

Abstract
During autophagy, LC3 and GABARAP proteins become covalently attached to phosphatidylethanolamine on the growing autophagosome. This attachment is also reversible. Deconjugation (or delipidation) involves the proteolytic cleavage of an isopeptide bond between LC3 or GABARAP and the phosphatidylethanolamine headgroup. This cleavage is carried about by the ATG4 family of proteases (ATG4A, B, C, and D). Many studies have established that ATG4B is the most active of these proteases and is sufficient for autophagy progression in simple cells. Here we examined the second most active protease, ATG4A, to map out key regulatory motifs on the protein and to establish its activity in cells. We utilized fully in vitro reconstitution systems in which we controlled the attachment of LC3/GABARAP members and discovered a role for a C-terminal LC3-interacting region on ATG4A in regulating its access to LC3/GABARAP. We then used a gene-edited cell line in which all four ATG4 proteases have been knocked out to establish that ATG4A is insufficient to support autophagy and is unable to support GABARAP proteins removal from the membrane. As a result, GABARAP proteins accumulate on membranes other than mature autophagosomes. These results suggest that to support efficient production and consumption of autophagosomes, additional factors are essential including possibly ATG4B itself or one of its proteolytic products in the LC3 family.
Keywords: autophagy, membrane reconstitution, liposome, cell biology, phosphorylation, autophagy-related protein 4A (ATG4), delipidation, GABARAP like-1, LC3-interacting region (LIR), MST4
Macroautophagy is a conserved pathway for intracellular degradation (1–4) and is highly induced under amino acid starvation (1, 5–8). After induction, portions of cytoplasm are captured as cargo into a double-membrane cup-shaped structure called the phagophore, which eventually closes upon itself, sequestering the cargo within and becoming the autophagosome. The process culminates in autophagosome–lysosome fusion, in which degradation of cargo is driven by lysosomal hydrolases (2, 9, 10). Disruption of autophagy is associated with a multitude of diseases related to neurodegeneration, aging, microbial infection, and cancer (11–13).
Atg8 is a peripheral ubiquitin-like protein that is crucial for phagophore expansion, autophagosome completion, cargo targeting, and regulation of autophagosome–lysosome fusion (2, 10, 14–17). Each of these activities depends upon the covalent attachment of Atg8 to the lipid phosphatidylethanolamine (PE) in a process we call lipidation (18–20). In yeast there is only one Atg8 gene, but in mammals there are six to eight genes spread across two families of ATG8 orthologs: LC3 and GABARAP, including LC3A/B/C, GABARAP, and GABARAPL1/2/3 (9, 21).
Atg8, LC3B, and GABARAP proteins are initially expressed in a pro-form that is not competent for conjugation. Before lipidation can proceed, this pro-form must be cleaved at its C terminus by a cysteine protease called Atg4 (yeast) or ATG4 (mammals) to expose the glycine residue (10, 22–25). This initial cleavage event is known as priming. Lipidation of Atg8/LC3B/GABARAP then proceeds through an enzymatic cascade very similar to ubiquitination, eventually coupling this free glycine to the amine group on PE (14, 26). Atg8/LC3B/GABARAP-PE stays bound to the autophagosome membrane through its formation but must be removed just prior to or concomitantly with fusion of the autophagosome into the lysosome or vacuole. The removal of external Atg8/LC3B/GABARAP from PE is also performed by Atg4/ATG4-mediated proteolysis, in a process known as delipidation (18, 27). Defects in delipidation of Atg8–PE by Atg4 prevents recycling of Atg8 off membranes, which both limits the available pool for future rounds of autophagy and prevents fusion of the autophagosome and the lysosome (27, 28).
Thus, ATG4 family proteins act constitutively to prime newly expressed Atg8/LC3B/GABARAP proteins but also must function under temporal and spatial regulation to release lipid-conjugated forms of Atg8/LC3/GABARAP from fully formed mature autophagosomes. We and others have begun to study how priming and delipidation are independently regulated to achieve these drastically different kinetics in the cell (29, 30). ATG4B is an extremely fast priming enzyme in vitro (29, 31) and is also the major driver of priming for both the LC3 and GABARAP subfamilies in cells. KO of ATG4B, but not of the other three mammalian ATG4 proteases, eliminates priming of all LC3 proteins and dramatically impairs priming of the GABARAP family proteins (29, 30). Delipidation is more complicated. Our laboratory has established that although ATG4A, ATG4C, and ATG4D are very poor at priming, consistent with many previous studies, they are surprisingly just as efficient as ATG4B in vitro at delipidation (29), suggesting the possibility that they play a key role in deciding when and which LC3 or GABARAP proteins are released from the membrane. Thus far, however, the study of ATG4A, ATG4C, and ATG4D function in cells has largely been confounded by the redundancy of the family. In particular, how and whether ATG4A-mediated priming and delipidation are independently controlled has not been known.
Here, we explore the limits of ATG4A-mediated delipidation in a gene-edited cell line we created in which all four mammalian ATG4 genes are missing, which allows us to specifically rescue with ATG4A and various engineered mutants. We discover that like ATG4B, the C-terminal LC3-interaction region (LIR) of ATG4A is essential to its function, but unlike ATG4B, this motif controls both priming and delipidation on ATG4A. Furthermore, we show that ATG4A is not sufficient to support autophagy in our cell lines and, more surprisingly, does not appear to support any delipidation of its substrate, GABARAPL1 (from now on referred to as GL1), in this context. We show that instead, GL1 accumulates on a variety of membranes and remains membrane-associated even if the normal cues for activating the lipidation pathway are shut off for as long as 24 h. This strongly implies that ATG4A does not have access to GL1–PE (also known as GL1-II) under these conditions, suggesting more complex intracellular regulation of its delipidation activity.
Results
Macroautophagic degradation is fully stalled in cells expressing only ATG4A
The redundancy of function across the ATG4 family limits the useful interpretation of single knockouts, and thus we have created a gene-edited HEK293 cell line that we call our ATG4 quadruple knockout (ATG4QKO). In this cell line, there is no detectable expression of any of the ATG4 proteins by Western blotting (Fig. S1A and Ref. 29), and we have confirmed genetic alteration at each locus (Fig. S1B). Without any ATG4 expression, these cells are completely incapable of priming the ATG8 protein family, and thus all lipidation of the ATG8 proteins is also inhibited (29). We previously showed that re-expression of ATG4B in this background is sufficient to rescue LC3 and GABARAP protein priming and lipidation and even to support flux of these proteins in a bafilomycin A1 (Baf A1)–dependent way, suggesting restoration of autophagy (29). In contrast, re-expression of ATG4A is unable to rescue LC3 processing, consistent with many previous publications, and also leads to anomalous processing of GABARAP proteins. In particular, these cells continue to have a large pool of unprimed pro-GL1 and accumulate fully lipidated GL1-II to high levels but do not show any significant levels of free-floating processed GL1 (GL1-I) (Ref. 29 and Fig. S2). This result suggests that ATG4A can prime a portion of the GABARAP family protein pool, and this entire population of primed GL1-I is utilized for lipidation. Should any delipidation take place, newly released protein is again rapidly lipidated. Thus, we have an opportunity to consider how autophagy progresses when only the GABARAP family is able to become lipid-attached and to assess how ATG4A contributes to this process.
Recent papers have suggested that the GABARAPs can be fully sufficient for at least some forms of macroautophagy and that even in the absence of all LC3 and GABARAP proteins, some macroautophagy persists (32–34). Therefore, we first established whether the delivery of cargo is possible in our ATG4QKO cells, where no primed LC3 or GABARAP is available to be lipidated. To look at starvation-mediated turnover of bulk cytoplasm, we used a long-lived protein degradation (LLPD) assay in which cells are incubated in media supplemented with [14C]valine and then chased with medium supplemented with cold valine. These cells were then incubated in either normal or nutrient-reduced (EBSS) medium for 4 h to look for starvation-dependent changes in bulk protein turnover. Consistent with previous results (35, 36), starvation induced an ∼50% increase in long-lived protein degradation in WT cells (Fig. 1A). Critically, when these cells were treated with the PI3K inhibitor 3-MA to block initiation of the macroautophagy pathway, starvation-dependent LLPD is mostly prevented. In contrast to WT cells, ATG4QKO cells were incapable of starvation-dependent LLPD. Thus, starvation-dependent flux depends on ATG4 proteins in this cell line.
Figure 1.

Both starvation-induced bulk autophagy and p62 homeostasis are impaired in ATG4QKO cells and are rescued only by ATG4B expression. A, long-lived protein degradation assay reveals ATG4QKO cells are unable to degrade long-lived proteins under starvation conditions. The cells were maintained under DMEM or incubated in starvation medium (EBSS) and treated without or with 10 mm 3-MA for 4 h. B, immunoblotting of cells after DMEM or EBSS without or with 0.1 μm bafilomycin A1 (Baf) for 2 h revealed p62 levels are elevated in ATG4QKO cells. C, under the same conditions as in B, ATG4B expression was able to restore p62 levels back to WT in the ATG4QKO background, whereas expression of ATG4A, ATG4C, or ATG4D could not. Quantifications for p62 levels of ATG4QKO cells with single ATG4 isoform rescues are done by dividing p62 density over GAPDH density and normalizing all values to the highest value in the experiment. Raw data for p62 quantification are shown in Fig. S4. Statistical significance is assessed with two-sample Student's t test. *, p < 0.05. **, p < 0.01. ***, p < 0.001.
Lentiviral-mediated rescue of individual ATG4A, B, C, or D genes in the ATG4QKO restored ATG8 protein family processing to varying degrees (29). We observed in these rescues that high expression of ATG4A drove significant lipidation of GL1, whereas re-expression of ATG4C or ATG4D resulted in more modest levels of lipidated GABARAP family proteins, and only ATG4B re-expression restored any LC3 processing (Fig. S2). To establish whether the ATG4A-, ATG4C-, or ATG4D-mediated processing of GABARAP proteins alone can drive full autophagy, we looked at LLPD in these lines. Surprisingly, only ATG4B re-expression was sufficient to support a starvation-mediated LLPD and did so at essentially WT levels. Cells expressing only ATG4A, ATG4C, or ATG4D behaved exactly like the ATG4QKO (Fig. 1A). Thus, bulk cargo delivery depends absolutely on the presence of ATG4B, consistent with other reports (36–40).
To look at how a specific autophagic cargo is handled, we followed levels of the adaptor proteins p62/SQSTM1 and NBR1 and a potential autophagic cargo lactate dehydrogenase (LDH) across various cellular stress conditions. Neither NBR1 nor LDH levels depended upon ATG4 expression, and neither changed in response to small molecule treatments in WT HEK293 cells (Fig. S3) and so were not further examined. We also could not detect changes in p62 levels in WT cells following starvation, even when the lysosomal inhibitor Baf A1 was included to accumulate lysosome-directed proteins (Fig. 1B and Fig. S4). Note that the levels of LC3B-II did increase in both Baf A1 and starvation conditions, indicating that only p62 flux in these cells was undetectable. As expected, there was also no flux of p62 (or of pro-LC3) in our ATG4QKO cells. Interestingly, however, overall p62 levels were elevated ∼2-fold higher in ATG4QKO cells relative to WT cells (Fig. 1B), indicating that loss of the ATG4 family alters the overall homeostasis of p62 (29). Re-expression of ATG4B in the ATG4QKO but not re-expression of ATG4A, C, or D restored p62 to levels similar to WT (Fig. 1C and Figs. S2 and S4). Thus, ATG4B is able to restore essentially normal processing of the LC3 and GABARAP families, fully support LLPD, and maintain normal p62 homeostasis, whereas expression of any other ATG4 alone can prime GABARAP protein families (to varying levels) but cannot otherwise support autophagy.
In cells expressing only ATG4A, GL1-II does not appear to undergo delipidation
Some studies have observed that in cells depleted of LC3, the GABARAP family is sufficient to drive autophagosome formation and even cargo delivery (32, 41). Thus, in principle, the ATG4QKO-ATG4A cell line that primes GABARAP, GL1, and GL2 and accumulates lipidated forms of all three proteins might also support macroautophagy. However, the absence of both starvation-mediated LLPD and normal p62 homeostasis indicates that instead there are unexpected detriments to autophagy.
To that end, we suspected that the strong accumulation of GL1-II in cells expressing only ATG4A (Fig. S2) reflected a breakdown in the normal GL1 cycle in cells that might underlie a larger problem with autophagosome progression (Fig. 2A, cartoon). In particular, the complete absence of GL1-I could arise because the total pool of primed LC3 and GABARAP proteins is very small, and thus all free GL1-I formed from either priming or from delipidation becomes immediately relipidated. Alternatively, GL1-I depletion could indicate a general failure of ATG4A to support delipidation in these cells.
Figure 2.

Delipidation of GL1 is not detectable in ATG4QKO-ATG4A rescue cells. A, cartoon of GL1 states in WT and ATG4QKO-ATG4A rescue cells with a model suggesting how the lipidation/delipidation cycle might work to set those states. B, suppression of autophagy initiation by wortmannin (Wort) does not reduce GL1-II levels in ATG4QKO-ATG4A rescue cells. Immunoblot of LC3B and GL1 lipidation in cells incubated in EBSS and treated with/without 0.1 μm wortmannin and with/without 0.1 μm bafilomycin A1 (Baf) for 2 h. Quantification of GL1-II as a percentage of total GL1 signals in WT and ATG4QKO-ATG4A rescue cells. C, GL1 lipidation is unchanged in ATG4QKO-ATG4A cells exposed to 0.1 μm wortmannin for up to 24 h. Quantification of GL1-II was done for the 3-, 6-, 12-, and 24-h time points. D, conditions as in B, except treatment with 10 mm 3-MA instead of wortmannin for 2 h also had no effect on delipidation extent of ATG4QKO-ATG4A rescue cells. E, kinases that alter LC3B-II or GL1-II levels in WT cells do not alter GL1-II levels in ATG4QKO-ATG4A rescue cells. 10 μm of Akt/MST4/ULK1 inhibitors were used to treat cells for 12 h and had no effect on delipidation kinetics in ATG4QKO-ATG4A rescue cells. On all blots, protein molecular masses are shown in kilodaltons on the left. Statistical significance is assessed with two-sample Student's t test. *, p < 0.05. **, p < 0.01. Ctrl, control.
To distinguish between these two possibilities, we reasoned that by blocking autophagosome biogenesis signals that generally drive lipidation, we might uncouple the two processes and measure delipidation rates. We treated cells with wortmannin, a potent phosphatidylinositol 3-kinase inhibitor that blocks autophagy initiation. In the absence of PI3P, the activation of autophagy in general and of the lipidation machinery in particular is largely inhibited. For example, although WT cells incubated in Baf A1 accumulate significant LC3-II and less but detectable GL1-II, 2 h of treatment with wortmannin blocks this accumulation entirely (Fig. 2B and Ref. 42). However, GL1-II levels in ATG4QKO-ATG4A cells stay the same whether these cells were treated or not with wortmannin, and GL1-I remains undetectable (Fig. 2B). Even prolonged wortmannin treatment of up to 24 h did not result in detectable GL1-I (Fig. 2C). Likewise, treatment with another PI3K inhibitor (3-MA) also did not reduce GL1-II levels or allow for any detection of GL1-I (Fig. 2D). These results strongly suggest that the pool of GL1-II in ATG4QKO-ATG4A cells is not dynamic.
Because the GABARAPs and LC3 proteins are thought to be only partially redundant and may specialize in different aspects of autophagosome formation (43), we considered that perhaps GL1-II accumulates because both LC3B-II and GL1-II are needed on autophagic membranes for autophagy to complete. To test whether lipidated LC3B would support the flux and/or delipidation of GL1-II, we transduced a FLAG-tagged LC3B protein in which the terminal glycine is already exposed (LC3BG120) into our ATG4QKO-ATG4A cells. We have previously shown that FLAG-tagged Atg8 family members undergo ordinary flux in WT cells (29), and confirmed that our FLAG-tagged LC3B construct also undergoes ordinary flux (Fig. S5A). Critically, LC3BG120 can be lipidated without the need of priming from ATG4B. We observed approximately half of the FLAG-tagged LC3BG120 population became lipidated in our ATG4QKO-ATG4A cells, whereas all of the endogenous LC3B molecules remained in the pro-form as expected (Fig. S5, A and B). However, the FLAG-tagged LC3B molecules did not exhibit any EBSS starvation or Baf A1–dependent flux, suggesting that LC3B-associated autophagosomes in these cells did not reach the lysosome. Endogenous GL1 did not exhibit any flux, and there was no evidence of GL1-I. Furthermore, LLPD results showed that starvation-mediated bulk autophagy was still absent (Fig. S5C). Collectively, these results imply that ATG4A alone is unable to support significant delipidation in these cells and is unable to support macroautophagy even when supplemented with primed LC3B-I.
The impact of kinase inhibition on GL1-II accumulation
Both mammalian ATG4B and yeast Atg4 can be regulated by kinases. Thus, one explanation for our results could be a specific down-regulation of ATG4A delipidation activity, but whether and how this protein is regulated is largely unknown. Furthermore, even where phosphorylation-dependent regulation of ATG4B is established, the regulation is generally inferred by following changes in LC3B proteins, but there is much less understanding of how and whether these kinases also impact the pool of GL1.
We next used small molecule inhibitors to manipulate the activity of three kinases previously implicated in either inhibiting or activating ATG4B to test whether they impact GL1-II accumulation in either WT or ATG4QKO-ATG4A cells. In WT cells, inhibition of ULK1 was largely without effect (Fig. 2E). Because ULK1 has been shown to inhibit ATG4B, it normally functions to increase LC3B-II levels. In our cells where LC3B-II/GL1-II are already at very low levels in the basal condition, it is unsurprising that further inhibition of ULK1 did not lead to a significant change. Inhibition of AKT1/2 in WT cells leads to significant increases of both LC3B-II and GL1-II, consistent with work showing that AKT2-mediated phosphorylation of ATG4B broadly activates this enzyme (44). In contrast, the inhibition of MST4 leads to a significant increase in LC3B-II but not GL1-II (Fig. 2E). MST4 was previously shown to limit the accumulation of LC3B-II (45), although whether that was a direct consequence of phosphorylation on ATG4B or resulted from a less direct intervention was not clear. In the next section, we consider how the MST4 locus on ATG4B influences protease activity in vitro to explore this result.
In contrast to WT cells, in ATG4QKO-ATG4A cells we were not able to detect significant changes following treatment with any of the small molecule inhibitors (Fig. 2E). GL1-II levels did not appreciably change, and there was no detectable GL1-I. We noted a modest change in pro-GL1 levels, particularly in the ULK1 inhibition, but again, this did not correlate with a change in the levels of either processing product. Thus, in cells expressing only ATG4A, these three kinases do not play a significant role in maintaining the GL1-I/GL-II ratio, and again, delipidation appears to be completely inhibited.
Regulation of ATG4A priming and delipidation by its C-terminal LIR
Delipidation by both Atg4 and ATG4B depends on motifs outside the catalytic pocket, which engage LC3 (LC3-interacting regions, or LIRs) or Atg8 (Atg8-interacting motifs, or AIMs) (29, 45–47). In particular, at the C terminus of ATG4B, there is an LIR motif that is involved in regulating protease activity (29, 45, 47). The C termini of ATG4A and ATG4B are very similar; the last 9 amino acids are either identical or have the same side-chain properties, and the core of the ATG4B C-terminal LIR (FEIL) is fully conserved in ATG4A (47). Importantly, in an in vitro reconstitution assay in which we can follow proteolysis over time, we previously showed that removal of the COOH-LIR dramatically slowed ATG4B-mediated delipidation but had no effect on the speed of priming (29). This surprising result implies that ATG4B on membranes can be regulated differently from its priming activity in solution and may offer an explanation for how ATG4A could support priming but not delipidation in our cells.
Thus, to better understand the relative specific activities of priming and delipidation on both ATG4A and ATG4B, here we have modified our in vitro reconstitution system to measure the extent of proteolysis at a single end point and titrated ATG4 proteins over several logs of concentration (Figs. 3 and 4; cartoon model in Fig. S6A). In this assay, priming is measured as the release of a GFP from a soluble LC3B–GFP or GL1–GFP construct, whereas delipidation is measured as the release of LC3B or GL1 from PE-conjugated forms anchored in proteoliposomes.
Figure 3.
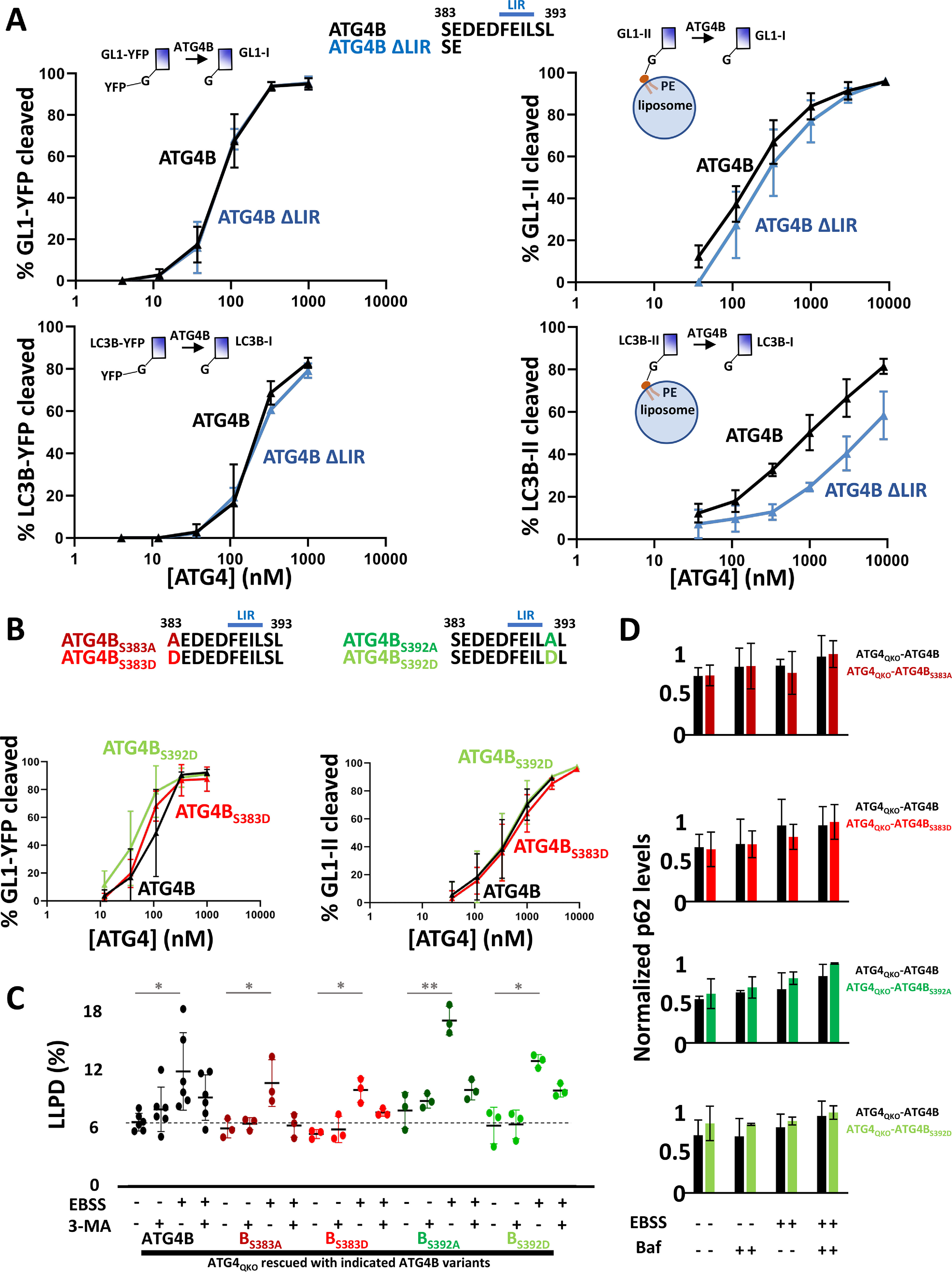
ATG4B C terminus is needed to support delipidation. A, in vitro reconstitution of GL1/LC3B priming and delipidation. For priming, recombinant purified GL1-YFP or LC3B-YFP was mixed with recombinant purified ATG4B or ATG4B ΔLIR for 16 min at 37 °C, and the extent of YFP release was measured by running samples on an SDS-PAGE gel. For delipidation, GL1 or LC3B was first coupled to PE on liposomes in an in vitro lipidation reaction, and these proteoliposomes were isolated on a density gradient. Then these samples were mixed with ATG4B or ATG4B ΔLIR for 16 min at 37 °C. Delipidation was assessed as a band shift of LC3B or GL1 on SDS-PAGE gels. B, phosphomimetic versions of ATG4B were tested in GL1 priming and delipidation assays as in A. C, rescue of ATG4QKO cells with phosphomimetic versions of ATG4B were tested for long-lived protein degradation (as in Fig. 1A). D, rescue of ATG4QKO cells with phosphomimetic versions of ATG4B tested for p62 homeostasis (as in Fig. 1, B and C). The raw data for p62 quantification are shown in Fig. S12A. Statistical significance is assessed with two-sample Student's t test. *, p < 0.05. **, p < 0.01. Baf, bafilomycin A1.
Figure 4.

ATG4A C terminus modulates GL1 priming and delipidation. A and B, in vitro reconstitution of GL1 priming (A) and delipidation (B) with various ATG4A constructs carried out as in Fig. 3A reveals that the C-terminal LIR on ATG4A is essential to both processes. C, rescue of ATG4QKO cells with expression of ATG4A LIR mutants reveals that the LIR is also essential to priming in cells and is potentially modulated by alteration at Ser397. Immunoblot of cells incubated in DMEM or EBSS, without or with 0.1 μm bafilomycin A1 (Baf) for 2 h. To quantify processing, densitometry was used to calculate the amount of pro-GL1 as a fraction of the total GL1 signal detected on the blot. Note that for values less than 100%, this represents a lower limit, because the antibody recognizes the GL1-II form more efficiently than the soluble proteins. D, rescue of ATG4QKO cells with C-terminal mutants of ATG4A were tested for long-lived protein degradation (as in Fig. 1A). E, rescue of ATG4QKO cells with C-terminal mutants of ATG4A were tested for p62 homeostasis (as in Fig. 1, B and C). Raw data for p62 quantification are shown in Fig. S12B. Statistical significance is assessed with two-sample Student's t test. *, p < 0.05. **, p < 0.01. ***, p < 0.001. ****, p < 0.0001.
ATG4B-mediated delipidation of GL1-II requires ∼8-fold as much enzyme as priming of a soluble pro-form of GL1–GFP (Fig. 3A and Fig. S6B), confirming our published results that ATG4B is naturally much better at priming (29). Deletion of the last 9 amino acids including the COOH-LIR (ATG4B ΔLIR) does not appreciably change the efficiency of priming but markedly increases the concentration of ATG4B needed to support delipidation (Fig. 3A and Fig. S7). Thus, on ATG4B, the C terminus is a delipidation-specific regulatory motif.
To test whether the ATG4A C terminus impacts proteolysis, we made a deletion mutant of ATG4A also removing the last 9 amino acids (ATG4A ΔLIR) (Fig. 4) and then subjected both the WT and deletion mutant protein to the same in vitro proteolysis assays (Fig. 4, A and B, and Fig. S8). Similarly to our previous kinetic measurements, we found that ATG4A priming and delipidation occurred with roughly equivalent efficiencies (half-maximal cleavage at 300 and 150 nm ATG4A, respectively). Like ATG4B, removal of the LIR dramatically impaired delipidation, requiring more than 2 mm protein to cleave half of the GL1-II. Unlike ATG4B, loss of the LIR also dramatically impaired ATG4A-mediated priming, reducing its specific activity to one-tenth of WT protein. Thus, in vitro, the ATG4A LIR is an essential component of the protease efficiency against all substrates.
To test the role of this motif in vivo, we rescued our ATG4QKO cell line with ATG4A LIR (ATG4QKO-ATG4A ΔLIR; Fig. 4C and Fig. S9). Despite being expressed at a much higher level than ATG4A in ATG4QKO-ATG4A cells, ATG4A LIR is unable to support any detectable priming of pro-GL1. These results strongly imply that the LIR motif on ATG4A is intimately associated with substrate capture for both priming and delipidation.
The interaction of LIR motifs with LC3 or GABARAP proteins can often be modulated by phosphorylation on immediately adjacent serine or threonine amino acids (45, 46). In both ATG4A and ATG4B, the COOH-LIR is adjacent to serines, and the serines at positions 383 and 392 are known sites of phosphorylation in ATG4B (29, 45, 46). Furthermore, MST4-mediated control of ATG4B delipidation is thought to occur through phosphorylation at Ser383 (45), although whether this phosphorylation directly modulates ATG4B activity or interferes with some other interaction in the cell is not known. Our in vitro reconstitution is an ideal system to test directly whether modifying these amino acids impacts protease activity and to explore whether ATG4A might also be subject to phosphorylation-dependent regulation at the LIR.
We expressed and purified recombinant phosphomimetic versions of ATG4A and ATG4B with the relevant serines mutated to aspartic acid (D) and tested these proteins in our in vitro assays. Mutation of one or both serines abutting the ATG4B LIR had no effect on either priming or delipidation in our assay (Fig. 3B and Fig. S10). In parallel, rescue of ATG4QKO cells with various ATG4B phosphomimetic mutants restored LLPD (Fig. 3C) and maintained p62 homeostasis levels (Fig. 3D and Figs. S11 and S12A) similar to WT ATG4B. These results suggest that any regulation via this motif is not due to a direct impact on protease–substrate interaction.
In contrast, mutation of the ATG4A S397 to D modestly activated delipidation but not priming in vitro, leading to a 5-fold increase in delipidation efficiency (Fig. 4B). Finally, to test whether aberrant phosphoregulation at this site might explain the accumulation of GL1-II in our ATG4A rescue, we created stable rescues of our ATG4QKO cells with either ATG4AS397D (phosphomimetic) or ATG4AS397A (phosphonull). The phosphomimetic had a slight but consistent increase in total pro-GL1 levels, but there was no apparent impact of either mutation on delipidation and no accumulation of any GL1-I (Fig. 4C and Fig. S9). In addition, none of the ATG4A mutants that we tested could support LLPD (Fig. 4D) or restore normal p62 homeostasis (Fig. 4E and Figs. S9 and S12B). Thus, ATG4A utilizes its COOH-LIR to support substrate proteolysis and is potentially subject to local regulation, but even with an activated phosphomimetic form, ATG4A alone is unable to support delipidation of the GABARAP proteins in ATG4QKO cells.
GL1-II accumulates on both incomplete autophagosomes and nonautophagic membrane structures in ATG4A-only rescues
GL1-II protein accumulates in our ATG4A rescues, but autophagy is inhibited. To establish whether classic autophagosomes are even forming, we next looked at which membranes become GL1-II–positive. To image GL1 distributions directly, we expressed a FLAG-tagged version of GL1 with a C-terminal deletion to expose the reactive glycine (FLAG-GL1G116), thus bypassing the already inefficient priming step in the ATG4A rescue. Like the endogenous protein, FLAG-GL1G116 is almost entirely GL1-I in WT cells and GL1-II in ATG4QKO-ATG4A (Fig. 5A). FLAG-GL1G116 accumulates in discrete puncta in both WT and ATG4QKO-ATG4A cells (Fig. 5 and Figs. S13–S16) but is diffuse across the cytoplasm in full ATG4 knockouts (29), suggesting the puncta represent accumulations of GL1-II. In WT cells, the numbers of puncta increase in response to starvation and bafilomycin. Furthermore, GL1-II puncta largely colocalize with the autophagosome marker WIPI-2 in the absence of bafilomycin, and with the lyosomal marker LAMP1 in the presence of bafilomycin (Figs. S13 and S15), consistent with its association to autophagosomes throughout their maturation. In contrast, GL1 puncta in ATG4QKO-ATG4A cells are apparent even in the absence of stress (Figs. S14 and S16). Some of these puncta are WIPI-2–positive, indicating that GL1 can be recruited into sites of autophagosome biogenesis, but much of the observable FLAG-GL1G116 is found in other WIPI2-negative structures (Fig. 5B and Fig. S14), and only a small fraction of the FLAG-GL1G116 colocalizes with LAMP1 (Fig. 5B and Fig. S16).
Figure 5.

GL1-II decorated membranes colocalize with WIPI2- and LAMP1-positive puncta. To image GL1 distributions in cells, a FLAG-tagged form of GL1 truncated at position 116 (FLAG-GL1116) was expressed in WT or ATG4QKO-ATG4A cells. A, immunoblot of cells grown in DMEM or EBSS without or with 0.1 μm bafilomycin A1 (Baf) for 4 h reveals that FLAG-GL1G116 behaves the same as endogenous GL1 and accumulates as GL1-I form in WT cells or GL1-II form in ATG4QKO-ATG4A cells. B, immunofluorescence microscopy of cells in A reveals that FLAG-GL1G116 forms puncta in ATG4QKO-ATG4A. B, top row, many WIPI2 puncta colocalize with FLAG-GL1G116 (white arrows), although some WIPI2 sites appear to be without FLAG-GL1G116 (yellow arrowheads). Bottom row, FLAG-GL1G116 frequently colocalizes with the lysosome-related marker LAMP1 (white arrowheads), although some FLAG-GL1G116 remains separate (yellow arrowheads). Scale bar, 10 μm.
We next assessed whether GL1-II becomes protected within a closed structure, as would be expected if half of the protein on the limiting membrane becomes encapsulated within an autophagosome. The cells were incubated in full medium with Baf A1 for 4 h to accumulate as many closed autophagic structures as possible, and then the total membranes were collected and fractionated by multiple rounds of varying speed centrifugation (48, 49) with fractions identified as “high-speed pellet”–containing larger membranes, including lysosomes, mitochondria, etc., and “low-speed pellet”–containing small vesicles. Each isolate was then subjected to incubation with or without proteinase K to proteolyze accessible outward-facing proteins.
Approximately 50% of GL1-II molecules are protected in WT pellets consistent with their recruitment to maturing autophagosomes (Fig. 6, A and B, black). In contrast, in membranes isolated from ATG4QKO-ATG4A cells, most GL1-II molecules were digested (Fig. 6, A and B, red) and thus not incorporated into fully closed mature autophagosomes. The autophagic cargo p62 exhibited similar trends, although the absolute changes in protease protection fell below statistical significance.
Figure 6.

GL1-II decorated membranes in ATG4QKO-ATG4A rescue cells are mostly unclosed structures localized to the ER. A, cell fractionation and protease protection assay of cell lysates. The indicated cells were grown in DMEM and treated with 0.1 μm bafilomycin A1 for 4 h before being subjected to multiple rounds of centrifugation as in Ref. 61. Isolated fractions were treated with 100 μg/ml proteinase K (protk) for 30 min, and the protection of proteins was evaluated by immunoblot. B, quantifications of A were conducted by dividing GL1-II density (or p62 density) of samples with proteinase K treatment over density of control samples (Ctrl). Statistical significance was assessed with two-sample Student's t test. *, p < 0.05. C, cell fractionation and ER/AV separation were performed by multiple density gradient isolations (cartoon and Ref. 62) from cells grown in DMEM and treated with 0.1 μm bafilomycin A1 for 4 h. To detect organelle enrichment in each final isolate (TL, total lysate; top, uncharacterized material at top of gradient; cyt, cytosol; upp, uncharacterized material in upper band; ER, endoplasmic reticulum; AV, autophagic vacuole; Mi, mitochondria/peroxisomes; each labeled in color in the cartoon), 3 μg of protein were run on SDS-PAGE gels and imaged by immunoblot. D, to determine the total amount of GL1-II or p62 localized to each organelle isolate, the intensity of the bands in C were normalized to the total mass collected of each fraction. Statistical significance is assessed with two-sample Student's t test. *, p < 0.05. **, p < 0.01. ***, p < 0.001.
To better establish the identity of these GL1-II–positive membranes in ATG4A-only rescues, we used a more comprehensive membrane fractionation protocol that has been well-established to segregate autophagosomes from other compartments (50). In WT cells, GL1-II is strongly enriched in the autophagosome fraction along with p62 and membrane-associated GAPDH (Fig. 6, C and D). In addition, all three of these proteins are largely resistant to added proteinase K, consistent with their accumulation inside mature closed autophagosomes. In contrast, in ATG4QKO-ATG4A cells much less p62, GAPDH, or GL1-II is associated with the autophagosome fraction. Instead, GL1-II and p62 are found in both the ER and autophagic vacuole (AV) (Fig. 6C), and when we account for the fact that there is far more ER in our sample than AV, it is apparent that the vast majority of these proteins are found in membranes copurifying with endoplasmic reticulum (Fig. 6D). Furthermore, proteins accumulating with the ER or mitochondrial fractions are completely digested by proteinase K, as expected if these are not autophagosomal membranes.
Collectively, our results show that ATG4A alone is not sufficient to support autophagy in these cells, and this failure likely derives from two primary defects. First, ATG4A-mediated priming is inefficient. In WT cells, unprimed pro-forms of the GABARAP and LC3 proteins are undetectable because priming occurs fast and constitutively (driven largely by ATG4B); however, in ATG4A-only rescues, approximately half of the GL1 pool remains as pro-GL1. Second, in cells expressing only ATG4A, there is a general inability to delipidate GL1 proteins that end up mistargeted to nonautophagic sites in the cell. These two defects combine to strongly limit the pool of soluble GL1-I protein needed to support nascent autophagosome formation.
Discussion
ATG4A is relatively understudied despite being the second most catalytically active mammalian ATG4 protein, after ATG4B (31, 51). Because ATG4B knockouts in mice are viable (52) and because GABARAP proteins, in the absence of LC3 proteins, are sufficient to support autophagy in cells (32), it seemed likely that ATG4A would play a significant and largely sufficient role in supporting autophagy in simple cellular systems. However, several earlier studies, including our own, have suggested the opposite: that cells lacking ATG4B were largely incapable of completing autophagy (21, 29, 47). Thus, here we explored the limits of ATG4A activity. Our primary discovery is that the turnover of lipid-attached GABARAP proteins is incredibly slow or nonexistent in cells expressing only ATG4A, despite the fact that intrinsic ATG4A delipidation activity measured in vitro is roughly equal to ATG4B-driven delipidation (Figs. 3 and 4 and Fig. 29). As a result, essentially all of the primed GABARAP family protein accumulates in a lipid-attached form in ATG4A-only cells. A small fraction of this protein copurifies at densities equivalent to autophagosomes and is protease-protected, suggesting that at least some closed autophagosomes are produced. The vast majority of the GL1-II, however, is found in our ER fraction and is not protease-protected. Thus, these cells may face two major challenges in completing autophagy; 1) the presence of GABARAP proteins on the outside of the mature autophagosome may hinder the delivery of these organelles into the lysosome because this step is thought to require removal of all LC3/GABARAP (53), but 2) equally problematic is that most of the available GABARAP protein in the cell accumulates at what may be nonautophagy sites and is unable to be recycled from these locales, limiting the available pool of GABARAP proteins for new autophagosome biogenesis (28).
Modulation of priming and delipidation by the C termini of ATG4A and ATG4B
Many groups have shown previously that ATG4A primes GL1 and other GABARAP proteins very slowly relative to ATG4B (29, 31, 51); however, the two proteases can remove GL1 and other GABARAPs from membranes in vitro with essentially equal kinetics (29). Further, an LIR in the C terminus of ATG4B (29, 47) is a critical determinant of its rate of delipidation but is dispensable for its priming (29). Because the C termini LIR motifs of ATG4A and ATG4B are highly conserved, it is curious that this study shows they appear to regulate GL1 processing differently, with essentially all of ATG4A's protease activities requiring an intact LIR (Fig. 4). We had previously speculated that a “delipidation-only” role for the LIR on ATG4B strongly implied a membrane-dependent activation of the enzyme perhaps through a simple model in which ATG4B recognizes one substrate at its active site and simultaneously recognizes a second LC3/GABARAP at its LIR, something that would be highly promoted at membranes where multiple lipidated substrates would be present. However, our results here that ATG4A requires this same motif for priming argue against that model and perhaps favor a conformational change that occurs when substrate first engages the LIR before moving into the active site. Details of this possible model will require further study.
There is mounting interest in understanding how the proteolytic activities of ATG4 proteins are regulated as a possible therapeutic node in diseases like cancer (54, 55). To that end, it is interesting that these C-terminal LIR sequences are flanked by serines of which the substrate interactions might be regulated by phosphorylation, as is commonly the case at other LIRs. Indeed, phosphorylation of Ser383 and Ser392 in ATG4B was previously shown to affect autophagic flux in cells through mechanisms interpreted as a change in delipidation (45, 46). Moreover, phosphorylation of Ser383 by MST4 enhances autophagic activity (measured as an increased conversion of LC3-I to LC3-II and more p62 degradation) and correlates with increased glioblastoma tumorigenicity (45). However, our results suggest the mechanism by which MST4 activates autophagy requires more investigation. First, when we tested the activity of ATG4B harboring phosphomimetic mutations, we did not observe a clear change in activity in vitro, and like a previous study by Rasmussen et al. (47), we also did not detect a significant difference between WT or phosphomimetic ATG4B in our knockout/rescue system. Thus, it appears unlikely that these serines directly impact the catalytic activity of ATG4B. In addition, small molecule inhibition of MST4 only appeared to impact LC3B-II levels, but not GL1-II, perhaps suggesting it works through a more precise effector of LC3 than ATG4B.
Considerably less is known about whether ATG4A is subject to kinase regulation, but our in vitro experiments with ATG4A phosphomimetics (S397D) were somewhat promising because we could detect a roughly 5-fold increase in specific activity. Whether this is meaningful in vivo is currently unclear, because we cannot detect delipidation at all in our KO-rescue.
Why is ATG4A alone insufficient when ATG4B KO mice are viable?
Knockout studies with mouse models revealed that although ATG3−/− (56), ATG5−/− (57), ATG7−/− (58) mice are neonatal lethal, ATG4B−/− (52) and ATG4C−/− (59) mice are viable with relatively mild phenotypes (including balance problems in ATG4B−/− and elevated vulnerability to fibrosarcoma in ATG4C−/−). Furthermore, LC3B and GABARAP KO mice are viable with no abnormalities (60). Thus, there is likely tremendous redundancy across the ATG4 and ATG8 protein families. In this background, it is somewhat surprising that ATG4A is not sufficient to support autophagy. Our long-lived protein degradation assay is a good measure of bulk starvation-dependent autophagy, which is completely absent in these ATG4A-only cells, but we did not search for autophagosomes by other criteria, such as EM, and thus we may have missed ATG5/ATG7-independent autophagy (33) and ATG8-independent autophagy (32) previously shown to function in mammalian cells.
An obvious limitation of removing ATG4B, ATG4C, and ATG4D from these cells is a loss of LC3 family processing; however, we could not rescue autophagy simply by adding LC3B back into the mix (Fig. 2E). Likewise, in cells still expressing ATG4C and ATG4D that can facilitate some LC3 family processing, we previously did not detect any autophagic flux at the level of GABARAP/LC3 turnover when ATG4B was missing (29). Thus, we conclude that ATG4A is insufficient on its own, and we suggest that the ATG4B−/− mouse is viable because of some compensation, perhaps through additional activation/expression of ATG4C and ATG4D.
The most surprising aspect of our study is the complete absence of detectable GL1 delipidation in cells expressing only ATG4A. Delipidation in cells is notoriously difficult to assess because LC3/GABARAP proteins are almost always in a steady-state of priming, lipidation, and delipidation, and thus changes in the relative amounts of form I and form II proteins can result from fluctuations in any of these parameters. In our case, however, there is no flux at all. All of the GL1, even when massively overexpressed as in our FLAG-tag experiments, is completely in the lipidated form. Furthermore, the lipidated protein appears to be collecting at sites other than the autophagosome, including on membranes that copurify with ER. This is the same phenotype observed in yeast when primed Atg8 protein is introduced into Atg4 KO cells (28, 53) and suggests that perhaps ATG4A is simply not competent to recycle GL1 from off-target sites, an activity that is considered to be essential to support LC3/GABARAP-dependent macroautophagy.
Experimental procedures
Materials
1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (850725C), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (850457C), l-α-phosphatidylinositol (bovine liver/phosphatidylinositol; 840042C) were purchased from Avanti Polar Lipids. Nycodenz (1002424) was purchased from Accurate Chemical and Scientific Corp. pCMV-VSV-G (8454) and psPAX2 (12260) were purchased from Addgene. BL21-Gold (DE3) competent cells (230132) were purchased from Agilent Technologies. ATP (AB00162), DTT (AB00490), isopropyl β-d-thiogalactopyranoside (AB00841), HEPES (AB00892), NaCl (AB01915), SDS (AB01920), sucrose (AB01900), Tris-HCl (AB02005), Triton X-100 (AB02025), Tween 20 (AB02038), were purchased from AmericanBio. Protein assay dye reagent (5000006) was purchased from Bio-Rad. Glass-bottomed 35-mm dishes (D35-20-1.5-N) were purchased from Cellvis. Lenti-X concentrator (631232) was purchased from Clontech. 16% Paraformaldehyde (15710) was purchased from Electron Microscopy Sciences. MAP/MST4/AMPK inhibitor (375680) was purchased from EMD Millipore. Bafilomycin A1 (BML-CM110) was purchased from Enzo. DMSO (D12345), Lipofectamine 3000 (L3000015), NuPAGETM, MOPS, and SDS running buffer 20× (NP0001) were purchased from Invitrogen. 100% glycerol, anhydrous (2136), CaCl2 (1332), EDTA (8993), MgCl2 (2444) were purchased from J. T. Baker. Immobilon-FL polyvinylidene difluoride membranes (IPFL00010) were purchased from MilliporeSigmaTM. l-[U-14C]Valine (NEC291EU050UC) was purchased from PerkinElmer. 3-MA (M9281), Akt1/2 inhibitor (A6730), BSA (A9647), carbenicillin (C1389), casein (C7078), cOmplete, EDTA-free protease inhibitor tablets (11873580001), d-mannitol (M4125), GSH beads (G4501), imidazole (I5513), octyl-glucoside (O8001), OptiPrepTM density gradient medium (D1556), Percoll® (P4937), proteinase K (3115887001), saponin (47036), NaN3 (S2002), thrombin (T6884), ULK1/2 inhibitor (SML1540), Whatman® NucleporeTM track-etched polycarbonate membranes (10417104), and wortmannin (W1628) were purchased from Sigma–Aldrich. 0.05% Trypsin-EDTA 1× (25300-054), 10% FBS (10438062), Coomassie Blue stain (ImperialTM protein stain), CyQUANTTM cell lysis buffer 20× concentrate (C7027), CyQUANTTM NF cell proliferation assay kit (C35006), DMEM (11965092), Dulbecco's Phosphate Buffered Saline (DPBS) 10× (14200075), ImperialTM protein stain (24615), NuPAGETM 12% Bis-Tris gels (NP0341BOX, NP0349BOX), NuPAGE® transfer buffer 20× (NP0006-1), ProLongTM Gold antifade reagent (P36935), ScintiVerseTM BD mixture (SX18-4), SeeBlue Plus2 prestained protein standard (LC5925), SuperSignalTM West Femto substrate (34096), and tris(2-carboxyethyl)phosphine (T2556) were purchased from Thermo Fisher Scientific. Borosilicate glass 10 × 75mm (47729-568) was purchased from VWR.
Antibodies used in this manuscript
For immunoblotting, the following primary antibodies were used: ATG4A: 7613S, Cell Signaling Technology; ATG4B: 13507S, Cell Signaling Technology; ATG4C: 5262S, Abcam; ATG4D: MA5-18110, Thermo Fisher Scientific; FLAG: F1804, Sigma–Aldrich; GABARAPL1 (GL1): 26632S, Cell Signaling Technology; GAPDH: ab9484, Abcam; LC3B: 3868S, Cell Signaling Technology; p62: 610832, BD Biosciences; LDH: ab47010, Abcam; and NBR1: ab126175, Abcam. For immunoblotting, the following secondary antibodies were used: ECL rabbit IgG, horseradish peroxidase–linked: NA934V, GE Life Sciences; IRDye® 680RD Donkey anti-Mouse IgG: 925–68072, LICOR; and IRDye® 800CW donkey anti-rabbit IgG: 925–32213, LICOR. For immunofluorescence imaging, the following primary antibodies were used: FLAG: F1804, Sigma–Aldrich; FLAG: 14793S, Cell Signaling Technology; LAMP1: 9091S, Cell Signaling Technology; and WIPI2: MABC91, EMD Millipore. For immunofluorescence imaging, the following secondary antibodies were used: Alexa FluorTM 488 donkey anti-Rabbit IgG: A11008, Thermo Fisher Scientific; and Alexa FluorTM 594 donkey anti-mouse IgG: A21203, Thermo Fisher Scientific.
Mutagenesis of ATG4s
The human ATG4A and ATG4B/C/D constructs were cloned in the pGEX-4T and pGEX-2T GST backbone vectors, respectively, as described (29). ATG4A and ATG4B were mutagenized to generate mutants using the QuikChange II site-Directed mutagenesis kit (Agilent Technologies) and verified by sequencing.
Recombinant protein expression and purification
Human ATG4, mouse ATG3, and the mammalian homologs of ATG8 (human GABARAPL1 (GL1) and human LC3B) were expressed and purified as previously described (29, 61). In summary, human GL1/LC3B were cloned in the PGEX-2T GST vector, mouse ATG3 was cloned into PGEX-6p GST vector. GL1/LC3B/ATG3/ATG4s were expressed in BL21-Gold (DE3) competent cells. The cells were cultured in 4 liters of Luria Bertani broth (LB) medium with 1:1000 carbenicillin (50 mg/ml) and induced with 0.5 mm (final concentration) isopropyl β-d-thiogalactopyranoside. Bacterial pellets were treated with EDTA-free protease inhibitor mixture tablets in either thrombin buffer (20 mm Tris, pH 7.5, 100 mm NaCl, 5 mm MgCl2, 2 mm CaCl2, 1 mm DTT) for GL1/LC3B/ATG4s or precision protease buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm DTT) for ATG3. The cells were broken in a cell disrupter, and the lysate was incubated with GSH beads for 3 h at 4 °C. The beads were washed several times and then incubated with GL1/LC3B/ATG4s cutting buffer (10 μl of thrombin + 500 μl of thrombin buffer + 0.5 μl of DTT + 500 μl of beads) or ATG3 cutting buffer (25 μl of precision protease + 500 μl of precision protease buffer + 0.5 μl of DTT + 500 μl of beads) to cut the proteins from GST tags overnight. Purified proteins were stored in 20% glycerol at −80 °C. GL1/LC3B were expressed in both a truncated form ending in the reactive C-terminal glycine (such that no ATG4-mediated preprocessing was needed for lipidation process) and a form with YFP (for GL1) or tag (for LC3B) to determine whether ATG4 priming activity of ATG8 took place.
Protein expression and purification for human ATG7
Human ATG7 was expressed and purified as previously described (35). In summary, the ATG7-containing plasmid was transformed into Bacmid DNA. 2 µg of DNA was used to infect 8 × 105 SF9 cells by using Cellfectin II two times to increase viral titer to 5 × 106 plaque-forming units/ml. 1 × 108 plaque-forming unit/ml of SF9 cells were infected with virus and they grew for 72 h. The cells were treated with EDTA-free protease inhibitor mixture tablets in the lysis buffer (20 mm Tris, pH 8, 500 mm NaCl, 20 mm imidazole, 1 mm DTT, 10% glycerol), sonicated with the Virsonic 600 (VirTis) microtip sonicator on for 3 min (30 s on, 30 s off; intensity 3.5), and centrifuged at 18,000 rpm for 1 h. The lysate was incubated with 1 ml of nickel resin (nickel–nitrilotriacetic acid–agarose) for 2 h at 4 °C. The beads were washed with the wash buffer (20 mm Tris, pH 8, 300 mm NaCl, 20 mm imidazole, 1 mm DTT) three times and eluted with the elution buffer (20 mm Tris, pH 7.5, 300 mm NaCl, 500 mm imidazole, 1 mm DTT). Purified proteins were stored in 20% glycerol at −80 °C.
Liposome and proteoliposome preparation
Liposomes and proteoliposomes were prepared as previously described (29). The composition of liposomes was 55% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, 35% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, and 10% l-α-phosphatidylinositol. Liposomes were extruded 21 times through polycarbonate membranes to 400 nm with the LipSoFast-Basic extruder (Avestin) and then were sonicated with the Virsonic 600 (VirTis) microtip sonicator to a size of 50 nm immediately prior to the lipidation reaction. Next, GL1 and LC3B underwent the lipidation process to be coupled to liposomes. In short, GL1/LC3B proteins (final concentration, 15 μm), ATG3 (2 μm), ATG7 (2 μm), and sonicated liposomes (3 mm) were mixed with DTT (1 mm) in SNH buffer (20 mm Tris, pH 8, 100 mm NaCl, and 5 mm MgCl2). Lipidation was initiated by adding ATP (1 mm), and reactions were incubated at 37 °C for 90 min.
Priming and delipidation assays
Priming and delipidation assays were done as previously described (29). The proteoliposomes were used to determine the delipidation activity, whereas GL1/LC3B-YFP proteins were used to determine priming activity. For delipidation samples, GL1/LC3B was lipidated onto liposomes at 37 °C for 90 min. Next, these samples were ran on a Nycodenz density gradient to purify the proteoliposomes. The bottom layer is composed of 150 μl of 80% Nycodenz and 150 μl of the lipidation reaction. The second layer is 250 μl of 30% Nycodenz. The top layer is 50 μl of SNH buffer. Gradients were spun at 48,000 rpm at 4 °C for 4 h in a Beckman SW55 rotor (Beckman Coulter). 50 μl of samples were extracted from the 30% Nycodenz/buffer interface and were stored at 4 °C. First, the range of concentrations for each individual ATG4 proteins stock in which the activity would go from 0% to 100% was determined. Several steps were chosen for every protein titration assay. Once evenly distributed, for each resulting step concentration, we calculated the volume needed to be added from stock to achieve that concentration with a volume of 25 μl. A portion of the initial 7.6-μl ATG4 mix was diluted with SNH buffer to make the next dilution, and this procedure is repeated several times. Then 7.6 μl from each of the ATG4 dilutions were placed in new tubes with SNH buffer. Finally, the substrates (proteoliposomes or GL1/LC3B-YFP) were added to these tubes (final volume, 25 μl), mixed, and incubated at 37 °C for 16 min.
Gel visualization and quantification
15-μl samples were mixed with 4 μl of 4× LDS loading buffer and boiled at 90 °C for 5 min. Electrophoreses of samples were run in 12% Bis-Tris gel with 1× MOPS buffer for 55 min at 200 V using the Hoefer EPS 2A200 power supply C. The samples were visualized with Coomassie Blue stain. Band intensities of samples were quantified with ImageJ software as described for densitometry (29). The gels were imaged with the VersaDoc imaging system (Bio-Rad) and analyzed for densitometry using the ImageJ software.
CRISPR/Cas9 knockout and rescue with lentiviral transfection
CRISPR/Cas9 knockout of ATG4A/B/C/D genes in HEK293T was described in the previous paper (29). 3× FLAG-tag GL1/LC3B and ATG4A constructs were cloned into high-expression pLVX-Puro vector. ATG4B constructs were cloned into low-expression pLenti-III-PGK vector. Lentiviral transfection to make stable cell lines was done as described previously (29). In short, HEK293FT cells were seeded for 24 h at 37 °C with 5% CO2 in DMEM (containing 10% FBS) until confluency reached 50%. Next, the cells were transfected with psPAX2, pCMV-VSV-G, and target plasmids using Lipofectamine 3000. After transfection, medium was collected every 24 h for 3 days and stored at 4 °C. The medium was then filtered with a 0.45-mm filter, and then Lenti-X concentrator was added at a ratio of 1:3 and mixed before incubation at 4 °C for 4 h. The mixture was centrifuged at 2700 rpm for 45 min at 4 °C. The pellet was gently resuspended in 1/250 of the original volume using complete DMEM and stored at −80 °C. ATG4 QKO HEK293T cells were transduced with 30 μl of the virus and 8 μl of Polybrene at 50% confluency for 48 h. Puromycin was used to a final concentration of 5 µg/ml for selection of successfully transduced cells every 24 h for 3 days.
The following CRISPR/cas9 guides were used for making the ATG4QKO: ATG4A, CCCAACCAGCATCTGATGAA; ATG4B, CTAGACTTTGGTTTACATAC; ATG4C, AATTCTCCTGTATTATTGCT; and ATG4D, ACCGTACTTGACGTTGTTCC. The following primers were used for making ATG4A/B constructs to rescue ATG4QKO cells: ATG4A S397A forward: GCT, GATTTTGAGATTCTGgctGTGTAGTCTAGATAATTCTAC; ATG4A S397A reverse: AGC, GTAGAATTATCTAGACTACACagcCAGAATCTCAAAATC; ATG4A S397D forward: GAT, GATTTTGAGATTCTGgatGTGTAGTCTAGATAATTCTAC; ATG4A S397D reverse: ATC, GTAGAATTATCTAGACTACACatcCAGAATCTCAAAATC; ATG4A ΔLIR forward: GAGGAGTTTGATCTGtAGGAAGATTTTGAGATT; ATG4A ΔLIR reverse: AATCTCAAAATCTTCCTaCAGATCAAACTCCTC; ATG4B S383A forward: GCA, GAAAGATTCTTCGACgcaGAAGATGAAGACTTTG; ATG4B S383A reverse: TGC, CAAAGTCTTCATCTTCtgcGTCGAAGAATCTTTC; ATG4B S383D forward: GAT, GAAAGATTCTTCGACgatGAAGATGAAGACTTTG; ATG4B S383D reverse: ATC, CAAAGTCTTCATCTTCatcGTCGAAGAATCTTTC; ATG4B S392A forward: GCC, GACTTTGAAATCCTGgccCTTTGAGCGGCCGCTG; ATG4B S392A reverse: GGC, CAGCGGCCGCTCAAAGggcCAGGATTTCAAAGTC; ATG4B S392D forward: GAC, GACTTTGAAATCCTGgacCTTTGAGCGGCCGCTG; and ATG4B S392D reverse: GTC, CAGCGGCCGCTCAAAGgtcCAGGATTTCAAAGTC.
Immunoblotting/Western blotting protocol
The cells were treated with DMEM or EBSS medium with or without 0.1 μm bafilomycin A1. The cell lysates were collected after reaching 80–90% confluency in 1× PBS and centrifuged at 2000 rpm for 5 min. The supernatant was aspirated and replaced in 250 μl of lysis buffer (1× PBS with protease inhibitor mixture tablet) and resuspended on ice for 5 min. The solution was centrifuged at 43,000 rpm for 10 min at 4 °C, and the supernatant was collected and stored at −80 °C. Protein concentration was determined with Bio-Rad protein assay. 45 µg of lysates were electrophoresed on 12% Bis-Tris gels for 70 min at 180 V. Transfer membranes were placed on gels in transfer buffer (5% NuPAGE transfer buffer, 10% methanol) and electrophoresed for 70 min at 30 V. For GABARAPL1, LDH, and NBR1, the membranes were washed with PBST (10% DPBS 10 ×, 0.05% Tween in 1 × PBS) and blocked with 1% BSA in PBST for 1 h. Then membranes were washed three times in PBST and placed in primary antibodies (1:500 dilution in 5% BSA) overnight. Next, the membranes were washed three times in PBST and placed in secondary antibodies (1:5000 dilution of horseradish peroxidase antibody in 1% BSA) for 1 h. The membranes were washed three times in PBST and incubated with SuperSignalTM West Femto substrate before imaging with VersaDoc imaging system (Bio-Rad). For other antibodies, the membranes were dried for 40 min and washed with methanol and PBST before incubation in LICOR blocking buffer (10% DPBS 10×, 0.5% casein, 0.1% NaN3) for 1 h. The membranes were washed in PBST and placed in primary antibodies overnight. Next, the membranes were washed in PBST and placed in secondary antibodies (1:10,000 dilution of antibody in LICOR secondary antibody solution (LICOR blocking buffer with 0.25% Tween 20 and 0.1% SDS)) for 1 h. The membranes were washed and incubated in substrate before imaging with LICOR Odyssey system. Densitometry quantifications of p62 and ATG8 proteins levels were done as described previously. Quantifications of p62 levels in a blot were done normalizing to the highest densitometry value by using the ImageJ software. Statistical significance is assessed with two-sample Student's t test.
Long-lived protein degradation assay
The experimental plan was adapted from previously published methods (36, 38, 40, 62). In summary, the cells were seeded to be grown overnight in DMEM so in 24 h the confluency is 80–90%. After 24 h, DMEM supplemented with l-[U-14C]valine (final concentration, 0.5 µCi ml−1) was added to cells and maintained for another 24 h. After this 24-h period, the cells were washed with fresh DMEM and maintained for 4 h in DMEM supplemented with cold valine to remove the degradation products of short-lived proteins. Next, the cells were washed and treated with DMEM or EBSS medium without or with 10 mm 3-methyladenine for another 4 h. Then supernatants and lysates of cells were treated separately the in-scintillation liquid and measured with the scintillation counter to determine acid-soluble (cpm released) and acid-precipitate radioactivity (cell cpm). Protein degradation assessment calculation is cpm released over total cpm × 100. Statistical significance is assessed with two-sample Student's t test.
Cell viability assays
The cells were plated to 80–90% confluency before being incubated with trypsin for 3 min. The cell suspension was spun down, and the cells were resuspended in 50 μl of fresh DMEM. 10 μl was mixed with 10 μl of Trypan blue, and this mixture was used to count total number of viable cells with the hetocytometer. The cells were also plated in 96-well plates with DMEM before being treated with the CyQuant cell proliferation fluorescence assay. Next, the SpectraMax M5 (Molecular Devices) was used to read fluorescence intensity (excitation, 485 nm; emission, 530 nm). The cells were plated on glass-bottomed dishes and visualized with light microscopy from the DeltaVision imaging system (GE Applied Precision) to observe for viability and morphology. Statistical significance is assessed with two-sample Student's t test.
Cell fractionation and protease protection assay
The experimental plan was adapted from that previously published (49) for experiments in Fig. 6 (A and B). In summary for cell fractionation, the cells were seeded to 80–90% confluency in DMEM and incubated with 0.1 μm bafilomycin A1 for 4 h. The cells were washed with PBS, centrifuged at 1995 rpm for 5 min at 4 °C, resuspended in 400 µl of EDTA-free protease inhibitor-containing homogenization buffer (10 mm HEPES, 0.22 m mannitol, and 0.07 m sucrose, pH 7.5) mixture, and lysed with a 2-ml Dounce homogenizer. The ;Lysates were centrifuged 1700 rpm for 5 min at 4 °C to get the nucleus pellets. Postnuclear supernatants were spun at 8500 rpm for 5 min to get the low-speed pellets, whereas supernatants were further centrifuged in a Beckman TLA-100.3 rotor at 43000 rpm for 30 min to collect the high-speed pellets and high-speed supernatants. Nucleus pellets, low-speed pellets, and high-speed pellets were resuspended in 40 µl of homogenization buffer. For the protease protection assay, 45 µg of each sample was incubated with 100 μg/ml proteinase K, or with 100 μg/ml proteinase K and 0.5% Triton X-100 on ice for 30 min. Next, the samples were boiled for 10 min to stop the reactions and then subjected to immunoblotting protocol. For each cell line, finding the percentage of protection entails measuring the densitometry of each protected sample and then dividing that over the densitometry of each control sample. Statistical significance is assessed with two-sample Student's t test.
ER and autophagosome (ER/AV) separation and protease protection assay
The cell fractionation and ER/AV separation assays were adapted from previously published methods (50) for experiments in Fig. 6 (C and D). In summary, the cells were seeded to 80–90% confluency in DMEM and incubated with 0.1 μm bafilomycin A1 for 4 h. The cells were resuspended in 1 ml of EDTA-free protease inhibitor-containing buffer (50 mm Tris, 150 mm NaCl, 1× PIC, and 10% sucrose, pH 7.5) mixture and lysed with a 2-ml Dounce homogenizer. The lysates were centrifuged 4000 rpm for 2 min at 4 °C to get the nucleus pellets. Postnuclear supernatants (PNSs) were loaded onto a Nycodenz gradient (105 μl of 22% Nycodenz at bottom, 270 μl of 9.5% Nycodenz in middle, and 225 μl of PNS on top) in an ultracentrifuge tube (Beckman 355090) and centrifuged at 38,600 rpm with the Beckman SW55 rotor for 1 h at 4 °C to get the following fractions: top of PNS layer, middle of PNS layer (Cytosol), interface between PNS and 9.5% Nycodenz layers, interface between 9.5% and 22.5% Nycodenz layers (ER/AV), and bottom of tube (mitochondria and peroxisomes). ER/AV fraction was loaded on a Percoll/Nycodenz gradient (110 μl of 22.5% Nycodenz at bottom, 330 μl of 33% Percoll in middle, and 160 μl ER/AV on top) and centrifuged at 27,600 rpm for 30 min at 4 °C to get these fractions: interface between ER/AV and 33% Percoll layers (ER), and interface between 33% Percoll and 22.5% Nycodenz layers (AV/Percoll). AV/Percoll fraction was mixed with 168 μl of 60% (w/v) OptiPrep in water, loaded onto an OptiPrep gradient (408 μl of AV/Percoll/OptiPrep at bottom, 72 μl of 30% OptiPrep in middle, and 120 μl of buffer on top), and centrifuged at 27,300 rpm for 30 min at 4 °C to get the AV fraction between the sucrose and 30% OptiPrep layers. All samples derived from cells were resuspended in buffer. For the protease protection assay, 3 µg of each sample was incubated with 100 μg/ml proteinase K, or with 100 μg/ml proteinase K and 0.5% Triton X-100 on ice for 30 min. Next, the samples were boiled for 10 min to stop the reactions and then subjected to immunoblotting protocol. To calculate the total proteins in each fraction, the densitometry of 3 µg from each sample was measured then normalized to the total volume collected for each respective fraction. Statistical significance is assessed with two-sample Student's t test.
Immunofluorescence and confocal microscopy
The cells were plated on coverslips in a 24-well plate at a density of 10,000 cells/well and were grown overnight at 37 °C. The next day, the media were aspirated from each well, and then 250 μl of 4% paraformaldehyde were added and incubated for 20 min. The coverslips were washed three times with 1 ml of 1× PBS for 5 min each. The coverslips were blocked with 500 μl of blocking solution (3% BSA, 0.1% saponin) diluted in 1× PBS at room temperature for 15 min. Then 40 μl of primary antibodies (1:500 dilution in blocking solution) was applied to coverslips and left overnight at 4 °C. The next day, the coverslips were placed back into a 24-well plate and washed three times with 1 ml of blocking solution for 5 min each time. 250 μl of secondary antibodies (1:600 dilution in blocking solution) was applied to the coverslips and incubated for 1 h in the dark. After 1 h, the coverslips were washed three times with 1 ml of blocking solution. Next, the coverslips were mounted with one drop of ProLongTM Gold per coverslip. The coverslips were dried in the dark overnight at 4 °C. The images were taken with the ZEISS LSM 880 microscope and analyzed with ImageJ.
Data availability
All raw data are available at reasonable request by contacting Dr. Thomas Melia (thomas.melia@yale.edu) from Yale University. All remaining data are contained within the article.
Supplementary Material
Acknowledgments
We are deeply grateful to Halle O'Brien for support and to Sunandini Chandra and Devin Fuller for discussion about this work.
This article contains supporting information.
Author contributions—N. N., J. J., and T. J. M. conceptualization; N. N., J. J., S. Y., L. L., S. N., K. J. K., and T. J. M. resources; N. N., T. J. O., and A. M. data curation; N. N. and T. J. M. supervision; N. N., T. J. O., and A. M. validation; N. N., T. J. O., and A. M. investigation; N. N., T. J. O., and A. M. visualization; N. N., T. J. O., A. M., and T. J. M. methodology; N. N. writing-original draft; N. N. and T. J. M. project administration; N. N., T. J. O., and T. J. M. writing-review and editing; T. J. M. funding acquisition.
Funding and additional information—This work was supported by National Institutes of Health Grants (GM100930 and NS063973 to T. J. M.), a predoctoral training grant from the China Scholarship Council (to S. Y.), Chinese Scholarship Council Student Exchange Fellowship Grant 201406180015 (to J. J.), National Institutes of Health Predoctoral Training Grant T32 GM007223 (to N. N., T. J. O., and K. J. K.), and funding from the National Agrarian University–La Molina (through the Research Experience for Peruvian Undergraduates (REPU) program to A. M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PE
- phosphatidylethanolamine
- LIR
- LC3-interacting region
- Baf A1
- bafilomycin A1
- LLPD
- long-lived protein degradation
- EBSS
- Earle's balanced salt solution
- 3-MA
- 3-methyladenine
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ER
- endoplasmic reticulum
- LDH
- lactate dehydrogenase
- DMEM
- Dulbecco's modified Eagle's medium
- PNS
- postnuclear supernatant
- AV
- autophagic vacuole
- DPBS
- Dulbecco's Phosphate Buffered Saline.
References
- 1. Deter R. L., Baudhuin P., and De Duve C. (1967) Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 35, C11–C16 10.1083/jcb.35.2.c11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tooze S., and Yoshimori T. (2010) The origin of the autophagosomal membrane. Nat. Cell Biol. 12, 831–835 10.1038/ncb0910-831 [DOI] [PubMed] [Google Scholar]
- 3. Kraft C., and Martens S. (2012) Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 24, 496–501 10.1016/j.ceb.2012.05.001 [DOI] [PubMed] [Google Scholar]
- 4. Lamb C. A., Yoshimori T., and Tooze S. A. (2013) The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 14, 759–774 10.1038/nrm3696 [DOI] [PubMed] [Google Scholar]
- 5. Mortimore G. E., and Schworer C. M. (1977) Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 270, 174–176 10.1038/270174a0 [DOI] [PubMed] [Google Scholar]
- 6. Klionsky D. J., and Emr S. D. (2000) Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levine B., and Klionsky D. J. (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477 10.1016/S1534-5807(04)00099-1 [DOI] [PubMed] [Google Scholar]
- 8. Jewell J. L., Russell R. C., and Guan K.-L. (2013) Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 14, 133–139 10.1038/nrm3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shibutani S. T., and Yoshimori T. (2014) A current perspective of autophagosome biogenesis. Cell Res. 24, 58–68 10.1038/cr.2013.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kirisako T., Baba M., Ishihara N., Miyazawa K., Ohsumi M., Yoshimori T., Noda T., and Ohsumi Y. (1999) Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 147, 435–446 10.1083/jcb.147.2.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levine B., and Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Z. J., Chee C. E., Huang S., and Sinicrope F. A. (2011) The role of autophagy in cancer: therapeutic implications. Mol. Cancer Ther. 10, 1533–1541 10.1158/1535-7163.MCT-11-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bishop E., and Bradshaw T. D. (2018) Autophagy modulation: a prudent approach in cancer treatment? Cancer Chemother. Pharmacol. 82, 913–922 10.1007/s00280-018-3669-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xie Z., Nair U., and Klionsky D. J. (2008) Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 19, 3290–3298 10.1091/mbc.e07-12-1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakatogawa H., Ichimura Y., and Ohsumi Y. (2007) Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178 10.1016/j.cell.2007.05.021 [DOI] [PubMed] [Google Scholar]
- 16. Lynch-Day M. A., and Klionsky D. J. (2010) The Cvt pathway as a model for selective autophagy. FEBS Lett. 584, 1359–1366 10.1016/j.febslet.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sawa-Makarska J., Abert C., Romanov J., Zens B., Ibiricu I., and Martens S. (2014) Cargo binding to Atg19 unmasks additional Atg8 binding sites to mediate membrane-cargo apposition during selective autophagy. Nat. Cell Biol. 16, 425–433 10.1038/ncb2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ichimura Y., Kirisako T., Takao T., Satomi Y., Shimonishi Y., Ishihara N., Mizushima N., Tanida I., Kominami E., Ohsumi M., Noda T., and Ohsumi Y. (2000) A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492 10.1038/35044114 [DOI] [PubMed] [Google Scholar]
- 19. Ohsumi Y. (2001) Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2, 211–216 10.1038/35056522 [DOI] [PubMed] [Google Scholar]
- 20. Klionsky D. J., and Schulman B. A. (2014) Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 21, 336–345 10.1038/nsmb.2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kabeya Y., Mizushima N., Yamamoto A., Oshitani-Okamoto S., Ohsumi Y., and Yoshimori T. (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117, 2805–2812 10.1242/jcs.01131 [DOI] [PubMed] [Google Scholar]
- 22. Kirisako T., Ichimura Y., Okada H., Kabeya Y., Mizushima N., Yoshimori T., Ohsumi M., Takao T., Noda T., and Ohsumi Y. (2000) The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 151, 263–275 10.1083/jcb.151.2.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., and Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumanomidou T., Mizushima T., Komatsu M., Suzuki A., Tanida I., Sou Y., Ueno T., Kominami E., Tanaka K., and Yamane T. (2006) The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J. Mol. Biol. 355, 612–618 10.1016/j.jmb.2005.11.018 [DOI] [PubMed] [Google Scholar]
- 25. Satoo K., Noda N. N., Kumeta H., Fujioka Y., Mizushima N., Ohsumi Y., and Inagaki F. (2009) The structure of Atg4B–LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 28, 1341–1350 10.1038/emboj.2009.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ichimura Y., Imamura Y., Emoto K., Umeda M., Noda T., and Ohsumi Y. (2004) In vivo and in vitro reconstitution of Atg8 conjugation essential for autophagy. J. Biol. Chem. 279, 40584–40592 10.1074/jbc.M405860200 [DOI] [PubMed] [Google Scholar]
- 27. Yu Z., Ni T., Hong B., Wang H., Jiang F., Zou S., Chen Y., Zheng X., Klionsky D. J., Liang Y., and Xie Z. (2012) Dual roles of Atg8–PE deconjugation by Atg4 in autophagy. Autophagy. 8, 883–892 10.4161/auto.19652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakatogawa H., Ishii J., Asai E., and Ohsumi Y. (2012) Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy 8, 177–178 10.4161/auto.8.2.18373 [DOI] [PubMed] [Google Scholar]
- 29. Kauffman K. J., Yu S., Jin J., Mugo B., Nguyen N., O'Brien A., Nag S., Lystad A. H., and Melia T. J. (2018) Delipidation of mammalian Atg8-family proteins by each of the four ATG4 proteases. Autophagy 14, 992–1010 10.1080/15548627.2018.1437341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Agrotis A., Pengo N., Burden J. J., and Ketteler R. (2019) Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 15, 976–997 10.1080/15548627.2019.1569925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li M., Hou Y., Wang J., Chen X., Shao Z.-M., and Yin X.-M. (2011) Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J. Biol. Chem. 286, 7327–7338 10.1074/jbc.M110.199059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nguyen T. N., Padman B. S., Usher J., Oorschot V., Ramm G., and Lazarou M. (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome–lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 215, 857–874 10.1083/jcb.201607039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nishida Y., Arakawa S., Fujitani K., Yamaguchi H., Mizuta T., Kanaseki T., Komatsu M., Otsu K., Tsujimoto Y., and Shimizu S. (2009) Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658 10.1038/nature08455 [DOI] [PubMed] [Google Scholar]
- 34. Ma T., Li J., Xu Y., Yu C., Xu T., Wang H., Liu K., Cao N., Nie B-M., Zhu S-y., Xu S., Li K., Wei W-G., Wu Y., Guan K-L., et al. (2015) Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat. Cell Biol. 17, 1379–1387 10.1038/ncb3256 [DOI] [PubMed] [Google Scholar]
- 35. Choy A., Dancourt J., Mugo B., O'Connor T. J., Isberg R. R., Melia T. J., and Roy C. R. (2012) The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338, 1072–1077 10.1126/science.1227026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lystad A. H., Carlsson S. R., Ballina L. R., De, Kauffman K. J., Nag S., Yoshimori T., Melia T. J., and Simonsen A. (2019) Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat. Cell Biol. 21, 372–383 10.1038/s41556-019-0274-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pattingre S., Petiot A., and Codogno P. (2004) Analyses of Gα-interacting protein and activator of G-protein–signaling-3 functions in macroautophagy. Methods Enzymol. 390, 17–31 10.1016/S0076-6879(04)90002-X [DOI] [PubMed] [Google Scholar]
- 38. Bauvy C., Meijer A. J., and Codogno P. (2009) Assaying of autophagic protein degradation. Methods Enzymol. 452, 47–61 10.1016/S0076-6879(08)03604-5 [DOI] [PubMed] [Google Scholar]
- 39. Wu Y.-T., Tan H.-L., Shui G., Bauvy C., Huang Q., Wenk M. R., Ong C.-N., Codogno P., and Shen H.-M. (2010) Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 285, 10850–10861 10.1074/jbc.M109.080796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dupont N., Leroy C., Hamaï A., and Codogno P. (2017) Long-lived protein degradation during autophagy. Methods Enzymol. 588, 31–40 10.1016/bs.mie.2016.09.074 [DOI] [PubMed] [Google Scholar]
- 41. Szalai P., Hagen L. K., Sætre F., Luhr M., Sponheim M., Øverbye A., Mills I. G., Seglen P. O., and Engedal N. (2015) Autophagic bulk sequestration of cytosolic cargo is independent of LC3, but requires GABARAPs. Exp. Cell Res. 333, 21–38 10.1016/j.yexcr.2015.02.003 [DOI] [PubMed] [Google Scholar]
- 42. Musiwaro P., Smith M., Manifava M., Walker S. A., Ktistakis N. T., Musiwaro P., Smith M., Manifava M., Walker S. A., Musiwaro P., Smith M., Manifava M., Walker S. A., and Ktistakis N. T. (2013) Characteristics and requirements of basal autophagy in HEK 293 cells. Autophagy 9, 1407–1417 10.4161/auto.25455 [DOI] [PubMed] [Google Scholar]
- 43. Weidberg H., Shvets E., Shpilka T., Shimron F., Shinder V., and Elazar Z. (2010) LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 29, 1792–1802 10.1038/emboj.2010.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pengo N., Prak K., Costa J. R., Luft C., Agrotis A., Freeman J., Gewinner C. A., Chan A. W. E., Selwood D. L., Kriston-Vizi J., and Ketteler R. (2018) Identification of kinases and phosphatases that regulate ATG4B activity by siRNA and small molecule screening in cells. Front. Cell Dev. Biol. 6, 148 10.3389/fcell.2018.00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang T., Kim C. K., Alvarez A. A., Pangeni R. P., Wan X., Song X., Shi T., Yang Y., Sastry N., Horbinski C. M., Lu S., Stupp R., Kessler J. A., Nishikawa R., Nakano I., et al. (2017) MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 32, 840–855 10.1016/j.ccell.2017.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang Z., Wilkie-Grantham R. P., Yanagi T., Shu C.-W., Matsuzawa S.-I., and Reed J. C. (2015) ATG4B (Autophagin-1) phosphorylation modulates autophagy. J. Biol. Chem. 290, 26549–26561 10.1074/jbc.M115.658088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rasmussen M. S., Mouilleron S., Shrestha B. K., Wirth M., Lee R., Larsen K. B., Abudu Y., Reilly N. O., Sjøttem E., Tooze S. A., Lamark T., Wirth M., Lee R., Larsen K. B., Princely Y. A., et al. (2017) ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy 13, 834–853 10.1080/15548627.2017.1287651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Velikkakath A. K. G., Nishimura T., Oita E., Ishihara N., and Mizushima N. (2012) Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 23, 896–909 10.1091/mbc.E11-09-0785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Valverde D. P., Yu S., Boggavarapu V., Kumar N., Lees J. A., Walz T., Reinisch K. M., and Melia T. J. (2019) ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 218, 1787–1798 10.1083/jcb.201811139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Strømhaug P. E., Berg T. O., Fengsrud M., and Seglen P. O. (1998) Purification and characterization of autophagosomes from rat hepatocytes. Biochem. J. 335, 217–224 10.1042/bj3350217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li M., Chen X., Ye Q.-Z., Vogt A., and Yin X.-M. (2012) A high-throughput FRET-based assay for determination of Atg4 activity. Autophagy 8, 401–412 10.4161/auto.18777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mariño G., Fernández A. F., Cabrera S., Lundberg Y. W., Cabanillas R., Rodríguez F., Salvador-Montoliu N., Vega J. A., Germanà A., Fueyo A., Freije J. M., and López-Otín C. (2010) Autophagy is essential for mouse sense of balance. J. Clin. Invest. 120, 2331–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nair U., Yen W., Mari M., Cao Y., Xie Z., Baba M., Reggiori F., and Klionsky D. J. (2012) A role for Atg8–PE deconjugation in autophagosome biogenesis. Autophagy 8, 780–793 10.4161/auto.19385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang L., Li J., Ouyang L., Liu B., and Cheng Y. (2016) Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 373, 19–26 10.1016/j.canlet.2016.01.022 [DOI] [PubMed] [Google Scholar]
- 55. Fu Y., Huang Z., Hong L., Lu J.-H., Feng D., Yin X.-M., and Li M. (2019) Targeting ATG4 in cancer therapy. Cancers (Basel) 11, 649 10.3390/cancers11050649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Allen G. F. G., Toth R., James J., and Ganley I. G. (2013) Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 14, 1127–1135 10.1038/embor.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cann G. M., Guignabert C., Ying L., Deshpande N., Bekker J. M., Wang L., Zhou B., and Rabinovitch M. (2008) Developmental expression of LC3α and β: absence of fibronectin or autophagy phenotype in LC3β knockout mice. Dev. Dyn. 237, 187–195 10.1002/dvdy.21392 [DOI] [PubMed] [Google Scholar]
- 58. O'Sullivan G. A., Kneussel M., Elazar Z., and Betz H. (2005) GABARAP is not essential for GABA receptor targeting to the synapse. Eur. J. Neurosci. 22, 2644–2648 10.1111/j.1460-9568.2005.04448.x [DOI] [PubMed] [Google Scholar]
- 59. Shirakabe A., Zhai P., Ikeda Y., Saito T., Maejima Y., Hsu C.-P., Nomura M., Egashira K., Levine B., and Sadoshima J. (2016) Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 133, 1249–1263 10.1161/CIRCULATIONAHA.115.020502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kuma A., Komatsu M., and Mizushima N. (2017) Autophagy-monitoring and autophagy-deficient mice. Autophagy 13, 1619–1628 10.1080/15548627.2017.1343770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nath S., Dancourt J., Shteyn V., Puente G., Fong W. M., Nag S., Bewersdorf J., Yamamoto A., Antonny B., and Melia T. J. (2014) Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat. Cell Biol. 16, 415–424 10.1038/ncb2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lavieu G., Scarlatti F., Sala G., Carpentier S., Levade T., Ghidoni R., Botti J., and Codogno P. (2006) Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J. Biol. Chem. 281, 8518–8527 10.1074/jbc.M506182200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are available at reasonable request by contacting Dr. Thomas Melia (thomas.melia@yale.edu) from Yale University. All remaining data are contained within the article.