

Abstract
Microfluidic lumen-based systems are microscale models that recapitulate the anatomy and physiology of tubular organs. These technologies can mimic human pathophysiology and predict drug response, having profound implications for drug discovery and development. Herein, we review progress in the development of microfluidic lumen-based models from the 2000s to the present. The core of the review discusses models for mimicking blood vessels, the respiratory tract, the gastrointestinal tract, renal tubules, and liver sinusoids, and their application to modeling organ-specific diseases. We also highlight emerging application areas, such as the lymphatic system, and close the review discussing potential future directions.
1. Introduction
Advances in modern healthcare in the last few decades have, in part, led to a better quality of life and higher life expectancy worldwide. However, mortality due to communicable and non-communicable diseases, such as drug-resistant tuberculosis, cardiovascular diseases, and cancers, continues to rise1. A concerted effort from multiple fronts is needed to lessen global disease burden, including better preventive care (e.g., lifestyle education), improved access to healthcare services, affordable medical diagnosis and treatment, and continued advancement of biomedical research. In the context of biomedical research, the costs of drug development have skyrocketed in the last couple of decades, applying an increasing burden in an already overwhelmed healthcare system. One of the leading causes behind the rise of such costs is the high attrition rate in our current drug development system2. Specifically, the vast majority of experimental drugs show promising results in the in vitro phase and then fail to demonstrate effectiveness in clinical trials. Thus, developing relevant tissue and organ models to better understand the pathophysiology of human diseases is of great importance to therapeutic discovery and drug development.
Two-dimensional (2D) in vitro culture models have formed the cornerstone of biomedical research for more than a century3. Despite their higher throughput, ease-of-use and experimental tractability, they often fail to recapitulate the three-dimensional (3D) tissue- and organ-level structure-function relationships observed in vivo4, 5 In this context, 3D cell culture techniques, including cell embedding and seeding on top of hydrogels, have been subsequently developed to address these limitations. Continued innovation has also led to organotypic culture, where self-assembled cellular clusters or organoids better mimic organ-level physiology5. Compared with 2D cell culture, 3D techniques can enable an improved in vitro mimicry of the complex mechanisms governing the pathophysiology of different diseases and can be valuable preclinical tools for drug testing. Nonetheless, current 3D models remain limited in their ability to capture crosstalk between multiple cell types and mechanical cues in the microenvironment that contribute to cellular mechanotransduction (e.g., fluid shear stress), which are important factors in disease progression. Mouse models could be suitable alternatives as they provide physiologically relevant microenvironments. However, they have low experimental tractability, and the interaction of human and mouse cells commonly leads to experimental artifacts that might bias the results observed, limiting the ability to model human disease6.
Microfluidic cell culture, i.e., the synergy of tissue engineering with microscale physics, has enabled the development of microphysiological systems or organs-on-chips. These systems enable multi-culture of different cell types mimicking the tissue organization, generation of stable biochemical gradients, and controlled fluid transport, among other unique capabilities. They have been used to mimic many human organ systems, including the heart, brain, kidney, liver, lung, pancreas, and circulation. Importantly, organs-on-chips have demonstrated utility for unraveling basic mechanisms of disease, drug screening, and precision medicine. Several reviews cover the development, fabrication, and application of various organs-on-chips7–9. However, the organ-on-a-chip field is increasingly developing and specializing. One of the most notable specializations is the development of models that mimic lumen-based physiological structures, yet the literature regarding these models is scarce. Therefore, this review focuses on organ systems comprising lumens as their fundamental functional unit, including blood and lymphatic vessels, mammary ducts, the respiratory tract, renal tubules, liver sinusoids, and the gastrointestinal tract (Figure 1).
Figure 1.
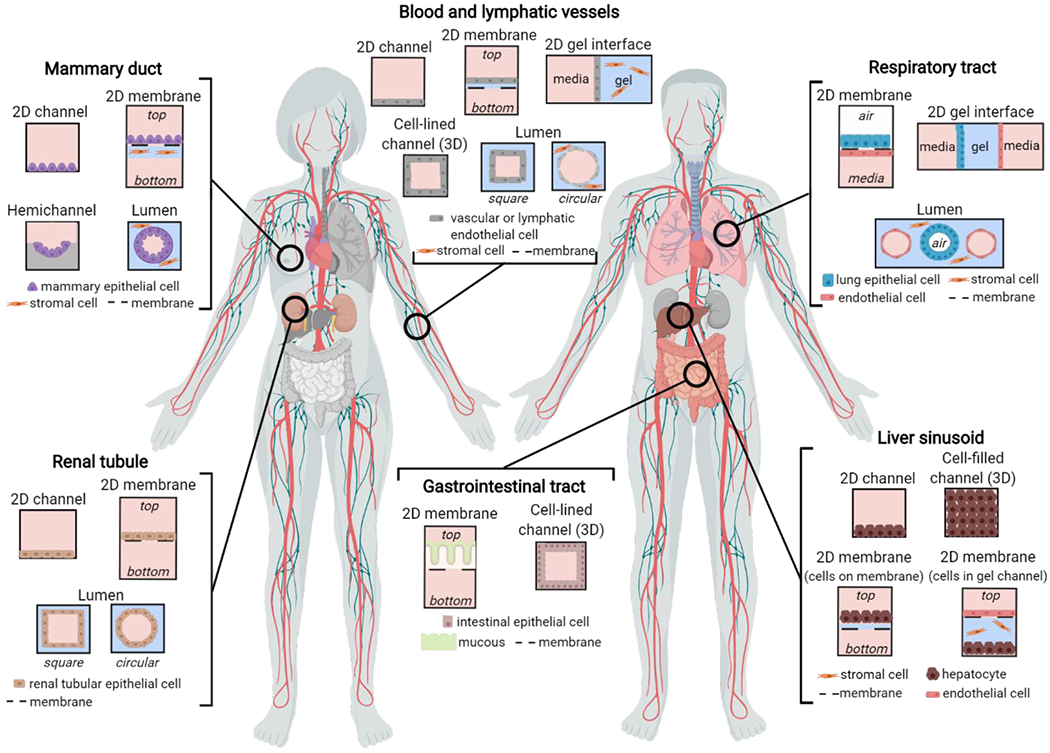
Overview of microfluidic device designs to mimic lumen-structured organs. Two-dimensional (2D) organ geometries include cells cultured on microchannel surfaces, porous membranes, and gel interfaces. Three-dimensional (3D) organ geometries include cell-lined channels, cell-filled channels, and lumens with square and circular cross-sections.
Lumens are tubular structures that are ubiquitous throughout all body systems, including the blood vessels that regulate nutrient and waste exchange in tissues, mammary ducts to produce milk or the gastrointestinal tract10. Further, luminal structures are also involved in multiple human diseases such as cancer. In the context of cancer progression, ductal dysfunction in the breast and prostate are early hallmarks of the tumorigenic process. In later stages, the conditioning of normal blood and lymphatic vessels to a tumor-associated phenotype enables the spread of cancer cells to secondary sites in the body11. Similarly, the stiffening and narrowing of blood vessels in atherosclerotic vascular disease can lead to complications in major organs, including the heart, brain, and kidney10. Therefore, the importance of capturing luminal geometry in disease models goes beyond aesthetics to mimicking in vivo structure-function relationships4. Previous research has demonstrated substantial differences in cell behavior and signaling when cultured as monolayers on 2D substrates compared with lumens, including changes in cytokine secretion, cell proliferation, and gene expression4, 12–16. Thus, the goal of this review is to provide a comprehensive evaluation of microfluidic lumen-based models of the vasculature, respiratory tract, gastrointestinal tract, renal tubules, and liver sinusoids from the 2000s to the present. The core of the review summarizes models recapitulating both the normal physiology and pathophysiology of these tubular organs. Furthermore, we highlight application areas where microfluidic luminal models have emerging utility, such as the lymphatic system, and close the review with an outlook on remaining limitations and possible future directions for the field.
2. Blood vessels
The cardiovascular system comprises the heart and a vast network of blood vessels that transport nutrients, oxygen, and waste products throughout the body. The vascular tree consists of arteries and veins (0.1-10+ mm in diameter) that branch into smaller arterioles and venules, which are interconnected by capillaries (5-10 μm in diameter). Networks of capillaries supply oxygen and nutrients to each major organ in the body, resulting in structurally and functionally diverse in different organs to accommodate their specific physiological and metabolic needs17. Major cardiovascular diseases such as atherosclerosis (i.e., the narrowing of blood vessels due to buildup of a lipid-based plaque) lead to other severe pathologies, including myocardial infarction and stroke. Blood vessels also have a critical role in cancer progression, where tumors hijack normal vasculature to produce structurally and functionally abnormal vessels that support tumor growth and dissemination18, 19. In this section, we review different approaches to mimic the vasculature in microfluidic devices and their application to modeling normal vascular function and associated diseases, including atherosclerosis and cancer metastasis. A summary of microfluidic vascular models is provided in Table 1.
Table 1.
Summary of microfluidic vascular models.
| Organ geometry | Cell type | Targeted application | Refs |
|---|---|---|---|
| 2D channel | HUVEC | -Effect of immunosuppressants on T cell adhesion to endothelium | 37 |
| 2D membrane | bEND HaVEC hAoSMC HMVEC HUVEC PAEC |
-Transendothelial electrical resistance -Laser-induced fluorescence measurement of endothelial permeability -Vascular permeability in co-fi with lymphatics -Early stage atherosclerosis model -Effect of mechanical strain and shear stress on endothelial-smooth muscle cell crosstalk -Leukocyte-endothelial interactions in a stenotic vessel |
23, 24, 44, 47, 50 |
| 2D gel interface | HUVEC HMVEC |
-Perfusable microvascular network generated by angiogenesis and vasculogenesis -Effect of shear stress on angiogenic sprouting -Microvascular network formation in co-culture with human mesenchymal stem cells derived from bone marrow -Transendothelial migration of neutrophils under chemotaxis -Breast cancer cell intravasation and extravasation -Breast cancer metastasis to a bone microenvironment -Adenoid cystic carcinoma cell intravasation -Tumor angiogenesis and evaluation of anti-angiogenic therapy |
13, 25, 26, 39, 57–62 |
| Cell-lined channel | HLMVEC HMVEC HUVEC |
-Effect of shear stress and inflammatory cytokines on microvascular occlusion and thrombosis -Effect of growth factors and hemodynamic forces on coagulation in a ‘bleeding’ channel |
49, 53 |
| Lumen (squarea) | HUVEC | -Vascular network for studying angiogenesis and thrombosis -Neutrophil transendothelial migration under chemotaxis -Stenotic vessel for studying atherosclerosis |
16, 48 |
| Lumen (circularb) | hEPC HMVEC HUVEC iPSC-EC NEnC TEnC |
-Perfusable luminal vessels -Effect of vessel geometry on cytokine secretion -Effect of various growth factors on angiogenic sprouting -Real-time monitoring of leukocyte transendothelial migration -Effect of endothelium on neutrophil lifetime and migration towards bacteria -Live imaging of breast cancer cell invasion and intravasation -Screening anti-angiogenic drugs using patient-derived vessels |
4, 27, 29, 30, 32, 36, 40, 41, 63, 65 |
Lumen with a square cross-section,
Lumen with a circular cross-section
bEND – brain-derived endothelial cell
HaVEC – human aortic vascular endothelial cell
hAoSMC – human aortic smooth muscle cell
hEPC – human endothelial progenitor cell
HLMVEC – human lung microvascular endothelial cell
HMVEC – human microvascular endothelial cell
HUVEC – human umbilical vein endothelial cell
iPSC-EC – induced pluripotent stem cell-derived endothelial cell
PAEC – primary porcine aorta endothelial cell
NEnC – normal endothelial cell from kidney cancer patient
TEnC – tumor endothelial cell from kidney cancer patient
2.1. Modeling normal vascular function
Establishing basic structure.
Cell-patterning and hydrogel molding have been the primary methods for generating vascular endothelium in microdevices13, 20, with the generation of 2D lumen interfaces and 3D printing gaining in popularity21 (Figure 2). Cell-patterning typically involves the seeding of endothelial cells onto a 2D surface within a microchannel, such as a porous membrane or hydrogel interface13, 22–25. Membrane-based vascular models, much like conventional Transwell assays, are ideally suited for examining endothelial barrier function under various conditions, whereas endothelium models formed on hydrogel interfaces have been typically used to study angiogenesis.
Figure 2.
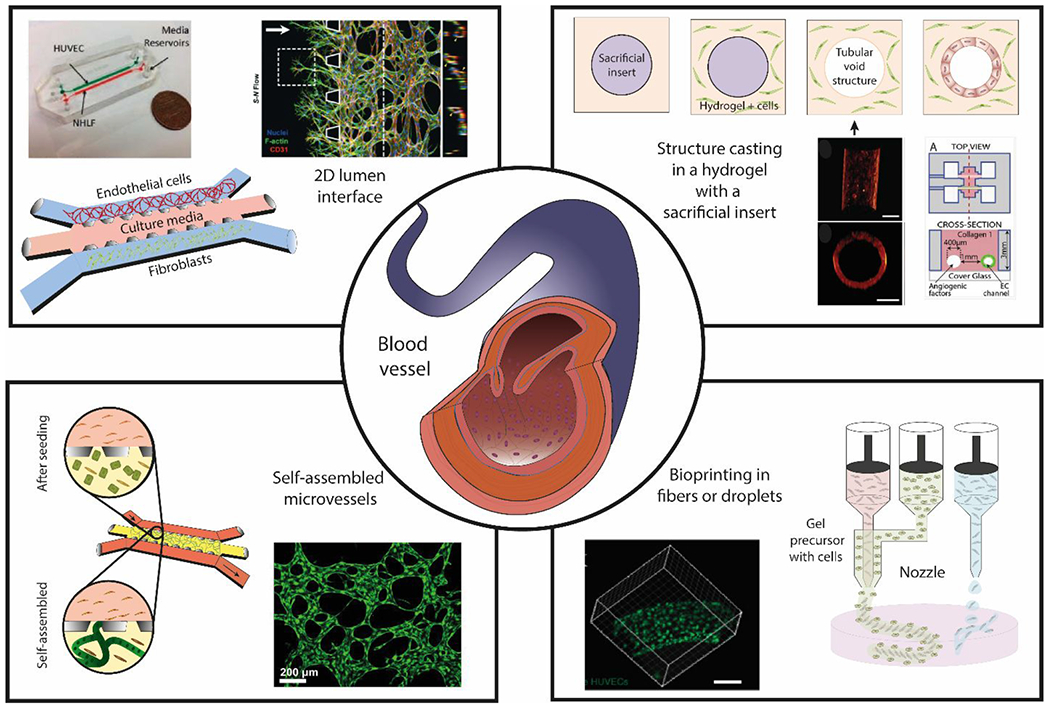
Overview of blood vessel structure and different approaches to generate microfluidic lumen-based systems. (Center) Schematic representation of a blood vessel in vivo. (Top-left) generation of 2D lumen interfaces using 3 parallel channels in contact with each other defined by capillary valves. Cells are seeded in the channels without a supporting matrix. (Top-right) Structure casting of luminal structures embedded in a hydrogel with a sacrificial insert29. (Bottom-left) Microvessel self-assembly of endothelial cells within a fibrin hydrogel in the presence of supporting cells. (Bottom-right) Bioprinting of fibers or droplets. Copyright (2013) National Academy of Sciences for top-right images, ref 32. Microscopy image at bottom-right is reproduced from ref. 182 with permission from the WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2014. Top left is reproduced from ref 59 with permission from Oxford University Press, copyright 2013. 21, 29, 32, 59, 225
One of the most popular designs that leverages cell-patterning on a hydrogel interface was the popular 3-channel device reported by Jeon et al., (among others) a modification of the design also initially developed by the Kamm Laboratory at MIT (Figure 2). The device consisted of a central hydrogel channel flanked by two parallel channels for cell culture or media perfusion26. Seeding endothelial cells into one flanking channel generates a vessel wall at the hydrogel interface. The opposite flanking channel can be used to generate molecular cues via co-culture with stromal cells (e.g., fibroblasts) or perfusion of growth factors (e.g., VEGF). Interstitial flow through the hydrogel channel can also be generated by perfusing media at different flow rates through the two flanking channels. The presence of shear stress and mechanical cues is known to affect endothelial cell differentiation and function, and therefore is considered a crucial set of cues to include in models. However, this device design relies on seeding cells on the 2D hydrogel interphase, limiting its capacity to generate 3D tubular structures. Moving towards 3D tubular structure, several groups have devised hydrogel molding techniques to generate 3D lumens in different hydrogels (e.g., collagen, fibrin) 16, 27–29. Bischel et al. leveraged viscous finger patterning (i.e., displacement of a more viscous fluid by a less viscous fluid) to mold lumens in collagen type I hydrogel, which could then be lined with endothelial cells to form 3D tubular blood vessels4, 28. Although more physiologically relevant then 2D cell-patterning, the success of the approach was highly dependent on user skill. Recently, de Graaf et al. further optimized the method to improve its reproducibility and scalability30. A more repeatable method for generating lumens involves the use of a sacrificial pin to cast a lumen template within a hydrogel (Figure 2). Chrobak et al. developed the needle molding technique, whereby a needle could be threaded and suspended within a microchannel27(Figure 3). After polymerizing a collagen hydrogel in the channel, the embedded needle is removed to form a hollow lumen that can be lined with endothelial cells. Similarly, Jimenez-Torres et al. demonstrated the formation of lumens in a hydrogel with removable PDMS rods (Figure 3)29. An advantage of using flexible PDMS rods over rigid needles is the ability to create templates with multiple branches to mimic the structural complexity of the vascular network better. Importantly, cultured blood vessels exhibited in vivo-like barrier function, as demonstrated by the regulation of dextran transport across their endothelium.
Figure 3.
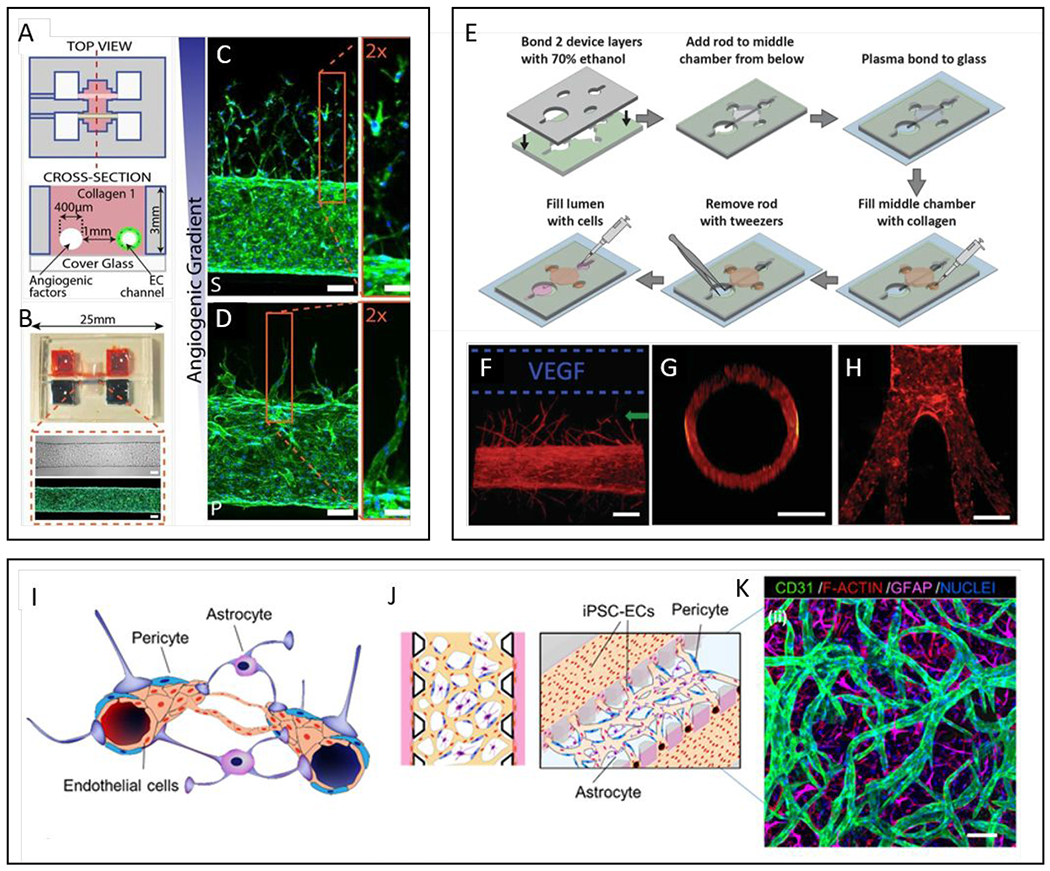
Commonly used models of endothelial lumens embedded in a hydrogel. (A-D) Two parallel luminal structures cast within a hydrogel using needles. (A) Schematics of the device showing two parallel lumens. One lumen is lined with endothelial cells and the other used as a source for angiogenic growth factors. (B) Representative images of the device and a cultured vessel. F-actin is stained with phalloidin in green and nucleus with DAPI in blue. (C-D) Confocal images of angiogenic sprouting in response to different cocktails of angiogenic growth factors, HFMVS and MVPS. F-actin (phalloidin, green), nuclei (DAPI, blue). Scale bars are 100 μm. (E-H) A single luminal structure embedded in a hydrogel generated using flexible PDMS rods with variable structures as templates. (E) Schematic of LumeNEXT device construction and operation. (F) Cultured vessels respond to VEGF stimulation and sprout in the direction of the source (F-actin, red). (G) Vessels have circular cross-sections. (H) Demonstration of non-linear geometries achievable with the method. (I-K) Blood-brain barrier and in vitro microvascular network model. (I) Schematic representation of the blood-brain barrier (BBB), composed of brain Endothelial cells (ECs) vessels overlapped by pericytes (PCs) and astrocytes (ACs). (J) Schematic representation of proposed 3D BBB microvascular network (μVN) model that mimics the microvascular structure present in the brain environment. (K) Confocal image of self-assembled BBB μVN model including iPSC-ECs (CD31, green), PCs (F-actin, red) and ACs (GFAP, magenta), and nuclei (DAPI, blue) 29, 32, 206, 226. Copyright (2013) National Academy of Sciences for top-right images, ref 32. Reproduced from ref. 226 with permission from Elsevier Science & Technology Journals, copyright 2017.
Vasculogenesis and angiogenesis.
Vasculogenesis and angiogenesis are the fundamental processes by which new blood vessels are formed, and are vital to tissue homeostasis and recovering normal tissue function after wounding. These two processes are also integral steps in disease progression, such as cancer metastasis31. Thus, a significant focus of developing microfluidic vascular models has been to recreate robust vasculogenesis and angiogenesis in vitro. Using the fabrication methods described above, researchers have demonstrated the formation of 3D perfusable vascular networks amenable to studying the molecular and biophysical mechanisms of these two processes. Jeon et al. developed a vasculogenesis model based on the 3-channel design by co-culturing bone marrow-derived human mesenchymal stem cells and human umbilical vein endothelial cells (HUVECs) in fibrin matrices26. Vessel networks formed after six days of culture and were perfusable as indicated by fluorescent dextran assays. The authors investigated the role of different growth factors on the formation of the networks, noting that the addition of vascular endothelial growth factor (VEGF) and angiopoietin promoted functional networks as opposed to VEGF plus transforming growth factor-beta. In a comparable study, Kim et al. employed a 5-channel design to demonstrate both vasculogenesis and angiogenic sprouting13.
Similarly to the 3-channel device, their model enabled co-culture of endothelial cells with stromal fibroblasts, either directly with HUVECs in the ECM gel channel to induce self-assembly, or in a flanking channel as a source of growth factors to induce angiogenic sprouting (Figure 3). Importantly, the microvessels had luminal structure and supported intraluminal fluid flow. Endothelial cells reorganized their cytoskeleton under fluid flow, which is a normal response observed in vivo. Moreover, the authors demonstrated the applicability of their model to study vascular-pericyte and vascular-tumor interactions by co-culturing the HUVECs with human placenta pericytes and human glioblastoma cells, respectively.
To study angiogenic sprouting from tubular structures, Jimenez-Torres et al. leveraged their hydrogel molding method (i.e., using PDMS rods as sacrificial mold) to create a double-lumen device, where one lumen was lined with endothelial cells, and the other was perfused with VEGF29. The two adjacent lumens were generated in collagen type I by removing PDMS rods suspended in parallel in a microchamber. Directional sprouting was observed towards the VEGF source. In a more extensive study, Nguyen et al. leveraged the needle templating method to create a similar double-lumen microdevice to assess the angiogenic potential of six common growth factors, including basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF), monocyte chemotactic protein-1 (MCP-1), sphingosine 1-phosphate (S1P), phorbol 12-myristate 13-acetate (PMA), and VEGF (Figure 3) 32. They observed that a cocktail of MCP-1, VEGF, PMA, and S1P (MVPS) promoted the most angiogenic sprouting, as compared to a cocktail of HGF, bFGF, MCP-1, VEGF, and S1P (HFMVS). Interestingly, angiogenic sprouting was reduced when vessels were treated with the HFMVS cocktail and VEGFR-2 inhibitor. However, the same inhibitory effect was not observed after treatment with the MVPS cocktail and VEGFR-2 inhibitor. These results suggest that specific molecular pathways outweigh others during angiogenesis and highlight the usefulness of organotypic vascular models for differentiating these critical pathways.
Immune cell trafficking.
Leukocyte trafficking is essential is involved in multiple processes such as infection, injury, and cancer. Leukocytes transit through endothelial vessels to sites of inflammation and facilitate their activation33. Trafficking of cells from the circulatory system to the interstitial space involves a multistep process, which begins with cytokine-mediated signaling, hemodynamic shear forces, and cell adhesion events. In this context, researchers have developed several microfluidic models to study immune cell trafficking34, 35. Using a human tissue-engineered blood vessel, Chen et al. studied how TNFa and NF-kB inhibitors attenuate endothelial activation, monocyte adhesion, and transendothelial migration (TEM)36.
Similarly, Park et al. developed a microfluidic flow device to study T cell-endothelial interactions37. The authors analyzed the binding of T cells from patients with systemic lupus erythematosus and found that the immunosuppressant significantly decreased T cell adhesion to the endothelium. With precision medicine on the rise, this work illustrates a potential application of microfluidic vascular models for diagnosis and treatment.
Once adhered to the endothelial cells, leukocytes exit the blood vessel and migrate towards sites of infection or injury. This complex process involves various cell types, matrix components, and cytokines38. Han et al. focused on understanding the factors that work together to coordinate leukocyte trafficking was the focus of work by Han et al. 39 and Hind et al.40 Han et al. developed a 3D model of neutrophil TEM and chemotaxis to characterize neutrophil migration to various cytokines39 (Figure 4). Instead of focusing on model development, Hind et al. leveraged existing organotypic blood vessel lumen models, such as that described by Jimenez-Torres et al.29, 41 to study how paracrine signaling between the endothelium and neutrophils alter their migration and lifespan in response to bacterial infection40. The authors found that IL-6 secreted by the endothelial cells was critical to enhancing neutrophil migration, but not their lifetime. While these examples show the utility of organotypic lumen models for studying immune cell trafficking, not much focus has been placed on ECM composition and how it can affect this process. Biophysical parameters such as ECM composition, porosity, stiffness, and fiber alignment remain largely understudied in the context of immune cell trafficking.
Figure 4.
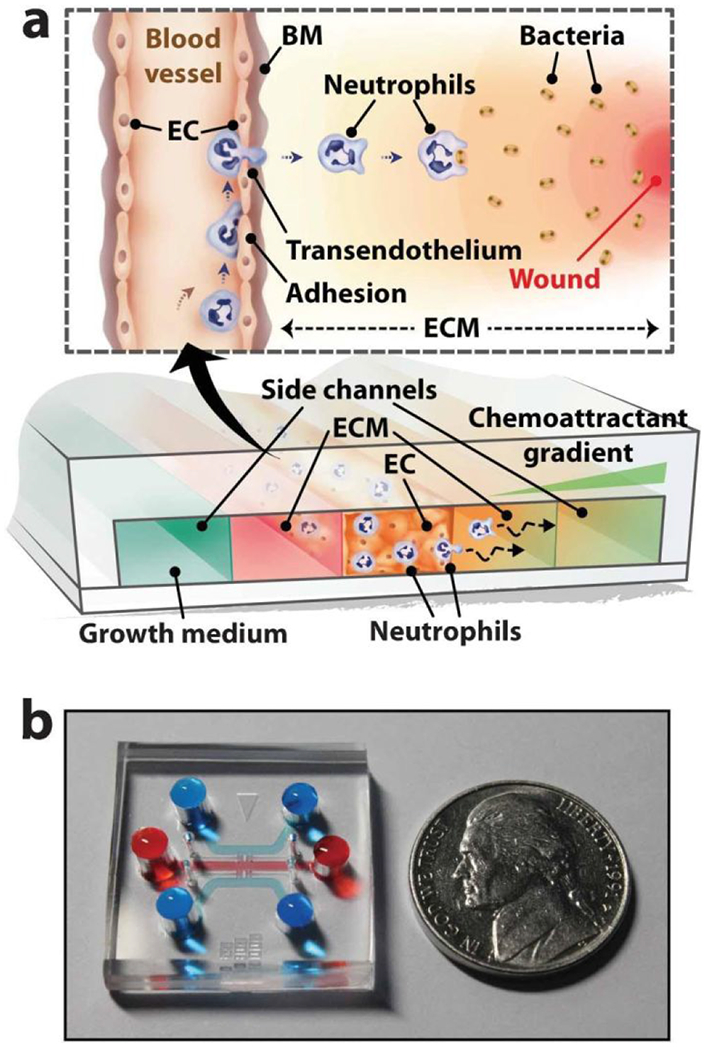
Microdevice for modeling neutrophil transendothelial migration. (a) Schematic of device and concept of neutrophil responses. A central channel for culturing an endothelial cell (EC) monolayer is flanked channels to perfuse media and chemoattractants. (b) Image of the device filled with dye for visualization of the channels. Reprinted with permission from Han et al., 201239.
2.2. Modeling vascular diseases
Atherosclerosis.
Atherosclerosis is a disease characterized by the narrowing of arteries due to the buildup of plaque42. Atherosclerotic plaques take years to form and are composed of foam cells, lipids, calcium deposits, and various ECM protein components that results in a thickening of the arterial wall and a loss of elasticity. Although atherosclerosis is one of the leading causes of death in developed countries, much is still unknown about how the disease initiates, progresses, and leads to pathologies like heart attack and stroke. The development of microfluidic atherosclerosis models is attractive to researchers due to the potential for higher-throughput in vitro models to study basic biology and screen potential treatments in a less costly manner than in vivo models.
The development of atherosclerotic plaques is still not fully understood but is believed to be mediated by an inflammatory process43. During the early stages of atherosclerosis, the upregulation of several cell adhesion proteins on the endothelial surface occurs in response to inflammatory signals in the blood. These events lead to monocyte adhesion and entry into the arterial tunica intima. While the full extent of these compounds is not known, several groups have begun to design and use microfluidic lumen-based vascular models to screen and test for these compounds. Zheng et al. used a 2D HUVEC lumen interface to model the effects of hyperglycemia and hyperlipidemia in atherosclerosis. To this end, they exposed their model to increased concentrations of glucose and cholesterol, both known inductors of atherosclerosis. The authors showed that these preconditions activated an inflammatory cascade, including an increase in the production of reactive oxygen species (ROS) and a decrease in the expression of blood vessel-specific cell-cell junction proteins (e.g. VE-cadherin). Atherosclerosis occurs preferentially at sites where the vessel walls have higher endothelial permeability. Therefore, their model reproduced hallmarks present in an atherogenic environment, consistent with clinical reports.44 These authors also tested a clinically used drug (i.e., probucol) to reduce the ROS production in their model, and demonstrated its effectiveness, but also a cytotoxic side effect previously reported in the clinic but poorly understood. Conversely, treatment with platinum nanoparticles decreased ROS production in this system, highlighting the potential of the platform for drug testing. In another study, Chen et al. investigated the role of oxidized low-density lipoprotein (i.e., a common biomolecule that is both a source of lipid and of oxidative stress) and the overall progression of atherosclerosis45. The authors found that oxidized low-density lipoprotein significantly increased both monocyte adhesion and macrophage activation, resulting in an increased secretion of pro-inflammatory molecules (i.e., TNF-α). This effect, in turn, resulted in a more pronounced increase in adhered monocytes, which contributed to arterial blockage and illustrates the chronic inflammatory pathways that occur during atherogenesis.
The progression of atherosclerosis entails the growth of the atheroma plaque by the infiltration of lipid-rich macrophages or “foam cells,” calcium deposition, the addition of connective tissue, and the hardening of the arterial wall due to smooth muscle cell proliferation42. Thus, the growth of the atheroma plaque is a slow process that involves multiple factors and cell types. Recapitulating this long-term growth of atheroma plaque in microfluidic devices is a challenging endeavor, in part due to the high cell-to-media ratio and maintenance of multiple cell types in a single device. However, several groups have demonstrated models capable of resolving the formation of atheroma plaque. Using a non-traditional microfluidic approach, Robert et al. coupled a microfluidic pump to a HUVEC-seeded bio-scaffold and perfused various inflammatory cytokines, lipids, and immune cells to this in vitro artery model46. With this device, the authors studied atherogenesis dynamics, including a quantification of the monocytes and macrophages infiltrating the bio-scaffold, and the amount of lipid absorption into the vessel wall. Beyond looking at the formation of the plaque, Van Engeland et al. developed a lumen-based system to elucidate the mechanisms of vessel hardening due to cell proliferation and deposition of connective tissue. This device allowed stiffness measurements of an aortic endothelium after co-culturing human aortic endothelial cells and human aortic vascular smooth muscle cells47.
In more advanced cases of atherosclerosis, the growth of the atheroma plaque and the hardening of the vessel leads to stenosis (i.e., a narrowing of the vessel). The combination of altered hemodynamic flow and plaque rupturing leads to thrombosis (i.e., clotting) and its associated pathologies, such as myocardial infarction, stroke, or ischemia. These pathophysiological events typically occur in vessels that have narrowed to micron-sized diameters. Thus, microfluidic models are well-suited for studying this process. Researchers have examined how different vessel geometries affect blood flow in situations of atherosclerotic thrombosis (i.e., blockage due to excessive clotting)48, 49. For instance, Westein et al. designed a microfluidic stenosis model using PDMS channels with varying degrees of stenosis and found that there was increased platelet adhesion downstream of the stenosis. The group further correlated those in vitro results in their findings in mice50. They associated this thrombus formation with an increase of von Willebrand factor (vWF). Improving on this design, Menon et al48. created a tunable stenosis model to measure the severity of the stenosis on thrombus formation46. They were able to visualize distinct platelet and leukocyte adherence patterns from whole blood as a result of the stenosis. These studies demonstrated that organotypic lumen models can accurately recapitulate a pathophysiology that occur during stenosis50, 51. Future stenosis models could address spatiotemporal control and include additional stromal components (e.g., pericytes or fibroblasts) known to play a key role in hemostasis52.
In addition to confined geometries, vessel injury is a significant cause of blood flow disruption. To study the effects of vessel injury, Sakurai et al. developed a specific microfluidic lumen-based device. This device used a perpendicular valve channel to puncture a hole into the side of an endothelial lumen (Figure 5)53. By flowing whole blood through the injured vessel, the authors observed and measured hydrostatic plug formation and studied the effect of hemodynamic factors, such as fluid shear stress and vWF (i.e., von Willebrand Factor, a critical factor in the clotting cascade), on the dynamics of clot formation. One of the significant downstream effects of these thromboses is myocardial infarction or heart attack. A heart attack occurs when a thrombus forms inside an artery, thereby blocking oxygen supply to cardiac muscles and eventually leading to ischemia and cell death. Using hydrodynamic focusing, an application of microscale fluid transport technologies, Su et al. were able to encapsulate endothelial cells within a mixture of gelatin methacrylamide, (PEG)-tetra-norbornene, PEG-tetra-thiol, and fibronectin. This mixture was photopolymerized, then woven into a “patch” and finally encapsulated by a cardiac stem cell-laden fibrin gel to create a vascularized cardiac patch54. Future work in this area should focus on assessing the long-term effects of devices like this used in vivo. Determining potential toxicity, and the adsorption of the ECM ad cellular components will be necessary for progression into human applications.
Figure 5.
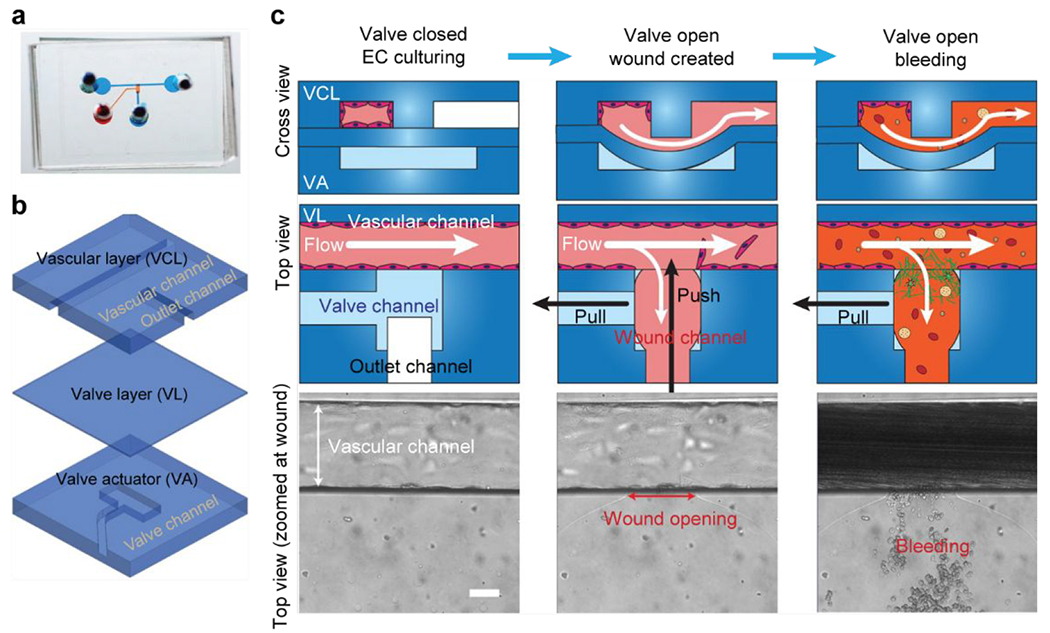
A microfluidic bleeding device for studying vascular injury and hemostasis. (a) Image of microfluidic bleeding device. Microchannels are filled with dye for visualization. (b) The device consists of three PDMS layers: a vascular layer (VCL) containing a vascular channel and outlet channel, a valve layer (VL), and a valve actuator (VA) layer. (c) Endothelial cells are cultured in the vascular channel to confluency while the valve is closed. A wound is created by opening the valve layer via negative pressure in the valve actuator channel (pull) and by applied fluid pressure through the outlet channel (push). Subsequently, whole blood is perfused through the vascular channel which leads to the outlet channel. Scale bar is 50 μm. Adapted with permission from Sakurai et al., 201853.
Cancer metastasis.
Cancer progression is a complex process involving many molecular signals (e.g., paracrine and autocrine signals from stromal fibroblasts, macrophages, and endothelial cells) and mechanical cues (e.g., interstitial flow, and ECM density and stiffness) in the tumor microenvironment (TME). The vasculature is an essential component of the TME, supplying nutrients and oxygen to the tumor and enabling cancer cells to metastasize to other organs. Tumor-associated blood vessels are different from their normal counterparts in organization, structure, and function. They are hyperpermeable to blood plasma and plasma proteins, have disorganized endothelium (i.e., randomly oriented endothelial cells), and are highly responsive to angiogenic growth factors55. To metastasize, tumor cells need to intravasate into the vasculature, and disseminate to distant organs to form secondary tumors. Tumor metastasis leads to poor patient prognosis and outcome. Therefore, anti-vascularization therapies that target tumor angiogenesis and aberrant vessel permeability form a large part of patient therapy for solid cancers, including breast cancer, prostate cancer, and renal cell carcinoma56.
Microfluidic tumor-vascular models have provided new biological insights into tumor biology and enabled preclinical assessment of cancer therapeutics50,51. Various microfluidic lumen-based models have been developed to study different steps of the metastatic cascade, such as cancer cell intravasation/extravasation and tumor angiogenesis. In a seminal paper, Zervantonakis et al. developed an intravasation model using the 3-channel design previously described by Jeon et al.57 (Figure 2–top left, and 19B-iii). This model consisted of a square cross-section tubular lumen lined with microvascular endothelial cells (i.e., MVECs) on a side channel, and an embedded 3D tumor cell culture component. This model was used to study intravasation of fibrosarcoma cells (HT1080) and breast cancer cells (MDA-MB-231) into the MVEC lumens.58 Macrophages were also added subsequently to study their effect in the vessel structure and in cancer cell extravasation. It is known that other cells in the microenvironment (i.e., stromal cells) play a role in extravasation, either preventing or facilitating the process. Their results showed that cancer cell intravasation was significantly enhanced in the presence of macrophages co-cultured in the device due to an increase in vessel permeability. A deeper analysis revealed that macrophage-secreted TNF-α induced the permeability changes and, thus, could be a potential therapeutic target. In subsequent work from the same group, the authors adapted the 3-channel system to study the basic mechanisms of cancer cell extravasation57, 59 and the organotropic (i.e., directed to a specific organ) extravasation of breast cancer cells to the bone microenvironment, which is a common site for breast cancer metastasis60, 61.
Figure 19.
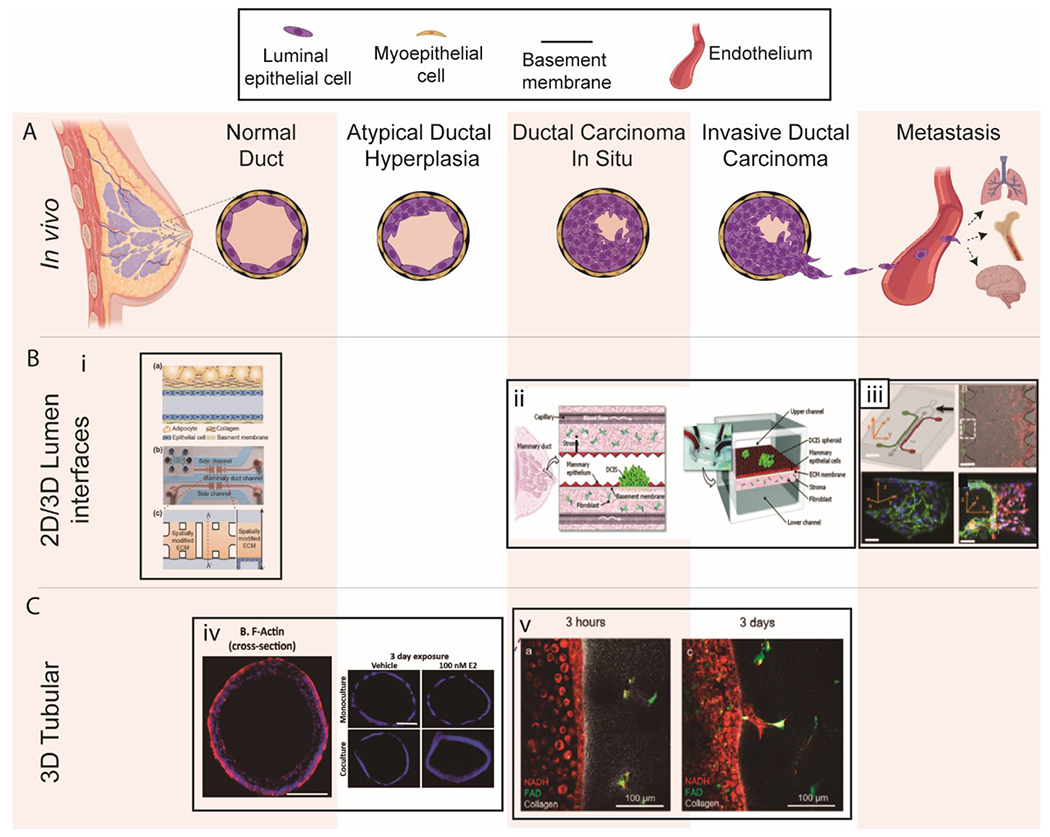
Overview of breast lumen models. (A) Schematic of breast cancer progression and metastasis. (B) 2D/3D lumen interface models. (i) a. Schematic illustration of a mammary duct b. Photograph of the 3D in vitro mammary duct remodeling model, c. Top and cross-sectional views of the microfluidic device203. (ii) Schematic of DCIS. This model used DCIS spheroids and a fibroblasts stromal layer on opposing sides of an ECM-derived membrane that mimics a basement membrane in vivo202. (iii) Microfluidic tumor-vascular interface model. (Top left) Endothelial channel(green), tumor channel (red), and 3D ECM (dark gray) between the two channels. (Scale bar: 2 mm) (Top right) Phase contrast images showing the fibrosarcoma cells (HT1080, red) invading through the ECM (grey) toward the endothelium (MVEC, green). (Bottom) A single 3D ECM hydrogel matrix region is outlined with the white dashed square. (Scale bar: 300μm). 58 (C) 3D tubular lumen models. (iv)Cross-sectional view of normal MCF7 ducts and hyperplastic lumens after exposure to E2 Scale bars are 100 μm14. (v) Transition from DCIS lumen models to invasive ductal carcinoma models. Copyright (2012) National Academy of Sciences for top-right images, ref 58. Reproduced from ref. 203 with permission from Springer Nature, copyright 2018.
Later, Zhang et al. developed a model to examine the extravasation of salivary gland adenoid cystic carcinoma cell aggregates62. CXCL12 signaling was found to enhance the transendothelial migration of the aggregates. This effect was effectively blocked after treatment with AMD3100, a CXCR4 antagonist (i.e., a receptor of CXCL12), highlighting its potential role as a therapeutic agent. Furthermore, Wong and Searson developed an organotypic blood vessel model for live-cell imaging of the intravasation process63 (Figure 6). The authors generated 3D lumens in collagen type I gel using Nitinol rods and subsequently lined with HUVECs. Importantly, continuous intraluminal fluid flow was applied to simulate in vivo circulation. Time-lapse imaging captured the invasion and intravasation dynamics of MDA-MB-231 and HT1080 cells co-cultured in the matrix surrounding the blood vessels.
Figure 6.
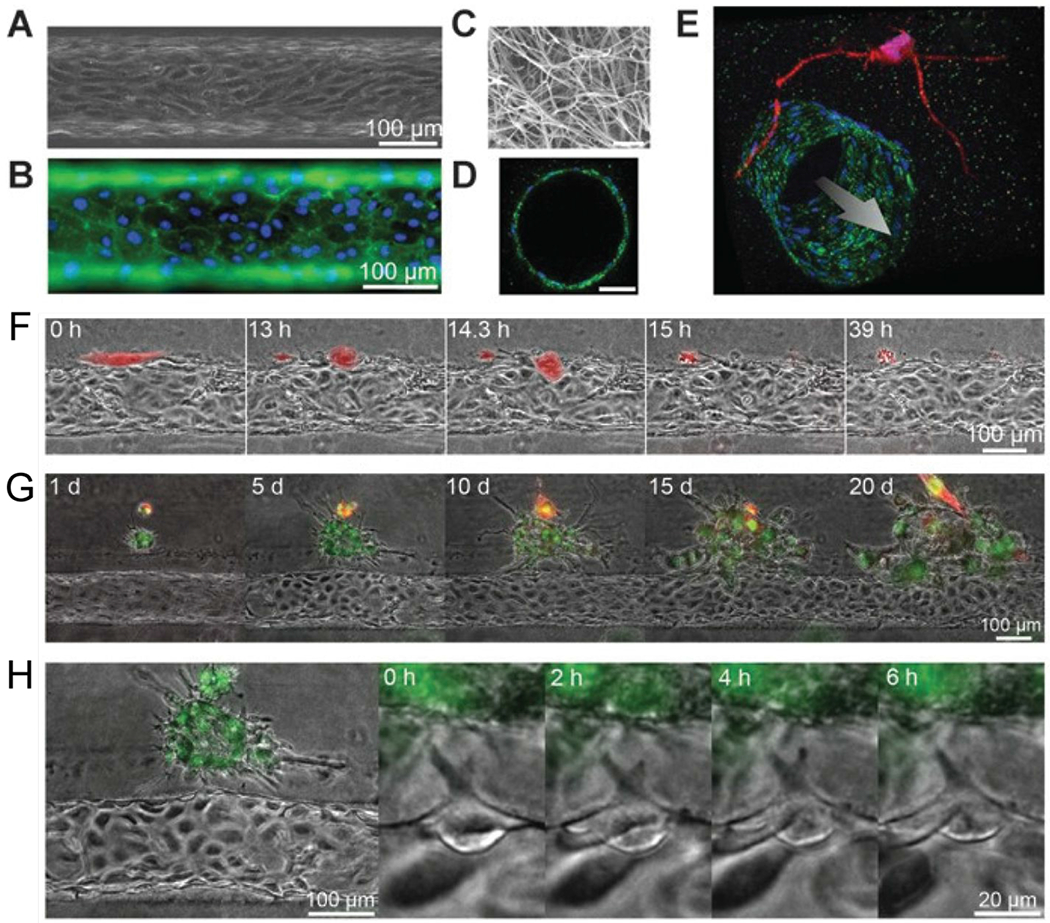
An organotypic blood vessel model for live-cell imaging of tumor invasion and intravasation. (A) Image of tubular HUVEC vessel formed in collagen type I gel. (B) Staining of CD31 (green) and nuclei (blue). (C) Scanning electron microscope image of collagen matrix. (D) Confocal image of vessel cross-section stained for CD31 (green) and nuclei (blue). Scale bar is 50 μm. (E) Confocal image of a vessel (green) co-cultured with HT1080 cells (red). Time-lapse images of (F) a single MDA-MB-231 cell (red) intravasating into a vessel, (G) a cluster of MDA-MB-231 cells (green) interacting with a vessel, and (H) angiogenic tip cell formation.63 Reproduced from ref. 63 with permission from the American Association for Cancer Research, copyright 2014.
In the context of tumor angiogenesis, the development of microfluidic models has primarily focused on platforms for screening anti-angiogenic drugs. Several studies have tested the efficacy of standard anti-angiogenic drugs, including bevacizumab, pazopanib, and sunitinib64. Despite their utility in assessing drug efficacy, most anti-angiogenic models continue to use non-primary cell sources. To improve the clinical translation capabilities of these models, Jimenez-Torres et al. recently developed a patient-specific organotypic blood vessel lumen model for testing standard of care anti-angiogenics for renal cell carcinoma65. Both normal and tumor-associated tubular vessels were generated using patient-derived endothelial cells. Vessels were treated with antiangiogenic drugs such as pazopanib and sunitinib, demonstrating highly variable drug responses including a compensatory increase in angiogenesis as a response to the treatment. This study exemplifies the value of taking a more personalized approach to drug testing, rather than using cell lines, to capture the heterogeneity in patient response. Overall, microfluidic tumor-vascular models continue to advance our understanding of tumor and vascular biology. While most vascular models still provide a simplified view of blood vessels (e.g., usually single endothelial layer, which is only accurate for capillaries, and a constant lumen diameter), the field is now turning toward organotypic vasculature and on pace with new advances in vascular biology. The physiological complexity of lumen-based microfluidic vascular models continues to increase (e.g., the addition of stromal and immune cells) and, in turn, their use for patient-specific drug testing. Leveraging these models could lead to the discovery and accurate testing of novel therapeutics.
3. Respiratory tract
The respiratory tract consists of the nose, throat, trachea, and lungs. The trachea gives rise to a network of increasingly branching and narrowing tubules within the lungs (i.e., bronchi and bronchioles), that end in clusters of microscopic air sacs (i.e., alveoli, 200-500 μm in diameter)66. The air-filled alveoli are surrounded by a capillary network, creating an ALI where the exchange between oxygen (from the lungs) and carbon dioxide (from the blood) occurs67. Structurally, the contact surface between alveoli and associated capillaries are separated by an ECM layer, which guarantees a high area of interchange, as well as structural support for both components. The structure-function correlation in the respiratory tract is especially highlighted in common pulmonary diseases, in which narrowing in the tubular geometry (or effective luminal diameter) of the airway results in multiple pathologies (e.g., chronic obstructive pulmonary disease (COPD), asthma, and cystic fibrosis (CF))68, 69. Mimicking the ALI and constant changes in the tensile stress of the lung in vitro presents many challenges70. Therefore, lung in vitro lumen-based models must capture the complexity required to provide physiologically-relevant responses68. In this section, we review microfluidic models applied to the respiratory tract. A summary of microfluidic respiratory tract models is provided in Table 2, and an overview of the models is shown in Figure 7.
Table 2.
Summary of microfluidic respiratory tract models.
| Organ geometry | Cell type | Targeted application | Refs |
|---|---|---|---|
| 2D membrane | HAEC HBEC |
-Asthmatic inflammation with IL-13 -Microbial injury with LPS -Evaluation of therapeutics |
78, 82, 98 |
| 2D gel interface | HAEC | -Nanoparticle toxicity on alveolar epithelium | 88 |
| Lumen (circulara) | LMVEC NPF Primary HBEC |
-Neutrophil extravasation toward fungal infection -Cytokine secretion response following exposure to volatile-producing fungi and bacteria |
92 |
Lumen with a circular cross-section
HAEC – human alveolar epithelial cell
HBEC – human bronchiole epithelial cell
LMVEC – human lung microvascular endothelial cell
NPF – normal pulmonary fibroblast
Figure 7.
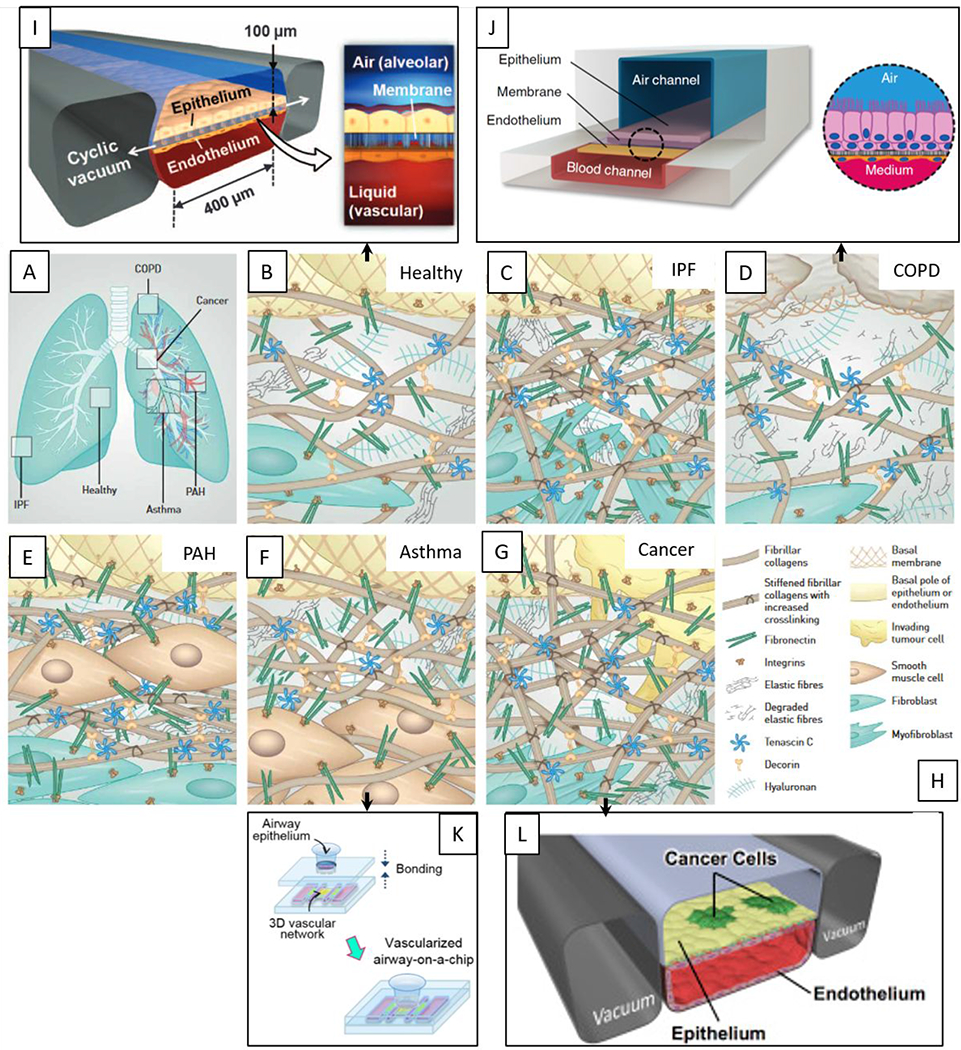
Overview of microfluidic device designs to mimic the lung. Pathological changes within the interstitial extracellular matrix (ECM) in the diseased lung. (A) Overview of the lung and sites of disease formation. (B) Healthy lung ECM is maintained by fibroblasts and represents a loose meshwork of protein anchored to the basal membrane of the epithelial cell layer. (C) In idiopathic pulmonary fibrosis (IPF), fibroblasts transdifferentiate to highly contractile myofibroblasts, depositing ECM and increasing its rigidity. (D) Chronic obstructive pulmonary disease (COPD) is characterized by the secretion of enzymes that degrade normal ECM, along with new ECM deposition by inflammatory cells. (E) In pulmonary arterial hypertension (PAH), remodeling of the ECM within the arterial wall is characterized by an increase in ECM proteins and hyperplasia of smooth muscle cells. (F) In asthma, the characteristic ECM changes take place beneath the bronchial epithelium and thickened basal membrane. (G) In cancer, tumors at primary and metastatic sides are surrounded by an extensive stiff stroma that contains highly crosslinked collagens, and specific proteins. (H) Legend depicting molecules and cell types. Reproduced with permission of the © ERS 2020: European Respiratory Journal 2017 50: 1601805; DOI: 10.1183/13993003.01805-2016.76 (I-L) Existing microfluidic models are linked to their specific ECM patterns. (I) Microfluidic organotypic model of normal lung function95, (J) COPD82, (K) asthma86, (L) lung cancer77. Reproduced from ref. 82 with permission from Springer Nature, copyright 2015. Reproduced from ref. 86 with permission from IOP Publishing, copyright 2018.
3.1. Modeling normal respiratory tract function
Establishing basic structure and function.
Most in vitro lung lumen-based models consist of lung epithelial cells, and human microvascular endothelial cells (HMVECs) seeded on opposing sides of a synthetic membrane. This setup allows the study of interactions between the lung epithelium and vascular endothelium, such as gas exchange across their interfaces68. Using this simple format, early organotypic microfluidic models integrated mechanical stimuli in different forms. Firstly, the model by Nalayanda et al. featured 2D lumen interfaces in a PDMS-glass serpentine device using A549 lung epithelial cell line and commercial HMVECs71. This model created a gas exchange interface when fluid flow was incorporated and first demonstrated lung-microvascular endothelial co-culture in a microfluidic platform. Sellgren et al72 took this model one step further and developed the first microfluidic Transwell-like model, where human microvascular endothelial cells and lung epithelial cells were seeded on opposing sides of a porous membrane. They achieved this by creating a three-layer PDMS device with membranes sandwiched between them, instead of the physiological ECM. The membranes were collagen IV-coated Polytetrafluoroethylene (PTFE) and Polyethylene Terephthalate (PET) membranes, both with 0.4 μm pore size. The reported pore size was consistent with that used for other devices, and, similarly to ECM, allowed the permeation of most solutes, including proteins; most of which have a hydrodynamic radius in the range of a few nm73. Their system also incorporated fluid flow, with minimal shear stress to prevent lung injuries described both in vitro and in vivo. Interestingly, this was one of the first studies to match lung cell layer thicknesses in their system to in vivo values from histologies, which is critical in vivo in ensuring efficient solute exchange in the ALI. They also observed mucus deposition on their lung epithelial layers and the appearance of specialized lung epithelial structures (i.e., cilia) both characteristic features observed in vivo, and responsible for stabilization of the ALI. Later, Humayun el al. developed a similar model using a polymethyl methacrylate (PMMA)-based device and in place of porous membranes, the authors generated the lung-vascular interface by culturing cells on a suspended fibronectin-coated collagen Type I hydrogel. Using this model, they reproduced similar physiological hallmarks as those described by Sellgren et al72 (i.e., mucus production, cilia). More recently, Zampagno et al. generated a lung-vascular interface using a thin gold mesh to support their collagen and elastin ECM74, similar to the suspended hydrogel approach reported by Humayun et al75. Both of these studies incorporated natural matrices to mimic the physiological stiffness of lung tissue (~200 Pa), which is critical to the mechanical deformation of the lung. Likewise, the incorporation of additional ECM proteins (i.e., fibronectin) increases the relevance of the model. However, further incorporation of additional ECM proteins, such as tenascin C, decorin, and hyaluronan, would provide a more complete model of the healthy lung microenvironment and help to elucidate the role of the ECM proteins in lung function76.
These two latest efforts traded the mechanical stimulation provided by the shear stress by generating an ECM-based microenvironment more accurate to that observed in the tissue. Therefore, another generation of 2D lumen interface studies included this tensile stress in their models.
Mimicking breathing tensile stress.
Recent reports indicate that the mechanical stimulus derived from breathing motions may play a more relevant key role in lung cell behavior and specialization than was initially thought77. In a seminal paper published in 2010, Huh et al. demonstrated their “breathing” lung-on-a-chip model (Figure 7 I)78. This model consisted of a multi-layered PDMS-based device with a collagen-coated PDMS-based porous membrane separating two vertically stacked microfluidic channels. An alveolar compartment was created by seeding an alveolar epithelial cell monolayer in the top channel and the bottom channel was patterned with a 2D HUVEC lumen interface to form a perfusable endothelial vessel. An air-liquid interface (ALI) was recreated in this device, mimicking the in vivo ALI in a microfluidic device for the first time. Vacuum applied in two flanking chambers allowed the authors to mimic “breathing” (i.e., cyclic straining of the alveolar epithelium). A similar model was later presented by Stucki et al. using a diaphragm-inspired pneumatic set up to study the effects of mechanical breathing on epithelial permeability, metabolic activity, and cytokine secretion79. In this study, the PDMS-based porous membrane was coated with gelatin and collagen, in an effort to include multiple ECM microenvironmental cues. While this model had a higher focus on the physiological effects of breathing motions in the epithelia than its predecessor, it lacked control over the deposition and durability of the coating, and did not include an ALI.
Mimicking 3D luminal structure.
The previously described models relied on seeding cells on 2D surfaces, which does not capture the 3D architecture of the lung. An early demonstration of an in vitro tubular lumen system is the ‘biofiber’ culture, which studied ion transport and volume flow in the lung80, 81. This model consisted of a permeable cellulose-ester tubular hollow biofiber coated with rat tail collagen type I. The biofiber had an inner and outer diameter of 0.65 mm and 1.22 mm (close to the physiological range of 0.2-0.5mm, thereby resembling in vivo transport ratios), respectively, and was collagen-coated to improve cell adhesion. This culture system was one of the first ‘micro’ scale demonstrations of an in vitro 3D luminal airway model, even if not traditionally considered microfluidics-based. The biofiber system was engineered to ensure epithelial cells (i.e., primary canine bronchial epithelial cells) lined the inside of the luminal structure. Grubb et al. later leveraged this model to study the relationship between ion transport and the airway surface liquid (ASL) volume flow in the normal airway epithelium80. The biofiber model offered two main advantages over traditional 2D methods (e.g., Transwell). First, the cylindrical geometry of the biofiber allowed for a larger cell surface area to volume ratio compared to 2D models, making it more suitable for measuring the flow volume. Second, the intra-luminal media was easily collected through a syringe for compositional analysis and less susceptible to evaporation as compared to the thin fluid layer above cells grown on planar surfaces. Finally, this system allowed for the detection of volume flow changes resulting from exposing the epithelium to different compounds, such as cholera toxin, amiloride, and forskolin. However, the relevance of the stiffness of this model and ECM microenvironmental cues is an unaddressed question about this model.
3.2. Modeling respiratory tract diseases
Chronic respiratory diseases: Chronic obstructive pulmonary disease (COPD), asthma, and cystic fibrosis.
In a similar model to the one demonstrated by Huh et al.,78 Benam et al. developed a small airway-on-a-chip device to study bronchiolar inflammation during asthma and COPD (Figure 7J)82. Here, the authors simulated asthmatic inflammation by stimulating the bronchial epithelial interface with IL-13, an inductor of biological responses related to asthma. After eight days of stimulation, epithelial cells upregulated the secretion of inflammatory cytokines, and on-site neutrophil recruitment increased as well. These responses demonstrate the ability of the model to elicit in vivo events in the presence of an pro-asthmatic agent. Moreover, to simulate pathogenic infection of the bronchial epithelium, normal or COPD epithelial cells lining the membrane were treated with LPS. The authors further tested the therapeutic response of the modeled small airways by successfully treating their model with relevant asthmatic and COPD drugs. In a follow-up study, Villenave et al. exacerbated the asthmatic response by using cells infected by a human rhinovirus83. This platform was further applied to evaluate responses to anti-inflammatory drugs. Overall, the question remains of the effect that these drugs would have in more complex asthmatic situations, including an inflamed tubular structure of the airway. An example of this is the different proinflammatory induction profiles secreted by fibroblasts, which are critical in COPD but were not included in this study.
A unique study published by Nesmith et al. mimicked only the airway musculature on a chip and its response to allergic asthmatic episodes, as induced with IL-1384. Their model consisted of a single contractile muscle band attached on a surface. The muscles in the airway are known to be the main component responsible for the severity of the asthmatic microenvironment76. With this model, they observed and measured bronchial muscle contraction and relaxation, which was successfully inhibited with a Rho kinase inhibitor, as observed in in vivo mouse models. Another interesting study published by Punde et al. mimicked the bronchial epithelium on a porous silicon membrane85. They used this setup to study fibrocyte (i.e., circulating cells that produce connective tissue) extravasation and their role in depositing ECM, leading to fibrosis. Their inclusion of fibrocytes is unique in the collection of studies we describe and is also representative of the physiological state that was mimicked in their study. However, a more integral view of the airway is necessary to accurately mimic asthmatic responses in lumen-based models.
An example in this direction is the recent asthma model described by Park et al., which used a classic PDMS device with stacked channels separated by pillars. In the central microchannel of this device, they generated a self-assembled 3D microvascular network and used the flanking channels for media exchange (Figure 7 K)86. To generate their lung compartment, on top of the device, they placed an additional module. This module, which generated the ALI, consisted of a printed 2D interface composed of fibroblasts and epithelial cells on top of decellularized porcine tracheal mucosa. Similar to previous studies, they used IL-13 to induce an asthmatic state, and they increased the response of the model by adding an inflammation exacerbating agent (i.e., dust mites). Their model exhibited mucus secretion and elicited an upregulation in the endogenous levels of TNFα upregulated. This study was one of the first ones to include a nativelike ECM with relevant cues and the presence of fibroblasts, recapitulating smooth muscle cells characteristic of the asthmatic lung microenvironment. Unlike muscle cells, fibroblasts actively secrete many proinflammatory molecules and modify the ECM87. The contribution of the fibroblasts could contribute to the phenotype observed in the model, yet it was not discussed in this work.
Cystic fibrosis (CF) is an autosomal recessive disease primarily characterized by chronic endobronchial airway infection69, which results in progressive blockage of the bronchi with mucus. This complication ultimately leads to respiratory failure, which is the primary cause of death in patients with CF. Matsui et al. used the biofiber platform alongside Transwell cultures to study whether ion transport is linked to ASL in the CF airway epithelium81. A challenge in this study was that less than 5% of cells harbored cilia after two weeks of culture, limiting the study of mucus transport in the CF epithelium. This lack of epithelial differentiation is a common challenge for luminal in vitro cultures of the lung. However, to the best of our knowledge, this is the only organotypic CF model currently published. Building upon this study, there is an opportunity to develop more physiologically complex lumen-based models that could be useful for both basic and applied CF studies.
Occupational lung disease/toxicity.
The increasing prevalence of environmental micro- and nano-particulates has prompted researchers to develop models to study the impact of toxic volatiles (e.g., cigarette smoke, asbestos) on respiratory health. Zhang et al. developed a 3D lung-on-a-chip model to assess the toxicity of nanoparticles on the alveolar epithelium88 (Figure 8A–D). The model leveraged the 3-channel microdevice design, where a middle ECM channel was flanked by an alveolar channel and an endothelial 2D lumen interface. Titanium dioxide and zinc oxide nanoparticles were added to the system as environmental irritants. Their toxicity was measured by evaluating alveolar barrier integrity and viability. In both cases, the addition of nanoparticles significantly increased the permeability of the alveolar epithelium by upregulating reactive oxygen species production and decreased cell viability. This model was the first to demonstrate the applicability of microfluidic lung models for environmental toxicology studies and provides a basis for future similar work.
Figure 8.
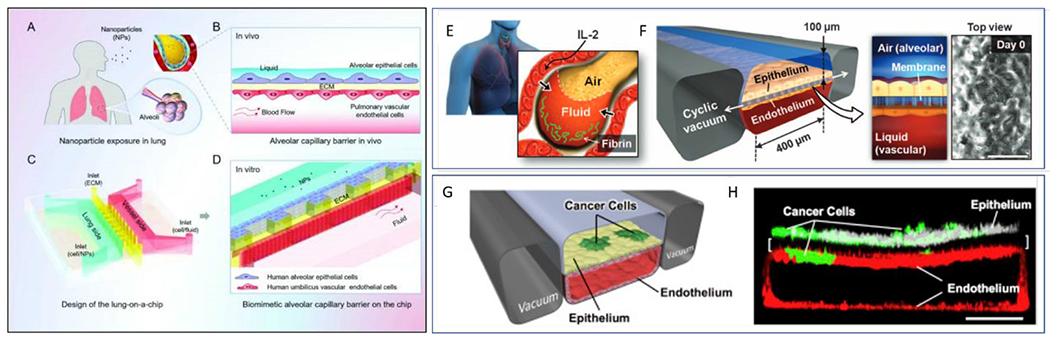
Overview of microfluidic device designs to mimic the lung-capillary interactions in the alveolus. (Left) (A) Diagram of the nanoparticle exposure to human lung. (B) Alveolar-capillary barrier in vitro. (C) Design and structure of the lung-on-a-chip. (D) Zoom-in showing the artificial alveolar-capillary barrier on the chip. Epithelial and endothelial cells are seeded on opposite sides of a Matrigel barrier. Media was flown through the endothelial channel88. (Top right) (E) Schematic of alveolus Lung-on-a-chip model comprising compartmentalized PDMS microchannels to form an alveolar-capillary barrier. Alveolar epithelial and endothelial cells are co-cultured on porous PDMS membrane coated with ECM. (F) Physiological cyclic strain or ‘breathing’ is generated by applying vacuum to the side chambers95. (G) Schematic diagram of device containing human lung epithelial cells and a low density of NSCLC tumor cells cultured on the upper surface of a porous ECM-coated membrane with an endothelial square cross-sectioned lumen. Physiological breathing motions were also mimicked in this device. (B) Confocal fluorescence micrograph of a cross-section of alveolus chip, showing lung cancer cells (green, anti-GFP), primary lung alveolar epithelial cells labeled with ZO-1 (white). A square cross-section endothelial cells lumen is created on the bottom microchannel and labeled with anti-VE-cadherin (red). (Scale bar, 200 μm)77. Reproduced from ref. 88 with permission from Oxford University Press, copyright 2018. Reproduced from ref. 77 with permission from The American Association for the Advancement of Science, copyright 2012.
Acute respiratory illnesses.
Respiratory infections are a major global concern of increasing interest and pandemic potential, especially among older adults and children89–91. This situation presents an opportunity for in vitro organotypic lumen-based models to help decrease the exceptional burden of respiratory infections in pre-clinical or personalized testing of new drugs. One of the most noteworthy efforts in this direction is the organotypic bronchiole model developed by Barkal et al. to study multi-kingdom and host-pathogen interactions within the human bronchioles92 (Figure 9). The model comprised three tubular lumens in a 3D hydrogel matrix: two lumens were lined with primary lung microvascular endothelial cells, and one lumen was lined with primary human bronchial epithelial cells. Normal pulmonary fibroblasts were embedded within the matrix to mimic the stromal compartment of lung tissue. Fluid from the center epithelial lumen was aspirated to allow air to fill the lumen and create an ALI. The result was a device that recapitulated the architecture of the bronchiole with lung vasculature. The authors incorporated the fungus Aspergillus fumigatus and the bacteria Pseudomonas aeruginosa due to their dominance as respiratory pathogens, as well as their cohabitation relationship in vivo93. After the fungi formed hyphal structures that penetrated through the epithelial layer into the collagen matrix, neutrophils extracted from whole human blood were injected within the endothelial lumen to study the immune response. This setup enabled real-time analysis of neutrophil extravasation and migration through the endothelial layer and collagen matrix towards the site of infection. The use of organotypic models in reproducing respiratory infections in vitro has thus far been minimal. Given the increasing burden of respiratory infections in the health systems around the world, and the recent advances made in lumen-based model technology we believe the development of more multi-kingdom models specific to other infections (e.g., tuberculosis, pneumonia or coronavirus infections) could be very beneficial in developing new treatments and precision medicine solutions68.
Figure 9.
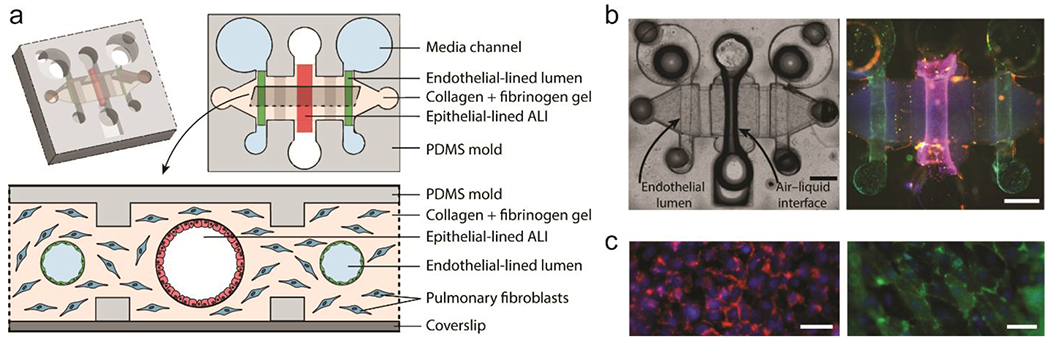
Organotypic bronchiole model to study multi-kingdom interactions in the human lung. (a) Schematic of organotypic bronchiole model. A central bronchiole epithelial lumen filled with air is flanked by two endothelial vessels. (b) Representative image of organotypic bronchiole model device with air in the epithelial lumen and media in the endothelial lumens. Staining of nuclei (Hoechst, blue), CD31 (green), and EpCAM (red). Scale bars are 500 μm. (c) Zoomed image of epithelium in the center lumen (left) and endothelium in the side lumens (right). Scale bars are 200 μm. Adapted with permission from Barkal, 201792.
Other efforts in mimicking acute respiratory illnesses include a few organotypic lumen interface models to mimic lung edema, thrombosis, and fibrosis. Felder et al. studied idiopathic pulmonary fibrosis (IPF) using a straightforward 2D lumen interface platform with hydrodynamic focusing capabilities94. IPF is thought to occur as a result of repeated alveolar epithelium micro-injuries. Therefore, these authors generated a gastric enzyme-induced microinjury tunable in size and successfully studied wound healing in a lung fibrosis model. In a follow-up model from their influential lung-on-a-chip model, Huh et al. developed a lung edema model to reproduce drug toxicity-induced pulmonary edema observed in human cancer patients treated with interleukin-2 (IL-2)95. They found that mechanical forces associated with physiological breathing motions play a crucial role in the development of increased vascular leakage that leads to pulmonary edema (Figure 8E–F).
On the other hand, Jain et al. used a PDMS device to mimic lung thrombosis, which is a lung obstruction due to a blood clot. Their device consisted of two stacked channels separated by a porous membrane. On the top channel, they had a 2D lumen epithelial interface to generate the ALI. In the bottom channel, the authors seeded endothelial cells to generate a 3D endothelial lumen with a square-shaped cross-section. Cells were seeded directly on the collagen and fibronectin-coated walls of the channel. Through this endothelial lumen, they perfused whole blood. The authors observed the effects of administering LPS or the pro-inflammatory molecule TNFα through the vascular lumen. Conversely, these effects were not observed when the treatments were administered directly onto the epithelium72. The authors observed a platelet accumulation consistent with thrombus formation, as well as a disruption of the endothelium. Finally, the potential of the model for pharmacological screening was demonstrated by testing a protease-activated receptor-1 (PAR-1) antagonist drug in their system.
Lung cancer.
To date, few studies have produced efforts toward mimicking lung cancer in microfluidic lumen models96. One model reported by Xu et al. This model used a PDMS-based device to create lumen 2D lumen interfaces with SPCA-1 cancer cells97. The authors also used primary lung cancer cells to produce patient-specific models and tested their response to commonly used chemotherapies (i.e., gefitinib, paclitaxel, and gemcitabine) by functionally measuring apoptosis of the cancer cells.
Recently, Hassell et al.,77 developed a lung cancer model with breathing motions as a means of mechanical stimulations, similarly to that of Jain et al98. Specifically, on both sides of a porous membrane they had a top channel containing a normal human lung epithelium and a bottom channel with an endothelial lumen channel. Non-small cell lung cancer cells were added to the normal lung epithelium channel to study tumor dormancy and clone growth. Interestingly, the study revealed that mechanical breathing could reduce cancer cell invasion of the vascular lumen by ~50% (Figures 7L and 8G–H). This result highlights the role of spatial cues in cancer progression and treatment, and the importance of including these cues in in vitro models. The results also suggested that mechanical stimuli affect the sensitivity of the cancer cells to two TKI therapies (i.e., erlotinib and rociletinib). Given the importance of the tissue microenvironment in cancer development and progression, newer models should tackle the role of microenvironmental factors, including hypoxia, angiogenesis, matrix stiffening, and immune infiltration, in the progression of lung cancer96. To accurately mimic this cancer for both basic and translational studies, the importance of including these events must be carefully considered. We expect that these and other questions will be explored soon.
Overall, microfluidic lung models have recapitulated multiple aspects of the structure-function relationships of the respiratory tract. However, only a few studies have incorporated the tubular geometry of the airway, as compared to the many existing vascular lumen models. Creative solutions to this challenge will be needed, given that the airway operates under unique biphasic conditions of air and liquid. Furthermore, airway geometry does not stop at simple cylinders but includes a rich architectural milieu of symmetric and asymmetric bifurcations of the airway tree. Future approaches may build on the ones presented here to generate organotypic models that are simple, and yet also represent the characteristic luminal geometry present in the respiratory system. Finally, we anticipate that more sophisticated applications of these models will arise soon, following the lead of the studies described in this section, that illustrated the potential of lumen-based models for biomedical applications.
4. Gastrointestinal tract
The gastrointestinal (GI) tract is a lumen-based organ system that includes the mouth, esophagus, stomach, and intestines. Except for a few unique reports, such as the recent “stomach-on-a-chip”99, efforts to mimic the GI tract have mainly focused on the intestine. The intestine or gut is composed of the large and small intestine (7.5 and 2.5 cm inner diameter, respectively). It presents several different layers: (1) the mucosa, composed of the intestinal epithelium, glands, and a layer of connective tissue called the lamina propria. Subsequent layers include (2) the submucosa, which consists of connective tissue, with larger blood vessels, lymphatics, and nerves; (3) smooth muscle layers and (4) a final connective tissue layer17. Given this complex structure, most efforts have focused on mimicking the intestine epithelium and other supporting components of the mucosa. Further, the intestinal epithelium is covered with a dense layer of mucus (i.e., organized glycoprotein network) to house gut microbiota (i.e., gut microorganisms). It is this same mucus layer that prevents the translocation of the gut microbiota into underlying tissues. The gut microbiota is key to intestinal health and immune modulation, among other emerging relationships, thereby receiving much attention lately100.
The gut carries out multiple and diverse functions by leveraging this specific structure, including nutrient digestion and absorption, and metabolism of certain drugs. As we have described in this section, the gut interacts closely with several organ systems. A noteworthy example is the interaction of the gut with the vascular and nervous system, forming the gut-brain axis101. Therefore, when mimicking the human intestinal physiology and pathophysiology, it is necessary to consider context-dependent factors and contributions from accessory organs. In this section, we will discuss efforts directed to mimic the basic structure and functions of the gut, as well as gut interactions with the microbiome and the immune system. Multi-organ systems, including a gut component (i.e., gut-kidney or gut-liver), will be discussed in a dedicated chapter. Finally, we will briefly discuss efforts to model gut-related pathologies. A summary of microfluidic models of the gastrointestinal tract is provided in Table 3.
Table 3.
Summary of microfluidic intestinal models.
| Organ geometry | Cell source | Targeted application | Refs |
|---|---|---|---|
| 2D membrane | Caco-2 | -Modeling gut structure and function -Microbial interactions with the gut epithelium -Modeling intestinal inflammatory diseases -Evaluation of anti-inflammatory probiotics and antibiotics |
103, 104, 109, 112, 115–117, 123 |
| Cell-lined channel | Caco-2 | -Drug-induced barrier dysfunction | 111, 126 |
| Lumen (circulara) | Caco-2 and myofibroblasts | -Modelling gut structure and function -Microbial interactions with the gut epithelium |
122 |
| Villi-shaped scaffold | Caco-2 | -Modelling gut structure and function | 106, 107, 110 |
Lumen with a circular cross-section
Caco-2 – Colorectal adenocarcinoma cell line
4.1. Modeling normal intestinal function
Establishing basic structure and function.
The gut has essential functions in drug absorption and metabolism. Therefore, producing models where these functions can be accurately reproduced is of great importance in preclinical drug development and toxicity screening. Many of the in vitro models used by the pharmaceutical industry use human intestinal cell lines (e.g., Caco-2 or HT-29 cells) cultured on ECM-coated 2D plates102. However, it is challenging to recapitulate key in vivo features of the gut using these platforms (e.g., villi formation, production of specific mucins), gut interactions with supporting tissues, and stable (i.e., over 24h) interactions with gut microbiota102. Microfluidic and organ-on-a-chip lumen-based models have emerged as alternative options to overcome many of these limitations. Early lumen interface in vitro models of the intestinal barrier consisted of a gut cell line cultured on top of a suspended synthetic porous membrane (i.e., polyester, polycarbonate, or PDMS)103. (Figure 10A–B) Imura et al. described an early model that used this microfluidic setup104. The authors used a glass-PDMS lumen interface microdevice with a PET permeable membrane (collagen I-coated polyester membrane with 1 μm diameter pores) and fluid flow. They used this setup to study gut absorption of cyclophosphamide (CPA, a common orally-administered chemotherapeutic). To quantify absorbed CPA, they performed a solvent extraction, and High-Performance Liquid Chromatography (HPLC) on media flowed through the system. Their model mimicked observations from previous literature, indicating that the gut is permeable to CPA. However, this effect was selective, as their model was not permeable to Lucifer yellow, a fluorescent compound often used in research and known not to be absorbed by the gut. Notably, while a membrane differs from a protein-based basal membrane in thickness and structure, the membrane pore size was adequate for this study, since it is much larger than the diameter of the molecules of interest. This study found limitations in acquiring enough sample volume for HPLC and proposed fluorescent determinations as an alternative method for future studies. While sample sizes remain a common limitation of microfluidic-based readouts, more modern Ultra-Performance Liquid Chromatography coupled with Mass Spectroscopy may be able to lower the sensitivity threshold enough to quantify diffusion coefficients in a similar setup. The question of a potential influence of PDMS in providing an accurate quantification also remains unanswered but overall, this model illustrated a useful application for more physiologically relevant gut models.
Figure 10:
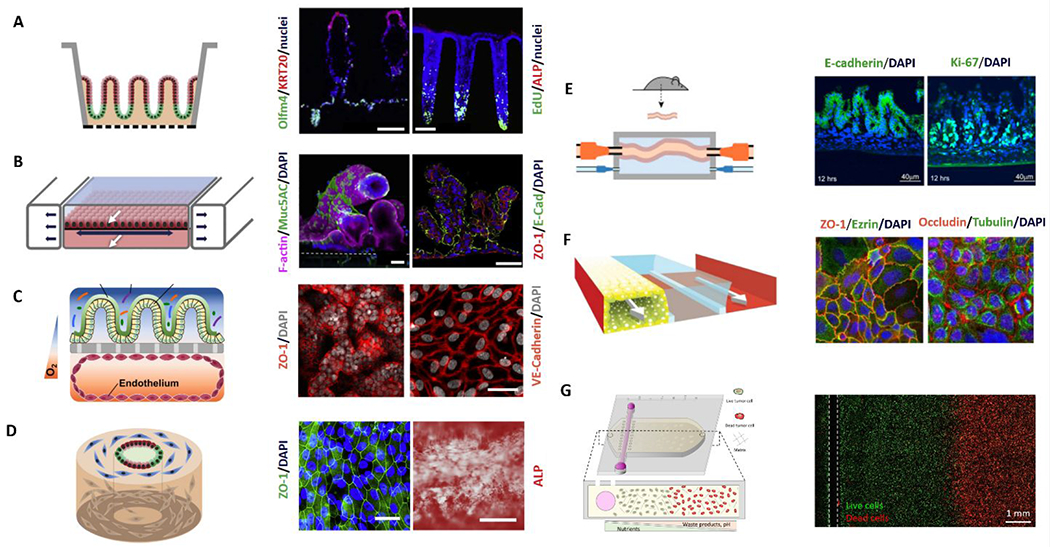
Overview of microfluidic device designs to mimic the gut. On the left of each panel, a schematic of the model is shown; and on the right, annotated representative microscopy images of the resulting model. (A) 2D monolayers on micro-molded scaffolds106. (B) Microfluidic lumen interface device with integrated peristalsis. White arrows mark fluid flow while dark arrows indicate the motion of the stretchable surface112. (C) Epithelial lumen interface on top of a microporous membrane cocultured with a square cross-sectioned endothelial lumen on the bottom. (D) Silk scaffolding tubular model with fibroblasts (blue), proliferative epithelial cells (green), and epithelial cells (red) 120, 122. (E) Microfluidic ex-vivo gut co-culture with immune and brain cells from rats. Intact intestinal tissue is connected to input and output ports of the chamber (top), pumps controlling medium flow inside the lumen and in the external medium chamber118. (F) Channel network from the Organoplate platform containing cultured gut epithelial tube, ECM gel, and perfusion111. (G) Gut tumor-on-a-chip gradient platform.127 Reproduced with permission from Elsevier, from ref. 106 copyright 2018; from ref 120 copyright 2019; from ref 118, copyright 2017. Ref. 111 adapted with permission of Springer Nature, copyright 2017.
Adding 3D extracellular matrix.
The studies mentioned in the previous section focused on the generation of 2D gut epithelial monolayers, whereas the gut is a 3D tubular organ, with many villi and crypts increasing its surface area for improved solute absorption. Specifically, intestinal villi have specialized transporters responsible for the absorption of macromolecules into the bloodstream, and therefore critical to correct gut function. To develop models that account for this 3D villi morphology, a small subset of studies have focused on using micromolding techniques, such as the early study by Sung et al105. These techniques can form scaffolds in the shape of microvilli structures, which can be lined with intestinal epithelial cells (e.g., Caco-2). Kim and Kim produced another early example of a villi template by 3D printing porcine collagen106. The authors then seeded Caco-2 cells to create a denticulated gut epithelial model with defined luminal spaces (Figure 10A).
Other authors have developed scaffolds to explore the influence of ECM in gut epithelial models. A popular application of scaffolds is exploring the response of cells to different stiffnesses. In the case of the gut, increased stiffness has been correlated with pathological states (e.g., inflammation or fibrosis). A noteworthy application of this concept was the study published by Gunasekara et al107. These authors developed an ECM stiffness gradient model by crosslinking a soft collagen hydrogel (1 mg/ml, with stiffness values around 20 - 2500 Pa for the highly crosslinked). The crosslinking was achieved via a carbodiimide crosslinking reaction with 1-ethyl-3-(-3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (Sulfo-NHS) to increase crosslinking efficiency108. Mouse colonic epithelial cells were seeded on the villi templates to create their denticulated gut epithelial models. While these authors did not characterize in-depth the resulting stiffness gradient of their hydrogels, they observed lower cell proliferation on the crosslinked scaffolds, which differs from previous 2D reports. These results suggest that topography or ECM-derived cues play a role in regulating cell proliferation. These authors also evaluated barrier function using gene expression and TEER (TransEpithelial Electrical Resistance), a functional measurement that uses impedance to evaluate barrier function109. In a follow-up study, they demonstrated a higher expression of transport proteins for cells cultured in the crosslinked scaffolds (i.e., BCRP and MRP2) compared to those seeded in non-crosslinked scaffolds, therefore facilitating substrate transport110.
Attempts to better mimic cell-ECM interactions within microfluidic systems include the model developed by Trietsch et al.111, which they called Organoplate (Figure 10F). This device consisted of three-channels, delimited by capillary valves (or phase guides). The central channel was filled with a collagen hydrogel. Caco-2 cells were then seeded in the collagen-defined channel (i.e., instead of on a membrane), generating a square cross-section lumen within a side channel. Fluid flow was incorporated through the Caco-2 luminal structure. One of the novelties of the platform was its higher throughput and compatibility with standard well plates. (Figure 12). Array-based culture systems present an efficient and cost-effective alternative to conventional culture techniques and are growing in use. However, the higher throughput usually results in simplified models, which do not incorporate peristalsis (i.e., rhythmic waves of contraction to move food down the gut) or villi differentiation of the epithelia, and rarely include a microbial component.
Figure 12.
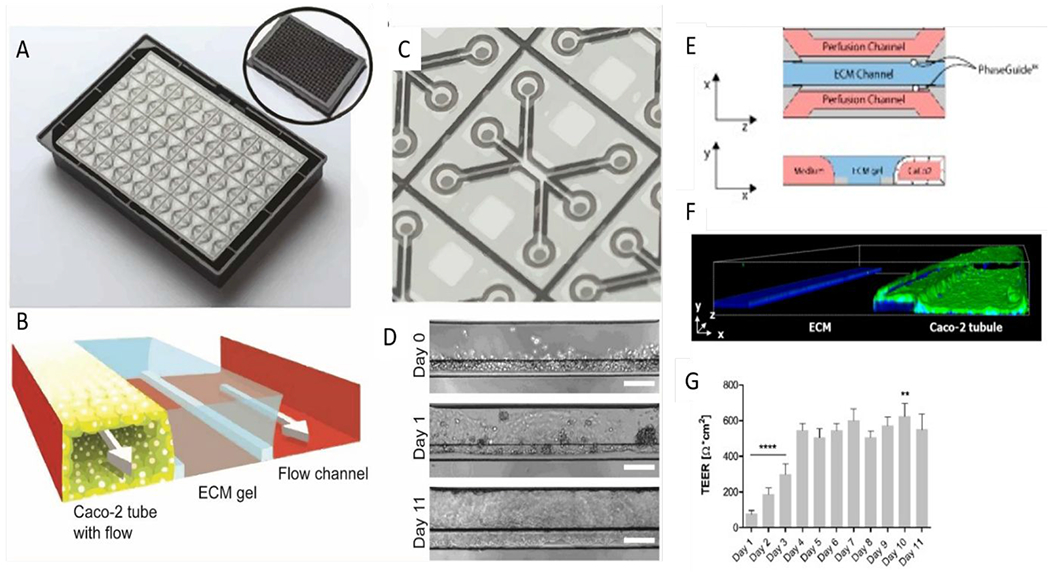
Studies using the Multiwell OrganoPlate intestinal platform. (A) Image of the bottom of an OrganoPlate showing 40 microfluidic channel networks. Inset shows top view of the 384-well plate device. (B) Zoomed view of a single microfluidic channel network comprising three channels. (C) Schematic of channel network containing cultured gut epithelial tube, ECM gel, and perfusion. (D) Brightfield images of Caco-2 tube formation over 11 days. Scale bars are 100 μm. (E) Transversal view of a microfluidic view; Caco-2 cells adhere to the ECM meniscus created by capillary valves. (F) 3D reconstruction image of a Caco-2 tubule at Day 4 stained for ACTIN (green) and DNA (blue), depicting the nuclei. (G) Transepithelial electrical resistance (TEER) values of Caco-2 tubules over time until Day 11. Data is represented as mean ± SEM. ** p < 0.01; **** p < 0.0001. Comparisons are performed against day 4. Ref. 111 adapted with permission of Springer Nature, copyright 2017. Reproduced from ref. 126 with permission from MDPI, copyright 2019.111, 126
Adding peristalsis and microbial components.
Peristalsis is known to be critical for normal human physiology102. Likewise, the microbiome is a component that contributes to the barrier function, metabolism, drug, and chemical absorption of the intestine. Together, these factors are critical for normal gut function. Therefore, the first models incorporating both factors were very influential in the field. Kim et al., from the same team that reported the seminal lung-on-a-chip model (i.e., Huh et al.78), developed a well-known organotypic gut model112, similar to their previous work. This model consists of two vertically stacked microchannels separated by a coated PDMS membrane and incorporates two flanking side channels for pneumatic application of cyclic strain that mimics the mechanical deformations during peristalsis. Unlike in previous models, the Caco-2 cells cultured in this model established crypt and villi structures and exhibited a barrier function. The authors also incorporated a normal intestinal microbe (Lactobacillus rhamnosus GG or LGG) in their model and maintained the co-culture for over a week, demonstrating β-galactosidase activity (Figure 10B, 11). A follow-up study demonstrated the expression of differentiation markers of Caco-2 cells for absorptive, goblet, enteroendocrine, and Paneth cells, as well as mucus production and higher cytochrome CYP3A4 activity (i.e., an enzyme implied in drug metabolism)113. This model constituted the start of incorporating microbiota-epithelial interactions. However, distinct microbiota have specific microenvironmental needs that remain to be explored in this model.
Figure 11.
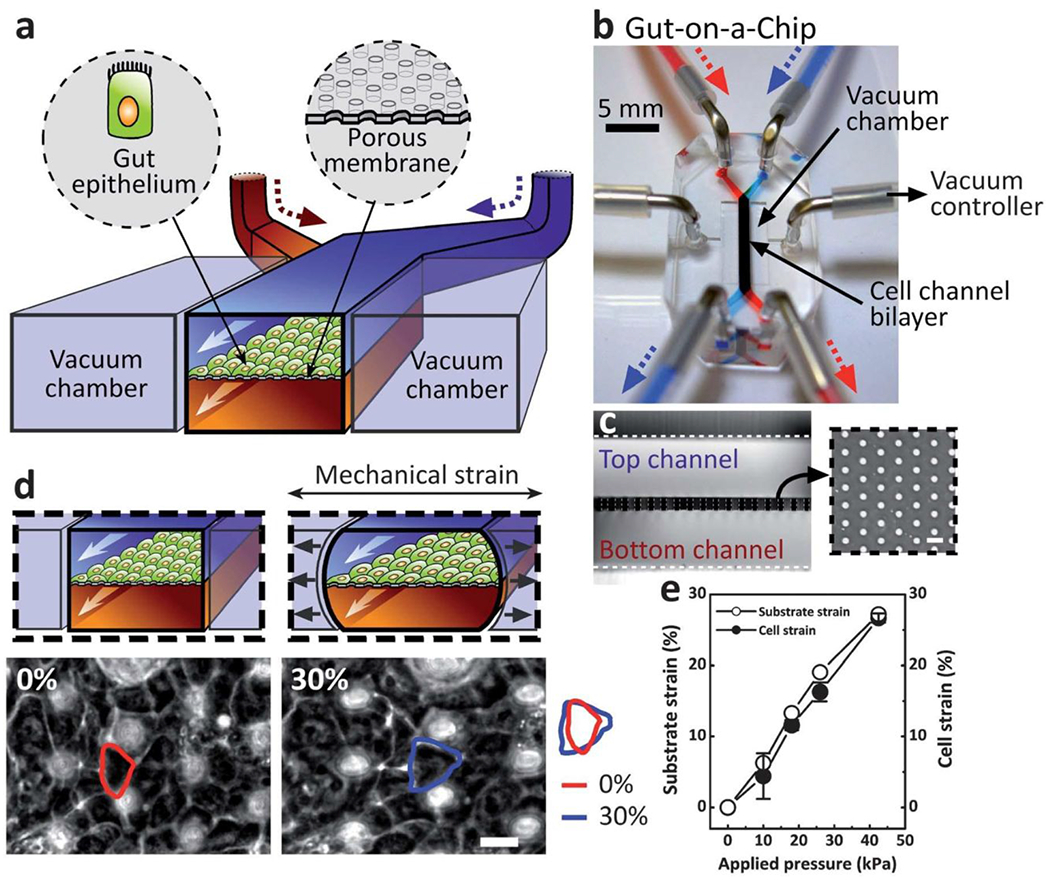
Gut-on-a-chip model to reconstitute intestinal structure and function. (a) A schematic of the gut-on-a-chip device showing ECM-coated membrane patterned with gut epithelial cells. Central microchannels allow perfusion and vacuum chambers enable peristaltic strain. (b) An image of the gut-on-a-chip device. A syringe pump controls fluid transport. (c) Cross-sectional view of the top and bottom channels. Inset shows a top view of the porous membrane (10 mm pores). (d) Intestinal monolayers cultured in the absence (left) or presence (right) of mechanical strain (30%; black arrows indicate direction). Red and blue outlines indicate the shape of a single Caco-2 cell before and after applied mechanical strain, respectively. The white circles are pores in the membrane beneath the epithelial monolayer. Measurement of strain in the PDMS membrane (open circles) and cells (closed circles) with respect to applied vacuum pressure. Scale bars are 20 mm. Reprinted with permission from Kim, 2012112.
Adding oxygen gradients.
It is well-known that oxygen levels inside human tissues can be lower than atmospheric oxygen pressures (i.e., physiologic hypoxia). Physiologic hypoxia is of particular relevance in the gut where the individual oxygen supply of each villus is consumed by the gut and microbiome, reducing oxygen pressure incrementally to physiological hypoxia levels (10 mmHg)114. The oxygen gradients established within the gut villi generate different environments that allow microbiota to grow, according to their oxygen tolerance and metabolic needs.
One of the first examples to mimic hypoxia in an organotypic model of the gut epithelium was a modified hypoxia workstation with a transwell insert, on which a Caco-2 monolayer was seeded. Ulluwishewa et al. demonstrated improved survival of Faecalibacterium prausnitzii in the anaerobic apical environment compared with culture in aerobic atmosphere115. They also demonstrated that live F. prausnitzii induced differences in barrier function and gene expression profile of the Caco-2 epithelial monolayer. While this model was more straightforward than previous studies, the authors were some of the first to illustrate the importance of integrating anaerobic bacteria in organotypic models. Several microfluidic organotypic models have since then included anaerobic bacteria in their setups.
One of the first microfluidic models to include physiological hypoxia levels is a multi-layered microfluidic device, called HuMIX116. Shah et al. developed this model, consisting of 3 elastomeric silicone rubber serpentine channels separated by two nanoporous membranes (1 μm and 50 nm pore sizes, the latter coated with a mucin/collagen solution). This setup was sandwiched in 2 polycarbonate layers and connected to fluid flow. This model used the membranes as cell culture support for epithelial gut cells and commensal bacteria. Specifically, this model facilitates the co-culture of Caco-2 cells with facultative and obligate anaerobic human gut bacteria (i.e., Lactobacillus rhamnosus GG and Bacteroides caccae). To generate both under aerobic and anaerobic conditions required by the bacteria, the authors used constant anoxic and oxygenated media perfusion. In this work, Shah et al. demonstrated the differential effects of co-culture on the transcriptional response of the intestinal cells. However, this model is limited in its ability to mimic direct contact host-microbe relationships and downstream functional responses resulting from these interactions.
Another follow-up study from Kim et al. built upon this concept to incorporate different strains of commensal bacterial microbiota117. The authors modified their original design to integrate a transluminal hypoxia gradient. Likewise, this model included an intestinal vascular endothelial lumen with a square cross-section additionally to the Caco-2 intestinal epithelium. Co-culture with stool samples under anaerobic conditions showed growth of obligate anaerobic bacteria that were not observed under aerobic conditions or conventional liquid culture conditions (e.g., Firmicutes and Bacteroidetes). The ratios of these bacteria were similar to those found in human stool samples.
A unique study by Yissachar et al. produced an ex vivo mouse gut lumen model in co-culture with commensal microbes and neurons118 (Figure 10E). The authors introduced commensal microbes (e.g., C. ramnosum) known to induce a specific subset of T cells exclusive to the gut with important, yet not fully known roles in gut homeostasis (Treg)119. The co-culture of the gut lumen model with C. ramnosum led to changes in genes associated with neurobiological processes, such as a nociceptor neuronal function. The authors observed the activation of neurons by Treg as a consequence of co-culturing their system with different bacterial strains and provided insight into the crosstalk mechanism between bacteria and neurons in the lumen gut environment. This biologically-oriented report illustrates how ex vivo microfluidic models can be used to identify previously unknown complex physiological tissue interactions. However, a caveat that remains in most models introduced to date is the use of mouse-derived cells or immortalized cell lines that may limit the applicability of the model for human research.
Towards patient-specific gut models.
Primary gut cultures derived from intestinal crypts from tissue explants have recently made breakthroughs in gastrointestinal research and are now extensively used for studying both basic and clinical biology120. Dawson et al. reported one of the first organotypic microfluidic gut models developed using primary patient-derived tissue121. The authors encased intestinal sections from patients undergoing bowel resection in a PDMS-glass dual-flow chamber. Tissue viability was demonstrated using the lactate dehydrogenase (LDH) colorimetric cytotoxicity assay. This assay uses the LDH (and, consequently, NADH) released from dead cells to estimate viability. This reaction measures the reduction of a yellow tetrazolium salt (INT), by NADH into a red, water-soluble formazan-class dye via absorbance. Tissue structure after culture retained its original organization, as observed in histological stains. This report is one of the few where ex vivo gut lumen sections have been cultured in microfluidic devices and illustrates that normal tissue structure and viability can be maintained in these models.
Chen et al. recently reported a primary cell-derived tubular model of the human small intestinal epithelium supported by myofibroblasts122. This model consisted of a small intestine cell-lined tubular structure within a silk-derived scaffolding matrix (Figure 10D). Co-culture with myofibroblasts was supported within this silk-based matrix. Leveraging this model, the authors demonstrated mucus deposition and generation of microvilli in their model. The tubular gut epithelium was also co-cultured with E. coli and exhibited an increase in antibacterial gene transcription (i.e., lysozyme and chromogranin). To our knowledge, this is the only tubular model of the gut epithelium and only co-culture with myofibroblasts to date, that captures both the native structure and stromal interactions in vivo.
Moreover, Kassendra et al. developed a patient-specific intestinal model analogous to that of Kim et al. using primary colon organoids derived from patient duodenal biopsies123. Villus morphogenesis and multilineage differentiation were demonstrated along with mucus production. The authors also obtained transcriptomic profiles of several models to illustrate patient-specific differences. While these models are in their early stages, the development of primary-derived tissue models is promising for personalized medicine applications.
4.2. Modeling intestinal diseases
Inflammatory Bowel Disease.
Building upon their established model, Kim et al. investigated the effects of inflammation in immune-microbiome interactions124. Their system was stimulated with LPS, which resulted in the release of proinflammatory cytokines (e.g., IL-1B, IL-6, IL-8, and TNF-α). This inflammatory response resulted in intestinal injury (i.e., villus blunting). Likewise, the expression of ICAM-1 on the endothelium increased, consistent with hallmarks observed in inflammatory bowel disease (IBD) patients125. Moreover, the cyclic mechanical strain of the gut epithelium was shown to contribute to bacterial overgrowth. Another study using this human intestine model examined the response of gut epithelium exposure to γ-radiation (Figure 10C). Radiation injury manifested in the form of cell death, villus blunting, and compromised barrier function. These responses were later shown to be mitigated by a potential prophylactic drug. A recent report has investigated IBD using the organotypic gut lumen model described by Trietsch et al111. These authors reported a similar IBD phenotype by stimulating individually with IL-1β, TNFα, and INF-γ during different periods of time126 instead of the LPS treatment used in other studies (Figure 11). Their results were consistent with those by Kim et al. and indicated an incremental effect of these cytokines in the IBD-like phenotype of the model.
Colon Cancer.
To date, we have only found two microfluidic colon cancer models in the literature. The first one, by Ayuso et al., leveraged a three-channel polystyrene-based microdevice to generate a confined 3D high-density (i.e., 40 million cells/ml) embedded culture of colon cancer cells127. Both flanking channels in the device acted as surrogate blood vessels (i.e., perfusable microchannels without endothelial cells). Gradient and glucose concentrations were demonstrated in this model, as well as their effects on cell proliferation and death. These authors also explored the migration of NK cells within the model and drug treatments targeting the tumor microenvironment. In a follow-up paper, these authors coupled their model with single-cell imaging flow cytometry to study cell death as a result of these gradients128. The second model was reported recently by Carvalho et al.,129, using a very similar microdevice design and model to that of Ayuso et al. 127; however, the flanking channels were used to coculture Human Colonic Microvascular Endothelial Cells. These authors explored angiogenic sprouting as a result of a VEGF gradient and nanoparticle-based therapies in their model. Specifically, the authors tested Gemtabicin-conjugated CMCht/PAMAM dendrimer nanoparticles, which diffused through their high-density co-cultures and induced apoptosis in the colon cancer cells in their model.
Overall, organotypic microfluidic gut models have made significant strides in mimicking the human intestine structure and function. However, several other features within the intestinal tissue may play a significant role in intestinal diseases and have, so far, not been modeled using these systems (e.g., pH variations in the stomach and gut, tubular structure of the gut). As discussed at the beginning of this chapter, the gut is a complex organ with many different cell types and interactions with other organs. Therefore it remains a challenge to determine what components will be necessary to include for each application. Notably, due to the large diameter of the gut (in the cm scale), the ability to mimic crypts in 2D lumen interfaces may provide enough of a 3D structure for most applications.
Significant interest in the field remains for these models, specifically for patient-derived tissue models, which could provide information on patient-specific disease mechanisms for applications in targeted therapies for genetic diseases like Crohn’s disease and ulcerative colitis130. Further studies are warranted to produce donor-specific chips that can help in designing and recommending effective treatment options. One area where microfluidic gut models can be impactful is in advancing our understanding of gut-microbiome interactions. We believe that these technologies hold great potential in unveiling the mechanisms of many pathologies in which the microbiome is thought to play a crucial role (e.g., allergies, degenerative disorders).
5. Renal tubules
The kidney filters blood by removing water-soluble metabolic waste from plasma, reabsorbs useful substances (e.g., glucose, amino acids), and maintains homeostasis of water and electrolytes17. Additionally, this organ has many essential functions, such as regulating the production of red blood cells, vitamin D, and glucose, and modulating arterial blood pressure. The functional unit of the kidney is the nephron, which comprises the glomerulus and the renal tubule (200 and 50 μm in diameter, respectively). The glomerulus is responsible for the filtration of blood plasma, and the renal tubule recovers water, nutrients, and electrolytes in the glomerular filtrate. These recovered substances are then reincorporated into the circulatory system. Early in vitro kidney models mimicked fluid transport and barrier function, especially the effect of shear stress on renal tubule cells131–134. In these models, a 2D renal tubule monolayer was recreated in a conventional cover slide-based flow chamber and subjected to different applied shear stress. The effects of shear stress were quantified via gene expression differences, immunofluorescence, and electrophoretic mobility shift assays. Most microfluidic kidney models have also focused on the filtration capabilities of the kidney and its transport of metabolites and exogenous solutes. The renal tubule and associated vessels have been of primary interest135. Conversely, the lack of stable relevant cells and the structural complexity of the glomerulus have hindered its model development136. In this section, we discuss the methods that have been used to generate renal tubules in microfluidic models. We also discuss the application of the models to kidney diseases, including nephrotoxicity, nephrolithiasis, and kidney cancer. Figure 13 highlights the milestones toward developing a kidney on a chip model, and a summary of microfluidic renal tubule models is provided in Table 4.
Figure 13.
Overview of microfluidic device designs to mimic the kidney. (A) Schematic of the structure of the nephron. (B) Magnification of a parallel renal tubule and renal capillary. Models depicted in this review mimic either one of the luminal structures or both. Reproduced structures and phenomena are delimited by the colored dashed lines. (C) Human kidney proximal tubule on a chip for drug transport and nephrotoxicity assessment.138 (D) Renal tubule model for screening nephrotoxic compounds in vitro152 (E) Fibrin-based model of the renal tubule for solute diffusion studies.141 (F) Intestine-kidney model to study drug absorption related nephrotoxicity.153 (G). Lumen-based microfluidic model of Renal Cell Carcinoma (RCC) for the study of tumor angiogenesis160 (H) Hydrogel-embedded 3D vascular networks to mimic transport between renal tubule and renal capillaries.139 (I) Organotypic kidney blood vessel models for anti-angiogenic drug response testing of RCC65. Reproduced from ref. 153 with permission from AIP Publishing, copyright 2017. Reproduced from ref. 141, with permission of Chee Ping Ng et al., copyright 2013. Reproduced from ref 160, with permission of Miller et al., Copyright 2019. Reproduced from ref. 152, with permission of the American Society of Nephrology, Copyright 2016.
Table 4.
Summary of microfluidic renal tubule models.
| Organ geometry | Cell type | Targeted application | Refs |
|---|---|---|---|
| 2D channel | HK-2 MDCK RPTEC |
-Effect of shear stress on renal epithelia morphology, cell adhesion, and ammonia metabolism -Kidney injury and inflammation due to excessive shear stress -Liver-kidney crosstalk in nephrotoxicity |
132, 133, 137, 150, 186 |
| 2D membrane | ARPC GEC HK-2 IMCD |
-Effect of oxidative stress on cell viability -Recapitulation of solute transport mechanisms -Artificial dialyzer for filtration of metabolites and reabsorption of nutrients -Nephrotoxicity induced by gentamicin -Intestine-kidney crosstalk in nephrotoxicity |
138, 142, 144, 146, 151, 153 |
| Cell-lined channel | MDCK | -Mimicry of passive diffusion in the nephron. | 139 |
| Cell-free channel | -Dynamics of calcium oxalate deposition in kidney stone formation | 156 | |
| Lumen (squarea) | RPTEC | -Solute transport and expression of relevant proximal tubule markers | 141 |
| Lumen (circularb) | ciPTEC NEnC TEnC RPTEC |
-Clearance of uremic toxins -Recapitulation of solute transport and homeostasis -Screening of nephrotoxicity compounds -Kidney stone formation -Drug testing against clear cell renal cell carcinoma |
65, 143, 148, 152, 157, 160 |
Lumen with a square cross-section
Lumen with a circular cross-section
ARPC – tubular adult renal stem/progenitor cell
ciPTEC – conditionally immortalized proximal tubule epithelial cell
GEC – glomerular endothelial cell
HK-2 – human kidney-2 cell line
IMCD – inner medullary collecting duct cell from rat kidney
MDCK – Madin-Darby canine kidney cell
NEnC – normal endothelial cell from kidney cancer patient
RPTEC – renal proximal tubule epithelial cell
TEnC – tumor endothelial cell from kidney cancer patient
5.1. Modelling normal kidney function
Establishing basic structure and function.
Baudoin et al. first adapted the conventional flow chamber format to PDMS and constructed a microchannel-based renal tubule model using the highly validated canine distal renal tubule cell line (MDCK) 132. The authors assessed the effect of shear stress of an ammonium chloride solution on the adhesion, morphology, proliferation, glucose uptake, and ammonia metabolism of the cells. Notably, their most relevant results are the decrease in proliferation of the cell line in vitro, which does not correlate to in vivo responses. In this context, the use of PDMS may lead to biased results, as it is known to absorb small molecules. Subsequent papers used a similar model design, as well as different chemical and protein surface treatments (e.g., octadecyltrichlorosilane and fibronectin) to improve the hydrophilicity of the devices, which resulted in higher protein and cell adhesion133, 137 Aiming to increase physiological function, Jang et al. developed a proximal tubule-on-a-chip model using vertically stacked microchannels, where a cell-lined membrane separated an upper luminal apical compartment and a lower basal interstitium138 (Figure 13A & 14 A–B). This format enabled mimicking both fluid flow through the luminal space and interstitial fluid transport to the apical side of the renal tubule interface. The authors assessed cisplatin toxicity and recovery from cisplatin aggression, demonstrating recapitulation of kidney injury observed in vivo. This paper describes the first study to use primary human renal proximal tubule epithelial cells (RPTECs) and physiologically relevant shear stress values to stimulate the cells. It was also an early demonstration of the use of microfluidics to assess chemotherapy-induced nephrotoxicity.
Figure 14.
Overview of most common organotypic kidney models. (A) The proximal tubule-on-a-chip consists of an apical luminal channel separated from a basolateral interstitial fluid channel by an ECM-coated porous membrane. Primary human proximal tubule epithelial cells line the membrane to form the renal epithelial interface. Both the luminal and interstitial channels can be perfused. The device mimics the natural tissue–tissue interface in the nephron. (B) Schematic of device layers and assembled device.138 (C) Schematic illustrating the formation of proximal tubules in the 3D lumen device. (i) Cells are isolated from human kidney cortex and (ii) expanded. (iii) Expanded cells are seed into the 3D lumen device. (iv) Image of a tubule and viability at day 28. (D) 3D projection images of the cultured proximal tubule. Staining for CD13 in B1 and B2, E-cadherin in B3 and B4, and aquaporin 1 in B5 and B6. Scale bars: Cii, 200 μM; Civ, 50 μM; D, all 20 μM148. Reproduced from ref. 148 with permission from Elsevier, copyright 2017 and ref 138 with permission of Oxford University press, copyright 2013.
The next generation of organotypic renal tubule models included 3D lumens instead of 2D lumen interfaces, which were used to explore molecular transport through lumen barriers. A noteworthy example is a report by Mu et al., that described a model of a 3D renal tubule with a square cross-section embedded in collagen hydrogel139 (Figure 13H). The renal tubule models consisted of canine MDCK cells and were co-cultured with lumens lined with human HUVEC cells, establishing the first renal tubule-vascular co-culture model. A diffusion assay demonstrated crosstalk between cells through the hydrogel. While we have found no papers directly comparing the effects of square and tubular morphologies on renal cell culture, cells are known to respond differently to geometrical differences in surface topography140. Therefore, cells can likely sense non-physiological geometries (i.e., square edges) and stiffnesses and, in turn, change their behavior accordingly. Therefore, differences in cell behavior (e.g., increased proliferation and migration) are expected to occur between these models and physiological conditions. In another study, Ng et al. created tubular proximal renal tubules within a synthetic hollow fiber and commercial human Renal Proximal Tubule Endothelial Cells (RPTECs)141 (Figure 13E). These tubules were housed in a microfluidic bioreactor to enable perfusion and collection of secreted factors (e.g., urea, creatinine, and glucose) from the RPTECs. Collectively, these studies demonstrated the capability of microfluidic approaches for mimicking the basic in vivo structure and fluid transport physiology of renal tubules, paving the way for more sophisticated models.
Renal filtration.
Zhu and collaborators presented a multilayer microfluidic dialyzer consisting of endothelial cell-lined (HUVECs) and human renal epithelial cell-lined (HK-2) permeable membranes sandwiched between fluidic layers with serpentine microchannels for perfusion 142. The reported design compartmentalized blood flow and dialysate layers. This design allowed the assessment of filtration of urea nitrogen and vitamin B12, and reabsorption of electrolytes. The materials used in building this microdevice comprise titanium (known for its biocompatibility), as well as dialysis membranes of different composition (polyester sulfones, mixed cellulose esters, and regenerated cellulose). These are uncommon materials for cell culture and could have an impact on the performance of the final device. Ammoniagenesis (i.e., ammonia formation) was further characterized by measuring the concentration of ammonia generated by the HK-2 cells under various pH conditions generated in the blood flow and dialysate compartments. In a recent study, Jansen et al.143 developed a renal tubule model to study uremic toxin excretion, using a similar model to that described by Ng et al141. The authors used human conditionally immortalized proximal renal tubule epithelial cells (ciPTEC) to build the tubule model. This model was used to assess the concentration-dependent inhibition of Organic Anion Transporters (OAT-1 and OAT-3), which are proteins implicated in the excretion of uremic toxins such as indoxyl sulfate and kynurenic acid. Additionally, the authors studied the filtration of inulin, a polysaccharide present in fruits and vegetables, considered as a standard to determine the filtration rate in the glomerulus. A PES membrane with a maximum pore size of 0.5 μm was used for the filtration experiments. The pore size should not affect the reported filtration results, since the hydrodynamic radius of most proteins is much smaller than 0.5 μm73. However, the stiffness and porosity of these membranes does not resemble in vivo basal membranes, and specific effects of this change in model performance should be evaluated. Despite the emphasis of the community to mimic filtration and excretion in lumen-based models, it remains a challenge to compare the rates achieved in these models to physiological values to validate their use for basic or applied research.
Aquaporin and ATPase function.
Aquaporin function and Na+/K+ ATPases are critical for the reabsorption of water and salts into the renal tubule, and eventually to form urine. Aquaporins (i.e., water transporters) are essential in maintaining chemical homeostasis in the body17. Therefore, there has been a high interest in developing organotypic models to assess aquaporin functionality. Jang and Suh144 published an early example of aquaporin functionality studies in organotypic lumen models. These authors presented a multilayer microchannel model with a porous membrane lined by primary rat inner medullary collecting duct (abbreviated IMCD) cells to form the renal epithelium. To demonstrate the model’s applicability for drug screening, they studied transport kinetics of aquaporin channels and Na+/K+ ATPases in response to vasopressin and aldosterone treatment, respectively. Treatment with vasopressin significantly increased water transport into the apical side of the renal epithelium. Conversely, the treatment with aldosterone decreased Na+ uptake.
Overall, these results demonstrated a simple microfluidic approach for assessing the response of renal epithelial cells to hormonal stimulation. In a subsequent study from the same group, Jang et al. used their model to evaluate the effect of luminal fluid shear stress on the translocation of aquaporin-2 and rearrangement of the actin cytoskeleton of the IMCD cells145. Using a similar approach, Sciancalepore et al. improved the human relevance of the multilayer microchannel model by using resident adult renal stem/progenitor cells to recreate the renal tubule epithelium146. Under applied shear stress, aquaporin-2 translocated to the apical side of the epithelium, and Na+K+ ATPase pumps localized to the basal region, consistent with in vivo epithelium. They were able to demonstrate the physiological recovery of urea and creatinine using this human renal tubule model. As previously discussed, the high permeability of PDMS to lipophiles and ions casts some doubt in the physiological relevance of their recovery. Likewise, the stiffness of the cell culture material is well above the physiologically relevant values, which reportedly modifies cell function147. However, there is no question in the relevance of shear stress-induced polarity (i.e., the polarization of cellular components within tissue organization) in their model. All these models offer a controlled system to test the effects of shear stress on a lumen interface, although these effects remain to be tested in a 3D environment.
Towards 3D tubular architecture.
More recently, Weber et al. created a 3D tubular model of the proximal renal tubule using primary human renal epithelial cells in a commercial microfluidic device from Nortis148 (Figure 14C&D). The proximal renal tubule phenotype of the recreated renal lumen was verified by the presence of aquaporin-1 and lotus lectin. Interestingly, the authors observed the expression of kidney injury molecule 1 (KIM-1), a marker of acute kidney injury, for the epithelial cells cultured in 2D but not in the 3D organotypic lumen culture. These results suggest that 3D tubular architecture provides a less injured, normal phenotype within the lumen. The authors assessed filtration and reabsorption in the model via quantification of glucose and the activity of the transporter γ-glutamyl transpeptidase. Moreover, the model was used to study secretion events, including ammoniagenesis and bioactivation of vitamin D. Overall, this model demonstrated the importance of capturing the physiological structure to elicit in vivo-like cell behavior and represents a milestone in the development of organotypic kidney models. Furthermore, this is one of the few models that explored pH-regulating capabilities of kidney models. Much progress has been made in modeling the normal renal tubule and mimicking its physiology; however, several essential functions have yet to be achieved. For example, the differential filtration and reabsorption processes coinciding along the renal tubule, as a result of salinity gradients in the interstitial space of the nephron, remain unexplored. Similarly, the interchange of substances between the nephron and the vasa recta (i.e., the blood capillaries surrounding the nephron) is yet to be modeled.
5.2. Modelling kidney diseases
Nephrotoxicity.
Kidney toxicity (also called nephrotoxicity) is one of the most frequent adverse events reported during drug development. Nephrotoxicity occurs when kidney detoxification and excretion are impaired due to exogenous or endogenous compounds. The general mechanisms that can cause nephrotoxicity include changes in glomerular hemodynamics, tubular cell toxicity, inflammation, crystal nephropathy, rhabdomyolysis, and thrombotic microangiopathy149. Researchers have approached the modeling of kidney toxicity in organotypic models from two different perspectives: (1) as direct nephrotoxicity on the kidney tissue and (2) as systemic toxicity in integrated systems with other organs. Material choice is critical since high biocompatibility and low molecule retention are required for reliable and reproducible results. In other words, toxicants in the microdevice or (most commonly) organic chemicals required for processing may be released in the cell culture media, therefore influencing cell behavior. Likewise, drug-derived toxicants and nutrients may be retained in the microdevice material or the hydrogel, therefore influencing nephrotoxicity results.
Direct nephrotoxicity models have focused on studying renal tubule inflammation due to urinary fluid shear stress and injury due to drug toxicity. Miravéte el al. used glass slides to build a 2D renal lumen interface on top of a gelatine-coated polymer. They used this device to model the effects of excessive shear stress in tubular aggression, which contributes to the development of inflammation150. They demonstrated that hour-long application (i.e., five hours) of fluid shear stress to the renal epithelium induced overexpression of inflammatory cytokines, such as TNF-α, which has been implicated in the initiation of renal disease and is consistent with in vivo observations. In a follow-up paper, the authors further correlated an excess of fluid shear stress with the appearance of kidney nephrotoxicity markers (e.g., KIM-1, lipocalin). The use of glass was an effective alternative to PDMS for biocompatibility and toxicity testing.
In the context of drug toxicity, Kim et al. 151 used a similar multilayer microchannel model reported by Jang et al.144 to test the nephrotoxicity of gentamicin on MDCK cells. Their building materials were PDMS and a polyester 0.4 μm membrane. While the PDMS is a drawback for toxicity analysis, the size of the membrane is consistent with prior literature, and polyester is adequately biocompatible and inert. Their data suggested that gentamicin disrupts cell-cell junctions, increases permeability, and decreases cell viability under prolonged exposure at lower dosages (~3mM). Interestingly, the authors observed differences in epithelial integrity and viability based on the method of drug delivery. For instance, a bolus injection of gentamicin versus continuous injection resulted in less injury to the renal epithelium, suggesting that the drug regimen may be relevant to avoiding drug toxicity. While the concentration values provided may not be reliable due to PDMS lipophile retention, their approach is still valid, and relevant for the community. More recently, Adler et al. used the 3D tubular device from Nortis to study the cytotoxic response of human primary tubule epithelial cells (HPTECs) to a broad panel of drugs and environmental chemicals152 (Figure 13D). This panel included cyclosporin A, cadmium chloride, aristolochic acid, gentamicin, and doxorubicin. The Nortis microdevice, like many other commercial devices, is composed of a cyclic olefin polymer (COP). COP is reportedly highly biocompatible, low molecule retaining, and highly hydrophilic, and therefore it has many desirable features for toxicity assays. By conducting a multiplexed gene expression profile of the HPTECs after exposure, the authors identified the heme oxygenase 1 (HO-1) enzyme as a potential biomarker for in vitro kidney injury. Further evaluation of HO-1 expression for 39 additional nephrotoxic compounds revealed the suitability of the enzyme as a biomarker for kidney injury. Their study demonstrates the unique capability of organotypic kidney models for broad screening of drug toxicity using valuable primary cell sources.
Regarding systemic toxicity, a few models have reported the interaction of kidney constructs with several other organ types and the implication of these interactions in nephrotoxicity, such as that by Li et al. (Figure 13F)153 These studies can be found in a later chapter, discussing multiorgan interactions exclusively.
Nephrolithiasis.
Urinary tract stones or nephrolithiasis is a common malady affecting nearly 1 in 11 individuals in the United States154. At least 50% of individuals will experience another stone within ten years of the first occurrence. The composition of urinary tract stones typically includes calcium oxalate (40-60%), calcium phosphate (20-60%), uric acid (5-10%), or combinations of these substances. The pathogenesis of nephrolithiasis can be environmental or genetic, generated from an excess of calcium or oxalate (i.e., derived from hypercalciuria or hyperoxaluria), resulting in precipitation of the calcium salts. A low urinary pH has also been correlated with an increased incidence of kidney stones. The treatment of nephrolithiasis often includes removal of the stones, dietary modifications, and pharmacological treatments. Models of the generation of mineral deposits are scarce in the literature, despite the clinical importance of nephrolithiasis155.
Laffite et al. reported a cell-free model that leveraged the laminar flow capabilities of microchannels to study the deposition of calcium oxalate in a Y-shaped channel156. The authors were able to observe the formation of calcium oxalate by flowing solutions of calcium chloride and sodium oxalate at physiological concentrations and flow rates. Similarly, Wei et al. demonstrated the real-time tracking of calcium phosphate deposition in a 3D proximal renal tubule generated with HK-2 cells by perfusing solutions of calcium chloride and sodium phosphate through the tubule157. These preliminary studies highlight the practicality of microfluidic models to study crystalline phases relevant to human pathological calcifications. They also emphasize the need for more in vitro nephrolithiasis models capable of producing stones of the different chemical compositions described above. Accurate mimicry of kidney stones may include careful screening of ionic concentrations and pH, which are also known to vary in different areas of the nephron17. This study of the kinetics of precipitation could be instrumental in developing drugs for nephrolithiasis. Additionally, the low occurrence of kidney stones in mice makes the use of in vivo models challenging. We believe further development of microfluidic lumen-based models of nephrolithiasis to study the kinetics of nephrolithiasis and salt precipitation could inform clinical practice in the treatment of this ailment. As previously discussed, careful material selection may be a critical factor in modeling nephrolithiasis accurately, as some materials can sequestrate inorganic ions and diminish their effective concentration.
Kidney cancer.
Kidney and renal pelvis malignancies are the 8th most common genitourinary malignancy in the US. Approximately 85% to 90% of kidney cancers are derived from the proximal tubular epithelial cells of the renal cortex and are classified as renal cell carcinoma (RCC)158. As RCC is a highly vascularized disease, the standard of care often relies on nephrectomy and treatment with anti-angiogenic therapies159. These therapies block the growth of blood vessels that supply the tumor with oxygen and nutrients, resulting in a decrease in tumor size. Currently, two microfluidic RCC models in the literature capture the 3D luminal structure of the vasculature65, 160. The first model leveraged the Nortis lumen microdevice to generate a tubular endothelial vessel to mimic blood capillaries surrounding the renal tubule160 (Figure 13G). Patient-derived RCC tumor spheroids were embedded into the same matrix to enable tumor-vascular crosstalk. Tumor-induced sprouting of the endothelial vessels was measured as a functional response to growth factors secreted by the tumor spheroids. Tumor angiogenesis was blocked by treating the endothelial vessels with media containing an antibody that inhibited the VEGF/VEGFR2 pathway, to demonstrate the applicability of the model for pharmacological testing. It is of interest that the composition of the hydrogel (as well as the proteins included in it) profoundly influences these last results. Hydrogel composition defines the resulting architecture and pore size, and therefore, in this case, they will impact the diffusion of the therapeutic antibody significantly. An in-depth study of the hydrogel and kidney cancer architecture may help solve some of these questions.
The second model takes advantage of a lumen microfabrication method previously developed by our group29. Using this approach, Jimenez-Torres et al. generated normal and tumor-associated blood vessels using primary endothelial cells isolated from clear cell RCC patients65 (Figures 14I &15). Vessels with the tumor-associated phenotype were structurally disorganized (i.e., random orientation of endothelial cells), leaky, and highly angiogenic as compared to their normal counterparts. These vessels also overexpressed pro-angiogenic growth factors, including VEGF-A and FGF-2. In an effort towards clinical translation, the patient tumor-associated vessels were treated with FDA-approved anti-angiogenic drugs (i.e., pazopanib and sunitinib). Interestingly, there was a heterogeneous response for different patients, where some patient-derived vessels responded to treatment, while others continued to sprout aggressively. These results highlight the heterogeneity that can manifest in patient response to therapy and demonstrate the power of organotypic microfluidic models for personalized medicine. While material choice (i.e., PDMS in this case) may indeed affect the final concentration of drug in the system, we believe this does not invalidate the approach of the article, and its bottom line, which is mimicking heterogeneous patient response in vitro. In this direction, we propose the use of retrospective studies to elucidate the accuracy of these models in reproducing patient response. Currently, the RCC field is undergoing many changes in standard of care, with reported discrepancies in the effectiveness and benefits of different treatment regimens159. With dozens of different drugs in clinical trials for kidney cancer, we believe that organotypic microfluidic RCC models can be informative tools to help clinicians find the best treatment strategy.
Figure 15.
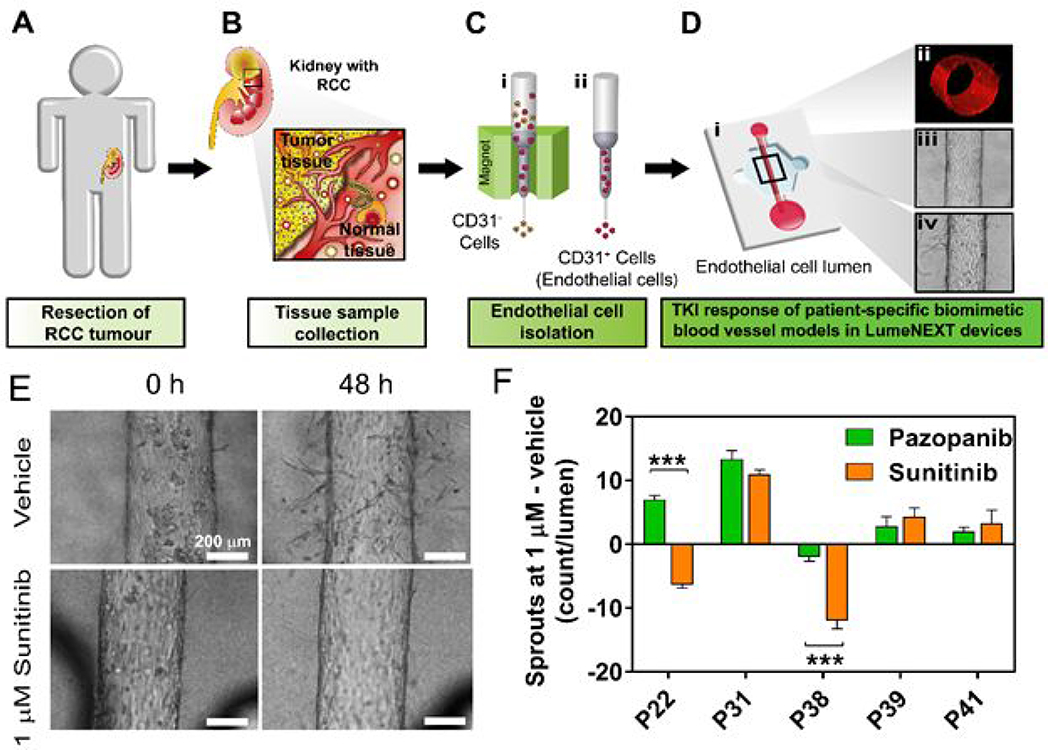
(A-C) Schematic of workflow for isolating kidney endothelial cells from normal and tumor patient samples. (D) (i) Isolated cells are used to generate 3D endothelial vessels. (ii) Vessels have tubular structure. Images of vessels before (iii) and after (iv) treatment with anti-angiogenic drugs. (E) Images of tumor-associated vessels treated with sunitinib showing less sprouting at 48 hours. (F) Angiogenesis of tumor-associated vessels to both pazopanib and sunitinib showing heterogeneity in patient responses. Adapted with permission from Jimenez-Torres, 201965.
6. Liver sinusoids
The liver is involved in nutrient metabolism, waste product and toxin breakdown, protein synthesis, and bile production. Structurally, the liver is divided into lobes containing thousands of functional units known as lobules. Hepatic lobules are hexagonal-like structures consisting of hepatocytes radiating from the central vein, which leads to the hepatic portal vein. In these lobules, blood flows from the portal triad, located at the corners of the lobules, to the central vein through liver capillaries called sinusoids. It is in these sinusoids where metabolic reactions mainly take place, including the breakdown of natural waste products (e.g., urea, bilirubin) and exogenous compounds (e.g., drugs, toxins). Many different proteins (e.g., albumin, clotting factors) and amino acids are also synthesized in the liver and distributed to other organs. In this section, we discuss the microfluidic approaches that have been employed to generate liver sinusoids and their application to modeling liver-specific and systemic drug toxicity17. Table 5 shows a summary of microfluidic liver sinusoid models.
Table 5.
Summary of microfluidic liver sinusoid models.
| Organ geometry | Cell type | Targeted application | Refs |
|---|---|---|---|
| 2D channel | HepG2/C 3A iPSC-Hep PHH PRH |
-Effect of spatial confinement on maintaining hepatocyte phenotype iPSC hepatocyte differentiation and transcriptomic analysis -Acetaminophen hepatotoxicity -Ammonia hepatotoxicity -Naphthalene hepatotoxicity -Hepatic clearance of common drugs (e.g., acetaminophen, caffeine, dextromethorphan, tolbutamide) -Real-time monitoring of cytochrome P450 activity -Integration of mass spectrometric analysis of drug metabolites -Effect of alcohol injury on hepatocyte-stellate cell TGF-β signaling |
161, 162, 166, 167, 172, 183–185, 187 |
| 2D membrane (cells on membrane) | FHH HC-04 HepG2 HLSEC PRH |
-Effect of spatial confinement on maintaining hepatocyte phenotype -Establishment of parenchymal and vascular interfaces -Hepatic serum protein synthesis (e.g., albumin production) and metabolism (e.g., cytochrome P450 activity) -Perfusable long-term hepatocyte culture (>7 days) -Hepatitis B viral replication |
164, 170, 179 |
| 2D membrane (cells in gel channel) | HepaRG LX-2 PHH PRH U937 |
-Effect of fluid shear stress on hepatocyte morphology and physiology -Establishment of parenchymal-vascular compartments in liver sinusoid with physiological functions |
165, 178–180 |
| Cell-filled channel | HepG2 PHH PRH |
-Perfusable long-term hepatocyte culture (> 7 days) -Effect of fluid shear stress on hepatocyte morphology and physiology -Lipid accumulation during the progression of non-alcoholic fatty liver disease |
169, 182, 190 |
| Multiorgan liver systems | HepaRG HepG2/C 3A |
-Nanoparticle toxicity on liver-intestine co-culture -Effect of TGF-β on crosstalk between liver, kidney, lung, and fat cells -Phenacetin transport through intestinal epithelium and metabolism by hepatocytes -Effect of neurotoxin on liver-neuron co-culture -Systemic repeated dose substance testing using liver-skin and liver-intestine co-cultures -First pass metabolism of flavonoids |
177, 192–198 |
FHH – FRG™-KO mouse-expanded human hepatocytes
HC-04 – hepatoblastoma cell line
HepaRG – human hepatic progenitor cell line
HepG2 – hepatocellular carcinoma cell line
HepG2/C3A – clonal derivative of HepG2
iPSC-Hep – induced pluripotent stem cell-derived hepatocytes
HLSEC – human liver sinusoid endothelial cell
LX-2 – human hepatic stellate cell line
PHH – primary human hepatocytes
PRH – primary rat hepatocytes
U937 – human macrophage cell line
6.1. Modeling normal liver function
Establishing basic structure and function.
Multiple studies have shown that when cultured in confined spaces like microfluidic channels, hepatocytes can dramatically change their behavior. A series of experiments revealed that hepatocytes secrete relatively low amounts of growth factors that are essential to keep them differentiated and active. In traditional 2D culture platforms, the vast culture medium-to-cell ratio dilutes growth factors, leading to early dedifferentiation. Conversely, the lower culture medium-to-cell ratio offered by microfluidic 2D interfaces enhances autocrine signaling by increasing effective growth factor concentrations. Therefore, hepatocytes can remain functional for more extended periods of time, retaining a differentiated phenotype161–163. Additionally, microfluidic approaches offer precise fluid control with a highly predictable flow. The earliest attempts to mimic liver function leveraged these properties to recapitulate blood flow through the sinusoids.
In early lumen interface models, hepatocyte cell lines were cultured in 2D monolayers within microchannels (i.e., 2D lumen interface), and culture media was perfused through the channel to mimic blood flow through the sinusoids. Using these design constraints, Carraro et al. combined two parallel PDMS-based arrays of microfluidic channels partitioned by a porous polycarbonate membrane (0.4 μm pore size, 12% porosity) to compartmentalize culture of human/rat hepatocytes and applied fluid shear stress164. Plasma-activated PDMS-PDMS bonding allowed the authors to immobilize the polycarbonate membrane between the two PDMS arrays without affecting the membrane integrity. The authors demonstrated protein synthesis and cytochrome P450 activity, a hemeprotein that plays a key role in the metabolism of drugs and other xenobiotics in the liver. Other early studies, such as the one published by Tan et al., cultured hepatocytes at physiological densities in a microfluidic chamber and applied continuous medium flow to maintain viability165. This approach produced levels of albumin secretion and urea synthesis similar to those found in vivo.
Recapitulating toxin metabolism.
Viravaidya et al. generated a PDMS device to study the metabolism of the toxin naphthalene166. Their model recapitulated the metabolic degradation routes of naphthalene observed in vivo, validating the platform as an early model to study liver function in microfluidic devices. This study showed the potential of microfluidic liver systems to study naphthalene and other polycyclic aromatic hydrocarbons (PAH). PAHs are abundant and ubiquitous pollutants found in thermally decomposed organic matter, highly liposoluble, and known to have multiple toxic and mutagenic effects in human physiology. However, PDMS-based microdevices are not amenable for mass production, and small hydrophobic molecules, including PAHs, are absorbed into the bulk material. In this context, polystyrene would be an interesting alternative since it is not permeable to hydrophobic molecules and is compatible with high-throughput fabrication techniques (i.e., injection molding). However, chemical treatments to bond polystyrene devices (e.g., thermocompression, UV-activated adhesives, etching) commonly compromise the integrity of the membrane or generate residues that damage the cells164. In a separate study, Chao et al. developed a device with multiple microchannels and microchambers to study liver clearance of different compounds (e.g., caffeine), observing similar clearance rates to those found in vivo167.
Similarly, Prot et al. measured the capacity of primary human hepatocytes, cultured under fluid flow conditions in microchannels, to metabolize a cocktail of common drugs containing acetaminophen, amodiaquine, caffeine, dextromethorphan, midazolam, omeprazole, and tolbutamide168. As compared to static cultures in well-plates, hepatocytes in the microchannels exhibited a more dynamic in vivo-like response. This study highlighted the impact of flow conditions in liver biology, a property that remains extremely challenging to mimic in traditional 2D culture systems. Additionally, the small dimensions of microfluidic systems allow researchers to mimic the laminar flow observed in liver capillaries.
Building the 3D liver microenvironment.
Several factors need to be considered to more accurately recapitulate liver structure-function relationships in vitro, such as physiologically-relevant cell types, densities, ECM, and 3D cellular spatial organization165. More recently, microfluidic liver models have focused on integrating these factors into their devices. An example was described by Goral et al.169. Their 3-channel device design was used to enable a high-density culture of liver cells without any ECM in the central channel, whereas medium flowed through the lateral channels. This study demonstrated the importance of fluid flow in ensuring oxygen supply, and therefore cell viability in microfluidic liver models with physiologically relevant cell densities. However, the use of external pumps makes microfluidic systems less accessible to a broad audience, thereby limiting their translational potential. Interestingly, Maher et al. demonstrated that controlling the spatial distribution of hepatocytes in the ECM enabled long-term culture (14 days) even in the absence of medium flow or other cell types170. In their model, cells exhibited albumin production, factor IX production, and cytochrome P450 3A4 drug metabolism. Interestingly, the morphogenesis and albumin production of hepatocytes were substantially different when confined within microchannels in a 2D lumen interface as compared to no confinement. This work demonstrated the importance of capturing relevant mechanical cues to achieve physiological cell behavior and removed the need for external pumps.
In a separate study, Hegde et al. used a 3 μm-pore PET membrane-based device to partition a perfusion channel and a culture channel171. In the culture channel, liver cells were sandwiched between a fibronectin layer and a collagen hydrogel. Liver ECM is limited, accounting for 3% of the area in liver slices, and the main components are collagens (primarily type I, III, IV, and V) and fibronectin. Thus, this sandwich structure mimicked the different ECMs observed in the liver sinusoids and enhanced liver response. Medium flow through the perfusion channel enhanced liver cell viability, as well as albumin secretion, cytochrome expression, and drug metabolism. While these studies presented exciting progress toward accurately mimicking liver function, they only included 2D lumen interfaces.
Including 3D environments.
A new generation of studies to mimic in vivo cellular spatial organization has relied on embedding liver cells in hydrogels (e.g., collagen, Matrigel). An example was reported by Sung et al., who embedded hepatocytes in a 3D Matrigel microfluidic chamber and perfused 3-Methylcholanthrene (i.e., a cytochrome P450 inducer) through a microchannel above the culture to study the enzymatic conversion of ethoxyresorufin to resorufin172. The results showed that hepatocytes had a cytochrome P450 induction response comparable to in vivo data in the microfluidic setup, more representative of in vivo data than traditional 2D platforms. However, this model cultured liver cells at significantly lower densities compared to in vivo tissue, whereas ECM content was overrepresented. Additionally, Matrigel is a mixture of mouse ECM proteins, growth factors, and chemokines that are not necessarily present in the human liver ECM composition and are known to vary batch-to-batch173.
To reduce batch-to-batch variability of naturally-derived ECMs in 3D cell cultures, researchers have explored the use of synthetic scaffolds for long-term, high-density cultures in microchannels.174–176 An example is the study reported by Lee et al. These authors encapsulated liver enzymatic fractions in a poly(ethylene glycol) diacrylate (PEGDA) hydrogel to study how liver cells metabolized antioxidants174. The microchannel was coupled to a second microchannel where a ROS scavenging reaction detected the presence of antioxidants generated or consumed by the liver cells. As a proof-of-concept, a typical plant pigment (quercetin) was added to the media, and after being metabolized by the liver cells, its ROS scavenging capacity was evaluated. However, this model evaluated liver metabolism using immobilized liver enzymes, which provides a limited capacity to study hepatocyte cell biology. Furthermore, synthetic hydrogels have the potential to generate chemically defined and controlled ECMs, offering a versatile alternative to natural ECMs (e.g., Matrigel, gelatin, collagen). Therefore, future models could explore the possibility of encapsulating liver organoids in synthetic hydrogels (e.g., PEGDA). In a similar model, Esch et al. trapped liver organoids containing liver cells and fibroblasts in a hydrogel within microfluidic chambers177. These microchambers were connected to a central microchannel where the medium with drugs was perfused. Culture media was retrieved, and the presence of metabolites generated by the liver organoids as a result of drug breakdown was analyzed by measuring albumin secretion and urea synthesis.
Including supporting cell types.
All the models previously described only included hepatocytes, whereas multiple cell types are observed in the human liver. In this context, various microfluidic liver models have attempted to capture the cellular heterogeneity of the liver stroma by including endothelial cells, stellate cells, and fibroblasts, among others.
One of those efforts is the report by Prodanov et al. In this study, stellate cells were embedded within collagen and cultured on top of hepatocytes seeded in the lower microchannel, while the endothelial-lined membrane formed the top vascular channel (Figure 16B) 178. Macrophages were also co-cultured on top of the vascular layer to recapitulate the presence of Kupfer cells. Continuous media transport through the perfusion channel maintained the culture for at least 28 days. Albumin secretion, urea synthesis, and cytochrome expression were monitored to demonstrate the capabilities of the system. Using a similar membrane-based device, Kang et al. co-cultured their stellate cells with endothelial cells lined on the membrane to form a vascular channel179 (Figure 16C). The media was perfused through the vascular channel to recapitulate blood flow through the sinusoid. Liver cells retained their normal morphology and synthesized urea for at least 30 days. The model was further used to study the viral replication of the hepatotropic hepatitis B virus. Other studies have leveraged membrane-based design to investigate hypoxia in the liver microenvironment under static and flow conditions, and the recruitment of neutrophils to the sinusoidal endothelium180, 181.
Figure 16.
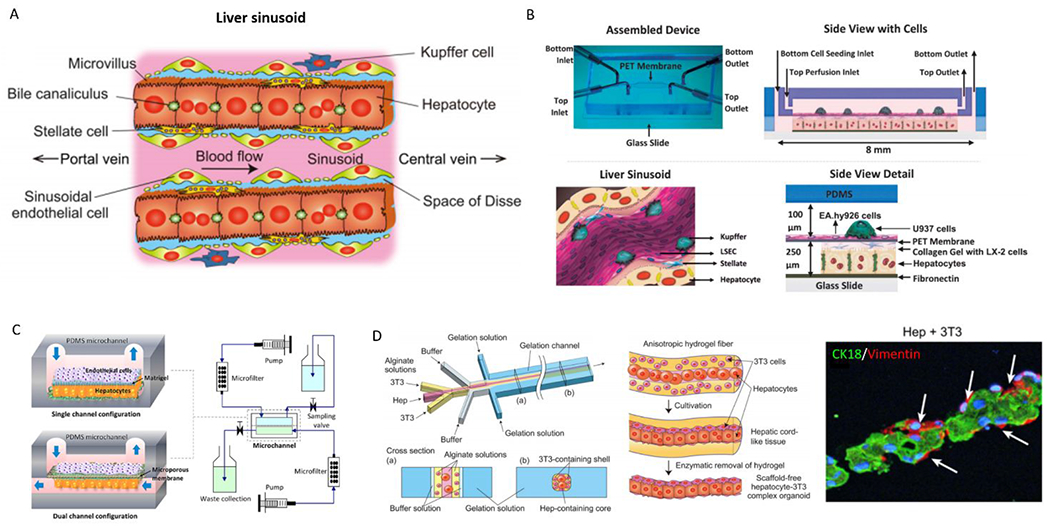
Overview of microfluidic device designs to mimic the liver. (A) Structure of the hepatic cord in the liver lobule. Parenchymal hepatocytes are separated from the sinusoid and are arranged in cord-like structures with a thickness of 1-hepatocyte layer. (B) Image of assembled microfluidic device (top left), schematic of device cross-section (top right). Representative schematic of the liver sinusoid microanatomy recapitulated by the model (bottom left). Hepatocytes are co-cultured with stellate cells (LX-2) in the bottom channel, endothelial cells line the PET membrane, and Kuppfer (U937) are cultured in the top channel (bottom right). (C) Two microfluidic platforms: single channel and dual channel configurations (left); and bioreactor circuit for continuous perfusion of media and waste collection (right). (D) Microfluidic system for fabricating sandwich-type alginate hydrogel microfibers that incorporate hepatocytes and 3T3 cells (left). Representative images of hydrogel fabrication (middle) and microscopy images of model (right). 178, 179, 182. Reproduced with permission from John Wiley and Sons, from ref. 178 copyright 2013; from Ref 179, copyright 2015. Reproduced from ref 182 with permission Elsevier, copyright 2012.
A unique example is the approach by Yamada et al. These authors generated a liver organoid in a microfluidic channel combining different cell types by 3D printing techniques182. Liver cells suspended in a sodium alginate solution were flowed through a microchannel flanked by two parallel streams containing feeder cells (i.e., 3T3 cells) in the same sodium alginate solution. Next, sodium alginate was polymerized, leading to the generation of a fiber-like organoid with a core of hepatocytes sandwiched by feeder cells. Finally, the hydrogel was degraded to generate a scaffold-free organoid where cell-cell interactions kept the fiber-like structure together. The width of this fiber mimicked the width of hepatocyte layers in the sinusoids, and the presence of feeder cells enhanced the hepatic function (i.e., increased albumin secretion, urea synthesis, as well as expression of hepatic-specific genes) compared with classic monolayers or fibers containing only hepatocytes.
Specifically focused on liver detoxification, Mao et al. reported a microdevice to study the response of hepatocytes to byproducts generated by liver microsomes (i.e., lipid vesicles with encapsulated liver enzymes) 183. In a follow-up study, the authors explored the potential of their system for high-throughput cytotoxicity screening of toxins at low concentration184. Taken together, these co-culture models offered a more holistic approach to liver pathophysiology. The models recapitulate the spatial organization observed in the liver, juxtaposing different cell types in 3D environments perfused with medium flow. However, there are several challenges these co-culture platforms must overcome. In this context, the use of a porous membrane to separate different compartments and cell types raises some concerns. In vivo, sinusoidal endothelial cells are separated from stellate cells and hepatocytes by a discontinuous basement membrane. This membrane is formed by a variety of structural and adhesion proteins that promote cell growth and attachment. Additionally, discontinuous basement membrane also allows cell trafficking by a selectively controlled process which enables immune cells to patrol the liver. Therefore, future studies should assess the capacity of porous membranes to mimic these functions and substitute them for other more biologically relevant membranes. Future developments in bioprinting techniques could offer an exciting alternative to generate basement membranes for microfluidic systems exogenously.
6.2. Modeling liver diseases
Hepatotoxicity.
Drug-induced liver injury can result in acute and chronic liver diseases. Several groups have developed microfluidic liver models to better understand the mechanisms of such injury. Prot et al. provided one of the first examples of this important application. The authors exposed HepG2/C3A cells cultured on the surface of microchannels to acetaminophen, and measured albumin production and gene expression168. Metabolism of acetaminophen was enhanced in the microchannels as compared to traditional 2D cultures and was comparable to in vivo observations. This model was then adapted to measure the clearance of a list of common drugs and toxicants by primary rat hepatocytes185. Another study examined the hepatotoxicity of prostate cancer prodrug flutamide. Liver cells metabolized this prodrug to the active compound (i.e., 2-hydroxiflutamide), which led to a reduction of the extracellular glucose and tricarboxylic acid cycle activity. As a response to the presence of flutamide and 2-hydroflutamide, hepatocytes exhibited high-energy demand and increased amino acid metabolism to clear the compound186.
Alcoholic liver disease.
Alcoholic liver disease is a result of overconsuming alcohol that damages the liver, leading to a buildup of fats, inflammation, scarring, and even death. Models of alcoholic liver disease could be of great importance in public health. An example of these models is the report by Zhou et al. In this study, liver and stellate cells were cultured in two parallel microchannels connected by a valve187. When the valve was closed, ethanol was selectively perfused through one of the microchannels with no effect on the other channel. After selective exposure, the valve was opened to enable crosstalk. In response to ethanol exposure, liver cells secreted TGF-β and, in turn, induced the secretion of excess TGF-β by the stellate cells. Collectively, these microfluidic liver models have demonstrated utility for mimicking hepatotoxic responses and could offer multiple clinical applications. However, the use of hepatocellular carcinoma cells (e.g., HepG2/C3A) and rat hepatocytes to generate ‘normal’ liver sinusoids undermines their physiological relevance. Thus, future models should move towards normal human hepatocytes, which are commercially available.
Additionally, most of these models still rely on PDMS, which can absorb small hydrophobic molecules. Since many hepatic toxins are hydrophobic (e.g., naphthalene), the material used to build the devices should be carefully considered. Finally, toxin pharmacokinetics is a complex process that is affected by a large variety of factors (e.g., hydrophobic toxins are trapped in fat tissue, endothelial permeability impacts toxin diffusion, and extravasation). Therefore, the device geometry, cell types, or media composition used in multiorgan liver systems should match the complexity required to render a biologically relevant response.
Non-alcoholic fatty liver disease.
Non-alcoholic fatty liver disease (NAFLD) encompasses a spectrum of liver diseases from steatosis (i.e., fat accumulation) to non-alcoholic steatohepatitis (i.e., fatty liver inflammation), cirrhosis, and hepatocellular carcinoma188. It is a growing cause of chronic liver injury, affecting about 25% of the global population189. Although factors that contribute to this disease are well-understood, the mechanisms of its progression remain unclear. To the best of our knowledge, only two microfluidic liver models have been developed to study NAFLD, which presents an opportunity in model development and, perhaps, can inspire the collaboration of biologists and engineers. Gori et al. developed a model to recapitulate steatosis, comprising a U-shaped perfusion microchannel connected by narrow capillary channels to a central hepatocyte channel190 (Figure 17). Liver cells were cultured at a high density in the central channel to generate a pseudo-3D organoid, and culture medium was perfused through the U-shaped channel. Due to the high fluidic resistance of the capillaries, medium transport was primarily through the U-shaped channel, not affecting the hepatocytes. This configuration closely mimicked the endothelial-parenchymal interface of the liver sinusoid and enabled the diffusion of nutrients and removal of waste products similar to the hepatic microvasculature. Palmitic and oleic acid were added to the media and perfused through the system for up to 48 h. Intracellular lipid accumulation in the microfluidic setup was more gradual as compared to static 2D cultures, which resembles the slow chronic onset of steatosis in vivo.
Figure 17.
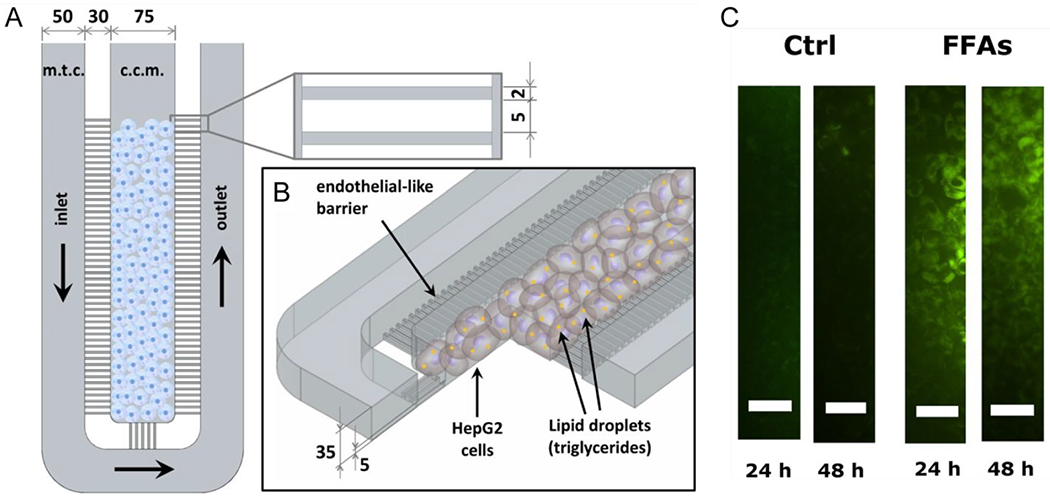
Microfluidic liver model for studying non-alcoholic fatty liver disease. (A) Top view of device showing a central channel for culturing hepatocytes and an outer curved channel for medium perfusion. Capillaries connect these two channels. (B) Zoomed view of the capillary channels. High fluidic resistance in these channels emulate that of the microvasculature in the liver sinusoid. HepG2 cells cultured in the central channel accumulate lipid droplets. (C) Treatment of HepG2 cells with medium containing free fatty acids (FFA). Cells accumulate lipids over 48 hours as indicated by staining with AdipoRed (green). Reproduced with permission from Gori, 2016190.
In the second study, Kostrzewski et al. generated an ex-vivo microfluidic model called LiverChip. The authors cultured liver microtissues in a 12-unit bioreactor system to measure their response to palmitic and oleic acid over 14 days191. Intracellular fat accumulation in the hepatocytes did not induce hepatotoxicity but reduced the metabolic activity of the CYP3A4 and CYP2C9 enzymes, which coincides with clinical observations. Moreover, several genes associated with NAFLD were upregulated, including insulin-like growth factor binding protein 1, fatty acid-binding protein 3, and CYP7A1. The authors further demonstrated the applicability of their model for screening anti steatotic drugs by treating hepatocytes, cultured under fatty conditions, with metformin. Treated cells had lower fat content and consumed less palmitic and oleic acid than untreated cells. Together, these two studies demonstrate the usefulness of microfluidic approaches for basic studies of NAFLD progression and offer potential strategies for screening relevant drugs.
Overall, microfluidic liver models have advanced rapidly and recapitulated liver morphogenesis, transport phenomena, and (patho)physiology. However, the development of lumen-based tubular models of sinusoidal capillaries is, strikingly, absent in the literature, along with a critical mass of studies recapitulating liver cancer or hepatitis. We expect that relevant tubular models of normal and pathophysiological situations will soon appear in the literature. In developing these and other models, there are challenges that remain. An example is the widespread use of PDMS, which is known to absorb small hydrophobic molecules. In gut, liver, and kidney studies, this issue is particularly important as many drugs or toxins can be absorbed by the PDMS and skew tissue response. In this context, other materials like polystyrene or PMMA are being explored.
7. Multi-organ platforms
In addition to single-organ models, multiorgan or body-on-a-chip systems were first introduced in 2004166 and remain a central focus of lumen model development. These systems aim to elucidate the interorgan crosstalk that is paramount to systemic processes, such as homeostasis and clearance of drugs and toxins. The latter process is of particular interest to drug development and testing. Therefore, most of the multi-organ platforms reported include the gut, kidney, and liver; organs implied in the absorption, excretion, and clearance of drugs and toxins.
Modelling multi-organ interactions of the kidney.
These models have overall featured lumen interfaces instead of tubular lumens, trading off structural complexity for cellular heterogeneity. Figure 18 shows a classification of multi-organ devices and highlights some relevant examples. An example by Li et al. was a gut-kidney model that enabled the compartmentalization of intestinal cells (Caco-2) and glomerular cells isolated from primary rat kidney tissue153 (Figure 18C). The toxicity of intestinally-absorbed drugs on the kidney cells was evaluated in terms of cell apoptosis, cell viability, and lactate dehydrogenase leakage. Drug regimens included single or combinatorial treatment with digoxin, cholestyramine, and Verapamil. Their results indicated that digoxin combined with Verapamil significantly damaged the glomerular cells, as compared to single treatments of digoxin and cholestyramine. While this approach is worthwhile, PDMS may also play a role in diminishing the effective drug concentration in the system in this study, which will require further investigation.
Figure 18.
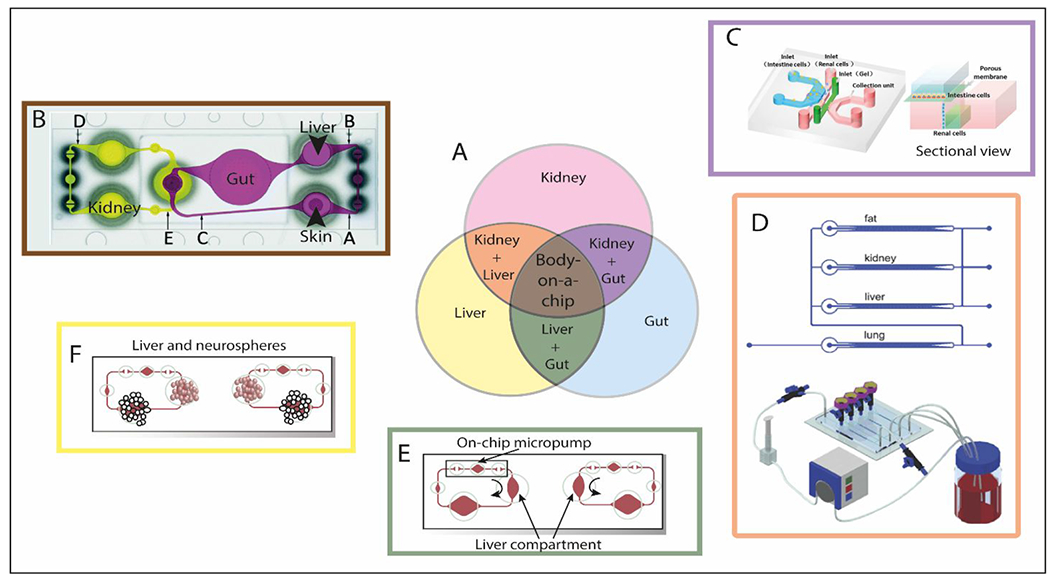
Multi-organ models: toward the body-on-a-chip. (A) Schematic of the most modeled organs and multi-organ interactions on chips. The color code of this schematic is used in the rest of the panels. (B) Top view of the four-organ-chip layout illustrating the positions of three measuring spots (depicted as A, B and C) in the surrogate blood circuit and two spots (depicted as D, E) (C) Schematic diagram of the design of intestine-kidney chip and sectional view. The top layer, porous membrane, and bottom layer were sealed together. The intestine cells were cultured in the chamber on the top layer, and renal cells were seeded in the chamber on the bottom layer. (D) Schematic representation of the multi-organ model called “multi-channel 3D-pFCCS” (top). Close-loop perfusion culture of cells (bottom). During culture, the A549 (lung) channel was closed to facilitate medium perfusion in a serial manner. (E) Experimental setup of the multi-organ liver-gut model for reproducing first-pass metabolism. (F) Experimental set up of single cultivation and co-cultivation of neurospheres and liver microtissues.153, 194–197. Reproduced from ref. 153 with permission from AIP Publishing, copyright 2017.
Using another multi-organ approach, Choucha-Snouber et al. studied systemic toxicity in a PDMS-fabricated liver-kidney model186. The liver component was modeled using HepG2/C3a and HepaRG cells and the kidney using MDCK cells. This model was used to test ifosfamide toxicity. Ifosfamide is metabolized in the liver, generating chloroacetaldehyde and acrolein, two nephrotoxic metabolites. Exposure to ifosfamide in the HepaRG-MDCK co-culture resulted in a decrease in the number of MDCK cells. This response was, however, not observed for MDCK mono-cultures or untreated co-cultures. This result illustrates the importance of incorporating organ-organ crosstalk in unraveling the mechanisms of nephrotoxicity, consistent with in vivo mechanisms. However, the role of the device material (i.e., PDMS) in the pharmacodynamics of ifosfamide remains unclear. Ifosfamide significantly reduced the viability of the kidney cells when co-cultured with liver cells in the microdevice187. Together, these two studies exemplify modeling organ-organ interactions for an accurate understanding of drug toxicity in the kidney and highlight an area of opportunity for future organotypic kidney models. It should be noted that drug toxicity occurs differently in 2D and 3D environments; therefore, we believe these studies should be validated in a 3D environment for more relevant conclusions.
Modeling multi-organ interactions of the liver.
Liver function in health and disease, like that of all major organs, depends on interorgan communication. Thus, recent advances in microfluidic liver models have aimed to capture these multiorgan interactions. One of the early examples of inter-organ interactions with applications towards toxicity testing was demonstrated by van Midwoud et al.192. The authors designed a two-microchamber microfluidic lumen-based device for ex vivo culture of rat intestinal and liver slices under fluid flow. The authors showed that by exposing the intestinal tissue to chenodeoxycholic acid (bile acid), FGF-15 expression was upregulated, resulting in a stronger downregulation of cytochrome P450 7A1 in the liver tissue. In another study, Maschmeyer et al. developed a multiorgan platform for culturing liver-intestine and liver-skin mimics, with vascularization of the liver-skin compartment193. The different tissues remained differentiated, as indicated by the expression of multiple tissue-specific markers: cytokeratin, Na+/K+-ATPase, and cytochrome expression for skin, intestine, and liver cells, respectively. The platform was applied to study oral and systemic exposure to toxins using Troglitazone as a model toxin.
Toward a human on a chip.
Zhang et al. demonstrated one of the earliest attempts to connect multiple cell types mimicking different organs within a single microfluidic system194 (Figure 18D). Human liver, lung, kidney, and adipose cells were cultured in individual microfluidic channels connected by a common microchannel delivering the culture media. The study highlighted the complexity of balancing media composition for different cell types. Interestingly, the authors showed that specific factors (e.g., TGF-β1 was needed for lung cells but inhibited liver cells) could be selectively released in the desired channel by using growth factor-containing gelatin microspheres fixed in that microchannel. Liver, lung, adipose, and kidney functions were analyzed by simultaneously measuring albumin secretion, PROD activity, GGT activity, and adiponectin secretion respectively.
Beyond the highly explored liver-intestine-gut triad, Materne et al. developed microscale models to emulate liver-brain interactions. In one example, liver microtissues and neurospheres were compartmentalized in separate microwells and connected by a vascularized microchannel to allow crosstalk195 (Figure 18F). Medium containing a neurotoxic compound, 2,5-hexanedione, was perfused through the system in a dose-dependent manner to elucidate the toxicity profiles of the neurons and liver cells. Interestingly, the liver-brain co-cultures were more sensitive to this compound in comparison to mono-cultures of each tissue, demonstrating the ability of the model to capture systemic interactions important to neurotoxicity.
In a follow-up study from Maschmeyer et al., the authors extended their model design to accommodate four organs, including the liver, skin, kidney, and intestine196. Similar models of the liver-intestine axis have been developed to enable cell retrieval after co-culture, and to assess the transport of drugs through the intestinal epithelium and their subsequent metabolism by hepatocytes197, 198 (Figure 18B). As we previously discussed Bricks etal. used the 2D membrane design to generate a perfusable liver-intestine co-culture system that recapitulated acetaminophen absorption and metabolism198. As compared to static 2D co-cultures in Transwells, hepatocytes cultured in the microfluidic system had a significantly better capacity for metabolizing phenacetin.
Ultimately, the goal of developing multi-organ platforms, such as those described above, is to provide predictive tools in place of conventional animal models. Current studies have demonstrated their potential preclinical analysis and their continued development will advance translational research. However, current multi-organ models still present several challenges that should be overcome. In particular, multi-organ systems often rely on culturing multiple cell types in several chambers, trading the inclusion of different cell types for in vivo tissue organization and complexity. Thus, future multi-organ liver systems should combine cell types from different organs preserving organ architecture to accurately mimic physiological functions.
8. Emerging applications
Organ-on-a-chip models are advancing at a rapid pace. Beyond the organ systems covered in this review, the community is developing luminal models of the bile duct,187 female reproductive tract,188 neural tube,189 mammary duct,15,16,190 and lymphatics21,191–194. These emerging areas present exciting opportunities for model development and knowledge translation. In the context of breast cancer, few in vitro models have been developed to study early-stage breast cancer and the influence of the breast tumor microenvironment towards metastatic disease. Organotypic mammary ducts could be enabling tools to fill this gap. Similarly, there are a limited number of lymphatic-on-a-chip models for studying lymphatic (patho)physiology, which is another area that could have a significant impact on the biomedical community. In this section, we provide illustrative examples of recent achievements in developing models of the mammary duct and lymphatics.
8.1. Modeling mammary duct physiology and breast cancer
The primary function of the mammary gland is to produce and secrete milk as nutrients for human offspring. The mammary gland is also subject to the development of breast cancer, which typically occurs in the lobules and terminal ducts of mammary glands, categorized as lobular or ductal, respectively. The ductal subtype is more prevalent, and results in both non-invasive and invasive breast cancers, accounting for ~80% of all cases199. Ductal breast cancer typically progresses from a benign lesion (e.g., atypical ductal hyperplasia or ductal carcinoma in situ) to invasive ductal carcinoma and, ultimately, metastasize (Figure 19A)200. Several microfluidic approaches have been developed to mimic the mammary epithelial interface with mammary epithelium in 2D201–203 (Figure 19B i–ii). However, these models do not recapitulate mammary epithelial structure and physicochemical cues present in the breast TME (e.g., matrix composition, matrix density, biochemical gradients, stromal cell interactions), which in turn influence function. Recently, 3D organotypic microfluidic models have started to emerge that recapitulate the tubular structure of the mammary duct and integrate components of the breast TME. Figure 21 highlights some examples of relevant breast cancer models and the different stages that were modeled.
Grafton et al. first demonstrated the difference in biological response of mammary epithelial cells cultured on traditional 2D assays and those cultured in 3D microfluidic mimics of the mammary duct 204 In their breast-on-a-chip model, the authors developed microchannels with semicircular cross-sections (i.e., hemichannels), on which basement membrane protein (e.g., laminin) and mammary epithelial cells were seeded. Both normal and tumor mammary epithelial cells cultured in the hemichannels exhibited basoapical polarity, as observed in vivo, whereas polarity was not achieved in a traditional 2D platform. Expanding on their initial work, the authors used the model to screen commonly prescribed anti-tumor drugs205. They found that cells in the hemichannels were significantly less sensitive to bleomycin and doxorubicin when compared to their 2D monolayer counterparts, suggesting that breast tumor geometry affected the observed biology within the models. Although this study highlighted drug efficacy differences between models with ductal geometry and traditional 2D platforms, the effect of hemichannel edges were not described. Edge effects have been described to produce functional changes in other cell types (e.g., migration patterns), and the question of their impact in organotypic models remains unsolved. Additionally, while the authors did include relevant proteins for cell adhesion (laminin), the effect of surface stiffness, which has been shown to profoundly impact cell function (e.g., migration and proliferation) was not characterized.
An influential paper by Zervantonakis et al. also used hemichannel geometry to model the extravasation of breast cancer cells into a nearby endothelial vessel model58 (Figure 19B iii). Towards a tubular model of the mammary duct, Bischel et al. further demonstrated the importance of ductal geometry using their “viscous finger patterning” approach to generate mammary epithelial tubular lumens in collagen hydrogels. The selection of collagen as a supporting matrix was appropriate since collagen is the bulk protein in the mammary duct microenvironment28. The authors compared cytokine secretion for various culture methods, including mammary cells cultured in conventional 2D plastic wells, embedded in a 3D gel, on top of the 3D gel, and patterned in lumens. Mammary cells cultured in lumen models produced significantly higher levels of secreted growth factors as compared to the other culture configurations, which was hypothesized to be a result of different mechanical stresses in the lumen microenvironment and in the spatial distribution of the cultured cells. While these results were remarkable, their relevance compared to in vivo models is still an open question. Using an approach similar to Bischel et al.28, but with higher reproducibility of the luminal dimensions, Morgan et al. developed 3D tumorigenic mammary ducts using estrogen-receptor (ER) positive (i.e., a breast cancer subtype) MCF7 cells14 (Figure 19 iv). The mammary epithelium of these ducts exhibited in vivo-like properties, with cells achieving basoapical polarity and proliferation rates (3%) similar to those found in vivo. The authors treated the MCF7 ducts with 17β-estradiol (E2), an ER agonist, to model hyperplasia. Results were similar to breast cancer progression in vivo, with E2-induced growth of MCF7 cells into the center of the duct. Additionally, the response to E2 was significantly enhanced by molecular crosstalk with mammary fibroblasts (HMFs) co-cultured in the surrounding collagen matrix. In follow-up studies, Morgan et al. and Ayuso et al, explored the link between breast cancer progression and obesity206 and microenvironmental factors (e.g., hypoxia), respectively207 (Figure 19 v). Notably, the latter study also leveraged analysis techniques to characterize metabolic changes occurring during breast cancer development and progression with nuclear magnetic resonance (NMR). The organotypic mammary duct models described above can mimic key biological and physicochemical cues of the breast tumor microenvironment and have paved the way for a new generation of biology-focused studies with improved modeling of in vivo situations. However, there is room for continued innovation of these models to expand their capabilities, such as incorporating different stromal cell types, protein compositions, and mechanical cues (e.g. collagen fiber alignment and matrix stiffness are critical factors in tumor cell migration stiffness208).
Looking forward, 3D microfluidic models of the mammary gland could be used to explore factors outside of oncogenic pathologies. Development and normal function of the mammary gland has primarily been studied in mouse models and traditional 2D culture, but organotypic 3D in vitro models have not yet been established. We believe these models could leverage the capabilities of microfluidics to incorporate stable gradients of signaling factors. Signaling gradients are crucial to gland development, and if perturbed, could result in severe pathologies. Likewise, the use of analytical techniques (e.g., NMR, MS/MS) could be coupled to the organotypic models, and provide an accurate snapshot of cell metabolism, function, and toxicity. Ultimately, the field of organotypic models is shifting toward predictive modeling of patient response, but this aspect remains to be modeled for the breast.
8.2. Modeling lymphatics in health and disease
The lymphatic system, which complements the circulatory system, is integral to fluid homeostasis, immune surveillance, and fat absorption, among other physiological functions202. Its dysfunction impacts health on a systemic level and is implicated in multiple diseases, including lymphedema, inflammation, and cancer. An overview of existing lumen-based lymphatic models is shown in Figure 20. Kim et al. developed a lymphangiogenesis model using a 2D interface multi-microchannel design (Figure 20 ii)209. The model incorporated multiple microenvironmental factors required for the robust sprouting of new lymphatic vessels, including interstitial flow, stromal fibroblasts, and pro-lymphangiogenic growth factors (e.g., VEGF-C). Among these factors, interstitial flow served as a primary regulatory cue for lymphangiogenesis, even in the absence of biochemical cues. In the presence of pro-lymphangiogenic growth factors, interstitial flow significantly enhanced lymphatic vessel sprouting. The authors further tested the effect of small molecule inhibitors and inflammatory cytokines in their model and observed significant vessel outgrowth after treatment with FTY720(S)-P, an agonist for S1P receptors. Overall, this study illustrated the importance of mechanical cues in regulating lymphangiogenesis and the applicability of microfluidic models for modeling normal lymphatic vessel physiology.
Figure 20.
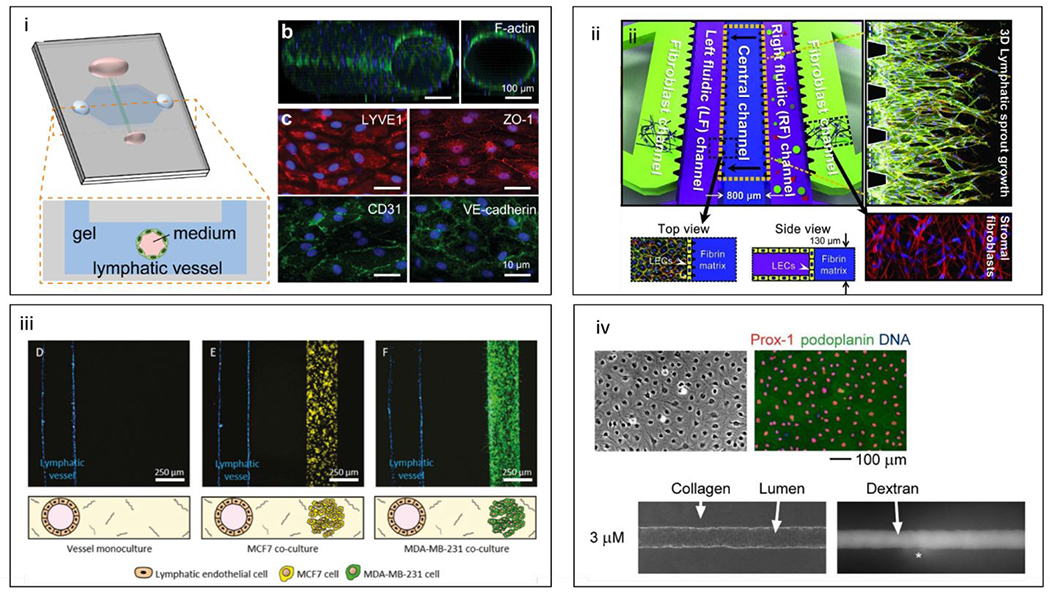
Microfluidic lymphatic models, (i) Schematic of organotypic lymphatic vessel model. A lymphatic endothelial tube is formed in natural hydrogel by removing a sacrificial mold. (b) Confocal image of a cultured lymphatic vessel with patent tubular structure. (c) Lymphatic vessels express lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1) and endothelial junctional proteins (ZO-1, CD31, and VE-cadherin).211 (ii) Schematic of multi-microchannel device comprising outer channels for stromal fibroblasts, a central channel for ECM, and two inner channels for lymphatic endothelial patterning or biochemical gradient generation. Top and side views of the lymphatic and ECM channels are included for clarification, along with immunofluorescent images of lymphatic vessel sprouting and stromal fibroblasts.209 (iii) Adaptation of the microfluidic lumen model shown above for lymphatic-breast cancer co-cultures. Representative images of vessel monoculture, coculture with MCF7 cells (in yellow), and coculture with MDA-MB-231 cells (in green).212 (iv) LEC cultures and tubes. Lymphatic markers Prox-1 (red) and podoplanin (green) and DNA (blue)(top). Phase-contrast image of LEC tubes cultured under 3 μM db-cAMP and fluorescence images after 26 minutes of perfusion with Alexa 488-conjugated 10 kDa dextran (bottom).210 Reproduced with permission from Elsevier: from ref. 209, copyright 2016; from reference 210, copyright 2008.
Towards recapitulating the luminal structure of lymphatic vessels, Price etal. developed the first organotypic lymphatic vessel model using human dermal lymphatic microvascular endothelial cells to generate lymphatic endothelial tubules (Figure 20 iv).210 The authors examined the effect of cyclic AMP (cAMP) on the barrier function of the lymphatic vessels to elucidate how soluble agonists regulate their permeability. Exposure of the vessels to high doses of cAMP (over 80 μM) decreased permeability and increased cell-cell junctions (i.e., VE-cadherin expression) compared with controls. These results indicated that the lymphatic endothelium responds similarly to cAMP as the blood vessel endothelium. In more recent work, Gong et al. further characterized the physiology of organotypic lymphatic vessels in normal, inflammatory, and tumor microenvironments211 (Figure 20 i). The authors were able to recapitulate the natural leakiness and draining function of lymphatic vessels, which were significantly different from blood vessels cultured in the same model. In co-culture with breast cancer-associated fibroblasts, vessel permeability significantly increased as a result of increased IL-6 secreted in the simulated tumor microenvironment. The lymphatic vessels were rescued, however, when IL-6 was neutralized with an anti-IL-6 antibody (an FDA approved therapy). In follow-up studies, the authors expanded this work to study the cross-talk between lymphatic vasculature and breast cancer cells, revealing the conditioning of the lymphatic vessels exerted by the tumor212, 213 (Figure 20 iv). Collectively, the results from the study demonstrated the capability of organotypic modeling to capture differential vessel response in varied microenvironments and unveiled the potential of such models for testing or identifying new targeted therapeutics.
Furthermore, a recent microfluidic model integrated ex vivo tumor and lymph node tissue slices into a single device to elucidate tumor-lymphatic interactions214. A unique feature of this approach was the ability to recirculate media throughout the device, enabling dynamic and continuous crosstalk between the tumor and lymph node. The authors examined T cell stimulation in lymph node slices co-cultured with tumor and healthy tissues. They observed that T cells were more immunosuppressed in the co-culture with the tumor, suggesting a role for the device in studying tumor-immune interactions. Indeed, the lymphatics play a critical role in cancer progression, where they are routes for tumor dissemination and modulators of immunoresistance in the tumor microenvironment. Therefore, continued development of organotypic lymphatic models for cancer, especially in the context of immunotherapy and immunoresistance, could have profound value for basic and translational cancer research.
9. Conclusions and outlook
Microfluidic lumen-based systems have advanced rapidly since their inception. There is now an extensive toolbox of models that can recapitulate many aspects of the normal and diseased states of major organs. These models have demonstrated value for therapeutic discovery, drug efficacy testing, and clinical translation. The recent interest in these models has been such, that major journals215–217 and US scientific funding mechanisms (e.g., “Clinical trials on a chip” RFA from the NIH or STAR initiative from EPA)218 have recently acknowledged the milestones accomplished by these models, highlighted their potential in the healthcare pipeline and encouraged further research on this topic. While the promise these models hold is high, the remaining work to deliver this promise is also considerable.
Toward wider adoption of lumen models into the clinic, several barriers remain, including a lack of (1) patient-specific models, and (2) standardized materials and device designs. Given that isolating patient cells is a nontrivial process and typically results in low cell numbers, microscale organ systems are particularly well-suited for culturing these small sample sizes as compared to conventional approaches. In this context, a handful of models have recently focused on using patient-derived cells (or iPSC-derived cells), in place of human cell lines and animal cells, to simulate patient-specific responses to drug treatment65 and have demonstrated that responses elicited by the models can correlate to clinical results206. However, the high variability associated with patient-derived samples hinders the reproducibility of models and constitutes a severe challenge to the field in general. We anticipate that further development and refinement of these patient-specific models will accelerate their translation to the clinic.
Next, a standardization of the models is required. As we have discussed in previous sections, successful models will require to achieve a balance between complexity (i.e., including all the necessary components and biological/physical cues) and utility for a given application. To this last point, as model complexity increases, sufficient throughput for applications like drug discovery and testing becomes a significant challenge for these models. Therefore, it will be necessary to elucidate for each application the optimal balance between complexity and utility. Along with standardization of components comes the standardization of fabrication materials and designs, which not only increases the potential for clinical translation but also impacts commercialization. Currently, lumen-based models rely much on PDMS as a prototyping material to develop new models and provide a basic characterization of their performance. Beyond proof-of-concept, devices need to transition to more scalable and clinically appropriate materials, such as polystyrene219, which does not suffer from the absorption issues typically ascribed to PDMS, undesirable for drug testing and cell culture applications219, 220. Conversely, alternative polymers (e.g., poly (methyl methacrylate and polycarbonates), are not compatible with soft lithography and have lower gas permeability than PDMS, required for constant O2/CO2 exchange into cell culture. Many concrete solutions have been presented to improve the physical properties of PDMS, aiming to improve the extrapolation of in vitro results in precise estimates of molecule absorption and bioavailability 219, 221. However, the transition away from PDMS would, ultimately, improves the utility and scalability of the models for drug testing. Several organ-on-a-chip companies have embraced this reality to generate successful products, including Hesperos, Inc., CN-BIO Innovations, MIMETAS, Nortis, and AIM Biotech among others222.
Along with scalability, the need for specialized equipment or training to operate current models drives their use away from biologists and clinicians and therefore hinders the integration of lumen models in the healthcare pipeline. Engineers should consider designs that can be streamlined with standard laboratory equipment (e.g., micropipettes for fluid handling223). In addition to scalable materials, these companies also developed their devices to integrate with standard equipment in biology laboratories, such as micropipettes and plate readers; thus, rendering them more approachable to new users and reducing the technical barrier to adoption. Researchers have started to create innovative device designs to bridge the gap between microfluidics engineering and biology224. Although still considered a fledgling industry, the organ-on-a-chip market is projected to grow to 220 million USD by 2025 (Market Study Report, LLC) and, thus, is well-positioned to revitalize the current drug development pipeline. Ultimately, we are optimistic that the continued efforts to develop lumen-based microfluidic technologies will have profound implications for basic and translational biomedical research.
11. Acknowledgements
The authors acknowledge funding support from the University of Wisconsin Carbone Cancer Center (AAB7173), Morgridge Institute for Research, National Institutes of Health (R01EB010039), and the Wisconsin Head & Neck Cancer SPORE (P50DE026787).
Footnotes
10. Conflicts of interest
David J. Beebe is a board member and stockowner of Tasso, Inc., and a stockowner of Bellbrook Labs, LLC. David J. Beebe is a founder, stockowner, and consultant of Salus Discovery LLC. David J. Beebe is an advisor and stockowner of Lynx Biosciences, LLC, Onexio Biosystems, LLC, and Stacks to the Future, LLC. David J. Beebe holds equity in Bellbrook Labs, LLC, Tasso Inc., Salus Discovery LLC, Stacks to the Future, LLC and Onexio Biosystems, LLC.
12. References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA and Jemal A, CA: A Cancer Journal for Clinicians, 2018, 68, 394–424. [DOI] [PubMed] [Google Scholar]
- 2.Hutchinson L and Kirk R, Journal, 2011. [Google Scholar]
- 3.Simian M and Bissell MJ, Journal of Cell Biology, 2017, 216, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bischel LL, Sung KE, Jiménez-Torres JA, Mader B, Keely PJ and Beebe DJ, The FASEB Journal, 2014, 28, 4583–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker BM and Chen CS, J Cell Sci, 2012, 125, 3015–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, McFarland JM, Wong B, Boehm JS, Beroukhim R and Golub TR, Nature genetics, 2017, 49, 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhatia SN and Ingber DE, Nature biotechnology, 2014, 32, 760–772. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B, Korolj A, Lai BFL and Radisic M, Nature Reviews Materials, 2018, 3, 257–278. [Google Scholar]
- 9.Huh D, Torisawa YS, Hamilton GA, Kim HJ and Ingber DE, Lab Chip, 2012, 12, 2156–2164. [DOI] [PubMed] [Google Scholar]
- 10.Bryant DM and Mostov KE, Nature reviews Molecular cell biology, 2008, 9, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaffer CL and Weinberg RA, Science, 2011, 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- 12.Bischel LL, Young EW, Mader BR and Beebe DJ, Biomaterials, 2013, 34, 1471–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim S, Lee H, Chung M and Jeon NL, Lab Chip, 2013, 13, 1489–1500. [DOI] [PubMed] [Google Scholar]
- 14.Morgan MM, Livingston MK, Warrick JW, Stanek EM, Alarid ET, Beebe DJ and Johnson BP, Sci Rep, 2018, 8, 7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grafton MM, Wang L, Vidi PA, Leary J and Lelievre SA, Integr Biol (Camb), 2011, 3, 451–459. [DOI] [PubMed] [Google Scholar]
- 16.Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, Lopez JA and Stroock AD, Proceedings of the National Academy of Sciences of the United States of America, 2012, 109, 9342–9347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Widmaier EP, Raff H and Strang KT, The Mechanisms of Body Function, 2011. [Google Scholar]
- 18.Folkman J, New England Journal of Medicine, 1971, 285, 1182–1186. [DOI] [PubMed] [Google Scholar]
- 19.Hanahan D and Weinberg RA, Cell, 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 20.Bogorad MI, DeStefano J, Karlsson J, Wong AD, Gerecht S and Searson PC, Lab Chip, 2015, 15, 4242–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA and Lewis JA, Advanced materials, 2014, 26, 3124–3130. [DOI] [PubMed] [Google Scholar]
- 22.Douville NJ, Tung YC, Li R, Wang JD, El-Sayed ME and Takayama S, Anal Chem, 2010, 82, 2505–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young EW, Watson MW, Srigunapalan S, Wheeler AR and Simmons CA, Analytical chemistry, 2010, 82, 808–816. [DOI] [PubMed] [Google Scholar]
- 24.Sato M, Sasaki N, Ato M, Hirakawa S, Sato K and Sato K, PLoS One, 2015, 10, e0137301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song JW and Munn LL, Proceedings of the National Academy of Sciences of the United States of America, 2011, 108, 15342–15347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeon JS, Bersini S, Whisler JA, Chen MB, Dubini G, Charest JL, Moretti M and Kamm RD, Integr Biol (Camb), 2014, 6, 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chrobak KM, Potter DR and Tien J, Microvascular research, 2006, 71, 185–196. [DOI] [PubMed] [Google Scholar]
- 28.Bischel LL, Lee S-H and Beebe DJ, Journal of laboratory automation, 2012, 17, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jimenez-Torres JA, Peery SL, Sung KE and Beebe DJ, Adv Healthc Mater, 2016, 5, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Graaf MNS, Cochrane A, van den Hil FE, Buijsman W, van der Meer AD, van den Berg A, Mummery CL and Orlova VV, APL Bioeng, 2019, 3, 026105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barlow HR and Cleaver O, Cells, 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen D-HT, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA and Chen CS, Proceedings of the National Academy of Sciences, 2013, 110, 6712–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benias PC, Wells RG, Sackey-Aboagye B, Klavan H, Reidy J, Buonocore D, Miranda M, Kornacki S, Wayne M and Carr-Locke DL, Scientific reports, 2018, 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lam RH, Kim M-C and Thorsen T, Analytical chemistry, 2009, 81, 5918–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bianchi E, Molteni R, Pardi R and Dubini G, Journal of biomechanics, 2013, 46, 276–283. [DOI] [PubMed] [Google Scholar]
- 36.Chen Z, Tang M, Huang D, Jiang W, Li M, Ji H, Park J, Xu B, Atchison LJ, Truskey GA and Leong KW, Lab Chip, 2018, DOI: 10.1039/c81c00202a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JY, Kim HO, Kim KD, Kim SK, Lee SK and Jung H, The Analyst, 2011, 136, 2831–2836. [DOI] [PubMed] [Google Scholar]
- 38.Nourshargh S and Alon R, Immunity, 2014, 41, 694–707. [DOI] [PubMed] [Google Scholar]
- 39.Han S, Yan JJ, Shin Y, Jeon JJ, Won J, Jeong HE, Kamm RD, Kim YJ and Chung S, Lab Chip, 2012, 12, 3861–3865. [DOI] [PubMed] [Google Scholar]
- 40.Hind LE, Ingram PN, Beebe DJ and Huttenlocher A, Blood, 2018, 132, 1818–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ingram PN, Hind LE, Jiminez-Torres JA, Huttenlocher A and Beebe DJ, Advanced healthcare materials, 2018, 7, 1700497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Libby P, Ridker PM and Hansson GK, Nature, 2011, 473, 317–325. [DOI] [PubMed] [Google Scholar]
- 43.Hansson GK and Hermansson A, Nat Immunol, 2011, 12, 204–212. [DOI] [PubMed] [Google Scholar]
- 44.Zheng W, Huang R, Jiang B, Zhao Y, Zhang W and Jiang X, Small, 2016, 12, 2022–2034. [DOI] [PubMed] [Google Scholar]
- 45.Chen C and Khismatullin DB, PLoS One, 2015, 10, e0123088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robert J, Weber B, Frese L, Emmert MY, Schmidt D, von Eckardstein A, Rohrer L and Hoerstrup SP, PLoS One, 2013, 8, e79821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Engeland NC, Pollet AM, den Toonder JM, Bouten CV, Stassen OM and Sahlgren CM, Lab on a Chip, 2018, 18, 1607–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menon NV, Tay HM, Wee SN, Li KHH and Hou HW, Lab Chip, 2017, 17, 2960–2968. [DOI] [PubMed] [Google Scholar]
- 49.Tsai M, Kita A, Leach J, Rounsevell R, Huang JN, Moake J, Ware RE, Fletcher DA and Lam WA, The Journal of clinical investigation, 2011, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Westein E, van der Meer AD, Kuijpers MJ, Frimat JP, van den Berg A and Heemskerk JW, Proceedings of the National Academy of Sciences of the United States of America, 2013, 110, 1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ayuso JM, Monge R, Martinez-Gonzalez A, Virumbrales-Munoz M, Llamazares GA, Berganzo J, Hernandez-Lain A, Santolaria J, Doblare M, Hubert C, Rich JN, Sanchez-Gomez P, Perez-Garcia VM, Ochoa I and Fernandez LJ, Neuro Oncol, 2017, 19, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Versteeg HH, Heemskerk JW, Levi M and Reitsma PH, Physiological reviews, 2013, 93, 327–358. [DOI] [PubMed] [Google Scholar]
- 53.Sakurai Y, Hardy ET, Ahn B, Tran R, Fay ME, Ciciliano JC, Mannino RG, Myers DR, Qiu Y and Carden MA, Nature communications, 2018, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Su T, Huang K, Daniele MA, Hensley MT, Young AT, Tang J, Allen TA, Vandergriff AC, Erb PD, Ligler FS and Cheng K, Acs Appl Mater Inter, 2018, 10, 33088–33096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagy J, Chang S, Dvorak A and Dvorak H, British journal of cancer, 2009, 100, 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Al-Abd AM, Alamoudi AJ, Abdel-Naim AB, Neamatallah TA and Ashour OM, Journal of advanced research, 2017, 8, 591–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeon JS, Zervantonakis IK, Chung S, Kamm RD and Charest JL, PLoS One, 2013, 8, e56910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB and Kamm RD, Proceedings of the National Academy of Sciences of the United States of America, 2012, 109, 13515–13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen MB, Whisler JA, Jeon JS and Kamm RD, Integr Biol (Camb), 2013, 5, 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bersini S, Jeon JS, Dubini G, Arrigoni C, Chung S, Charest JL, Moretti M and Kamm RD, Biomaterials, 2014, 35, 2454–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M and Kamm RD, Proceedings of the National Academy of Sciences of the United States of America, 2015, 112, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Q, Liu T and Qin J, Lab Chip, 2012, 12, 2837–2842. [DOI] [PubMed] [Google Scholar]
- 63.Wong AD and Searson PC, Cancer Res, 2014, 74, 4937–4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lupo G, Caporarello N, Olivieri M, Cristaldi M, Motta C, Bramanti V, Avola R, Salmeri M, Nicoletti F and Anfuso CD, Frontiers in pharmacology, 2017, 7, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jimenez-Torres JA, Virumbrales-Munoz M, Sung KE, Lee MH, Abel EJ and Beebe DJ, EBioMedicine, 2019, 42, 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bajaj P, Harris JF, Huang J-H, Nath P and Iyer R, ACS Biomaterials Science & Engineering, 2016, 2, 473–488. [DOI] [PubMed] [Google Scholar]
- 67.Lalley PM, Respiratory physiology & neurobiology, 2013, 187, 199–210. [DOI] [PubMed] [Google Scholar]
- 68.Shrestha J, Razavi Bazaz S, Aboulkheyr Es H, Yaghobian Azari D, Thierry B, Ebrahimi Warkiani M and Ghadiri M, Critical Reviews in Biotechnology, 2020, 1–18. [DOI] [PubMed] [Google Scholar]
- 69.Goetz D and Ren CL, Pediatr Ann, 2019, 48, e154–e161. [DOI] [PubMed] [Google Scholar]
- 70.Suki B, Ito S, Stamenovic D, Lutchen KR and Ingenito EP, J Appl Physiol (1985), 2005, 98, 1892–1899. [DOI] [PubMed] [Google Scholar]
- 71.Nalayanda DD, Puleo CM, Fulton WB, Wang T-H and Abdullah F, Experimental lung research, 2007, 33, 321–335. [DOI] [PubMed] [Google Scholar]
- 72.Sellgren KL, Butala EJ, Gilmour BP, Randell SH and Grego S, Lab on a Chip, 2014, 14, 3349–3358. [DOI] [PubMed] [Google Scholar]
- 73.Dorshow R and Nicoli D, The Journal of Chemical Physics, 1981, 75, 5853–5856. [Google Scholar]
- 74.Zamprogno PGV, Wuthrich S, Achenbach S, Stucki J, Hobi N, Schneider-Daum N, Lehr C-M, Huwer H, Geiser T and Schmid R, BioRxiv, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Humayun M, Chow CW and Young EWK, Lab Chip, 2018, 18, 1298–1309. [DOI] [PubMed] [Google Scholar]
- 76.Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB and Eickelberg O, European Respiratory Journal, 2017, 50, 1601805. [DOI] [PubMed] [Google Scholar]
- 77.Hassell BA, Goyal G, Lee E, Sontheimer-Phelps A, Levy O, Chen CS and Ingber DE, Cell reports, 2017, 21, 508–516. [DOI] [PubMed] [Google Scholar]
- 78.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY and Ingber DE, Science, 2010, 328, 1662–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stucki AO, Stucki JD, Hall SR, Felder M, Mermoud Y, Schmid RA, Geiser T and Guenat OT, Lab on a Chip, 2015, 15, 1302–1310. [DOI] [PubMed] [Google Scholar]
- 80.Grubb BR, Schiretz FR and Boucher RC, The American journal of physiology, 1997, 273, C21–29. [DOI] [PubMed] [Google Scholar]
- 81.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW and Boucher RC, Cell, 1998, 95, 1005–1015. [DOI] [PubMed] [Google Scholar]
- 82.Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee HH, Alves SE, Salmon M, Ferrante TC, Weaver JC, Bahinski A, Hamilton GA and Ingber DE, Nature methods, 2016, 13, 151–157. [DOI] [PubMed] [Google Scholar]
- 83.Villenave R, Lucchesi C, Cheng D, Lee H, Nguyen J, Varone A, Karalis K, Alves S, Salmon M and Hamilton GA, Am J Resp Crit Care, 2017, 195. [Google Scholar]
- 84.Nesmith AP, Agarwal A, McCain ML and Parker KK, Lab Chip, 2014, 14, 3925–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Punde TH, Wu W-H, Lien P-C, Chang Y-L, Kuo P-H, Chang MD-T, Lee K-Y, Huang C-D, Kuo H-P and Chan Y-F, Integrative Biology, 2015, 7, 162–169. [DOI] [PubMed] [Google Scholar]
- 86.Park JY, Ryu H, Lee B, Ha DH, Ahn M, Kim S, Kim JY, Jeon NL and Cho DW, Biofabrication, 2018, 11, 015002. [DOI] [PubMed] [Google Scholar]
- 87.Alkasalias T, Moyano-Galceran L, Arsenian-Henriksson M and Lehti K, International journal of molecular sciences, 2018, 19, 1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang M, Xu C, Jiang L and Qin J, Toxicol Res (Camb), 2018, 7, 1048–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Childs A, Zullo AR, Joyce NR, McConeghy KW, van Aalst R, Moyo P, Bosco E, Mor V and Gravenstein S, BMC geriatrics, 2019, 19, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Everard ML, European Respiratory Review, 2016, 25, 36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lipsitch M, Swerdlow DL and Finelli L, New Englandjournal of medicine, 2020, 382, 1194–1196. [DOI] [PubMed] [Google Scholar]
- 92.Barkal LJ, Procknow CL, Álvarez-García YR, Niu M, Jiménez-Torres JA, Brockman-Schneider RA, Gern JE, Denlinger LC, Theberge AB and Keller NP, Nature communications, 2017, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Briard B, Heddergott C and Latgé J-P,MBio, 2016, 7, e00219–00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Felder M, Stucki A, Stucki J, Geiser T and Guenat OT, Integrative Biology, 2014, 6, 1132–1140. [DOI] [PubMed] [Google Scholar]
- 95.Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, Thorneloe KS, McAlexander MA and Ingber DE, Science translational medicine, 2012, 4, 159ra147–159ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sontheimer-Phelps A, Hassell BA and Ingber DE, Nature Reviews Cancer, 2019, DOI: 10.1038/s41568-018-0104-6. [DOI] [PubMed] [Google Scholar]
- 97.Xu Z, Gao Y, Hao Y, Li E, Wang Y, Zhang J, Wang W, Gao Z and Wang Q, Biomaterials, 2013, 34, 4109–4117. [DOI] [PubMed] [Google Scholar]
- 98.Jain A, Barrile R, van der Meer AD, Mammoto A, Mammoto T, De Ceunynck K, Aisiku O, Otieno MA, Louden CS, Hamilton GA, Flaumenhaft R and Ingber DE, Clin Pharmacol Ther, 2018, 103, 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee KK, McCauley HA, Broda TR, Kofron MJ, Wells JM and Hong CI, Lab on a Chip, 2018, 18, 3079–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schroeder BO, Gastroenterology report, 2019, 7, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Carabotti M, Scirocco A, Maselli MA and Severi C, Annals of gastroenterology: quarterly publication of the Hellenic Society of Gastroenterology, 2015, 28, 203. [PMC free article] [PubMed] [Google Scholar]
- 102.Bein A, Shin W, Jalili-Firoozinezhad S, Park MH, Sontheimer-Phelps A, Tovaglieri A, Chalkiadaki A, Kim HJ and Ingber DE, Cellular and molecular gastroenterology and hepatology, 2018, 5, 659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gao D, Liu H, Lin JM, Wang Y and Jiang Y, Lab Chip, 2013, 13, 978–985. [DOI] [PubMed] [Google Scholar]
- 104.Imura Y, Asano Y, Sato K and Yoshimura E, Analytical sciences : the international journal of the Japan Society for Analytical Chemistry, 2009, 25, 1403–1407. [DOI] [PubMed] [Google Scholar]
- 105.Sung JH, Yu J, Luo D, Shuler ML and March JC, Lab Chip, 2011, 11, 389–392. [DOI] [PubMed] [Google Scholar]
- 106.Kim W and Kim GH, Chemical Engineering Journal, 2018, 334, 2308–2318. [Google Scholar]
- 107.Gunasekara DB, Speer J, Wang Y, Nguyen DL, Reed MI, Smiddy NM, Parker JS, Fallon JK, Smith PC, Sims CE, Magness ST and Allbritton NL, Anal Chem, 2018, 90, 13331–13340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hermanson GT, Bioconjugate techniques, Academic press, 2013. [Google Scholar]
- 109.Odijk M, van der Meer AD, Levner D, Kim HJ, van der Helm MW, Segerink LI, Frimat JP, Hamilton GA, Ingber DE and van den Berg A, Lab Chip, 2015, 15, 745–752. [DOI] [PubMed] [Google Scholar]
- 110.Speer JE, Gunasekara DB, Wang Y, Fallon JK, Attayek PJ, Smith PC, Sims CE and Allbritton NL, Journal of biological engineering, 2019, 13, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Trietsch SJ, Naumovska E, Kurek D, Setyawati MC, Vormann MK, Wilschut KJ, Lanz HL, Nicolas A, Ng CP, Joore J, Kustermann S, Roth A, Hankemeier T, Moisan A and Vulto P, Nature communications, 2017, 8, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim HJ, Huh D, Hamilton G and Ingber DE, Lab on a Chip, 2012, 12, 2165–2174. [DOI] [PubMed] [Google Scholar]
- 113.Kim HJ and Ingber DE, Integrative Biology, 2013, 5, 1130–1140. [DOI] [PubMed] [Google Scholar]
- 114.Zheng L, Kelly CJ and Colgan SP, American journal of physiology. Cell physiology, 2015, 309, C350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ulluwishewa D, Anderson RC, Young W, McNabb WC, van Baarlen P, Moughan PJ, Wells JM and Roy NC, Cellular microbiology, 2015, 17, 226–240. [DOI] [PubMed] [Google Scholar]
- 116.Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, Niegowska M, Estes M, Jager C, Seguin-Devaux C, Zenhausern F and Wilmes P, Nature communications, 2016, 7, 11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jalili-Firoozinezhad S, Gazzaniga FS, Calamari EL, Camacho DM, Fadel CW, Bein A, Swenor B, Nestor B, Cronce MJ and Tovaglieri A, Nature biomedical engineering, 2019, 3, 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yissachar N, Zhou Y, Ung L, Lai NY, Mohan JF, Ehrlicher A, Weitz DA, Kasper DL, Chiu IM and Mathis D, Cell, 2017, 168, 1135–1148. e1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K and Ohkura N, Annual Review of Immunology, 2020, 38. [DOI] [PubMed] [Google Scholar]
- 120.Dutton JS, Hinman SS, Kim R, Wang Y and Allbritton NL, Trends Biotechnol, 2019, 37, 744–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dawson A, Dyer C, Macfie J, Davies J, Karsai L, Greenman J and Jacobsen M, Biomicrofluidics, 2016, 10, 064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen Y, Zhou W, Roh T, Estes MK and Kaplan DL, PloS one, 2017, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kasendra M, Tovaglieri A, Sontheimer-Phelps A, Jalili-Firoozinezhad S, Bein A, Chalkiadaki A, Scholl W, Zhang C, Rickner H, Richmond CA, Li H, Breault DT and Ingber DE, Sci Rep, 2018, 8, 2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim HJ, Li H, Collins JJ and Ingber DE, Proceedings of the National Academy of Sciences, 2016, 113, E7–E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hanauer SB and Present DH, Reviews in gastroenterological disorders, 2003, 3, 81–92. [PubMed] [Google Scholar]
- 126.Beaurivage C, Naumovska E, Chang YX, Elstak ED, Nicolas A, Wouters H, van Moolenbroek G, Lanz HL, Trietsch SJ and Joore J, International journal of molecular sciences, 2019, 20, 5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ayuso JM, Virumbrales-Munoz M, McMinn PH, Rehman S, Gomez I, Karim MR, Trusttchel R, Wisinski KB, Beebe DJ and Skala MC, Lab Chip, 2019, 19, 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Virumbrales-Munoz M, Ayuso JM, Lacueva A, Randelovic T, Livingston MK, Beebe DJ, Olivan S, Pereboom D, Doblare M, Fernandez L and Ochoa I, Sci Rep, 2019, 9, 6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Carvalho M, Barata D, Teixeira L, Giselbrecht S, Reis R, Oliveira J, Truckenmuller R and Habibovic P, Science advances, 2019, 5, eaaw1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sartor RB, Nature clinical practice Gastroenterology & hepatology, 2006, 3, 390–407. [DOI] [PubMed] [Google Scholar]
- 131.Essig M, Terzi F, Burtin M and Friedlander G, American journal ofphysiology. Renal physiology, 2001, 281, F751–762. [DOI] [PubMed] [Google Scholar]
- 132.Baudoin R, Griscom L, Monge M, Legallais C and Leclerc E, Biotechnol Prog, 2007, 23, 1245–1253. [DOI] [PubMed] [Google Scholar]
- 133.Frohlich EM, Zhang X and Charest JL, Integr Biol (Camb), 2012, 4, 75–83. [DOI] [PubMed] [Google Scholar]
- 134.Friedrich C, Endlich N, Kriz W and Endlich K, American journal of physiology. Renal physiology, 2006, 291, F856–865. [DOI] [PubMed] [Google Scholar]
- 135.Nieskens TT and Wilmer MJ, European journal of pharmacology, 2016, 790, 46–56. [DOI] [PubMed] [Google Scholar]
- 136.Desrochers TM, Palma E and Kaplan DL, Adv DrugDeliv Rev, 2014, 69-70, 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gao X, Tanaka Y, Sugii Y, Mawatari K and Kitamori T, Analytical sciences : the international journal of the Japan Society for Analytical Chemistry, 2011, 27, 907–912. [DOI] [PubMed] [Google Scholar]
- 138.Jang KJ, Mehr AP, Hamilton GA, McPartlin LA, Chung S, Suh KY and Ingber DE, Integr Biol (Camb), 2013, 5, 1119–1129. [DOI] [PubMed] [Google Scholar]
- 139.Mu X, Zheng W, Xiao L, Zhang W and Jiang X, Lab Chip, 2013, 13, 1612–1618. [DOI] [PubMed] [Google Scholar]
- 140.Rumpler M, Woesz A, Dunlop JW, Van Dongen JT and Fratzl P, Journal of the Royal Society Interface, 2008, 5, 1173–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ng CP, Zhuang Y, Lin AWH and Teo JCM, International Journal of Tissue Engineering, 2013, 2013, 1–10. [Google Scholar]
- 142.Zhu W, Li J and Liu J, BiomedMicrodevices, 2013, 15, 781–791. [Google Scholar]
- 143.Jansen J, Fedecostante M, Wilmer MJ, Peters JG, Kreuser UM, van den Broek PH, Mensink RA, Boltje TJ, Stamatialis D, Wetzels JF, van den Heuvel LP, Hoenderop JG and Masereeuw R, Sci Rep, 2016, 6, 26715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jang KJ and Suh KY, Lab Chip, 2010, 10, 36–42. [DOI] [PubMed] [Google Scholar]
- 145.Jang KJ, Cho HS, Kang DH, Bae WG, Kwon TH and Suh KY, Integr Biol (Camb), 2011, 3, 134–141. [DOI] [PubMed] [Google Scholar]
- 146.Sciancalepore AG, Sallustio F, Girardo S, Gioia Passione L, Camposeo A, Mele E, Di Lorenzo M, Costantino V, Schena FP and Pisignano D, PLoS One, 2014, 9, e87496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Discher DE, Janmey P and Wang Y.-l., Science, 2005, 310, 1139–1143. [DOI] [PubMed] [Google Scholar]
- 148.Weber EJ, Chapron A, Chapron BD, Voellinger JL, Lidberg KA, Yeung CK, Wang Z, Yamaura Y, Hailey DW and Neumann T, Kidney international, 2016, 90, 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zager RA, 1997.
- 150.Miravete M, Klein J, Besse-Patin A, Gonzalez J, Pecher C, Bascands JL, Mercier-Bonin M, Schanstra JP and Buffin-Meyer B, Biochem Biophys Res Commun, 2011. 407, 813–817. [DOI] [PubMed] [Google Scholar]
- 151.Kim S, LesherPerez SC, Kim BC, Yamanishi C, Labuz JM, Leung B and Takayama S, Biofabrication, 2016, 8, 015021. [DOI] [PubMed] [Google Scholar]
- 152.Adler M, Ramm S, Hafner M, Muhlich JL, Gottwald EM, Weber E, Jaklic A, Ajay AK, Svoboda D, Auerbach S, Kelly EJ, Himmelfarb J and Vaidya VS, Journal of the American Society of Nephrology : JASN, 2016, 27, 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li Z, Su W, Zhu Y, Tao T, Li D, Peng X and Qin J, Biomicrofluidics, 2017, 11, 034114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Scales CD Jr, Smith AC, Hanley JM, Saigal CS and U. D. i. A. Project, European urology, 2012, 62, 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Alelign T and Petros B, Advances in urology, 2018, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Laffite G, Leroy C, Bonhomme C, Bonhomme-Coury L, Letavernier E, Daudon M, Frochot V, Haymann JP, Rouziere S, Lucas IT, Bazin D, Babonneau F and Abou-Hassan A, Lab Chip, 2016, 16, 1157–1160. [DOI] [PubMed] [Google Scholar]
- 157.Wei Z, Amponsah PK, Al-Shatti M, Nie Z and Bandyopadhyay BC, Lab Chip, 2012, 12, 4037–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Grignon DJ and Che M, Clin Lab Med, 2005, 25, 305–316. [DOI] [PubMed] [Google Scholar]
- 159.Rini BI, Battle D, Figlin RA, George DJ, Hammers H, Hutson T, Jonasch E, Joseph RW, McDermott DF, Motzer RJ, Pal SK, Pantuck AJ, Quinn DI, Seery V, Voss MH, Wood CG, Wood LS and Atkins MB, J Immunother Cancer, 2019, 7, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Miller CP, Tsuchida C, Zheng Y, Himmelfarb J and Akilesh S, Neoplasia (New York, N.Y.), 2018, 20, 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Haque A, Gheibi P, Gao Y, Foster E, Son KJ, You J, Stybayeva G, Patel D and Revzin A, Scientific reports, 2016, 6, 33980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Leclerc E, Kimura K, Shinohara M, Danoy M, Le Gall M, Kido T, Miyajima A, Fujii T and Sakai Y, Genomics, 2017, 109, 16–26. [DOI] [PubMed] [Google Scholar]
- 163.Beckwitt CH, Clark AM, Wheeler S, Taylor DL, Stolz DB, Griffith L and Wells A, Experimental cell research, 2018, 363, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Carraro A, Hsu WM, Kulig KM, Cheung WS, Miller ML, Weinberg EJ, Swart EF, Kaazempur-Mofrad M, Borenstein JT, Vacanti JP and Neville C, Biomed Microdevices, 2008, 10, 795–805. [DOI] [PubMed] [Google Scholar]
- 165.Tan GD, Toh GW, Birgersson E, Robens J, van Noort D and Leo HL, Biotechnol Bioeng, 2013, 110, 1663–1673. [DOI] [PubMed] [Google Scholar]
- 166.Viravaidya K, Sin A and Shuler ML, Biotechnol Prog, 2004, 20, 316–323. [DOI] [PubMed] [Google Scholar]
- 167.Chao P, Maguire T, Novik E, Cheng KC and Yarmush ML, Biochemical pharmacology, 2009, 78, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Prot JM, Videau O, Brochot C, Legallais C, Benech H and Leclerc E, Int J Pharm, 2011, 408, 67–75. [DOI] [PubMed] [Google Scholar]
- 169.Goral VN, Hsieh YC, Petzold ON, Clark JS, Yuen PK and Faris RA, Lab Chip, 2010, 10, 3380–3386. [DOI] [PubMed] [Google Scholar]
- 170.Maher SP, Crouse RB, Conway AJ, Bannister EC, Achyuta AK, Clark AY, Sinatra FL, Cuiffi JD, Adams JH, Kyle DE and Saadi WM, Biomed Microdevices, 2014, 16, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hegde M, Jindal R, Bhushan A, Bale SS, McCarty WJ, Golberg I, Usta OB and Yarmush ML, Lab Chip, 2014, 14, 2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sung JH, Choi JR, Kim D and Shuler ML, Biotechnol Bioeng, 2009, 104, 516–525. [DOI] [PubMed] [Google Scholar]
- 173.Vukicevic S, Kleinman HK, Luyten FP, Roberts AB, Roche NS and Reddi AH, Experimental cell research, 1992, 202, 1–8. [DOI] [PubMed] [Google Scholar]
- 174.Lee J, Choi JR, Ha SK, Choi I, Lee SH, Kim D, Choi N and Sung JH, Lab Chip, 2014, 14, 2948–2957. [DOI] [PubMed] [Google Scholar]
- 175.Hongo T, Kajikawa M, Ishida S, Ozawa S, Ohno Y, Sawada J, Umezawa A, Ishikawa Y, Kobayashi T and Honda H, J Biosci Bioeng, 2005, 99, 237–244. [DOI] [PubMed] [Google Scholar]
- 176.Livingston MK, Morgan MM, Daly WT, Murphy WL, Johnson BP, Beebe DJ and Virumbrales-Muñoz M. a., ACS Biomaterials Science & Engineering, 2019, 5, 6089–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Esch MB, Mahler GJ, Stokol T and Shuler ML, Lab on a Chip, 2014, 14, 3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Prodanov L, Jindal R, Bale SS, Hegde M, McCarty WJ, Golberg I, Bhushan A, Yarmush ML and Usta OB, Biotechnol Bioeng, 2016, 113, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kang YB, Sodunke TR, Lamontagne J, Cirillo J, Rajiv C, Bouchard MJ and Noh M, Biotechnol Bioeng, 2015, 112, 2571–2582. [DOI] [PubMed] [Google Scholar]
- 180.Rennert K, Steinborn S, Groger M, Ungerbock B, Jank AM, Ehgartner J, Nietzsche S, Dinger J, Kiehntopf M, Funke H, Peters FT, Lupp A, Gartner C, Mayr T, Bauer M, Huber O and Mosig AS, Biomaterials, 2015, 71, 119–131. [DOI] [PubMed] [Google Scholar]
- 181.Yang H, Li N, Du Y, Tong C, Lu S, Hu J, Zhang Y and Long M, Exp Cell Res, 2017, 351, 91–99. [DOI] [PubMed] [Google Scholar]
- 182.Yamada M, Utoh R, Ohashi K, Tatsumi K, Yamato M, Okano T and Seki M, Biomaterials, 2012, 33, 8304–8315. [DOI] [PubMed] [Google Scholar]
- 183.Mao S, Gao D, Liu W, Wei H and Lin J-M, Lab on a Chip, 2012, 12, 219–226. [DOI] [PubMed] [Google Scholar]
- 184.Wu Q, Gao D, Wei J, Jin F, Xie W, Jiang Y and Liu H, Chem Commun (Camb), 2014, 50, 2762–2764. [DOI] [PubMed] [Google Scholar]
- 185.Baudoin R, Legendre A, Jacques S, Cotton J, Bois F and Leclerc E, J Pharm Sci, 2014, 103, 706–718. [DOI] [PubMed] [Google Scholar]
- 186.Choucha-Snouber L, Aninat C, Grsicom L, Madalinski G, Brochot C, Poleni PE, Razan F, Guillouzo CG, Legallais C, Corlu A and Leclerc E, Biotechnol Bioeng, 2013, 110, 597–608. [DOI] [PubMed] [Google Scholar]
- 187.Zhou Q, Patel D, Kwa T, Haque A, Matharu Z, Stybayeva G, Gao Y, Diehl AM and Revzin A, Lab Chip, 2015, 15, 4467–4478. [DOI] [PubMed] [Google Scholar]
- 188.Pierantonelli I and Svegliati-Baroni G, Transplantation, 2019, 103, e1–e13. [DOI] [PubMed] [Google Scholar]
- 189.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L and Wymer M, Hepatology, 2016, 64, 73–84. [DOI] [PubMed] [Google Scholar]
- 190.Gori M, Simonelli MC, Giannitelli SM, Businaro L, Trombetta M and Rainer A, PLoS One, 2016, 11, e0159729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kostrzewski T, Cornforth T, Snow SA, Ouro-Gnao L, Rowe C, Large EM and Hughes DJ, World J Gastroenterol, 2017, 23, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.van Midwoud PM, Merema MT, Verpoorte E and Groothuis GM, Lab on a Chip, 2010, 10, 2778–2786. [DOI] [PubMed] [Google Scholar]
- 193.Maschmeyer I, Hasenberg T, Jaenicke A, Lindner M, Lorenz AK, Zech J, Garbe LA, Sonntag F, Hayden P, Ayehunie S, Lauster R, Marx U and Materne EM, European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V, 2015, 95, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang C, Zhao Z, Abdul Rahim NA, van Noort D and Yu H, Lab Chip, 2009, 9, 3185–3192. [DOI] [PubMed] [Google Scholar]
- 195.Materne EM, Ramme AP, Terrasso AP, Serra M, Alves PM, Brito C, Sakharov DA, Tonevitsky AG, Lauster R and Marx U, J Biotechnol, 2015, 205, 36–46. [DOI] [PubMed] [Google Scholar]
- 196.Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hubner J, Lindner M, Drewell C, Bauer S, Thomas A, Sambo NS, Sonntag F, Lauster R and Marx U, Lab Chip, 2015, 15, 2688–2699. [DOI] [PubMed] [Google Scholar]
- 197.Choe A, Ha SK, Choi I, Choi N and Sung JH, Biomed Microdevices, 2017, 19, 4. [DOI] [PubMed] [Google Scholar]
- 198.Bricks T, Paullier P, Legendre A, Fleury M-J, Zeller P, Merlier F, Anton PM and Leclerc E, Toxicology in Vitro, 2014, 28, 885–895. [DOI] [PubMed] [Google Scholar]
- 199.Eheman CR, Shaw KM, Ryerson AB, Miller JW, Ajani UA and White MC, Cancer Epidemiol Biomarkers Prev, 2009, 18, 1763–1769. [DOI] [PubMed] [Google Scholar]
- 200.Polyak K, J Clin Invest, 2007, 117, 3155–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Huang X, Li L, Tu Q, Wang J, Liu W, Wang X, Ren L and Wang J, Microfluidics and Nanofluidics, 2011, 10, 1333–1341. [Google Scholar]
- 202.Choi Y, Hyun E, Seo J, Blundell C, Kim HC, Lee E, Lee SH, Moon A, Moon WK and Huh D, Lab Chip, 2015, 15, 3350–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cho Y, Moon WK, Kim HS, Na K, Yang JH, Huh YH, Kim JA, Chung S and Lee SH, NPG Asia Materials, 2018, 10, 970–981. [Google Scholar]
- 204.Grafton MM, Wang L, Vidi P-A, Leary J and Lelievre SA, Integrative Biology, 2011, 3, 451–459. [DOI] [PubMed] [Google Scholar]
- 205.Vidi PA, Maleki T, Ochoa M, Wang L, Clark SM, Leary JF and Lelievre SA, Lab Chip, 2014, 14, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Morgan MM, Arendt LM, Alarid ET, Beebe DJ and Johnson BP, The FASEB Journal, 2019, fj. 201802347RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ayuso JM, Gillette A, Lugo-Cintrón K, Acevedo-Acevedo S, Gomez I, Morgan M, Heaster T, Wisinski KB, Palecek SP and Skala MC, EBioMedicine, 2018, 37, 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG and Keely PJ, BMC medicine, 2006, 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kim S, Chung M and Jeon NL, Biomaterials, 2016, 78, 115–128. [DOI] [PubMed] [Google Scholar]
- 210.Price GM, Chrobak KM and Tien J, Microvascular research, 2008, 76, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gong MM, Lugo-Cintron KM, White BR, Kerr SC, Harari PM and Beebe DJ, Biomaterials, 2019, 214, 119225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ayuso JM, Gong MM, Skala MC, Harari PM and Beebe DJ, Advanced Healthcare Materials, 2020, 1900925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lugo-Cintron KM, Ayuso JM, White BR, Harari PM, Ponik SM, Beebe DJ, Gong MM and Virumbrales-Munoz M, Lab Chip, 2020, DOI: 10.1039/d0lc00099j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shim S, Belanger MC, Harris AR, Munson JM and Pompano RR, Lab on a Chip, 2019, 19, 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.NatBiotechnol, 2013, 31, 85. [Google Scholar]
- 216.Sontheimer-Phelps A, Hassell BA and Ingber DE, Nat Rev Cancer, 2019, 19, 65–81. [DOI] [PubMed] [Google Scholar]
- 217.van den Berg A, Mummery CL, Passier R and van der Meer AD, Lab on a chip, 2019, 19, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.N. I. o. H.-R. c. t. o. a. chip, https://grants.nih.gov/grants/guide/rfa-files/RFA-TR-19-014.html, (accessed 5/27/2020). [Google Scholar]
- 219.Berthier E, Young EW and Beebe D, Lab Chip, 2012, 12, 1224–1237. [DOI] [PubMed] [Google Scholar]
- 220.Regehr KJ, Domenech M, Koepsel JT, Carver KC, Ellison-Zelski SJ, Murphy WL, Schuler LA, Alarid ET and Beebe DJ, Lab on a Chip, 2009, 9, 2132–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ren K, Chen Y and Wu H, Current opinion in biotechnology, 2014, 25, 78–85. [DOI] [PubMed] [Google Scholar]
- 222.Zhang B and Radisic M, Lab Chip, 2017, 17, 2395–2420. [DOI] [PubMed] [Google Scholar]
- 223.Walker G and Beebe DJ, Lab Chip, 2002, 2, 131–134. [DOI] [PubMed] [Google Scholar]
- 224.Berry SB, Zhang T, Day JH, Su X, Wilson IZ, Berthier E and Theberge AB, Lab on a Chip, 2017, 17, 4253–4264. [DOI] [PubMed] [Google Scholar]
- 225.Kim S, Chung M, Ahn J, Lee S and Jeon NL, Lab on a Chip, 2016, 16, 4189–4199. [DOI] [PubMed] [Google Scholar]
- 226.Campisi M, Shin Y, Osaki T, Hajal C, Chiono V and Kamm RD, Biomaterials, 2018, 180, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]