

SUMMARY
Cellular responses to stimuli can evolve over time, resulting in distinct early and late phases in response to a single signal. DNA damage induces a complex response that is largely orchestrated by the transcription factor p53, whose dynamics influence whether a damaged cell will arrest and repair the damage or will initiate cell death. How p53 responses and cellular outcomes evolve in the presence of continuous DNA damage remains unknown. Here, we have found that a subset of cells switches from oscillating to sustained p53 dynamics several days after undergoing damage. The switch results from cell cycle progression in the presence of damaged DNA, which activates the caspase-2-PIDDosome, a complex that stabilizes p53 by inactivating its negative regulator MDM2. This work defines a molecular pathway that is activated if the canonical checkpoints fail to halt mitosis in the presence of damaged DNA.
In Brief
Tsabar et al. show that a subset of cells switches from oscillatory to sustained p53 dynamics more than 24 h after irradiation-induced DNA damage. Switching is maximal at intermediate radiation doses, requires escape from irradiation-induced cell cycle arrest, and is facilitated by caspase-2-PIDDosome-mediated degradation of p53’s inhibitor MDM2.
Graphical Abstract
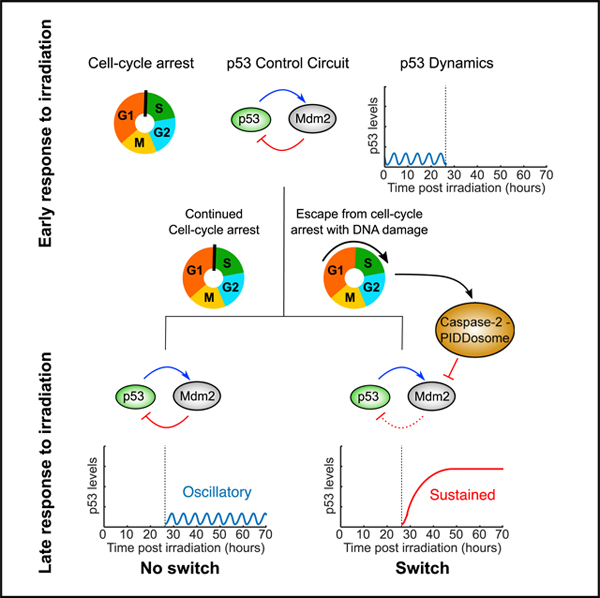
INTRODUCTION
In response to internal or external stimuli, cells execute a coordinated set of responses to maintain homeostasis. The dynamics of signaling pathways have recently been shown to play an important role in executing appropriate responses across many systems and organisms (Purvis and Lahav, 2013). However, in most systems, signaling dynamics are studied over periods of time that exhibit a single dynamical pattern (e.g., oscillations) or allow steady state to be reached. The evolution of dynamical responses to signals, how they change over long periods of time, and the molecular mechanisms underlying these changes remain poorly understood. Here, we investigated how the response to persistent DNA damage evolves over time and determined the cellular events and molecular mechanisms leading to a switch in the dynamics of the tumor suppressor protein p53, a key regulator of the response to DNA damage.
The DNA damage response (DDR) is activated in response to a variety of DNA lesions, such as crosslinks or breaks, and is orchestrated by the transcription factor (TF) p53. Within the first few hours of inducing DNA double-strand breaks (DSBs), p53 activates several hundred genes including genes that facilitate seemingly opposing fates such as cell survival via cell cycle arrest and DNA repair, cellular senescence, and apoptosis (Hafner et al., 2017; Kannan et al., 2001; Madden et al., 1997; Mirza et al., 2003). The p53 target gene critical for mediating cell cycle arrest is CDKN1A, which encodes the cyclin-dependent kinase inhibitor p21 (Brugarolas et al., 1995; el-Deiry et al., 1993; Harper et al., 1993). Cell cycle arrest promotes long-term survival by preventing division in the presence of damaged DNA, which could result in mitotic catastrophe and massive chromosome missegregation, leading to cell death or oncogenesis (Deckbar et al., 2011; Paulovich et al., 1997). Following repair, p53 levels, and subsequently p21 levels, decrease, allowing cell cycle progression.
The p53 response is shaped by multiple regulatory mechanisms. Under non-stressed conditions, p53 levels are kept low by mouse double minute 2 (MDM2), an E3 ubiquitin ligase (Haupt et al., 1997; Kubbutat et al., 1997; Momand et al., 1992; Oliner et al., 1992). Following DNA damage, PI3-like kinases, such as ataxia telangiectasia mutated (ATM) or ataxia telangiectasia and Rad3-related (ATR) (Ciccia and Elledge, 2010; Zhou and Elledge, 2000), stabilize p53 and inhibit MDM2 by phosphorylating each protein (Banin et al., 1998; Canman et al., 1998; Cheng and Chen, 2010; Khosravi et al., 1999; Shieh et al., 1997), and they also act on other targets that promote DNA repair. Phosphorylation of MDM2 and p53 disrupts their interaction, leading to p53 stabilization and elevation of p53 protein levels.
The dynamics of p53 protein levels have also emerged as key regulators of the response to DNA damage. The dynamics are largely controlled by MDM2, which not only degrades p53, but is also transcriptionally activated by it (Haupt et al., 1997; Honda et al., 1997; Kubbutat et al., 1997; Wu et al., 1993). This negative-feedback loop cooperates with other aspects of p53 regulation, resulting in pulsatile dynamics following damage that activates ATM (such as ionizing irradiation) and a single broad pulse following damage that activates ATR (such as UV) (Batchelor et al., 2011; Figure 1A). Pulsatile p53 dynamics are associated with cell survival and repair of DNA damage, whereas sustained p53 dynamics result in senescence and cell death (Purvis et al., 2012). The different outcomes result from different patterns of mRNA accumulation, depending on p53 dynamics, due to differences in mRNA half-lives of p53 target genes (Hafner et al., 2017; Paek et al., 2016; Porter et al., 2016). p53 also regulates target genes involved in positive-feedback loops, such as PIDD1. Induction of PIDD1 by p53 promotes the formation of the caspase-2-PIDDosome, leading to MDM2 cleavage and inhibition followed by p53 stability (Oliver et al., 2011). How PIDD1 controls p53 dynamics is presently unknown.
Figure 1. p53 Protein Levels Exhibit a Late-Phase Increase Following Irradiation.

(A) Schematic of p53 dynamics following ionizing irradiation or UV treatment.
(B) p53 protein levels at the indicated time points following 10 Gy ionizing irradiation. Tubulin is shown as a loading control.
(C) Schematic of possible p53 dynamics in single cells. All cells initiate a p53 response in a synchronous manner but later lose synchrony, resulting in signal decay as measured in population assays.
(D) p53 levels up to 72 h following 10 Gy ionizing irradiation. Actin is shown as a loading control.
We have previously shown that the initial response to irradiation-induced DSBs results in oscillations in p53 levels (Lahav et al., 2004; Purvis et al., 2012). Recently, we found that a subset of cells undergoes divisions at late time points after DNA damage (Reyes et al., 2018), suggesting that unique molecular events may be occurring at this stage. Here, we followed the evolution of the p53 response over multiple days of DNA damage signaling and the molecular mechanisms underlying these decisions. Our work reveals a PIDD1-dependent stabilization of p53 in cells that escape from DSB-induced arrest and undergo cell division. We suggest that this mechanism prevents cells from undergoing subsequent divisions in the presence of DNA damage.
RESULTS
p53 Protein Levels Exhibit a Late-Phase Increase Following Irradiation
Radiation-induced DSBs trigger a series of undamped p53 oscillations with a frequency of approximately 5.5 h that lasts for at least 24 h (Lahav et al., 2004; Purvis et al., 2012). The responses of p53 and its regulators several days post-radiation remained unknown. We irradiated human breast cancer cells expressing wild-type (WT) p53 and first confirmed the previously described p53 oscillations (Figure 1B). Oscillations in p53 levels appeared dampened when assayed by western blotting due to a loss of synchrony between p53 pulses in individual cells (Batchelor et al., 2008; Lahav et al., 2004; Purvis et al., 2012) (Figure 1C). Continuous loss of synchrony between cells is predicted to result in a steady state that is lower than the maximum amplitude when assessing p53 levels by population measurements. Indeed, we found that the p53 protein levels were lower 24 h post-irradiation than they were 2 h post-irradiation (Figure 1D). However, when we probed p53 levels beyond 24 h (“late-phase response”), we observed a substantial change: p53 protein levels rose at 48 h to a level comparable with that of the first peak, and they remained elevated up to 72 h following irradiation (Figure 1D). This result suggests that p53 dynamics may switch from an oscillatory behavior into elevated levels in the late-phase response to irradiation.
Only a Subset of Cells Switches p53 Protein Levels from Oscillatory to Elevated
The increase in p53 levels as detected in a western blot could be a result of a moderate increase in all cells or a more significant increase in a subset of cells. To examine the penetrance of the late-phase p53 increase, we used live-cell imaging and monitored individual cells expressing p53 tagged with a Venus fluorescent reporter for 72 h post-irradiation. As was previously shown (Batchelor et al., 2008; Lahav et al., 2004), during the first 24 h, p53 exhibited a pulsatile dynamic with a gradual loss of synchrony between cells (Figure 2A). Subsequently, individual cells showed differing patterns of p53 behavior: the majority of cells showed a decrease in p53 levels or continuous oscillations, while only a small subset of cells (~10%) exhibited an increase in p53 levels (Figures 2A and 2B). This fr"action of cells, which we termed “switchers,” dominated the p53 levels observed in western blots (Figure 1D), masking the behavior of the rest of the population. Note that switchers were also observed in two additional lines—in the lung cancer line A549 and in non-transformed RPE1 cells (Figures S1A and S1B)—suggesting that late-phase p53 elevation does not result from unique genetic or epigenetic features of MCF7 cells.
Figure 2. Only a Subset of Cells Switches p53 Protein Levels from Oscillatory to Elevated.

(A) Heatmap of scaled p53-Venus fluorescence intensity in single cells following 10 Gy irradiation. Cells were tracked for 72 h (x axis) and sorted by mean p53 intensity (y axis). Each row on the y axis represents a single cell. Fluorescence intensity is color coded from blue (low intensity) to yellow (high intensity). Red dotted rectangle highlights the subset of cells that switches from low to high p53 intensity.
(B) Example p53 trajectories of oscillating and switching cells.
(C) Heatmaps of scaled p53-Venus fluorescence intensity in single cells following increasing doses of irradiation, plotted as described in (A).
(D) Percent of cells that exhibit p53 switching as determined by k-means clustering. All doses were clustered together (approximately 400 cells per dose; see C), and only cells with 95% confidence were counted (see Materials Availability and Method Details). Error bars represent standard error of the proportions.
See also Figure S1.
We next tested if the fraction of switchers depends on the amount of DNA damage. It was previously shown that the number of irradiation-induced DSBs is linearly correlated with the radiation dose (Rothkamm and Löbrich, 2003). We therefore irradiated cells with increasing doses and monitored p53 levels using live-cell imaging (Figure 2C). The frequency and amplitude of oscillations in the early phase were independent of radiation dose (Figure 2C), as previously reported (Lahav et al., 2004). However, the fraction of switchers depended on radiation dose in a non-monotonic manner: the proportion of switchers increased with dose up to 5 Gy and then decreased at higher doses (Figure 2C). Notably, no cell death was observed in response to irradiation across all doses tested here; therefore, the decrease in the fraction of switchers at higher irradiation doses cannot be attributed to radiation-induced cell death. To further characterize and quantify the fraction of switchers and oscillating cells in an unbiased way, we used k-means clustering to group cells into switcher and non-switcher categories. This clustering strategy resulted in no overlap between the first and third quartiles of the two populations (Figure S1C). Importantly, the fraction of switchers determined by k-means clustering was also highest at intermediate doses of irradiation and decreased at higher doses (Figure 2D). Applying the same clustering approach to the non-cancerous RPE1 cells uncovered a similar non-monotonic dose response (Figure S1D). Taken together, these observations show that the late-phase p53 increase occurs only in a subset of cells that switches from oscillatory to elevated p53 levels, and the fraction of switchers is dose dependent, with maximum switching occurring at an intermediate dose of irradiation.
Failure to Maintain DNA-Damage-Induced Arrest Leads to the Switch in p53 Dynamics
We next sought to determine the cellular events that lead to a switch in p53 dynamics. We noted that the frequency and number of micronuclei were greater in switchers compared to non-switchers (Figures 3A and S2A). Micronuclei are a hallmark of irradiation treatment and are thought to arise when mitosis occurs prior to completion of repair (Fenech et al., 2011; Huang et al., 2011). At 5 Gy, over 91% of switchers contained micronuclei at the end of the time course, whereas only 69% of the non-switchers showed micronuclei (Figure S2A). Moreover, when we quantified the number of micronuclei in individual dividing cells, we found that switchers had significantly larger numbers of micronuclei than non-switchers did (Figure S2B). Taken together, these results suggest that switchers might result from division in the presence of DNA damage. Indeed, recently, we have shown that cells can initiate division following irradiation, despite failing to repair the irradiation-inflicted DNA damage (Reyes et al., 2018). These studies were conducted in RPE1 cells, which show a switching response similar to MCF7 cells (Figures S1B and S1D). We therefore sought to determine whether progression through the cell cycle affects the switch in p53 dynamics by monitoring the number and timing of mitoses in our time-lapse movies. Similar to previous studies (Reyes et al., 2018), the number of mitotic events per cell, as well as the number of cells that had undergone mitosis, decreased with increasing radiation dose (Figures S2C–S2E). Linking divisions with p53 dynamics in MCF7 and RPE1 cells, we observed that non-switchers displayed cell cycle behavior that depended on the dose of irradiation. At low doses, many non-switchers executed multiple cell divisions, whereas at higher doses, they largely remained arrested (Figures 3B and S2D–S2F). These results could arise if the non-switchers that divided at lower doses repaired their DNA and resumed normal proliferation, whereas at higher doses, they fail to completely repair the DNA damage, thus remaining arrested in G1 throughout the time course. By contrast, at high and low doses, switchers in MCF7 and RPE1 lines tended to divide just once (e.g., over 75% of switchers divided once at 10 Gy) (Figures 3B, S2D, right panel, and S2F). We further found that mitosis preceded switching by ~8 h with a high correlation when calculated across all doses (Figures 3C, S1C, and S2G; r = 0.71, p = 2.4173E–31), suggesting that progression through the cell cycle following irradiation might lead to the switch in p53 dynamics. Because most cells arrest in G1 after irradiation, these studies cannot distinguish whether progression through S and/ or M phases is required.
Figure 3. Failure to Maintain DNA-Damage-Induced Arrest Leads to the Switch in p53 Dynamics.

(A) Representative cells showing non-switching (top) and switching (middle and bottom) behavior following 10 Gy irradiation. Middle and bottom panels show the same cells with (middle) and without (bottom) linear signal enhancement. p53-Venus intensity is shown in yellow, and white arrows mark the tracked cells.
(B) Distribution of the number of divisions undergone by switching and non-switching cells during the 72-h time course following 10 Gy irradiation.
(C) Scatterplot of the time of division (y axis) versus the time of switching (x axis). Only switching cells that divided were plotted (r = 0.71; p = 2.4173E – 31).
(D) Heatmaps of scaled p53-Venus fluorescence intensity in cells subjected to 10 Gy irradiation with (right panel) or without (left panel) ribociclib (10 μM, 12 h prior to irradiation, right panel).
(E) Heatmaps of scaled p53-Venus fluorescence intensity in cells subjected to 10 Gy irradiation followed by treatment with control (left) or CDKN1A (right) siRNA 2 h after irradiation.
In (D) and (E), heatmaps are plotted as described in Figure 2A.
See also Figures S2 and S3.
To further examine the potential causality between cell cycle progression and the switch in p53 dynamics, we added the CDK4/CDK6 inhibitor ribociclib 12 h prior to irradiation to maintain cells in G1 (Kim et al., 2018) throughout the time course and tested the resultant p53 dynamics after irradiation. Ribociclib treatment abolished cell divisions throughout the time course (72 h; Figure S3A) and eliminated the population of cells showing a late-phase switch in p53 dynamics (Figure 3D) without affecting its oscillatory dynamics during the first 24 h post-irradiation (Figure 3D). Note that there were no cell divisions in the first 24 h in the presence or absence of ribociclib due to radiation-induced cell cycle arrest. We next performed the inverse experiment and tested if increasing the number of cells escaping arrest would increase the fraction of switchers. In response to DSBs, p53 imposes arrest mainly by upregulating the expression of CDKN1A, which encodes for the cyclin-dependent kinase inhibitor p21 (Brugarolas et al., 1995; Harper et al., 1993). Knockdown of CDKN1A is expected to alleviate the p53-dependent G1/S arrest and increase the number of dividing cells. Indeed, we found that knockdown of CDKN1A by small interfering RNA (siRNA) increased the frequency of division from 22% to 70% (Figures S3B and S3C), accompanied by an increase in switching (Figure 3E). Taken together, our results suggest that failure to maintain the G1/S arrest imposed by DSBs results in cell cycle progression that leads to a switch in p53 dynamics from oscillatory to elevated levels.
The Caspase-2-PIDDosome Contributes to Switching in p53 Dynamics
We next sought to determine the upstream molecular mechanisms that lead to the switch in p53 dynamics. ATM is the primary kinase that controls p53 activity in response to DNA DSBs (Batchelor et al., 2011; Shiloh and Ziv, 2013). Indeed, inhibition of ATM 1 h prior to irradiation completely abolished the early-phase increase of p53 levels and prevented p53-dependent upregulation of Mdm2 (Figure 4A). To investigate the potential role of ATM in regulating the switch in p53 dynamics during the late phase of the response, we irradiated cells and treated them with an ATM inhibitor (ATMi) 16 h following irradiation. We found that late inhibition of ATM did not alter the late-phase p53 increase, despite efficient inhibition of the kinase, as gauged by diminished CHK2 phosphorylation (Figure 4B). We also tested the role of another DNA damage checkpoint kinase, ATR, which was previously shown to dominate the p53 response to other types of damage, including that induced by UV (Batchelor et al., 2011). Inhibition of ATR did not prevent the late-phase p53 increase despite efficiently inhibiting the enzyme, as gauged by phosphorylation of CHK1 (Figure S4A). We conclude that the late-phase switch in p53 dynamics following irradiation does not require the continuous activity of the canonical DNA damage checkpoint kinases ATM and ATR.
Figure 4. The Caspase-2-PIDDosome Contributes to the Switch in p53 Dynamics.

(A) Western blot showing p53 and Mdm2 levels with or without ATM inhibitor (KU55933 10 μM) added 1 h prior to irradiation. Mdm2 levels represent p53 activity. Actin is shown as a loading control.
(B) Western blot showing p53 levels with or without ATM inhibition 16 h following irradiation. Chk2 phosphorylation is used to measure ATM activity. Actin is shown as a loading control.
(C) Schematic of the caspase-2-PIDDosome interaction with the p53-Mdm2 network following irradiation.
(D) p53 levels following CRISPR/Cas9 KO of PIDD1. Actin is shown as a loading control.
See also Figure S4.
The fact that the late-phase switch in p53 dynamics following radiation is independent of p53’s canonical upstream kinases prompted us to search for other proteins known to regulate p53 by controlling MDM2 stability. One such potential regulator is the PIDDosome, a protein complex comprising the p53-induced death domain (PIDD1) and the receptor interacting protein-associated ICH-1/CED-3 homologous protein with a death domain (RAIDD). These proteins can complex with one of several additional binding partners, including caspase-2 (Baliga et al., 2004; Berube et al., 2005; Janssens and Tinel, 2012; Tinel and Tschopp, 2004), which plays a central role in maintaining genomic integrity and promoting mitotic catastrophe (Dawar et al., 2017; Dorstyn et al., 2012; Fava et al., 2017; Sladky et al., 2020). PIDD1 and RAIDD act as an activation platform to caspase-2, forming a complex termed the caspase-2-PIDDosome, which assembles following genotoxic stress (Ando et al., 2012, 2017; Fava et al., 2017; Thompson et al., 2015; Tinel and Tschopp, 2004). The caspase-2-PIDDosome complex was shown to stabilize p53 by cleaving and inhibiting MDM2 (Fava et al., 2017; Lim et al., 2018; Oliver et al., 2011; Terry et al., 2015; Figure 4C). The ability of the PIDDosome to regulate p53 in response to chromosomal abnormalities makes the complex an attractive candidate for facilitating the switch in p53 dynamics. To test the role of the caspase-2-PIDDosome in p53 switching, we generated a PIDD1 deletion by CRISPR/Cas9. PIDD1 knockout (KO) led to inhibition of MDM2 cleavage and a strong reduction in p53 levels during the late phase of the response without affecting p53 dynamics during the early phase (Figures 4D, S4B, and S4C), suggesting that PIDD1 is indeed required for the switch in p53 dynamics (Figure 4D). Similar results were obtained by siRNA-mediated knockdown of PIDD1 (Figures S4D and S4E).
We further tested the role of PIDD1 on p53 in single cells by quantifying the dynamics of the p53 fluorescent reporter in irradiated cells with and without PIDD1. PIDD1 KO cells showed a decreased percentage of switchers post-radiation (Figures 5A and 5B). Of note, PIDD1 deletion had only a minimal effect on the ability of the cells to undergo mitosis following irradiation (Figure 5C) and did not significantly affect the number of micronuclei formed following mitosis (Figure S4F), suggesting that PIDD1 acts downstream of mitosis to induce switching in p53 dynamics.
Figure 5. Manipulating the Caspase-2-PIDDosome Alters Switching.

(A) Heatmaps of scaled p53 intensity in the PIDD1 KO cell line (right panel) and control (left panel).
(B) Percent of switchers in control (n = 418 cells) and PIDD1 KO (n = 454 cells) populations as determined by k-means clustering.
(C) Percent of dividing cells in control (n = 418 cells) and PIDD1 KO populations (n = 454).
(D) Heatmaps of scaled p53 intensity following caspase-2 inhibition with 100 μM NH-23-C2 (right panel) or DMSO (control, left panel).
(E) Percent of switchers in control (n = 203 cells) and NH-23-C2 (n = 210 cells) populations as determined by k-means clustering.
(F) Percent of dividing cells in control (n = 203 cells) and NH-23-C2-treated (Casp2i) populations (n = 210).
(G) Heatmaps of scaled p53 intensities in PIDD1-overexpressing cells. Left panel is the uninduced control. In the right panel, PIDD1 was induced with doxycycline addition 24 h following irradiation (white dashed line).
(H) Percent of switchers in the control (n = 253 cells) and PIDD1-overexpressing cells (n = 153 cells) as determined by k-means clustering.
(I) Percent of cells that divided in the control (n = 253 cells) and PIDD1-overexpressing cells (n = 153 cells).
(B, C, E, F, H, I) Error bars represent standard error of the proportions.
See also Figure S4.
We next tested the role of caspase-2 in promoting p53 dynamics switching. Inhibition of caspase-2 with inhibitor Z-VDVAD-FMK showed a dose-dependent inhibition of MDM2 cleavage and, accordingly, a decrease in p53 levels 48 h following irradiation (Figure S4G), as observed for PIDD1-KO (Figure 4D). Live-cell fluorescence imaging in the presence of inhibitor NH-23-C2 (Poreba et al., 2019), which is more specific for caspase-2 and does not exhibit the auto-fluorescence of Z-VDVAD-FMK, revealed diminished p53 switching without affecting the oscillatory phase (Figures 5D and 5E) and only minimally reducing the frequency of mitosis (Figure 5F). The similar effects of PIDD1 and caspase-2 on MDM2 cleavage and p53 levels support the suggestion that the caspase-2-PIDDosome is indeed required for cleaving MDM2, thus promoting the switch in p53 dynamics.
To further investigate the causal relationship between PIDD1 and the switch in p53 dynamics, we artificially increased PIDD1 levels at 24 h following irradiation using a doxycycline-inducible PIDD1 (Figure S4H) and followed p53 dynamics in single cells. Importantly, we found that artificial elevation of PIDD1 increased the fraction of switchers post-irradiation (Figures 5G and 5H). Moreover, we noted that overexpression of PIDD1 decoupled switching from division, as PIDD1 overexpression led to a decrease in the fraction of dividing cells (Figures 5I and S4I), as was previously observed by others (Lin et al., 2000; Oliver et al., 2011). Taken together, these observations suggest that following division of damaged cells, the caspase-2-PIDDosome leads to a switch in p53 dynamics from oscillatory to sustained. Since sustained p53 levels were previously associated with the activation of terminal fates (Purvis et al., 2012), we suggest that the switch in p53 dynamics observed here serves to prevent subsequent divisions of damaged cells during the late phase response to irradiation.
Modeling Suggests That Enhanced MDM2 Degradation by the Caspase-2-PIDDosome Is Sufficient to Explain p53 Switch after Mitosis
Our results suggested that PIDDosome activity is necessary for p53 elevation in switchers, but they did not address its sufficiency for causing this phenomenon. It remained possible that other events linked to cell division—such as changes in cell cycle regulators or acquisition of additional DNA damage during cell cycle progression—might also be necessary for p53 elevation. To explore this computationally, we implemented a minimal, validated model of p53 dynamics and asked whether degradation of MDM2 upon mitosis (e.g., by the PID-Dosome) would be sufficient to recapitulate the switching dynamics that we observed experimentally. We used a time-delay model of the p53-MDM2 feedback loop, recently published by
Heltberg et al. (2019), in which p53-induced transcription of MDM2 coupled with MDM2-mediated degradation of p53 yields p53 oscillations with a period and amplitude that match experimentally observed p53 dynamics. To this model, we added PIDDosome-dependent Mdm2 degradation after mitosis (below and Figure 6A):
where θ(t – tmitosis) is the Heaviside step function (stepping from 0 to 1 at the time of mitosis), and ε is the rate of MDM2 degradation by the PIDDosome. Values for all parameters are given in Table S1. We found that post-mitotic switching was recapitulated by this model (Figure 6B). Systematically varying ε showed that the magnitude of p53 levels in switchers was proportional to the rate of MDM2 degradation by the PIDDosome (Figure S5). This model predicts that degradation of MDM2 by the PIDDo-some upon mitosis can be sufficient to trigger a switch to a high p53 level.
Figure 6. Modeling of Caspase-2-PIDDosome Activity Captures the Switch in p53 Dynamics after Mitosis.

(A) Depiction of the minimal model used to computationally test for switching behavior following mitosis; equations governing this model are described in the text. The model is based on Heltberg et al. (2019), with the addition of PIDDosome-dependent degradation of Mdm2 following mitosis.
(B) Simulated p53 levels over time when running this computational model with the parameters in Table S1. A post-mitotic rise in p53 levels is observed when PIDDosome-dependent MDM2 degradation occurs after mitosis.
DISCUSSION
Cellular responses to a single signal can evolve over time, leading to distinct outcomes at early and late phases of the response. The study of p53 dynamics has unearthed different types of dynamics and their relationships to different cell fates, yet the evolution of the response over time has been poorly studied. By monitoring p53 dynamics for several days after DNA damage, we have shown that a subset of cells can switch from oscillatory to sustained dynamics. Switching shows a non-monotonic dose dependence on radiation levels: switching is maximal in MCF7 cells at 5 Gy and decreases at lower and higher levels of radiation. The heterogeneity in p53 dynamics and the non-monotonic response results from the ability of some cells to escape cell cycle arrest. Following cell division, the caspase-2-PIDDosome stabilizes p53. Modeling of the p53-PIDDosome network revealed that PIDDosome activation after mitosis is sufficient to elevate p53 levels. We propose that switching p53 dynamics serves a functional role in promoting arrest in the next cell cycle following escape from the DNA damage checkpoint. Of note, switching from oscillatory p53 to sustained p53 also occurs in the lung cancer A549 and non-cancerous RPE1 cell lines (Figures S1A and S1B). This suggests that switching is a general phenomenon, not specific only to MCF7 cells. It remains to be determined whether the same mechanism sustaining p53 in MCF7 cells also operates in these and other cell lines. However, since caspase-2-PIDDosome formation and function are well conserved across multiple cell lines, this complex likely regulates switching in other lines as well.
Although p53 levels in almost all cells initially oscillated in response to irradiation across all doses we tested, only a small subset of cells switched from oscillatory to sustained dynamics. Switchers corresponded to cells that had undergone division in the presence of DNA damage, as evidenced by micronuclei in daughter cells. Interestingly, Geva-Zatorsky et al. (2006) previously reported that in some MCF7 cells that divided following irradiation, Mdm2 dynamics (which can serve as a proxy for p53 dynamics) were oscillatory. However, these cells had divided soon after irradiation, which may indicate that they were already committed to division. In our study, most switchers divided at later time points after establishing prolonged oscillatory p53 dynamics, which indicates that they established, and escaped, a p53-dependent G1 arrest. Similarly, our previous work (Reyes et al., 2018) showed that a subset of cells that stochastically experiences low amplitudes of p53 oscillations following prolonged arrest is prone to dividing with damaged DNA, bypassing the canonical checkpoints that kept them arrested for several days. We hypothesize that switchers represent this class of cells post-division. The relatively low frequency of switching is likely due to the efficiency of the canonical DNA damage response, which largely suppresses cell division in the presence of damaged DNA.
The ability to undergo mitosis in the presence of DNA DSBs has been documented in several systems, yet the consequences in mammalian cells have not been well studied. The yeast Saccharomyces cerevisiae can undergo several divisions in the presence of a single irreparable DSB, a process termed adaptation (Lee et al., 1998; Toczyski et al., 1997), but it remains arrested in the presence of additional DSBs (Lee et al., 1998). In contrast, mammalian cells are able to escape the G1/S arrest with numerous DSBs (Lee et al., 1998; Reyes et al., 2018). Our results demonstrate that this escape results in a switch from oscillatory to sustained p53 dynamics. The change in p53 dynamics suggests that there is a signaling consequence to the failure to maintain the DSB-induced G1/S arrest. Pulsatile p53 dynamics are associated with survival of cells, while sustained dynamics are associated with terminal cell fates such as senescence and apoptosis; these correlations are independent of the extent of DNA damage, as they can be observed by pharmacologically manipulating p53 (Purvis et al., 2012). We found that most switchers divide only once during our time course (Figures 3B, S2D, right, and S2F). Interestingly, even at low irradiation doses that allow cell division, most RPE1 switchers divide only once during a 5-day time course, while non-switchers divide significantly more (Figures S2E and S2F). Given these observations, it is tempting to speculate that switching of p53 dynamics acts as a “fail-safe” mechanism for the DNA damage checkpoint, being activated when the canonical DNA damage checkpoint fails in order to promote arrest in the following cell cycle. Although switchers did not divide a second time during the course of our experiments, we could not establish their terminal long-term fate, since assays that determine terminal senescence (e.g., β-gal assays) are largely incompatible with live-cell imaging. Further studies of transcriptional programs activated in switchers and their ultimate cell fates will help elucidate the physiological outcomes of the p53 elevation in these cells.
There are a few ways by which switching in p53 dynamics could prevent further cell division. The first is a cell-intrinsic mechanism of promoting terminal cell fates such as death or senescence. The stabilization of p53 would result in rapid accumulation of p53-dependent factors that support terminal fates, as has been previously published (Purvis et al., 2012). Of note, the caspase-2-PIDDosome has been shown to promote p53-dependent cell death in response to genotoxic stress (Ando et al., 2012, 2017; Janssens and Tinel, 2012; Tinel and Tschopp, 2004). Supporting the cell-intrinsic model, we find that most switching cells fail to divide more than once (Figures 3B, S2D, and S2F). Nevertheless, a small fraction of switching cells continue dividing even after p53 has switched to sustained dynamics (compare Figures 2C and S2C). This raises the possibility of a cell-extrinsic pathway that eliminates the switching cells in vivo. Recent work has demonstrated that micronuclei trigger the innate immune response (Harding et al., 2017; Mackenzie et al., 2017; Vanpouille-Box et al., 2017). The abundance of micronuclei in switching cells (Figures 3A, S2A, and S2B) might indicate that these cells are eliminated by the immune system following switching.
The role of the caspase-2-PIDDosome in upregulating p53 following escape from DSB-induced arrest is consistent with our model that switching represents a fail-safe mechanism to prevent additional cell divisions after cells escape arrest in the presence of damaged DNA. Such a mechanism should be able to elicit a p53 response independently of continuous ATM-dependent p53 activation, which presumably failed to prevent cell cycle progression with unresolved DSBs. The PIDDosome has been shown to activate p53 in response to cytokinesis failure (Fava et al., 2017; Thompson et al., 2015), and it is therefore poised to integrate mitotic errors into a p53 response.
Our results suggest the existence of an ATM-independent mechanism of caspase-2-PIDDosome activation leading to p53 stabilization in switcher cells a few days after DNA damage. It is interesting to discuss how our findings fit with recent studies showing that ATM phosphorylates PIDD1 at Thr788 (the death domain), which facilitates its interaction with RAIDD, a prerequisite for caspase-2 activation (Ando et al., 2012; Terry et al., 2015). PIDD1 phosphorylation by ATM, and subsequent caspase-2 cleavage, occurred 24 h following treatment with a combination of γ-irradiation and Chk1 inhibition. γ-irradiation alone or γ-irradiation combined with Chk1 inhibition in p53 WT cells did not engage this pathway or activate caspase-2 (Sidi et al., 2008). Notably, the switching phenotype described in our work occurs following mitotic failures that take place at time points beyond 24 h. It is possible that this cellular event activates the caspase-2-PIDDosome independently of ATM. In addition, several lines of evidence suggest that ATM-mediated phosphorylation of PIDD1 may not be strictly necessary for caspase-2 cleavage. First, overexpression of p53 (Seth et al., 2005) or PIDD1 (Berube et al., 2005; Tinel and Tschopp, 2004) led to PIDD1-dependent caspase-2 activation in the absence of DNA damage and, therefore, presumably in the absence of ATM activity. Similarly, UV treatment, which primarily activates ATR rather than ATM (Batchelor et al., 2011), induced caspase-2 cleavage in various cell lines (O’Reilly et al., 2002). In addition, γ-irradiation alone led to caspase-2-dependent apoptosis in mouse embryonic fibroblasts (MEFs) (Ho et al., 2009), and H1299 lung cancer cells expressing WT p53 showed robust activation of the caspase 2-PIDDosome pathway following daunorubicin-induced DNA damage (Baptiste-Okoh et al., 2008). Finally, a crystal structure of a complex of the death domains of PIDD1 and RAIDD showed extensive interactions that were not dependent on ATM-mediated phosphorylation of PIDD1 (Park et al., 2007). These results support the existence of an ATM-independent PIDD1 activation mechanism and suggest that the requirements for ATM-mediated PIDD1 phosphorylation may depend on the cellular context and type of DNA damage.
Our data suggest that the optimum dose for switching is achieved by two opposing trends: (1) the DNA damage inflicted by irradiation that is positively correlated to dose and (2) the likelihood of dividing that is negatively correlated to dose. At low doses, cells can repair the DNA damage and resume a normal cell cycle, with p53 returning to basal levels. At very high doses, the extensive DNA damage likely triggers a stringent checkpoint that prevents mitosis. At intermediate doses, however, cells do not elicit a sufficiently robust checkpoint despite retaining DNA damage, resulting in progression through mitosis with damaged DNA. Interestingly, a recent study showed that irradiation elicits an abscopal effect when moderate doses are administered, but the effect diminished at higher doses (Vanpouille-Box et al., 2017). The abscopal effect was mediated by the irradiation-induced innate immune response. This study showed that the expression of the exonuclease TREX1 is induced by irradiation in a dose-dependent manner, and at high irradiation doses, TREX1 clears the cytoplasmic DNA, preventing the induction of the cyclic GMP-AMP synthase (cGAS)-STING pathway and subsequently reducing interferon-β secretion (Vanpouille-Box et al., 2017). Our study provides further insight into the differences in the activation of p53-dependent signaling at different irradiation doses. We propose that a moderate dose might be more effective in leading to failed mitosis attempts and accumulation of cytosolic DNA, thus activating p53 and perhaps other micronuclei-dependent pathways such as innate immune pathways to clear damaged cells and resulting in mitotic catastrophe.
In this study, we have uncovered that p53 dynamics in response to DSBs can switch from oscillatory to sustained following escape from cell cycle arrest, and this switch results from caspase-2-PIDDosome-dependent degradation of MDM2. Taken together with our knowledge that sustained p53 dynamics are associated with terminal outcomes in cells, the data presented here afford us potential new pathways to target in order to stimulate a sustained p53 response to halt the growth of damaged cancer cells.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Galit Lahav (galit@hms.harvard.edu).
Materials Availability
All unique reagents generated in this study are available upon request with Material Transfer Agreement.
Data and Code Availability
Custom MATLAB scripts are available upon request. The ModelSimulator code used to model p53 activation through degradation of MDM2 by the PIDDosome can be downloaded at https://github.com/MTsabar/PIDDosome/tree/PIDDosome
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell-lines and plasmids
MCF7 and A549 cells were grown in RPMI supplemented with 10% FBS, 100U/mL penicillin, 100mg/mL streptomycin and 250ng/mL fungizone (Gemini Bio-Products). RPE1 cells were grown in DMEM/F12 supplemented with 10% FBS, 100U/mL penicillin, 100mg/mL streptomycin and 250ng/mL fungizone (Gemini Bio-Products). Cells were grown in 37oC, 5% CO2 and 80% humidity.
Exogenous expression of p53 in MCF7 was achieved either by infecting MCF7 cells with lentivirus containing exogenous p53-Venus under either ubiquitin promoter (Stewart-Ornstein and Lahav, 2017), or by transfection of MCF7 cells with a p53-Venus under a metallothionine promoter (Purvis et al., 2012). Both p53-Venus constructs contained neomycin resistance gene. Cells were subsequently cloned and single cell clones were expanded and screened for marker expression.
Overexpression of PIDD1 was achieved by transfecting MCF7 cell-line containing the metallothionine induced p53-Venus with doxycycline inducible PIDD1 with a c-terminus ECFP fluorescent reporter.
Exogenous expression of p53 in RPE1 cells was achieved by infecting RPE1 cells containing a p21-mKate reporter with a lentivirus carrying the p53-NeonGreen reporter under ubiquitin promoter (Reyes et al., 2018). Cells were subsequently cloned and single cell clones were expanded and screened for marker expression.
Exogenous expression of p53 in A549 cells was achieved by infecting A549 cells with a lentivirus carrying the p53-NeonGreen reporter under ubiquitin promoter. Cells were subsequently cloned and single cell clones were expanded and screened for marker expression, and clones were then infected with a lentivirus containing H2B-mCherry reporter (Stewart-Ornstein and Lahav, 2017).
PIDD1 was knocked out by Crispr/CAS9 as described in Ran et al. (2013) and Sanjana et al. (2014). The PIDD1 sgRNA was introduced into lentiCRISPR v2 (a gift from Feng Zhang (Addgene plasmid # 52961; http://addgene.org/52961; RRID:Addgene_52961)). The constructed Cas9-PIDD1-sgRNA-puro vector was introduced into MCF7 cells containing metallothionine induced p53-Venus. Knockout of PIDD1 was verified by sequencing.
Oligonucleotides for constructing PIDD1 sgRNA:
Fwd: CACCGGCGGGAAACCACAAGGAACC
Rev: AAACGGTTCCTTGTGGTTTCCCGC
PIDD1 deletion was verified by sequencing the PIDD1 gene. One allele had 1 nt insertion 2400 nt from the start codon, the second allele had a 7 nt deletion 2395 nt from the start codon. Both mutations generate frameshifts on exon 9 (out of 16) of the gene. The mutations generate stop-codons on exon 9 directly downstream of the mutations. This abolishes the C terminus of the gene which contains the death domain and is essential for Caspase-2 – PIDDosome formation (Park et al., 2007).
PIDD1 exon9 WT
cggctctgggctcactgccaggtgccccacttctcctggttccttgtggtttcccgccctgtgtccaatgcctgcctggtgccaccggaggggacactgctgtgctcctcgggtc atcctggggtcaaagtcatcttcccccctggggccactgaggagcctcgtcgagtctccatgcag
PIDD1 exon9 mut1 (single nt insertion)
cggctctgggctcactgccaggtgccccacttctcctggTttccttgtggtttcccgccctgtgtccaatgcctgcctggtgccaccggaggggacactgctgtgctcctcggg tcatcctggggtcaaagtcatcttcccccctggggccactgaggagcctcgtcgagtctccatgcag
PIDD1 exon9 mut2 (seven nt deletion)
cggctctgggctcactgccaggtgccccacttctcctgg______tggtttcccgccctgtgtccaatgcctgcctggtgccaccggaggggacactgctgtgctcctcgggtcatcctggggtcaaagtcatcttcccccctggggccactgaggagcctcgtcgagtctccatgcag
METHOD DETAILS
Ionizing irradiation
Cells were treated with γ-irradiation in a 60CO irradiator or X-ray irradiation in a RS-2000 irradiator, both administer time-controlled doses of irradiation.
Western blots
Cells were harvested with RIPA buffer (Cold Springs Harbor protocols) containing protease and phosphatase inhibitors, and proteins were separated on a 4%–12% Bis-Tris gradient gels (Thermo-Fisher). and transferred to Pure Nitrocellulose Blotting Membrane (Life Sciences) or Amersham™ Hybond™ PVDF membrane (0.45 μM GE Healthcare Life Science). Membranes were blocked with 4% BSA, incubated with primary antibody overnight (1:1000–1:2000 dilution), washed, and incubated with secondary fluorescent conjugate anti-bodies or HRP, followed by a last wash. Membranes were scanned using LI-COR Odyssey CLX, Bio-rad Chemidoc™ or exposed to film.
Primary antibodies used are: Mouse monoclonal anti-p53 (Santa Cruz Biotechnology Cat# sc-126, RRID:AB_628082), mouse monoclonal anti-MDM2 (Ab-1, Millipore Cat# OP46T-10UG, RRID:AB_564805), rabbit anti-pChk2 (T68) (Cell Signaling Technology), rabbit anti-pChk1 (S317) (Cell Signaling Technology), mouse anti-tubulin (E7) (Developmental Studies Hybridoma Bank), mouse monoclonal anti-β-Actin (AC-74) (Sigma-Aldrich Cat# A2228, RRID:AB_476697). Info on Secondary antibodies?
RNAi of PIDD1 and CDKN1A
PIDD1 knockdown by siRNA was done by reverse transfection using RNAiMAX reagent (Thermo Fischer). In brief Optimem (500ul/sample) and siRNA (10mM, 1uL/sample) were mixed and put in wells. 5uL RNAiMax was added to each well and incubated for 30 min. 1.5 mL MCF7 cells were added to each well (150K cells) and incubated for 2 days prior to irradiation.
The PIDD1 siRNA sequences: rCrArGrArCrUrGrUrUrCrCrUrGrArCrCrUrCrArGrArUrU – sense rCrArGrArCrUrGrUrUrCrCrUr GrArCrCrUrCrArGrArUrU – antisense
Negative control for the siPIDD1 was purchased from Invitrogen
For CDKN1A knockdown we used siGenome Smart pool Human CDKN1A (Dharmacon™, Cat M-003471–00-0005 5nmol Lot 140909), control was Non-Targeting #2, Cat D-001206–14-05 5nmol Lot 1476981.
Inhibitors
ATM inhibitor (KU55933, Sigma) was used at a 10 μM concentration. ATR inhibitor (VE822, Cayman Chemical) was used at 50 nM concentration. Caspase-2 inhibitor Z-VDVAD-FMK (R&D systems) was used at the concentrations indicated in Figure S3D. Caspase-2 inhibitor NH-23-C2 was used at the concentration indicated in Figure 5D.
Live-cell microscopy
For microscopy, RPMI (for MCF7 and A549) or DMEM/F12 (for RPE1) without phenol-red was supplemented with 5% FBS and 100mg/mL streptomycin and 250ng/mL fungizone. 20K to 50K cells were seeded in a 35 mm poly-D-lysine-coated glass bottom plates (MatTek Corporation) 2 days prior to imaging. Cells were imaged using a Nikon Eclipse TE-2000 inverted microscope with a 20X Plan Apo objective and a Hammamatsu Orca ER camera, equipped with environmental chamber controlling temperature, atmosphere (5% CO2) and humidity. Images were acquired every 15 min using the MetaMorph Software.
Tracking and image-analysis was performed as previously published (Reyes et al., 2018).
Computational Modeling
We implemented the model of ordinary differential equations in MATLAB (MathWorks), as previously described by Batchelor et al. (2008) and Heltberg et al. (2019). The code used to implement this model is available for download in the online supplemental information.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell tracking
Cells were tracked using a semi-automated tracking method, as described in Reyes et al. Software is available at github (see data and software availability)
Quantifying switching cells by k-means Clustering
To determine if a cell switched we used bootstrapping approach combined with k-means clustering. We ran 100 iterations of k-means clustering, sampling with replacement 70% of the population at each iteration using MATLAB. We next assigned each cell a confidence score based on how many times it appeared in a certain cluster. Only cells with 95% confidence or above were included in the analysis. Confidence was determined by the fraction of times a trajectory was clustered in the same cluster (non-switching cells clustered > 50 times in p53 low, switching clustered >950 times in p53 high).
Determining time of switching
We determined the time of switching by manually creating a threshold of p53 intensity that would allow detection of the point of inflection in the p53 signal. The threshold was determined by using 10 traces as a training set. When the threshold succeeded in detecting the inflection point of the p53 signal in all 10 traces it was applied to all traces.
We first discounted the first 4 h to avoid cells with abnormally high intensity at the beginning of the time course. We defined time of switching (tS) by first finding a time point (tc) that met the following criteria:
The derivative of the smoothed signal (smoothed(dS)) exhibited a positive derivative for all time points between tc – 1 hr to tc +2.5 hr, and;
The signal S at tc was more than 2 standard deviations over the mean signal intensity from t0 to tc, and;
The maximum signal in tC to tC +2.5 hr was higher than the maximum signal in tc−2.5 hr to tc, and;
The signal at tc was higher than 0.8 standard deviations of the median peak intensity for all peaks (P) prior to tc, when peak (P) is defined as a local maximum of the smoothed signal (smoothed(S)).
When a time point tc satisfied all these conditions, we defined the switching time point (ts) as the time point between tc and tc + 5 hr with the maximal derivative.
Let S(ti, tj) denote the signal S in the time interval between ti and tj
Let P(ti, tj) denote the set of peaks of S (local maxima of smoothed S) in the time interval between ti and tj
Let Tc denote a time point that meets the following criteria
Let ts denote the time of switching
For all times tc to tc+5
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-p53 | Santa Cruz Biotechnology | Cat# sc-126; RRID: AB_628082 |
| mouse monoclonal anti-MDM2 (Ab-1) | Millipore | Cat# OP46T-10UG; RRID: AB_564805 |
| rabbit anti-pChk2 (T68) | Cell Signaling Technology | Cat# 2661; RRID: AB_331479 |
| rabbit anti-pChk1 (S317) | Cell Signaling Technology | Cat# 2344; RRID: AB_331448 |
| mouse anti-tubulin (E7) | Developmental Studies Hybridoma Bank | N/A |
| mouse monoclonal anti-β-Actin (AC-74) | Sigma-Aldrich | Cat# A2228; RRID: AB_476697 |
| HRP donkey anti-rabbit | VWR | Cat# 95017–556 |
| HRP goat anti-mouse | Thermo-Fisher | Cat# G21040; RRID: AB_2536527 |
| IRDye® 680RD donkey anti-rabbit | Li-COR | Cat# 925–68073; RRID: AB_2716687 |
| IRDye® 800CW goat anti-rabbit | Li-COR | Cat# 926–32211; RRID: AB_621843 |
| IRDye® 680RD goat anti-mouse | Li-COR | Cat# 926–68070; RRID: AB_10956588 |
| IRDye® 800CW donkey anti-mouse | Li-COR | Cat# 925–32212; RRID: AB_2716622 |
| Bacterial and Virus Strains | ||
| lentiCRISPR v2 | Feng Zhang | Addgene plasmid # 52961; http://addge.neorg/52961; RRID:Addgene_52961 |
| leniGuide-puro | Feng Zhang | Addgene plasmid # 52963; http://addgene.org/52963; RRID:Addgene_52963 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ATM inhibitor (KU55933) | Sigma-Aldrich | Cat# SML1109 |
| ATR inhibitor (VE822) | Cayman Chemical | Cat# 24198 |
| Caspase 2 inhibitor (Z-VDVAD-FMK) | R&D Systems | Cat# 2165 (discontinued) |
| Caspase 2 inhibitor (NH-23-C2) | Poreba et al., 2019 | N/A |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| Ribociclib | MedChem Express | Cat# HY-15777 |
| SMARTpool siGenome CDKN1A (p21) | Dharmacon | Cat M-003471–00-0005 Lot 140909 |
| Critical Commercial Assays | ||
| RNAi MAX transfection reagent | Thermo Fisher Scientific | Cat# 13778075 |
| Experimental Models: Cell Lines | ||
| Human: MCF7 pMT-p53-mVenus | Purvis et al., 2012 | N/A |
| Human: MCF7 pMT-p53-mVenus pCW22TRE-PIDD-mCer | This work | N/A |
| Human: MCF7 pMT-p53-mVenus PIDD1 KO | This work | N/A |
| Human: MCF7 pUb-p53-mVenus | Stewart-Ornstein and Lahav, 2017 | N/A |
| Human: RPE p21-mKate2 + pUBp-p53-mNeonGreen | Reyes et al., 2018 | N/A |
| Human: A549 pUb-p53-mNeonGreen H2B-mCherry | Stewart-Orenstein et al., 2017 | N/A |
| Oligonucleotides | ||
| PIDD1 siRNA sequences - sense | rCrArGrArCrUrGrUrUrCrCrUrG rArCrCrUrCrArGrArUrU |
|
| PIDD1 siRNA sequences - antisense | rCrArGrArCrUrGrUrUrCrCrUr GrArCrCrUrCrArGrArUrU |
|
| PIDD1 sgRNA forward | CACCGGCGGGAAACC ACAAGGAACC |
|
| PIDD1 sgRNA reverse | AAACGGTTCCTTGTG GTTTCCCGC |
|
| Recombinant DNA | ||
| pCW22TRE-PIDD1-mCer | This study | N/A |
| PIDD1-sgRNA-puro | This study | N/A |
| Software and Algorithms | ||
| P53Cinema Single Cell Tracking Software | Reyes et al., 2018 | https://github.com/balvahal/p53CinemaManual |
| Model for p53 regulation through MDM2 degradation by the PIDDosome | This study | https://github.com/MTsabar/PIDDosome/tree/PIDDosome |
Highlights.
Post-irradiation, a subset of cells switches from oscillatory to sustained p53 levels
Switching is a late response that occurs more than 24 h following irradiation
Switching requires escape from irradiation-induced cell cycle arrest
The caspase-2-PIDDosome sustains p53 levels by degrading p53’s inhibitor MDM2
ACKNOWLEDGMENTS
We thank Dr. Jacob Stewart-Orenstein for their help with data analysis and tracking pipelines. We thank Dr. Alba Jimenez for help and advice with developing the mathematical models. We thank Dr. Marcin Poreba and Dr. Marcin Drag for their kind gift of the caspase-2 inhibitor NH-23-C2. This work was supported by National Institutes of Health (NIH) grants R01 GM 083303 and R01 GM 116864. M.T. was supported by NIH grant RM1HG006193 and the American Cancer Society—New England Pay-if Group postdoctoral fellowship, PF-18–126-01-DMC. J.R. was supported by CONACyT/Fundacion Mexico en Harvard grant 404476) and Harvard graduate merit fellowship. T.G.O. was supported by American Cancer Society (ACS) grant RSG-13–300-01-TBG.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107995.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ando K, Kernan JL, Liu PH, Sanda T, Logette E, Tschopp J, Look AT, Wang J, Bouchier-Hayes L, and Sidi S (2012). PIDD death-domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling. Mol. Cell 47, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Parsons MJ, Shah RB, Charendoff CI, Paris SL, Liu PH, Fassio SR, Rohrman BA, Thompson R, Oberst A, et al. (2017). NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J. Cell Biol. 216, 1795–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliga BC, Read SH, and Kumar S (2004). The biochemical mechanism of caspase-2 activation. Cell Death Differ. 11, 1234–1241. [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, and Ziv Y (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- Baptiste-Okoh N, Barsotti AM, and Prives C (2008). A role for caspase 2 and PIDD in the process of p53-mediated apoptosis. Proc. Natl. Acad. Sci. USA 105, 1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor E, Mock CS, Bhan I, Loewer A, and Lahav G (2008). Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell 30, 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor E, Loewer A, Mock C, and Lahav G (2011). Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 7, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berube C, Boucher L-M, Ma W, Wakeham A, Salmena L, Hakem R, Yeh W-C, Mak TW, and Benchimol S (2005). Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc. Natl. Acad. Sci. USA 102, 14314–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, and Hannon GJ (1995). Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377, 552–557. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, and Siliciano JD (1998). Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- Cheng Q, and Chen J (2010). Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 9, 472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawar S, Lim Y, Puccini J, White M, Thomas P, Bouchier-Hayes L, Green DR, Dorstyn L, and Kumar S (2017). Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene 36, 2704–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckbar D, Jeggo PA, and Löbrich M (2011). Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 46, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorstyn L, Puccini J, Wilson CH, Shalini S, Nicola M, Moore S, and Kumar S (2012). Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ. 19, 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, and Vogelstein B (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, and Villunger A (2017). The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 31, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD, and Thomas P (2011). Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 26, 125–132. [DOI] [PubMed] [Google Scholar]
- Geva-Zatorsky N, Rosenfeld N, Itzkovitz S, Milo R, Sigal A, Dekel E, Yarnitzky T, Liron Y, Polak P, Lahav G, and Alon U (2006). Oscillations and variability in the p53 system. Mol. Syst. Biol. 2, 0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A, Stewart-Ornstein J, Purvis JE, Forrester WC, Bulyk ML, and Lahav G (2017). p53 pulses lead to distinct patterns of gene expression albeit similar DNA-binding dynamics. Nat. Struct. Mol. Biol. 24, 840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, and Elledge SJ (1993). The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, and Oren M (1997). Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299. [DOI] [PubMed] [Google Scholar]
- Heltberg ML, Chen SH, Jiménez A, Jambhekar A, Jensen MH, and Lahav G (2019). Inferring Leading Interactions in the p53/Mdm2/Mdmx Circuit through Live-Cell Imaging and Modeling. Cell Syst. 9, 548–558.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, and Kumar S (2009). A tumor suppressor function for caspase-2. Proc. Natl. Acad. Sci. USA 106, 5336–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, and Yasuda H (1997). Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 420, 25–27. [DOI] [PubMed] [Google Scholar]
- Huang Y, Hou H, Yi Q, Zhang Y, Chen D, Jiang E, Xia Y, Fenech M, and Shi Q (2011). The fate of micronucleated cells post X-irradiation detected by live cell imaging. DNA Repair (Amst.) 10, 629–638. [DOI] [PubMed] [Google Scholar]
- Janssens S, and Tinel A (2012). The PIDDosome, DNA-damage-induced apoptosis and beyond. Cell Death Differ. 19, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan K, Kaminski N, Rechavi G, Jakob-Hirsch J, Amariglio N, and Givol D (2001). DNA microarray analysis of genes involved in p53 mediated apoptosis: activation of Apaf-1. Oncogene 20, 3449–3455. [DOI] [PubMed] [Google Scholar]
- Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, and Shkedy D (1999). Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl. Acad. Sci. USA 96, 14973–14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Tiedt R, Loo A, Horn T, Delach S, Kovats S, Haas K, Engstler BS, Cao A, Pinzon-Ortiz M, et al. (2018). The potent and selective cyclin-dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget 9, 35226–35240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MHG, Jones SN, and Vousden KH (1997). Regulation of p53 stability by Mdm2. Nature 387, 299–303. [DOI] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, and Alon U (2004). Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 36, 147–150. [DOI] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, and Haber JE (1998). Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94, 399–409. [DOI] [PubMed] [Google Scholar]
- Lim Y, De Bellis D, Dorstyn L, and Kumar S (2018). p53 accumulation following cytokinesis failure in the absence of caspase-2. Cell Death Differ. 25, 2050–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Ma W, and Benchimol S (2000). Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Net. Genet. 26, 124–127. [DOI] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden SL, Galella EA, Zhu J, Bertelsen AH, and Beaudry GA (1997). SAGE transcript profiles for p53-dependent growth regulation. Oncogene 15, 1079–1085. [DOI] [PubMed] [Google Scholar]
- Mirza A, Wu Q, Wang L, McClanahan T, Bishop WR, Gheyas F, Ding W, Hutchins B, Hockenberry T, Kirschmeier P, et al. (2003). Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene 22, 3645–3654. [DOI] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, and Levine AJ (1992). The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- O’Reilly LA, Ekert P, Harvey N, Marsden V, Cullen L, Vaux DL, Hacker G, Magnusson C, Pakusch M, Cecconi F, et al. (2002). Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of cas-pase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ. 9, 832–841. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Kinzler KW, Meltzer PS, George DL, and Vogelstein B (1992). Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358, 80–83. [DOI] [PubMed] [Google Scholar]
- Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, Hubbard D, Bhutkar A, and Jacks T (2011). Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol. Cell 43, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek AL, Liu JC, Loewer A, Forrester WC, and Lahav G (2016). Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell 165, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, and Wu H (2007). Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128, 533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Toczyski DP, and Hartwell LH (1997). When checkpoints fail. Cell 88, 315–321. [DOI] [PubMed] [Google Scholar]
- Poreba M, Rut W, Groborz K, Snipas SJ, Salvesen GS, and Drag M (2019). Potent and selective caspase-2 inhibitor prevents MDM-2 cleavage in reversine-treated colon cancer cells. Cell Death Differ. 26, 2695–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JR, Fisher BE, and Batchelor E (2016). p53 Pulses Diversify Target Gene Expression Dynamics in an mRNA Half-Life-Dependent Manner and Delineate Co-regulated Target Gene Subnetworks. Cell Syst. 2, 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, and Lahav G (2013). Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, and Lahav G (2012). p53 dynamics control cell fate. Science 336, 1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes J, Chen JY, Stewart-Ornstein J, Karhohs KW, Mock CS, and Lahav G (2018). Fluctuations in p53 Signaling Allow Escape from Cell-Cycle Arrest. Mol. Cell 71, 581–591.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothkamm K, and Löbrich M (2003). Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl. Acad. Sci. USA 100, 5057–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth R, Yang C, Kaushal V, Shah SV, and Kaushal GP (2005). p53-dependent caspase-2 activation in mitochondrial release of apoptosis-inducing factor and its role in renal tubular epithelial cell injury. J. Biol. Chem. 280, 31230–31239. [DOI] [PubMed] [Google Scholar]
- Shieh S-Y, Ikeda M, Taya Y, and Prives C (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, and Ziv Y (2013). The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210. [PubMed] [Google Scholar]
- Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, et al. (2008). Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 133, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladky VC, Knapp K, Soratroi C, Heppke J, Eichin F, Rocamora-Reverte L, Szabo TG, Bongiovanni L, Westendorp B, Moreno E, et al. (2020). E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Dev. Cell 52, 335–349.e7. [DOI] [PubMed] [Google Scholar]
- Stewart-Ornstein J, and Lahav G (2017). p53 dynamics in response to DNA damage vary across cell lines and are shaped by efficiency of DNA repair and activity of the kinase ATM. Sci. Signal. 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry MR, Arya R, Mukhopadhyay A, Berrett KC, Clair PM, Witt B, Salama ME, Bhutkar A, and Oliver TG (2015). Caspase-2 impacts lung tumorigenesis and chemotherapy response in vivo. Cell Death Differ. 22, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R, Shah RB, Liu PH, Gupta YK, Ando K, Aggarwal AK, and Sidi S (2015). An Inhibitor of PIDDosome Formation. Mol. Cell 58, 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel A, and Tschopp J (2004). The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304, 843–846. [DOI] [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ, and Hartwell LH (1997). CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90, 1097–1106. [DOI] [PubMed] [Google Scholar]
- Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, and Demaria S (2017). DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 8, 15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, and Levine AJ (1993). The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7 (7A), 1126–1132. [DOI] [PubMed] [Google Scholar]
- Zhou BB, and Elledge SJ (2000). The DNA damage response: putting checkpoints in perspective. Nature 408, 433–439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Custom MATLAB scripts are available upon request. The ModelSimulator code used to model p53 activation through degradation of MDM2 by the PIDDosome can be downloaded at https://github.com/MTsabar/PIDDosome/tree/PIDDosome