP4 +), the two T cell subsets (T cell 1: CCL7 +, CD62L +; T cell 2: CCL7 −, CD62L −), B cells, and sensory neurons consisted almost entirely of cells from CD CrF. In contrast, the only cell types to distinguish H MAT were P2 (CD34 +, FABP4 +, and PPARG +) and P4 (ICAM1 +). This comparison to healthy tissues reveals that CrF is clearly defined by an abundance of distinct immune cells. When CrF was compared to its adjacent CD MAT in a separate experiment (Figure 5D, left), 11 cell types emerged, but nearly all consisted of cells from both tissue sites. Of these 11, three were progenitor cell types (P1–P3) that phenotypically differed from the previous analysis in that they were not significantly enriched in FABP4 or PPARG. While T cells were still the most abundant immune cell type, they were less heterogeneous than in the comparison to H MAT, and the B cells were notably expanded. When the clusters were distinguished by CrF or CD MAT (Figure 5D, right), the same adipose-derived fibroblast cell cluster that distinguished H MAT from CrF also distinguished CD MAT (P2, cluster 1). Interestingly, this cell type also distinguished CrF from UC MAT (cluster P2, Figure S5 A), suggesting this progenitor cell type is specific to MAT in the ileal region. P1, P3, and macrophages were represented by a higher frequency of cells from CrF. The macrophage population became increasingly of interest due to their expression of TLR2, which recognizes lipoproteins on gram-positive bacteria such as C. innocuum, among its top ten most highly expressed genes (p < 1.75 e-251), as well as NLRP3 (p < 1.88 e-259) (Table S8), both of which are acutely responsive to PAMPs. This, combined with abundant pro-inflammatory cytokine expression such as IL1β as well as significant TGFβ expression (p < 6.58 e-139), indicates both an M1 and M2 presence in this cluster. Given that macrophages were the primary innate immune cells present in our dataset, and highly expressed genes for bacterial recognition, this suggested that macrophages are likely orchestrating the initial communication of bacterial presence in the MAT, resulting in a dual polarization to both optimize bacterial cell killing and promote wound healing. Overall, the consistent feature across these two experiments is that CrF appears to have a dramatic reduction in adipose-derived fibroblasts and a higher frequency of immune cells compared to H MAT. However, these distinctions are lost when compared to CD MAT, suggesting that CD MAT is likely a transitionary state between H MAT and CrF.
Figure S5.

UMAP Plots from scRNA-Seq for Ulcerative Colitis Tissues and for Each Individual Patient, Related to Figure 5
(A) Total cell clusters from UC involved MAT and uninvolved MAT (left), and colored by tissue source (involved or uninvolved). n = 2 UC patients.
(B) Individual plots for each patient included in experiment 1 comparing CD to UC adipose tissues.
(C) Individual plots for each patient included in experiment 2 comparing CD creeping fat to healthy tissue controls.
Gene set enrichment analysis was used to identify pathways significantly enriched in CrF compared to H MAT and CD MAT (Figures 6A and 6B. These data show that the progenitor cells are significantly enriched for pathways related to ECM production. This is consistent with the bulk RNA sequencing data that shows that, while CrF is enriched for adipogenesis genes, it is the ECM and immune-related pathways that are most significant (Figure S4). Significant pathways enriched in the macrophage populations largely related to microbial pattern recognition and signaling to other cell types. However, the macrophages were also the only other cell type outside of endothelial cells that upregulated pathways for lipid catabolic and biosynthetic processes, indicating that they play a role in tissue remodeling in CrF.
Figure 6.

Upregulated ECM and Anti-MMicrobial-Related Pathways Are Dominant Cellular Phenotypes of CrF
(A) GSEA of pathways differentially expressed in CrF compared to H MAT. Top five significant pathways for each cell cluster are listed on the y axis.
(B) GSEA of pathways differentially expressed in CrF compared to adjacent CD MAT.
NES = normalized enrichment score. Size = size of the gene set for each pathway listed.
C. innocuum Promotes Pro-fibrotic Phenotypes in Primary Macrophages and Progenitor Cells In Vitro
The data thus far characterize CrF as a tissue cellularly dominated by pro-fibrotic progenitor cells and an influx of adaptive and innate immune cells that collectively appear to be directed by bacterial translocation. To test whether C. innocuum specifically can direct these responses, we first performed an immunogenicity assay whereby fresh lysates from either a CrF- or CD MAT-derived strain of C. innocuum were co-cultured with PBMC-derived macrophages from healthy volunteers, following the approach of Schirmer et al. (Schirmer et al., 2016), and measured cell surface markers and cytokines indicative of M1 or M2 polarized cells. The CrF strain of C. innocuum used in these experiments was the same strain used in the gnotobiotic mouse experiments. In parallel, we also tested whether C. innocuum, as part of a consortium of selected CD-specific cultivable bacteria (E. ramosum, P. distasonis, and B. pseudolongum; Figure 3B), would polarize macrophages similarly to C. innocuum alone. Macrophages have been shown to exhibit morphology changes when differentiated to M1 or M2 subtypes (McWhorter et al., 2013), with M1s displaying a typical rounded, spiked morphology and M2s forming elongated spindles. Indeed, these respective morphologies were observed when exposed to LPS and IL-4, M1 and M2 positive controls (Figure 7 A, representative images shown). When the cells were exposed to C. innocuum alone, they exhibited a pronounced elongated morphology, while macrophages exposed to the CD-associated consortium were heterogenous.
Figure 7.

C. innocuum Promotes M2 Macrophage Polarization and Wound-Healing Response in Progenitor Cells In Vitro
(A) Representative images of PBMC-derived macrophages after 24 h exposure to LPS, IL-4, C. innocuum lysates alone, and lysates from the consortium of CD-associated organisms including C. innocuum.
(B) Immunogenicity assay determining polarization of PBMC-derived macrophages upon exposure to C. innocuum lysates sourced from either CrF or CD MAT (strain CD-CrF B and CD-MAT C from Figure 3D) or a consortium of CD-associated organisms without C. innocuum.
(C) Co-culture of CrF-derived progenitor cells with C. innocuum lysate directly, or with conditioned media from macrophages exposed to C. innocuum lysates from (B). Gene expression was measured by qRT-PCR.
Error bars ± SEM. One-way ANOVA with Tukey’s multiple comparison test (D); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
We measured polarization state using canonical M1 macrophage markers that were also co-expressed in our scRNA-seq dataset, as well as M2 markers that have been indicated to substratify M2a/M2b/M2c subsets. This distinction, while highly context dependent, may be important in CrF, as M2a macrophages have been described as wound-healing/pro-fibrotic (measured here by the mannose receptor CD206), M2b macrophages have been described as either pro- or anti-inflammatory (CD86), and M2c macrophages have been described for their tissue remodeling properties (CD206/CD163). In these experiments, we also removed C. innocuum from the CD consortium group so as to more clearly delineate which bacteria were driving the M1 and M2 morphologies observed. We found that C. innocuum, both isolates, stimulated a minimal M1 response significantly lower than the LPS positive control in all instances and not significantly different from the unstimulated or IL-4 negative control (Figure 7B). The CD consortium, however, elicited the highest IL1β and TNFα response of all the treatment groups. The M2 marker analysis revealed that M2b, the inflammation-modulating M2 macrophages, were stimulated by LPS and the CD consortium, but not by the C. innocuum isolates. However, C. innocuum significantly increased CD206 but not CD163 expression, suggesting that C. innocuum may selectively promote M2a, pro-fibrotic macrophages. While determining M2 subset polarization is dependent on many factors and would benefit from further protein-level analysis, what these data suggest is that C. innocuum translocation is likely not responsible for eliciting an overt pro-inflammatory response in macrophages. This may be due to its ability to evade these cells as evidenced by the conserved gene for arginase across all C. innocuum CrF strains. Instead, the presence of C. innocuum in CD MAT likely promotes an M2 phenotype that serves to remodel the adipose environment. To test this hypothesis, we isolated primary fibroblasts and adipose-derived stem cells from CD MAT, which receive cues from the local microenvironment to proliferate. We exposed these cells to either C. innocuum lysate directly or macrophage-conditioned media from the C. innocuum-exposed macrophages. We measured gene expression of collagens I (COL1A1) and VI (COL6A3), and hyaluronan synthase 1 (HAS1), which were among the most highly expressed genes in the CrF progenitor cell population and are directly involved in tissue fibrosis and remodeling. C. innocuum lysates alone were insufficient in modulating any of these genes; however, macrophage-conditioned media significantly increased expression of COL1A1 (p < 0.001) with a trend toward increased HAS1 expression (Figure 7C). The limited marker panel does not conclusively allow us to determine which progenitor cell type is directly involved in the adipose fibrosis we observe; however, it does tell us that C. innocuum needs an immune cell mediator to elicit its pro-fibrotic effects.
Discussion
While the subject of this study is a unique extra-intestinal phenomenon in CD, the findings herein lend new insights into the role of adipose tissue in the human body and how the gut microbiome may influence its behavior. The primary role of adipose as a storage form for excess calories, while necessary, suggests a passive role in the body. However, we show here that adipose plasticity may have another, equally important, purpose that involves protecting the body from dissemination of harmful antigens at sites of inflammation or injury. We hypothesized that this is the primary driver of CrF development in CD patients.
Here, we find that bacterial translocation from the gut to MAT occurs to a degree in healthy tissue controls, without acute consequence such as influx of immune cells nor activated phenotypes. However, in states of chronic intestinal inflammation such as in CD, the gut microbiota are perpetually perturbed, giving rise to opportunistic organisms able to take advantage of impaired host and microbiota defenses. We demonstrate that bacterial translocation occurs to an even greater degree in this instance, and with a different set of microbial travelers than is seen in healthy states. Given that adipose does not only consist of adipocytes, but also a rich community of innate and adaptive immune cells, endothelial cells, and progenitor cells, the presence of opportunistic bacteria in this tissue must elicit a cellular response.
Indeed, we identify a microbial mechanism explaining the expansion of CrF in CD, driven by a gut-derived bacterium, C. innocuum, that translocates and survives in MAT. We find, within IBD, its ability to translocate and remain viable is unique to CD patients, and exhibits a genetic strain variation that distinguishes mucosa and adipose, and further functional variation that distinguishes CrF and MAT. Core genomic features of C. innocuum include type IV pili and twitching motility, a preference for lipid-derived metabolic substrates, and multiple genes for lipid catabolism, as well as a functional substrate preference for β-hydroxybutyrate, a byproduct of fatty acid oxidation. This suggests that C. innocuum is well suited for, and perhaps prefers, a lipid-rich environment and seeks these out when the opportunity arises. Distinguishing features of adipose isolates compared to mucosal isolates include genes for evading killing strategies by innate immune cells. This highlights the importance of accounting for strain variation in microbiome studies, particularly the concept that even genetically identical strains may behave differently depending on tissue source.
CrF tissues colonized by C. innocuum were characterized by T and B cell populations that were nearly absent from H MAT, whereas CD adjacent uninvolved MAT presented cellular features of both H MAT and CD CrF, suggesting that CD MAT likely represents an intermediate transitionary stage between healthy and CrF. We also find that while CrF is a visually distinct state of adipose expansion, ECM-related genes and genes related to microbial sensing and killing were the most highly expressed in the collective transcriptome. We therefore were particularly intrigued by the mixed M1/M2 macrophage population identified by scRNA-seq. These were the dominant innate immune cell type across all MAT tissues we analyzed; therefore, if bacteria were translocating to the degree we were observing, then we expected these macrophages to be the primary cell communicating their presence. Given that CrF is characterized by increased expression of fibrosis, and M2 subtypes are involved in promoting fibrosis and tissue repair, we wanted to understand whether C. innocuum was polarizing macrophages toward this phenotype. The finding that C. innocuum could not elicit any signs of M1 polarization, but did significantly increase expression of CD206 + M2 macrophages, indicates that C. innocuum may directly interact with a pro-fibrotic wound-healing M2a subtype. The consortium of other CD-specific bacteria did elicit M1 polarization, which may explain the heterogeneous macrophage phenotypes. It has been shown in rodents that a degree of adipose inflammation is required for adipose expansion, and this adipose expansion serves to manage underlying intestinal inflammation. Knockout mice that lack the ability to expand their mesenteric adipose exhibit an impaired intestinal barrier (Wernstedt-Asterholm et al., 2014), which is consistent with our gnotobiotic and human data showing that the presence of MAT expansion is correlated with decreased circulating LBP and soluble CD14.
Many interesting questions still remain. For example, lymphedema, a chronic state of lymphatic fluid stasis, has been shown to also lead to adipogenic and fibrotic adipose expansion surrounding the lymphatics in response to injury or bacterial antigen (Mattacks et al., 2003; Zampell et al., 2012). Given that mesenteric lymph nodes are normally embedded throughout the MAT but removed from our samples, we suspect that they may play a role in CrF development. This warrants further investigation. We also cannot conclude that C. innocuum alone causes MAT expansion, nor do we believe this to be the case. Rather, it is likely the presence of a critical mass of microbial load of which, in the context of CD at least, C. innocuum is the most abundant.
Nonetheless, the data presented here help illuminate the long-standing question of whether CrF in human CD is harmful or beneficial. It is likely both. What begins as a reaction to intestinal injury and bacterial dissemination, aiding the body’s protective response and limiting the collateral damage of systemic antigen exposure, appears to have no off switch in the presence of continued microbial exposure. This wound-healing response, in turn, leads to significantly fibrotic mesenteric adipose encasing the underlying ileum which, by the time of resection, is also significantly fibrotic. This may protect the body from systemic inflammation, attempting to keep the inflammation localized; however, unmitigated expansion has consequences for the underlying tissue, including CrF encroachment into the bowel wall. Therefore, strategies to therapeutically target or outcompete the intestinal reservoir of C. innocuum in high-risk patients may offer an avenue for preventing or attenuating the fibrotic cascade.
Limitations of the Study
The gnotobiotic mouse experiment presented (Figures 4C–4H) was meaningfully impacted by COVID-19 lab closures. We acknowledge that our mouse numbers are far below the standard for an animal experiment, which was unintentional. Subsequent litters were earmarked for repeat experiments and unfortunately reached the age-matched window just as lab closures were implemented. Lack of facility access and required culling of animals to reduce staffing load prevented us from running the repeat experiments. As a result, the data are presented as a small proof-of-concept that we, and we hope others, will build upon.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Altered Schaedler Flora (ASF) (bacteria, fecal pellet) | Taconic | N/A |
| Bifidobacterium pseudolongum | This study | N/A |
| Clostridium innocuum strain 2959 | NCBI | RefSeq: NZ_AGYV00000000.1 |
| Clostridium innocuum strain CD-CrF B | This study | Strain: CD-CrF B |
| Clostridium innocuum strain CD-MAT C | This study | Strain: CD-MAT C |
| Clostridium innocuum strain DSM1286 | DSMZ | Strain: DSM1286 |
| Erysipeloclostridium ramosum | This study | N/A |
| Parabacteroides distasonis | This study | N/A |
| Biological Samples | ||
| Human surgical specimen | Cedars-Sinai Medical Center | N/A |
| Human peripheral blood | Cedars-Sinai Medical Center | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Agar | Fisher Scientific | Cat.# BP-1423 |
| Agencourt AMPure XP beads | Beckman Coulter | Cat.# A63881 |
| Ammonium iron citrate | Sigma-Aldrich | Cat.# F5879 |
| Bacteroides bile esculin agar | Anaerobe Systems | Cat.# AS-144 |
| Bovine serum albumin | MP Biomedicals | Cat.# 216006980 |
| Brain heart infusion media | Sigma-Aldrich | Cat.# 53286 |
| Brucella media | Hardy Diagnostics | Cat.# C5311 |
| Ceramic beads | Omni International | Cat.# 19-628 |
| Chloroform-isoamyl alcohol (24:1) | Sigma-Aldrich | Cat.# C0549 |
| Chocolate blood agar | BD | Cat.# BD 221267 |
| Collagenase II | Invitrogen | Cat.# 17101015 |
| CryoStor cell preservation media | Sigma-Aldrich | Cat.# 2874 |
| Dextran sulfate sodium salt | MP Biomedicals | Cat.# 0216011090 |
| DMEM | Corning | Cat.# MT10013CV |
| DMEM F12 50/50 | Corning | Cat.# 10-090-CV |
| DMSO | Sigma-Aldrich | Cat.# D2650 |
| DNase I | Sigma-Aldrich | Cat.# DN25 |
| EDTA | Invitrogen | Cat.# AM9260G |
| FBS | Omega | Cat.# FB-02 |
| Ficoll-Paque Premium | GE Healthcare | Cat.# 17-5442-02 |
| Hemin | BeanTown Chemical | Cat.# 138155 |
| Human recombinant IL-4 | PeproTech | Cat.# 200-04 |
| Human recombinant M-CSF | PeproTech | Cat.# 300-25 |
| Lactobacilli MRS media | BD | Cat.# BD 288130 |
| LPS | Sigma-Aldrich | Cat.# L3024 |
| Lyticase | Sigma-Aldrich | Cat.# L4025 |
| PBS | Corning | Cat.# 21-031-CV |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat.# 15140122 |
| Proteinase K | Life Technologies | Cat.# 25530049 |
| RBC lysis buffer | Invitrogen | Cat.# 00-4333-57 |
| Reinforced Clostridial media | BD | Cat.# BD 218081 |
| RPMI 1640 | Corning | Cat.# 10-041-CV |
| Sabouraud dextrose agar | Hardy Diagnostics | Cat.# W70 |
| Sodium pyruvate | VWR | Cat.# VWRV0342 |
| Taurine | Alfa Aesar | Cat.# AAA12403 |
| Tris | Bio-Rad | Cat.# 1610719 |
| TRIzol | Invitrogen | Cat.# 15596026 |
| Vitamin K | Alfa Aesar | Cat.# L10575-06 |
| β-mercaptoethanol | Sigma-Aldrich | Cat.# M3148 |
| Critical Commercial Assays | ||
| AN inoculating fluid | Biolog | Cat.# 72007 |
| AN MicroPlate | Biolog | Cat.#1007 |
| Chromium Chip B Single Cell Kit | 10X Genomics | Cat.# 1000154 |
| Chromium Single Cell 3ʹ GEM, Library and Gel Bead Kit v3 | 10X Genomics | Cat.# 1000092 |
| DNeasy PowerSoil Kit | QIAGEN | Cat.# 12888-100 |
| E.Z.N.A. MicroElute RNA Clean Up Kit | Omega Bio-tek | Cat.# R6247-01 |
| E.Z.N.A. Total RNA Kit II | Omega Bio-tek | Cat.# R6934-01 |
| EasySep Human Monocyte Enrichment Kit | STEMCELL Technologies | Cat.# 19059 |
| High sensitivity DNA Kit | Agilent | Cat.# 5067-4626 |
| Human IL-1β ELISA | R&D Systems | Cat.# DLB50 |
| Human LBP ELISA | Hycult Biotech | Cat.# HK315; RRID: AB_10989638 |
| Human sCD14 ELISA | R&D Systems | Cat.# DC140 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat.# 1708891 |
| iTaq DNA polymerase | Bio-Rad | Cat.# 1708870 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat.# 1725121 |
| KAPA HyperPlus Kit | Kapa Biosystems | Cat.# KK8514 |
| MagAttract PowerSoil DNA Kit | QIAGEN | Cat.# 27000-4-KF |
| NEBNext Library Quant Kit for Illumina | New England BioLabs | Cat.# E7630 |
| QIAseq FastSelect rRNA Removal Kit | QIAGEN | Cat.# 334386 |
| RNeasy Mini Kit | QIAGEN | Cat.# 74106 |
| SureCell Whole Transcriptome Analysis 3¢ Library Prep Kit | Illumina | Cat.# 20014279 |
| TruSeq Stranded Total RNA Library Prep kit | Illumina | Cat.# 20020596 |
| Deposited Data | ||
| 16S, ITS, metagenomics and WGS datasets | NCBI BioProject | Accession # PRJNA659515 |
| Bulk RNA sequencing datasets | GEO | Accession # GSE156044 |
| Single-cell RNA sequencing datasets | GEO | Accession # GSE156776 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6NTac Germ-free, then ASF-colonized | Taconic | Cat.# B6 GF |
| Oligonucleotides | ||
| See Table S9 for primers used | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| Atropos | Didion et al., 2017 | https://github.com/biocore/mg-scripts/blob/master/atropos_filter_parallel.sh |
| bcbio-nextgen Python toolkit v1.1.1a0-06f3c2a9 | Blue Collar Bioinformatics | https://bcbio-nextgen.readthedocs.io |
| bcl2fastq v2.20 | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Bowtie2 | Langmead and Salzberg, 2012 | https://github.com/BenLangmead/bowtie2 |
| CD-HIT | Fu et al., 2012 | http://weizhongli-lab.org/cd-hit/ |
| Cell Ranger v3.1.0 | 10X Genomic | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| DAVID v6.8 | Huang et al., 2009 | https://david.ncifcrf.gov/ |
| FinchTV v1.4 | Geospiza | https://finchtv.software.informer.com/1.4/ |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com |
| MetaCyc | Caspi et al., 2018 | https://metacyc.org/ |
| PATRIC | Wattam et al., 2017 | https://www.patricbrc.org/ |
| progressiveMauve | Darling et al., 2010 | http://darlinglab.org/mauve/user-guide/progressivemauve.html |
| QIIME2 | Bolyen et al., 2019 | https://qiime2.org/ |
| Qurro | Fedarko et al., 2020 | https://github.com/biocore/qurro |
| R package acidplots v0.2.29 | Acid Genomics | https://acidplots.acidgenomics.com/ |
| R package bcbioSingleCell v0.4.12 | Harvard Chan Bioinformatics Core | https://bioinformatics.sph.harvard.edu/bcbioSingleCell |
| R package Chromium v0.1.5 | Acid Genomics | https://chromium.acidgenomics.com |
| R package DADA2 v1.5.8 | Callahan et al., 2016 | https://github.com/benjjneb/dada2 |
| R package decontam | Davis et al., 2018 | https://github.com/benjjneb/decontam |
| R package DESeq2 v.1.26.0 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R package edgeR v3.30.3 | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| R package gplots v3.0.3 | Warnes et al., 2009 | https://github.com/talgalili/gplots |
| R package phyloseq v1.22.3 | McMurdie and Holmes, 2013 | https://github.com/joey711/phyloseq |
| R package pointillism v0.4.11 | Acid Genomics | https://pointillism.acidgenomics.com/ |
| R package Seurat v3.1.5 | Satija et al., 2015 | https://github.com/satijalab/seurat |
| Rapmap v0.5.0 | Srivastava et al., 2016 | https://github.com/COMBINE-lab/RapMap |
| RAST tool kit | Brettin et al., 2015 | http://rast.nmpdr.org |
| RSEM v1.2.28 | Li and Dewey, 2011 | https://github.com/deweylab/RSEM |
| SHOGUN | Hillmann et al., 2020 | https://github.com/knights-lab/SHOGUN |
| Songbird | Morton et al., 2019 | https://github.com/biocore/songbird |
| STAR v2.6.1 | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| umis toolkit v1.0.0 | Smith et al., 2017 | https://umi-tools.readthedocs.io/en/latest/index.html |
| Woltka | Zhu et al., 2019 | https://github.com/qiyunzhu/woltka |
| Other | ||
| 2100 Bioanalyzer | Agilent | Cat.# G2939BA |
| 4200 TapeStation System | Agilent | Cat.# G2991AA |
| Bead Ruptor 24 Homogenizer | Omni International | Cat.# 19-040 |
| C1000 Touch Thermal Cycler | Bio-Rad | Cat.#1851148 |
| CFX Connect System | Bio-Rad | Cat.# 1855201 |
| Chromium Controller | 10X Genomics | Cat.# 1000202 |
| CLARIOstar Plus Microplate Reader | BMG LABTECH | N/A |
| ddSEQ Single-Cell Isolator | Bio-Rad | Cat.# 12004336 |
| GasPak EZ Pouch System | BD | Cat.# BD 260683 |
| NextSeq 550 | Illumina | N/A |
| NovaSeq 6000 | Illumina | N/A |
| Qubit Fluorometer | Thermo Fisher Scientific | N/A |
| Standing mortar and pestle | Fisher Scientific | Cat.# 02-911-526 |
| Vinyl anaerobic chamber | Coy Lab Products | Cat.# Type A |
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Suzanne Devkota (Suzanne.Devkota@cshs.org).
Materials Availability
This study did not generate new reagents.
Data and Code Availability
All of the DNA and RNA sequencing datasets generated in this study have been deposited to NCBI. The accession number for the microbial sequencing files reported in this paper is BioProject: PRJNA659515. The accession numbers for the Bulk RNA sequencing files and scRNA-seq files are GSE156044 and GSE156776, respectively. scRNA-seq codes are available on Github (https://github.com/DevkotaLab/ha-et-al-2020-cell).
Experimental Model and Subject Details
Human subjects
Surgical resection specimens were collected from healthy ileal controls and patients with a diagnosis of Crohn’s disease or ulcerative colitis undergoing intestinal resection. Patients provided informed consent during their pre-operative visit, and the study was approved by the Cedars-Sinai Medical Center Institutional Review Board. Exclusion criteria included patients under 18, unwilling to provide informed consent, antibiotic or antifungal use in the six weeks prior to surgery, or individuals with colorectal cancer or undergoing chemoradiation therapy. Clinical characteristics and demographics of the patient cohort are detailed in Table S1.
Mice
Altered Schaedler Flora (ASF) mice were generated by colonizing germ-free mice with ASF stool (Taconic). ASF mice were kept under sterile conditions in flexible film isolators, exposed to a 14/10 h light/dark cycle and provided standard, autoclaved water and gamma irradiated mouse chow ad libitum. Animal experiment was conducted according to Cedars-Sinai Medical Center Institutional Animal Care and Use Committee guidelines. Male ASF mice at 30 weeks of age were used in this study.
Bacteria and fungi
For cultivation of oxygen-sensitive organisms, media listed in the Method Details were pre-reduced in the anaerobic chamber (90% N2, 5% CO2, 5% H2) for 48 h before inoculation. Bacterial cultures intended for in vitro assays were grown overnight at 37°C in the anaerobic chamber before use.
Primary cells
Human peripheral blood mononuclear cells (PBMCs) were freshly isolated from heparinized syringes using Ficoll density gradient centrifugation. All isolated PBMCs were cryopreserved in freezing media (CryoStor CS10, Sigma-Aldrich) and stored in liquid nitrogen until use. Revived cells were incubated at 37°C in humidified air containing 5% CO2. A fresh batch of culture media was made every 7 days. PBMCs were obtained under ethical approvals and with informed consent as described in the Human Subjects section.
Method Details
Tissue Collection
Whole blood, resected intestinal tissue, and attached mesenteric adipose were aseptically collected from healthy ileal controls via ileostomy take-down procedure, CD and UC patients undergoing bowel resection. Specimens were transferred directly from the operating room to the biosafety cabinet within 20 min of resection and transported in sterile containers. The ends of the resected bowel specimen were immediately sutured closed in the operating room to prevent any contamination from luminal contents onto the mesenteric adipose. Upon arrival to the biosafety cabinet, the specimen was rinsed in sterile PBS until all surface blood was cleared. Intestinal and adipose tissues were subsampled for i) microbial cultivation; ii) bacterial community profiling by metagenomics or 16S rRNA sequencing and iii) fungal community profiling by ITS sequencing and microbial cultivation. 0.5 g tissue was trimmed from various regions of the mesenteric adipose depot to capture any regional variability of translocation for each of the microbial experiments. 5mL of remaining adipose was reserved for stromal vascular cell isolation and for RNA isolation. We ensured these subsamples were free of blood clots and visible blood vessels. The lumen of the intestine was rinsed with sterile PBS. 0.1 g intestinal tissue was stored in TRIzol (Invitrogen) for qRT-PCR, and mucosal scrapings were collected for microbial characterization. Specimens designated for sequencing, qRT-PCR, or excess samples were stored at −80°C until use, others were processed within 2 h of surgery. Both adipose and intestinal samples were submitted for histology.
Microbial cultivation and identification
MAT and mucosal scrapings were homogenized by standing mortar and pestle (Fisher Scientific) in sterile PBS using aseptic techniques. Samples were serially diluted and plated on the following media with 1.2% agar in both aerobic and anaerobic conditions: chocolate blood (CBA;BD), Lactobacilli MRS (BD), brain heart infusion media (BHI; Sigma-Aldrich, USA), Brucella (BRU; Hardy Diagnostics) with 0.5% pyruvate, 0.5% taurine and 0.05% ammonium iron citrate, reinforced Clostridial media (RCM; BD), Bacteroides bile esculin (Anaerobe Systems) and Sabouraud dextrose (SAB; Hardy Diagnostics) with and without the addition of olive oil post-inoculation. BHI, BRU and RCM were supplemented with 5 mg/L hemin (BeanTown Chemical) and 0.5 mg/L vitamin K (Alfa Aesar). All the plates were incubated at 37°C except for SAB, which was cultured at room temperature. Distinct colony forming units were re-steaked at day 4 and 7 post-incubation. Colony PCR was performed with full length 16S or ITS primers (Key Resources Table). Amplification was carried out using the iTaq DNA polymerase kit (Bio-Rad). Amplicons were submitted to Laragen for Sanger sequencing. Sequence traces were examined in FinchTV v1.4, and the resultant trimmed reads were identified by Microbial BLAST.
Metabolic properties of bacterial isolates
Bacteria resuspended in inoculating fluid (Biolog) were added to AN MicroPlate (Biolog) with 95 distinct carbon sources as per manufacturer’s instruction. Plates were incubated in GasPak EZ anaerobic pouch system (BD) at 37°C. Growth was measured colorimetrically by microplate reader (BMG LABTECH) after 48 h incubation.
DNA extraction for 16S and ITS sequencing
DNA was extracted from mucosal scrapings and adipose tissue using the DNeasy PowerSoil Kit (QIAGEN) with additional steps to maximize cell lysis. Samples for microbiota profiling were added to lysis tubes with 400 ug proteinase K (Invitrogen) and homogenized at 5 m/s for 2 min. This was followed by heat treatment at 95°C for 15 min and centrifugation at 16,000 x g for 5 min at 4°C. Supernatant was transferred to a new tube and reserved for later use. 300 μL fresh lysis buffer was added back to the lysis tube for a second round of bead beating and heating. Supernatant from both rounds of cell lysis were pooled for DNA isolation as per manufacturer’s protocol. Tissue aliquots reserved for mycobiota profiling were first homogenized at 6 m/s for 1 min in tubes containing 2.8 mm ceramic beads (Omni), 50 mM Tris buffer (pH7.5), 1 mM EDTA and 0.2% β-mercaptoethanol. 1,000 U/mL lyticase (Sigma-Aldrich) was added to the mixture and incubated at 37°C for 30 min with gentle agitation every 5 min, followed by centrifugation at 16,000 x g for 5 min at 4°C. Cell pellet was processed in the same manner as samples for bacterial profiling. DNA extracts were then submitted to the High Throughput Sequencing and Genotyping Unit at the University of Illinois at Urbana-Champaign for bacterial 16S sequencing (V4 region) and the Genomics Core at Cedars-Sinai Medical Center for fungal ITS sequencing (ITS1 region), respectively.
16S rRNA and ITS sequence analysis
R packages were used to process and analyze 16S and ITS sequences. Paired-end reads were quality filtered, trimmed, merged, denoised, chimera filtered, and binned into sequence variants using DADA2 v1.5.8 (Callahan et al., 2016)(Callahan et al., 2016). Average number of 16S and ITS reads per sample was 5,760 and 2,369, respectively. Samples with less than 1,000 reads were removed from analysis. 16S sequence variants were aligned to the Greengenes reference database v13.8 and taxonomically assigned with a minimum bootstrap confidence level of 80. ITS sequence variants were classified using the Targeted Host Fungi ITS1 database v1.6 (Tang et al., 2015). Sequence variants unresolved for taxonomic classification and singletons were omitted from further analyses. Samples were rarefied to the minimum read count to account for uneven sampling effort. phyloseq v1.22.3 (McMurdie and Holmes, 2013) was used to assess α and β diversity measures. Bray-Curtis distance between samples were visualized by principal coordinate analysis.
DNA extraction and library preparation for deep shotgun sequencing
DNA was extracted with the QIAGEN MagAttract PowerSoil Kit following the manufacturer’s protocol with an additional 10 min incubation in 65°C water bath after vortexing with C1 solution (Marotz et al., 2017). Using the purified DNA, sequencing libraries were prepared using the HyperPlus library prep kit (KAPA Biosciences) according to the manufacturer’s instructions, and Illumina indexed adapters. These libraries were prepared with 1 ng of input DNA and 15 cycles of PCR library amplification, pooled and size selected (Sanders et al., 2019). Libraries were first characterized using the Agilent 4200 TapeStation System and its concomitant High Sensitivity DNA Kit, followed by qPCR quantification (NEBNext Library Quant Kit for Illumina; New England BioLabs). Triplicate technical replicates per biological sample were made to provide enough concentration and volume for ultra-deep sequencing; additionally, a total of four negative control blanks were processed alongside the samples during library preparation. Next, we performed paired-end 2x150bp sequencing using a S4 flow cell of a NovaSeq 6000 instrument (Illumina) across all samples and blanks, and these were equally pooled across all four lanes during sequencing.
Bioinformatic processing of shotgun sequencing samples
The NovaSeq run produced a total of 11,913,525,082 reads, of which 133,901,391 remained (1.12%) after demultiplexing, adaptor trimming, and human filtering with Atropos (Didion et al., 2017) (script at https://github.com/biocore/mg-scripts/blob/master/atropos_filter_parallel.sh). To account for two-color chemistry of the NovaSeq 6000 instrument, which can result in confident but erroneous polyG stretches in the case of no signal on the instrument, we additionally implemented polyG filtering in Atropos by using the (–nextseq-trim 30) option (Poore et al., 2020), using the (–nextseq-trim 30) option in Atropos. Further quality control filtering was implemented with Trimmomatic using the following settings: {ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:7, MINLEN:50, TRAILING:20, AVGQUAL:20, SLIDINGWINDOW:20:20} (Bolger et al., 2014). Read pairs were subsequently discarded if either mate mapped to the GRCh38.p7 human genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.33/) or the Phi X 174 viral genome using Bowtie2 and its (–fast-local) option (Langmead and Salzberg, 2012). Filtered paired and unpaired reads were then concatenated, converted to fasta format, and processed by the SHOGUN align function (Hillmann et al., 2020) and associated Web of Life phylogenetic database (Zhu et al., 2019). SHOGUN-aligned files were then utilized by Woltka for gOTU table generation and functional pathway characterization on a per-sample basis, the latter using the Woltka classify function in combination with MetaCyc protein, enzyme, reaction, and pathway information (Caspi et al., 2018). Briefly, for functional characterization, Woltka maps sequencing reads to microbial genes based on their concomitant genomic coordinates and then utilizes this mapping information toward enumerating microbial functional units (e.g., MetaCyc pathways); collectively, this permits functional profiling of the sample’s microbial constituents without relying on the presence or absence of predefined marker genes. Lastly, before further data analysis, the gOTU table was filtered to remove microbial features with less than 0.01% relative abundance, leaving 2,992 unique gOTUs out of 4,088 original gOTUs (73.2% left) found across all samples.
Decontamination and microbial taxonomic and functional analyses
Taxonomic (gOTU) counts per sample were then pooled (summated) across four sequencing lanes and subsequently stringently decontaminated using the decontam package in R (Davis et al., 2018). Putative contaminants were identified using both final library DNA concentrations (collected independently for every sample and technical replicate) and negative blank samples (that is, “method=combined” in decontam; P∗ = 0.5); in this manner, taxa were labeled as putative contaminants for either being more abundant in negative blanks than in biological samples or for having a strong negative correlation between read fraction and DNA concentration across many biological samples (Davis et al., 2018). The main two assumptions of this decontamination framework is (i) that contaminants are consistently added across samples (e.g., from technician handling or reagents) and (ii) that contaminants are overall more lowly abundant than authentic microbial constituents. Collectively, 189 taxa were removed as putative contaminants out of 2,992 total gOTUs, accounting for 13.96% of the total read count and leaving 2,803 gOTUs available for downstream analyses across all samples. After decontamination, technical replicates were pooled on a per-sample basis. After removing blanks and one healthy MAT sample that had no remaining microbial reads, leaving 23 out of 24 total patient samples, the data were saved as a biom table for later processing in QIIME 2 (Bolyen et al., 2019).
QIIME 2 was then used to calculate alpha and beta rarefaction curves in order to determine the ideal rarefaction depth for subsequent alpha and beta diversity analyses. To prevent the loss of too many samples while still capturing substantial intra- and inter-sample diversity, a rarefaction depth of 1,700 reads/sample was chosen, leaving 22 out of 24 total patient samples. Standard phylogenetic (α: Faith’s PD; β: unweighted UniFrac, weighted UniFrac) and non-phylogenetic (α: Shannon, Simpson, inverse Simpson; β: Bray-Curtis) measures of alpha and beta diversity were then determined in QIIME 2, using the Web of Life tree (Zhu et al., 2019) for phylogenetic-based calculations (Faith, 1992; Lozupone and Knight, 2005; Lozupone et al., 2011). Adonis PERMANOVA was then used to estimate differences between sample types while blocking by participant (that is, across the four samples originating from each CD patient and across the two samples originating from each healthy, non-CD subject), based on weighted and unweighted UniFrac distances (Anderson, 2001; Oksanen et al., 2015).
Next, gOTU counts were summarized at the species level and inputted into Songbird (Morton et al., 2019) for multinomial regression with the following parameters: {formula = ”C(SampleTypeAbbreviated, Treatment(‘H MAT’))+C(CaseControl, Diff, levels = [‘Control’, ‘Case’])+ParticipantID+Gender+Age,” epochs = 40000, differential-prior = 0.5, summary-interval = 1, random-seed = 42}. Note that the formula structure follows Patsy formatting (https://patsy.readthedocs.io/en/latest/), such that (i) “C(SampleTypeAbbreviated, Treatment('H MAT'))” denotes a categorical variable of Sample Type with “H MAT” (that is, healthy mesenteric adipose tissue) as the baseline reference group, (ii) “C(CaseControl, Diff, levels=['Control', 'Case'])” denotes an ordered factor where cases (defined as all tissue sections directly affected by Crohn’s disease [CrF and involved MUC samples]) are ‘worse’ than controls (defined as tissue sections not affected by Crohn’s disease [uninvolved MUC and MAT samples from CD patients, as well as H MUC and H MAT samples from healthy subjects]), (iii) and where “ParticipantID+Gender+Age” denotes two categorical (ParticipantID, Gender) covariates and one numerical (Age) covariate. To ensure proper model fit while guarding against potential model overfitting, a null model was generated using the same parameters except that {formula = ”Gender+Age”} to account for gender and age effects. Comparing this null model to the fitted model demonstrated better fit for the latter (pseudo-Q2 = 0.147237), enabling further utilization of the differentials (Morton et al., 2019). Of note, a pseudo-Q2>0 is sufficient enough to utilize the ranked differentials (Morton et al., 2019). These differentials were then inputted into Qurro for visualization (Fedarko et al., 2020), which revealed Parabacteroides merdae CAG:48 as the species most associated with “H MAT” when compared against “CD CrF” samples. In order to calculate statistical differences across samples in a compositionally coherent manner (Morton et al., 2019), the natural log-ratio of microbial abundances was calculated of samples with the numerator of the log-ratio including microbial species identified via culturing to be selectively associated with CD (Figure 3A) and the denominator being the abundance of P. merdae CAG:48, based on Songbird results associating it with H MAT samples. Then, the approximately log-normal distribution of microbial abundance ratios permitted Student t test hypothesis testing, using one-sided for tests comparing CD samples to healthy subject samples, given a priori information that the numerator contained microbial species selectively cultured in CD samples and that the denominator contained a species most associated with healthy MAT samples in the multinomial regression.
After MetaCyc pathway counts were summarized per biological sample using Woltka, as described above, samples with 0 pathway hits were dropped (7 out of 24 samples) and this resultant feature table was inputted into Songbird for multinomial regression (Morton et al., 2019). Since these pathway counts were generated on compositional microbial abundance data, it was considered necessary to treat them with compositionally coherent statistics. The Songbird parameters were: {formula = ”C(DiseaseStatus, Diff, levels = [‘Healthy’, ‘Crohns’])+Age,” min-sample-count = 10, min-feature-count = 1, differential-prior = 0.5, summary-interval = 1, epochs = 40000, random-seed = 42}. Due to dropping out a number of samples with 0 pathway hits, leaving just 17 samples on which to fit the model, a simpler parameterization was required, particularly for the formula. Again, per Patsy formatting, “C(DiseaseStatus, Diff, levels=['Healthy', 'Crohns'])” denotes an ordered factor, whereby CD samples are ‘worse’ than Healthy samples. To ensure proper fitting and guard against overfitting, a null model was generated with the same parameterization except that {formula = ”Age”}. Comparing the fitted model to the null model resulted in a pseudo-Q2 = 0.057106, which, being greater than 0, enabled downstream utilization of the differentials. Using Qurro to visualize the differentials (Fedarko et al., 2020), the top 10 ranked pathway features associated with Crohn’s disease status and the top 10 ranked pathway features associated with Healthy status were selected for further log-ratio analysis (Figure 2C). Of note, the Songbird-generated differentials do not impact the subsequent log-ratio calculation, which relies on the actual pathway abundances among samples; additionally, any samples having 0 hits among these 20 pathways would not appear in the concomitant log-ratio analysis, as the logarithm of 0 is undefined. For statistical testing, the pathways associated with CD status were used for the numerator and those associated with Healthy status for the denominator; moreover, since there is no a priori information suggesting that these pathways are inherently implicated in either status, a two-sided t test was used to compare them.
Whole genome sequencing and analysis
C. innocuum isolates were grown on pre-reduced chocolate blood agar at 37°C for 36 h. Genomic content was extracted from scraped colonies using the DNeasy PowerSoil Kit (QIAGEN). Purified DNA was sent to the Microbial Genome Sequencing Center at University of Pittsburgh for library preparation (Baym et al., 2015), followed by whole genome sequencing on the Illumina NextSeq 550 flow cell. Paired-end reads were assembled by the PATRIC pipeline (Wattam et al., 2017), which involved preprocessing by BayesHammer (Nikolenko et al., 2013), assemblers Velvet (Zerbino and Birney, 2008), SPAdes (Bankevich et al., 2012) and IDBA (Peng et al., 2012) on reads followed by ARAST quality evaluation procedure. After assembly, genomes were annotated using the RAST tool kit (Brettin et al., 2015). Patient-derived C. innocuum genomes were compared against two reference stains, including type strain C. innocuum DSM 1286 and C. innocuum 2959 (from the HMP). Phylogenetic relatedness of the C. innocuum genomes was determined by progressiveMauve (Darling et al., 2004; Darling et al., 2010). To determine the gene presence across C. innocuum genomes, coding genes were binned on the basis of their protein sequences using CD-HIT (Li et al., 2001) with 80% identity threshold. Genes were identified as core genes if they were shared by all the genomes in comparison. Coding genes were also annotated with KEGG orthologous groups (KOs) by mapping them to the KEGG database (version 54) (Kanehisa et al., 2016) using BLAST. The pathway abundance in the genome was calculated by summing all the genes involved in each pathway. For genes involved in multiple pathways, they were split evenly among the involved ones to avoid over-counting. Statistical analysis was performed to calculate the differences in pathways among clinical categories using the Kruskal-Wallis test.
Metabolic properties of bacterial isolates
Freshly cultured bacteria were first resuspended in inoculating fluid (Biolog) and then added to AN MicroPlate (Biolog) with 95 distinct carbon sources as per manufacturer’s instruction. Plates were incubated in GasPak EZ anaerobic pouch system (BD) at 37°C. Growth was measured colorimetrically by microplate reader (BMG LABTECH) after 24 h incubation. Data shown here is the average of two independent runs.
Plasma protein assays
Whole blood collected at the time of surgery were separated for plasma by centrifugation at 1,200 x g for 10 min at 4°C. Plasma at two different dilutions were used for ELISA quantification of lipopolysaccharide-binding protein (1:1,000; Hycult Biotech) and soluble CD14 (1:500 dilution; R&D Systems). Assays were carried out as per manufacturer’s instructions and measured colorimetrically by a microplate reader (BMG LABTECH).
Gnotobiotic animal study
ASF mice were either untreated and served as the control cohort, or they were gavaged with a single dose of 1 × 109 live C. innocuum cells. Stool was collected and cultivated daily to ensure colonization of C. innocuum in the treatment group. At day 4, a subset of gavaged mice was supplemented with 3% DSS (MP Biomedicals) in their drinking water for 7 days to induce intestinal barrier disruption, followed by 3 days of recovery on regular water. Body weight was monitored daily for all animals. Stool, plasma, mesenteric adipose tissue, and colon were collected aseptically at day 14, and stored at −80°C for downstream analysis. A portion of the mesenteric adipose tissue was homogenized in the anaerobic chamber immediately, and serially plated on pre-reduced chocolate blood agar.
Bulk RNA sequencing of human mesenteric adipose
RNA extraction was performed using aliquots of 50mg tissue, and up to 250 mg tissue was processed from each specimen. E.Z.N.A.® Total RNA Kit II (Omega Bio-tek) was used to isolate total RNA. Some modifications were made due to the high-lipid content of the specimens. Chloroform-isoamyl (24:1) instead of plain chloroform was used to separate the organic and aqueous phases, and an extra wash step was performed with wash buffer II. Aliquots of extracted RNA from the same specimen were pooled and further purified using the E.Z.N.A.® MicroElute RNA Clean Up Kit (Omega Bio-tek). Library construction was performed using the TruSeq Stranded Total RNA library preparation kit (Illumina). Briefly, total RNA samples were assessed for concentration using a Qubit fluorometer (Thermo Fisher Scientific) and for quality using the 2100 Bioanalyzer (Agilent Technologies). Ribosomal RNA depletion on 240 ng total RNA was performed using QIAseq FastSelect rRNA Removal Kit (QIAGEN). cDNA was synthesized from enriched and fragmented RNA using reverse transcriptase (Invitrogen) and random primers. The cDNA was further converted into double-stranded DNA (dsDNA), and the resulting dsDNA was enriched with PCR for library preparation. The PCR-amplified library was purified using Agencourt AMPure XP beads (Beckman Coulter). The concentration of the amplified library was measured with a Qubit fluorometer and an aliquot of the library was resolved on a Bioanalyzer. Sample libraries were multiplexed and sequenced on a NovaSeq 6000 platform (Illumina) using 75bp single-end sequencing. More than 50 million reads were generated per sample.
Raw sequencing data was demultiplexed and converted to fastq format by using bcl2fastq v2.20 (Illumina). Reads were then aligned to the transcriptome using STAR (version 2.6.1) (Dobin et al., 2013) / RSEM (version 1.2.28) (Li and Dewey, 2011) with default parameters, using a custom human GRCh38 transcriptome reference (http://www.gencodegenes.org), containing all protein coding and long non-coding RNA genes based on human GENCODE version 33 annotation. Expression counts for each gene in all samples were normalized by a modified trimmed mean of the M-values normalization method and the unsupervised principal component analysis was performed with DESeq2 (version 1.26.0) (Love et al., 2014). Each gene was fitted into a negative binomial generalized linear model, and the Wald test was applied to assess the differential expressions between CrF and healthy ileal controls by DESeq2. Benjamini and Hochberg procedure was applied to adjust for multiple hypothesis testing, and differential expression gene candidates were selected with a false discovery rate less than 0.05. The pathway enrichment analysis was performed on these candidates with DAVID v6.8 (Huang et al., 2009). For visualization of coordinated gene expression in samples, a two-way hierarchical clustering with Pearson correlation distance matrix was performed with samples and differentially expressed gene candidates using the g-plots package (version 3.0.3) in R.
Stromal vascular cell (SVC) isolation from adipose tissue for single-cell RNA sequencing (scRNA-seq)
Adipose tissue was minced into 3 mm pieces and subjected to collagenase digestion for 50 min at 37°C with continuous rotation. Collagenase buffer consisted of 1X PBS containing calcium and magnesium, 2% bovine serum albumin (BSA; MP Biomedicals), 0.2 mg/mL DNase I (Sigma-Aldrich), and 1mg/mL collagenase II (Invitrogen). Following digestion, cells were incubated with 0.01 M EDTA for an additional 10 min then filtered through a 100 μM cell strainer (Fisher Scientific) and centrifuged at 1500 rpm for 5 min. The pellet was resuspended in 1X RBC Lysis Buffer (Invitrogen) as per manufacturer’s instruction. The samples were centrifuged as above, and the final pellet was converted to a single-cell suspension and frozen for later analysis such that all samples could be run at the same time. The cell-freezing protocol for scRNA-seq was developed in conjunction with Bio-Rad Genomics. Briefly, the cell pellet was thoroughly mixed by pipetting up and down 10 times. Cells were counted a total of 4 times for each cell preparation to ensure accuracy of total cell count and viability. Aliquots of 5∗10∧6 cells/mL were prepared in chilled cryopreservation medium (DMEM + 20% FBS + 10% DMSO) and placed in a 4°C pre-chilled CoolCell FTS30 (Corning) and placed in a −80C freezer for at least 4 h. After 4 h the cryovials were transferred to liquid nitrogen for long-term storage.
Single-cell RNA sequencing library preparation
CrF versus H MAT
Single-cell libraries of the dissociated SVCs were prepared using the Chromium Single Cell 3ʹ GEM, Library & Gel Bead Kit v3 (10X Genomics) and the Chromium Chip B Single Cell Kit protocols, aiming for recovery of 1200 cells for each sample. Briefly, cells were individually partitioned and encapsulated into subnanoliter oil droplets in the Chromium Controller instrument for cell lysis and barcoded reverse transcription of mRNA, followed by amplification, shearing and Illumina library construction. Single-cell-barcoded cDNA libraries were sequenced on the Illumina NextSeq platform.
CrF versus CD MAT and UC MAT
Dissociated SVCs were first diluted to 2500 cells/uL in PBS with 0.1% BSA. Cells were individually partitioned and co-encapsulated with barcodes into subnanoliter oil droplets using the ddSEQ single-cell isolator (Bio-Rad) as per manufacturer’s instructions in the Illumina Bio-Rad SureCell WTA 3¢ Library Prep Kit. Following cell isolation, the droplets were transferred to a 96-well PCR plate for cell lysis, barcoding, reverse transcription using a thermal cycler. The droplet emulsion was then disrupted for generation of second strand cDNA, followed by fragmentation, tagging and amplification of cDNA. Single-cell-barcoded cDNA libraries were sequenced on the Illumina NextSeq platform at an average read depth of 175,000 reads/sample.
Single-cell RNA sequencing analysis
10X Genomics Cell Ranger sample processing
Cell counts were generated using the 10X Genomics Cell Ranger v3.1.0 pipeline against the GRCh38 reference genome (10X genome version 3.0.0; Ensembl release 93, July 2018; http://jul2018.archive.ensembl.org). The filtered count matrices generated by Cell Ranger were used as input. The Chromium R package v0.1.5 was used to perform additional cell quality control analysis and filtering prior to clustering. Similar to the approach used for the SureCell samples, UMIs per cell, genes per cell, and mitochondrial ratio cutoffs were applied to remove additional low quality cells from analysis.
Bio-Rad SureCell sample processing
Cell counts were generated with the bcbio-nextgen Python toolkit v1.1.1a0-06f3c2a9. Reads were assigned per cell via the cellular barcodes, and per gene via the UMIs, using the umis toolkit v1.0.0 [@Svensson2017-hp]. Reads were quasi-mapped to the Ensembl GRCh38 transcriptome (Release 90, August 2017; https://aug2017.archive.ensembl.org) using Rapmap v0.5.0 [@Srivastava2016-tm]. Only cells containing at least 1,000 reads were analyzed. The bcbioSingleCell R package v0.4.12 was used to perform cell quality control analysis and filtering prior to clustering. The distributions of reads per cell, UMIs per cell, genes per cell, and mitochondrial ratio per cell were used to remove low quality cells from analysis. Analysis was performed on 1,200 filtered cells per specimen, analyzed at a read depth of 175,000 reads/cell, with ∼200 unique genes per cell.
Clustering analysis
Clustering analysis was performed with the Seurat R package v3.1.5. [@Satija2015-rr]. Counts were log normalized and scaled per cell to account for variations in sequencing depth. Linear dimensional reduction was performed using PCA on the most variable genes detected; these are determined via binned z-scores based on the average expression and dispersion for each gene (Satija et al., 2015). Non-linear dimensional reduction was performed using t-SNE [@Maaten2008-qy] and UMAP [@McInnes2018-aa, @Becht2018-jn]. We defined cell cluster specific marker genes using the Findmarkers function in Seurat across all samples using a Wilcoxon rank sum test. Differential expression analysis was performed using the edgeR package v3.30.3 [@Robinson2010-eu]. Plots were generated using the acidplots v0.2.29 and pointillism v0.4.11 R packages. GSEA analysis was run as described in (Korotkovich et al., 2019). Code is available at: https://github.com/DevkotaLab/ha-et-al-2020-cell
Immunogenicity assays with monocyte-derived macrophages
Macrophages were generated from peripheral blood monocytes isolated from healthy donors without any diagnosed GI disorders or on antibiotics in the six weeks prior to blood draw. Blood fractions were first separated by density gradient using Ficoll-Paque Premium (GE Healthcare). Buffy coat was collected, and platelets were discarded by 3 washing steps in PBS. Monocytes were negatively selected using EasySep Human Monocyte Enrichment Kit (STEMCELL Technologies). Monocytes were seeded in RPMI with 10% FBS, 4 mM L-Glutamine, 100 I.U./mL penicillin and streptomycin in tissue culture treated plates for 7 days in the presence of 50 ng/mL M-CSF (PeproTech). Macrophages were incubated with 25μg/mL lysate from bacterial isolates for 24 h. For M1 and M2 polarization, macrophages were stimulated with 50 ng/mL LPS (Sigma-Aldrich) and 25 ng/mL IL-4 (PeproTech), respectively. Phase contrast images of macrophages were acquired on an inverted microscope (Echo Labs). Supernatant was reserved for further co-culture experiments or quantification of IL-1β by ELISA (R&D Systems). Macrophages were lysed and stored in TRIzol (Invitrogen) for RT-qPCR.
Co-culture assays involving adipose-derived stem cells (ADSCs) and fibroblasts
Dissociated SVCs were cultured in DMEM F-12 (Corning) supplemented with 10% FBS (Omega) and 1% penicillin/streptomycin (Sigma-Aldrich, Munich, Germany) (Ge et al., 2016). After 24 h cell culturing, non-adherent cells were removed and adherent progenitor cells were washed with PBS. Progenitor cells were cultured in fresh media until 70% confluence. At day 7, cells were co-cultured with 25μg/mL bacterial lysates or supplemented with 20% macrophage supernatant (collected after bacterial stimulation) from CD patients. Cells were lysed after 36 h incubation and stored in TRIzol (Invitrogen) for qRT-PCR.
Quantitative real-time PCR
Total RNA was extracted from adipose tissue, intestinal tissue and cell culture homogenate in TRIzol (Invitrogen), and purified using the RNeasy Mini Kit (QIAGEN). The iScript cDNA Synthesis kit (Bio-Rad) was used to reverse transcribe RNA for qRT-PCR. SYBR Green Supermix (Bio-Rad) and the CFX Connect System were used to detect amplification of target genes (Key Resources Table), using the following protocol: 95°C for 3 min; 40 cycles of 95°C for 15 s and 58°C for 40 s. Relative expression of target genes was normalized to GAPDH, ACTB, RPL37A or Hprt1 and quantified by 2ΔΔCT.
Quantification and Statistical Analysis
Statistical analysis was performed using GraphPad Prism software v 8. Data were assessed for normal distribution and plotted in the figures as mean ± SEM. For each figure, n = the number of subjects. Two-sided Mann-Whitney U test, paired Wilcoxon signed rank test, Kruskal-Wallis test, t test and PERMANOVA were used for metagenomic analysis in Figures 2 and S1. One-way ANOVA with Tukey’s post hoc test was used for taxonomic comparisons from ITS sequencing analysis in Figure 2, expression of tight junction genes and gnotobiotic mouse experiment in Figure 4, and co-culture assays in Figure 7. Significant differences emerging from the above tests are indicated in the figures by ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Acknowledgments
We would like to thank all of the patients who consented to donating their tissues for our study. We also thank the clinical coordinators and surgical staff who assisted with procurement and collection of these samples, and the MIRIAD Biobank for providing consenting and patient database oversight. The authors give thanks to Jongsoon Lee, Philip Scherer, and Jeffrey F. Miller for their advisement on aspects of this project. This study was funded by the Leona M. and Harry B. Helmsley Charitable Trust (G-2019PG-CD012) and the National Institutes of Health (R01DK123446). The MIRIAD Biobank is supported by U01DK062413. D.P.B.M. is supported by P01DK046763. G.D.S.-P. is supported by 1F30CA243480-01A1.
Author Contributions
Conceptualization, C.W.Y.H., P.F., and S.D.; Investigation, C.W.Y.H., A.M., K.G., G.H., K.S., G.H., J.R.C., C.A., and J.E.M.; Formal Analysis, C.W.Y.H., S.D., G.D.S.-P., B.S., Y.W., K.K., and M.J.S.; Methodology, C.W.Y.H., S.D., G.D.S.-P., K.K., H.L., M.J.S., and R.K.; Resources, P.F., D.U., and D.P.B.M.; Visualization, C.W.Y.H., S.D., G.D.S.-P., B.S., Y.W., M.J.S.,Y.R, and K.S.L.C.; Supervision, S.D., S.J.H.S., S.K., H.L., and R.K.; Data Curation, S.Y., M.J.S., and S.D.; Writing – Original Draft, C.W.Y.H. and S.D.; Writing – Review & Editing, C.W.Y.H, A.M., G.D.S.-P., B.S., Y.W., K.G., G.H., K.S., G.H., J.C., C.A., J.M., S.Y., S.J.H.S., D.U., M.B., S.K., K.K., H.L., M.J.S., D.P.B.M., R.K., P.F., and S.D.; Funding acquisition, S.D.
Declaration of Interests
S.D. and C.W.Y.H. are inventors on US patent application #62/679,624.
Published: September 28, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.09.009.
Supplemental Information
Table S1: CD and UC patient metadata (related to Figure 1). Table S2: Metagenomic read counts per sample (related to Figure 2). Table S3: Adonis PERMANOVA of beta diversity measures from metagenomic sequencing identifies sample type (tissue source—MAT or mucosa and CD or H) and participant ID (inter-individual differences) as most significant parameters defining bacterial diversity (related to Figure 2). Table S4: Bacterial isolates recovered from CD and UC mesenteric adipose (related to Figure 3). Table S9: Primers used in the study (related to Key Resources Table in STAR Methods).
Whole Genome Analysis of Clostridium innocuum Isolates, Related to Figure 3
Bulk RNA Sequencing of Mesenteric Adipose Tissue, Related to Figures 5 and S4
Single-Cell RNA Sequencing Gene List, CD CrF + H MAT, Related to Figure 5
Single-Cell RNA Sequencing Gene List, CD CrF + CD MAT, Related to Figure 5
References
- Amar J., Chabo C., Waget A., Klopp P., Vachoux C., Bermúdez-Humarán L.G., Smirnova N., Bergé M., Sulpice T., Lahtinen S. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011;3:559–572. doi: 10.1002/emmm.201100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J., Pan G., Poncz M., Wei J., Ran M., Zhou Z. Serpin functions in host-pathogen interactions. PeerJ. 2018;6:e4557. doi: 10.7717/peerj.4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M., Kryazhimskiy S., Lieberman T.D., Chung H., Desai M.M., Kishony R. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS ONE. 2015;10:e0128036. doi: 10.1371/journal.pone.0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettin T., Davis J.J., Disz T., Edwards R.A., Gerdes S., Olsen G.J., Olson R., Overbeek R., Parrello B., Pusch G.D. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015;5:8365. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J., Holmes S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell B.J., Yu L.G., Rhodes J.M. Altered glycosylation in inflammatory bowel disease: a possible role in cancer development. Glycoconj. J. 2001;18:851–858. doi: 10.1023/a:1022240107040. [DOI] [PubMed] [Google Scholar]
- Caspi R., Billington R., Fulcher C.A., Keseler I.M., Kothari A., Krummenacker M., Latendresse M., Midford P.E., Ong Q., Ong W.K. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018;46(D1):D633–D639. doi: 10.1093/nar/gkx935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthorn W.P., Scheller E.L., MacDougald O.A. Adipose tissue stem cells meet preadipocyte commitment: going back to the future. J. Lipid Res. 2012;53:227–246. doi: 10.1194/jlr.R021089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Leong R.W., Wasinger V.C., Ip M., Yang M., Phan T.G. Impaired Intestinal Permeability Contributes to Ongoing Bowel Symptoms in Patients With Inflammatory Bowel Disease and Mucosal Healing. Gastroenterology. 2017;153:723–731.e1. doi: 10.1053/j.gastro.2017.05.056. [DOI] [PubMed] [Google Scholar]
- Chia J.H., Feng Y., Su L.H., Wu T.L., Chen C.L., Liang Y.H., Chiu C.H. Clostridium innocuum is a significant vancomycin-resistant pathogen for extraintestinal clostridial infection. Clin. Microbiol. Infect. 2017;23:560–566. doi: 10.1016/j.cmi.2017.02.025. [DOI] [PubMed] [Google Scholar]
- Crewe C., An Y.A., Scherer P.E. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J. Clin. Invest. 2017;127:74–82. doi: 10.1172/JCI88883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crohn B.B., Ginzburg L., Oppenheimer G.D. Regional ileitis; a pathologic and clinical entity. J. Am. Med. Assoc. 1932;99:1323–1329. [Google Scholar]
- Darling A.C., Mau B., Blattner F.R., Perna N.T. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A.E., Mau B., Perna N.T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P., Lahiri A., Lahiri A., Chakravortty D. Modulation of the arginase pathway in the context of microbial pathogenesis: a metabolic enzyme moonlighting as an immune modulator. PLoS Pathog. 2010;6:e1000899. doi: 10.1371/journal.ppat.1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis N.M., Proctor D.M., Holmes S.P., Relman D.A., Callahan B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didion J.P., Martin M., Collins F.S. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ. 2017;5:e3720. doi: 10.7717/peerj.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992;61:1–10. [Google Scholar]
- Fedarko M.W., Martino C., Morton J.T., González A., Rahman G., Marotz C.A., Minich J.J., Allen E.E., Knight R. Visualizing ‘omic feature rankings and log-ratios using Qurro. NAR Genom. Bioinform. 2020;2 doi: 10.1093/nargab/lqaa023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X., Leow S., Sathiakumar D., Stünkel W., Shabbir A., So J., Lomanto D., McFarlane C. Isolation and Culture of Human Adipose-derived Stem Cells from Subcutaneous and Visceral White Adipose Tissue Compartments. Bio Protoc. 2016;6 doi: 10.21769/BioProtoc.2027. https://bio-protocol.org/e2027 [DOI] [Google Scholar]
- Hillmann B., Al-Ghalith G.A., Shields-Cutler R.R., Zhu Q., Knight R., Knights D. SHOGUN: a modular, accurate and scalable framework for microbiome quantification. Bioinformatics. 2020;36:4088–4090. doi: 10.1093/bioinformatics/btaa277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huh J.Y., Park Y.J., Ham M., Kim J.B. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol. Cells. 2014;37:365–371. doi: 10.14348/molcells.2014.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C., Gevers D., Knight R., Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S., Stange E.F., Wehkamp J. Inflammatory bowel disease: an impaired barrier disease. Langenbecks Arch. Surg. 2013;398:1–12. doi: 10.1007/s00423-012-1030-9. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44(D1):D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw E.E., Flier J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- Knights D., Kuczynski J., Charlson E.S., Zaneveld J., Mozer M.C., Collman R.G., Bushman F.D., Knight R., Kelley S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods. 2011;8:761–763. doi: 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkovich G., Sukhov V., Sergushichev A. Fast Gene Set Enrichment Analysis. bioRxiv. 2019 doi: 10.1101/060012. [DOI] [Google Scholar]
- Kredel L.I., Siegmund B. Adipose-tissue and intestinal inflammation - visceral obesity and creeping fat. Front. Immunol. 2014;5:462. doi: 10.3389/fimmu.2014.00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Jaroszewski L., Godzik A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics. 2001;17:282–283. doi: 10.1093/bioinformatics/17.3.282. [DOI] [PubMed] [Google Scholar]
- Limon J.J., Tang J., Li D., Wolf A.J., Michelsen K.S., Funari V., Gargus M., Nguyen C., Sharma P., Maymi V.I. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe. 2019;25:377–388.e6. doi: 10.1016/j.chom.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J., Arze C., Ananthakrishnan A.N., Schirmer M., Avila-Pacheco J., Poon T.W., Andrews E., Ajami N.J., Bonham K.S., Brislawn C.J., IBDMDB Investigators Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C., Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C., Lladser M.E., Knights D., Stombaugh J., Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankertz J., Schulzke J.D. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 2007;23:379–383. doi: 10.1097/MOG.0b013e32816aa392. [DOI] [PubMed] [Google Scholar]
- Marotz C., Amir A., Humphrey G., Gaffney J., Gogul G., Knight R. DNA extraction for streamlined metagenomics of diverse environmental samples. Biotechniques. 2017;62:290–293. doi: 10.2144/000114559. [DOI] [PubMed] [Google Scholar]
- Mattacks C.A., Sadler D., Pond C.M. The cellular structure and lipid/protein composition of adipose tissue surrounding chronically stimulated lymph nodes in rats. J. Anat. 2003;202:551–561. doi: 10.1046/j.1469-7580.2003.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick J.S. Type IV pili and twitching motility. Annu. Rev. Microbiol. 2002;56:289–314. doi: 10.1146/annurev.micro.56.012302.160938. [DOI] [PubMed] [Google Scholar]
- McMurdie P.J., Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter F.Y., Wang T., Nguyen P., Chung T., Liu W.F. Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. USA. 2013;110:17253–17258. doi: 10.1073/pnas.1308887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton J.T., Marotz C., Washburne A., Silverman J., Zaramela L.S., Edlund A., Zengler K., Knight R. Establishing microbial composition measurement standards with reference frames. Nat. Commun. 2019;10:2719. doi: 10.1038/s41467-019-10656-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto V., Kroken A.R., Grosser M.R., Smith B.E., Metruccio M.M.E., Hagan P., Hallsten M.E., Evans D.J., Fleiszig S.M.J. Type IV Pili Can Mediate Bacterial Motility within Epithelial Cells. MBio. 2019;10 doi: 10.1128/mBio.02880-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolenko S.I., Korobeynikov A.I., Alekseyev M.A. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics. 2013;14(Suppl 1):S7. doi: 10.1186/1471-2164-14-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J., Blanchet F.G., Kindt R., Legendre P., Minchin P., O’Hara B., Simpson G., Solymos P., Stevens H., Wagner H. Vegan: Community Ecology Package. R Package Version 22-1 2, 1-2. 2015 [Google Scholar]
- Peng Y., Leung H., Yiu S., Chin F. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- Poore G.D., Kopylova E., Zhu Q., Carpenter C., Fraraccio S., Wandro S., Kosciolek T., Janssen S., Metcalf J., Song S.J. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020;579:567–574. doi: 10.1038/s41586-020-2095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes J.M. Unifying hypothesis for inflammatory bowel disease and associated colon cancer: sticking the pieces together with sugar. Lancet. 1996;347:40–44. doi: 10.1016/s0140-6736(96)91563-9. [DOI] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samad T., Billings N., Birjiniuk A., Crouzier T., Doyle P.S., Ribbeck K. Swimming bacteria promote dispersal of non-motile staphylococcal species. ISME J. 2017;11:1933–1937. doi: 10.1038/ismej.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders J.G., Nurk S., Salido R.A., Minich J., Xu Z.Z., Zhu Q., Martino C., Fedarko M., Arthur T.D., Chen F. Optimizing sequencing protocols for leaderboard metagenomics by combining long and short reads. Genome Biol. 2019;20:226. doi: 10.1186/s13059-019-1834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M., Smeekens S.P., Vlamakis H., Jaeger M., Oosting M., Franzosa E.A., Ter Horst R., Jansen T., Jacobs L., Bonder M.J. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell. 2016;167:1125–1136.e8. doi: 10.1016/j.cell.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seksik P., Rigottier-Gois L., Gramet G., Sutren M., Pochart P., Marteau P., Jian R., Doré J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut. 2003;52:237–242. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan A.L., Warren B.F., Gear M.W., Shepherd N.A. Fat-wrapping in Crohn’s disease: pathological basis and relevance to surgical practice. Br. J. Surg. 1992;79:955–958. doi: 10.1002/bjs.1800790934. [DOI] [PubMed] [Google Scholar]
- Smith L.D., King E. Clostridium innocuum, sp. n., a sporeforming anaerobe isolated from human infections. J. Bacteriol. 1962;83:938–939. doi: 10.1128/jb.83.4.938-939.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T., Heger A., Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A., Sarkar H., Gupta N., Patro R. RapMap: a rapid, sensitive and accurate tool for mapping RNA-seq reads to transcriptomes. Bioinformatics. 2016;32:i192–i200. doi: 10.1093/bioinformatics/btw277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J., Iliev I.D., Brown J., Underhill D.M., Funari V.A. Mycobiome: Approaches to analysis of intestinal fungi. J. Immunol. Methods. 2015;421:112–121. doi: 10.1016/j.jim.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Ishibashi J., Trefely S., Shao M., Cowan A.J., Sakers A., Lim H.W., O’Connor S., Doan M.T., Cohen P. A PRDM16-Driven Metabolic Signal from Adipocytes Regulates Precursor Cell Fate. Cell Metab. 2019;30:174–189.e5. doi: 10.1016/j.cmet.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes G.R., Bolker B., Bonebakker L., Gentleman R., Huber W., Liaw A., Lumley T., Maechler M., Magnusson A., Moeller S. gplots: Various R programming tools for plotting data. R package version 2, 1. 2009 [Google Scholar]
- Wattam A.R., Davis J.J., Assaf R., Boisvert S., Brettin T., Bun C., Conrad N., Dietrich E.M., Disz T., Gabbard J.L. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017;45(D1):D535–D542. doi: 10.1093/nar/gkw1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernstedt Asterholm I., Tao C., Morley T.S., Wang Q.A., Delgado-Lopez F., Wang Z.V., Scherer P.E. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20:103–118. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampell J.C., Aschen S., Weitman E.S., Yan A., Elhadad S., De Brot M., Mehrara B.J. Regulation of adipogenesis by lymphatic fluid stasis: part I. Adipogenesis, fibrosis, and inflammation. Plast. Reconstr. Surg. 2012;129:825–834. doi: 10.1097/PRS.0b013e3182450b2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D.R., Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q., Mai U., Pfeiffer W., Janssen S., Asnicar F., Sanders J.G., Belda-Ferre P., Al-Ghalith G.A., Kopylova E., McDonald D. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat. Commun. 2019;10:5477. doi: 10.1038/s41467-019-13443-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulian A., Cancello R., Ruocco C., Gentilini D., Di Blasio A.M., Danelli P., Micheletto G., Cesana E., Invitti C. Differences in visceral fat and fat bacterial colonization between ulcerative colitis and Crohn’s disease. An in vivo and in vitro study. PLoS ONE. 2013;8:e78495. doi: 10.1371/journal.pone.0078495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: CD and UC patient metadata (related to Figure 1). Table S2: Metagenomic read counts per sample (related to Figure 2). Table S3: Adonis PERMANOVA of beta diversity measures from metagenomic sequencing identifies sample type (tissue source—MAT or mucosa and CD or H) and participant ID (inter-individual differences) as most significant parameters defining bacterial diversity (related to Figure 2). Table S4: Bacterial isolates recovered from CD and UC mesenteric adipose (related to Figure 3). Table S9: Primers used in the study (related to Key Resources Table in STAR Methods).
Whole Genome Analysis of Clostridium innocuum Isolates, Related to Figure 3
Bulk RNA Sequencing of Mesenteric Adipose Tissue, Related to Figures 5 and S4
Single-Cell RNA Sequencing Gene List, CD CrF + H MAT, Related to Figure 5
Single-Cell RNA Sequencing Gene List, CD CrF + CD MAT, Related to Figure 5
Data Availability Statement
All of the DNA and RNA sequencing datasets generated in this study have been deposited to NCBI. The accession number for the microbial sequencing files reported in this paper is BioProject: PRJNA659515. The accession numbers for the Bulk RNA sequencing files and scRNA-seq files are GSE156044 and GSE156776, respectively. scRNA-seq codes are available on Github (https://github.com/DevkotaLab/ha-et-al-2020-cell).