

Abstract
Strong evidence suggests that dysregulated lipid metabolism involving dysfunction of the retinal pigmented epithelium (RPE) underlies the pathogenesis of age-related macular degeneration (AMD), the leading cause of irreversible blindness in the elderly. A hallmark of AMD is the overproduction of lipid- and protein-rich extracellular deposits that accumulate in the extracellular matrix (Bruch's membrane (BrM)) adjacent to the RPE. We analyzed apolipoprotein A-1 (ApoA-1)-containing lipoproteins isolated from BrM of elderly human donor eyes and found a unique proteome, distinct from high-density lipoprotein (HDL) isolated from donor plasma of the same individuals. The most striking difference is higher concentrations of ApoB and ApoE, which bind to glycosaminoglycans. We hypothesize that this interaction promotes lipoprotein deposition onto BrM glycosaminoglycans, initiating downstream effects that contribute to RPE dysfunction/death. We tested this hypothesis using two potential therapeutic strategies to alter the lipoprotein/protein profile of these extracellular deposits. First, we used short heparan sulfate oligosaccharides to remove lipoproteins already deposited in both the extracellular matrix of RPE cells and aged donor BrM tissue. Second, an ApoA-1 mimetic, 5A peptide, was demonstrated to modulate the composition and concentration of apolipoproteins secreted from primary porcine RPE cells. Significantly, in a mouse model of AMD, this 5A peptide altered the proteomic profile of circulating HDL and ameliorated some of the potentially harmful changes to the protein composition resulting from the high-fat, high-cholesterol diet in this model. Together, these results suggest that targeting HDL interactions with BrM represents a new strategy to slow AMD progression in humans.
Keywords: lipoprotein, heparan sulfate, complement, retinal degeneration, aging, high-density lipoprotein (HDL), apolipoprotein, glycosaminoglycan, oligosaccharide, age-related macular degeneration, complement factor H, heparan sulfate proteoglycans, retinal pigmented epithelium
Age-related macular degeneration (AMD) is the most common cause of blindness among the elderly in the developed world (1). It is a progressive, degenerative disease influenced by both genetic and environmental factors and depends upon advanced age (2). Lipid metabolism is one of the major pathways involved in AMD pathogenesis (3). Genetic associations in ABC transporter A1 (ABCA1), apolipoprotein E (APOE), cholesteryl-ester transport protein (CETP), and hepatic lipase (LIPC) have all been shown to influence the risk of developing AMD (4, 5). These gene products all impact HDL homeostasis. Although there are conflicting results from human studies, several publications have shown an association between plasma HDL-cholesterol levels and the risk for AMD, including a large study from Europe that showed that HDL-cholesterol is positively associated with increased risk of AMD (6–11). The basis for the contradictory results may come from the effects of diet and medications, which are difficult to control for in patient populations. In addition, most studies investigating AMD risk association rely on plasma levels of lipoproteins, and it is possible that locally synthesized and secreted lipoproteins in the eye are more consequential (12, 13).
One of the most replicated genetic variants associated with AMD risk is the tyrosine to histidine substitution at amino acid 402 (Y402H) in complement factor H (FH) (14–17). FH is the soluble regulator of the alternative complement pathway (18, 19). The Y402H amino acid lies outside of the complement-regulating short consensus repeats (SCRs) 1–4 of FH, and thus it is not surprising that no differences in regulation of fluid phase complement activation between the Tyr-402 and His-402 variants have been detected (20–23). Instead, the Y402H polymorphism is located in SCR 7, a region that is known to mediate FH binding to polyanions such as heparin, GAGs, and C-reactive protein (24, 25). The Y402H polymorphism decreases the binding of FH to heparin (22, 26), M6 protein of Streptococcus pyogenes (26, 27), C-reactive protein (22, 26–28), Bruch's membrane (BrM) (29), malondialdehyde epitopes (30), and oxidized phospholipids (31).
We developed transgenic mouse models that express equal concentrations of the normal human FH Tyr-402 (CFH-Y/0) or the AMD risk–associated FH His-402 (CFH-H/H), in place of mouse Fh. These mice were aged over 90 weeks and fed a high-fat, cholesterol-enriched (HFC) diet to test the in vivo effect of the His-402 FH risk variant on AMD pathobiology (32, 33). Only the old CFH-H/H fed an HFC diet (CFH-H/H∼HFC), develop an AMD-like phenotype (33). This is true despite the fact that complement activation is equivalent in these two mouse models (33). Interestingly, the variant specific differences detected in these mice were associated with changes in lipid homeostasis. Aged CFH-H/H∼HFC mice have increased ApoA-1 and ApoB48 in the retinal pigmented epithelium (RPE)/BrM/choroid following the HFC diet when compared with the age-matched, control CFH-Y/0 mice fed the HFC diet (CFH-Y/0∼HFC) (33). ApoA-1 is the major, defining protein associated with HDL, accounting for ∼70% of the protein on the particle, and each HDL particle typically contains 2–5 ApoA-1 molecules (34). In addition, CFH-H/H∼HFC mice had lower levels of plasma LDL-cholesterol than the CFH-Y/0∼HFC mice (33). Taken together, the CFH-H/H∼HFC AMD model provides evidence that there is an intersection between lipid metabolism and the human His-402 “risk”-associated variant of FH.
This paper details experimental evidence demonstrating that the lipoproteins present in aged, human donor BrM are HDL-like based on density and size. The protein composition of these HDL-like lipoproteins is very different from the composition found on HDL isolated from the plasma of the same donor, and these BrM HDL-like particles are likely made by the RPE cells. A 96-well plate binding assay was developed that measures the binding of proteins to primary porcine RPE cell culture extracellular matrix (ECM), which we show has a heparan sulfate fingerprint similar to that of human and porcine BrM. This assay was used to demonstrate the binding characteristics of various lipoproteins in the presence or absence of FH and heparan sulfate molecules. In an ex vivo system, using human BrM tissue from aged donors, we demonstrate the potential for therapeutic use of short HS oligosaccharides to remove lipoproteins from deposits in BrM.
The effects of an ApoA-1 mimetic peptide, 5A peptide, were also examined in RPE cells and were shown to increase the secretion of HDL-associated proteins when exposed to the peptide. In addition, the 5A peptide was administered to a cohort of the old CFH-H/H∼HFC mice, by intraperitoneal injection for the duration of the HFC diet. HDL was then isolated from the plasma of these mice (CFH-H/H∼HFC + 5A), mice fed an HFC diet alone (CFH-H/H∼HFC), and mice fed a normal diet (CFH-H/H∼ND). The proteomes of these HDL particles were analyzed, and, as shown below, the HDL proteome of the 5A peptide–treated animals was modified to more closely resemble the normal control CFH-H/H∼ND plasma HDL proteome than the HDL proteome in the CFH-H/H∼HFC animals. This is significant because the proteins attached to HDL particles have been shown to affect the properties of HDL (35). In certain disease states or even with aging, there is evidence that the HDL proteome can dictate whether the HDL particle is an anti-inflammatory, antioxidant particle or is more harmful, becoming inflammatory and promoting oxidation (35–37).
Currently, there are no good therapeutic options for those suffering from the early to intermediate forms of AMD, characterized by the appearance of medium-sized sub-RPE drusen (63–125 μm) in early AMD and large drusen (>125 μm) and/or pigmentary abnormalities in intermediate AMD (38). The ideal therapy would ameliorate the early symptoms and prevent the progression to the late, more impactful disease. Age is the strongest risk factor in developing AMD, with small drusen being a normal finding in aged patients. Understanding the mechanisms that give rise to pathogenic changes in individuals with drusen accumulation that progresses to late AMD is likely to be a key to developing successful therapies for the disease. In this paper, we highlight two potential therapeutic options, one that could reduce already established deposits and the other that may prevent the initial build-up of lipids and proteins.
Results
The majority of cholesterol in aged human donor BrM is present in HDL-like lipoproteins
BrM-enriched tissue was isolated from aged human donor eyes. BrM lysates were prepared from 6-mm punches of the tissue using RIPA buffer with protease inhibitors. Lysates from three central “near macula” punches were pooled and fractionated by FPLC, and the total cholesterol in each fraction was analyzed. The majority of cholesterol was detected in HDL-sized lipoproteins (Fig. 1A), although the amounts found in BrM from individual donors were variable. Substantial amounts of cholesterol (>2 μg of HDL-cholesterol per lysate from three 6-mm diameter biopsy punches of BrM-rich tissue) were detected in 11 of 30 eyes (37%) from donors >70 years of age (Table S1). Cholesterol in the FPLC fractions where VLDL and LDL would elute was rarely detected. Human plasma (30 μl) was fractionated in a similar manner (Fig. 1B). Comparison with the fractions containing cholesterol in the BrM tissue lysate confirmed that the cholesterol eluted in the same size fractions as HDL in the plasma (Fig. 1, A and B).
Figure 1.
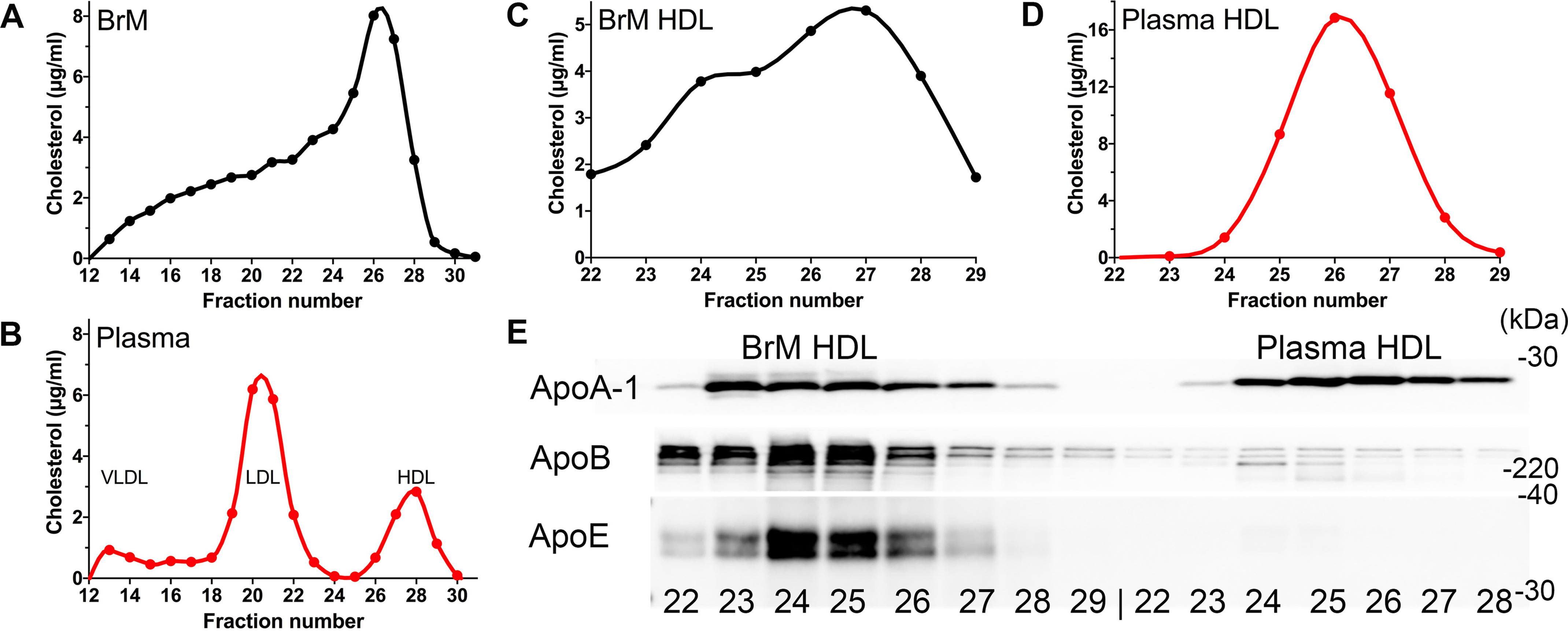
Characterization and comparison of the lipoproteins isolated from BrM tissue and from plasma. A, FPLC fractionated lysates of BrM tissue isolated from an 84-year-old male human donor (described in Table S1A) reveal that the majority of cholesterol (μg/ml) is found in the same fractions (fractions 24–29), that contain the HDL lipoproteins from 30 μl of fractionated normal human plasma (American Red Cross) (B). HDL purified from both BrM tissue lysate (C) and plasma from the same donor (D) (an 88-year-old male, Table S1C), using a combination of density gradient ultracentrifugation followed by size separation by FPLC, shows that the majority of cholesterol is in fractions 24–29. E, Western blots of these fractions revealed approximately equal amounts of ApoA-1, whereas substantially more ApoB and ApoE were present in the fractions of HDL-sized lipoproteins from BrM tissue HDL (left) compared with plasma HDL (right).
Eyes that had appreciable amounts (>2 μg of cholesterol/3 punches, Table S1) of HDL-cholesterol were used to isolate highly purified HDL particles, using the method described by Holzer et al. (39) that uses a 5-h (relatively short) density gradient ultracentrifugation followed by size fractionation on FPLC. This method allows for the reliable quantification and analysis of the HDL proteome. We purified HDL from both BrM (Fig. 1C) and plasma (Fig. 1D) from the same donor and measured cholesterol in each of the FPLC fractions. Most of the cholesterol in the BrM tissue HDL sample was detected in fractions 22–29, and all of the cholesterol in plasma HDL was in fractions 24–28. ApoA-1, ApoE, and ApoB were assayed in each of these fractions by Western blotting (Fig. 1E). With the amount of ApoA-1 normalized, considerably more ApoB and ApoE were found in the HDL-like lipoproteins isolated from the BrM tissue than in the HDL purified from the plasma (Fig. 1E).
The LC–MS/MS–based proteomic analysis of 25 μg of protein from each of the isolated BrM HDL and plasma HDL confirmed these findings and revealed other interesting protein changes (Table 1). The amounts of proteins are displayed as the percentage of each protein (fmol) relative to ApoA-1 (fmol) in each sample. In total, three sets of HDL from BrM and plasma were isolated and analyzed. Two of the sets were BrM and plasma HDL from the same donor (Table 1, sets A and B), and one set was plasma HDL from a normal American Red Cross plasma donor and BrM HDL from an aged human donor eye (Table 1, set C). In all three comparisons, ApoB, ApoE, ApoA-IV, and ApoC-III were higher in the BrM HDL than in the plasma HDL. In addition, there was more C3, clusterin, and vitronectin in the BrM HDL. Albumin, fibrinogen, and hemoglobin β were also consistently higher in the BrM HDL (Table 1). The extent of the changes was highly variable; however, two of the most prevalent proteins increased on the BrM HDL are ApoB and ApoE, and they, unlike most of the other proteins identified by proteomics, are known to bind to GAGs.
Table 1.
Comparison of the HDL proteomes isolated from human BrM tissue and plasma of the same donors
| Accession no. Uniprot Hu | Set Aa |
Set Ba |
Set Ca |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BrM | Plasma | Unique peptides | BrM | Plasma | Unique peptides | BrM | Plasma | Unique peptides | ||
| % protein relative to ApoA-1 | ||||||||||
| Apolipoprotein A-I | P02647 | 100 | 100 | 41 | 100 | 100 | 73 | 100 | 100 | 49 |
| Apolipoprotein D | P05090 | 9.76 | 8.95 | 7 | 3.67 | 8.07 | 13 | 11.82 | 26.08 | 6 |
| APOL1 protein | O14791 | 13.2 | 12.47 | 18 | 8 | 1.78 | 3.9 | 9.12 | 11 | |
| Apolipoprotein E | P02649 | 19.5 | 6.31 | 25 | 11.16 | 0.77 | 33 | 37.83 | 8.83 | 20 |
| Apolipoprotein B-100 | A8MUN2 | 15.3 | 0.72 | 41 | 2.55 | 0.08 | 30 | 10.77 | 0.47 | 29 |
| Apolipoprotein C-I | P02654 | 4.4 | 3.53 | 4 | 0.99 | 1.4 | 7 | 7.42 | 4.85 | 4 |
| Apolipoprotein A-II | P02652 | 3.56 | 3.29 | 5 | 3.5 | 2.97 | 9 | 4.2 | 8.07 | 4 |
| Apolipoprotein C-III | P02656 | 6.06 | 3 | 2 | 13.88 | 4.38 | 6 | 3.48 | 1.97 | 2 |
| Apolipoprotein A-IV | P06727 | 8.44 | 0.78 | 28 | 0.27 | 0.11 | 9 | 13.76 | 0.98 | 24 |
| Apolipoprotein C-II | P02655 | 3.15 | 0.15 | 7 | 2.04 | 0.62 | 2 | |||
| Apolipoprotein M | O95445 | 0.5 | 0.32 | 3 | 2.52 | 0.54 | 6 | |||
| Serum amyloid A-2 | P0DJI9 | 14 | 8.67 | 2 | 17.39 | 35.17 | 21 | 13.95 | 6.39 | |
| Serum amyloid A-1 | P0DJI8 | 7.04 | 6.48 | 2 | ||||||
| Serum amyloid A | B2R5G8 | 41.4 | 29.37 | 10 | 1.08 | 12.51 | 13 | |||
| Serum amyloid A SAA2-SAA4 | P35542 | 2.38 | 0.69 | 6 | ||||||
| Serum amyloid P | P02743 | 1.16 | 0.03 | 3 | 7 | 1.82 | 0.07 | 2 | ||
| Serum paraoxonase/arylesterase 1 | P27169 | 25.1 | 5.1 | 14 | 2.11 | 20.53 | 26 | 17.3 | 24.72 | 11 |
| Serum paraoxonase/lactonase 3 | Q15166 | 1.13 | 0.56 | 5 | 0.23 | 0.24 | 5 | |||
| C4b-binding protein α chain | P04003 | 5.67 | 0.02 | 12 | 1.38 | 0.02 | ||||
| Complement C3 | P01024 | 2.61 | 1.05 | 29 | 0.44 | 0.27 | 28 | 4.61 | 1 | 35 |
| Complement C4-B | P0C0L5 | 1.71 | 0.61 | 24 | 0.14 | 0.47 | 8 | 2.47 | 0.25 | 12 |
| Complement component 9 | P02748 | 0.96 | 0.06 | 6 | 1.75 | 0.11 | 3 | |||
| Complement C1s subcomponent | P09871 | 0.26 | 0.04 | 4 | ||||||
| Complement component 1r subcomponent (C1R) | P00736 | 0.05 | 0.14 | 11 | ||||||
| Clusterin OS | P10909 | 8.99 | 0.47 | 13 | 1.6 | 0.1 | 7 | 11.63 | 0.13 | 12 |
| Vitronectin OS | P04004 | 4.98 | 0.05 | 6 | 0.49 | 0.03 | 4 | 4.46 | 0.13 | 4 |
| Carboxypeptidase D | O75976 | 13 | 0.02 | 2 | ||||||
| α1-Antichymotrypsin SERPINA3 | P01011 | 2.31 | 0.14 | 10 | 0.27 | 0.03 | 5 | 1.79 | 0 | 6 |
| α2-Macroglobulin | P01023 | 1.5 | 0.09 | 10 | 5.84 | 0 | 21 | |||
| Phospholipid transfer protein | P55058 | 7.92 | 0.99 | 17 | 0.4 | 0.85 | 23 | 4.69 | 0.51 | 7 |
| Serum albumin | P02768 | 17.8 | 2.5 | 33 | 5.3 | 2.68 | 29 | 15.24 | 4.67 | 25 |
| CP protein | A5PL27 | 0.72 | 0.12 | 11 | 1.99 | 0.07 | 5 | |||
| Hemopexin | P02790 | 13.1 | 0.08 | 20 | ||||||
| Haptoglobin | P00738 | 6.99 | 0.54 | 18 | 1.46 | 1.85 | 20 | 4.06 | 0.41 | 7 |
| Hemoglobin β | Q6J1Z7 | 35 | 0.52 | 6 | 18.78 | 0.49 | 11 | 9.49 | 1.09 | 11 |
| Lipopolysaccharide-binding protein | P18428 | 10.2 | 0.07 | 10 | 0.61 | 0.04 | 5 | |||
| Actin cytoplasmic 1 | P60709 | 23.7 | 0.28 | 21 | 15.5 | 0.6 | 25 | |||
| Coagulation factor VIII | P00451 | 4.3 | 2.69 | 2 | 4.84 | 8.85 | 6 | |||
| Gelsolin | P06396 | 0.96 | 0 | 8 | 0.26 | 0.01 | 3 | 1.37 | 0 | 2 |
| Fibrinogen β chain | P02675 | 26.9 | 0.05 | 34 | 1.97 | 0.16 | 9 | 11.53 | 0.3 | 18 |
| Fibrinogen γ chain | P02679 | 11.5 | 0.21 | 24 | 1.66 | 0.03 | 7 | 13.89 | 0.06 | 11 |
| Fibrinogen α chain | P02671 | 10.5 | 0.01 | 25 | 0.34 | 0.01 | 4 | 8.23 | 0.49 | 14 |
| Ig α-1 chain C region | P01876 | 7 | 3.67 | 1.76 | 4 | |||||
| IGK@ protein | Q6PIL8 | 6.76 | 1.74 | 6 | ||||||
| Ig γ-1 chain C region | Q0ZCF9 | 28.1 | 12.58 | 11 | 13.57 | 3.81 | ||||
| Immunoblobulin light chain | Q0KKI6 | 9.48 | 3.22 | 6 | ||||||
| Ig κ chain V-I region CAR | P01596 | 1.53 | 0.48 | 2 | 1.02 | 0.15 | 5 | |||
| Ig γ-3 chain C region (fragment) | P01860 | 0.79 | 0.49 | 8 | 6.67 | 2.99 | 3 | |||
a Set A, BrM and plasma from the same donor: an 88-year-old white male, cause of death bacteremia and a diagnosis of hypercholesterolemia with a death to recovery time of 4 h 56 min. Set B, BrM and plasma from the same donor: an 82-year-old white male, coronary artery disease and pulmonary hypertension with a death to recovery time of 7 h 40 min. Set C, BrM from an 80-year-old white female, coronary artery disease and respiratory failure and plasma from a sample of American Red Cross–recovered frozen plasma. The amounts are expressed as (amount of protein (fmol)/amount of ApoA-1 in sample (fmol)) × 100 or as percentage of protein relative to ApoA-1. The analysis uses LC–MS/MS MS analysis, with Progenesis QI for protein identification and quantitation.
This analysis required HDL isolated from the BrM from an entire eye. Ideally, HDL isolated from the macula punch from an AMD-donor eye would be more informative. As better sensitivity from new instrumentation develops, an even more interesting insight into the HDL lipoproteins in macular drusen will emerge.
Validation of primary pig RPE-derived ECM-coated plastic plates for modeling lipid and protein binding to BrM
A 96-well plate binding assay using primary pig RPE cells was developed to model lipoprotein and protein binding to GAGs found in human BrM. Because GAGs, particularly heparan sulfate, will not adhere directly to plastic (40, 41), we chose to grow primary porcine RPE cells into well-differentiated monolayers (42) for extended periods (6–8 weeks) to form an ECM on plastic and determine whether, following decellularization, the HS-GAGs remaining in the RPE-derived ECM on the plate were similar to the GAGs isolated from either human or porcine BrM. The HS fingerprints of GAGs isolated from human and porcine BrM, from the ECM produced by ARPE-19 cells (an immortalized human RPE cell line (43)), and from the ECM of primary porcine RPE cells were analyzed by the Glycobiology Core Facility at the University of California, San Diego (La Jolla, CA, USA). This method provides an analysis of the relative amounts of all of the disaccharides present in the HS-GAGs in the sample. The HS-GAGs isolated from human and porcine BrM and from the ECM produced by the primary porcine RPE cells were very similar, whereas the most noticeable differences were seen with the ARPE-19 cell ECM (Table 2 and Table S2). This is not entirely surprising because ARPE-19 is an RPE-like cell line lacking several important hallmarks of RPE cells (44, 45). These results establish that HS-GAGs in the ECM from primary porcine RPE cells are comparable with those isolated from human and porcine BrM and that this ECM should be a useful platform and model to perform binding studies. To assess the reproducibility of this ECM plate binding assay, we compared the binding of FH to the plate at six different concentrations with eight replicates at each concentration, which showed high reproducibility within the plate (Fig. S1).
Table 2.
Comparison of the HS composition, in terms of the percentage of each type of disaccharide present, in GAGs isolated from human donor BrM, porcine BrM, primary porcine RPE cell ECM and ARPE-19 cell ECM (see Table S2 for a more comprehensive analysis)
| Mole percentages of the number of sulfates or the type of sulfation |
Average SO3/disaccharide | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 SO3 | 1 SO3 | 2 SO3 | 3 SO3 | % | Unsulfated | N-SO3 | 2-O-SO3 | 6-O-SO3 | ||
| Human BrM | 56.51 | 27.17 | 14.01 | 2.31 | 100 | 56.51 | 35.08 | 12.86 | 14.18 | 0.62 |
| Porcine BrM | 59.95 | 26.63 | 10.45 | 2.97 | 100 | 59.95 | 29.91 | 10.19 | 16.34 | 0.56 |
| Porcine RPE ECM | 43.19 | 33.7 | 23.1 | 0 | 100 | 43.19 | 37.98 | 15.84 | 26.1 | 0.8 |
| ARPE-19 cell ECM | 23.2 | 16.32 | 28 | 32.47 | 100 | 23.2 | 69.17 | 48.29 | 52.28 | 1.7 |
Identification of proteoglycans found in human and porcine BrM
Proteoglycans can be intracellular, cell-surface, pericellular, or extracellular. Analysis of the proteoglycans found in porcine and human BrM identifies a number of HS proteoglycans present in BrM. Perlecan and collagen XVIII are pericellular and found in the basement membrane zone, and syndecan-1 is transmembrane (46). These HS proteoglycans were also isolated from the porcine ECM. Proteoglycans can have HS, chondroitin sulfate, dermatan sulfate, or keratin sulfate GAGs. We identified extracellular proteoglycans with each type of GAG in the human and porcine BrM. (Table S3A). Their presence supports the possibility of their participation in lipoprotein and FH binding because each type of GAG has been shown to bind lipoproteins and FH (47–49). Other potentially relevant proteins found in the porcine ECM include ApoE and ApoA-1, C3, vitronectin, and clusterin, all of which were also identified in both sources of BrM samples (Table S3B).
Lipoprotein binding to RPE ECM is determined by its protein composition
Lipoprotein binding was measured using the binding assay described above, which utilizes 96-well plates coated with primary porcine RPE cell ECM. After blocking the 96-well plates, increasing concentrations of plasma VLDL or LDL, in the presence of 0 or 0.5 μg of FH (His-402 or Tyr-402) were added. Both VLDL and LDL binding (as measured by the amount of ApoB100 detected on the plate) were inhibited to the same extent by either His-402 or Tyr-402 FH (Fig. 2, A and B). This confirms our previous findings that FH and lipoproteins likely compete for the same binding sites on the ECM (50). Plasma-derived HDL binding to this porcine ECM was measured by ApoA-1 binding and revealed very little binding and no effect of the addition of FH (Fig. 2C). When binding of HDL isolated from plasma and BrM from the same donor were compared, the plasma HDL showed very little binding, whereas HDL from BrM bound significantly better (Fig. 2, D and E). This was observed when assaying for both ApoA-1 (Fig. 2D) and ApoB (Fig. 2E) binding, indicating that ApoA-1 and ApoB are likely on at least some of the same lipoprotein molecules and that the ApoB is facilitating HDL binding to the ECM.
Figure 2.
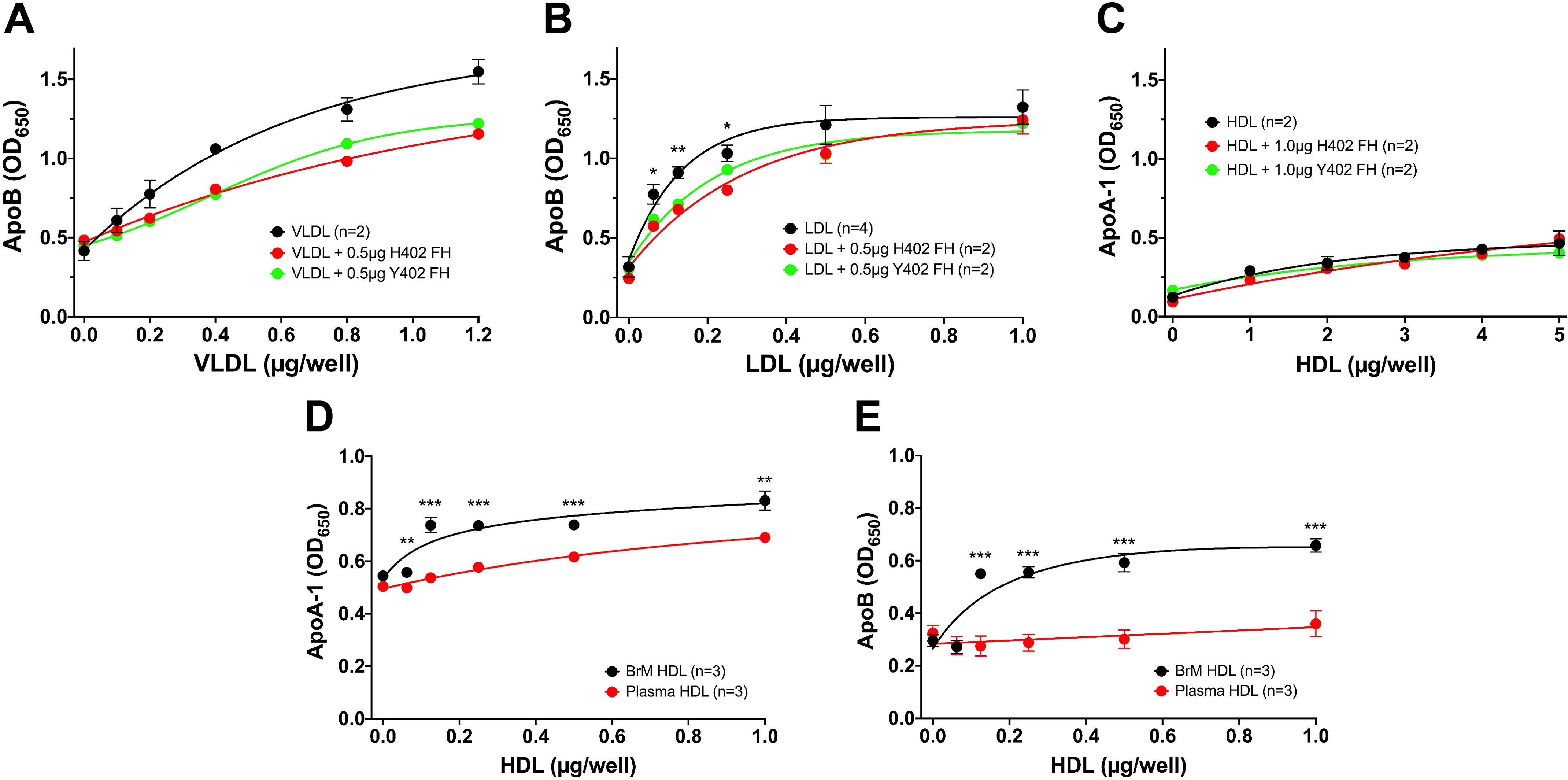
Measurement of lipoprotein binding to porcine RPE-derived ECM. A 96-well binding assay with primary porcine RPE cell ECM-coated wells was used to measure plasma lipoprotein binding to porcine RPE-derived ECM. A, VLDL binds well to the ECM, as measured by the ApoB bound on the plate. Each concentration point of VLDL alone was measured in duplicate, and the VLDL binding when mixed with 0.5 μg of each variant of FH was an individual point. This assay was repeated with similar results using 1 μg of each FH variant (see Fig. S2). B, LDL also binds well to this ECM (n = 4), and this binding can also be inhibited equally by both the “risk” variant of FH, His-402 (n = 2), and the normal Tyr-402 (n = 2). This experiment was repeated with 1.0 μg of FH with similar results. C, plasma-derived HDL does not bind well to the ECM, and FH has no effect on the binding as measured by ApoA-1 binding. This assay had n = 2 for each point. This assay was repeated with HDL at a concentration range of 0–1 μg and 1 μg FH, and similar results were seen. D and E, comparisons of HDL isolated from BrM tissue and from plasma from the same donor reveal that the BrM-derived HDL bound better to the RPE-derived ECM compared with the same concentration of plasma-derived HDL, as measured by both ApoA-1 (D) and ApoB (E) binding, suggesting that at least some of the BrM lipoprotein particles contain both ApoA-1 and ApoB. Error bars, S.D. Very small errors did not produce visible error bars. *, p < 0.05; **, p < 0.01; ***, p < 0.001. OD, optical density.
Short HS oligomers are sufficient to inhibit VLDL and LDL binding, whereas inhibition of FH binding requires longer-chain HS species
It is technically challenging to determine the exact structures of the sugar moieties that bind to proteins. Instead, short HS oligomers of defined structure and molecular weight (Fig. S3) were used to inhibit LDL, VLDL, and FH binding to the ECM. VLDL and LDL binding to porcine RPE ECM were inhibited with the highly sulfated 12-sugar oligosaccharide, NS2S 12-mer (Fig. 3 (A and B) and Figs. S4 and S5). The nonsulfated NAC 12-mer was not as effective at inhibiting VLDL binding but somewhat inhibited LDL binding. (Fig. 3 (A and B) and Figs. S4 and S5). Neither of these short 12-mers had any effect on either His-402 or Tyr-402 FH binding (Fig. 3, C and D). However, in the presence of longer HS GAGs (∼40 kDa), FH binding to porcine RPE ECM was decreased. (Fig. 3 (E and F) and Fig. S6). This could be because FH has more than one binding site for heparin and heparan sulfate (SCR 7 and SCR 19-20) (51) and may require simultaneous inhibition at both sites to block attachment to the ECM. The GAG-binding sites on ApoB100 and ApoE have been described (48, 52). On ApoE, the region spanning amino acids 136–150 with many positively charged arginine and lysine residues has both LDLR– and HS GAG–binding capacity (52). On ApoB100, two short positively charged regions come together in the tertiary structure (amino acids 3145–3157 and 3359–3367), forming overlapping LDLR- and GAG-binding regions (48). These shorter regions may be blocked by a small HS oligosaccharide.
Figure 3.

Short HS oligosaccharides can inhibit ECM binding of VLDL and LDL but not FH to porcine RPE-derived ECM. The short 12-mer HS oligosaccharides, NAC and NS2S, can inhibit binding of VLDL (A) and LDL (B) to porcine RPE-derived ECM. These graphs are representative of five separate experiments with both VLDL and LDL treated with different oligosaccharides (see Figs. 4 and 5). C and D, these short oligosaccharides had no effect on either His-402 FH (C) or Tyr-402 FH (D) binding to the ECM. E and F, 20 μg of longer HS GAGs (∼40 kDa) had an inhibitory effect on FH binding. This experiment was repeated with similar results (see Fig. S6). Error bars, S.D. Very small errors did not produce visible error bars.
Short HS oligosaccharides can remove lipoproteins from aged, human BrM tissue
Overnight incubation of 6-mm-diameter BrM tissue explants isolated from six pairs of elderly human donor eyes (with substantial amounts of BrM HDL) with sulfated HS 12-mers removed endogenous lipoproteins. Tissue lysates were prepared following overnight incubation of human tissue with either the buffer control or the HS 12-mer. The samples were pooled from five 6-mm tissue punch lysates for each treatment and fractionated by FPLC. Cholesterol was measured in each fraction. Fig. 4A shows the effect of the NS2S 12-mer, whereas Fig. 4B shows similar results obtained independently with the NS6S2S3S 12-mer (average of 3 experiments in each case). ApoA-1, ApoB, and ApoE were measured using immunoblots of corresponding fractions from an NS2S-treated and nontreated sample (Fig. 4C). The cholesterol in all of the fractions was significantly decreased following treatment (Fig. 4, A and B), and ApoA-1, ApoB, and ApoE were all depleted following incubation with the NS2S 12-mer (Fig. 4C). These results suggest that there may be therapeutic potential for this type of molecule in removing and/or preventing lipoproteins from accumulating in this particular tissue.
Figure 4.
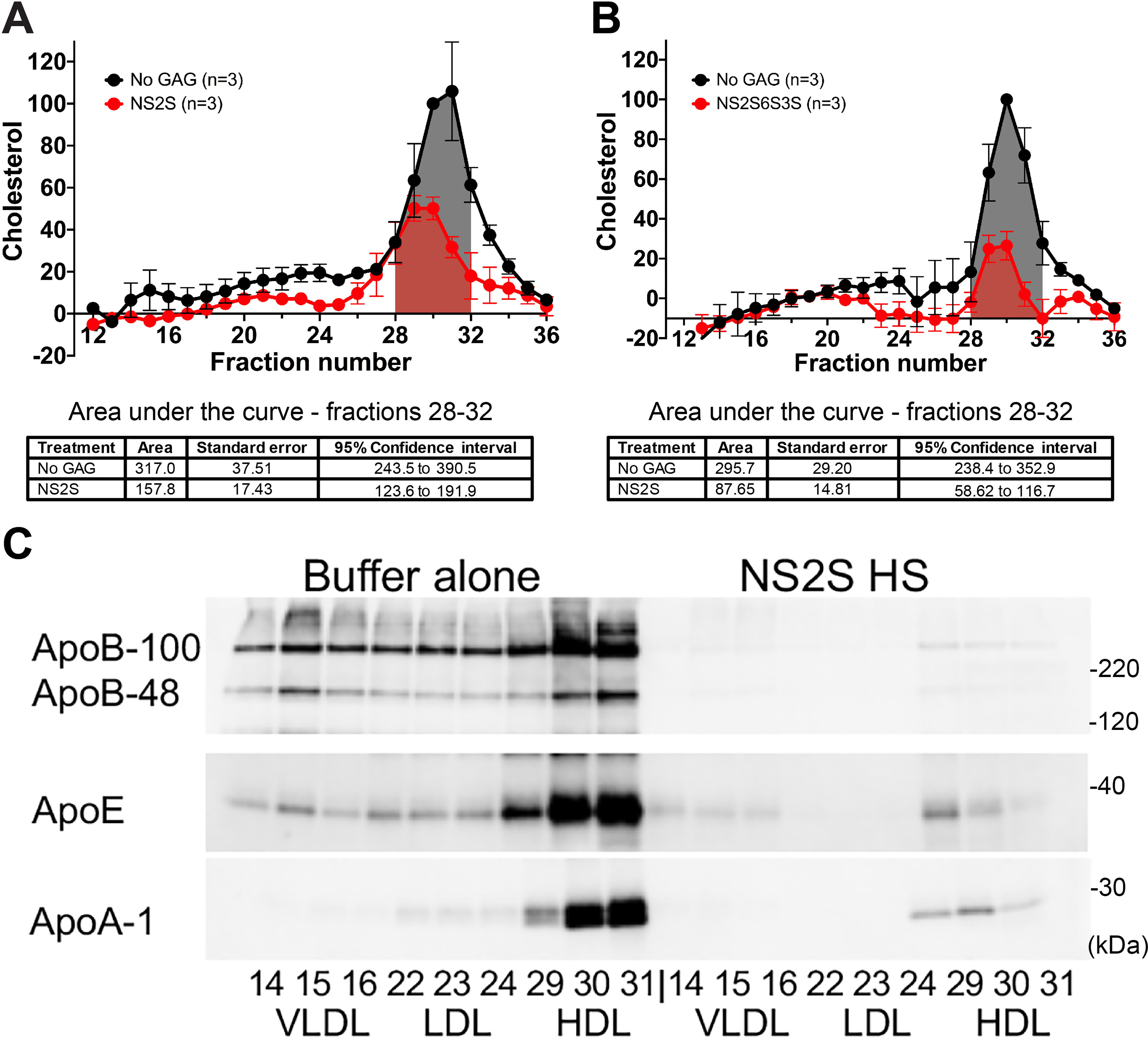
Short glycosaminoglycans (NS2S or NS2S6S3S 12-mers) can be used to remove cholesterol from aged human BrM donor tissue. A, 6-mm diameter BrM tissue explants (five per treatment) were incubated in a hypotonic solution overnight at +4 °C to lyse any cells, washed thoroughly, and then incubated overnight at +4 °C in a well of a 96-well plate with either 100 μl of buffer or buffer containing 16 μg of the HS NS2S 12-mer. B, tissue from a different donor was treated with 16 μg of the HS NS2S6S3S 12-mer or buffer alone. A and B, after extensive washing, each tissue sample was homogenized in 80 μl of RIPA buffer. Forty μl of samples from each treatment group was pooled and separated by FPLC on a Superose 6HR 10/30 column with PBS + 1 mm EDTA, and cholesterol was measured in each fraction. These experiments were repeated with tissue from three different donors (Table S1E) for each type of HS 12-mer. The cholesterol content was normalized in each experiment to the HDL peak fraction (fraction 30) in the No GAG sample to compare experiments. Mean and S.E. bars for three separate experiments are indicated. Area under the curve was measured for the HDL-containing fractions 28–32 using the AUC function in GraphPad Prism 8.4.2 (San Diego, CA) and is shown in tables below the graphs. Treatments with short GAGs removed cholesterol in a statistically significant manner as demonstrated by the nonoverlapping 95% confidence intervals. C, a representative Western blotting analysis of the fractions containing VLDL (14–16), LDL (22–24), and HDL (29–31) revealed that most of the apolipoproteins were removed after treatment with both the NS2S and NS2S6S3S 12-mer.
The ApoA-1 mimetic peptide 5A increases reverse cholesterol transport (RCT) and apolipoproteins found in the media of primary pig RPE
We next tested whether an ApoA-1 mimetic peptide, 5A, could facilitate excess cholesterol clearance from RPE cells, similar to how it has been reported to reduce atherosclerosis and inflammation in animal models (53–56). The 5A peptide mimetic consists of a 37-amino acid peptide (DWLKAFYDKVAEKLKEAF-P-DWAKAAYDKAAEKAKEAA) that was reconstituted with sphingomyelin in a 1:8 molar ratio (Table F of Ref. Ref. 53). The peptide is bihelical, with the first amphipathic helix having a high lipid affinity and the other loop displaying 5 alanines (5A) on the hydrophobic face, which appears to reduce its cytotoxicity but increases its specificity to enhance the efflux of cholesterol from cells by the ABCA1 route (56), thus increasing RCT from the cells. It has also been shown to display both anti-inflammatory and antioxidant properties in vivo and in vitro (53). These properties of the mimetic led us to hypothesize that an increased number of HDL particles would be produced by the RPE in the presence of this 5A peptide, but these HDLs might be smaller, with lower amounts of apolipoproteins other than ApoA-1. This would maintain their anti-inflammatory and antioxidant profile, and they would potentially be less prone to depositing on BrM.
Primary pig RPE were grown to confluence on Transwells, as we have described previously (42), and then increasing concentrations of 5A peptide in serum-free media (0, 8, 40, and 200 μg/ml) were added for 48 h. Apical and basolateral media were collected and concentrated 10-fold. A portion of the sample was used for Western blotting analysis of ApoA-1, ApoE, clusterin, and FH (Fig. 5A). We saw a marked increase in all these proteins, in particular in the basolateral media, even at the lowest concentration of the 5A peptide. The cells maintained normal morphology after the 48-h treatment. The cell lysates were analyzed by Western blotting analysis for ABCA1, and this protein was clearly affected by the mimetic (Fig. 5B). The decrease in the amount of the lower-molecular weight, normal-sized ABCA1 band and corresponding increase in the upper ABCA1 band could be due to dimerization (57, 58). These results confirm that RPE cells can secrete ApoA-1, the essential component of HDL, and can use ABCA1 in RCT to eliminate cholesterol from the cells.
Figure 5.
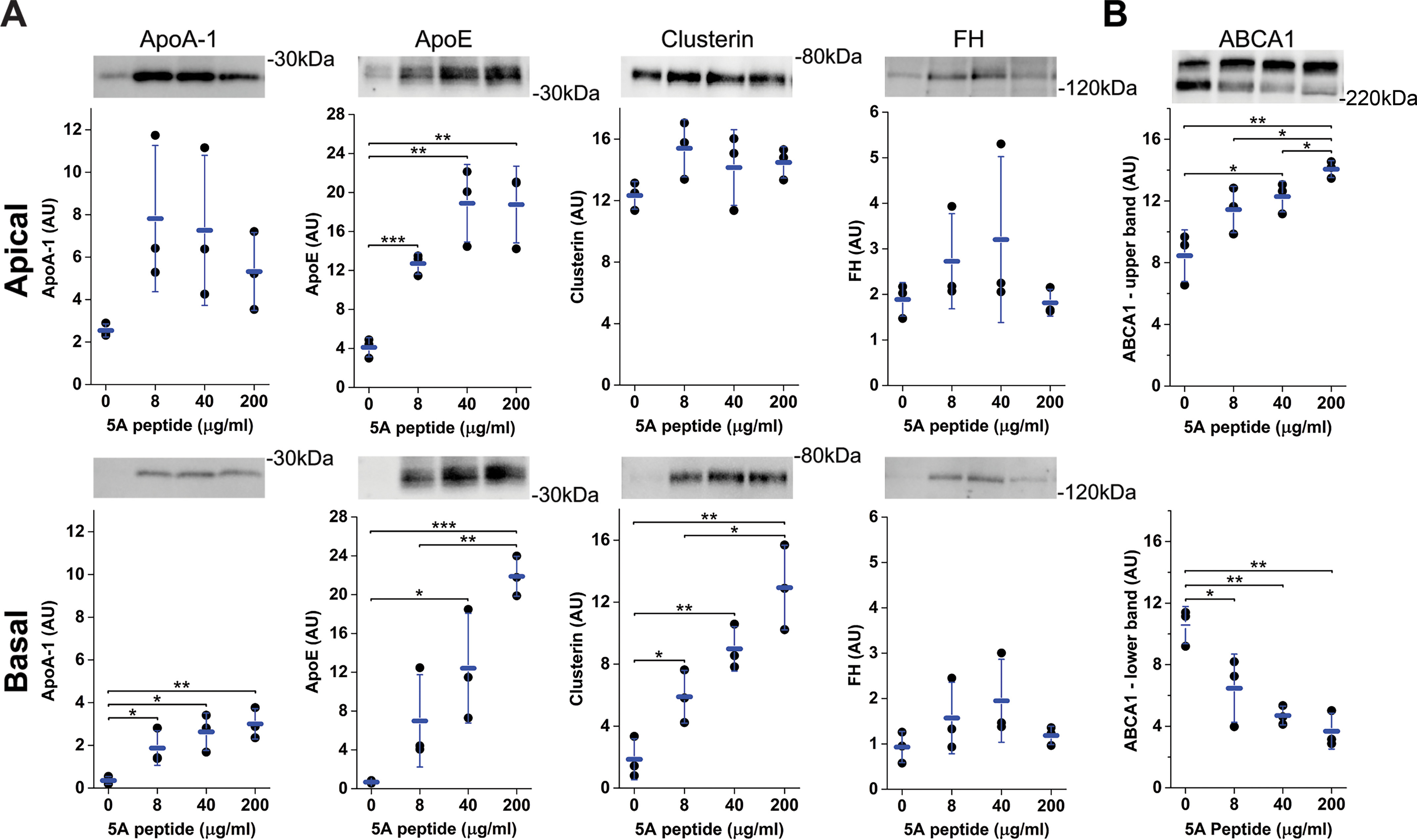
Primary Pig RPE cells secrete ApoA-1, ApoE, clusterin, and FH and increase RCT in response to the 5A ApoA-1 mimetic peptide. Well-established RPE cells, grown for 6 weeks on 12-mm Transwells, were incubated with serum-free media with 0, 8, 40, or 200 μg/ml 5A peptide. A and B, quantification of immunoblots of ApoA-1, ApoE, clusterin, and FH in the apical (top row) and basolateral media (bottom row) (A) and ABCA1 in cell lysate (B) following 48 h of growth in serum-free media with increasing concentrations of 5A peptide. The immunoblot images shown are representative of the findings in triplicate wells of the effect of the 5A peptide on each protein measured. Error bars, S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001. AU, Arbitrary units.
The ApoA-1 mimetic can ameliorate some of the changes to the HDL proteome caused by a high-fat and cholesterol diet
ApoA-1 mimetic 5A peptide was delivered systemically to our AMD mouse model of old CFH-H/H mice fed an HFC diet (33). At ∼90 weeks of age, one group of CFH-H/H mice remained on a normal diet (ND), and one was switched to an HFC diet for 8 weeks. The treatment group was switched to an HFC diet plus 30 mg of 5A peptide/kg of body weight by intraperitoneal injections, three times a week, for the duration of the 8-week HFC diet. Three mice were in each group. At the completion of the experiment, all the animals were fasted for 5 h, blood was obtained, and the plasma was pooled from mice in the same treatment group. 250 μl of plasma was used to isolate and purify HDL, by density gradient ultracentrifugation followed by size-exclusion chromatography by FPLC (39). The cholesterol was measured in each fraction and was found exclusively in the HDL-sized fractions. HDL fractions 26–28 were pooled and concentrated, and 25 μg protein for each sample was prepared and analyzed using LC–MS/MS analysis with Progenesis QI for protein identification and quantitation. The HFC diet caused many more proteins to be associated with the HDL, and the 5A peptide had a moderating effect on this, resulting in a protein profile more similar to the HDL proteome in the normal diet control group (Table 3 and Table S4). Of particular interest was the fact that both ApoE and ApoB were substantially higher in the HFC diet–fed plasma HDL, whereas the HFC with 5A peptide–derived plasma HDL showed levels much closer to those on a normal diet. This supports our hypothesis that the 5A peptide can produce HDL particles that are less likely to bind to BrM.
Table 3.
Summary of proteomic analysis of HDL isolated from plasma from CFH-H/H mice aged 90 weeks and then continued on an ND for an additional 8 weeks, fed an HFC diet, or fed HFC + 5A peptide for 8 weeks
The 5A peptide had an effect on several potentially important proteins; especially of interest are ApoE and ApoB. Results are expressed as percentage abundance relative to ApoA-1 (fmol). (See Table S4 for a more extensive list of HDL proteins identified in these samples.)
| Protein on HDL | ND | HFC | HFC + 5A |
|---|---|---|---|
| % | % | % | |
| ApoA-1 | 100 | 100 | 100 |
| ApoE | 4.1 | 11.2 | 6.3 |
| ApoA-II | 0.6 | 2.1 | 0.2 |
| ApoD | 0.7 | 3.6 | 0.2 |
| ApoA-IV | 0.3 | 0.14 | 0.4 |
| ApoB | 0.01 | 0.2 | 0.03 |
| Paraoxonase-1 | 15.2 | 11.5 | 12.7 |
| Haptoglobin | 0.7 | 13.5 | 5.8 |
| Hemopexin | 1.2 | 7.6 | 1.3 |
Discussion
The RPE, which forms the outer blood-retina barrier, handles the influx of large quantities of lipids and cholesterol from the daily phagocytosis of photoreceptor outer segments and from endocytosis of lipoproteins from the choriocapillaris (59). Cholesterol in the RPE is then transferred apically to the photoreceptors, retained in the cell, or eliminated from the basal side (12, 13). RPE cells use RCT, an ABCA1-dependent mechanism, to eliminate excess phospholipid and cholesterol from cells via ApoA-1 and HDL (60). As stated in the Introduction, there is evidence that four genes involved with lipid metabolism are associated with AMD: ABCA1, APOE, LIPC, and CETP (4, 5). Although there have been conflicting results from population studies looking for an association between HDL and AMD, the most recent studies suggest that higher plasma concentrations of HDL-cholesterol are associated with an increased incidence of AMD (6, 7, 10, 61).
We isolated lipoproteins from aged, human donor eye BrM and found that the cholesterol was mainly carried by lipoproteins, which had the same density and size as HDL (Fig. 1). The amount of HDL in this tissue is variable: eyes from young donors have very little, and some (about 37%) of the 30 aged donor eyes (from donors aged 70 years and older) examined had substantial amounts (Table S1). This is similar to the findings of variable amounts of drusen detected in eyes of individuals in this age range.
One of the main functions of HDL is to facilitate cholesterol and phospholipid efflux from cells and transport it for eventual elimination by the liver. HDL particles additionally have anti-inflammatory (62) and antioxidant properties (63, 64). Increasingly, it has become apparent that under certain conditions, such as cardiovascular disease (36), aging (37), or an acute phase response, HDL is converted from an anti-inflammatory to a pro-inflammatory molecule (35). This is largely due to the dynamic changes in the HDL proteome found in these different conditions. It is unclear whether the alterations seen in the plasma HDL proteome contribute to the condition or if they are changes that occur as a result of the disease. Several groups are now monitoring changes that are detected in the plasma HDL proteome in different disease states, including coronary artery disease (65, 66), type II diabetes (67), renal disease (68), and psoriasis (69).
One study looked at selected HDL-associated proteins in serum of AMD compared with control serum and found increased plasma serum amyloid A (SAA) (70). It is, however, possible that it is not the circulating levels of HDL-cholesterol but rather the local concentrations in the eye and in particular the composition of the HDL particle that results in a damaging phenotype.
We isolated and purified the HDL lipoproteins from three separate samples of both BrM tissue and plasma: plasma and BrM HDL from the same donor were compared in two of the sets, and the third set compared a normal plasma HDL sample with BrM HDL from a third aged donor. The proteomic profile of these purified HDL samples was examined, and the data showed an increase in several apolipoproteins, including ApoE, ApoB, ApoC-III, and ApoA-IV, associated with the HDL from BrM compared with the plasma HDL; this was also true for the complement pathway–related proteins C3, clusterin, and vitronectin (Table 1). HDL has a dynamic protein composition, so increases in proteins involved in lipid metabolism and complement regulation may be reflective of the environment in the eyes from which they are isolated (71). AMD-donor eyes were not available for HDL isolation, and we used the entire BrM from an eye for each HDL isolation described in this paper. As more sensitive instrumentation becomes available and isolation techniques improve, it should be possible to analyze the HDL proteome of an isolated macula versus periphery BrM punch and from AMD versus control donor.
It is a tenet of the “response to retention” hypothesis of atherosclerosis that ApoB100 and ApoE on lipoproteins from the plasma can interact with the GAGs on the ECM in the arterial wall in the initial stages of the disease (72, 73). It follows that the increases in ApoB and ApoE found on the human BrM-derived HDL would increase its ability to bind to the GAGs on BrM. We show that VLDL and LDL containing ApoB and ApoE bind to the RPE ECM, whereas plasma-derived HDL, which has very low concentrations of ApoB and lower ApoE content compared with BrM-HDL, does not bind to the RPE ECM to the same extent (Fig. 2, A–C). The increase in BrM-HDL binding can be measured by an increase in both ApoB binding and ApoA-1 binding (Fig. 2, D and E), indicating that at least some of the lipoprotein particles contain both ApoB and ApoA-1, likely resulting in an increased deposition of lipoprotein particles in BrM. In atherosclerosis, lipoprotein binding then leads to modification by oxidation or glycation, which induces an inflammatory response in the artery wall (74). It is possible that similar downstream events could occur in the eye.
The current study describes two classes of molecules, HS oligosaccharides and ApoA-1 mimetic peptides, which have the potential to therapeutically limit HDL binding to BrM and thus reduce the buildup of lipids and proteins in the eye. Specifically, we present evidence that FH, heparin, short well-defined HS oligosaccharides, and longer HS moieties can compete with and inhibit binding of ApoB-containing lipoproteins to the ECM produced by primary porcine RPE cells (Figs. 2 (A and B) and 3 (A, B, E, and F)). We also demonstrate that the short HS oligosaccharides may have the advantage of inhibiting lipoprotein deposition while retaining FH on the surface, thus maintaining the regulatory properties of inhibiting local complement activation (Figs. 3, C and D). We also demonstrate that short HS oligosaccharides can remove the ApoB-containing HDL on human BrM explants (Fig. 4, A and B). This work supports the idea that FH, heparin, or heparan sulfate could inhibit the initial deposition or remove deposited lipoproteins from BrM at an early stage of AMD and prevent the continuous buildup and subsequent visual damage due to the accumulation of this material.
We then tested the effect of an ApoA-1 mimetic peptide (5A) on primary pig RPE cells and on the proteome of plasma HDL in vivo in our AMD mouse model (aged CFH-H/H∼HFC). Describing ApoA-1 mimetics in great detail is beyond the scope of this discussion; please see Islam et al. (75) for a review. Briefly, these peptides mimic the three-dimensional structure of the lipid-accepting portion of the ApoA-1 molecule and remove phospholipids and cholesterol from cells in conjunction with ABCA-1, creating a discoidal HDL-like particle. In primary RPE cell cultures, the 5A peptide increased ApoA-1, ApoE, clusterin, and FH in the media (Fig. 5). This may be due to the peptide modulating and/or stabilizing ABCA1, or it could be releasing HDL or other lipoproteins already bound to the RPE cells. This facility to increase lipid efflux from RPE cells could be beneficial to RPE cells in older individuals where build-up of lipids inside RPE cells can be harmful (76). In an in vivo environment, HDL was isolated from plasma from old CFH-H/H mice fed an ND, an HFC diet, or an HFC diet + 5A peptide intraperitoneally. We show that the HFC diet alone had a considerable effect compared with the plasma HDL proteome from age-matched control mice fed a normal diet. Of particular interest in this study, the HFC diet caused a 3-fold increase in ApoE content and a 20-fold increase in ApoB, and both of these changes were reduced substantially in the mice that were given the 5A peptide along with the HFC diet (Table 3 and Table S4). This supports the idea that if given locally, there could also be a reduction of the potentially damaging protein content of HDL in the eye. This is corroborated by a study described by Rudolf et al. (77) in which a single intravitreal injection of an ApoA-1 mimetic in 10-month-old ApoE−/− mice appeared to ameliorate thickening of BrM.
We hypothesize that under conditions that contribute to AMD pathobiology (advanced age, diet, smoking, complement activation, CFH variants, and/or alterations in the RPE metabolic state), the concentration and content of the sub-RPE HDL-like lipoproteins increase. Furthermore, the changes in HDL protein composition cause the HDL to switch from being anti-inflammatory and antioxidant to being inflammatory and subsequently sequestered locally. This switch could be due to protein and lipid modifications resulting in increased oxidation, inflammatory responses, and less efficient cholesterol and lipid removal from cells. These pathophysiological changes ultimately damage the RPE and the surrounding milieu, likely contributing to AMD development and progression.
The incidence of deposited HDL in the back of the eye appears to be age-dependent; however, the initiating factors are unknown. It is possible that aging BrM, which becomes less permeable to plasma proteins, lipids, and oxygen, can prevent the passage of locally synthesized lipoproteins to the circulation. It has also been shown that the GAGs in BrM change with age (78), and this might facilitate lipoprotein binding in older individuals. One thing is clear: the buildup of lipid- and protein-rich material in soft drusen, basal linear deposits, and subretinal drusenoid deposits is harmful to the health of the RPE and photoreceptors in the eye. Reducing the load of these deposits would be beneficial to the health of the eye and potentially postpone the worst outcomes of late-stage AMD. The results reported in this study support this hypothesis and constitute a proof of concept for targeting HDL as a therapy for AMD.
Experimental procedures
Analysis of size and type of lipoproteins present in human BrM tissue
Human donor eyes were obtained from Miracles in Sight (Winston-Salem, NC, USA) and were distributed by BioSight (Duke Core Service) under the Institutional Review Board protocol PRO-00050810. Human donor eye tissue was obtained within 10 h of death. Posterior eyecups were processed as described previously (20, 50). Briefly, after removal of the retina from the eyecup, 6-mm biopsy punches (Sklar Instruments, West Chester, PA, USA) were taken, and the RPE/BrM/choroid was removed from the sclera. From each eye, one macular punch, five or six “near macula” punches, and the “rest” of the RPE/BrM/choroid were isolated. The tissue was treated with 0.1 m 6-aminohexanoic acid/5 mm benzamidine hydrochloride in H2O overnight at +4 °C to lyse any cells. Any remaining RPE or choroidal cell material was removed. To determine whether the tissue had a significant amount of lipoprotein, 80 μl of RIPA buffer with protease inhibitors was added to three “near macula” tissue samples, and a hand-held pestle motor mixer (Argos Technologies) was used to homogenize the samples, followed by 30 min on ice with vigorous vortexing every 10 min. After centrifugation for 15 min at 14,000 rpm at +4 °C, 50 μl of each supernatant was pooled and fractionated by size-exclusion chromatography using FPLC (AKTA Pure, GE Healthcare, Danderyd, Sweden) on a Superose 6 Increase 10/300 GL column (GE Healthcare) in a PBS + 1 mm EDTA buffer. The cholesterol content of each fraction (25 μl of a total 500-μl fraction) was analyzed (Molecular Probes Amplex Red kit). 30 μl of human plasma was fractionated using the same procedure as a control for sizes of VLDL, LDL, and HDL.
Isolation of HDL for proteomic analysis
BrM was isolated from the entire posterior pole of the eye as described above and solubilized in RIPA buffer with protease inhibitors. After centrifugation, the lysate was made up to 4 ml with PBS. Similarly, 250 μl of plasma was diluted in PBS to 4 ml. Using the method described by Holzer et al. (39), the solutions were brought to a density of 1.24 g/ml by adding KBr (1.56g/4 ml) and added to Beckman Ultra-Clear centrifuge tubes (#344322; 16 × 76 mm). The tubes were then carefully filled with 140 mm NaCl made to a density of 1.063 g/ml (3.76 g of KBr/40 ml of 140 mm NaCl), and the tops were sealed. The tubes were spun in a Type 70.1 Ti rotor for 5 h at 65,000 rpm (289,835 × gavg), in an Optima XE-90 Ultracentrifuge (Beckman-Coulter). The HDL fraction was removed using a syringe with a 21-gauge needle in a volume of 2.5 ml and concentrated to ∼300 μl using Amicon Ultra-4 centrifuge filters (Ultracel-10K #UFC801024, Merck Millipore, Carrigtwohill, Ireland). The sample was then fractionated by FPLC as described in the previous section, and the same five ApoA-1–containing fractions were pooled and concentrated.
MS for protein isolation, identification, and quantitation
The protein concentration was measured (Pierce BCA protein assay kit), and 25 μg of protein from both the plasma and the tissue HDL was used for proteomics. A paramagnetic bead protocol for protein sample cleavage for MS analysis described by Hughes et al. (79) was used to prepare the samples for analysis. This uses both hydrophilic and hydrophobic SeraMag SpeedBead carboxylate-modified magnetic particles (part 45152105050250 and part 65152105050250, GE Healthcare). Briefly, proteins solubilized in 2% SDS, 100 mm Tris·HCl (pH 8.0) were reduced with 10 mm DTT (D0632, Sigma–Aldrich), alkylated with 25 mm iodoacetamide (I1149, Sigma-Aldrich), and subjected to tryptic hydrolysis using the HILIC bead SP3 protocol (79). The resulting peptides were analyzed with a nanoAcquity UPLC system coupled to a Synapt G2 HDMS mass spectrometer (Waters Inc.) employing the LC–MS/MS protocol in a data-independent acquisition mode complemented with ion mobility separation (HDMSE). The peptides were separated on a 75 μm × 150-mm 1.7-μm C18 BEH column (Waters) using a 90-min gradient of 5–30% of acetonitrile in 0.1% formic acid at a flow rate of 0.3 ml/min at 45 °C. Eluting peptides were sprayed into the ion source of the Synapt G2 using the 10 μm PicoTip emitter (Waters) at a voltage of 2.75 kV. PLGS 2.5.1 software was used to assign peptides to the features and generate searchable files, which were submitted to the IdentityE search engine incorporated into Progenesis QI Proteomics software 3.1 (Waters). For peptide identification, we searched against either UniProt 2016 mouse (16,806 entries), human (20,206 entries), or pig databases (7222 entries) using carbamidomethyl at Cys as a fixed modification and Met oxidation as a variable modification. Only proteins identified by at least two peptides and with confidence scores of >95% were included in Tables 1 and 3 and Table S3 (A and B). Trypsin was the only protease used for cleavage. Only one missed and/or nonspecific cleavage was permitted, and there was a 20-ppm mass tolerance for precursor ions and a 0.1-ppm mass tolerance for fragment ions. The threshold score for accepting individual spectra was 6, and there was a >95% confidence level. Calculated protein decoy false discovery rate was 0.1%, and peptide decoy false discovery rate was 0.1%. All calculations were made from an average of technical duplicates. For protein quantification (Table 3), we used a sum of the intensity of the top three peptides for each protein to compare their amounts in each sample, essentially as described by Silva et al. (80).
An aliquot of each fraction from the FPLC was retained and analyzed by immunoblotting, using Bio-Rad 10% XT Criterion gels with MOPS buffer and nonreduced samples, transferred to nitrocellulose, blocked, incubated with primary antibody overnight at +4 °C, followed by horseradish peroxidase–conjugated secondary antibodies (Jackson ImmunoResearch; 1:10,000), and visualized with ECL Plus Substrate (Thermo Fisher Scientific). Immunoblots were imaged using a Bio-Rad ChemiDoc imaging system. Membranes were probed for ApoE (goat anti-ApoE, #178479, Calbiochem; 1:5000), ApoB (goat anti-ApoB, #AB742, Millipore; 1:5000), and ApoA-1 (goat anti-ApoA-1, #11A-G2b, Academy Bio-Medical Co; 1:2500.).
Creating 96-well ECM-coated plates
Primary pig RPE were isolated from freshly obtained pig eyes (City Packing Co., Burlington, NC). Whole globes were immersed in +4 °C, 0.2% povidone-iodine for 10 min, and then 1000 units/ml penicillin-streptomycin for 5 min. After removal of the cornea, lens, and vitreous, the eyecup was filled with PBS + 1 mm EDTA and incubated for 30 min at 37 °C. The retina was gently removed, and the eyecups were then incubated with 0.25% trypsin + 0.9 mm EDTA for 30 min at 37 °C. The RPE were removed from BrM by repeated pipetting of the trypsin over the inside of the eyecup. The RPE cells were added to DMEM/F-12 medium supplemented with 10% FBS and washed. The cells were seeded into 75-cm2 flasks (∼4 × 106 cells/flask) and grown to confluence (3–4 days). The cells were then trypsinized and passaged into 96-well plates (∼3.5 × 104 cells/well). The cells achieved confluence and were then grown for 6–8 weeks in DMEM/F-12 medium with 10% FBS. The plates were decellularized using the method of Vlodavsky et al. (81). Briefly, after washing with PBS, 20 mm ammonium hydroxide and 0.5% Triton X-100 was added to the plate for 30 min at room temperature, followed by four 10-min washes with PBS. The plates were covered and kept at +4 °C for use within 14 days.
ECM-binding assays
The ECM-coated plates were blocked overnight with 0.4% gelatin (Sigma, #G2625) in 50 mm sodium acetate, 50 mm NaCl, 0.2% Tween 20, pH 7.2 After washing with the buffer without gelatin, increasing amounts of FH, heparan sulfate, or lipoprotein, or a combination, were added to the wells in 200 μl of 50 mm sodium acetate, 50 mm NaCl, 0.2% Tween 20, 0.2% gelatin. Plates were incubated for 2 h at 37 °C and washed four times in buffer, and then the appropriate antibody was added as follows: anti-FH (goat anti-human FH, 1:1000; Quidel #A312,), anti-ApoB100 (goat anti-human ApoB100, 1:2000; Millipore #AB742) or anti-ApoA-1 (goat anti-human ApoA-1, 1:1000) incubated for 1 h at 37 °C, followed by horseradish peroxidase–conjugated bovine anti-goat IgG (Jackson ImmunoResearch, 1:5000) for 1 h. Antibody binding was detected using TMB reagent (BD Biosciences). The plate was read at 650 nm in a SpectraMax M5 microplate reader (Molecular Devices). The short HS oligomers (NS2S, NS2S6S3S, and NAC 12-mers) were prepared by Dr. Liu at the University of North Carolina (Chapel Hill, NC, USA) (82). The following GAGs were purchased as follows: heparin (Sigma, #H3393), porcine mucosa HS (PI-HS, Celsus Laboratories), 6-O-desulfated heparin (#DSH002/6, Iduron, distributed by Galen Laboratory Supplies). FH was isolated, using an affinity column, from American Red Cross frozen plasma, which we had determined was homozygous for either the His-402 or Tyr-402 polymorphism, as we have described previously (83). The FH was kept at 4 °C and used within 14 days. The FH and lipoproteins were mixed with these GAGs before adding to the 96-well plate.
Isolation of VLDL, LDL, and HDL used in binding assays
Using the method of Aviram et al. (84), plasma was made to d = 1.25 g/ml using KBr, and 4 ml of this sample was placed in Beckman 14 × 89-mm Ultra-Clear centrifuge tubes (#344059). The samples were overlaid with 4 ml of 2 m NaCl, 1 mm EDTA, pH 8.6, followed by a final overlay of 4 ml of 150 mm NaCl, 1 mm EDTA, pH 8.6. Tubes were centrifuged in an SW41 swing-out rotor for 48 h at 40,000 rpm (200,000 × gavg) at +4 °C in a Beckman-Coulter Optima XE-90 ultracentrifuge. The VLDL was removed from the top of the tube, and it was concentrated with an Amicon Ultra 3K cutoff filter unit. The LDL was recovered at the interface of the 150 mm NaCl and the 2 m NaCl and the HDL in the 2 m NaCl fraction. The lipoproteins were dialyzed, kept at +4 °C, and used within 14 days. Coomassie-stained gels of these preparations showed only one dominant band per preparation, corresponding to ApoB100 for VLDL and LDL and ApoA-1 for HDL. Thus, a protein estimation (Pierce BCA protein assay kit) was used to determine amounts of lipoprotein added to the assays in terms of protein concentrations.
Identification of proteins in porcine and human BrM and isolation of GAGs from these tissues
BrM tissue was prepared from a pair of porcine eyes and from tissue from an 80-year-old male donor, using the method described by McHarg et al. (85). Briefly, 8 m guanidine hydrochloride was added to the BrM for 6 h at room temperature with shaking, and then samples were centrifuged at 10,000 × g. The supernatant was dialyzed against 20 mm acetate, 150 mm NaCl, pH 6.2, overnight at +4 °C. Twenty-four μl of this 1.5-ml sample was run on a gel, and bands were cut out, trypsinized, and prepared for protein identification by MS. GAGs were then isolated from the remainder of this tissue lysate using the method described by Foley (86). Briefly, 2 mg/ml Pronase (Sigma, #10165921001) was added to the sample and incubated for 24 h at 37 °C. The GAGs were then captured on DEAE-Sepharose (GE Healthcare, #17-0709). After washing, the GAGs were eluted with 2 m NaCl and then desalted on a PD-10 column (GE Healthcare, #17-0851-01) and concentrated to dryness by vacuum centrifugation. Likewise, Pronase was added to the pellet resulting from the 10,000 × g centrifugation of the guanidine hydrochloride solution and was processed for GAG analysis alongside the supernatant sample.
Isolation of GAGs from the ECM from primary porcine and ARPE-19 cells
Two 75-cm2 flasks of pig RPE or ARPE-19 cells (ATCC #CRL-2302) were grown to confluence and kept an additional 10 days. The cells were lysed and washed off as described above for the 96-well plates. The plates were washed three times in PBS, and then 8 m guanidine hydrochloride was added, and a cell scraper was used to ensure that all of the ECM was recovered. This lysate was then processed in the same way as the BrM lysate described above.
HS fingerprinting of human and porcine BrM and the ECM produced by ARPE-19 cells and porcine RPE cells
The Glycobiology Core Facility at the University of North Carolina (Chapel Hill, NC, USA) analyzed the GAG samples. They used isotopic aniline tagging for GAG disaccharide analysis by MS (GRIL; glycan reductive isotope labeling) and an LTQ Orbitrap-MS. Briefly, samples were homogenized in PBS and treated with Pronase (Sigma) at 37 °C for 24 h, followed by anion-exchange chromatography using DEAE-Sephacel (GE Healthcare). After loading the samples, the reaction tube was rinsed with ultrapure water and loaded on the column. The column was washed with 10 ml of DEAE-wash buffer (50 mm NaOAc containing 150 mm NaCl, pH 6.0), and bound GAG was eluted with DEAE-elution buffer (50 mm NaOAc containing 1 m NaCl, pH 6.0). Finally, the samples were desalted by gel-filtration chromatography using a PD-10 column (GE Healthcare) and lyophilized. The GAG samples were digested with a mixture of Heplyase I, II, and III for 16 h at 37 °C in HS digestion buffer (40 mm ammonium acetate and 3.3 mm calcium acetate, pH 7.0). The sample was then passed over 3K centrifugal filters (Pall Life Sciences), and the flow-through containing HS disaccharides was dried and used for aniline tagging.
Reductive amination and acylation of disaccharides
Samples were dissolved in 15 μl of 12C6-aniline and freshly prepared 15-μl solution of 1 m sodium cyanoborohydride in DMSO/acetic acid (7:3, v/v). The reaction was carried out at 37 °C for 16 h, followed by drying down excess reagent in a SpeedVac. The samples were then spiked with a known concentration of 13C6-aniline–tagged standard disaccharide (HS-DP2) mixture prior to analysis on an LTQ-Orbitrap mass spectrometer.
LC–MS analysis
An LTQ-Orbitrap Discovery mass spectrometer (Thermo Scientific) attached with an Ultimate-3000 (Thermo-Dionex) instrument was used for profiling of HS-disaccharides, and the data were quantified after comparing with the internal standards. The aniline-tagged HS-DP2 were separated on a Targa C18 column (150 mm × 1.0 mm × 5 μm; Higgins Analytical) using a running buffer containing dibutylamine as ion-paring reagent (87). To minimize the in-source fragmentation of the sulfated HS-DP2, the capillary temperature and ion-spray voltage were kept at 140 °C and 4.75 kV, respectively.
HS treatment of human BrM tissue
After preparing 6-mm explant punches of human BrM tissue from aged donor eyes as described above, 10 near-macula punches were taken from the pair of posterior poles. Five were used for controls (incubated with buffer only), and the other five were incubated overnight at +4 °C in wells of a 96-well plate, with 16 μg of NS2S 12-mer or NS2S6S3S 12-mer (Fig. S3) in 100 μl of 10 mm PO4 buffer. After three 10-min washes with PBS, each 6-mm tissue explant was homogenized in 80 μl of RIPA buffer, incubated on ice for 30 min, and then centrifuged at 14,000 rpm. Supernatants from the controls and the treated samples were pooled separately and fractionated by FPLC on a Superose 6HR 10/30 column with PBS + 1 mm EDTA. Twenty-five μl of each 500-μl fraction was analyzed for cholesterol (Molecular Probes Amplex Red Kit). Western blotting analysis was performed as described under “Isolation of HDL for proteomic analysis.”
Cell culture experiments using primary porcine RPE cells
Pig RPE cells were isolated and seeded at passage p1 onto laminin/entactin (Corning, #354259)-coated 12-mm diameter cell culture inserts with pore sizes of 0.4 μm (Corning Transwell™, #3460) exactly as detailed by Klingeborn et al. (42). Once the cells were established, they were grown for a total of 6 weeks in DMEM/F-12 with 10% FBS. The cells were washed three times and then switched to serum– and phenol red–free DMEM with 4.5 g/liter glucose for 48 h. A range of concentrations of 5A peptide mixed with sphingomyelin in a 1:8 molar ratio (Alan Remaley, NHLBI, National Institutes of Health, Bethesda, MD, USA (88)) were added in triplicate wells: 0, 8, 20, and 200 μg of 5A peptide/ml of medium in 0.5 ml of medium on the apical side and 1.5 ml on the basal side. After 48 h, apical and basal media were collected from each well, and each was concentrated to ∼70 μl using a spin filter (Amicon Ultra 0.5 ml for apical medium or Amicon Ultra 4 ml for basal medium). Samples were used for Western blotting analysis as described above. The cells were still confluent and displayed the characteristic hexagonal cobblestone RPE cell shape at the end of this treatment.
Animals and diet
CFH-H/H mice were generated and genotyped as described previously (32). No mice used in this study have the rd8 mutation (89). Old (88–98 weeks) CFH-H/H mice were continued on an ND (Isopurina 5001; Prolab) or switched to an HFC diet (TD 88051; Envigo) for 8 weeks. All mice were housed conventionally on a middle rack in the same mouse facility under ambient light conditions to control for environmental factors and microbiome fluctuations. The care and use of these mice adhered to the Institutional Animal Care and Use Committee guidelines at Duke University.
Treatment of CFH-H/H∼HFC mice with HFC and 5A peptide
The CFH-H/H mouse expresses the human His-402 FH AMD-risk variant and no mouse Fh (on a Cfh−/− background) and develops an AMD phenotype when aged to 90 weeks and fed an HFC diet for 8 weeks (33). Nine CFH-H/H mice were aged to 90 weeks and then either kept on normal mouse chow, switched to an HFC diet for 8 weeks, or switched to an HFC + 5A peptide regime of HFC diet for 8 weeks with 30 mg of 5A peptide/kg of body weight by intraperitoneal injections, 3 times a week, for the duration of the 8-week HFC diet. Three mice were in each group. At the end of the study, blood was collected after 5 h of fasting, and the plasma for each group was pooled. 250 μl of plasma was used to isolate and purify HDL, using density gradient ultracentrifugation followed by size-exclusion chromatography by FPLC as described above. Samples were then prepared for proteomics as described above. All of the proteins in fmol amounts were expressed as a percentage of fmol of ApoA-1 in the sample, thus giving an indication of how many HDL particles contained a given protein.
Statistical analyses
In experiments with replicate data points, mean and S.D. are shown unless indicated otherwise. In cases where representative experiments are shown, additional experiments are shown in the supporting information. Where indicated, data sets were tested for statistical significance using a two-sample two-tailed Student's t test assuming equal variance. p values of <0.05 were considered statistically significant. Comparisons that were statistically significant to the following degrees are indicated: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (90) partner repository with the data set identifier PXD020251.
Supplementary Material
Author contributions—U. L. K. and C. B. R. conceptualization; U. L. K. and C. B. R. data curation; U. L. K., M. K., and C. B. R. formal analysis; U. L. K., D. G., M. A. C., and N. P. S. methodology; U. L. K. writing-original draft; U. L. K., D. G., M. K., and C. B. R. writing-review and editing; D. G., M. L., N. P. S., and M. K. investigation; J. L. and A. T. R. resources; C. B. R. supervision; C. B. R. funding acquisition; C. B. R. project administration.
Funding and additional information—This work was supported by NEI, National Institutes of Health, Grants R01 EY026161 (to C. B. R.) and P30 EY005722 (to Duke Eye Center); an Edward N. and Della L. Thome Memorial Foundation Award in Age-Related Macular Degeneration Research (to C. B. R.); a Research to Prevent Blindness/International Retinal Research Foundation Catalyst award for Innovative Research Approaches for AMD (to C. B. R.); and an unrestricted grant from Research to Prevent Blindness (to the Duke Eye Center). A. R. is supported by intramural NHBLI-DIR funds from the National Institutes of Health. J. L. is supported by National Institutes of Health Grants 1R01HL094463 and R01 HL144970. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—J. L. is a founder and the chief scientific officer for Glycan Therapeutics, LLC. A. R. is listed on a patent related to the 5A peptide.
- AMD
- age-related macular degeneration
- HDL
- high-density lipoprotein
- LDL
- low-density lipoprotein
- VLDL
- very-low-density lipoprotein
- FH
- complement factor H
- HS
- heparan sulfate
- ECM
- extracellular matrix
- GAG
- glycosaminoglycan
- RPE
- retinal pigmented epithelium
- BrM
- Bruch's membrane
- ApoA-1
- apolipoprotein A-1
- FPLC
- fast protein liquid chromatography
- HFC
- high-fat, cholesterol-enriched
- ND
- normal diet
- RIPA
- radioimmune precipitation assay
- RCT
- reverse cholesterol transport
- SAA
- serum amyloid A.
References
- 1. Flaxman S. R., Bourne R. R. A., Resnikoff S., Ackland P., Braithwaite T., Cicinelli M. V., Das A., Jonas J. B., Keeffe J., Kempen J. H., Leasher J., Limburg H., Naidoo K., Pesudovs K., Silvester A., et al. (2017) Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. Lancet Glob. Health 5, e1221–e1234 10.1016/S2214-109X(17)30393-5 [DOI] [PubMed] [Google Scholar]
- 2. Bowes Rickman C., Farsiu S., Toth C. A., and Klingeborn M. (2013) Dry age-related macular degeneration: mechanisms, therapeutic targets, and imaging. Invest. Ophthalmol. Vis. Sci. 54, ORSF68–ORSF80 10.1167/iovs.13-12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pennington K. L., and DeAngelis M. M. (2016) Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. (Lond.) 3, 34 10.1186/s40662-016-0063-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Y. F., Han Y., Zhang R., Qin L., Wang M. X., and Ma L. (2015) CETP/LPL/LIPC gene polymorphisms and susceptibility to age-related macular degeneration. Sci. Rep. 5, 15711 10.1038/srep15711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fritsche L. G., Igl W., Bailey J. N., Grassmann F., Sengupta S., Bragg-Gresham J. L., Burdon K. P., Hebbring S. J., Wen C., Gorski M., Kim I. K., Cho D., Zack D., Souied E., Scholl H. P., et al. (2016) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 48, 134–143 10.1038/ng.3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burgess S., and Davey Smith G. (2017) Mendelian randomization implicates high-density lipoprotein cholesterol-associated mechanisms in etiology of age-related macular degeneration. Ophthalmology 124, 1165–1174 10.1016/j.ophtha.2017.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fan Q., Maranville J. C., Fritsche L., Sim X., Cheung C. M. G., Chen L. J., Gorski M., Yamashiro K., Ahn J., Laude A., Dorajoo R., Lim T. H., Teo Y. Y., Blaustein R. O., Yoshimura N., et al. (2017) HDL-cholesterol levels and risk of age-related macular degeneration: a multiethnic genetic study using Mendelian randomization. Int. J. Epidemiol. 46, 1891–1902 10.1093/ije/dyx189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kersten E., Paun C. C., Schellevis R. L., Hoyng C. B., Delcourt C., Lengyel I., Peto T., Ueffing M., Klaver C. C. W., Dammeier S., den Hollander A. I., and de Jong E. K. (2018) Systemic and ocular fluid compounds as potential biomarkers in age-related macular degeneration. Surv. Ophthalmol. 63, 9–39 10.1016/j.survophthal.2017.05.003 [DOI] [PubMed] [Google Scholar]
- 9. Cheung C. M. G., Gan A., Fan Q., Chee M. L., Apte R. S., Khor C. C., Yeo I., Mathur R., Cheng C. Y., Wong T. Y., and Tai E. S. (2017) Plasma lipoprotein subfraction concentrations are associated with lipid metabolism and age-related macular degeneration. J. Lipid Res. 58, 1785–1796 10.1194/jlr.M073684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Colijn J. M., Hollander A. I. D., Demirkan A., Cougnard-Grégoire A., Verzijden T., Kersten E., Meester M. A., Merle B. M. J., Papageorgiou G., Ahmad S., Mulder M. T., Costa M. A., Benlian P., Bertelsen G., Bron A., et al. (2018) Increased high-density lipoprotein levels associated with age-related macular degeneration: evidence from the EYE-RISK and E3 consortia. Ophthalmology 126, 393–406 10.1016/j.ophtha.2018.09.045 [DOI] [PubMed] [Google Scholar]
- 11. van Leeuwen E. M., Emri E., Merle B. M. J., Colijn J. M., Kersten E., Cougnard-Gregoire A., Dammeier S., Meester-Smoor M., Pool F. M., de Jong E. K., Delcourt C., Rodrigez-Bocanegra E., Biarnés M., Luthert P. J., Ueffing M., et al. (2018) A new perspective on lipid research in age-related macular degeneration. Prog. Retin. Eye Res. 67, 56–86 10.1016/j.preteyeres.2018.04.006 [DOI] [PubMed] [Google Scholar]
- 12. Storti F., Raphael G., Griesser V., Klee K., Drawnel F., Willburger C., Scholz R., Langmann T., von Eckardstein A., Fingerle J., Grimm C., and Maugeais C. (2017) Regulated efflux of photoreceptor outer segment-derived cholesterol by human RPE cells. Exp. Eye Res. 165, 65–77 10.1016/j.exer.2017.09.008 [DOI] [PubMed] [Google Scholar]
- 13. Pikuleva I. A., and Curcio C. A. (2014) Cholesterol in the retina: the best is yet to come. Prog. Retin. Eye Res. 41, 64–89 10.1016/j.preteyeres.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Klein R. J., Zeiss C., Chew E. Y., Tsai J. Y., Sackler R. S., Haynes C., Henning A. K., Sangiovanni J. P., Mane S. M., Mayne S. T., Bracken M. B., Ferris F. L., Ott J., Barnstable C., and Hoh J. (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 10.1126/science.1109557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haines J. L., Hauser M. A., Schmidt S., Scott W. K., Olson L. M., Gallins P., Spencer K. L., Kwan S. Y., Noureddine M., Gilbert J. R., Schnetz-Boutaud N., Agarwal A., Postel E. A., and Pericak-Vance M. A. (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308, 419–421 10.1126/science.1110359 [DOI] [PubMed] [Google Scholar]
- 16. Edwards A. O., Ritter R. 3rd, Abel K. J., Manning A., Panhuysen C., and Farrer L. A. (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–424 10.1126/science.1110189 [DOI] [PubMed] [Google Scholar]
- 17. Hageman G. S., Anderson D. H., Johnson L. V., Hancox L. S., Taiber A. J., Hardisty L. I., Hageman J. L., Stockman H. A., Borchardt J. D., Gehrs K. M., Smith R. J., Silvestri G., Russell S. R., Klaver C. C., Barbazetto I., et al. (2005) A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 102, 7227–7232 10.1073/pnas.0501536102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DiScipio R. G. (1992) Ultrastructures and interactions of complement factors H and I. J. Immunol. 149, 2592–2599 [PubMed] [Google Scholar]
- 19. Sharma A. K., and Pangburn M. K. (1996) Identification of three physically and functionally distinct binding sites for C3b in human complement factor H by deletion mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 93, 10996–11001 10.1073/pnas.93.20.10996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kelly U., Yu L., Kumar P., Ding J. D., Jiang H., Hageman G. S., Arshavsky V. Y., Frank M. M., Hauser M. A., and Rickman C. B. (2010) Heparan sulfate, including that in Bruch's membrane, inhibits the complement alternative pathway: implications for age-related macular degeneration. J. Immunol. 185, 5486–5494 10.4049/jimmunol.0903596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez de Cordoba S., Esparza-Gordillo J., Goicoechea de Jorge E., Lopez-Trascasa M., and Sanchez-Corral P. (2004) The human complement factor H: functional roles, genetic variations and disease associations. Mol. Immunol. 41, 355–367 10.1016/j.molimm.2004.02.005 [DOI] [PubMed] [Google Scholar]
- 22. Skerka C., Lauer N., Weinberger A. A., Keilhauer C. N., Sühnel J., Smith R., Schlötzer-Schrehardt U., Fritsche L., Heinen S., Hartmann A., Weber B. H., and Zipfel P. F. (2007) Defective complement control of factor H (Y402H) and FHL-1 in age-related macular degeneration. Mol. Immunol. 44, 3398–3406 10.1016/j.molimm.2007.02.012 [DOI] [PubMed] [Google Scholar]
- 23. Zipfel P. F., and Skerka C. (2009) Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 9, 729–740 10.1038/nri2620 [DOI] [PubMed] [Google Scholar]
- 24. Fearon D. T. (1978) Regulation by membrane sialic acid of β1H-dependent decay-dissociation of amplification C3 convertase of the alternative complement pathway. Proc. Natl. Acad. Sci. U. S. A. 75, 1971–1975 10.1073/pnas.75.4.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mold C., Kingzette M., and Gewurz H. (1984) C-reactive protein inhibits pneumococcal activation of the alternative pathway by increasing the interaction between factor H and C3b. J. Immunol. 133, 882–885 [PubMed] [Google Scholar]
- 26. Ormsby R. J., Ranganathan S., Tong J. C., Griggs K. M., Dimasi D. P., Hewitt A. W., Burdon K. P., Craig J. E., Hoh J., and Gordon D. L. (2008) Functional and structural implications of the complement factor H Y402H polymorphism associated with age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 49, 1763–1770 10.1167/iovs.07-1297 [DOI] [PubMed] [Google Scholar]
- 27. Yu J., Wiita P., Kawaguchi R., Honda J., Jorgensen A., Zhang K., Fischetti V. A., and Sun H. (2007) Biochemical analysis of a common human polymorphism associated with age-related macular degeneration. Biochemistry 46, 8451–8461 10.1021/bi700459a [DOI] [PubMed] [Google Scholar]
- 28. Molins B., Fuentes-Prior P., Adán A., Antón R., Arostegui J. I., Yagüe J., and Dick A. D. (2016) Complement factor H binding of monomeric C-reactive protein downregulates proinflammatory activity and is impaired with at risk polymorphic CFH variants. Sci. Rep. 6, 22889 10.1038/srep22889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clark S. J., Perveen R., Hakobyan S., Morgan B. P., Sim R. B., Bishop P. N., and Day A. J. (2010) Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch's membrane in human retina. J. Biol. Chem. 285, 30192–30202 10.1074/jbc.M110.103986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weismann D., Hartvigsen K., Lauer N., Bennett K. L., Scholl H. P., Charbel Issa P., Cano M., Brandstätter H., Tsimikas S., Skerka C., Superti-Furga G., Handa J. T., Zipfel P. F., Witztum J. L., and Binder C. J. (2011) Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 478, 76–81 10.1038/nature10449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shaw P. X., Zhang L., Zhang M., Du H., Zhao L., Lee C., Grob S., Lim S. L., Hughes G., Lee J., Bedell M., Nelson M. H., Lu F., Krupa M., Luo J., et al. (2012) Complement factor H genotypes impact risk of age-related macular degeneration by interaction with oxidized phospholipids. Proc. Natl. Acad. Sci. U. S. A. 109, 13757–13762 10.1073/pnas.1121309109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ding J. D., Kelly U., Landowski M., Toomey C. B., Groelle M., Miller C., Smith S. G., Klingeborn M., Singhapricha T., Jiang H., Frank M. M., and Bowes Rickman C. (2015) Expression of human complement factor H prevents age-related macular degeneration-like retina damage and kidney abnormalities in aged Cfh knockout mice. Am. J. Pathol. 185, 29–42 10.1016/j.ajpath.2014.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Landowski M., Kelly U., Klingeborn M., Groelle M., Ding J. D., Grigsby D., and Bowes Rickman C. (2019) Human complement factor H Y402H polymorphism causes an age-related macular degeneration phenotype and lipoprotein dysregulation in mice. Proc. Natl. Acad. Sci. U. S. A. 116, 3703–3711 10.1073/pnas.1814014116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kontush A., Lindahl M., Lhomme M., Calabresi L., Chapman M. J., and Davidson W. S. (2015) Structure of HDL: particle subclasses and molecular components. Handb. Exp. Pharmacol. 224, 3–51 10.1007/978-3-319-09665-0_1 [DOI] [PubMed] [Google Scholar]
- 35. Van Lenten B. J., Hama S. Y., de Beer F. C., Stafforini D. M., McIntyre T. M., Prescott S. M., La Du B. N., Fogelman A. M., and Navab M. (1995) Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response: loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J. Clin. Invest. 96, 2758–2767 10.1172/JCI118345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heinecke J. W. (2009) The HDL proteome: a marker—and perhaps mediator—of coronary artery disease. J. Lipid Res. 50, S167–S171 10.1194/jlr.r800097-jlr200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holzer M., Trieb M., Konya V., Wadsack C., Heinemann A., and Marsche G. (2013) Aging affects high-density lipoprotein composition and function. Biochim. Biophys. Acta 1831, 1442–1448 10.1016/j.bbalip.2013.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ferris F. L. 3rd, Wilkinson C. P., Bird A., Chakravarthy U., Chew E., Csaky K., and Sadda S. R., and Beckman Initiative for Macular Research Classification Committee (2013) Clinical classification of age-related macular degeneration. Ophthalmology 120, 844–851 10.1016/j.ophtha.2012.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holzer M., Kern S., Birner-Grünberger R., Curcic S., Heinemann A., and Marsche G. (2016) Refined purification strategy for reliable proteomic profiling of HDL2/3: impact on proteomic complexity. Sci. Rep. 6, 38533 10.1038/srep38533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marson A., Robinson D. E., Brookes P. N., Mulloy B., Wiles M., Clark S. J., Fielder H. L., Collinson L. J., Cain S. A., Kielty C. M., McArthur S., Buttle D. J., Short R. D., Whittle J. D., and Day A. J. (2009) Development of a microtiter plate-based glycosaminoglycan array for the investigation of glycosaminoglycan-protein interactions. Glycobiology 19, 1537–1546 10.1093/glycob/cwp132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mahoney D. J., Whittle J. D., Milner C. M., Clark S. J., Mulloy B., Buttle D. J., Jones G. C., Day A. J., and Short R. D. (2004) A method for the non-covalent immobilization of heparin to surfaces. Anal. Biochem. 330, 123–129 10.1016/j.ab.2004.03.055 [DOI] [PubMed] [Google Scholar]
- 42. Klingeborn M., Dismuke W. M., Skiba N. P., Kelly U., Stamer W. D., and Bowes Rickman C. (2017) Directional exosome proteomes reflect polarity-specific functions in retinal pigmented epithelium monolayers. Sci. Rep. 7, 4901 10.1038/s41598-017-05102-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dunn K. C., Aotaki-Keen A. E., Putkey F. R., and Hjelmeland L. M. (1996) ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 62, 155–169 10.1006/exer.1996.0020 [DOI] [PubMed] [Google Scholar]
- 44. Beebe D. C. (2013) The use of cell lines to “model” ocular tissues: cautionary tales. Invest. Ophthalmol. Vis. Sci. 54, 5720 10.1167/iovs.13-12873 [DOI] [PubMed] [Google Scholar]
- 45. Rizzolo L. J. (2014) Barrier properties of cultured retinal pigment epithelium. Exp. Eye Res. 126, 16–26 10.1016/j.exer.2013.12.018 [DOI] [PubMed] [Google Scholar]
- 46. Iozzo R. V., and Schaefer L. (2015) Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol. 42, 11–55 10.1016/j.matbio.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stanford K. I., Bishop J. R., Foley E. M., Gonzales J. C., Niesman I. R., Witztum J. L., and Esko J. D. (2009) Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J. Clin. Invest. 119, 3236–3245 10.1172/JCI38251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Flood C., Gustafsson M., Pitas R. E., Arnaboldi L., Walzem R. L., and Borén J. (2004) Molecular mechanism for changes in proteoglycan binding on compositional changes of the core and the surface of low-density lipoprotein-containing human apolipoprotein B100. Arterioscler. Thromb. Vasc. Biol. 24, 564–570 10.1161/01.ATV.0000117174.19078.85 [DOI] [PubMed] [Google Scholar]
- 49. Zaferani A., Vivès R. R., van der Pol P., Navis G. J., Daha M. R., van Kooten C., Lortat-Jacob H., Seelen M. A., and van den Born J. (2012) Factor h and properdin recognize different epitopes on renal tubular epithelial heparan sulfate. J. Biol. Chem. 287, 31471–31481 10.1074/jbc.M112.380386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toomey C. B., Kelly U., Saban D. R., and Bowes Rickman C. (2015) Regulation of age-related macular degeneration-like pathology by complement factor H. Proc. Natl. Acad. Sci. U. S. A. 112, E3040–E3049 10.1073/pnas.1424391112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blackmore T. K., Hellwage J., Sadlon T. A., Higgs N., Zipfel P. F., Ward H. M., and Gordon D. L. (1998) Identification of the second heparin-binding domain in human complement factor H. J. Immunol. 160, 3342–3348 [PubMed] [Google Scholar]
- 52. Jiang J., Cun W., Wu X., Shi Q., Tang H., and Luo G. (2012) Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 86, 7256–7267 10.1128/JVI.07222-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tabet F., Remaley A. T., Segaliny A. I., Millet J., Yan L., Nakhla S., Barter P. J., Rye K. A., and Lambert G. (2010) The 5A apolipoprotein A-I mimetic peptide displays antiinflammatory and antioxidant properties in vivo and in vitro. Arterioscler. Thromb. Vasc. Biol. 30, 246–252 10.1161/ATVBAHA.109.200196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sviridov D. O., Andrianov A. M., Anishchenko I. V., Stonik J. A., Amar M. J., Turner S., and Remaley A. T. (2013) Hydrophobic amino acids in the hinge region of the 5A apolipoprotein mimetic peptide are essential for promoting cholesterol efflux by the ABCA1 transporter. J. Pharmacol. Exp. Ther. 344, 50–58 10.1124/jpet.112.198143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sethi A. A., Stonik J. A., Thomas F., Demosky S. J., Amar M., Neufeld E., Brewer H. B., Davidson W. S., D'Souza W., Sviridov D., and Remaley A. T. (2008) Asymmetry in the lipid affinity of bihelical amphipathic peptides: a structural determinant for the specificity of ABCA1-dependent cholesterol efflux by peptides. J. Biol. Chem. 283, 32273–32282 10.1074/jbc.M804461200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Amar M. J., D'Souza W., Turner S., Demosky S., Sviridov D., Stonik J., Luchoomun J., Voogt J., Hellerstein M., Sviridov D., and Remaley A. T. (2010) 5A apolipoprotein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. J. Pharmacol. Exp. Ther. 334, 634–641 10.1124/jpet.110.167890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Phillips M. C. (2018) Is ABCA1 a lipid transfer protein? J. Lipid Res. 59, 749–763 10.1194/jlr.R082313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nagata K. O., Nakada C., Kasai R. S., Kusumi A., and Ueda K. (2013) ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc. Natl. Acad. Sci. U. S. A. 110, 5034–5039 10.1073/pnas.1220703110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Curcio C. A. (2018) Soft drusen in age-related macular degeneration: biology and targeting via the oil spill strategies. Invest. Ophthalmol. Vis. Sci. 59, AMD160–AMD181 10.1167/iovs.18-24882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Duncan K. G., Hosseini K., Bailey K. R., Yang H., Lowe R. J., Matthes M. T., Kane J. P., LaVail M. M., Schwartz D. M., and Duncan J. L. (2009) Expression of reverse cholesterol transport proteins ATP-binding cassette A1 (ABCA1) and scavenger receptor BI (SR-BI) in the retina and retinal pigment epithelium. Br. J. Ophthalmol. 93, 1116–1120 10.1136/bjo.2008.144006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cougnard-Grégoire A., Delyfer M. N., Korobelnik J. F., Rougier M. B., Le Goff M., Dartigues J. F., Barberger-Gateau P., and Delcourt C. (2014) Elevated high-density lipoprotein cholesterol and age-related macular degeneration: the Alienor study. PLoS ONE 9, e90973 10.1371/journal.pone.0090973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Calabresi L., Gomaraschi M., Villa B., Omoboni L., Dmitrieff C., and Franceschini G. (2002) Elevated soluble cellular adhesion molecules in subjects with low HDL-cholesterol. Arterioscler. Thromb. Vasc. Biol. 22, 656–661 10.1161/hq0402.105901 [DOI] [PubMed] [Google Scholar]
- 63. Parthasarathy S., Barnett J., and Fong L. G. (1990) High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein. Biochim. Biophys. Acta 1044, 275–283 10.1016/0005-2760(90)90314-N [DOI] [PubMed] [Google Scholar]
- 64. Navab M., Hama S. Y., Anantharamaiah G. M., Hassan K., Hough G. P., Watson A. D., Reddy S. T., Sevanian A., Fonarow G. C., and Fogelman A. M. (2000) Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J. Lipid Res. 41, 1495–1508 [PubMed] [Google Scholar]
- 65. Vaisar T., Mayer P., Nilsson E., Zhao X. Q., Knopp R., and Prazen B. J. (2010) HDL in humans with cardiovascular disease exhibits a proteomic signature. Clin. Chim. Acta 411, 972–979 10.1016/j.cca.2010.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shah A. S., Tan L., Long J. L., and Davidson W. S. (2013) Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 54, 2575–2585 10.1194/jlr.R035725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hoofnagle A. N., Becker J. O., Oda M. N., Cavigiolio G., Mayer P., and Vaisar T. (2012) Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin. Chem. 58, 777–781 10.1373/clinchem.2011.173856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Weichhart T., Kopecky C., Kubicek M., Haidinger M., Döller D., Katholnig K., Suarna C., Eller P., Tölle M., Gerner C., Zlabinger G. J., van der Giet M., Hörl W. H., Stocker R., and Säemann M. D. (2012) Serum amyloid A in uremic HDL promotes inflammation. J. Am. Soc. Nephrol. 23, 934–947 10.1681/ASN.2011070668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Holzer M., Wolf P., Curcic S., Birner-Gruenberger R., Weger W., Inzinger M., El-Gamal D., Wadsack C., Heinemann A., and Marsche G. (2012) Psoriasis alters HDL composition and cholesterol efflux capacity. J. Lipid Res. 53, 1618–1624 10.1194/jlr.M027367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pertl L., Kern S., Weger M., Hausberger S., Trieb M., Gasser-Steiner V., Haas A., Scharnagl H., Heinemann A., and Marsche G. (2016) Correction: High-density lipoprotein function in exudative age-related macular degeneration. PLoS ONE 11, e0157210 10.1371/journal.pone.0157210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Murphy A. J. (2013) High density lipoprotein: assembly, structure, cargo, and functions. ISRN Physiol. 2013, 1–20 10.1155/2013/186365 [DOI] [Google Scholar]
- 72. Fogelstrand P., and Borén J. (2012) Retention of atherogenic lipoproteins in the artery wall and its role in atherogenesis. Nutr. Metab. Cardiovasc. Dis. 22, 1–7 10.1016/j.numecd.2011.09.007 [DOI] [PubMed] [Google Scholar]
- 73. Boren J., and Williams K. J. (2016) The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr. Opin. Lipidol. 27, 473–483 10.1097/MOL.0000000000000330 [DOI] [PubMed] [Google Scholar]
- 74. Packard R. R., and Libby P. (2008) Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin. Chem. 54, 24–38 10.1373/clinchem.2007.097360 [DOI] [PubMed] [Google Scholar]
- 75. Islam R. M., Pourmousa M., Sviridov D., Gordon S. M., Neufeld E. B., Freeman L. A., Perrin B. S. Jr., Pastor R. W., and Remaley A. T. (2018) Structural properties of apolipoprotein A-I mimetic peptides that promote ABCA1-dependent cholesterol efflux. Sci. Rep. 8, 2956 10.1038/s41598-018-20965-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vives-Bauza C., Anand M., Shiraz A. K., Shirazi A. K., Magrane J., Gao J., Vollmer-Snarr H. R., Manfredi G., and Finnemann S. C. (2008) The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J. Biol. Chem. 283, 24770–24780 10.1074/jbc.M800706200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rudolf M., Mir Mohi Sefat A., Miura Y., Tura A., Raasch W., Ranjbar M., Grisanti S., Aherrahrou Z., Wagner A., Messinger J. D., Garber D. W., Anantharamaiah G. M., and Curcio C. A. (2018) ApoA-I mimetic peptide 4F reduces age-related lipid deposition in murine Bruch's membrane and causes its structural remodeling. Curr. Eye Res. 43, 135–146 10.1080/02713683.2017.1370118 [DOI] [PubMed] [Google Scholar]
- 78. Keenan T. D., Pickford C. E., Holley R. J., Clark S. J., Lin W., Dowsey A. W., Merry C. L., Day A. J., and Bishop P. N. (2014) Age-dependent changes in heparan sulfate in human Bruch's membrane: implications for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 55, 5370–5379 10.1167/iovs.14-14126 [DOI] [PubMed] [Google Scholar]
- 79. Hughes C. S., Foehr S., Garfield D. A., Furlong E. E., Steinmetz L. M., and Krijgsveld J. (2014) Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757 10.15252/msb.20145625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Silva J. C., Denny R., Dorschel C. A., Gorenstein M., Kass I. J., Li G. Z., McKenna T., Nold M. J., Richardson K., Young P., and Geromanos S. (2005) Quantitative proteomic analysis by accurate mass retention time pairs. Anal. Chem. 77, 2187–2200 10.1021/ac048455k [DOI] [PubMed] [Google Scholar]
- 81. Vlodavsky I., Folkman J., Sullivan R., Fridman R., Ishai-Michaeli R., Sasse J., and Klagsbrun M. (1987) Endothelial cell-derived basic fibroblast growth factor: synthesis and deposition into subendothelial extracellular matrix. Proc. Natl. Acad. Sci. U.S.A. 84, 2292–2296 10.1073/pnas.84.8.2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu R., Xu Y., Chen M., Weïwer M., Zhou X., Bridges A. S., DeAngelis P. L., Zhang Q., Linhardt R. J., and Liu J. (2010) Chemoenzymatic design of heparan sulfate oligosaccharides. J. Biol. Chem. 285, 34240–34249 10.1074/jbc.M110.159152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kelly U., Bowes Rickman C., Postel E. A., Hauser M. A., Hageman G. S., Arshavsky V. Y., and Skiba N. P. (2009) Rapid and sensitive method for detection of Y402, H402, I62, and V62 variants of complement factor H in human plasma samples using mass spectrometry. Invest. Ophthalmol. Vis. Sci. 50, 1540–1545 10.1167/iovs.08-2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Aviram M. (1983) Plasma lipoprotein separation by discontinuous density gradient ultracentrifugation in hyperlipoproteinemic patients. Biochem. Med. 30, 111–118 10.1016/0006-2944(83)90013-3 [DOI] [PubMed] [Google Scholar]
- 85. McHarg S., Brace N., Bishop P. N., and Clark S. J. (2015) Enrichment of Bruch's membrane from human donor eyes. J. Visualized Exp. 10.3791/53382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Foley E. M., Gordts P. L., Stanford K. I., Gonzales J. C., Lawrence R., Stoddard N., and Esko J. D. (2013) Hepatic remnant lipoprotein clearance by heparan sulfate proteoglycans and low-density lipoprotein receptors depend on dietary conditions in mice. Arterioscler. Thromb. Vasc. Biol. 33, 2065–2074 10.1161/ATVBAHA.113.301637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lawrence R., Olson S. K., Steele R. E., Wang L., Warrior R., Cummings R. D., and Esko J. D. (2008) Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem. 283, 33674–33684 10.1074/jbc.M804288200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bourdi M., Amar M., Remaley A. T., and Terse P. S. (2018) Intravenous toxicity and toxicokinetics of an HDL mimetic, Fx-5A peptide complex, in cynomolgus monkeys. Regul. Toxicol. Pharmacol. 100, 59–67 10.1016/j.yrtph.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mattapallil M. J., Wawrousek E. F., Chan C. C., Zhao H., Roychoudhury J., Ferguson T. A., and Caspi R. R. (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci. 53, 2921–2927 10.1167/iovs.12-9662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Perez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (90) partner repository with the data set identifier PXD020251.