

Abstract
An important context in which metabolism influences tumorigenesis is the genetic cancer syndrome hereditary leiomyomatosis and renal cell carcinoma (HLRCC), a disease in which mutation of the tricarboxylic acid cycle enzyme fumarate hydratase (FH) causes hyperaccumulation of fumarate. This electrophilic oncometabolite can alter gene activity at the level of transcription, via reversible inhibition of epigenetic dioxygenases, as well as posttranslationally, via covalent modification of cysteine residues. To better understand the potential for metabolites to influence posttranslational modifications important to tumorigenesis and cancer cell growth, here we report a chemoproteomic analysis of a kidney-derived HLRCC cell line. Using a general reactivity probe, we generated a data set of proteomic cysteine residues sensitive to the reduction in fumarate levels caused by genetic reintroduction of active FH into HLRCC cell lines. This revealed a broad up-regulation of cysteine reactivity upon FH rescue, which evidence suggests is caused by an approximately equal proportion of transcriptional and posttranslational modification–mediated regulation. Gene ontology analysis highlighted several new targets and pathways potentially modulated by FH mutation. Comparison of the new data set with prior studies highlights considerable heterogeneity in the adaptive response of cysteine-containing proteins in different models of HLRCC. This is consistent with emerging studies indicating the existence of cell- and tissue-specific cysteine-omes, further emphasizing the need for characterization of diverse models. Our analysis provides a resource for understanding the proteomic adaptation to fumarate accumulation and a foundation for future efforts to exploit this knowledge for cancer therapy.
Keywords: chemical biology, metabolism, inborn error of metabolism, posttranslational modification (PTM), proteomics, redox signaling, TCA cycle, mitochondria, tricarboxylic acid cycle (TCA cycle) (Krebs cycle)
Metabolites play diverse roles in cellular homeostasis, acting as transcription factor ligands, secondary messengers, feedback inhibitors, and allosteric effectors of enzyme function (1–3). An emerging function by which metabolites modulate cell signaling is via covalent protein modification due to their intrinsic reactivity (4, 5). For example, in the genetic cancer predisposition syndrome hereditary leiomyomatosis and renal cell carcinoma (HLRCC), mutation of the tricarboxylic acid (TCA) cycle enzyme fumarate hydratase (FH) causes the hyperaccumulation of fumarate, an electrophilic “oncometabolite.” Reaction of cysteines with fumarate's α,β-unsaturated double bond results in protein S-succination, a nonenzymatic posttranslational modification that can substantially modulate protein function (6–8). In addition to direct modification, fumarate accumulation can also indirectly alter cysteine activity, through redox stress that causes oxidative cysteine modifications (9–11) or transcriptional changes that up-regulate or down-regulate the expression of cysteine-containing proteins (Fig. 1a) (12–16).
Figure 1.
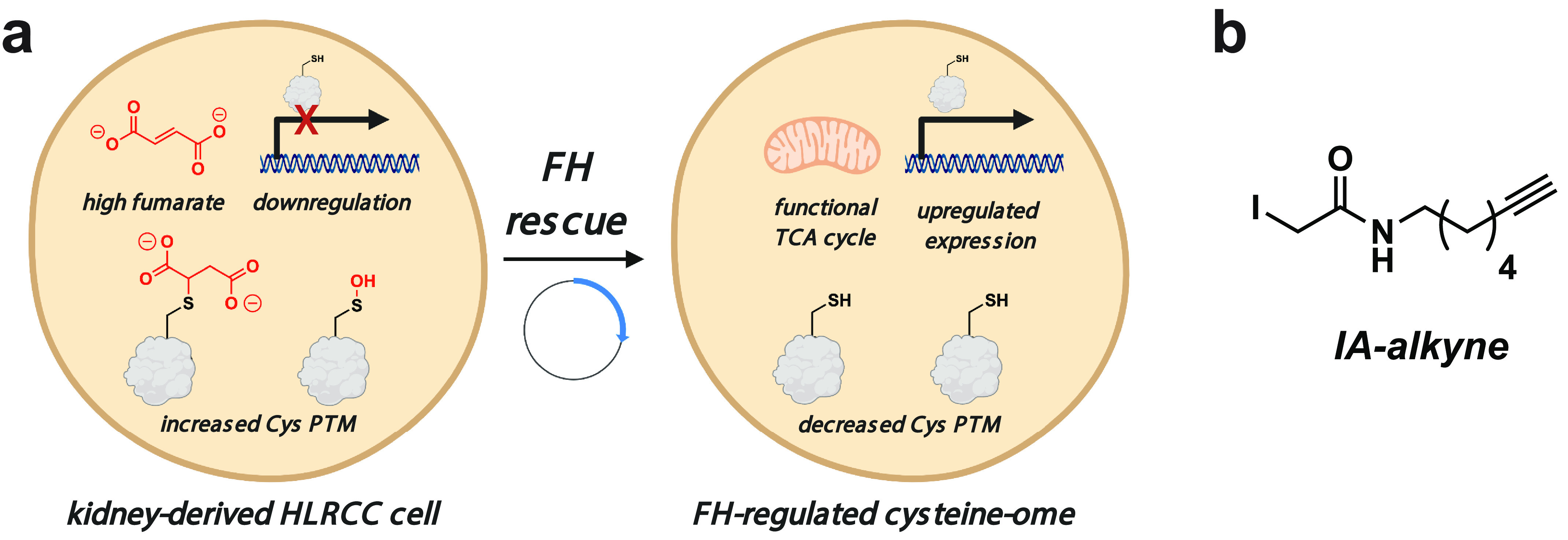
a, left, inactivation of FH in the genetic cancer syndrome HLRCC causes the accumulation of high levels of fumarate, which can decrease cysteine reactivity either by down-regulating the expression of cysteine-containing proteins or by increasing the level of cysteine posttranslational modifications. Right, FH rescue reverses many of these changes, which can be identified based on their altered cysteine reactivity. b, IA-alkyne, an enrichable reactive electrophile used to quantify cysteine reactivity in this study.
Results
To better understand fumarate's potential as a metabolic signal, our laboratory recently reported a quantitative chemoproteomic approach to study conditional cysteine reactivity in HLRCC (17). This approach uses a chemical probe, iodoacetamide alkyne (IA-alkyne), to substoichiometrically label cysteines across the proteome in isogenic FH-deficient (FH−/−) and WT (FH+/+) rescue cell lines (Fig. 1b) (18). Conjugation to azide-biotin tags using click chemistry followed by enrichment, cleavage, and quantitative LC–MS/MS analysis enables comparison of how the reactivity of individual cysteine residues are regulated by FH rescue and subsequent reduction in intracellular levels of fumarate. In our initial study, we applied this method to quantify FH-regulated cysteine residues in an immortalized cell line derived from an HLRCC metastasis. This led to the identification of new candidate targets of S-succination as well as evidence that protonation of fumarate is necessary for it to act as a covalent modifier in HLRCC (17).
Indirect readouts, such as hypoxia-inducible factor accumulation (19) and oxidative respiration (20), as well as experimental data, such as anti-S-succination immunoblotting (17), indicate that fumarate accumulates at different levels in cell lines derived from primary HLRCC tumors as compared with distal metastases (Fig. S1a). This suggests the FH-regulated cysteine-ome of different tumors may display substantial heterogeneity, potentially contributing to distinct biology, diagnostic markers, or therapeutic vulnerabilities. To investigate this hypothesis, we applied our chemoproteomic approach to an immortalized cell line derived from the kidney of an HLRCC patient (20). Briefly, a kidney-derived HLRCC cell line (UOK268, FH−/−) and a rescue line in which the FH gene had been reintroduced (UOK268, FH+/+) were cultured on a large scale, and proteomes were isolated from ∼30–40 million cells by mild sonication. Altered fumarate levels in the two cell lines were verified by NMR (Fig. S1b). Paired samples were IA-alkyne–labeled, conjugated to a UV-cleavable biotin azide via click chemistry, enriched, isotopically tagged, pooled, and analyzed by LC–MS/MS (Fig. 2a). Analysis of the intensity ratio (R) of MS1 spectra of light/heavy isotopic pairs in IA-alkyne–treated samples was used to determine relative cysteine reactivity between the two samples. An R value of 1 indicates a cysteine was equally reactive in FH−/− and FH+/+ samples, whereas an increased R value of 2 indicates a cysteine's reactivity was recovered by 50% upon FH rescue (based on the formula, relative reactivity (%) = [(1 − (1/R)] × 100%; Table S1).
Figure 2.
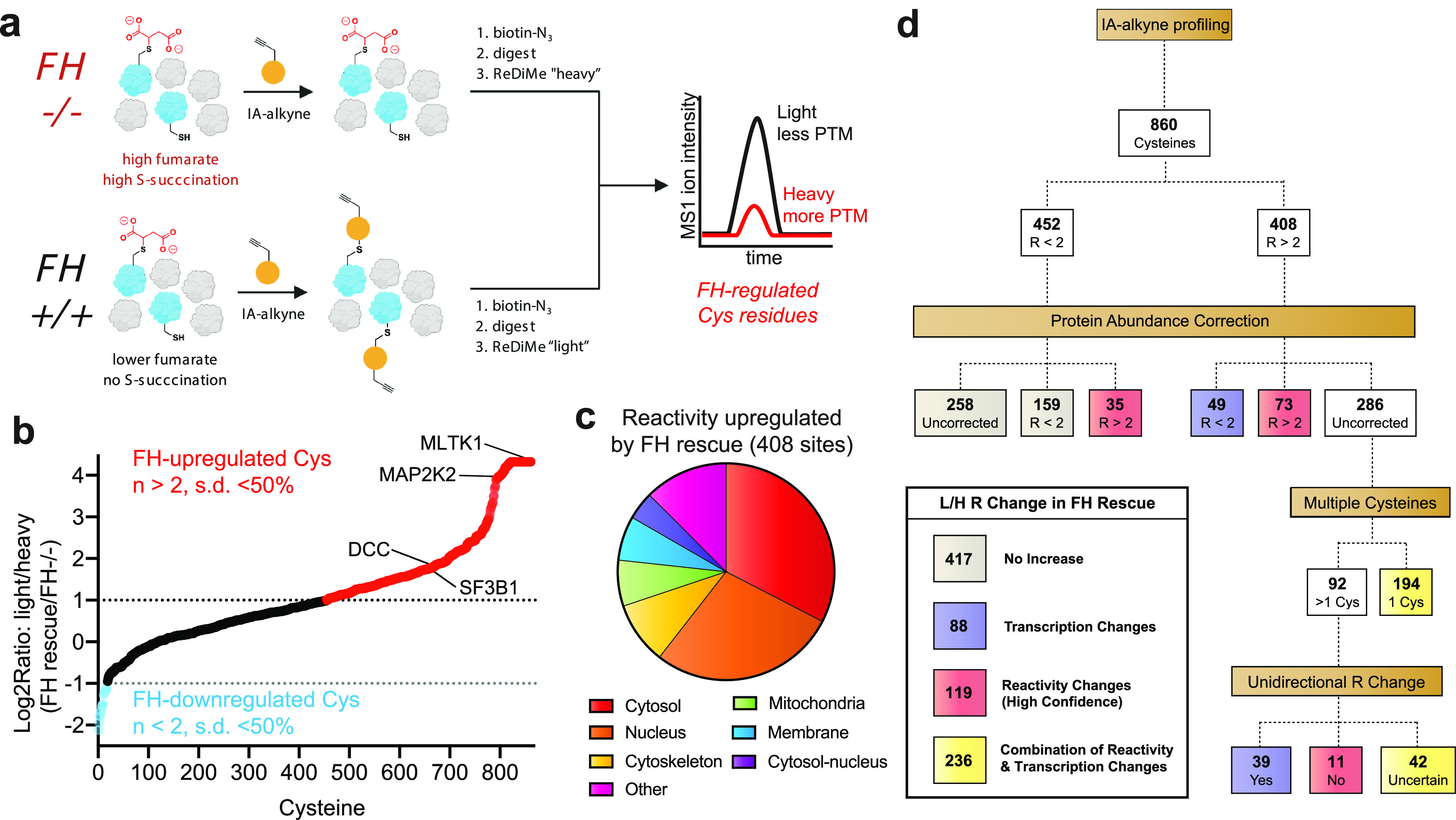
Defining the consequences of FH rescue in HLRCC via quantitative cysteine reactivity profiling. a, general workflow for reactive cysteine–profiling experiment. FH−/−, FH mutant cells; FH+/+, FH rescue cells. b, graph of quantified cysteine reactivity in FH−/− (heavy) and FH+/+ (light) UOK268 cell lines, sorted from high to low peptide R values. c, cellular localization of cysteine residues up-regulated by FH rescue (R ≥ 2, S.D. ≤ 50%). d, flow chart describing how protein abundance correction and the presence of multiple cysteine residues in a single protein were used to define cysteine reactivity changes likely to be driven by FH-dependent transcription (purple), FH-dependent reactivity (red), and a combination thereof (yellow).
Performing four independent replicate measurements of cysteine reactivity in paired FH+/+ and FH−/− kidney-derived UOK268 cell lines enabled the analysis of cysteine reactivity across 860 individual residues with high confidence (n ≥ 2, S.D. of R (reactivity) of ≤50%) (Fig. 2b and Table S2). Assessing the directionality of cysteine reactivity changes between the isogenic cell lines, we found that the reactivities of 408 cysteines were up-regulated ≥2-fold upon FH rescue (Fig. 2b, red), whereas the reactivities of only 17 cysteines were down-regulated to this extent (Fig. 2b, blue). This is consistent with our expectation of increased cysteine reactivity in FH rescue (+/+) due to decreased accumulation of the electrophilic oncometabolite fumarate.
Interestingly, the cysteines whose reactivity was most strongly increased upon FH rescue predominantly mapped to the nucleus and cytosol (Fig. 2c), whereas the relatively few cysteines whose reactivity was decreased by FH rescue were enriched in mitochondrial proteins (Fig. S2a). Hypothesizing that the latter effect may be due to compensatory up-regulation of mitochondrial proteins in FH−/− cells, we applied two complementary approaches to examine the relationship between protein expression and cysteine reactivity in our data set (Fig. 2d). First, we used reductive dimethyl (ReDiMe) labeling to quantitatively assess changes in overall protein abundance between the two cell lines (Table S3) (21). We obtained quantitative abundance data for the parent proteins of one-third of cysteines whose relative reactivity was measured in UOK268 FH−/− and rescue cell lines (Table S4). Overall, relative cysteine reactivity and protein abundance showed a strong correlation in the two HLRCC cell lines (Pearson's r = 0.57; Fig. S2b). Cysteines exhibiting the greatest reactivity increases upon FH rescue (R ≥ 2) also showed the most augmented expression in this condition, consistent with the overall trend (Fig. S2c). Focusing on cysteine residues with increased reactivity upon FH rescue (R ≥ 2), we found that 49 were also strongly up-regulated (≥2-fold decreased) at the protein level (Fig. 2d and Table S4), suggesting up-regulated protein expression. Conversely, the FH-dependent reactivities of 73 cysteines were relatively unchanged, whereas an additional 35 shifted from R ≤ 2 to R ≥ 2 when correcting for protein abundance, consistent with the potential for these residues to be targets of FH-dependent posttranslational modification (Fig. 2d and Table S4).
To extend this analysis to FH-regulated (R ≥ 2) cysteines whose proteomic abundance was not sampled by ReDiMe, we developed a second approach, examining cases where multiple IA-alkyne–quantifiable cysteine residues were identified within a single polypeptide (Fig. 2d and Table S5). Our hypothesis was that changes in expression levels should cause a consistent change in reactivity across multiple quantified cysteine residues of a protein, whereas changes in cysteine post-translational modifications, such as S-succination or cysteine oxidation, may alter the reactivity of only a subset of sites (22). Applying this approach, we observed consistent (S.D. ≤ 50%) unidirectional changes in cysteine reactivity for 39 of these FH-regulated (R ≥ 2) cysteines and distinct reactivity changes (S.D. ≥ 50%) for 11 additional cysteine residues, with the remainder (42 cysteines) falling in between (Fig. 2d and Table S5). For example, Cys-154, Cys-380, and Cys-410 of UQCRC1 display very similar R values (2.61, 2,07, and 2.73, respectively) despite residing in distinct domains of the protein. In contrast, two unique cysteine residues were quantified within the microsomal protein LRRC59, one of which displays distinct FH-dependent reactivity (Cys-277, R = 14.86; Cys-48, R = 1.95). These studies suggest that cysteine reactivity changes sampled by IA-alkyne in this isogenic HLRCC model are caused by a combination of gene expression changes (88 cysteines) and FH-dependent posttranslational protein modifications (119 cysteines).
Our comprehensive chemoproteomic data set affords a unique opportunity to define how cellular pathways respond to reintroduction of the FH tumor suppressor at both the level of protein expression and cysteine reactivity. Focusing first on protein expression (Fig. 3 and Table S3), gene ontology analysis using the informatic tool DAVID (23) found that FH rescue up-regulated the expression of proteins involved in cell-cell adhesion (FLNA, MYH9, PFN1, and KIF5B), protein translation (EIF4G2, RPL12, RPL13, RPS25, and RPLP1), and glycolysis (ALDOA/C, ENO1, GPI, GAPDH, PFKP, PGK1, PGAM1, TPI1, and LDHA/B) while down-regulating mitochondrial enzymes involved in the TCA cycle and redox homeostasis (Fig. 3, b and c). HLRCC tumors are well-known for their metastatic potential (16, 24), and the up-regulation of cell adhesion proteins suggests that FH rescue may reverse this phenotype. In contrast, the up-regulation of glycolytic enzymes upon reintroduction of FH activity was unexpected. Previous studies have established that glycolytic gene expression is up-regulated in HLRCC tumors and tumor-derived cells (20), and chemoproteomic profiling analysis also found that rescue of FH in the metastasis-derived UOK262 HLRCC cell line down-regulated many glycolytic enzymes at the level of protein expression, suggesting that UOK262 cells may show a greater degree of metabolic flexibility (17). The glycolytic up-regulation of UOK268 FH rescue cells highlights a potential caveat of our model, which is that reintroduction of FH into transformed cells may recapitulate only a subset of differences between HLRCC tumors and healthy tissues, due to irreversible signaling changes and genomic insults that accompany malignant transformation. Our results indicate that restoring the respiratory capability of kidney-derived UOK268 cells does not necessarily “cure” the heavy reliance of these cells on glycolytic enzymes. FH rescue disproportionately affected the levels of enzymes involved in oxidation-reduction processes, causing ≥2-fold down-regulation of eight mitochondrial NAD(P)H-producing enzymes: OGDH, MDH2, and IDH3A (all members of the TCA cycle) and ALDH2, GLUD1, FDXR, HIBADH, and ME3 (Table S3 and Fig. 3 (d and e)), consistent with previous observations of FH-regulated changes in redox homeostasis (9, 10, 25, 26). Our studies, together with the literature, are consistent with a model in which FH inactivation causes a dependence on enzymes involved in glycolytic metabolism and the enzymatic production of reducing equivalents (26), only the latter of which is reversed by reintroduction of functional FH activity.
Figure 3.
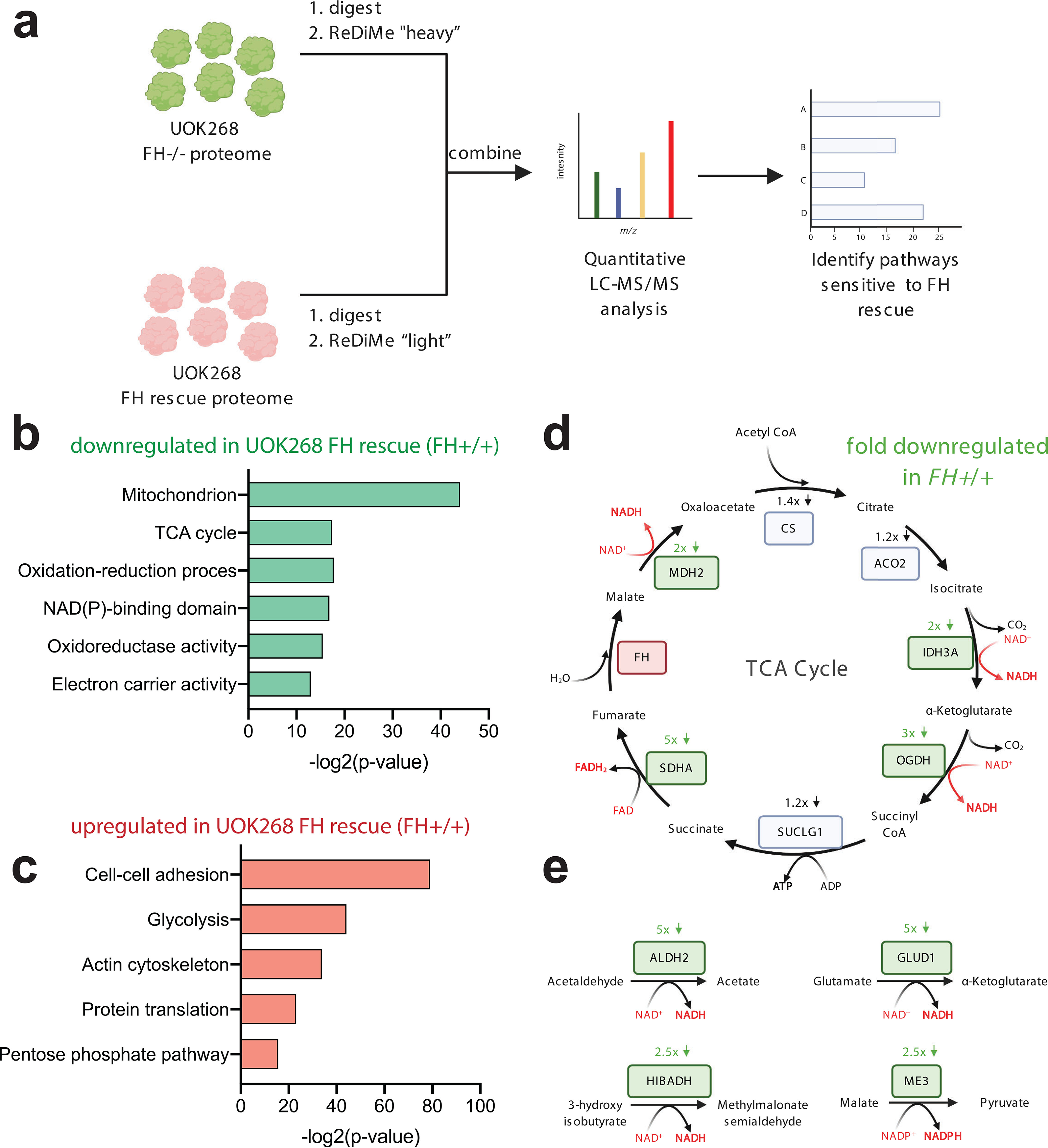
Defining the consequences of FH rescue in HLRCC at the whole-proteome level via quantitative ReDiMe labeling. a, workflow for whole-proteome ReDiMe analysis of UOK268 HLRCC cells. b, gene ontology terms encompassing proteins found to be down-regulated upon FH rescue in kidney-derived HLRCC cells. c, gene ontology terms encompassing proteins found to be up-regulated upon FH rescue in kidney-derived HLRCC cells. d, influence of FH rescue on TCA cycle enzyme expression. Genes boxed in green are down-regulated >2-fold in the FH rescue (FH+/+) UOK268 cell line relative to mutant (FH−/−). e, influence of FH rescue on NAD(P)H-producing mitochondrial metabolic enzymes. Genes boxed in green are down-regulated >2-fold in the FH rescue UOK268 cell line relative to mutant.
Next, we explored changes in cysteine reactivity not explained by altered protein abundance between the mutant and rescue UOK268 cell lines, which represent candidate sites of FH-dependent posttranslational modification (Fig. 4a and Tables S4–S6). Here, we focused on 119 “high-confidence” residues, which displayed an R ≥ 2 following correction for protein abundance or multiple cysteines (Fig. 2d, red), as well as an additional 236 “lower-confidence” cysteines whose parent proteins were not quantified by whole-proteome LC–MS/MS analysis and whose reactivity changes thus may be driven by either altered expression or posttranslational modifications (Fig. 2d (yellow) and Tables S4–S6). To identify functional cysteine modifications with potential cancer relevance, we employed a two-step analysis. First, the parent proteins of these 355 candidate FH-regulated cysteine residues were queried for evidence of involvement in human cancer using the Network of Cancer Genes database (27, 28). Hits from this analysis were then assessed for conservation of the candidate modified cysteine using the online informatic tool Mutation Assessor (29). Applying this strategy, we identified 55 genes known to be deleted, overexpressed, or amplified in human cancer that harbored a total of 69 candidate FH-regulated cysteine residues. In eight instances, mutation of the FH-regulated cysteine was predicted to highly perturb the function of the parent protein, whereas mutation of a further 23 cysteine residues was hypothesized to exert a moderate effect (Fig. 4b and Table S7). Conserved FH-regulated cysteine residues were found to lie within functional domains of the following.
Figure 4.
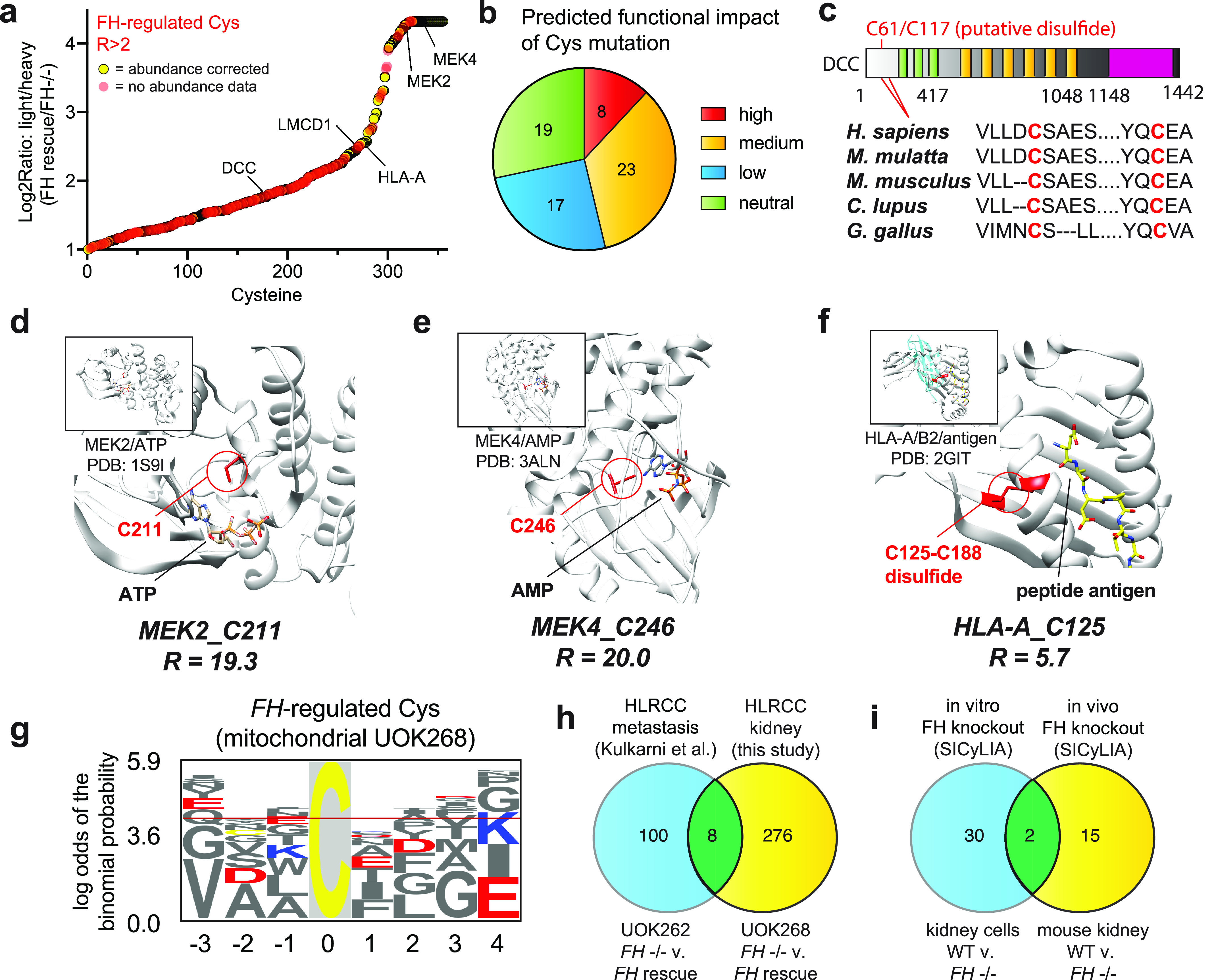
a, cysteines showing increased occupancy in FH rescue cells. Red, high-confidence abundance-corrected cysteine reactivity changes; yellow, non-abundance-corrected cysteine reactivity changes that potentially arise from either altered transcription or posttranslational modification. b, predicted functional impact of R > 2 cysteines mapping to annotated cancer genes based on Mutation Assessor analysis. c, domain map of DCC, indicating conservation of Cys-61 in metazoans. d, crystal structure of MEK2, showing the position of FH-regulated cysteine near the ATP-binding pocket. e, crystal structure of MEK4, showing the position of FH-regulated cysteine near the ATP-binding pocket. f, crystal structure of HLA-A, showing the position of FH-regulated cysteine involved in a disulfide bond. g, motif analysis of FH-regulated cysteines. The logo is derived from mitochondrial cysteines, where fumarate concentrations are expected to be highest. Logos from cytosolic and nuclear FH-regulated cysteines are provided in Fig. S3. h, overlap of FH-regulated cysteine residues identified in UOK262 and UOK268 cells. i, overlap of FH-regulated cysteine residues identified in mouse kidney cells and in vivo mouse kidney by van der Reest et al. (34).
Tumor suppressors. Examples include deleted in colon cancer (DCC), in which FH-regulated Cys-61 (R = 3.4), lies within a highly conserved region predicted to form a disulfide with Cys-117 (Fig. 4c), and LIM and cysteine-rich domain-containing protein 1 Cys-52 (LCRDCP, Cys-52, R=5.7), which is found in the Cys-rich region of the protein that is required for interaction and repression of GATA transcription factors (30).
Signaling enzymes. FH-regulated cysteines were identified within the ATP-binding sites of the mitogen-activated protein kinases MEK2 (Cys-211, R = 19.3; Fig. 4d) and MEK4 (Cys-246, R = 20; Fig. 4e) as well as within the catalytic subunit of AMPK (PRKAA2 Cys-174, R =15.7; Table S6) (31). The latter cysteine maps to a previously characterized site of functional oxidative modification in AMPK, suggesting a novel mechanistic input that may further reduce the AMPK activity of HLRCC tumors (19).
Components of the immune system. FH-dependent decreases in cysteine reactivity were observed in two major histocompatibility complex (MHC) proteins: HLA-A (Cys-125, R = 3.0) and HLA-B (Cys-188, R = 3.8). Each maps to an extracellular disulfide critical to the folding and structure of its respective MHC molecule (Fig. 4f) (32), and the differential reactivity in the two cell lines indicates the potential of these proteins to be transcriptionally or posttranslationally regulated by FH.
Motif analysis of cysteine-containing peptides whose reactivity was decreased by ≥2-fold by FH inactivation found a high occurrence of flanking glutamate and aspartate residues, independent of protein localization (Fig. 4g, Fig. S3, and Table S10). These carboxylate amino acids are not commonly found proximal to hyperreactive cysteines and may contribute to fumarate reactivity via hydrogen bonding with hydrogen fumarate (17) or by stabilization of surface-exposed α-helices (33). Overall, our survey of cysteine reactivity provides a novel resource for the identification of functional protein activity changes involved the development, progression, or treatment of HLRCC.
Concurrent with our initial chemoproteomic survey of FH-regulated cysteines in patient-derived UOK262 cells, van der Reest et al. (34) reported a related method for quantifying cysteine reactivity termed stable isotope cysteine labeling with iodoacetamide (SICyLIA) and applied it to map cysteine occupancy changes in Fh1 (termed FH here for brevity) knockout mice. SICyLIA differs from our approach in that it does not use an enrichment step to increase the coverage of low-abundance cysteine residues but similarly provides a quantitative and mechanism agnostic readout of FH-regulated cysteine reactivity. To understand how covalent S-succination and oxidative cysteine modifications vary across diverse models of FH inactivation, we examined overlap between four complementary data sets: UOK262 (FH−/− and FH rescue), UOK268 (FH−/− and FH rescue), mouse kidney cells (WT and FH−/−), and in vivo mouse kidney (WT and FH−/−) (17, 34). Starting with analysis of FH-regulated cysteines in human UOK262/UOK268 cells, we found only eight sites with R ≥ 2 overlap (Table S8 and Fig. 4h). Included among these hits was Chromobox 5 (CBX5 Cys-188, R = 4.8), whose cysteine-dependent capture we have previously shown is sensitive to fumarate. Intrigued by the limited overlap in these HLRCC models, we performed a similar analysis of peptides found to exhibit 2-fold decreased reactivity (log2 -fold change >1) upon FH knockout in mouse kidney cells and mouse kidney tissues by SICyLIA (Table S9). Here again, FH-regulated cysteines between these two models demonstrated a very modest overlap, with only two specific cysteine residues (cathepsin D Cys-288 and thioredoxin domain–containing protein 5 Cys-293) displaying coordinate regulation by FH (Fig. 4i). No common residues were found to be regulated among all four models. The finding that distinct cysteine-omes are altered in these four models of FH perturbation has precedent in the literature, where cysteine occupancy has been found to exhibit similar cell- and tissue-specific disruption in response to oxidative stress (35, 36). Overall, these data demonstrate that considerable heterogeneity may be observed in the adaptive response of reactive cysteines to fumarate accumulation in different HLRCC models.
Discussion
Here we have reported a quantitative chemoproteomic analysis of cysteine reactivity in the kidney-derived HLRCC UOK268 tumor cell line. Application of this approach enabled the identification of new candidate reactive cysteine residues potentially subject to posttranslational regulation by FH activity. Integrating this approach with quantitative whole-proteome analyses of kidney-derived FH−/− and FH+/+ rescue cells identified additional pathways up-regulated at the level of protein expression in UOK268, including those related to the generation of mitochondrial NADH by the TCA cycle (26). An additional notable target identified in these studies was two variants of the MHC complex (HLA-A and HLA-B), which were found to undergo specific losses of reactivity in their extracellular regions. Previous studies have found that the redox modification of HLA cysteines can result in altered antigen presentation (32). Our studies raise the possibility that reducing fumarate levels by FH rescue may augment the immune response, whereas FH inactivation may limit it. Together, these data provide a fertile mechanistic resource from which to formulate biological hypotheses regarding the development and progression of this genetically defined kidney cancer.
An unexpected finding of our study was the limited overlap of FH-dependent cysteines between metastatic (UOK262) (17) and kidney-derived (UOK268) HLRCC cell lines. Conceivably, this observation may reflect either biological heterogeneity or a technical limitation of our method. Our findings are consistent with the literature, in which an independent group using a distinct cysteine-profiling technology observed limited overlap in reactivity changes caused by in vitro and in vivo FH loss in mice (34). This suggests that heterogeneity in the adaptive response of reactive cysteine residues to fumarate accumulation may represent a bona fide biological phenomenon. Supporting the plausibility of this observation, other groups have found in proteome-wide profiling studies that cells and tissues exhibit distinct and nonoverlapping changes in cysteine occupancy upon exposure to identical stimuli, such as redox stress (35, 36). The distinct cohort of down-regulated reactive cysteines in UOK262 and UOK268 cells potentially reflects differences in the underlying biology of the tissues of origin, as well as the magnitude and spatial and temporal generation of fumarate in these different models. Comparative metabolomic analyses of these different models and the development of improved tools to assess subcellular metabolite levels should aid in the future definition of this phenomenon (37, 38). An ancillary insight from our analysis is the distinct information available from studying the consequences of FH rescue (as was done in this study) versus studying the conditions of FH-dependent transformation. For example, our whole-proteome analyses unexpectedly found that glycolytic enzymes become even more highly expressed in UOK268 cells upon FH rescue, suggesting that, at least in this model, the Warburg effect may not be completely reversible by reintroduction of FH alone. In the future, we anticipate that combining chemoproteomic profiling with methods to modulate FH levels with high temporal precision (39, 40) will greatly aid the identification of specific oncometabolite-driven cysteine reactivity changes that contribute to tumorigenesis.
Several recent studies have demonstrated that proteomic profiling of oncogene-induced tumorigenesis can aid the design of new treatment strategies (41–43), and two therapeutic hypotheses are raised by our data. The first is that targeting pathways that demonstrate a compensatory up-regulation upon FH inactivation (either at the level of protein abundance or cysteine reactivity) may serve to limit adaptation of cancer cells and trigger cell death. The second is that inhibiting pathways found to be “damaged” by S-succination (R ≥ 2) or down-regulated upon FH loss at the whole-proteome level may push essential pathways already poised on the precipice of failure over the edge, inducing cytotoxicity (44). Our resource should have substantial utility in this regard, as it represents one of the first catalogues of FH-dependent protein expression and cysteine reactivity in a model of human HLRCC. Future studies integrating proteomics data with screens of pathway-targeted inhibitors (45) and polypharmacological cysteine-reactive fragments (46, 47) should enable the rapid testing of these hypotheses and facilitate the identification of new FH-dependent biology, therapeutic targets, and preclinical drug candidates. It will also be important to understand how proteomic remodeling by electrophilic (fumarate) and nonelectrophilic (succinate, 2-hydroxyglutarate) oncometabolites compares and whether distinct or overlapping biology (and therapeutic vulnerabilities) may exist. Overall, these studies extend our quantitative knowledge of the FH-dependent proteome and provide a novel resource to aid therapeutic design in HLRCC.
Experimental procedures
Full experimental procedures including cell culture, preparation of proteomes, LC–MS/MS, data analysis, Tables S1–S3, and Figs. S1–S3 are provided in the supporting information.
Data availability
MS data has been uploaded to the ProteomeXchange consortium via the PRIDE partner repository with identifier PXD009378. All other data generated and analyzed in this study are included in the article or can be obtained from the authors upon reasonable request. Please direct all requests to Dr. Jordan L. Meier (jordan.meier@nih.gov).
Supplementary Material
Acknowledgments
Figures were created in part using BioRender.
This article contains supporting information.
Author contributions—M. P., D. W. B., S. E. B., E. W., and J. L. M. conceptualization; M. P., D. W. B., E. W., W. M. L., and J. L. M. data curation; M. P., D. W. B., D. R. C., E. W., W. M. L., and J. L. M. formal analysis; M. P., D. W. B., S. E. B., E. W., W. M. L., and J. L. M. investigation; M. P., D. W. B., S. E. B., D. R. C., B. S. A., Y. Y., E. W., W. M. L., and J. L. M. methodology; M. P., D. W. B., and J. L. M. writing-original draft; M. P., D. W. B., D. R. C., E. W., W. M. L., and J. L. M. writing-review and editing.
Funding and additional information—This work was supported by the Intramural Research Program of the NCI, National Institutes of Health, Center for Cancer Research (ZIA BC011488 and ZIA BC011038) and the CCR FLEX Program. Support for E. W. was provided by National Institutes of Health Grants 1R01GM117004 and 1R01GM118431-01A1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- HLRCC
- hereditary leiomyomatosis and renal cell carcinoma
- TCA
- tricarboxylic acid
- FH
- fumarate hydratase
- IA-alkyne
- iodoacetamide alkyne
- ReDiMe
- reductive dimethyl
- AMPK
- 5'-AMP-activated protein kinase
- MHC
- major histocompatibility complex
- SICyLIA
- stable isotope cysteine labeling with iodoacetamide.
References
- 1. Liu J. Y., and Wellen K. E. (2020) Advances into understanding metabolites as signaling molecules in cancer progression. Curr. Opin. Cell Biol. 63, 144–153 10.1016/j.ceb.2020.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meier J. L. (2013) Metabolic mechanisms of epigenetic regulation. ACS Chem. Biol. 8, 2607–2621 10.1021/cb400689r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schvartzman J. M., Thompson C. B., and Finley L. W. S. (2018) Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 217, 2247–2259 10.1083/jcb.201803061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kulkarni R. A., Montgomery D. C., and Meier J. L. (2019) Epigenetic regulation by endogenous metabolite pharmacology. Curr. Opin. Chem. Biol. 51, 30–39 10.1016/j.cbpa.2019.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harmel R., and Fiedler D. (2018) Features and regulation of non-enzymatic post-translational modifications. Nat. Chem. Biol. 14, 244–252 10.1038/nchembio.2575 [DOI] [PubMed] [Google Scholar]
- 6. Alderson N. L., Wang Y., Blatnik M., Frizzell N., Walla M. D., Lyons T. J., Alt N., Carson J. A., Nagai R., Thorpe S. R., and Baynes J. W. (2006) S-(2-Succinyl)cysteine: a novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch. Biochem. Biophys. 450, 1–8 10.1016/j.abb.2006.03.005 [DOI] [PubMed] [Google Scholar]
- 7. Bardella C., El-Bahrawy M., Frizzell N., Adam J., Ternette N., Hatipoglu E., Howarth K., O'Flaherty L., Roberts I., Turner G., Taylor J., Giaslakiotis K., Macaulay V. M., Harris A. L., Chandra A., et al. (2011) Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J. Pathol. 225, 4–11 10.1002/path.2932 [DOI] [PubMed] [Google Scholar]
- 8. Merkley E. D., Metz T. O., Smith R. D., Baynes J. W., and Frizzell N. (2014) The succinated proteome. Mass Spectrom. Rev. 33, 98–109 10.1002/mas.21382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sullivan L. B., Martinez-Garcia E., Nguyen H., Mullen A. R., Dufour E., Sudarshan S., Licht J. D., Deberardinis R. J., and Chandel N. S. (2013) The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 51, 236–248 10.1016/j.molcel.2013.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng L., Cardaci S., Jerby L., MacKenzie E. D., Sciacovelli M., Johnson T. I., Gaude E., King A., Leach J. D., Edrada-Ebel R., Hedley A., Morrice N. A., Kalna G., Blyth K., Ruppin E., et al. (2015) Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat. Commun. 6, 6001 10.1038/ncomms7001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paulsen C. E., and Carroll K. S. (2013) Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem. Rev. 113, 4633–4679 10.1021/cr300163e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. MacKenzie E. D., Selak M. A., Tennant D. A., Payne L. J., Crosby S., Frederiksen C. M., Watson D. G., and Gottlieb E. (2007) Cell-permeating α-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell Biol. 27, 3282–3289 10.1128/MCB.01927-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tennant D. A., Frezza C., MacKenzie E. D., Nguyen Q. D., Zheng L., Selak M. A., Roberts D. L., Dive C., Watson D. G., Aboagye E. O., and Gottlieb E. (2009) Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene 28, 4009–4021 10.1038/onc.2009.250 [DOI] [PubMed] [Google Scholar]
- 14. Kinch L., Grishin N. V., and Brugarolas J. (2011) Succination of Keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell 20, 418–420 10.1016/j.ccr.2011.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adam J., Hatipoglu E., O'Flaherty L., Ternette N., Sahgal N., Lockstone H., Baban D., Nye E., Stamp G. W., Wolhuter K., Stevens M., Fischer R., Carmeliet P., Maxwell P. H., Pugh C. W., et al. (2011) Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537 10.1016/j.ccr.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sciacovelli M., Gonçalves E., Johnson T. I., Zecchini V. R., da Costa A. S., Gaude E., Drubbel A. V., Theobald S. J., Abbo S. R., Tran M. G., Rajeeve V., Cardaci S., Foster S., Yun H., Cutillas P., et al. (2016) Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547 10.1038/nature19353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kulkarni R. A., Bak D. W., Wei D., Bergholtz S. E., Briney C. A., Shrimp J. H., Alpsoy A., Thorpe A. L., Bavari A. E., Crooks D. R., Levy M., Florens L., Washburn M. P., Frizzell N., Dykhuizen E. C., et al. (2019) A chemoproteomic portrait of the oncometabolite fumarate. Nat. Chem. Biol. 15, 391–400 10.1038/s41589-018-0217-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weerapana E., Wang C., Simon G. M., Richter F., Khare S., Dillon M. B., Bachovchin D. A., Mowen K., Baker D., and Cravatt B. F. (2010) Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 10.1038/nature09472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tong W. H., Sourbier C., Kovtunovych G., Jeong S. Y., Vira M., Ghosh M., Romero V. V., Sougrat R., Vaulont S., Viollet B., Kim Y. S., Lee S., Trepel J., Srinivasan R., Bratslavsky G., et al. (2011) The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell 20, 315–327 10.1016/j.ccr.2011.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang Y., Valera V., Sourbier C., Vocke C. D., Wei M., Pike L., Huang Y., Merino M. A., Bratslavsky G., Wu M., Ricketts C. J., and Linehan W. M. (2012) A novel fumarate hydratase-deficient HLRCC kidney cancer cell line, UOK268: a model of the Warburg effect in cancer. Cancer Genet. 205, 377–390 10.1016/j.cancergen.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsu J. L., Huang S. Y., Chow N. H., and Chen S. H. (2003) Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 75, 6843–6852 10.1021/ac0348625 [DOI] [PubMed] [Google Scholar]
- 22. Bar-Peled L., Kemper E. K., Suciu R. M., Vinogradova E. V., Backus K. M., Horning B. D., Paul T. A., Ichu T. A., Svensson R. U., Olucha J., Chang M. W., Kok B. P., Zhu Z., Ihle N. T., Dix M. M., et al. (2017) Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell 171, 696–709.e23 10.1016/j.cell.2017.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 24. Grubb R. L. 3rd, Franks M. E., Toro J., Middelton L., Choyke L., Fowler S., Torres-Cabala C., Glenn G. M., Choyke P., Merino M. J., Zbar B., Pinto P. A., Srinivasan R., Coleman J. A., and Linehan W. M. (2007) Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J. Urol. 177, 2074–2079 10.1016/j.juro.2007.01.155 [DOI] [PubMed] [Google Scholar]
- 25. Xu Y., Andrade J., Ueberheide B., and Neel B. G. (2019) Activated thiol Sepharose-based proteomic approach to quantify reversible protein oxidation. FASEB J. 33, 12336–12347 10.1096/fj.201900693R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang Y., Lane A. N., Ricketts C. J., Sourbier C., Wei M. H., Shuch B., Pike L., Wu M., Rouault T. A., Boros L. G., Fan T. W., and Linehan W. M. (2013) Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS ONE 8, e72179 10.1371/journal.pone.0072179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Repana D., Nulsen J., Dressler L., Bortolomeazzi M., Venkata S. K., Tourna A., Yakovleva A., Palmieri T., and Ciccarelli F. D. (2019) The Network of Cancer Genes (NCG): a comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol. 20, 1 10.1186/s13059-018-1612-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reva B., Antipin Y., and Sander C. (2011) Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39, e118 10.1093/nar/gkr407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rath N., Wang Z., Lu M. M., and Morrisey E. E. (2005) LMCD1/Dyxin is a novel transcriptional cofactor that restricts GATA6 function by inhibiting DNA binding. Mol. Cell Biol. 25, 8864–8873 10.1128/MCB.25.20.8864-8873.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shao D., Oka S., Liu T., Zhai P., Ago T., Sciarretta S., Li H., and Sadoshima J. (2014) A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 19, 232–245 10.1016/j.cmet.2013.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trujillo J. A., Croft N. P., Dudek N. L., Channappanavar R., Theodossis A., Webb A. I., Dunstone M. A., Illing P. T., Butler N. S., Fett C., Tscharke D. C., Rossjohn J., Perlman S., and Purcell A. W. (2014) The cellular redox environment alters antigen presentation. J. Biol. Chem. 289, 27979–27991 10.1074/jbc.M114.573402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kortemme T., and Creighton T. E. (1995) Ionisation of cysteine residues at the termini of model α-helical peptides: relevance to unusual thiol pKa values in proteins of the thioredoxin family. J. Mol. Biol. 253, 799–812 10.1006/jmbi.1995.0592 [DOI] [PubMed] [Google Scholar]
- 34. van der Reest J., Lilla S., Zheng L., Zanivan S., and Gottlieb E. (2018) Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 9, 1581 10.1038/s41467-018-04003-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fu L., Liu K., Sun M., Tian C., Sun R., Morales Betanzos C., Tallman K. A., Porter N. A., Yang Y., Guo D., Liebler D. C., and Yang J. (2017) Systematic and quantitative assessment of hydrogen peroxide reactivity with cysteines across human proteomes. Mol. Cell. Proteomics 16, 1815–1828 10.1074/mcp.RA117.000108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao H., Jedrychowski M. P., Schweppe D. K., Huttlin E. L., Yu Q., Heppner D. E., Li J., Long J., Mills E. L., Szpyt J., He Z., Du G., Garrity R., Reddy A., Vaites L. P., et al. (2020) A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 180, 968–983.e24 10.1016/j.cell.2020.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wellen K. E., and Snyder N. W. (2019) Should we consider subcellular compartmentalization of metabolites, and if so, how do we measure them? Curr. Opin. Clin. Nutr. Metab. Care 22, 347–354 10.1097/MCO.0000000000000580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kulkarni R. A., Briney C. A., Crooks D. R., Bergholtz S. E., Mushti C., Lockett S. J., Lane A. N., Fan T. W., Swenson R. E., Marston Linehan W., and Meier J. L. (2019) Photoinducible oncometabolite detection. Chembiochem 20, 360–365 10.1002/cbic.201800651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nabet B., Roberts J. M., Buckley D. L., Paulk J., Dastjerdi S., Yang A., Leggett A. L., Erb M. A., Lawlor M. A., Souza A., Scott T. G., Vittori S., Perry J. A., Qi J., Winter G. E., et al. (2018) The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441 10.1038/s41589-018-0021-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Natsume T., Kiyomitsu T., Saga Y., and Kanemaki M. T. (2016) Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 15, 210–218 10.1016/j.celrep.2016.03.001 [DOI] [PubMed] [Google Scholar]
- 41. Santana-Codina N., Chandhoke A. S., Yu Q., Małachowska B., Kuljanin M., Gikandi A., Stańczak M., Gableske S., Jedrychowski M. P., Scott D. A., Aguirre A. J., Fendler W., Gray N. S., and Mancias J. D. (2020) Defining and targeting adaptations to oncogenic KRAS(G12C) inhibition using quantitative temporal proteomics. Cell Rep. 30, 4584–4599.e4 10.1016/j.celrep.2020.03.021 [DOI] [PubMed] [Google Scholar]
- 42. Biancur D. E., Paulo J. A., Małachowska B., Quiles Del Rey M., Sousa C. M., Wang X., Sohn A. S. W., Chu G. C., Gygi S. P., Harper J. W., Fendler W., Mancias J. D., and Kimmelman A. C. (2017) Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 8, 15965 10.1038/ncomms15965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kohnz R. A., Mulvihill M. M., Chang J. W., Hsu K. L., Sorrentino A., Cravatt B. F., Bandyopadhyay S., Goga A., and Nomura D. K. (2015) Activity-based protein profiling of oncogene-driven changes in metabolism reveals broad dysregulation of PAFAH1B2 and 1B3 in cancer. ACS Chem. Biol. 10, 1624–1630 10.1021/acschembio.5b00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muller F. L., Aquilanti E. A., and DePinho R. A. (2015) Collateral lethality: a new therapeutic strategy in oncology. Trends Cancer 1, 161–173 10.1016/j.trecan.2015.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yohe M. E., Gryder B. E., Shern J. F., Song Y. K., Chou H. C., Sindiri S., Mendoza A., Patidar R., Zhang X., Guha R., Butcher D., Isanogle K. A., Robinson C. M., Luo X., Chen J. Q., et al. (2018) MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 10, eaan4470 10.1126/scitranslmed.aan4470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Backus K. M., Correia B. E., Lum K. M., Forli S., Horning B. D., González-Páez G. E., Chatterjee S., Lanning B. R., Teijaro J. R., Olson A. J., Wolan D. W., and Cravatt B. F. (2016) Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574 10.1038/nature18002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Counihan J. L., Wiggenhorn A. L., Anderson K. E., and Nomura D. K. (2018) Chemoproteomics-enabled covalent ligand screening reveals ALDH3A1 as a lung cancer therapy target. ACS Chem. Biol. 13, 1970–1977 10.1021/acschembio.8b00381 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MS data has been uploaded to the ProteomeXchange consortium via the PRIDE partner repository with identifier PXD009378. All other data generated and analyzed in this study are included in the article or can be obtained from the authors upon reasonable request. Please direct all requests to Dr. Jordan L. Meier (jordan.meier@nih.gov).