

Summary
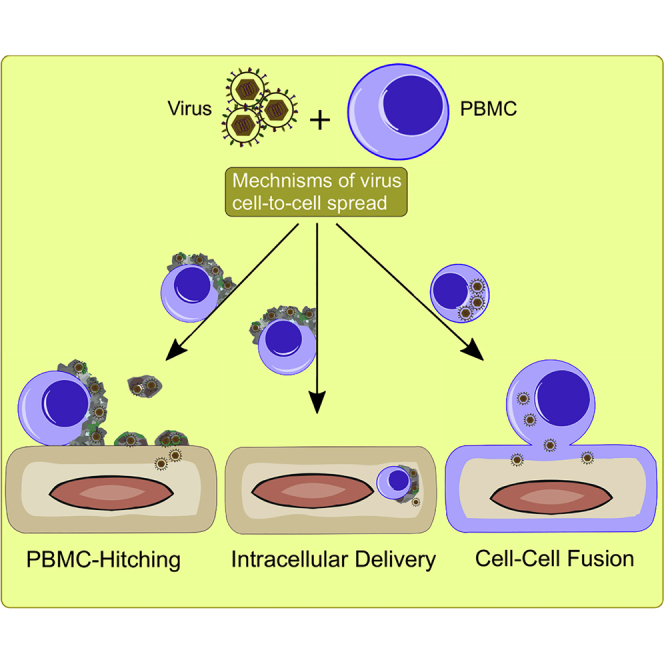
Mononuclear cells are the first line of defense against microbial infection. Yet, several viruses have evolved different mechanisms to overcome host defenses to ensure their spread. Here, we show unique mechanisms of how equid herpesvirus-1 manipulates peripheral blood mononuclear cells (PBMC) to travel further in the body. (1) “PBMC-hitching”: at the initial contact, herpesviruses lurk in the extracellular matrix (ECM) of PBMC without entering the cells. The virus exploits the components of the ECM to bind, transport, and then egress to infect other cells. (2) “Intracellular delivery”: transendothelial migration is a physiological mechanism where mononuclear cells can transmigrate through the endothelial cells. The virus was intangible and probably did not interfere with such a mechanism where the infected PBMC can probably deliver the virus inside the endothelium. (3) “Classical-fusion”: this process is well mastered by herpesviruses due to a set of envelope glycoproteins that facilitate cell-cell fusion and virus spread.
Subject Areas: Virology, Cell Biology
Graphical Abstract
Highlights
-
•
Several viral proteins facilitate rapid and efficient virus cell-to-cell spread
-
•
Viruses can evade face-off engagement with host defenses via cell-to-cell spread
-
•
The ECM of PBMC ensures safe transmission of viruses to target cells
-
•
Viruses can be transmitted during a transendothelial migration process of PBMC
Virology; Cell Biology
Introduction
Cell-to-cell transmission is one of the strategies adopted by several viruses to overcome host barriers and to establish successful infection (Zhong et al., 2013). Several advantages have been documented for this mode of transmission that center mainly on avoiding face-off engagement with host defenses (Gupta et al., 1989; Mothes et al., 2010; Sattentau, 2008). Viruses have evolved several mechanisms, including cell-cell fusion, synapse formation, and induction of actin tails, filopodial bridges, or nanotubes, and are equipped with the necessary information to facilitate a secure and rapid cell-to-cell dissemination (Aubert et al., 2009; Bennett et al., 2009; Sherer et al., 2007; Sowinski et al., 2008). Other modes of cell-to-cell transmission involve the utilization of the carbohydrate-rich extracellular matrix (ECM) (Mothes et al., 2010). The ability of viruses to utilize and hijack cell-cell contact machineries contributes to the success of viral infection and spreading.
The ECM is a complex three-dimensional structure present in all tissues that is generally composed of well-organized networks of macromolecules (including polysaccharides and proteins) with distinct biochemical and biomechanical properties (Stavolone and Lionetti, 2017). Moreover, besides providing a physical support for tissue integrity and elasticity, the ECM contributes functionally to the regulation of cell growth, survival, motility, intercellular adhesion and communication, as well as differentiation (Bonnans et al., 2014; Stavolone and Lionetti, 2017). The ECM is a dynamic structure that is constantly remodeled and specifically tuned in response to different physiological cell conditions as well as in response to diseases and infections (Bonnans et al., 2014; Humphrey et al., 2014). Although ECM represents a formidable barrier against pathogen invasion, many viruses have evolved diverse mechanisms to overcome and even exploit the ECM for entry and spread (Akhtar and Shukla, 2009; DiGiuseppe et al., 2017; Dimitrov, 2004). Human T cell leukemia virus type 1 (HTLV-1) was shown to infect T lymphocytes and then bud into the ECM, which carries the virus through virus-induced ECM components allowing virus spread and infection of other T lymphocytes (Pais-Correia et al., 2010).
Infection of peripheral blood mononuclear cells (PBMC) plays a key role in disease outcomes because PBMC have access to all body tissues. Although non-adherent, PBMC can adhere to endothelial cells (EC) after stimulation, as is presumably the case after certain viral infections, where viruses can attach to, enter, and replicate in EC (Keller et al., 2003; Valbuena and Walker, 2006). The PBMC-EC interface thus represents a crucial spot that can influence virus pathogenesis and disease outcomes. Infection of EC through infected PBMC has been documented for members of several virus families, including bunya, herpes, filo, flavi, toga, and arena viruses (Charrel and de Lamballerie, 2010; Chu et al., 2016; Gowen and Hickerson, 2017; Kunz and de la Torre, 2017; Ma et al., 2013; Mackow and Gavrilovskaya, 2009). However, the mechanism of how these viruses spread from infected PBMC to EC is only poorly understood. Disease outcomes of these viral infections are mostly fatal (to humans and animals) due to hemorrhagic fevers, severe respiratory diseases, as well as neurological and/or congenital disorders.
Herpesviruses represent a large group of DNA viruses that infect a wide variety of hosts. However, the mechanism(s) used by herpesviruses to spread from cell to cell differs greatly depending on virus species and their dedicated target cells or tissues. Cell-cell fusion, synapse formation, filopodial bridges, and nanotubes were documented for cytomegaloviruses (CMV), varicella zoster virus (VZV), pseudorabies virus (PRV), and herpes simplex virus (HSV-1) (Cole and Grose, 2003; Dingwell et al., 1994; Favoreel et al., 2005; Gerna et al., 2000; Sweet, 1999; van Leeuwen et al., 2002). Infection of PBMC with PRV, CMV, and VZV and subsequently their transfer to EC represents a critical step for viral pathogenesis. CMV and PRV were documented to spread between PBMC and EC via cell-cell (micro)fusion (Digel et al., 2006; Gerna et al., 2000; Van de Walle et al., 2003). Equine herpesvirus type 1 (EHV-1) is another example of herpesviruses infecting PBMC and then being transferred to EC via cell-associated viremia causing severe outcomes including abortion, perinatal mortality, neurological disorders, and death (Chowdhury et al., 1986; Van Maanen, 2002). Primary virus replication occurs in the respiratory epithelia of infected horses. The virus rapidly reaches the lymphoid tissue associated with the upper respiratory tract, infects mononuclear cells that enter bloodstream, and carry the virus within a short time frame to the vasculature of tissues, such as the pregnant uterus or the central nervous system. The virus can spread and replicate in EC causing different pathological injuries such as vasculitis, thrombosis, and ischemia (Smith et al., 1996; Wilson, 1997). However, the exact mechanism of virus spread is still unknown. EHV-4 infection, on the other hand, is mainly restricted to the upper respiratory tract with very rare events of leukocyte-associated viremia (Osterrieder and Van de Walle, 2010; Vandekerckhove et al., 2011). It is likely that this clear difference in EHV-1 and EHV-4 pathogenesis and disease outcomes is associated with differences in gene products, despite the significant genetic and antigenic similarity, that facilitate virus spread from cell to cell (Sattentau, 2008). Using dynamic in vitro models, we recently showed that EHV-1 was able to maintain tethering and rolling of infected PBMC on EC, which resulted in virus transfer from PBMC to EC (Spiesschaert et al., 2015a). Most remarkably, no EHV-1-productive infection in PBMC was observed, which, however, does not exclude unambiguously restricted productive virus replication albeit at low levels (Drebert et al., 2015; Laval et al., 2015; Spiesschaert et al., 2015a).
Here, we combine confocal imaging, live-cell imaging, and electron microscopy analyses together with functional assays to study virus cell-to-cell spread between PBMC and EC. Our data unravels unique mechanisms of cell-to-cell transmission exploited by herpesviruses, in which the virus is embedded in the ECM of PBMC without entering or infecting the cells. The embedded viruses were protected against circulating neutralizing antibodies until the PBMC reached the EC, where the virus was released to infect the endothelium. We were also able to document several transendothelial migration events of mononuclear cells through EC, where infected PBMC might be able to deliver the virus directly inside the EC.
Results
Virus Embedding in the Carbohydrate-Rich Extracellular Matrix Structures
Confocal microscopy was performed to localize virus particles with respect to the plasma membrane and the ECM of PBMC. We used an EHV-1 strain with a red fluorescent (mRFP) protein fused to the small capsid protein VP26 (EHV-1RFP; to facilitate virus particle tracking) and the fluorescein isothiocyanate (FITC)-labeled plant lectins (ConA and WGA) to stain glycan-rich carbohydrate components of the ECM. EHV-1RFP (multiplicity of infection [MOI] = 0.5) was added to PBMC for different time periods (5 min, 1 h, 24 h, 2 days, 3 days, 5 days, and 7 days) at 37°C, treated with ice-cold citrate buffer (pH 3) for 1.5 min to get rid of ECM-unbound viruses, and then fixed with paraformaldehyde 4%.
Interestingly, we found that virus signals (either single viruses or clusters) were colocalizing with the ECM at all time points, even after 7 days (5 min: Figures 1A and 1B; 1 and 24 h: Figures S1A and S1B; 1–7 days: Figure S2). The 3D image with virus particles colocalizing with the ECM after 5 min (Figure S1D) showed embedding of EHV-1 viral particles in these structures. We only detected virus particles inside the infected cells after 24 h of infection and up to 7 days (Figures S1C and S2).
Figure 1.

Colocalization of Virus Particles with the Carbohydrate-Rich Extracellular Matrix
(A–E) PBMC were infected with EHV-1 RFP (red; MOI = 0.5) for 5 min. Cell surface glycoproteins of the ECM were stained green with FITC-labeled ConA (A), lectin from Triticum vulgaris (wheat germ agglutinin; WGA) (B), anti-collagen (C), anti-agrin (D), or anti-ezrin (E). PBMC nucleus was stained with DAPI (blue). Data are representatives of three independent experiments. Scale bar, 10 μm, and scale bar of magnification, 7 μm. Image stacks (number of stacks = 17 with 0.75 μm z stack step size) were photographed using VisiScope Confocal FRAP microscope. Presented here is a single optical section of the stacks. See also Figures S1–S5.
To further confirm that virus particles were embedded in the ECM and not just bound to cell plasma membrane, EHV-1RFP (MOI = 0.5) was added to PBMC for 5 min at 37°C. The cells were stained with CellVue dye to stain plasma membrane and FITC-labeled ConA to stain ECM. It was clear that virus particles were colocalizing only with the ECM (Figure S3).
Comparison between infected (Figures 1A and 1B) and non-infected cells (Figures S4A and S4B) showed no significant differences with respect to organization of the ECM glycans in infected and non-infected cells (Figure S5).
We next looked at the components of the ECM and their colocalization with EHV-1RFP. Confocal microscopy analyses were carried out after 5 min infection of PBMC with EHV-1RFP (MOI = 0.5). The cells were stained for several surface proteins including collagen, agrin, and ezrin (Pais-Correia et al., 2010). We found that virus particles were colocalized and clustered with collagen and agrin; however, there was no significant reorganization of these proteins among infected and non-infected cells (Figures 1C, 1D, S4C, and S4D). There was almost no colocalization of the virus particles with ezrin with no reorganization (Figures 1E and S4E). Heparinase enzyme treatment that hydrolyzes heparan sulfate (HS) proteoglycans resulted in destruction of the ECM-bearing viral particles (Figure 4C) indicating their involvement as traps and/or carriers of viruses.
Figure 4.

Relevance of Extracellular Carbohydrate-Rich Matrix in EHV-1 Cell-to-Cell Spread
(A and B) PBMC were infected with EHV-1 RFP + GFP (MOI = 0.1) for 5 min. The ECM were disrupted mechanically (extensive pipetting; EHV-1_5M_Cell_pip), chemically (heparin treatment; EHV-1_5M_Cell_hep), or both (EHV-1_5M_Cell_pip_hep). The disrupted cells (A) or the supernatant (EHV-1_5M_Sup_pip, EHV-1_5M_Sup_hep, EHV-1_5M_Sup_pip_hep) (B) were added to EC. As a control, the ECM was left undisrupted (EHV-1_5M_Cell or EHV-1_5M_Sup). The data represent the mean ± standard deviation of three independent and blinded experiments. Significant differences in plaque numbers on EC were seen between the different treatment procedures. n = 3; one-way ANOVA test and Dunnett's multiple comparisons test; ∗∗p < 0.01, ∗∗∗p < 0.001.
(C and D) Treatment with heparinase III or heparin fractionates and elution of ECM-viral assemblies. Confocal microscopy showing infected PBMC with EHV-1 RFP + GFP (red) treated with heparinase III (C) or heparin (D) and stained for cell surface carbohydrate-rich matrix with ConA (green). PBMC nucleus was stained with DAPI (blue). Data are representative of three independent experiments. Scale bar, 10 μm, and scale bar of magnification, 7 μm. Image stacks (number of stacks = 17 with 0.75 μm z stack step size) were photographed using VisiScope Confocal FRAP microscope. Presented here is a single optical section of the stacks.
Spread of ECM-Associated Viruses from Infected PBMC to Endothelial Cells
To test virus transmission from infected PBMC to EC and the role of extracellular carbohydrate-rich matrix structures, overlay assays (under either static or dynamic conditions) were performed. EHV-1GFP virus (EHV-1 expresses the enhanced green fluorescent protein [EGFP] for rapid identification of infected cells) was added to PBMC for different time periods (5 min, 1 h, or 6 h) followed by citrate treatment as mentioned above. Infected PBMC were then overlaid on EC in “contact” model (Figure 2A) for 2 h in the presence of virus-neutralizing antibodies to prevent the infection of free virus particles, followed by extensive washing several times to get rid of unbound PBMC. The “no-contact” model was used as a control to exclude cell-free virus transmission (Figure 2B). Our data showed EHV-1 transmission from infected PBMC to EC at all time points in the presence of neutralizing antibodies, including the early time point (5 min; Figure 2D). This indicates that ECM-embedded viruses on the surface of PBMC were able to spread and replicate in the target EC without previous entry into PBMC. In all cases, no virus transfer from infected PBMC to EC was observed under “no-contact” conditions at all time points (Figure 2D).
Figure 2.

Virus Transmission from Infected PBMC to EC under “Static” and “Dynamic” States
(A–F) Schematic depiction of contact (A), transwell (B), and flow chamber assays (C) is shown. PBMC were infected with EHV-1 GFP for different time periods (5min, 1 h, and 6 h). Infected PBMC were overlaid on EC under “static” conditions (D) or allowed to flow over EC “dynamic” (E and F) in the presence of neutralizing antibodies. Under dynamic conditions, whole PBMC population (E) and/or each PBMC subpopulation (T-lymphocyte, B-lymphocyte, monocyte); (F) were infected for 5 min either at 37°C (E and F) or at 4°C (E). After 24 h, virus spread was assessed by counting the plaques on EC excluding the inlet and outlet of the slide. As a control, infected PBMC or PBMC subpopulation were placed into a transwell insert without direct contact between EC and PBMC “no contact.” The data represent the mean ± standard deviation of three independent and blinded experiments. Significant differences in plaque numbers were seen between different infection points under “static” conditions (∗) (n = 3; one-way ANOVA test followed by multiple comparisons tests; ∗p < 0.05; ∗∗p < 0.01). PBMC in (F) refers to whole PBMC population.
(G) Confocal microscopy of overlaid infected PBMC on endothelial cells after 2 h. ECM was stained green with FITC-labeled ConA (green), EHV-1 RFP viral particles are red (arrowhead), and nucleus was stained with DAPI (blue). Data are representative of three independent experiments. Scale bar, 10 μm, and scale bar of magnification, 7 μm. Image stacks (number of stacks = 17 with 0.75 μm z stack step size) were photographed using VisiScope Confocal FRAP microscope. Presented here is a single optical section of the stacks.
(H) Confocal live-cell imaging showing the transfer of EHV-1 RFP + GFP viral particles (red; arrowhead) from overlaid 5-min-infected PBMC to the endothelial cells. Virus replication in EC is indicated by GFP expression (green), whereas RFP signals (at time points 03:44 and 05:36 min) represent new progeny RFP-labeled virus production (red) in the infected cells. Image stacks (number of stacks = 15 with 0.5 μm z stack step size) were photographed using VisiScope Confocal FRAP microscope. Presented here is a single optical section of the stacks.
See also Figures S6–S8.
As was shown in the stationary setup, we further expanded our investigation to test EHV-1 transfer under dynamic conditions using a fluidic setup (flow chamber assay; Figure 2C). In this assay, EHV-1GFP was added to PBMC for 5 min (at 37°C or at 4°C). At 4°C, viruses are attached to the cells without entry or penetration. Cells were allowed to flow over the confluent EC monolayer in the presence of virus-neutralizing antibodies to neutralize free virus particles. Our data showed that the 5-min-infected PBMC were able to transfer the virus to EC under flow condition (Figure 2E). To determine which PBMC subpopulation might be responsible for virus spread, PBMC were sorted and different subpopulations (T lymphocytes, B lymphocytes, or monocytes) were infected with EHV-1 for 5 min and the cells were allowed to flow over EC in the presence of virus-neutralizing antibodies. All PBMC subpopulations were able to transfer the virus to EC (Figure 2F). In all cases, no virus transfer from infected PBMC or the subpopulations to EC was observed under “no-contact” conditions (Figures 2E and 2F).
To further visualize virus transfer from PBMC to EC, EHV-1RFP was added to PBMC for 5 min, overlaid on EC for 2 h, and analyzed using confocal microscopy. The z stack immunofluorescence confocal images came in-line with the other infection assays showing PBMC in close proximity to EC with viral particles on the surface of infected PBMC and the underneath EC (Figures 2G and S6).
To track EHV-1 transmission from the surface of infected PBMC to EC, time-lapse live cell imaging was performed. After 5 min infection of PBMC with EHV-1RFP+GFP, the cells were added to EC and visualized by time-lapse live cell imaging confocal microscopy. Interestingly, virus movement (represented by red signals) from infected PBMC to EC was tracked at the early time points (within the first hour after adding infected PBMC) and virus replication in EC (represented by GFP expression and production of progeny RFP-labeled viruses) was visualized 2–3 h post overlay (Figures 2H and S7 and Video S1). No virus replication (no GFP expression) was detected in PBMC carrying the virus. The free red signals found in the field might represent free virus particles or cell debris. It was clear that free virus particles, if present, were not able to infect EC due to the presence of neutralizing antibodies in the medium. This conclusion was confirmed by the absence of GFP-expressing EC (except the one with PBMC on top) and the transwell control assay (Figures 2D and 2E).
PBMC were infected with EHV-1RFP+GFP (MOI=3, red) for 5 min and overlaid on EC. Virus replication in EC is indicated by GFP expression, which is integrated in the viral genome (green), whereas RFP signals represent new progenies of RFP-labeled virus production (red) in the infected cells.
Taking all previous results together, the data display the role of extracellular carbohydrate-rich matrix in virus PBMC-EC transmission that physically protects the virus against virus neutralizing antibodies and possibly other immune reaction. This carbohydrate-rich ECM not only has a role in virus transmission between PBMC and EC but also probably has a role in viral transfer between PBMC themselves as the PBMC tend to aggregate together in the infection than the non-infection state (Spiesschaert et al., 2015a; van der Meulen et al., 2001) (Figure S8).
Correlative Fluorescent and Electron Microscopy Identifies Virus Particles in the Interface between PBMC and EC
Compared with fluorescence microscopy, transmission electron microscopy (TEM) provides a significantly higher resolution, however, at the price of a much smaller field of view and elaborate sample preparation. To further elucidate the mechanism of virus transfer between PBMC and EC at ultrastructural level, we applied a correlative workflow to identify and target individual virus-loaded PBMC in close proximity to EC. EC were grown on gridded coverslips, incubated with EHV-1RFP-infected PBMC, and imaged using an automated wide-field fluorescence microscope (CellDiscoverer7, Zeiss). To unambiguously identify PBMC comprising a more compact, nearly spherical nuclei compared with EC, Hoechst was used to counterstain the nucleus. PBMC showing a discrete, punctual RFP signal (indicating individual virus particles) located in close proximity to EC were targeted during TEM sample preparation and ultrathin sectioning based on their position relative to the coverslip grid (Figure 3A). We observed the presence of enveloped EHV-1 particles on the surface of infected PBMC that are approaching the resting EC (Figures 3B, 3C, and S9). Interestingly, virus particles were found in the intercellular space between tightly interacting infected PBMC and EC (Figures 3D–3F). We did not find evidence of virus budding from the PBMC or fusion events between infected PBMC and EC plasma membranes at this time point (5 min infection of PBMC and 1 h overlaying incubation period, see Transparent Methods). This further indicates that virus particles might be transported embedded on the surface of PBMC and do not necessarily need to enter them.
Figure 3.

Virus Particles in the Interface between Infected-PBMC and EC
(A–F) (A) Fluorescence image of EC grown on gridded transmission electron microscope (TEM) coverslips and overlaid with infected PBMC. PBMC were infected with EHV-1 RFP (MOI = 5) for 5 min and overlaid on EC. Red: mRFP-labeled EHV-1; blue: Hoechst-stained nuclei. The insert shows one PBMC (red arrow) carrying a virus load and attached to one endothelial cell. The dashed line indicates the direction of the targeted plane for ultrathin sectioning. TEM images of targeted PBMC show virus particles (white arrows) on the surface of infected PBMC (B and C) as well as one virus particle (white arrow) in the contact zone between infected PBMC and EC (D–F). Marked parts with rectangles in (B and E) are magnified in (C and F), respectively. Scale bar, 1 μm. See also Figure S9.
Destruction of ECM-Associated Virus Clusters Reduces Virus Spread
To unravel the prominence of the extracellular carbohydrate-rich matrix in virus transmission from PBMC to EC, extracellular viral clusters were disrupted mechanically by vigorous pipetting, chemically by heparin or heparinase treatment, or both (pipetting and heparin treatment). Heparin has been shown to compete with HS for binding to protein components of the ECM leading to its removal from the cell surface (Pais-Correia et al., 2010; Tarasevich et al., 2015). Heparinase enzyme has been shown to cleave HS proteoglycans (Ernst et al., 1995). Infected PBMC were mechanically and/or chemically treated, washed several times, and added to EC monolayers. Treatment with heparinase III or heparin leads to the detachment of ECM-viral assemblies (Figures 4C and 4D). Partial destruction of the ECM clearly leads to a significant reduction in virus transfer between PBMC and EC accompanied by reduction of EC infection rates (Figure 4A). On the other hand, supernatants from treated PBMC contained more ECM-viral assemblies and gained a significant increase in infectivity after adding to EC, even in the presence of virus-neutralizing antibodies (Figure 4B). Although PBMC treatment significantly reduced virus spread, it did not completely abolish virus transmission due to the remaining ECM-virus clusters on the surface of PBMC that can still transfer the virus (Figures 4C and 4D). These results indicate that integrity of ECM is essential in virus transmission from PBMC to EC.
Transendothelial Migration of PBMC through the Endothelial Cell
Mononuclear cell migration across the endothelial barrier is a distinct process that can be through either the endothelial junctions (paracellular) or the EC body (transcellular) (Muller, 2013; Schimmel et al., 2017). The process of transendothelial migration is usually preceded by orchestrated steps starting with rolling, firm adhesion, and microvillar projections invaginating the endothelium that might trigger transmigration (Schmidt et al., 2011). While running TEM analysis, we noticed several PBMC behaving as they are preparing for transmigration; they firmly attached to EC and having projections into the EC (Figures 5A and 5B). We further detected intact PBMC transmigrating through the EC (Figures 5C–5F). Interestingly, we could identify in several occasions incorporated infected PBMC (with virus particles) inside EC; however, they seem to be partially digested (Figures 5G–5L and S10). We further detected virus particle on the surface of a digested PBMC within EC (Figures 5G–5I). This pathway might represent a novel strategy and a mechanism of direct intracellular cell-to-cell spread of viruses.
Figure 5.

Transcellular Migration of Infected-PBMC through Endothelial Cells
(A–L) Ultrathin cross sections of PBMC adherent to and crossing endothelial cells. PBMC were infected for 5 min with EHV-1 RFP and incubated over adherent EC for 3 h. Multiple intracellular protrusions invaginating the apical endothelial membrane (A and B; black arrows) and PBMC migrating through the endothelial cells (C) are shown. Intact PBMC is shown within the cytoplasm of EC (D–F; red arrow). Migrating PBMC (E) is marked with a rectangle and further magnified (F, red arrow). Partially digested infected PBMC is shown (G–L) and marked with a rectangle, which is magnified (H–I and K–L). Virus particles attached to migrating PBMC (either outside, H and I, or inside, K and L) are marked with white arrows. Scale bar, 1 μm. See also Figures S10 and S13.
Cell-to-Cell Fusion Events between Infected PBMC and EC
To test the possible incidence of classical fusion events between infected PBMC and EC, PBMC were transfected with mRFP-expressing plasmid and infected with EHV-1GFP for 24 h to allow virus replication and expression of virus fusion machinery (namely, gD, gB, gH-gL) on the surface of infected PBMC (Figure S11). PBMC were then co-cultured with the target EC for 3 h. Upon adhesion, infected PBMC fused with the underlying uninfected EC, which leads to the transfer of their red cytoplasm as well as intracellular virions between the two cells. EC expressed GFP due to virus replication after transfer through the fusion canal (Figure 6A).
Figure 6.

Cell-Cell Membrane Fusion Induced by EHV-1
(A) Fusion between PBMC and EC was visualized using confocal laser scanning microscope. Expression of mRFP protein is indicated by red and EHV-1 GFP replication is represented by green after GFP expression, which is integrated in the viral genome. Colocalization of both colors is shown in the merge panel. Data are representative of two independent experiments. Scale bar, 10 μm, and scale bar of magnification, 7 μm. Image stacks (number of stacks = 17 with 0.75 μm z stack step size) were photographed using VisiScope Confocal FRAP microscope. Presented here is a single optical section of the stacks.
(B) Fusion between infected PBMC and EC was further detected by luciferase activity. Results are shown as means of three independent and blinded experiments with standard errors and are presented as luciferase luminescence unit (RLU). The asterisk indicates p value <0.05 using Kruskal-Wallis test followed by Dunns test for multiple comparisons. Infected PBMC only (Inf_PBMC + Trans_EC) and transfected PBMC only (Trans_PBMC + Trans_EC) were used as negative controls. Control positive is represented by transfected PBMC with both plasmids (double Trans_PBMC). Trans_inf_PBMC + Trans_EC:PBMC was transfected with T7 RNA polymerase-encoding plasmid, then infected with EHV-1 GFP, and overlaid on EC transfected with a plasmid encoding luciferase gene under a T7 promoter. See also Figure S11.
In another assay, PBMC were transfected with T7 RNA polymerase-encoding plasmid and then infected with EHV-1GFP (Trans_inf_PBMC). PBMC were overlaid on EC transfected with a plasmid encoding luciferase gene under a T7 promoter (Trans_EC). As negative controls, PBMC were either transfected with the T7 RNA polymerase-encoding plasmid without infection or infected with EHV-1GFP without transfection. In all cases, the EC were transfected with a plasmid encoding luciferase under a T7 promoter. As a positive control, PBMC were transfected with the two plasmids encoding T7 RNA polymerase and luciferase under a T7 promoter. Luciferase activity was recorded as a measure of cell-cell fusion. PBMC-EC fusion events were only detected in the setup where PBMC were first transfected and infected and then overlaid on transfected EC. Transfection of PBMC with both plasmids gave the same luciferase activity (Figure 6B).
No Transmission Events for ECM-Clustered EHV-4
We have shown before that EHV-4 was unable to spread (under contact or dynamic conditions) from infected PBMC to EC (Spiesschaert et al., 2015a). Here, we show that EHV-4, like EHV-1, was clustered with the extracellular carbohydrate-rich matrix of infected PBMC (Figure S12A). However, infected PBMC were not able to transfer EHV-4GFP to EC as shown in the contact assay (Figure S12B) and the flow chamber assay (Figure S12C). Destruction of the extracellular carbohydrate-rich structures from infected PBMC by extensive pipetting and heparin treatment released ECM-virus assemblies into the medium; however, these assemblies were not infectious to EC as shown for EHV-1 (Figure S12D). It is worth mentioning that EC preparations are permissive for EHV-4 infection using free infectious virus produced by equine dermal (ED) cells (Figure S12E).
Viral gB and gD Influence ECM-Associated Virus Cell-to-Cell Transmission
From our results, it was clear that both EHV-1 and EHV-4 are embedded in the ECM of infected PBMC before entering the cell itself. However, only EHV-1 was able to step forward and spread to infect EC at early time point. Several factors can be involved in this privilege of EHV-1; of interest are the envelope glycoproteins involved in virus entry (Azab and Osterrieder, 2017b). The role of two envelope proteins gB and gD in virus transmission was tested. The two genes were swapped between EHV-1 and EHV-4 (Azab and Osterrieder, 2012; Spiesschaert et al., 2015b), PBMC were infected for 5 min with either EHV-1_gB4 and EHV-1_gD4 or EHV-4_gB1 and EHV-4_gD1, and infected PBMC were added to EC in “contact” and “flow chamber” models. Interestingly, swapping the two genes significantly affected ECM-associated virus spread from infected PBMC to EC. The recombinant viruses (EHV-4_gB1 and gD1) were able to spread and infect EC under “contact” condition only (Figures 7B and 7D). On the other hand, EHV-1_gD4 and EHV-1_gB4 showed a significant reduction in virus transfer when compared with parental EHV-1, under both static and dynamic conditions (Figures 7A and 7C).
Figure 7.

Role of Viral gB and gD in Virus Transmission
(A–D) PBMC were infected with the indicated viruses (MOI = 0.1) for 5 min. Infected PBMC were overlaid on EC under “static” conditions (A and B) or allowed to flow over EC “dynamic” (C and D) in the presence of neutralizing antibodies. After 24 h, virus spread was assessed by counting the plaques on EC. As a control, infected PBMC were placed into a transwell insert without direct contact between EC and PBMC “no contact.” The data represent the mean ± standard deviation of three independent and blinded experiments. Significant differences in plaque numbers were seen between the different viruses as indicated. One-way ANOVA test followed by Dunnett's multiple comparisons test (B) or Kruskal-Wallis test with Dunns multiple comparisons test (A and C); ∗p < 0.05. See also Figure S12.
Discussion
As successful parasites, viruses have evolved several and diverse mechanisms to facilitate rapid cell-to-cell dissemination and to promote immune evasion (Mothes et al., 2010; Sattentau, 2008). Different pathways of cell-to-cell spread have been identified that fall mainly into two main categories; (1) cell-free viruses: where intracellular viruses are released into the extracellular space and infected new cells; (2) direct cell-to-cell contact: where intracellular viruses spread from one cell to another without being extracellularly egressed (Marsh and Helenius, 2006; Mothes et al., 2010; Sattentau, 2011). Here, we document two unique pathways of virus transmission that include (1) the embedding of virus particles in the ECM without entering the cells and (2) a possible intracellular delivery during transendothelial migration of infected PBMC into EC. The ECM-associated viruses are protected from the immune attack and efficiently spread from infected PBMC to other immune cells or to EC. Furthermore, infected PBMC can transmigrate through the EC where ECM-associated viruses can be delivered directly inside EC.
The portal of entry is not the ultimate goal of herpesviruses, and they need to travel (through either blood or nerve) to their preferred tissues where they can establish latency or induce disease (Adler et al., 2017). To achieve this goal, different mechanisms of direct cell-to-cell spread have been documented including cell-cell fusion, synapse formation, or tunneling nanotubes (Gerna et al., 2000; Jansens et al., 2017; Saksena et al., 2006). The pathogenesis of EHV-1 is distinct from other members of the Herpesviridae family. Infection starts first at the respiratory epithelium where PBMC contracts the virus and delivers it to the endothelium (Ma et al., 2013; Smith et al., 2001). Infection of PBMC and subsequent virus spread to the endothelium are still to be understood. We and others have shown that virus replication in PBMC is “restricted” without obvious viral protein production or expression on the cell surface. However, when infected PBMC reach the endothelium, the virus finds its way outside the PBMC to infect new cells, probably through cell-cell fusion (Laval et al., 2015; Spiesschaert et al., 2015a). This pathway provides a secure trip for the virus without being attacked by the immune system. In the mechanism that we provide here for the first time, PBMC (during the first hour of infection) carry the virus on its surface associated with the ECM; the virus does not enter the cells. Although appeared to be exposed to the surrounding environment, viruses are fully protected against neutralizing antibodies that prevented free virus spread. Upon contact with EC either under “static” or “dynamic” conditions, the virus can spread and infect the endothelium. We further showed that all three subpopulations of PBMC (T lymphocytes, B lymphocytes, and monocytes) could transfer the virus to EC early after infection. It would be interesting, for future studies, to stain ECM on the surface of each subpopulation to determine virus localization on the surface of these cells.
We next asked the question whether this mechanism is spontaneous and any virus can be stuck to the surface of PBMC and then spread. EHV-4, a close relative to EHV-1 and having a restricted pathogenesis to the upper respiratory tract, was not able to spread to the endothelium, although it was also stuck to the ECM. This indicates that EHV-1 exploits the existing natural ECM and hides in this complex network until it reaches the endothelium where it can exit the surrounding matrix and infect EC. ECM-associated viruses can also be spread to other non-infected PBMC in the circulation or in secondary lymphoid organs. It was shown that dendritic cells contract EHV-1 infection from the respiratory epithelium (Vandekerckhove et al., 2011); in turn, other cells may get infected after contacting EHV-1 clusters on the surface of dendritic cells. A similar but distinct mechanism was described before for HTLV-1 virus (Pais-Correia et al., 2010). It has been shown that HTLV-1-infected lymphocytes produce and store viral particles as extracellular assemblies attached to cell surface extracellular components through a virus-induced mechanism. HTLV-1 seems to hijack certain host cell proteins and enhance their expression to build the extracellular assemblies. In that case, virus infection takes place first within the cells where the virus can modify the carbohydrate compositions and/or their spatial organization. The virus then buds into and accumulates in the extracellular structures that may locally increase the infectious titer and facilitate HTLV-1 cell-to-cell spread (Pais-Correia et al., 2010).
In the natural situation where horses are infected with EHV-1, PBMC transfer the virus from the site of primary infection in respiratory epithelia to the endothelium. In this pathway, PBMC might take hours to reach lymph nodes and days until viremia develops. We show here that PBMC can keep the virus on its surface (associated with the ECM) for up to 7 days, which may point to their ability to transfer the extracellular viruses to EC. However, transfer of intracellular virus particles through cell-cell fusion or any other mechanism cannot be excluded.
Interestingly, we detected enveloped EHV-1 in the interface between tightly interacting infected PBMC and EC. Our correlative approach enables us to roughly target infected PBMC in close proximity to EC during TEM sample preparation, whereas visualizing virus particles with ∼180 nm diameter within 50- to 70-nm ultrathin section through an entire cell is still a rare event and thus remains challenging. Fluorescence signals from single labeled virus particles are weak, and most fluorophores are not compatible with resin-embedding procedures. Using fluorescence imaging is thus difficult to target rare events on ultrathin sections before TEM imaging. Further investigating the mechanism of virus transfer at the ultrastructural level would need serial sectioning techniques such as SEM array tomography or block face imaging to reconstruct large volumes, hence increasing the probability to capture rare events within their cellular context.
The composition of ECM is diverse but falls into two classes of macromolecules, namely, proteoglycans and fibrous proteins (Järveläinen et al., 2009; Schaefer and Schaefer, 2010; Stavolone and Lionetti, 2017). Glycosaminoglycan (GAG) chains composed of HS and chondroitin sulfate fill the majority of the ECM (Stavolone and Lionetti, 2017). Several viruses, including herpesviruses, have been shown to interact with HS in a charge-based and a relatively non-specific manner (Azab and Osterrieder, 2017a; Barth et al., 2003; Chen et al., 1997; Tyagi et al., 2001). Herpesvirus envelope glycoproteins (gC and gB) interact reversibly with HS, which lead to virus concentration at the cell surface (Azab and Osterrieder, 2017a) and can explain the embedding of EHV-1 within the ECM. We further showed that virus particles are colocalizing with agrin and collagen of the ECM. The HS proteoglycan, agrin, plays a key role in efficient cell-to-cell communication at the synaptic zone (Alfsen et al., 2005; Dustin and Dustin, 2001; Khan et al., 2001) and in virus cell-to-cell transmission (Alfsen et al., 2005; Dityatev and Schachner, 2006; Pais-Correia et al., 2010). It is worthy to mention that the role of synapse formation in EHV-1 spread has not yet been elucidated. Collagens are the most abundant structural element of the ECM and play a role in the regulation of cell adhesion, providing tensile strength and supporting chemotaxis and migration (Rozario and DeSimone, 2010). The actual interaction of the virus with the two proteins and their exact role in virus transmission need to be elucidated. We barely detected a colocalization with ezrin protein, which is a member of the ECM protein family and plays a key role in cell surface structure adhesion, migration, and organization (Vaheri et al., 1997).
The chemical compositions (GAGs and fibrous proteins) of ECM endowed this matrix with its gelatinous properties that play important physiological roles (Rhodes and Simons, 2007). To investigate the functional relevance of ECM integrity on virus spread, we applied mechanical and/or chemical (through heparin or heparinase enzyme) shearing forces to the infected cells. Partial destruction of EHV-1-associated ECM, although did not completely remove virus-ECM structures, significantly reduced virus cell-to-cell spread and the overall infectious capacity of PBMC. Cell supernatants obtained after ECM destruction, on the other hand, contained more virus assemblies and gained more infectivity even in the presence of neutralizing antibodies. Interestingly, destruction of EHV-4-associated ECM did not result in any infectivity at both levels (cell-to-cell or supernatants). Taken together, this indicates that the integrity of ECM is essential for EHV-1 transfer at the PBMC-EC interface. Further studies will be needed to unravel the composition, structure, regulation, and function of the ECM of PBMC. EHV-1 was able to egress the ECM at certain time point, probably when reaching the EC, through a virus-specific mechanism, which is not available for its close relative (EHV-4).
EHV-1 interaction with the ECM components of PBMC and its subsequent egress when reaching the EC is enigmatic. Several herpesvirus glycoproteins have been implicated in cell-to-cell spread (Azab and Osterrieder, 2017a). Two candidates (gB and gD) have been shown to have essential roles in virus entry, penetration, and tissue tropism. In all members of Herpesviridae family, gB is a highly conserved fusogenic protein and it interacts with HS or other specific cell receptors. gD, when present, is the main receptor-binding protein that triggers the entry steps (Azab and Osterrieder, 2017a). Swapping gB and gD between EHV-1 and EHV-4 resulted in a dramatic change in virus infectious ability. ECM-associated EHV-1 with either gD4 or gB4 showed a significant reduction in cell-to-cell spread. On the other hand, EHV-4 with gB1 or gD1 obtained, for the first time, the ability to spread from infected PBMC to EC. We surmise that EHV-1 gD and gB were able to interact with certain structures of the ECM, such as HS or yet unidentified cell receptors, that keep the virus attached during the whole trip. How the virus breaks this interaction to infect the EC is unknown. It is possible that the virus interacts with EC while attaching to the ECM of PBMC. Another explanation could be that the virus adheres avidly to the ECM deposited by EC before penetration. It is possible that following virus binding to the ECM of PBMC, the ECM could be remodeled resulting in the virus having greater affinity for the ECM of EC. Such interaction with EC can be stronger and specific to influence virus transfer from a temporary situation on the surface of PBMC to a stable one on EC. The clear difference between the fusogenic functions of EHV-1 and EHV-4 gBs can also explain the difference of infection outcomes (Spiesschaert et al., 2015b). The interaction of EHV-1 gD with new cell receptors on the surface of EC cannot be excluded. Detection of EHV4 associated with the ECM indicates that the virus can interact or, at least, be captured by the gelatinous nature of the ECM. However, the failure of virus egress to infect EC can be interpreted by the absence of suitable genetic makeup that can support its release. Being stuck longer may lead to its inactivation on the surface of PBMC as a defense mechanism as previously shown with Newcastle disease virus (Yaacov et al., 2012). This interpretation is supported by the ability of EHV-4 to egress and infect EC after supporting its genome with more powerful genes (gD and gB) from EHV-1.
Transcellular and paracellular pathways have been described to illustrate PBMC migration through the endothelial barrier (Engelhardt and Wolburg, 2004; Muller, 2011). Although the process of transcellular migration and the influencing factors are not fully understood, the entire process of trafficking through the endothelium requires three basic steps: rolling, adhesion, and transmigration (Schmidt et al., 2011). We have recently shown that EHV-1-infected PBMC have the ability to roll and then adhere to the endothelium to facilitate virus spread, probably after the upregulation of certain adhesion molecules and cytokines (Holz et al., 2017; Proft et al., 2016; Spiesschaert et al., 2015a). Here, we document the final step of trafficking and show that infected PBMC can migrate through the endothelium in a process that might lead to a safe delivery of the virus inside the cell. Although such a mechanism may not represent the default pathway of virus transmission, the possibility to be an “alternative” pathway cannot be excluded. Transmigration of peripheral mononuclear cells containing bacteria through epithelial cells was previously reported (Wewer et al., 2011). It was shown that infection with Streptococcus suis enhanced the process of transcellular migration. Whether EHV-1 has an influencing effect remains to be investigated. We noticed that the transmigrating infected PBMC was digested within a cytoplasmic vacuole. It seems that lysis of naive PBMC during transmigration is an expected process (Figure S13) (Bedoya et al., 1969; Herman et al., 2019). However, it is also possible that virus particles on the surface of PBMC may trigger cell lysis. As transendothelial migration is a rare event and cannot be easily captured by TEM, we could not detect intact PBMC with virus particles on the surface; however, we detected virus particle on the surface of digested PBMC. We surmise that virus particles are able to escape from the vacuole before complete lysis through fusion with the vacuole membrane; a process that is well mastered by herpesviruses (Atanasiu et al., 2010).
Cell-cell fusion is considered the classical and most investigated event of herpesvirus transmission (Cole and Grose, 2003). EHV-1-infected PBMC were capable of transferring infected cytoplasmic material into uninfected EC through fusion events. These results are described before for other herpesviruses including HCMV, PRV, and a VZV (Digel et al., 2006; Gerna et al., 2000; Van de Walle et al., 2003) as well as other viruses (Duprex et al., 1999; Takeuchi et al., 2003). In general, herpesviruses are equipped with a set of glycoproteins (gD, gB, gH, and gL) that carry out all fusion events either between the virus and cells or between infected and non-infected cells (Ambrosini and Enquist, 2015; Atanasiu et al., 2010). During viremia, infected PBMC slow down, roll over the endothelium, and finally adhere to EC. Such a process gives the infected cells a chance to establish cell-cell fusion due to replication of intracellular virus particles and expression of viral envelope glycoproteins on the surface of infected PBMC. Interestingly, all detected fusion events were in PBMC infected for 24 h, where viruses already entered the cells and produced the needed glycoproteins for fusion (Van de Walle et al., 2003).
In conclusion, among all viruses, herpesviruses have proven themselves as “masters of co-evolution.” It is surprising how herpesviruses manipulate infected PBMC with different mechanisms and pathways to ensure a safe and secure spread in the same scene. (1) “PBMC-hitching”: at the first hit, herpesviruses attach to the ECM, hide, transport, and then egress to infect other cells. (2) “Intracellular delivery”: transendothelial migration of infected PBMC may lead to a direct delivery of the virus inside the cytoplasm of EC. (3) “Classical-fusion”: this event is well controlled by herpesviruses and always occurs to facilitate virus spread.
Limitations of the Study
Animal study is the main limitation of this study. It is difficult to determine virus spread from PBMC to EC in vivo. One possible animal study could be the infection of horses with EHV-1RFP and collecting PBMC at different time points during viremia and check whether the virus could be present on the surface of the cells. However, this will not prove that the virus was stuck from the beginning on the surface of PBMC. Another limitation is the confirmation that viruses delivered intracellularly with migrating PBMC are infectious. To conduct such experiment, we will need to isolate EC with infected PBMC inside and propagate them separately to track if they will develop infection; however, this is technically very difficult. Finally, characterization of the ECM of PBMC and determining their composition, structure, regulation, and function would add significantly to our understanding of virus spread. However, conducting this experiment was severely affected by the current COVID-19 pandemic, which hindered our regular access to the laboratories.
Resource Availability
Lead Contact
Further information and requests for resources and reagents or data should be directed to and will be fulfilled by the Lead Contact, Walid Azab: walid.azab@fu-berlin.de.
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate/analyze datasets/code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by a grant from Deutsche Forschungsgemeinschaft (AZ 97/3-2) to W.A.. M.K. was supported by a scholarship from the Egyptian Ministry of Higher Education (MoHE) and the German Academic Exchange Service (DAAD). We thank Prof. Dr. Klaus Osterrieder for the useful discussion. We thank Prof. Dr. Heidrun Gehlen, Horse Clinic, Freie Universitaet Berlin, for providing horse blood for PBMC isolation. We thank Prof. Dr. Johanna Plendl, Institut für Veterinär-Anatomie, Freie Universitaet Berlin for providing endothelial cells. We thank Oleksandr Kolyvushko for helping with the live cell imaging and Michaela Zeitlow for technical assistance.
Author Contributions
First author M.K. designed and conducted most of the experiments, designed the figures, and wrote the manuscript. Coauthor S.P. designed and conducted the live cell imaging and prepared the samples for EM, designed live cell imaging figures and movie, and wrote part of the manuscript. Coauthor B.F. designed and conducted the EM experiment. Coauthor T.M. designed and conducted the EM experiment, conducted data analysis of the EM pictures, and wrote part of the manuscript. Corresponding author W.A. designed the project, discussed the results at different stages of work, and wrote and corrected the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101615.
Supplemental Information
References
- Adler B., Sattler C., Adler H. Herpesviruses and their host cells: a successful liaison. Trends Microbiol. 2017;25:229–241. doi: 10.1016/j.tim.2016.11.009. [DOI] [PubMed] [Google Scholar]
- Akhtar J., Shukla D. Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009;276:7228–7236. doi: 10.1111/j.1742-4658.2009.07402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfsen A., Yu H., Magérus-Chatinet A., Schmitt A., Bomsel M. HIV-1-infected blood mononuclear cells form an integrin-and agrin-dependent viral synapse to induce efficient HIV-1 transcytosis across epithelial cell monolayer. Mol. Biol. Cell. 2005;16:4267–4279. doi: 10.1091/mbc.E05-03-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini A.E., Enquist L.W. Cell-fusion events induced by α-herpesviruses. Future Virol. 2015;10:185–200. [Google Scholar]
- Atanasiu D., Saw W.T., Cohen G.H., Eisenberg R.J. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 2010;84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert M., Yoon M., Sloan D.D., Spear P.G., Jerome K.R. The virological synapse facilitates herpes simplex virus entry into T cells. J. Virol. 2009;83:6171–6183. doi: 10.1128/JVI.02163-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azab W., Osterrieder K. Initial contact: the first steps in herpesvirus entry. Adv. Anat. Embryol. Cell Biol. 2017;223:1–27. doi: 10.1007/978-3-319-53168-7_1. [DOI] [PubMed] [Google Scholar]
- Azab W., Osterrieder K. Cell Biology of Herpes Viruses. Springer; 2017. Initial contact: the first steps in herpesvirus entry; pp. 1–27. [DOI] [PubMed] [Google Scholar]
- Azab W., Osterrieder N. Glycoproteins D of equine herpesvirus type 1 (EHV-1) and EHV-4 determine cellular tropism independently of integrins. J. Virol. 2012;86:2031–2044. doi: 10.1128/JVI.06555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth H., Schäfer C., Adah M.I., Zhang F., Linhardt R.J., Toyoda H., Kinoshita-Toyoda A., Toida T., van Kuppevelt T.H., Depla E. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003;278:41003–41012. doi: 10.1074/jbc.M302267200. [DOI] [PubMed] [Google Scholar]
- Bedoya V., Grimley P.M., Rabson A.S. Ultrastructural evidence of in vitro interaction among burkitt lymphoma cells - possible relevance to phagocytic activity of starry sky histiocytes in vivo. Cancer Res. 1969;29:753. [PubMed] [Google Scholar]
- Bennett A.E., Narayan K., Shi D., Hartnell L.M., Gousset K., He H., Lowekamp B.C., Yoo T.S., Bliss D., Freed E.O. Ion-abrasion scanning electron microscopy reveals surface-connected tubular conduits in HIV-infected macrophages. PLoS Pathog. 2009;5:e1000591. doi: 10.1371/journal.ppat.1000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C., Chou J., Werb Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014;15:786. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrel R.N., de Lamballerie X. Zoonotic aspects of arenavirus infections. Vet. Microbiol. 2010;140:213–220. doi: 10.1016/j.vetmic.2009.08.027. [DOI] [PubMed] [Google Scholar]
- Chen Y., Maguire T., Hileman R.E., Fromm J.R., Esko J.D., Linhardt R.J., Marks R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997;3:866. doi: 10.1038/nm0897-866. [DOI] [PubMed] [Google Scholar]
- Chowdhury S., Kubin G., Ludwig H. Equine herpesvirus type 1 (EHV-1) induced abortions and paralysis in a Lipizzaner stud: a contribution to the classification of equine herpesviruses. Arch. Virol. 1986;90:273–288. doi: 10.1007/BF01317376. [DOI] [PubMed] [Google Scholar]
- Chu H., Zhou J., Wong B.H., Li C., Chan J.F., Cheng Z.S., Yang D., Wang D., Lee A.C., Li C. Middle East respiratory syndrome coronavirus efficiently infects human primary T lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J. Infect. Dis. 2016;213:904–914. doi: 10.1093/infdis/jiv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole N.L., Grose C. Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella-zoster virus. Rev. Med. Virol. 2003;13:207–222. doi: 10.1002/rmv.377. [DOI] [PubMed] [Google Scholar]
- Digel M., Sampaio K.L., Jahn G., Sinzger C. Evidence for direct transfer of cytoplasmic material from infected to uninfected cells during cell-associated spread of human cytomegalovirus. J. Clin. Virol. 2006;37:10–20. doi: 10.1016/j.jcv.2006.05.007. [DOI] [PubMed] [Google Scholar]
- DiGiuseppe S., Bienkowska-Haba M., Guion L.G., Sapp M. Cruising the cellular highways: how human papillomavirus travels from the surface to the nucleus. Virus Res. 2017;231:1–9. doi: 10.1016/j.virusres.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov D.S. Virus entry: molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004;2:109. doi: 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingwell K.S., Brunetti C.R., Hendricks R.L., Tang Q., Tang M., Rainbow A.J., Johnson D.C. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 1994;68:834–845. doi: 10.1128/jvi.68.2.834-845.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev A., Schachner M. The extracellular matrix and synapses. Cell Tissue Res. 2006;326:647–654. doi: 10.1007/s00441-006-0217-1. [DOI] [PubMed] [Google Scholar]
- Drebert Z., Golke A., Cymerys J., Słońska A., Chmielewska A., Tucholska A., Bańbura M. Equid herpesvirus type 1 (EHV-1) disrupts actin cytoskeleton during productive infection in equine leukocytes. Polish J. Vet. Sci. 2015;18:107–112. doi: 10.1515/pjvs-2015-0014. [DOI] [PubMed] [Google Scholar]
- Duprex W.P., McQuaid S., Hangartner L., Billeter M.A., Rima B.K. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J. Virol. 1999;73:9568–9575. doi: 10.1128/jvi.73.11.9568-9575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin M.L., Dustin L.B. The immunological relay race: B cells take antigen by synapse. Nat. Immunol. 2001;2:480–483. doi: 10.1038/88666. [DOI] [PubMed] [Google Scholar]
- Engelhardt B., Wolburg H. Mini-review: transendothelial migration of leukocytes: through the front door or around the side of the house? Eur. J. Immunol. 2004;34:2955–2963. doi: 10.1002/eji.200425327. [DOI] [PubMed] [Google Scholar]
- Ernst S., Langer R., Cooney C.L., Sasisekharan R. Enzymatic degradation of glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 1995;30:387–444. doi: 10.3109/10409239509083490. [DOI] [PubMed] [Google Scholar]
- Favoreel H.W., Van Minnebruggen G., Adriaensen D., Nauwynck H.J. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. U S A. 2005;102:8990–8995. doi: 10.1073/pnas.0409099102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerna G., Percivalle E., Baldanti F., Sozzani S., Lanzarini P., Genini E., Lilleri D., Revello M.G. Human cytomegalovirus replicates abortively in polymorphonuclear leukocytes after transfer from infected endothelial cells via transient microfusion events. J. Virol. 2000;74:5629–5638. doi: 10.1128/jvi.74.12.5629-5638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowen B.B., Hickerson B.T. Hemorrhagic fever of bunyavirus etiology: disease models and progress towards new therapies. J. Microbiol. 2017;55:183–195. doi: 10.1007/s12275-017-7029-8. [DOI] [PubMed] [Google Scholar]
- Gupta P., Balachandran R., Ho M., Enrico A., Rinaldo C. Cell-to-cell transmission of human immunodeficiency virus type 1 in the presence of azidothymidine and neutralizing antibody. J. Virol. 1989;63:2361–2365. doi: 10.1128/jvi.63.5.2361-2365.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman H., Fazakas C., Hasko J., Molnar K., Meszaros A., Nyul-Toth A., Szabo G., Erdelyi F., Ardelean A., Hermenean A. Paracellular and transcellular migration of metastatic cells through the cerebral endothelium. J. Cell Mol. Med. 2019;23:2619–2631. doi: 10.1111/jcmm.14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz C.L., Nelli R.K., Wilson M.E., Zarski L.M., Azab W., Baumgardner R., Osterrieder N., Pease A., Zhang L., Hession S. Viral genes and cellular markers associated with neurological complications during herpesvirus infections. J. Gen. Virol. 2017;98:1439–1454. doi: 10.1099/jgv.0.000773. [DOI] [PubMed] [Google Scholar]
- Humphrey J.D., Dufresne E.R., Schwartz M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014;15:802. doi: 10.1038/nrm3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansens R.J., Van den Broeck W., De Pelsmaeker S., Lamote J.A., Van Waesberghe C., Couck L., Favoreel H.W. Pseudorabies virus US3-induced tunneling nanotubes contain stabilized microtubules, interact with neighboring cells via cadherins, and allow intercellular molecular communication. J. Virol. 2017;91 doi: 10.1128/JVI.00749-17. e00749–00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järveläinen H., Sainio A., Koulu M., Wight T.N., Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol. Rev. 2009;61:198–223. doi: 10.1124/pr.109.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller T.T., Mairuhu A.T., de Kruif M.D., Klein S.K., Gerdes V.E., ten Cate H., Brandjes D.P., Levi M., van Gorp E.C. Infections and endothelial cells. Cardiovasc. Res. 2003;60:40–48. doi: 10.1016/s0008-6363(03)00354-7. [DOI] [PubMed] [Google Scholar]
- Khan A.A., Bose C., Yam L.S., Soloski M.J., Rupp F. Physiological regulation of the immunological synapse by agrin. Science. 2001;292:1681–1686. doi: 10.1126/science.1056594. [DOI] [PubMed] [Google Scholar]
- Kunz S., de la Torre J.C. Breaking the barrier: host cell invasion by lujo virus. Cell Host Microbe. 2017;22:583–585. doi: 10.1016/j.chom.2017.10.014. [DOI] [PubMed] [Google Scholar]
- Laval K., Favoreel H.W., Poelaert K.C., Van Cleemput J., Nauwynck H.J. Equine herpesvirus type 1 enhances viral replication in CD172a+ monocytic cells upon adhesion to endothelial cells. J. Virol. 2015;89:10912–10923. doi: 10.1128/JVI.01589-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma G., Azab W., Osterrieder N. Equine herpesviruses type 1 (EHV-1) and 4 (EHV-4)--masters of co-evolution and a constant threat to equids and beyond. Vet. Microbiol. 2013;167:123–134. doi: 10.1016/j.vetmic.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Mackow E.R., Gavrilovskaya I.N. Hantavirus regulation of endothelial cell functions. Thromb. Haemost. 2009;102:1030–1041. doi: 10.1160/TH09-09-0640. [DOI] [PubMed] [Google Scholar]
- Marsh M., Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothes W., Sherer N.M., Jin J., Zhong P. Virus cell-to-cell transmission. J. Virol. 2010;84:8360–8368. doi: 10.1128/JVI.00443-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W. Getting leukocytes to the site of inflammation. Vet. Pathol. 2013;50:7–22. doi: 10.1177/0300985812469883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W.A. Mechanisms of leukocyte transendothelial migration. Annu. Rev. Pathol. Mech. Dis. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterrieder N., Van de Walle G.R. Pathogenic potential of equine alphaherpesviruses: the importance of the mononuclear cell compartment in disease outcome. Vet. Microbiol. 2010;143:21–28. doi: 10.1016/j.vetmic.2010.02.010. [DOI] [PubMed] [Google Scholar]
- Pais-Correia A.-M., Sachse M., Guadagnini S., Robbiati V., Lasserre R., Gessain A., Gout O., Alcover A., Thoulouze M.-I. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 2010;16:83–89. doi: 10.1038/nm.2065. [DOI] [PubMed] [Google Scholar]
- Proft A., Spiesschaert B., Izume S., Taferner S., Lehmann M.J., Azab W. The role of the equine herpesvirus type 1 (EHV-1) US3-encoded protein kinase in actin reorganization and nuclear egress. Viruses. 2016;8:275. doi: 10.3390/v8100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes J.M., Simons M. The extracellular matrix and blood vessel formation: not just a scaffold. J. Cell Mol. Med. 2007;11:176–205. doi: 10.1111/j.1582-4934.2007.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozario T., DeSimone D.W. The extracellular matrix in development and morphogenesis: a dynamic view. Dev. Biol. 2010;341:126–140. doi: 10.1016/j.ydbio.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksena M.M., Wakisaka H., Tijono B., Boadle R.A., Rixon F., Takahashi H., Cunningham A.L. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J. Virol. 2006;80:3592–3606. doi: 10.1128/JVI.80.7.3592-3606.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattentau Q. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008;6:815–826. doi: 10.1038/nrmicro1972. [DOI] [PubMed] [Google Scholar]
- Sattentau Q.J. The direct passage of animal viruses between cells. Curr. Opin. Virol. 2011;1:396–402. doi: 10.1016/j.coviro.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Schaefer L., Schaefer R.M. Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 2010;339:237. doi: 10.1007/s00441-009-0821-y. [DOI] [PubMed] [Google Scholar]
- Schimmel L., Heemskerk N., van Buul J.D. Leukocyte transendothelial migration: a local affair. Small GTPases. 2017;8:1–15. doi: 10.1080/21541248.2016.1197872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt E.P., Lee W.L., Zemans R.L., Yamashita C., Downey G.P. On, around, and through: neutrophil-endothelial interactions in innate immunity. Physiology. 2011;26:334–347. doi: 10.1152/physiol.00011.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer N.M., Lehmann M.J., Jimenez-Soto L.F., Horensavitz C., Pypaert M., Mothes W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007;9:310. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D., Hamblin A., Edington N. Infection of endothelial cells with Equine herpesvirus-1 (EHV-1) occurs where there is activation of putative adhesion molecules: a mechanism for transfer of virus. Equine Vet. J. 2001;33:138–142. doi: 10.1111/j.2042-3306.2001.tb00591.x. [DOI] [PubMed] [Google Scholar]
- Smith K.C., Mumford J.A., Lakhani K. A comparison of equid herpesvirus-1 (EHV-1) vascular lesions in the early versus late pregnant equine uterus. J. Comp. Pathol. 1996;114:231–247. doi: 10.1016/s0021-9975(96)80045-4. [DOI] [PubMed] [Google Scholar]
- Sowinski S., Jolly C., Berninghausen O., Purbhoo M.A., Chauveau A., Köhler K., Oddos S., Eissmann P., Brodsky F.M., Hopkins C. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008;10:211. doi: 10.1038/ncb1682. [DOI] [PubMed] [Google Scholar]
- Spiesschaert B., Goldenbogen B., Taferner S., Schade M., Mahmoud M., Klipp E., Osterrieder N., Azab W. Role of gB and pUS3 in equine herpesvirus 1 transfer between peripheral blood mononuclear cells and endothelial cells: a dynamic in vitro model. J. Virol. 2015;89:11899–11908. doi: 10.1128/JVI.01809-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiesschaert B., Osterrieder N., Azab W. Comparative analysis of glycoprotein B (gB) of equine herpesvirus type 1 and type 4 (EHV-1 and EHV-4) in cellular tropism and cell-to-cell transmission. Viruses. 2015;7:522–542. doi: 10.3390/v7020522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavolone L., Lionetti V. Extracellular matrix in plants and animals: hooks and locks for viruses. Front. Microbiol. 2017;8:1760. doi: 10.3389/fmicb.2017.01760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet C. The pathogenicity of cytomegalovirus. FEMS Microbiol. Rev. 1999;23:457–482. doi: 10.1111/j.1574-6976.1999.tb00408.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi K., Miyajima N., Nagata N., Takeda M., Tashiro M. Wild-type measles virus induces large syncytium formation in primary human small airway epithelial cells by a SLAM (CD150)-independent mechanism. Virus Res. 2003;94:11–16. doi: 10.1016/s0168-1702(03)00117-5. [DOI] [PubMed] [Google Scholar]
- Tarasevich A., Filatov A., Pichugin A., Mazurov D. Monoclonal antibody profiling of cell surface proteins associated with the viral biofilms on HTLV-1 transformed cells. Acta Virol. 2015;59:247–256. doi: 10.4149/av_2015_03_247. [DOI] [PubMed] [Google Scholar]
- Tyagi M., Rusnati M., Presta M., Giacca M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2001;276:3254–3261. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- Vaheri A., Carpén O., Heiska L., Helander T.S., Jääskeläinen J., Majander-Nordenswan P., Sainio M., Timonen T., Turunen O. The ezrin protein family: membrane-cytoskeleton interactions and disease associations. Curr. Opin. Cell Biol. 1997;9:659–666. doi: 10.1016/s0955-0674(97)80119-6. [DOI] [PubMed] [Google Scholar]
- Valbuena G., Walker D.H. The endothelium as a target for infections. Annu. Rev. Pathol. Mech. 2006;1:171–198. doi: 10.1146/annurev.pathol.1.110304.100031. [DOI] [PubMed] [Google Scholar]
- Van de Walle G.R., Favoreel H.W., Nauwynck H.J., Mettenleiter T.C., Pensaert M.B. Transmission of pseudorabies virus from immune-masked blood monocytes to endothelial cells. J. Gen. Virol. 2003;84:629–637. doi: 10.1099/vir.0.18796-0. [DOI] [PubMed] [Google Scholar]
- van der Meulen K.M., Nauwynck H.J., Pensaert M.B. Mitogen stimulation favours replication of equine herpesvirus-1 in equine blood mononuclear cells by inducing cell proliferation and formation of close intercellular contacts. J. Gen. Virol. 2001;82:1951–1957. doi: 10.1099/0022-1317-82-8-1951. [DOI] [PubMed] [Google Scholar]
- van Leeuwen H., Elliott G., O'Hare P. Evidence of a role for nonmuscle myosin II in herpes simplex virus type 1 egress. J. Virol. 2002;76:3471–3481. doi: 10.1128/JVI.76.7.3471-3481.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Maanen C. Equine herpesvirus 1 and 4 infections: an update. Vet. Q. 2002;24:57–78. [PubMed] [Google Scholar]
- Vandekerckhove A.P., Glorieux S., Gryspeerdt A., Steukers L., Van Doorsselaere J., Osterrieder N., Van de Walle G., Nauwynck H. Equine alphaherpesviruses (EHV-1 and EHV-4) differ in their efficiency to infect mononuclear cells during early steps of infection in nasal mucosal explants. Vet. Microbiol. 2011;152:21–28. doi: 10.1016/j.vetmic.2011.03.038. [DOI] [PubMed] [Google Scholar]
- Wewer C., Seibt A., Wolburg H., Greune L., Schmidt M.A., Berger J., Galla H.J., Quitsch U., Schwerk C., Schroten H. Transcellular migration of neutrophil granulocytes through the blood-cerebrospinal fluid barrier after infection with Streptococcus suis. J. Neuroinflamm. 2011;8:51. doi: 10.1186/1742-2094-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson W.D. Equine herpesvirus 1 myeloencephalopathy. Vet. Clin. North Am. Equine Pract. 1997;13:53–72. doi: 10.1016/s0749-0739(17)30255-9. [DOI] [PubMed] [Google Scholar]
- Yaacov B., Lazar I., Tayeb S., Frank S., Izhar U., Lotem M., Perlman R., Ben-Yehuda D., Zakay-Rones Z., Panet A. Extracellular matrix constituents interfere with Newcastle disease virus spread in solid tissue and diminish its potential oncolytic activity. J. Gen. Virol. 2012;93:1664–1672. doi: 10.1099/vir.0.043281-0. [DOI] [PubMed] [Google Scholar]
- Zhong P., Agosto L.M., Munro J.B., Mothes W. Cell-to-cell transmission of viruses. Curr. Opin. Virol. 2013;3:44–50. doi: 10.1016/j.coviro.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PBMC were infected with EHV-1RFP+GFP (MOI=3, red) for 5 min and overlaid on EC. Virus replication in EC is indicated by GFP expression, which is integrated in the viral genome (green), whereas RFP signals represent new progenies of RFP-labeled virus production (red) in the infected cells.
Data Availability Statement
This study did not generate/analyze datasets/code.