

Abstract
Yes-associated protein (YAP) signaling has emerged as a crucial pathway in several normal and pathological processes. Although the main upstream effectors that regulate its activity have been extensively studied, the role of the endosomal system has been far less characterized. Here, we identified the late endosomal/lysosomal adaptor MAPK and mTOR activator (LAMTOR) complex as an important regulator of YAP signaling in a preosteoblast cell line. We found that p18/LAMTOR1-mediated peripheral positioning of late endosomes allows delivery of SRC proto-oncogene, nonreceptor tyrosine kinase (SRC) to the plasma membrane and promotes activation of an SRC-dependent signaling cascade that controls YAP nuclear shuttling. Moreover, β1 integrin engagement and mechano-sensitive cues, such as external stiffness and related cell contractility, controlled LAMTOR targeting to the cell periphery and thereby late endosome recycling and had a major impact on YAP signaling. Our findings identify the late endosome recycling pathway as a key mechanism that controls YAP activity and explains YAP mechano-sensitivity.
Keywords: late endosomes, LAMTOR complex, Yes-associated protein (YAP), cell adhesion, SRC kinase, vesicular trafficking, mechanosensing, cell signaling, extracellular matrix (ECM)
The integration of biological and mechanical signals coming from the surrounding cells and the extracellular matrix (ECM) is crucial for development, tissue homeostasis, and tumor progression. In adherent cells, external cues are involved in the control of numerous processes, including the adhesion-regulated formation of signaling platforms at the cell surface (1), endocytosis and trafficking of signaling receptors (2–4), and consequently the control of specific transcription factors and chromatin remodelers that shuttle into the nucleus to modulate gene expression (5–7).
The nuclear shuttling of Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) is directly controlled through ECM compliance and composition and also through cell shape and confluence (8). In the nucleus, YAP and TAZ interact with TEAD family members to drive or modulate gene expression (6). In turn, the expression of specific genes affects cell behavior, such as proliferation/differentiation and migration, thus integrating external cues for cells to adapt to their extracellular environment (9). Over the last decade, the core signaling pathway leading to YAP and TAZ nuclear translocation has been extensively studied, giving important insights into the cell physiology (9, 10). Integrin-dependent cell adhesion and particularly β1 integrins are crucial for YAP nuclear translocation (11, 12). Indeed, β1 integrin–dependent cell adhesion, through the SRC family kinases, leads to Rac1 recruitment and activation at protrusive cell borders to stimulate a PAK1-dependent cascade resulting in merlin phosphorylation. Phosphorylated merlin releases YAP from a merlin/LATS/YAP inhibitory complex, allowing its nuclear translocation (12).
Vesicular trafficking is emerging as an important process involved in cell signaling. Whereas receptor activation mostly takes place at the plasma membrane, receptor endocytosis and sorting between recycling and degradation compartments also are important for receptor signaling output. Late endosome (LE) subcellular positioning is crucial for controlling the cell anabolic/catabolic status (13–16). LE peripheral or perinuclear positioning depends on kinesin and dynein activities, respectively. In addition, the LAMTOR complex, a LE/lysosomal scaffolding protein complex that integrates several pathways, such as the mitogen-activated protein kinase and mTORC1 signaling cascades (16–18), participates in LE positioning by inhibiting the Arl8/BORC complex involved in LE peripheral targeting (19–20). At the cell periphery, LEs target focal adhesions (FAs) and regulate their dynamics during cell migration, although the exact mechanism involved is still puzzling (21). However, the role of this vesicular compartment in the regulation of the signaling pathway that controls YAP activation has not been described yet.
Here, we found that β1 integrin–mediated cell adhesion and mechanical inputs regulate LE peripheral dispersion concomitantly with YAP activation. By knocking down p18/LAMTOR1, a major LAMTOR complex subunit, we demonstrated that LAMTOR recruitment to LEs is required for YAP nuclear shuttling and for LE targeting to FAs. Finally, we found that β1 integrins, through integrin-linked protein kinase (ILK), allows the organization of a functional microtubule network that transports SRC-positive LEs to the cell periphery, a crucial process in the regulation of YAP nuclear shuttling.
Results
LE distribution is mechano-sensitive
β1 integrins integrate mechano-dependent inputs from the ECM and control vesicular trafficking (3, 22). These receptors also specifically regulate the nuclear translocation of YAP, a bona fide mechano-sensitive regulator of gene expression (8, 11, 12, 23). Therefore, we asked whether LAMTOR-positive LE distribution and dynamics were mechano-sensitive and could act as a molecular link between integrins and YAP activation. First, we verified that YAP activation was mechano-sensitive in the preosteoblast cell line used for this study. YAP expression was mainly nuclear in cells cultured on fibronectin-coated polydimethylsiloxane (PDMS) medium and stiff hydrogels (10 and 30 kPa). Lowering compliance to 2 kPa led to a significant YAP redistribution in the cytoplasm (Fig. 1, A and B). Next, we analyzed LAMTOR complex distribution under the same experimental conditions by monitoring the localization of p18/LAMTOR1 fused to GFP (p18-GFP), one of the main subunits of this complex. In cells grown under high-stiffness conditions, p18-positive vesicles were both perinuclear and peripheral, whereas in cells grown in low-stiffness conditions, the amount of peripheral p18-positive vesicles was significantly reduced (Fig. 1, C and D). We observed a similar effect of the matrix rigidity on LE distribution when GFP–Rab-7 was used as an LE marker (Fig. 1C). These data demonstrate that YAP mechano-sensitive response is correlated with LAMTOR-positive LE dispersion toward the plasma membrane. As β1 integrins are key mechano-receptors that control YAP nuclear translocation (11, 12, 24), we asked whether β1 integrins also control LE subcellular distribution. After transduction of viral particles that express p18-GFP and RFP-VASP (to reveal FAs) in parental (β1f/f) preosteoblasts (control) and in preosteoblasts that lack the β1 integrin subunit (β1−/−) (25), we observed that p18/LAMTOR1-positive vesicles were at the cell periphery in control cells. Conversely, in β1−/− cells, the density of peripheral LEs as well as the number of LEs targeting FA were significantly reduced (Fig. 1, E and F). This defect in LE distribution appears to be specific because when early endosome distribution was analyzed we did not notice any significant difference in their distribution between both genotypes (Fig. 1, G and H). Altogether, these data show that β1 integrins are crucial mechano-receptors controlling LE peripheral dispersion and YAP activation.
Figure 1.

Mechanical and β1 integrin–mediated regulation of YAP and LE subcellular localization in preosteoblasts. A, comparison of YAP cytoplasmic/nuclear ratios in different stiffness conditions. Preosteoblast cells that stably express mCherry-YAPwt were seeded and grown on fibronectin-coated PDMS hydrogels of different stiffness (30, 10, and 2 kPa) for 2 h. YAP subcellular localization was then analyzed by fluorescence imaging with a confocal microscope. Intensity values were obtained using Fiji software (data are represented on a logarithmic scale). Data were compared with the two-tailed unpaired Student's t test and are representative of two independent experiments with n > 30 cells analyzed (***, p < 0.0001; ns, not significant). B, subcellular localization of mCherry-YAPwt (red) in preosteoblast cells spread on fibronectin-coated PDMS hydrogels of different stiffness (Young moduli are 30, 10, and 2 kPa) for 2 h. Scale bar, 10 μm. C, subcellular localization of p18/LAMTOR1 (top panels) and Rab-7 (bottom panels) in preosteoblast cells spread on fibronectin-coated PDMS hydrogels of different stiffness (Young moduli are 30, 10, and 2 kPa) for 2 h. Scale bar, 10 μm. D, comparison of p18/LAMTOR1-GFP subcellular distribution in preosteoblast cells spread on fibronectin-coated PDMS hydrogels of different stiffness (30, 10, and 2 kPa) for 2 h. GFP fluorescence was imaged, and p18/LAMTOR1 distribution was analyzed using Icy software. The histogram shows the localization of p18/LAMTOR1-positive vesicles from the cell edges to the nucleus (expressed as percentage of all vesicles). Data are the mean ± S.D. (error bars) of two independent experiments. The localization of p18/LAMTOR1-positive vesicles was significantly (E) different only between the 20- and 2-kPa conditions for 0–1 (***) and 1–2 (**) µm (two-tailed unpaired Student's t test). F, control (β1f/f) and β1 integrin–deficient preosteoblasts (β1−/−) that stably express p18/LAMTOR1-GFP (green) and mRFP-VASP (red) were grown overnight on glass coverslips and then imaged by fluorescence microscopy. Scale bar, 10 μm. G, comparison of p18/LAMTOR1-GFP subcellular distribution in control (β1f/f, green) and in β1-deficient osteoblasts (β1−/−, blue) cells quantified using Icy software. Histograms represent the localization from the cell edges to the cell nucleus, which is expressed as a percentage of all vesicles. The localization of p18/LAMTOR1-positive vesicles was significantly different only for 0–1 (***) and 1–2 (**) µm (two-tailed unpaired Student's t test). H, control (β1f/f) and β1 integrin–deficient preosteoblasts (β1−/−) were grown overnight on glass coverslips and stained for EEA1 to visualized early endosomes. Scale bar, 10 μm. I, comparison of the subcellular distribution of EEA1-positive vesicles in control (β1f/f, green) and in β1-deficient osteoblasts (β1−/−, blue) cells quantified using Icy software. Histograms represent the localization from the cell edges to the cell nucleus, which is expressed as a percentage of all vesicles.
LAMTOR controls YAP nuclear translocation
As the LAMTOR complex is implicated in the regulation of LE positioning in the cell (18, 26), we investigated its role in YAP signaling. To this goal, preosteoblast cells expressing stable shRNAs against p18/LAMTOR1 (sh-p18 cells) were generated and characterized. It was reported that p18/LAMTOR1 is required for docking the LAMTOR complex to LE surface, and consequently its loss leads to the complete inactivation of the complex (18). Indeed, the LAMTOR complex that was properly detected at LE surface in control cells or in rescue cells (sh-p18 expressing p18-GFP) was no longer observed upon p18/LAMTOR1 silencing (Fig. S1, A and B).
Having validated the functional loss of the LAMTOR complex upon p18/LAMTOR1 silencing, next we investigated whether this complex is involved in YAP signaling by analyzing its subcellular localization. With cells cultured on stiff substrate, the silencing of p18/LAMTOR1 significantly reduced YAP nuclear staining compared with control cells (sh-ctl; scramble shRNA) (Fig. 2, A and B). Although not being specifically addressed in this experimental setting, the re-expression of GFP-p18 restored the defective endogenous YAP nuclear localization observed upon p18 silencing (see Fig. 5G), indicating that the effect of p18-directed shRNAs was not due to an off-target silencing. In agreement with its reduced nuclear localization, YAP was reduced but hyperphosphorylated in sh-p18 cells compared with sh-ctl and rescued cells (resc) (Fig. 2C). The reduced level of YAP is likely reflecting its phosphorylation-dependent degradation as reported previously (27).
Figure 2.

p18/LAMTOR1 is involved in YAP nuclear signaling. A, comparison of the YAP cytoplasmic/nuclear ratios (logarithmic scale) in preosteoblast cells that stably express mCherry-YAPwt and scramble (sh-ctl) or shRNAs against p18/LAMTOR1 (sh-p18) grown on glass coverslips overnight. YAP subcellular localization was analyzed by confocal microscopy and quantified with the Fiji software. Data are the mean ± S.D. (error bars) of two independent experiments with n > 30 (two-tailed unpaired Student's t test). B, subcellular localization of mCherry-YAPwt (red) in preosteoblast cells that stably express scramble (sh-ctl) or sh-RNAs against p18/LAMTOR1 (sh-p18). Scale bar, 10 μm. C, Western blot analysis of YAP, YAPpS127, and p18/LAMTOR1 expression in the indicated preosteoblast cell lines. Band intensities were quantified with a Chemidoc CCD camera (Bio-Rad), and ratios were calculated with Image Laboratory software (Bio-Rad). Actin was used as an internal loading control. Results are representative of three independent experiments. resc, sh-p18 cells that express exogenous p18/LAMTOR1-GFP. D, comparison of YAP cytoplasmic to nuclear ratios (logarithmic scale) in preosteoblast cells (β1f/f) that express p18-GFP or GFP-p18 after overnight growth on glass coverslips. YAP subcellular localization was analyzed by indirect immunofluorescence, and signal intensity was quantified from confocal images with the Fiji software. Data (mean ± S.D.) are representative of two independent experiments with n > 30 cells analyzed (two-tailed unpaired Student's t test). E, in vitro osteogenic differentiation of preosteoblast cells (β1f/f) that stably express scramble (sh-ctl) or shRNAs against p18/LAMTOR1 (sh-p18). Alkaline phosphatase (ALP) and Alizarin Red S (ARS; to detect calcium deposition) staining were performed to monitor differentiation status at day (D) 0, 4, and 15, as indicated. ***, p < 0.0001.
Figure 5.

p18/LAMTOR-dependent SRC delivery to the plasma membrane controls YAP nuclear shuttling. A, control preosteoblast cells were transiently transfected with p18-GFP and SRC-mCherry. 24 h post-transfection, cells were seeded on glass coverslips, and GFP and mCherry signals were acquired by confocal microscopy. Scale bar, 10 μm. B, histogram representing the Pearson coefficient and thresholded Manders (tM1, tM2) values obtained from confocal images of p18-GFP and SRC-mCherry co-transfected preosteoblast cells. Images were analyzed using the Fiji Jacop plugin; data are the mean ± S.D. of >35 cells/condition (two-tailed unpaired Student's t test). C, control (sh-ctl) and sh-p18 preosteoblast cells were transiently transfected with GFP–Rab-7 and SRC-mCherry. 24 h post-transfection, cells were seeded on glass coverslips, and the GFP and mCherry signals were acquired by confocal microscopy. Due to the lack of an appropriate tagged p18/LAMTOR1 construct, rescued cells were not investigated. Scale bar, 10 μm. D, histogram representing the Pearson coefficient and thresholded Manders (tM1, tM2) values obtained from confocal images of sh-ctl (red) and sh-p18 cells (blue) co-transfected with GFP–Rab-7 and mCherry-SRC. Images were analyzed using the Fiji Jacop plugin. Due to the lack of an appropriate tagged p18/LAMTOR1 construct, rescued cells were not investigated. Data are the mean ± S.D. of >35 cells/condition (two-tailed unpaired Student's t test). E, Western blotting analysis of SRC, phosphorylated (p) SRCY146, p130CAS, pp130CAS, paxillin, and ppaxillinY31 from sh-ctl, sh-p18, and resc (sh-p18 with p18/LAMTOR1-GFP expression) cell lysates. Actin was used as a loading control. Results are representative of three independent experiments. F, sh-ctl cells incubated or not (ctl) with everolimus (evero.), sh-p18 cells, and sh-p18 cells that express p18-GFP (resc) were grown overnight on glass coverslips and stained for SRC and pSRCY416. Scale bar, 10 μm. G, comparison of YAP cytoplasmic/nuclear ratio (logarithmic scale) in sh-p18 cells and resc cells (sh-p18 cells that express p18-GFP) that express or not the active form of SRC (SRCYF) seeded overnight on glass coverslips. YAP subcellular localization was quantified from confocal images using Fiji software. Data are the mean ± S.D. of two independent experiments with >30 cells/condition (two-tailed unpaired Student's t test). Subcellular localization of YAP in sh-p18 preosteoblast cells and in sh-p18 cells that express constitutively active SRCYF (sh-p18+SRCYF). Scale bar, 10 μm. ***, p < 0.0001; ns, not significant.
As mentioned above, the LAMTOR complex docks at LE surfaces using p18/LAMTOR1, in particular its N-terminal moiety. Therefore, the addition of a tag to the p18/LAMTOR1 N terminus should interfere with its recruitment to the LE surface, and this fusion protein should act as a dominant negative form. Therefore, to complement the silencing strategy, we generated a p18/LAMTOR1 chimeric protein in which the GFP protein was fused to its N terminus (GFP-p18). As expected for a dominant negative construction, GFP-p18 displayed a diffuse staining within the cells, and the LAMTOR complex was no longer detected at the LE surface (Fig. S1, A and B). Further supporting the role of p18/LAMTOR1 in YAP signaling, p18/LAMTOR1 silenced cells displayed a significant reduction in YAP nuclear localization (Fig. 2D and Fig. S1B). These results not only confirmed that LAMTOR mediates YAP nuclear localization but also suggest that this process requires LAMTOR docking at the LE surface.
YAP signaling was reported to support osteoblast differentiation (8), and consequently sh-p18 cells are expected to present a defect in this process. Therefore, we investigated whether osteogenic differentiation was affected in sh-p18 preosteoblasts. Control and sh-p18 cells were cultured in osteogenic medium, and alkaline phosphatase activity (ALP) was analyzed to visualize the initial stage of osteogenesis. At day 4 of differentiation, ALP staining was significantly reduced in sh-p18 cells compared with sh-ctl cells. At later differentiation stages (day 15), mineralization also was defective in sh-p18 cells, but not in control cells (Fig. 2E). Altogether, these findings show that the LAMTOR complex has an important role in well-characterized YAP signaling regulations.
LAMTOR controls YAP nuclear translocation and LE targeting to FAs independently of the mTOR pathway
Next, we aimed at understanding how the LAMTOR complex regulates YAP nuclear translocation. Because this later complex controls mTORC1 signaling (17, 28), we investigated the potential role of mTORC1 in YAP signaling. Supporting previous findings, p18/LAMTOR1 silencing induced a decrease in the phosphorylation level of the S6 ribosomal protein, a well-known downstream target of mTORC1 (Fig. 3A). Then we tested whether the defect in YAP nuclear localization observed in sh-p18 cells was caused by down-regulation of the mTOR pathway by inhibiting mTORC1 activity with everolimus. Upon incubation with everolimus, phosphorylated S6 was almost undetectable in β1f/f preosteoblasts cultured on a stiff substrate, confirming the strong inhibitory effect of this molecule (Fig. 3A). However, YAP subcellular localization was comparable in everolimus-treated and control cells (not treated), showing that mTORC1 activation is dispensable for YAP nuclear translocation on a stiff substrate (Fig. 3, B and C).
Figure 3.
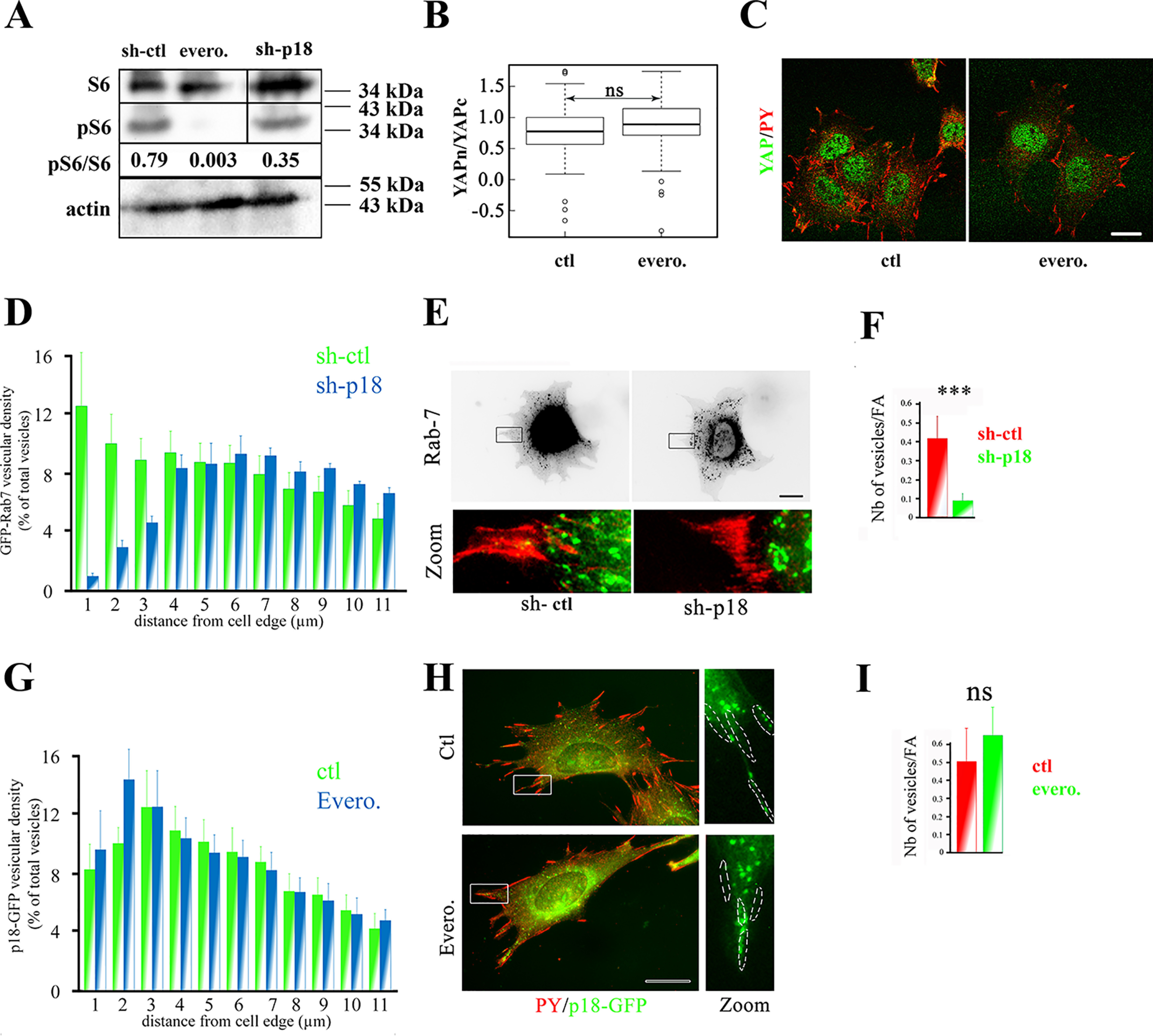
p18/LAMTOR1 controls LE peripheral distribution and YAP signaling in a mTORC1-independent manner. A, Western blotting analysis of the ribosomal protein S6 (S6) and its phosphorylated form (pS6). Preosteoblasts (β1f/f) were incubated (evero.) or not (ctl) with everolimus (10 nm, 3 h). Band intensity was quantified with a Chemidoc CCD camera (Bio-Rad) and Image Laboratory software (Bio-Rad). Actin was used as internal loading control. Results are representative of three independent experiments. B, comparison of YAP cytoplasmic to nuclear ratio (logarithmic scale) in preosteoblasts (β1f/f) grown overnight and then incubated (evero.) or not (ctl) with everolimus (10 nm, 3 h) and stained for YAP. YAP subcellular localization was analyzed by confocal microscopy, and intensity values were obtained using Fiji software. Data are the mean ± S.D. (error bars) of two independent experiments with n >30 cells analyzed (two-tailed unpaired Student's t test). C, immunostaining of YAP (green) in preosteoblasts (β1f/f) incubated (evero.) or not (ctl) with everolimus (10 nm, 3 h). Cells were incubated with antibodies against YAP and phosphorylated tyrosines (PY) to label focal adhesions. Images were obtained with a confocal microscope. Bar, 10 μm. D, comparison of Rab-7–GFP subcellular distribution in sh-ctl (green histograms) and sh-p18 (blue histograms) cells. Rab-7–GFP distribution was analyzed using Icy software. Data are the mean ± S.D. (two experiments) of Rab-7–positive vesicle localization from the cell edges to the cell nucleus (percentage of all vesicles). Vesicular distribution was significantly different for : 0–3 μm (p < 0.0001), 10–12 μm (p < 0.001) and 8–10 μm (p < 0.01). Due to the lack of an appropriate tagged p18/LAMTOR1 construct, rescued cells were not investigated in this experiment. E, sh-ctl and sh-p18 cells that stably express mRFP-paxillin (red) were transiently transfected with GFP–Rab-7 (green). 24 h post-transfection, cells were seeded on glass coverslips and fixed overnight. Due to the lack of an appropriate tagged p18/LAMTOR1 construct, rescued cells were not investigated. Scale bar, 10 μm. F, quantification of GFP–Rab-7 targeting to focal adhesions in control (sh-ctl, red) and sh-p18 cells (green). Two-tailed unpaired Student's t test was used with n = 20 cells/condition. *** p < 0.0001. G, comparison of p18-GFP subcellular distribution in control (ctl, red) and everolimus-treated cells (evero., green). p18-GFP distribution was analyzed using Icy software. A histogram represents the stepwise localization of p18-GFP–positive vesicles (percentage of all vesicles) from the cell edges to the cell nucleus, and data are the mean ± S.D. of three independent experiments (n >30 cells/condition; two-tailed unpaired Student's t test). H, preosteoblast cells were incubated (evero) or not (ctl) with everolimus (10 nm, 3 h), and p18-GFP distribution was visualized. Cells were labeled with the anti-phosphotyrosine (PY) antibody to FAs. Bar, 10 μm. I, quantification of p18-GFP targeting to FAs in control (red) and everolimus (10 nm, 3 h)-treated cells. Data were compared with the two-tailed unpaired Student's t test (n = 15 cells/condition). ns, not significant.
Having excluded the involvement of a LAMTOR-dependent mTOR signaling axis, we hypothesized that this complex might regulate YAP by controlling LE distribution. In particular, it was reported that LEs are targeted to adhesive structures, such as FAs and fibrillar adhesions (21, 29), from where they could initiate or modulate some signaling pathways. To further investigate this putative connection, we co-expressed GFP–Rab-7 and RFP-paxillin (to label LEs and FAs, respectively) in both sh-ctl and sh-p18 cells. First, we confirmed that p18/LAMTOR1 silencing significantly affected LE subcellular positioning (Fig. 3, D and E). We also observed that LE vesicles (green in Fig. 3E) were in close contact with FAs (red in Fig. 3E) in control cells. Conversely, the number of vesicles targeted to FAs was significantly reduced in sh-p18 cells (Fig. 3E; for quantification, see Fig. 3F). Altogether, these data show that p18/LAMTOR1 is an important player in LE targeting to FAs. Importantly, LE positioning and targeting to FAs were not modified by incubation of β1f/f preosteoblasts with everolimus, showing that mTORC1 is not involved in these processes (Fig. 3 (G–I) and Movies S1 and S2). Collectively, these data indicated that LE dynamics and targeting to FAs are controlled by the LAMTOR complex in an mTORC1-independent manner.
LAMTOR-positive LE subcellular distribution and targeting to FAs is β1 integrin/ILK– and microtubule–dependent
β1 integrin–mediated adhesion is crucial for YAP nuclear translocation (11, 12, 24); therefore, the p18/LAMTOR1 role in YAP nuclear translocation could be a consequence of a defective β1 integrin delivery to the plasma membrane and/or its recruitment to FAs. To monitor the β1 integrin amount at the cell surface, we included Rab-11-silenced cells (sh-Rab-11) as a positive control because Rab-11 regulates β1 integrin recycling (30–32). First, we analyzed β1 integrin cell-surface expression in sh-ctl, sh-Rab-11, and sh-p18 cells by FACS. Compared with sh-ctl cells, the cell-surface expression of β1 integrins was decreased in sh-Rab-11 cells and increased in sh-p18 cells (Fig. 4A). Quantification of the projected cell surface after 3 h of spreading on fibronectin supported these findings. Indeed, Rab-11 silencing promoted the partial rounding up of cells, but not p18/LAMTOR1 silencing (Fig. 4B). Finally, immunostaining revealed that β1 integrins were localized in phosphorylated paxillin (pPaxillin)-positive FAs in both sh-ctl and sh-p18 cells, but not in sh-Rab-11 cells (Fig. 4C). These results strongly suggest that p18/LAMTOR1 controls YAP localization not by simply down-regulating β1 integrin expression at the cell surface.
Figure 4.

LE subcellular distribution and targeting to FAs are β1 integrin/ILK- and microtubule-dependent. A, FACS analysis of β1 integrin surface expression. Preosteoblast cells (β1f/f) that stably express scramble (sh-ctl, green) or shRNAs against p18/LAMTOR1 (sh-p18, red) or Rab-11a (sh-Rab-11, blue) were stained with the monoclonal anti-mouse β1 integrin MB1.2 antibody. B, quantification of spreading of sh-ctl, sh-p18, and sh-Rab11 preosteoblast cells (β1f/f). Cells were seeded and grown on fibronectin (5 µg/ml) for 2 h, and cell spreading was quantified using ImageJ. C, immunostaining of β1 integrins (MB1.2, green) and phosphorylated paxillin (anti-pTyr-31, red) of preosteoblast cells (β1f/f) that stably express sh-ctl, sh-p18, or sh-Rab-11 spread overnight on glass coverslips. Due to the lack of an appropriate tagged p18/LAMTOR1 construct, rescued cells were not investigated. Bar, 10 μm. D, control (β1f/f) and β1−/− preosteoblasts that stably express p18/LAMTOR1-GFP (green) and mRFP-VASP (red) were seeded and grown overnight on glass coverslips, and GFP and mRFP distribution was visualized. Scale bar, 10 μm. E, quantification of p18/LAMTOR1 targeting to FAs in control (β1f/f, green), sh-Rab-11 (red), and β1−/− preosteoblasts (blue). Data (mean ± S.D. (error bars) of n > 20 cells/condition) were compared with the two-tailed unpaired Student's t test. F, β1−/− preosteoblasts that stably express the β3-GFP (green, left) or β3-ILK-GFP (green, right) fusion proteins were seeded on glass coverslips and stained for microtubules (red, top panels) or YAP (red, bottom panels). Scale bar, 10 μm. G, comparison of YAP cytoplasmic/nuclear ratio (logarithmic scale) in β1f/f and β1−/− preosteoblasts that stably express or not the β3-GFP or β3-ILK-GFP fusion protein after overnight growth on glass coverslips. YAP subcellular localization was quantified from confocal microscopy images with Fiji software; shown are data (mean ± S.D.) from two independent experiments (n > 30 cells/condition) (two-tailed unpaired Student's t test). *, p < 0.01; **, p < 0.001; ***, p < 0.0001; ns, not significant.
Together with the previous findings showing that β1 integrins regulate LE positioning, this suggested that β1 integrins are required for LE targeting to FAs. Indeed, the targeting of p18/LAMTOR–GFP-positive LEs to FAs was significantly decreased in β1−/− cells compared with control β1f/f cells (Fig. 4, D and E). Similarly, dynamic analyses by total internal reflection fluorescence (TIRF)-based video microscopy demonstrated that p18/LAMTOR1 targeting to paxillin-labeled FAs was defective in β1−/− cells (Movies S3 and S4). All of these findings indicate that β1 integrin–dependent cell adhesion controls p18/LAMTOR1-positive LE trafficking and targeting to FA sites.
In addition to β1 integrins, ILK also has been implicated in YAP signaling (12, 33). One of the functions of ILK is to scaffold proteins to allow microtubule anchoring to FAs (Fig. S2A) (22, 34). This suggests that the β1 integrin/ILK complex might be required for LE trafficking along the microtubule network. Indeed, we frequently observed p18/LAMTOR1-positive vesicles close to microtubules. Moreover, incubation with nocodazole (a microtubule inhibitor) led to their relocation toward a perinuclear region (Fig. S2A). In β1−/− cells, ILK expression at FAs was reduced, and consequently, microtubule targeting to FAs was reduced as well (Fig. S2, B–D). Therefore, these data show that β1 integrin/ILK–positive FAs are required for microtubule anchoring that ultimately drives LE dynamics.
To more directly assess whether the microtubule-targeting defect at FAs affected YAP nuclear translocation, we expressed a β3 integrin-ILK-GFP chimeric protein in β1−/− cells to force ILK localization to FAs. The chimeric protein was expressed at a similar level as β3-GFP used as control (Fig. S2E) and correctly incorporated in FAs, as reported previously (35). Microtubule targeting to FAs was restored in β3 integrin-ILK-GFP– but not in β3 integrin-GFP–expressing cells (Fig. 4F, top panels). Immunostaining and quantification of the cytoplasmic/nuclear YAP ratio revealed a significant increase in YAP nuclear translocation in β3 integrin-ILK-GFP–expressing β1−/− cells when compared with β3-GFP–expressing β1−/− or β1−/− cells (Fig. 4, F (bottom panels) and G). These data show that microtubule targeting to FAs is crucial for YAP nuclear translocation to control LAMTOR-positive LE targeting to FAs.
p18/LAMTOR1 regulates SRC signaling
We next hypothesized that these LE vesicles could carry signaling proteins involved in YAP regulation. The nonreceptor tyrosine kinase SRC, which is emerging as an important regulator of YAP nuclear shuttling upon integrin activation, is dynamically translocated from a vesicular pool toward the plasma membrane, where it activates its downstream targets (36–37). First, to determine whether SRC could be detected on LE vesicles, we transiently co-expressed fluorescently tagged SRC (SRC-mCherry) and p18/LAMTOR1 (p18-GFP) in preosteoblasts. As reported previously (37), confocal microscopy imaging revealed that the SRC-mCherry signal was not only diffused in the cytoplasm, but also appeared as punctuate staining. These vesicular-like structures were also p18/LAMTOR1-positive (Fig. 5A). Quantification analysis revealed a Spearman coefficient >0.5 (significant) and thresholded Manders (tM1) values up to 0.7 when the green channel (p18/LAMTOR1) overlapped with the red channel (SRC-mCherry). The smaller tM2 value (red overlapping with green) might reflect a SRC fraction more broadly located to membranes (Fig. 5B). Then, to assess whether SRC and p18/LAMTOR1 were dynamically coupled, we transiently transfected SRC-mCherry and p18-GFP in SRC, Yes, and Fyn (SYF) triple knockout cells. Time-lapse video microscopy confirmed SRC and p18/LAMTOR1 co-localization and co-trafficking in these cells (Movie S5). Altogether, these data showed that a pool of SRC is recruited to p18/LAMTOR1-positive vesicles. As we reported previously that p18/LAMTOR1 controls LE distribution, we asked whether it also regulates SRC distribution. Analysis of SRC distribution in sh-p18 and sh-ctl cells that express GFP–Rab-7 (to visualize LEs) showed a significant co-localization of SRC-mCherry and GFP–Rab-7 in control cells, further confirming SRC localization on LE. Conversely, in sh-p18 cells, SRC recruitment on LE was significantly reduced (Fig. 5, C and D).
Next, we investigated whether p18/LAMTOR1 silencing affects SRC activity by monitoring SRC phosphorylation at Tyr-416 (pSRCY416) by Western blotting. In sh-p18 cells, total SRC and pSRCY416 levels were strongly reduced compared with sh-ctl and resc cells (Fig. 5E). This suggests that LE dynamics might regulate SRC sorting between recycling LEs (signaling) and the degradative endolysosomal compartment. Immunofluorescence analysis did not allow detection of endogenous total SRC and pSRCY416 on vesicles. Conversely, they were clearly present at FAs in sh-ctl cells but barely visible in sh-p18 cells (arrows in Fig. 5F and Fig. S3A). In sh-p18 cells, total SRC and pSRCY416 signal in FAs was strongly increased upon expression of exogenous p18 (resc cells). Incubation with the mTORC1 inhibitor everolimus, which did not have any effect on LE distribution (Fig. 3G), did not modify total SRC and pSRCY416 signal at FAs (Fig. 5F).
Next, we monitored the phosphorylation and localization of two well-established SRC downstream targets: p130CAS and paxillin. In agreement with the reduced level of activated SRC in FAs of sh-p18 cells, p130CAS phosphorylation also was reduced upon p18/LAMTOR1 silencing (Fig. 5E). In sh-ctl cells, we observed p130CAS phosphorylation mainly at FA sites, as expected, whereas it was markedly reduced in sh-p18 (Fig. S3B). As previous studies showed that p130CAS is phosphorylated at FAs upon stretching, these data strongly suggest that p18/LAMTOR1 controls SRC delivery and activity at these sites (38). Conversely, paxillin phosphorylation was not modified in sh-p18 cells (Fig. 5E and Fig. S3C), suggesting that paxillin phosphorylation is not dependent on FA-associated SRC activity or alternatively involves another SRC family member, such as Fyn or Yes. Together, these data highlight an important role for the LAMTOR complex in regulating SRC association with and expression in LEs.
Finally, to determine whether the defect in YAP nuclear translocation in sh-p18 cells was mediated by the lack of SRC activity, we transduced sh-p18 cells with the constitutive activated form of SRC (sh-p18+SRCYF) and analyzed YAP subcellular localization (Fig. 5, G and H). Expression of activated SRC restored YAP nuclear localization. We previously reported that SRC-dependent regulation of YAP nuclear shuttling relies on the local activation of a Rac1/PAK1 axis that inhibits the formation of the inhibitory complex merlin/LATS/YAP (12). Accordingly, either the silencing of p18/LAMTOR1 or the pharmacological inhibition of SRC reduces Rac1 recruitment at cell edges (Fig. S4). Altogether, these data showed that the LAMTOR complex is required for SRC localization on LE and its trafficking toward the plasma membrane, and specifically at FA sites, to drive YAP nuclear translocation, likely via a Rac1/PAK1 axis.
Discussion
In this study, we identified the role of LE vesicular trafficking in the control of YAP activation. YAP is a well-known co-transcription factor that plays a key role in delivering information on the mechanical environments surrounding the cell to the nuclear transcription machinery. Along with TAZ, YAP is implicated in multiple cellular functions in tissue homeostasis and pathology. YAP and TAZ are both regulated by mechanical cues, and so far, little is known about their differential regulation. Although it is an interesting hypothesis, whether TAZ also depends on LE for its regulation is still an open question. Here, we demonstrated that β1 integrin–dependent cell adhesion allows the recycling of p18/LAMTOR1-positive LEs at adhesion sites by ILK-mediated anchoring of microtubules to FAs. This is required for the local delivery of the tyrosine kinase SRC at FAs, a necessary mechanism for YAP nuclear translocation (12, 39). These findings identify p18/LAMTOR1-mediated LE recycling as an important player in YAP signaling regulation.
The LAMTOR complex controls LE-dependent delivery of SRC to FAs
This work adds to the growing evidence of LE's role as a major cell signaling compartment (17–18). Its signaling function is tightly correlated to its dynamics/positioning and more specifically to LE's capacity to recycle back to the plasma membrane near or at FAs (26, 40). Indeed, LE localization (perinuclear versus peripheral) determines its function (catabolic versus anabolic) (13, 14, 16, 17). Our present work supports this idea by showing that FA-associated LEs promote YAP nuclear translocation, a well-known cell growth promoter.
The LAMTOR complex, which was isolated from late endosomal detergent–resistant membranes, is involved in the regulation of LE dynamics and signaling (16, 18, 19). In agreement, we observed that p18/LAMTOR1 has a critical role in LE targeting to the plasma membrane and FAs. Our data suggest that this is independent of its signaling function in the mTORC1 pathway. It was reported that LAMTOR restricts LE distribution to the perinuclear area by inhibiting the Arl8/BORC complex (19). We propose that LAMTOR is also required for the peripheral delivery of LEs that are targeted to adhesive structures. Although in apparent contradiction, these discrepant observations may be explained by the methods used to assess LE distribution and/or by the different cell types used. Indeed, in previous reports, LE distribution was analyzed by quantifying LE markers from the nuclear barycenter or the microtubule-organizing center as the origin, without delimiting the cell borders. Here, we accurately delimited the cell borders and quantified the vesicle densities from this position. This is particularly important in cells that generate large lamellipodia, such as mesenchymal cells. Indeed, we observed that upon p18/LAMTOR1 silencing, LE density was reduced mostly within the lamellipodial region.
One important LAMTOR function is to regulate mTORC1 activation. In agreement with previous reports, mTORC1 signaling appears to be dispensable for YAP nuclear translocation and also for LE positioning. Our present findings support the idea that, conversely, mTORC1 signaling is regulated by LE positioning and LATS1/2 (15, 16, 41).
One of our key findings is the identification of the LAMTOR complex role in the regulation of SRC local delivery to adhesion sites. We propose that SRC local delivery to FAs at the plasma membrane regulates signaling pathways involved in YAP nuclear translocation downstream of β1 integrin–mediated cell adhesion, as reported previously (11, 12, 39). The connection between LE and the adhesive sites is known, but the functional consequences were poorly understood. It was reported previously that LE docking at FAs regulates their turnover and β1 integrin endocytosis (21, 29). Here, we unraveled another function of these vesicles. Indeed, their targeting to FAs regulates downstream integrin-dependent signaling, as exemplified here by YAP regulation downstream of SRC. Our data extend and support the emerging role of SRC in regulating YAP nuclear translocation (11, 12, 42). Whereas SRC involvement in this pathway has been well-documented, its exact role is still the subject of debate, and different alternative mechanisms have been proposed. Indeed, SRC was shown to directly interact and phosphorylate YAP but also its upstream kinase LATS1/2, thus promoting YAP nuclear translocation (43, 44). Whereas we focused here on the role of LE trafficking in YAP nuclear translocation, it remains to be clarified in the future whether this trafficking could also control YAP/SRC interaction and/or its phosphorylation. This is even more important, considering that SRC is a well-known activator of Rac1 that is also involved in YAP nuclear translocation (45). Several lines of evidence support this latter finding: (i) v-SRC–induced cell transformation relies on the PAK1/Rac1-binding protein β-Pix/Cool 1, which promotes Rac1 recruitment at membrane ruffles (45–47), and (ii) β-Pix/Cool 1 was identified in a high-throughput screen as a regulator of YAP nuclear translocation (48). Importantly, merlin phosphorylation by the Rac1 effector PAK-1 inhibits its interaction with both LATS and YAP, thus promoting YAP dephosphorylation and nuclear translocation (12). Accordingly, SRC activity reduction by p18/LAMTOR1 silencing or by pharmacological inhibition affected Rac1 localization at protrusive borders (Fig. S4). Although SRC might regulate YAP nuclear translocation through different pathways, it is clear that its translocation to the plasma membrane at or near FA sites is of critical importance. Interestingly, the plasma membrane is emerging as an important compartment for YAP regulation (49, 50). Therefore, this pathway appears to be an alternative mechanism to a direct role of SRC on YAP and LATS1/2. It will be important to investigate the relative contribution of these specific pathways in the control of YAP activity.
β1 integrin–dependent cell adhesion regulates LE subcellular positioning and FA targeting
Firm cell adhesion to the ECM is required for the optimal occurrence of many cellular processes. For instance, loss of cell adhesion promotes lipid raft endocytosis, whereas cell adhesion favors their return to the surface. This tight connection between the adhesive system and membrane trafficking might play a major role in several signaling pathways, such as differentiation, proliferation, and anoikis (2, 3, 51). How exactly cell adhesion is involved is still puzzling, but our data fully support previous studies highlighting the role of microtubules. Part of the microtubule network is anchored at the cell periphery through β1 integrins and ILK (22, 34). In line with these data, loss of the β1 integrin subunit resulted in a defect of ILK localization at FAs that correlates with reduced microtubule anchoring to FAs. Moreover, forcing ILK localization into FAs restored not only microtubule anchoring, but also YAP nuclear shuttling. Interestingly, we also observed an increase in YAP nuclear localization upon forced expression of the β3 integrin subunit (although still being less efficient than the β3-ILK construct). This could be of importance in pathological situations when β3-containing integrins are overexpressed. This could be largely explained by the fact that ILK recruitment to FAs is only facilitated by β1 integrins under normal levels of expression (ILK presence in FAs is weaker but still detectable even in the absence of β1 integrins), whereas overexpression of β3 should also lead to an increase in ILK localization at FAs and thereby promote microtubule targeting. The loss of integrins is associated with defective microtubule targeting to FAs, which induces a defect in LE subcellular positioning and dynamics. This is in line with previous data showing that LEs move along the microtubular network (52). In addition, we rarely observed LEs associated with FAs in the absence of β1 integrins. As IQGAP1 interacts with ILK and LAMTOR (p14/LAMTOR2) (21), it is tempting to hypothesize that the β1 integrin/ILK axis regulates both LE trafficking and docking to FAs.
This relationship between integrin, YAP signaling, and the extracellular environment provides a rational framework for solid tumor cell progression. Although it is broadly accepted that YAP signaling is regulated by cell contractility and substrate stiffness, the mechanical cues affecting the vesicular trafficking involved in this process have attracted less attention. For instance, caveolin is involved in the mechano-dependent response of YAP and also in detergent-resistant membrane trafficking (53, 54). Moreover, it might promote Rac1 withdrawal from the plasma membrane upon cell detachment or reduced matrix stiffness (3, 51, 55). Therefore, vesicular trafficking, by controlling Rac1 internalization or accumulation through LE recycling, appears as a powerful mechanism to control YAP nuclear shuttling and, thereby, anchorage-dependent growth.
Experimental procedures
Cell lines
The generation and characterization of the β1f/f and β1−/− preosteoblastic cell lines were presented previously (25). SYF fibroblasts, derived from Fyn, Yes, and SRC triple knockout mice, were obtained from ATCC. From these original cell lines, the sh-p18, sh-Rab-11a, and sh-ctl cell lines were generated by transduction of lentivirus particles that express the shRNAs against p18 (Santa Cruz Biotechnology (Heidelberg, Germany), sc-36146-v), Rab-11a (Addgene no. 26710, Dr. K. Mostov) and scramble (Addgene no. 17920, Dr. S. Stewart). Cells were maintained in medium with puromycin. All other cell lines were generated by retroviral transduction, and transgene expression was verified by Western blotting and/or immunostaining.
Antibodies and expression vectors
Anti-YAPS127, -Rab-11, -SRC, -SRCY416, -p130CASY410, -S6, and -pS6 antibodies were from Cell Signaling (Ozyme, Saint-Quentin-en-Yvelines, France). Anti-YAP, and -β-tubulin (clone 2.1) antibodies were from Santa Cruz Biotechnology. Antibodies against mouse β1 integrin (MB1.2), mouse/human β1 integrin (9EG7), Rac1, and p130CAS were from BD Biosciences (Le Pont-de-Claix, France). The anti-actin antibody was from Sigma–Aldrich (L'Isle-d'Abeau, France). Anti-LAMP1 and -p-PAK antibodies were from Abcam. The anti-paxillinY31 antibody was from Invitrogen, and the anti-paxillin antibody was from Millipore (Fontenay-sous-Bois, France). The anti-phosphorylated tyrosine mAb 4G10 (hybridoma supernatant) was produced in our laboratory. Rabbit polyclonal antibodies against p18/LAMTOR1 were a generous gift by Dr. S. Manié (Lyon, France).
The human β1-expressing construct was based on the pCL-MFG retroviral vector, as described previously (25). pCL-MFG-β3-GFP-ILK, pCL-MFG-β3-GFP, and pCL-MFG-hILK-EGFP were a gift from Drs. E. Van Obberghen and R. Fässler. pBABE-puro-FLAGYAP2 was from Dr. M. Sudol (Addgene no. 27472). FLAG-tagged YAP25SA was from Dr. K. L. Guan (Addgene no. 27371). The mCherry-YAP constructs were produced from these initial plasmids and cloned into pCL-MFG retroviral vectors. The pEGFP-Rac1G12V plasmid was a gift from Dr. C. Gauthier-Rouvière. The GFP-Rac1G12V insert was subcloned into the retroviral vector pBaba-puro. WT and dominant negative mutants of Rab proteins were from Dr. M. McCaffrey. Human WT paxillin cDNA in the pBABE vector was generously provided by Dr. M. Hiraishi (Osaka Bioscience Institute, Osaka, Japan), and the mRFP-paxillin construct was generated from this initial vector. The pEGFP-N1-p18 construct was a gift from Dr. Masado Okada (Osaka University) and was subcloned into the pCLM-FG retroviral vector. The pmCherryN1-SRC construct was from Dr. S. Roche (CRBM, Montpellier, France). Everolimus and nocodazole were purchased from Sigma–Aldrich.
Transfections and infections
HEK GP 293 cells (Takara–Clontech, Saint-Germain-en-Laye, France) were transfected with plasmid DNA using the TurboFect Transfection reagent (Thermo Fisher Scientific, Courtabeuf, France) according to the manufacturer's instructions. Preosteoblasts were transduced with retroviral particles as described previously (56).
Immunohistochemistry
Cells grown on glass coverslips were fixed with 4% paraformaldehyde (PFA) and 5% sucrose in PBS at room temperature (RT) for 10 min and then permeabilized in 0.1% Triton X-100 in PBS for 5 min. Coverslips were washed twice with PBS, blocked in 1% BSA in PBS, and incubated at RT with primary antibodies for 1 h. Cells were rinsed in PBS, and secondary antibodies were added at RT for 1 h. Coverslips were mounted in Mowiol from Calbiochem (VWR International, Strasbourg, France) containing 4′,6-diamidino-2-phenylindole. Fixed cells were examined using a confocal laser-scanning microscope (LSM 510, Zeiss, Le Pecq, France), equipped with a plan-apochromat ×60 oil immersion objective, NA 1.4, with ×2 zoom. The pinhole was adjusted to 1 Airy unit.
Quantification of YAP nuclear localization
Cells were immunostained with an anti-YAP and images acquired with a confocal laser-scanning microscope (Zeiss LSM510) equipped with a ×63 plan-apochromat oil immersion objective (NA 1.4) and a pinhole set to 1 Airy. On each cell image, a region of interest (ROI) was defined either within the nucleus or in the cytoplasmic area next to the nuclear envelope. As the ROI thickness in the two positions was likely to be identical, the average fluorescence intensity should be proportional to YAP concentration in that area and was estimated using Fiji public software. Within the same cell, the ratio of the fluorescence intensities in the nucleus versus the cytoplasmic area reflects the YAP concentration ratio in the two compartments. This ratio was represented with a logarithmic scale to have an identical range of positive and negative ratios. Measurements were performed with n ≥ 50 (unless otherwise indicated), and differences were compared with Student's t test. Box plots were generated with R public software.
Video microscopy and TIRF
For live imaging, cells were seeded at subconfluent densities on Labtech chambers and grown overnight in Dulbecco's modified Eagle's medium supplemented with 10% FCS prior to imaging on a 37 °C heated stage in 5% CO2 atmosphere (Carl Zeiss Microimaging GmbH, Gottingen, Germany) with an Zeiss Axiovert 200M microscope equipped with a CoolSNAP HQ2, ×100 (NA 1.4) plan-apochromat objective and filters set to specifically detect Alexa 488/GFP or Alexa 546/pTRFP. Time lapse was 5 s. TIRF microscopy was carried out with the same set-up equipped with the TIRF 1 slider (Carl Zeiss Microimaging).
Vesicle distribution
Cell images were acquired with an Axioimager Z.1 microscope (Carl Zeiss Microimaging) equipped with a ×63/NA 1.4 plan-apochromat oil objective and an Axiocam Mrm CCD camera controlled by Axiovision software. Images were then analyzed using Icy software (http://icy.bioimageanalysis.org) with graphical programming tools for protocol editing. Channels were separated and processed to automatically detect cell borders with the best threshold, (HK-Means and Active Contours plugins). In parallel, vesicles were detected using the wavelet spot detector block, and the distance between the vesicle cell borders was quantified using the ROI inclusion analysis plugin. The detailed protocol will be provided upon request.
PDMS hydrogels
PDMS hydrogels of different stiffness (2, 10, and 30 kPa) were purchased from Excellness Biotech SA (Lausanne, Switzerland). Hydrogels were coated with bovine plasma fibronectin (2.5 µg/ml) according to the company's protocol. Cells were seeded on such hydrogels and left at 37 °C in CO2 and a humidified incubator for 2–3 h. After washing, cells were fixed with 3% PFA, 4% sucrose for 15 min, and then images were acquired as described previously.
Colocalization analysis
Images were obtained with a Zeiss Axiovert LSM510 biphoton confocal microscope equipped with a plan-apochromat ×63/NA 1.4 oil objective controlled by LSM510 acquisition software. Optical sectioning was set to 0.8 μm. Images were then analyzed using the Fiji software to run the JACOP plugin. Except when indicated, whole-cell bodies were considered as ROI for analysis.
Quantification of vesicle targeting
Cells were imaged using an Axioimager Z.1 (Carl Zeiss Microimaging) microscope equipped with a ×63/NA 1.4 plan-apochromat oil objective and an Axiocam Mrm CCD camera controlled by the Axiovision software. Images were then processed using Fiji software. First, the background was subtracted using a rolling ball of 50 pixels. The p18-GFP and mRFP-VASP channels were merged, and vesicles in direct contact with FAs were manually counted using the counter plugin. Then the mean value of the attached vesicles was divided by the number of FAs within the cell to obtain the vesicle/FA ratio. Twenty cells were analyzed for each experimental condition.
Cell spreading
Cells were spread on fibronectin-coated (5 µg/ml) Petri dishes for 3 h, fixed in methanol, and stained with the Coomassie dye. Thresholded digital images were then processed using Fiji software to quantify the cell area.
FACS
Cells were trypsinized and fixed in 3% PFA/PBS, and β1 integrins were detected by incubation with the MB1.2 antibody (1/100) at 4 °C for 1 h. β1−/− cells were the negative control.
Statistics
Cell analyses were carried out using at least 20 cells/experimental condition (for most of the presented experiments, n ranged between 50 and 100). Statistical significance was estimated with Student's t test with bilateral distribution and unequal variance. A p value of <0.01 was considered significant.
Data availability
All data are contained with the article.
Supplementary Material
Acknowledgments
We thank Drs. Manié, Zerial, Roche, Van Obberghen, Okada, Gertler, Fässler, McCaffrey, and Hiraishi for sharing tools and reagents. We also thank Ms Shalini Chandrashekhar for reading and editing the manuscript and Mr J. Mazzega for technical assistance with cell imaging.
This article contains supporting information.
Author contributions—M. R. B., M. B., M. P., G. C., and D. B. conceptualization; M. R. B., D. L., M. P., P. R., C. G.-R., B. W.-H., and D. B. resources; M. R. B., M. B., T. Z., and D. B. data curation; M. R. B., M. B., T. Z., G. C., P. R., and D. B. formal analysis; M. R. B. and D. B. supervision; D. B. funding acquisition; M. R. B., M. B., T. Z., M. P., G. C., P. R., C. G.-R., B. W.-H., and D. B. validation; M. R. B., M. B., T. Z., D. L., G. C., P. R., and D. B. investigation; M. R. B., M. B., C. G.-R., and B. W.-H. visualization; M. R. B., M. B., T. Z., M. P., G. C., and D. B. methodology; M. R. B., M. B., and D. B. writing-original draft; D. B. project administration; M. R. B., M. B., D. L., M. P., P. R., C. G.-R., B. W.-H., and D. B. writing-review and editing; M. P. and G. C. software.
Funding and additional information—This work was supported by Société Française de lutte contre les Cancers et les leucémies de l'Enfant et de l'Adolescent (SFCE) Grant CRAUFESD16, the Finovi Foundation, and Agence nationale de la recherche (ANR) Grant 15-CE14-0010-03.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ECM
- extracellular matrix
- LAMTOR
- late endosomal/lysosomal adaptor MAPK and mTOR activator
- mTOR
- mechanistic target of rapamycin
- YAP
- Yes-associated protein
- TAZ
- PDZ-binding motif
- LE
- late endosome
- FA
- focal adhesion
- ILK
- integrin-linked protein kinase
- PDMS
- polydimethylsiloxane
- kPa
- kilopascals
- ctl
- control
- resc
- rescue
- ALP
- alkaline phosphatase activity
- TIRF
- total internal reflection fluorescence
- PFA
- paraformaldehyde
- RT
- room temperature
- ROI
- region of interest.
References
- 1. Humphries J. D., Chastney M. R., Askari J. A., and Humphries M. J. (2019) Signal transduction via integrin adhesion complexes. Curr. Opin. Cell Biol. 56, 14–21 10.1016/j.ceb.2018.08.004 [DOI] [PubMed] [Google Scholar]
- 2. Alanko J., Mai A., Jacquemet G., Schauer K., Kaukonen R., Saari M., Goud B., and Ivaska J. (2015) Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 17, 1412–1421 10.1038/ncb3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balasubramanian N., Scott D. W., Castle J. D., Casanova J. E., and Schwartz M. A. (2007) Arf6 and microtubules in adhesion-dependent trafficking of lipid rafts. Nat. Cell Biol. 9, 1381–1391 10.1038/ncb1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mosesson Y., Mills G. B., and Yarden Y. (2008) Derailed endocytosis: an emerging feature of cancer. Nat. Rev. Cancer 8, 835–850 10.1038/nrc2521 [DOI] [PubMed] [Google Scholar]
- 5. Le H. Q., Ghatak S., Yeung C. Y., Tellkamp F., Günschmann C., Dieterich C., Yeroslaviz A., Habermann B., Pombo A., Niessen C. M., and Wickström S. A. (2016) Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 18, 864–875 10.1038/ncb3387 [DOI] [PubMed] [Google Scholar]
- 6. Chang L., Azzolin L., Di Biagio D., Zanconato F., Battilana G., Lucon Xiccato R., Aragona M., Giulitti S., Panciera T., Gandin A., Sigismondo G., Krijgsveld J., Fassan M., Brusatin G., Cordenonsi M., et al. (2018) The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 563, 265–269 10.1038/s41586-018-0658-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Halder G., Dupont S., and Piccolo S. (2012) Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 13, 591–600 10.1038/nrm3416 [DOI] [PubMed] [Google Scholar]
- 8. Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., Elvassore N., and Piccolo S. (2011) Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- 9. Yu F. X., Zhao B., and Guan K. L. (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163, 811–828 10.1016/j.cell.2015.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Totaro A., Panciera T., and Piccolo S. (2018) YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 20, 888–899 10.1038/s41556-018-0142-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elbediwy A., Vincent-Mistiaen Z. I., Spencer-Dene B., Stone R. K., Boeing S., Wculek S. K., Cordero J., Tan E. H., Ridgway R., Brunton V. G., Sahai E., Gerhardt H., Behrens A., Malanchi I., Sansom O. J., et al. (2016) Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 143, 1674–1687 10.1242/dev.133728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sabra H., Brunner M., Mandati V., Wehrle-Haller B., Lallemand D., Ribba A. S., Chevalier G., Guardiola P., Block M. R., and Bouvard D. (2017) β1 integrin-dependent Rac/group I PAK signaling mediates YAP activation of Yes-associated protein 1 (YAP1) via NF2/merlin. J. Biol. Chem. 292, 19179–19197 10.1074/jbc.M117.808063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Korolchuk V. I., Saiki S., Lichtenberg M., Siddiqi F. H., Roberts E. A., Imarisio S., Jahreiss L., Sarkar S., Futter M., Menzies F. M., O'Kane C. J., Deretic V., and Rubinsztein D. C. (2011) Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 13, 453–460 10.1038/ncb2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Poüs C., and Codogno P. (2011) Lysosome positioning coordinates mTORC1 activity and autophagy. Nat. Cell Biol. 13, 342–344 10.1038/ncb0411-342 [DOI] [PubMed] [Google Scholar]
- 15. Jia R., and Bonifacino J. S. (2019) Lysosome positioning influences mTORC2 and AKT signaling. Mol. Cell 75, 26–38.e3 10.1016/j.molcel.2019.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colaco A., and Jäättelä M. (2017) Ragulator—a multifaceted regulator of lysosomal signaling and trafficking. J. Cell Biol. 216, 3895–3898 10.1083/jcb.201710039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Araujo M. E. G., Naschberger A., Fürnrohr B. G., Stasyk T., Dunzendorfer-Matt T., Lechner S., Welti S., Kremser L., Shivalingaiah G., Offterdinger M., Lindner H. H., Huber L. A., and Scheffzek K. (2017) Crystal structure of the human lysosomal mTORC1 scaffold complex and its impact on signaling. Science 358, 377–381 10.1126/science.aao1583 [DOI] [PubMed] [Google Scholar]
- 18. Nada S., Hondo A., Kasai A., Koike M., Saito K., Uchiyama Y., and Okada M. (2009) The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 28, 477–489 10.1038/emboj.2008.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Filipek P. A., de Araujo M. E. G., Vogel G. F., De Smet C. H., Eberharter D., Rebsamen M., Rudashevskaya E. L., Kremser L., Yordanov T., Tschaikner P., Fürnrohr B. G., Lechner S., Dunzendorfer-Matt T., Scheffzek K., Bennett K. L., et al. (2017) LAMTOR/Ragulator is a negative regulator of Arl8b- and BORC-dependent late endosomal positioning. J. Cell Biol. 216, 4199–4215 10.1083/jcb.201703061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pu J., Keren-Kaplan T., and Bonifacino J. S. (2017) A Ragulator-BORC interaction controls lysosome positioning in response to amino acid availability. J. Cell Biol. 216, 4183–4197 10.1083/jcb.201703094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schiefermeier N., Scheffler J. M., de Araujo M. E., Stasyk T., Yordanov T., Ebner H. L., Offterdinger M., Munck S., Hess M. W., Wickström S. A., Lange A., Wunderlich W., Fässler R., Teis D., and Huber L. A. (2014) The late endosomal p14-MP1 (LAMTOR2/3) complex regulates focal adhesion dynamics during cell migration. J. Cell Biol. 205, 525–540 10.1083/jcb.201310043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wickström S. A., Lange A., Hess M. W., Polleux J., Spatz J. P., Krüger M., Pfaller K., Lambacher A., Bloch W., Mann M., Huber L. A., and Fässler R. (2010) Integrin-linked kinase controls microtubule dynamics required for plasma membrane targeting of caveolae. Dev. Cell 19, 574–588 10.1016/j.devcel.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao B., Li L., Wang L., Wang C. Y., Yu J., and Guan K. L. (2012) Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 26, 54–68 10.1101/gad.173435.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang Y., Rowe R. G., Botvinick E. L., Kurup A., Putnam A. J., Seiki M., Weaver V. M., Keller E. T., Goldstein S., Dai J., Begun D., Saunders T., and Weiss S. J. (2013) MT1-MMP-dependent control of skeletal stem cell commitment via a β1-integrin/YAP/TAZ signaling axis. Dev. Cell 25, 402–416 10.1016/j.devcel.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brunner M., Millon-Frémillon A., Chevalier G., Nakchbandi I. A., Mosher D., Block M. R., Albigès-Rizo C., and Bouvard D. (2011) Osteoblast mineralization requires β1 integrin/ICAP-1-dependent fibronectin deposition. J. Cell Biol. 194, 307–322 10.1083/jcb.201007108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Soma-Nagae T., Nada S., Kitagawa M., Takahashi Y., Mori S., Oneyama C., and Okada M. (2013) The lysosomal signaling anchor p18/LAMTOR1 controls epidermal development by regulating lysosome-mediated catabolic processes. J. Cell Sci. 126, 3575–3584 10.1242/jcs.121913 [DOI] [PubMed] [Google Scholar]
- 27. Liu C. Y., Zha Z. Y., Zhou X., Zhang H., Huang W., Zhao D., Li T., Chan S. W., Lim C. J., Hong W., Zhao S., Xiong Y., Lei Q. Y., and Guan K. L. (2010) The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFβ-TrCP E3 ligase. J. Biol. Chem. 285, 37159–37169 10.1074/jbc.M110.152942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., and Sabatini D. M. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 10.1016/j.cell.2010.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rainero E., Howe J. D., Caswell P. T., Jamieson N. B., Anderson K., Critchley D. R., Machesky L., and Norman J. C. (2015) Ligand-occupied integrin internalization links nutrient signaling to invasive migration. Cell Rep. 10, 398–413 10.1016/j.celrep.2014.12.037 [DOI] [PubMed] [Google Scholar]
- 30. Caswell P. T., Chan M., Lindsay A. J., McCaffrey M. W., Boettiger D., and Norman J. C. (2008) Rab-coupling protein coordinates recycling of α5β1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J. Cell Biol. 183, 143–155 10.1083/jcb.200804140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nader G. P., Ezratty E. J., and Gundersen G. G. (2016) FAK, talin and PIPKIγ regulate endocytosed integrin activation to polarize focal adhesion assembly. Nat. Cell Biol. 18, 491–503 10.1038/ncb3333 [DOI] [PubMed] [Google Scholar]
- 32. Powelka A. M., Sun J., Li J., Gao M., Shaw L. M., Sonnenberg A., and Hsu V. W. (2004) Stimulation-dependent recycling of integrin β1 regulated by ARF6 and Rab11. Traffic 5, 20–36 10.1111/j.1600-0854.2004.00150.x [DOI] [PubMed] [Google Scholar]
- 33. Serrano I., McDonald P. C., Lock F., Muller W. J., and Dedhar S. (2013) Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat. Commun. 4, 2976 10.1038/ncomms3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akhtar N., and Streuli C. H. (2013) An integrin-ILK-microtubule network orients cell polarity and lumen formation in glandular epithelium. Nat. Cell Biol. 15, 17–27 10.1038/ncb2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stanchi F., Grashoff C., Nguemeni Yonga C. F., Grall D., Fässler R., and Van Obberghen-Schilling E. (2009) Molecular dissection of the ILK-PINCH-parvin triad reveals a fundamental role for the ILK kinase domain in the late stages of focal-adhesion maturation. J. Cell Sci. 122, 1800–1811 10.1242/jcs.044602 [DOI] [PubMed] [Google Scholar]
- 36. Fincham V. J., Unlu M., Brunton V. G., Pitts J. D., Wyke J. A., and Frame M. C. (1996) Translocation of SRC kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J. Cell Biol. 135, 1551–1564 10.1083/jcb.135.6.1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sandilands E., Brunton V. G., and Frame M. C. (2007) The membrane targeting and spatial activation of SRC, Yes and Fyn is influenced by palmitoylation and distinct RhoB/RhoD endosome requirements. J. Cell Sci. 120, 2555–2564 10.1242/jcs.003657 [DOI] [PubMed] [Google Scholar]
- 38. Geiger B. (2006) A role for p130Cas in mechanotransduction. Cell 127, 879–881 10.1016/j.cell.2006.11.020 [DOI] [PubMed] [Google Scholar]
- 39. Lamar J. M., Xiao Y., Norton E., Jiang Z. G., Gerhard G. M., Kooner S., Warren J. S. A., and Hynes R. O. (2019) SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J. Biol. Chem. 294, 2302–2317 10.1074/jbc.RA118.004364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takahashi Y., Nada S., Mori S., Soma-Nagae T., Oneyama C., and Okada M. (2012) The late endosome/lysosome-anchored p18-mTORC1 pathway controls terminal maturation of lysosomes. Biochem. Biophys. Res. Commun. 417, 1151–1157 10.1016/j.bbrc.2011.12.082 [DOI] [PubMed] [Google Scholar]
- 41. Gan W., Dai X., Xie J., Yin S., Zhu J., Wang C., Liu Y., Guo J., Wang M., Liu J., Hu J., Quinton R. J., Ganem N. J., Liu P., Asara J. M., et al. (2020) LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat. Cell Biol. 22, 246–256 10.1038/s41556-020-0463-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Calvo F., Ege N., Grande-Garcia A., Hooper S., Jenkins R. P., Chaudhry S. I., Harrington K., Williamson P., Moeendarbary E., Charras G., and Sahai E. (2013) Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 15, 637–646 10.1038/ncb2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shanzer M., Adler J., Ricardo-Lax I., Reuven N., and Shaul Y. (2017) The nonreceptor tyrosine kinase c-SRC attenuates SCF(β-TrCP) E3-ligase activity abrogating Taz proteasomal degradation. Proc. Natl. Acad. Sci. U. S. A. 114, 1678–1683 10.1073/pnas.1610223114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Si Y., Ji X., Cao X., Dai X., Xu L., Zhao H., Guo X., Yan H., Zhang H., Zhu C., Zhou Q., Tang M., Xia Z., Li L., Cong Y. S., et al. (2017) SRC inhibits the Hippo tumor suppressor pathway through tyrosine phosphorylation of Lats1. Cancer Res. 77, 4868–4880 10.1158/0008-5472.CAN-17-0391 [DOI] [PubMed] [Google Scholar]
- 45. Feng H., Hu B., Liu K. W., Li Y., Lu X., Cheng T., Yiin J. J., Lu S., Keezer S., Fenton T., Furnari F. B., Hamilton R. L., Vuori K., Sarkaria J. N., Nagane M., et al. (2011) Activation of Rac1 by SRC-dependent phosphorylation of Dock180(Y1811) mediates PDGFRα-stimulated glioma tumorigenesis in mice and humans. J. Clin. Invest. 121, 4670–4684 10.1172/JCI58559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Feng Q., Baird D., Peng X., Wang J., Ly T., Guan J. L., and Cerione R. A. (2006) Cool-1 functions as an essential regulatory node for EGF receptor- and SRC-mediated cell growth. Nat. Cell Biol. 8, 945–956 10.1038/ncb1453 [DOI] [PubMed] [Google Scholar]
- 47. ten Klooster J. P., Jaffer Z. M., Chernoff J., and Hordijk P. L. (2006) Targeting and activation of Rac1 are mediated by the exchange factor β-Pix. J. Cell Biol. 172, 759–769 10.1083/jcb.200509096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sero J. E., and Bakal C. (2017) Multiparametric analysis of cell shape demonstrates that β-PIX directly couples YAP activation to extracellular matrix adhesion. Cell Syst. 4, 84–96.e6 10.1016/j.cels.2016.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yin F., Yu J., Zheng Y., Chen Q., Zhang N., and Pan D. (2013) Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 154, 1342–1355 10.1016/j.cell.2013.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tei R., and Baskin J. M. (2020) Spatiotemporal control of phosphatidic acid signaling with optogenetic, engineered phospholipase Ds. J. Cell Biol. 219, e201907013 10.1083/jcb.201907013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Balasubramanian N., Meier J. A., Scott D. W., Norambuena A., White M. A., and Schwartz M. A. (2010) RalA-exocyst complex regulates integrin-dependent membrane raft exocytosis and growth signaling. Curr. Biol. 20, 75–79 10.1016/j.cub.2009.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Loubéry S., Wilhelm C., Hurbain I., Neveu S., Louvard D., and Coudrier E. (2008) Different microtubule motors move early and late endocytic compartments. Traffic 9, 492–509 10.1111/j.1600-0854.2008.00704.x [DOI] [PubMed] [Google Scholar]
- 53. Moreno-Vicente R., Pavón D. M., Martín-Padura I., Català-Montoro M., Díez-Sánchez A., Quílez-Álvarez A., López J. A., Sánchez-Álvarez M., Vázquez J., Strippoli R., and Del Pozo M. A. (2019) Caveolin-1 modulates mechanotransduction responses to substrate stiffness through actin-dependent control of YAP. Cell Rep. 26, 1679–1680 10.1016/j.celrep.2019.01.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parton R. G., and Richards A. A. (2003) Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic 4, 724–738 10.1034/j.1600-0854.2003.00128.x [DOI] [PubMed] [Google Scholar]
- 55. Cerezo A., Guadamillas M. C., Goetz J. G., Sánchez-Perales S., Klein E., Assoian R. K., and del Pozo M. A. (2009) The absence of caveolin-1 increases proliferation and anchorage-independent growth by a Rac-dependent, Erk-independent mechanism. Mol. Cell Biol. 29, 5046–5059 10.1128/MCB.00315-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bouvard D., Aszodi A., Kostka G., Block M. R., Albigès-Rizo C., and Fässler R. (2007) Defective osteoblast function in ICAP-1-deficient mice. Development 134, 2615–2625 10.1242/dev.000877 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained with the article.