

Abstract
C–H Functionalization has become widely recognized as an exciting new strategy for the synthesis of complex molecular targets. Instead of relying on functional groups as the controlling elements of how molecules are assembled, it offers a totally different logic for organic synthesis. For this type of strategy to be successful, reagents and catalysts need to be developed that generate intermediates that are sufficiently reactive to functionalize C–H bonds but still capable of distinguishing between the different C–H bonds and other functional groups present in a molecule. The most well-established approaches have tended to use substrates that have inherently a favored site for C–H functionalization or rely on intramolecular reactions to control where the reaction will occur. A challenging but potentially more versatile approach would be to use catalysts to control the site-selectivity without requiring the influence of any directing group. One example that is capable of achieving such transformations is the C–H insertion chemistry of transient metal carbenes. Dirhodium tetracarboxylates have been shown to be especially effective catalysts for these reactions. This review will highlight the development of these dirhodium catalysts and illustrate their effectiveness to control both site-selective and stereoselective C–H functionalization of a wide variety of substrates.
Introduction:
Numerous strategies have been developed to achieve site-selective C–H functionalization and this area of research continues to be a very active field of study.1–12 Some have a long history such as the venerable Hofmann–Löffler-Freytag reaction, which achieves site-selective radical-induced hydrogen abstraction to occur by means of intramolecular reactions.13–20 Numerous modern radical-induced C–H functionalization reactions have been developed, including radical processes, but the site selectivity in these reactions is usually controlled by the inherent reactivity profile of the substrate (Fig. 1a).4,21–33 Enzymatic C–H oxidations are capable of exquisite site selectivity and bio-inspired approaches to replicate such selectivity for a broad array of substrates is of considerable interest.34 The use of a directing group to coordinate to a metal catalyst, which thus, places the metal in close proximity to a specific C–H bond has become a very popular method for C–H functionalization and has been broadly applied for the synthesis of pharmaceutical and other complex targets (Fig. 1b).35–44 This approach can also be applied to enantioselective C–H functionalization by using relatively weak directing groups and chiral ligands on the catalyst (Fig. 1c).45–47
Figure 1: Introduction to the most significant strategies for site selective C–H functionalization.
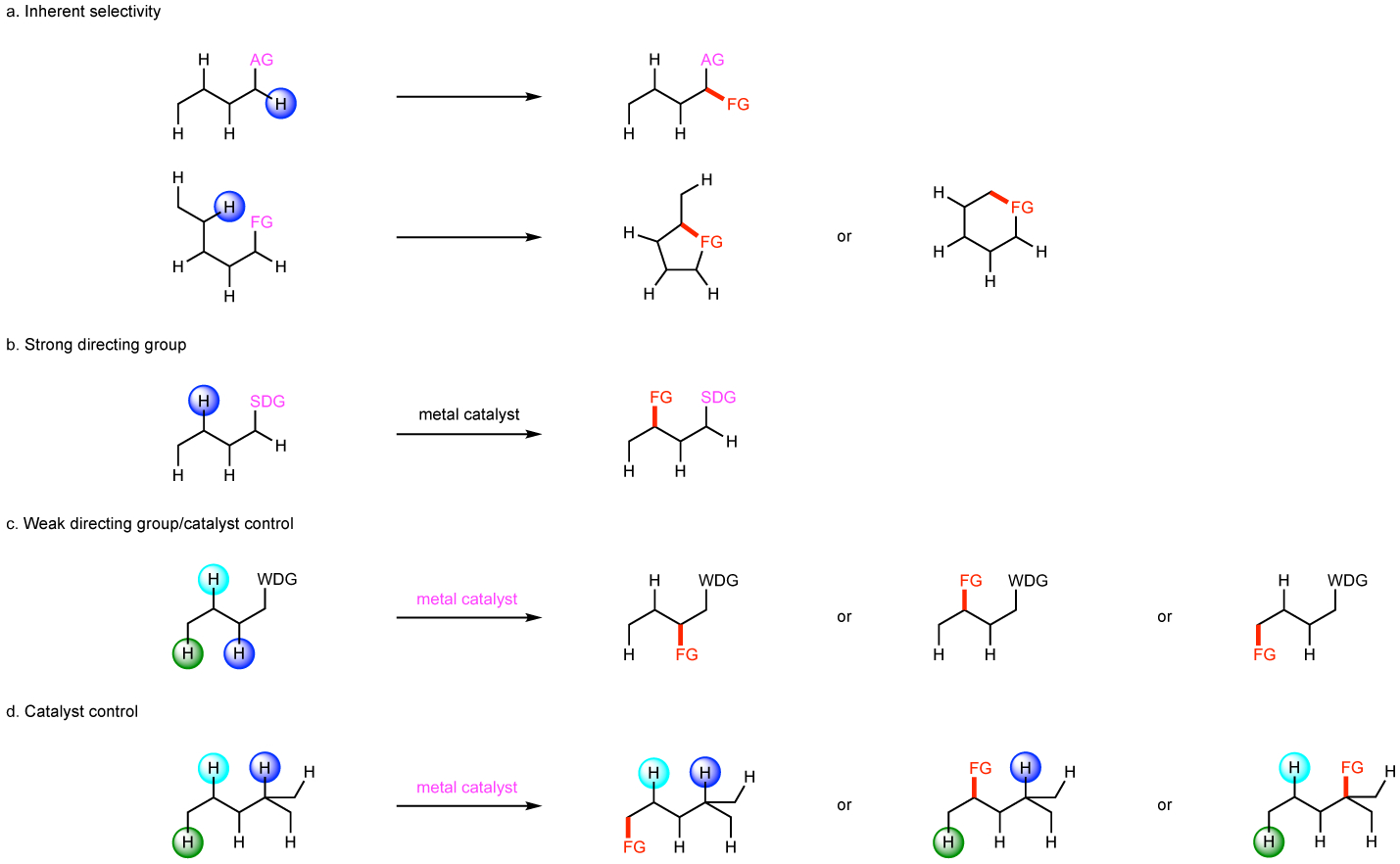
a: radical type reaction in which an activating group (AG) in the substrate causes a specific C–H bond to be most reactive and the introduction of a functional group (FG) at that specific position. b: Strong directing group (SDG) that coordinates to the metal catalyst and places it in close proximity to a specific C–H bond. c: Weak directing group (WDG) that coordinated to the metal catalyst but the selectivity can be varied depending on the nature of the ligands around the catalyst. d: Catalyst controlled C–H functionalization without prior coordination to the catalyst, in which site selectivity is dependent on which catalyst is used.
Considerable interest has also been shown in developing catalyst-controlled C–H functionalization methodologies, which avoid having defined directing groups within the substrate or prior coordination of the catalyst to the substrate (Fig. 1d).48–56 The most extensively studied reactions have been C–H oxidation,57 C–H borylation,58 C–H amination,59 and C–H alkylation by metal carbenes.29 This review will describe recent progress in site-selective intermolecular sp3 C–H functionalization using rhodium carbene intermediates.52–54,560−65 Dirhodium tetracarboxylates are the optimum catalysts for these reactions, and this review will describe the evolution of these catalysts to the current stage in which catalyst-controlled intermolecular functionalization of unreactive primary, secondary or tertiary C–H bonds can be achieved by simply choosing the appropriate catalyst.9,66–70
Background on Rhodium Carbene-Induced C–H Functionalization
The first example of the metal carbene-induced intermolecular C–H functionalization was the copper oxide-catalyzed reaction of cyclohexane (1) with ethyl diazoacetate (2) to form the functionalized product 3, reported by Scott in 1974 (Fig. 2a).71 This reaction is proposed to proceed via a copper carbene intermediate 4, and the carbene inserts into the C–H bond. A few years later, Teyssié demonstrated that dirhodium tetracarboxylates were far superior as catalysts compared to copper for intermolecular C–H functionalization of alkanes with ethyl diazoacetate (Fig. 2b).72 The yields were generally high for both cyclic and acyclic alkanes but the reaction generated mixtures of products with substrates containing different C–H bonds, as illustrated in the reaction with pentane (5), which generated all three possible products 6–8. The rhodium catalyst 9 is highly electrophilic and the electron-withdrawing ester group further reinforces this characteristic. Consequently, the metal carbene is sufficiently reactive to insert into C–H bonds but lacks the selectivity to distinguish effectively between different C–H bonds. Since this period, efforts have been made to design catalysts to improve on the site selectivity with ethyl diazoacetate. Silver complexes with bulky ligands such as 10, developed by Pérez73 and rhodium complexes developed by Callott74–76have been shown to direct the C–H functionalization towards less crowded C–H bonds, but still, the selectivity is relatively modest.77 Iridium carbenes have modulated reactivity compared to their silver- and rhodium-carbene counterparts, and enantioselective C–H functionalization has been reported by Katsuki using the chiral iridium catalyst 11 but the range of substrates applicable to these reactions have been limited to cycloalkanes and cyclic ethers.27 An example is the functionalization of tetrahydrofuran 12 with ethyl diazopropionate 13 to form 14 in 90% enantiomeric excess (e.e.) (Fig. 2c).
Figure 2: Background to carbene-induced intermolecular C–H insertion with diazoacetates.
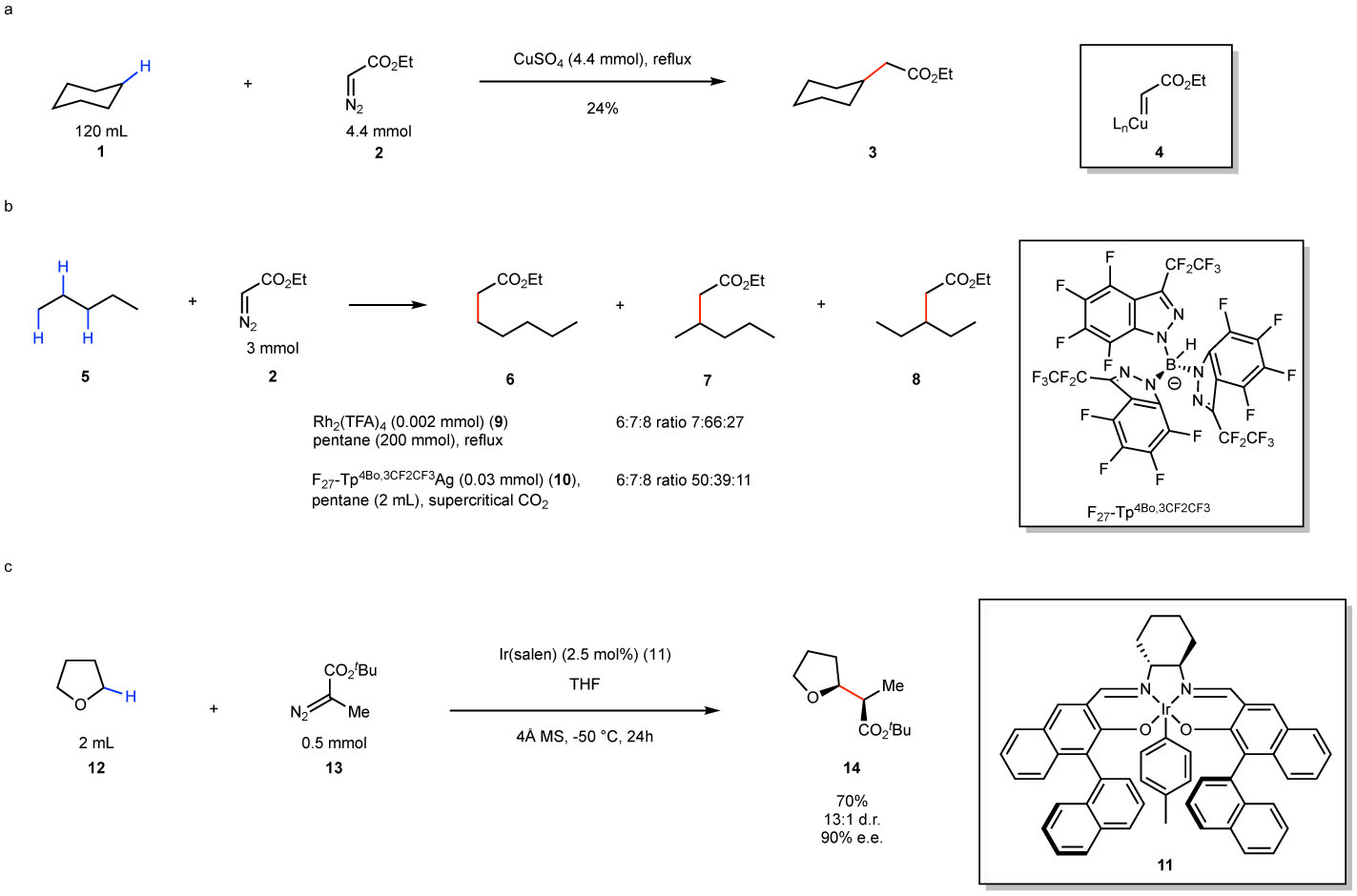
a: Seminal studies with copper catalysis. b: Enhanced reactivity with dirhodium tetracarboxylates as catalysts and enhanced site-selectivity with bulky silver catalysts. c: Enhanced enantioselectivity with an iridium catalyst.
In order to overcome the site-selectivity challenges of the carbene-induced C–H functionalization reaction, early studies in the 1980’s focused on the intramolecular version, as illustrated in the rhodium acetate-catalyzed reaction of the diazoacetoacetate 15 to form the bicycle 16 (Fig. 3a).78 Cyclization to form five-membered rings is generally preferred, although with certain substrates four- or six-membered ring products are the preferred.70,79–82The utility of the process was further enhanced with the development in the 1990’s of enantioselective variants with the highly effective chiral dirhodium tetracarboxylates, such as Rh2(S-PTTL)4 and dicarboxamides catalysts, such as Rh2(4S-MACIM)4, developed by Hashimoto83 and Doyle84, respectively (Fig. 3b). Representative examples of the use of these catalysts, is the cyclization of the diazoacetoacetate 17 to form the bicycle 18, which after deacylation, generated 19 in 53% e.e., and the desymmetrization of the diazoacetate 20 to from 21 in 97% e.e.. The synthetic potential of these intramolecular reactions is broad and the reaction has been applied to the synthesis of a variety of complex targets as illustrated in the conversion of 22 to 23 as a key step in the synthesis of (+)-isolauricerisinol 24 (Fig. 3c).85 The intramolecular C–H functionalization is now well established and has been extensively reviewed70 and will not be discussed further here.
Figure 3. Overview of intramolecular C–H insertion reactions.
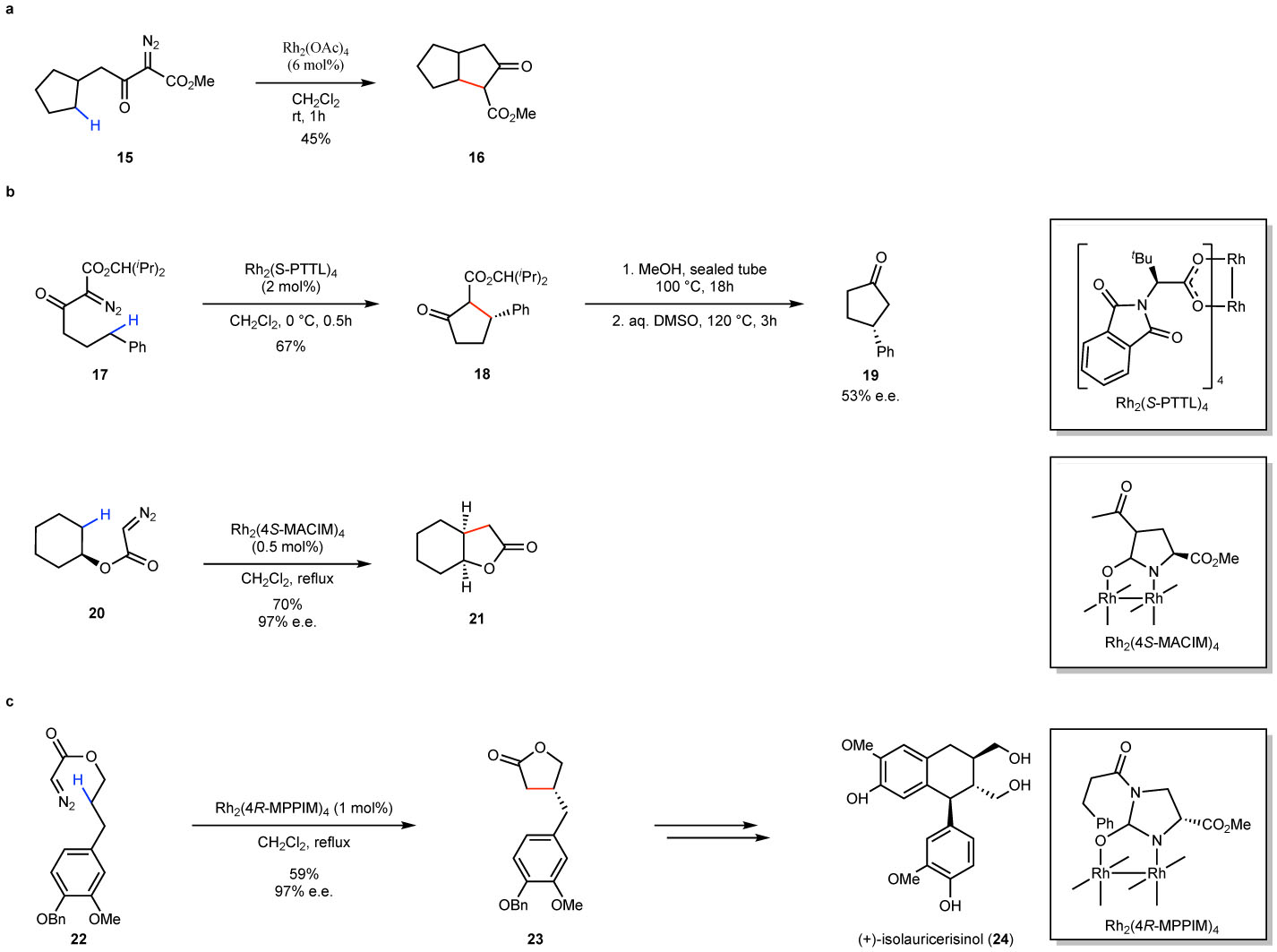
a: Early example using an achiral catalyst. b: Enantioselective C–H functionalization examples using chiral dirhodium catalysts. c: Example of a key carbene C–H functionalization step in the total synthesis of (+)-isolauricerisinol.
Development of Donor/Acceptor Carbenes for C–H Functionalization
The carbene generated from ethyl diazoacetate has been characterized as an “acceptor carbene” (Fig. 4a), and these carbenes are generally too reactive for exceptional control of site selectivity in intermolecular reactions.86 Copper and rhodium carbenes have limited back-bonding from the metal and thus are highly electrophilic. Addition of further electron-withdrawing groups to the carbene increases the electrophilic character of the carbene, which explains why the established acceptor and acceptor/acceptor carbenes have had most success in intramolecular reactions. In order to achieve selectivity in intermolecular reactions, less reactive and more selective types of carbenes would be needed. The Davies group developed a new class of metal carbenes in the 1980’s, the so called “donor/acceptor” carbenes, which contain both an acceptor group, such as an ester, and a donor group, such as aryl or vinyl (Figure 4).87,88 During extensive early studies on cyclopropanation reactions, his group discovered that these carbenes are much more selective than the conventional carbenes lacking a donor group. Furthermore, these carbenes will undergo highly enantioselective cyclopropanation when the reactions are catalyzed by the rhodium prolinate catalysts Rh2(DOSP)4, as illustrated in the cyclopropanation of styrene 25 with the styryldiazoacetate 26 to form 27 in 98% e. e.89 The n-dodecyl group was incorporated into the catalyst to make it soluble in hydrocarbon solvents, even at low temperature, which are the optimized conditions for high asymmetric induction. In 1997, highly enantioselective C–H functionalization of cycloalkanes catalyzed by Rh2(DOSP)4 was reported, as illustrated in the reaction of cyclohexane 2 with methyl aryldiazoacetate 28 to form 29 in 95% e. e..22 These studies represented the first examples of highly enantioselective intermolecular C–H functionalization by rhodium carbene intermediates. Recently, there have been some interesting C–H functionalization studies going beyond donor/acceptor carbenes to donor/donor carbenes. So far, the reported examples have been limited to intramolecular versions,90–93 presumably because the carbenes are no longer sufficiently reactive to undergo effective intermolecular C–H functionalization reactions.
Figure 4. Early development of the donor/acceptor carbenes.
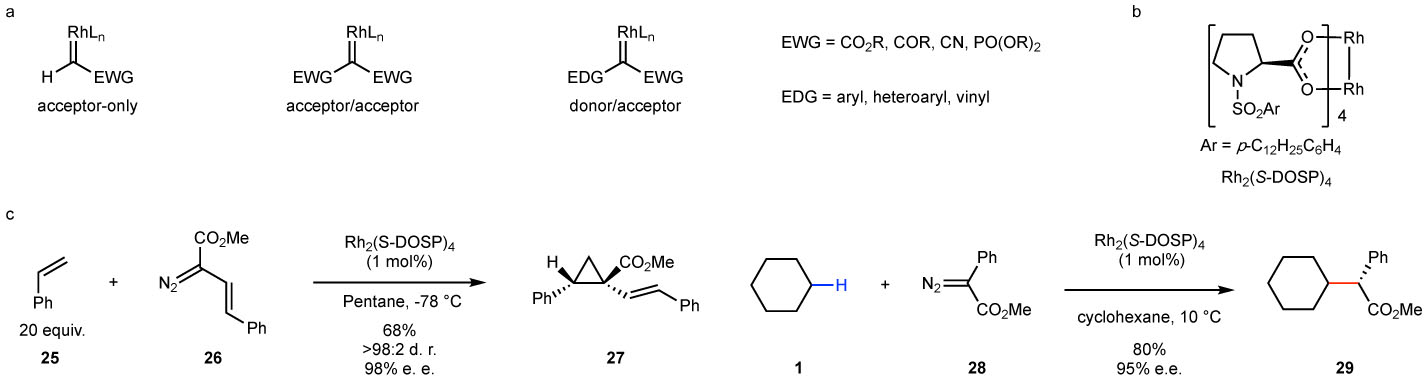
a: Introduction to the three major types of reactive carbene intermediates. b: Rh2(DOSP)4 – a highly effective chiral catalyst for the reactions of donor/acceptor carbenes. c: Early examples of Rh2(DOSP)4-catalyzed asymmetric cyclopropanation and C–H functionalization with donor/acceptor carbenes.
The Rh2(DOSP)4/aryldiazoacetate combination was effective for enantioselective intermolecular C–H insertion of a wide variety of substrates.66,68 Even though relatively moderate site selectivity was found with alkanes, the reactions with substrates containing electronically activated sites were very site-selective. The most favored groups for C–H functionalization are summarized in Fig. 5a. C–H Functionalization next to groups that can stabilize positive charge build up in the transition state for the C–H functionalization are favored. Thus benzylic, allylic and C–H bonds adjacent to nitrogen or oxygen are electronically favored. C–H Functionalization generally occurs at secondary C–H bonds because even though tertiary C–H bonds are electronically favored the rhodium carbene is sterically demanding. If the two groups adjacent to the secondary site are distinctly different in size, the reaction can be highly diastereoselective. Non-crowded tertiary sites such as adamantane can be readily functionalized as well as primary C–H bonds that are electronically activated. However, in general if there is competition between electronically activated primary and secondary sites, functionalization of the secondary site is preferred unless there is extreme steric crowding. The mechanism of the C–H functionalization has been studied computationally and was proposed to be a concerted asynchronous process as shown in Fig 5b.94 The reaction begins more as a hydride transfer event before finally the carbene inserts into the C–H bond. Based on this computational analysis, a model was developed that is predictive of both the relative and absolute configurations of the C–H functionalization products. The substrate is considered to approach the carbene with the large group pointing away from the catalyst, and the medium group resides on the side of the donor group rather than the acceptor group, because the acceptor group lies orthogonal to the carbene and would point towards the approaching substrate.
Figure 5. Overview of Rh2(DOSP)4-catalyzed intermolecular C–H functionalization.
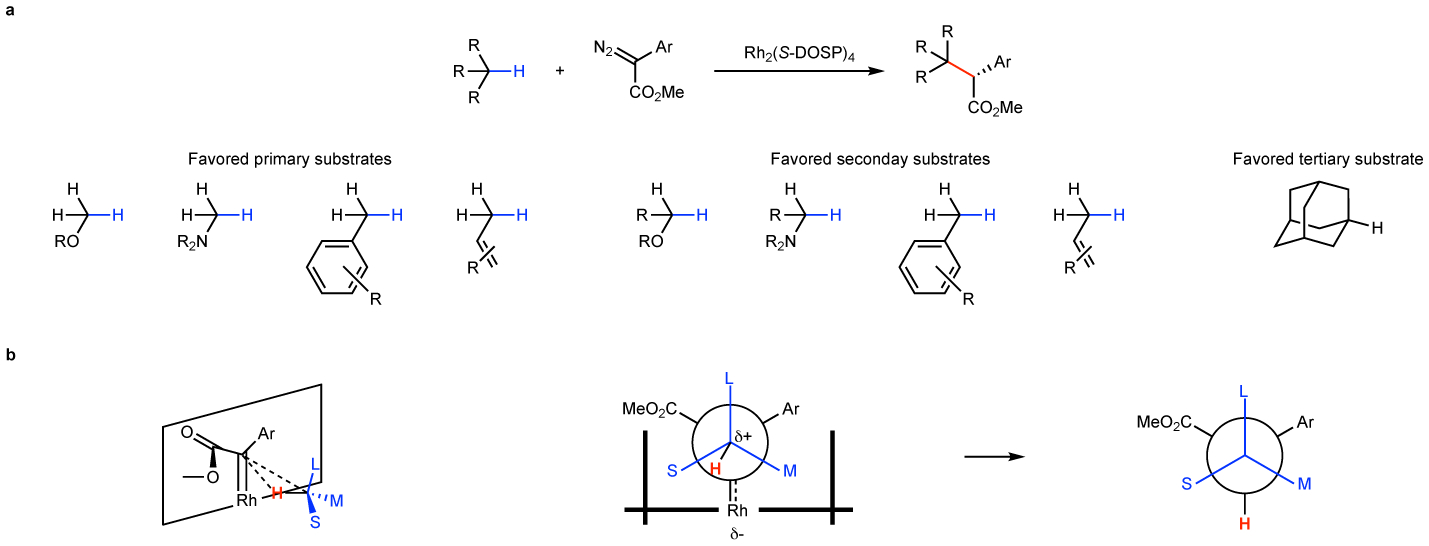
a: Favored sites for C–H Functionalization. b: Predictive model for the relative and absolute configuration for Rh2(DOSP)4-catalyzed C–H functionalization. The large (L) group points away from the catalyst, the medium (M) group is on the side of the aryl group, which is in the same plane as the rhodium-carbene, and the small (S) group is on the side on the esters group which is orthogonal to the rhodium carbene-aryl plane and points towards the approaching substrate.
The general scope of the Rh2(DOSP)4-catalyzed intermolecular C–H functionalization has been reviewed elsewhere66,68 and only the highlights will be discussed here. The C–H functionalization can be considered as equivalent to some of the classic strategic reactions of organic synthesis.66 Particularly effective substrates are those with electronically activated methylene sites in which the two methylene substituents are distinctly different in size because the reactions with these substrates are highly diastereoselective and enantioselective as illustrated in Fig. 6. The reaction with various siloxy derivatives generated protected beta-hydroxy esters, as illustrated for the reaction with tetraethoxysilane 30 to form 31. The reaction is a C–H functionalization equivalent to an aldol reaction as illustrated in the retrosynthetic scheme from the acetaldehyde 32 and the enolate 33.23,22 Reaction with N-Boc-pyrrolidine 34 generated the beta-amino ester 35, containing the functionality typically generated by a Mannich reaction derived from the imine 36 and the enolate 33.95 Various other examples of surrogates of classic reactions have been reported, such as the Claisen rearrangement96, Michael addition97 and the Claisen condensation98.
Figure 6. C–H Functionalization as a strategic equivalent of classic strategic reactions of organic synthesis.
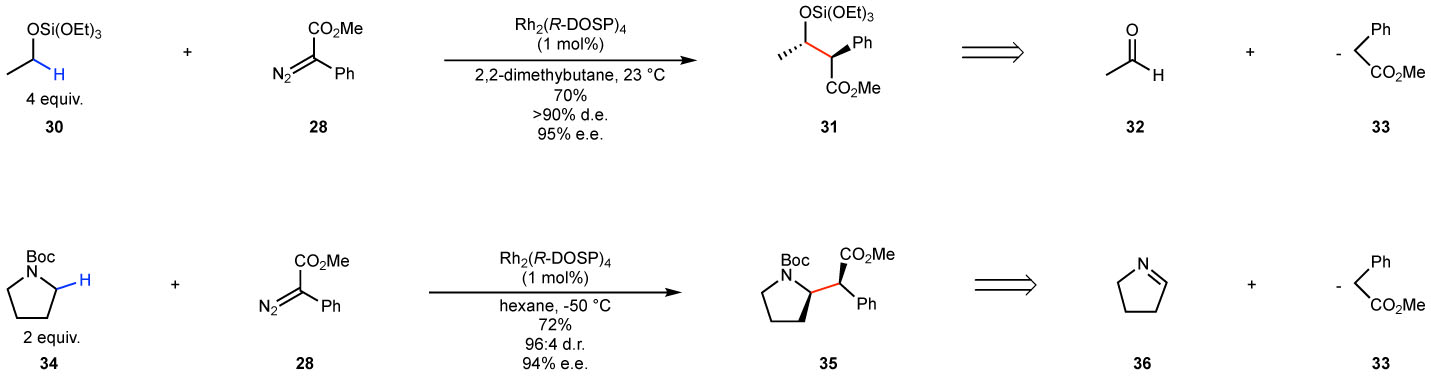
C–H functionalization adjacent to oxygen is a surrogate equivalent of an aldol reaction. C–H functionalization adjacent to nitrogen is equivalent to a Mannich reaction
A related C–H functionalization process of great synthetic utility is the reaction of vinyldiazoacetates 37 with allylic substrates 38, the so-called combined C–H functionalization/Cope rearrangement. The product 40 of this reaction is not the normal C–H functionalization product but one in which it appears as if the C–H functionalization product has undergone a Cope rearrangement (Fig. 7a). However, this is not the case because the C–H functionalization product is the thermodynamic product. Computational studies revealed that the reaction begins with a hydride transfer event to generate a complex 39, consisting essentially of an allyl cation and a rhodium-bound allyl anion, which then immediately forms a C-C bond at the distal position before any bond rotation can occur.99 The synthetic transformation has been reviewed100 and so, only two representative examples will be discussed here.101–103 In most cases, the products are formed with exceptionally high levels of asymmetric induction and diastereocontrol. Typically, the high diastereocontrol is caused by the reaction proceeding through a chair-like transition state as illustrated for 39, but in certain cases, steric interference with the face of the catalyst forces the reaction to proceed through a boat transition state. The C–H functionalization process offers some interesting synthetic possibilities. For example, the reaction of the vinyldiazoacetate 41 with the alkene 42 generates 43 with two new stereogenic centers (Fig 7b).102 The transformation can be considered as a surrogate of the vinylogous Mukaiyama aldol surrogate, equivalent to the Lewis acid catalyzed reaction of the aldehyde 44 with the diene 45. The process is applicable as a powerful strategic reaction in total synthesis as illustrated in the synthesis of colombiasin A (46, Fig 7c).104 The reaction of the racemic dihydronaphthalene 47 with the vinyldiazoacetate 48 generates the C–H functionalization product 49 with control of stereochemistry at three stereogenic centers. This key step is enantiodivergent as only one enantiomer of the dihydronaphthalene 47 undergoes the desired C–H functionalization to form 49 whereas the other enantiomer undergoes a cyclopropanation reaction. Previous efforts at the total synthesis of colombiasin A had struggled to control these stereogenic centers but with 49 in hand, the completion of the total synthesis was readily achieved in six additional steps.
Figure 7. Combined C–H functionalization/Cope rearrangement.
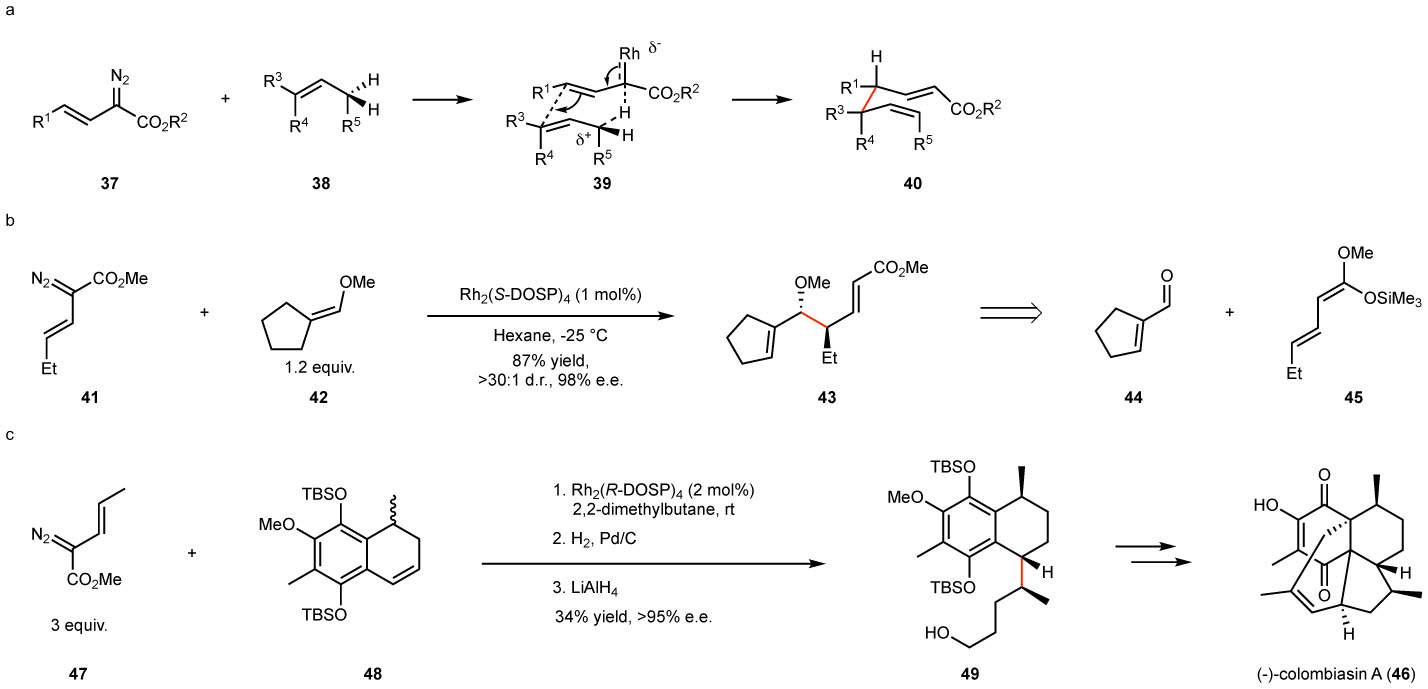
a: General scheme of the transformation. Due to the ordered transition state, the reactions are generally highly enantioselective and diastereoselective. b: Representative example illustrating the highly stereoselective nature of these transformations. c: Application to the total synthesis of colombiasin A. Even though the dihydronaphthalene 47 is racemic, the resulting C–H functionalization product 49 is generated with high
Catalyst-Controlled C–H functionalization with Donor/Acceptor Carbenes
One of the grand challenges in the field of C–H functionalization is to be able to use catalyst control to overcome the inherent reactivity profile of a substrate. Rh2(DOSP)4 was a good start as it is an effective catalyst for many carbene-induced C–H functionalization reactions. Certain types of substrates with the right balance of steric and electronic influences work well with Rh2(DOSP)4. In order to move from substrate control to the era of catalyst control, new classes of chiral catalysts were required with varied steric demand. Che demonstrated that bulky rhodium-porphyrin catalysts tended to favor C–H functionalization of alkanes preferentially at less crowded sites.105 However, the major breakthrough in this area has been the use of bulky chiral dirhodium catalysts.
A critical design element for dirhodium tetracarboxylate catalysts has been to exploit the symmetry of the dirhodium tetracarboxylate core. A freely rotating achiral dirhodium tetracarboxylate with four identical ligands is D4h symmetric. Several years ago, we proposed that if the ligand contained a very large substituent, then that substituent would not be able to align in the periphery of the catalyst and would need to exist on either the top (α) or bottom (β) face of the catalyst. Depending on the specific orientations of the chiral ligands, the catalysts could adopt higher symmetry than the ligands themselves (Fig. 8a).106 Originally, we proposed, the complexes could be C4, D2 or C2 symmetric (50-52). More recently, complexes adopting an alternative C2 symmetric orientation 53 has been identified, in which all the bulky groups are on the same face of the catalysts, but two are orientated differently from the other two.
Figure 8. Chiral dirhodium catalysts for intermolecular C–H functionalization.
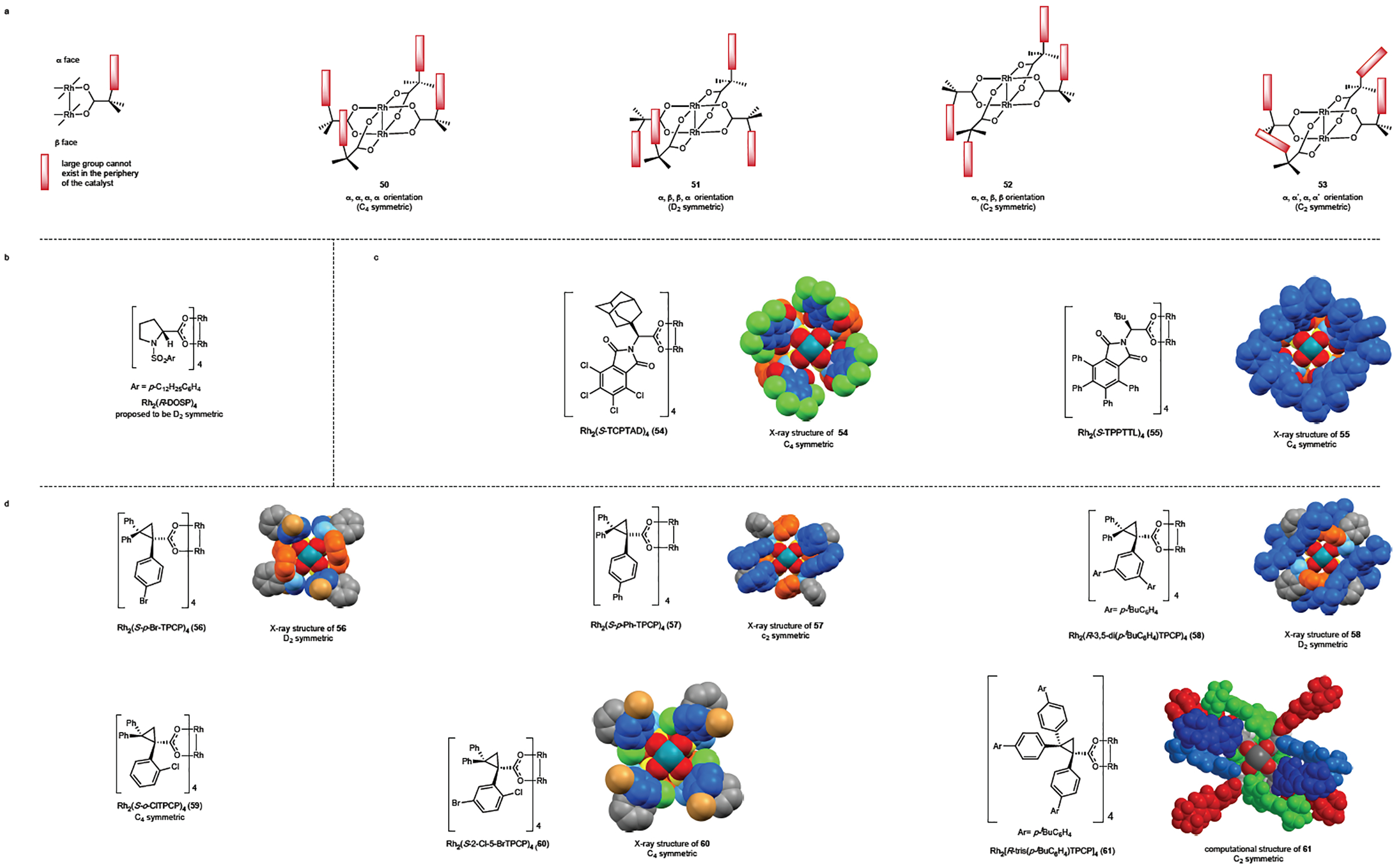
a: Design models for chiral catalysts, illustrating that if the ligands contain a large group (marked in red) that cannot exist in the periphery of the catalyst, then the resulting complex can have higher symmetry that the ligands themselves. b: The original catalyst, Rh2(DOSP)4, has been proposed to be D2 symmetric but this has not been verified experimentally. c: The phthalimido-derived catalyst Rh2(TCPTAD)4 (54) and Rh2(TPPTTL)4 (55) adopt a C4 symmetric structure and not especially sterically demanding, capable of effective C–H functionalization of tertiary and secondary C–H bonds. d: The TPCP catalysts 56-61 are sterically demanding and depending on the substitution pattern can adopt different orientations, leading to complexes that are D2, C4 or C2 symmetric.
The three major classes of chiral dirhodium catalysts for intermolecular C–H functionalization with donor/acceptor carbenes are shown in Figs 8b–d. The original chiral catalyst Rh2(DOSP)4 was proposed to behave as if it was D2 symmetric (Fig 8b). However, the D2 model for Rh2(DOSP)4 has not been confirmed experimentally. Since then two other classes of chiral catalysts have been developed and they dramatically influence the site-selectivity of the C–H functionalization. One class is related to the phthalimido amino acid catalysts originally developed by Hashimoto (Fig. 8c).107, 108 Rh2(TCPTAD)4 (54)109 and Rh2(TPPTTL)4 (55)65 both adopt a slightly distorted C4 symmetric structure. Previously, related catalysts have been considered as adopting a C2-symmetric chiral crown, rather than as a pseudo-C4 symmetric structure.110–113 However, we propose that considering the catalyst as essentially adopting a pseudo-C4 symmetric structure is more appropriate because these ligands adopt an induced fit when the carbene binds and the resulting rhodium complex is not altered by the initial carbene orientation to the dirhodium. An intriguing feature of Rh2(TPPTTL)4 is the orientation of the 16 phenyl groups, which adopt a preferred twist of each ring leading to an additional induced chiral influence due to a C4-propeller-like structure of the phenyl groups.
The second class of chiral dirhodium catalysts is based on triphenylcyclopropane carboxylate (TPCP) ligands (Fig. 8d), and they are sterically much more crowded that the other two classes of catalysts. They are readily synthesized by a 3 or 4-step synthesis: Rh-DOSP catalyzed cyclopropanation of 1,1-diarylethylene, followed by ester hydrolysis and ligand exchange.52,60 Furthermore, a library of catalysts with differing steric environment can be generated by means of a subsequent multifold Suzuki coupling reaction on the preformed catalyst.52,114
The first two catalysts developed were Rh2(p-BrTPCP)4 (56) and Rh2(p-PhTPCP)4 (57) and these catalysts were prepared directly by a ligand exchange reaction with dirhodium tetraacetate.60 X-ray crystallographic studies reveal that Rh2(p-BrTPCP)4 (56) adopts a D2 symmetric structure in the solid state whereas in (p-PhTPCP)4 (57), the biphenyl groups are on the same face but they are involved in π-stacking with each other, which breaks the C4 symmetry and leads to a C2 symmetric structure.54 More recently, the 3,5-diaryl-TPCP catalyst 58, formed by an 8-fold Suzuki coupling,52 the ortho-chloro-TPCP catalysts Rh2(o-ClTPCP)4 (59) and Rh2(2-Cl-5-BrTPCP)4 (60),54,65 and the fully-para-substituted TPCP catalyst 61, formed by 12-fold Suzuki coupling,114 have been developed and they all display their own distinctive selectivity characteristics for C–H functionalization. These catalysts adopt different shapes and these greatly influence their site selectivity characteristics. The 3,5-diaryl TPCP catalyst 58 adopts an up-down-up down arrangement of the large 3,5-diaryphenyl groups leading to a D2 symmetric structure, similar to Rh2(p-BrTPCP)4.52 The ortho-chloro-phenyl group in Rh2(o-ClTPCP)4 (59) and Rh2(2-Cl-5-BrTPCP)4 (60) also cannot align in the periphery of the catalyst but in this case they adopt an up-up-up-up arrangement leading to a C4 symmetric structure.54,65 A distinctive feature of 59 and 60 is the induced axial chirality observed between the cyclopropane and the ortho-chloro-phenyl group, and consequently, adoption of the C4-symmetric structure ensures that the ortho-chlorine groups are far away from each other. The 2-chloro-5-bromo catalyst 60 gives significantly higher asymmetric induction than the other ortho-chloro-TPCP catalysts because the bromine atoms are better positioned to help distinguish between the two faces of the rhodium-bound carbene. In the case of the fully-para-substituted TPCP catalyst 61 computational studies reveal that the biphenyl groups at the geminal carbon to the acid are all on the same face but are involved in π-stacking, analogous to Rh2(p-PhTPCP)4 (57), leading to a C2 symmetric structure.54
The distinction between the Rh2(TPCP)4 catalysts and the more established catalyst Rh2(DOSP)4 has been demonstrated in reactions at electronically activated benzylic sites (Fig. 9).61 The Rh2(DOSP)4 catalyzed reaction of aryldiazoacetate 63 with p-ethyltoluene (62) preferentially occurs at the secondary benzylic site to form 64 (Fig. 9a). In contrast, the Rh2(p-PhTPCP)4-catalyzed reaction of 62 with 63 preferentially occurs at the primary benzylic site to form 65. The site selectivity and enantioselectivity could be further improved by using the trichloroethyl ester of the aryldiazoacetate as illustrated in the formation of 66 (Fig. 9b). A similar switch in site selectivity was observed in competition studies between tertiary and primary benzylic sites as illustrated in the formation of 67. Rh2(p-PhTPCP)4 gave similar enhanced site selectivity for primary C–H bonds at other activated sites such as allylic and α to oxygen as shown for 68-70.61–63 These studies revealed that the Rh2(TPCP)4 catalysts are much more sterically demanding than the original catalyst Rh2(DOSP)4 and that this steric requirement can be used to control site selectivity.
Figure 9. Catalyst-controlled site selective functionalization at activated C–H bonds.
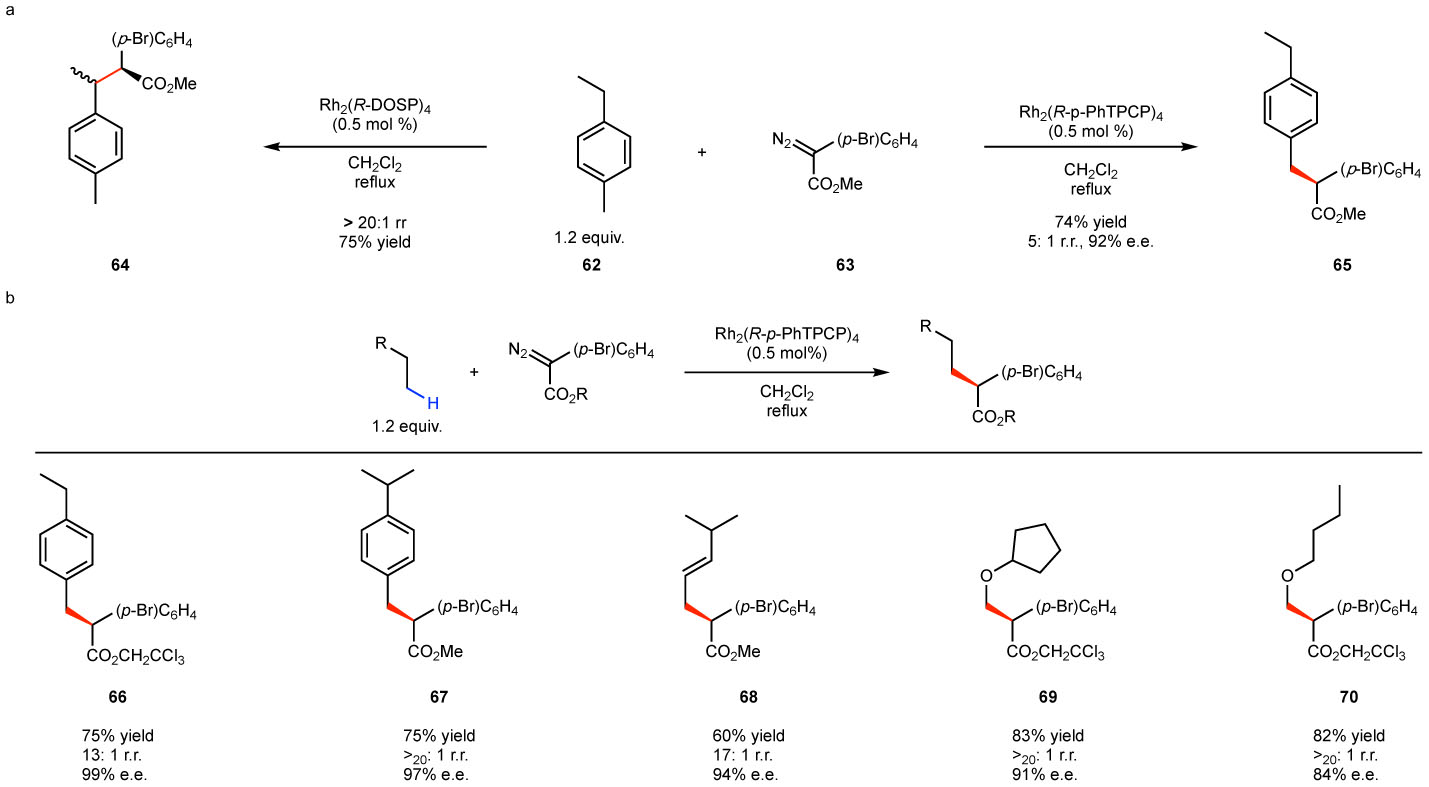
a: Rh2(DOSP)4 prefers C–H functionalization at secondary benzylic site whereas Rh2(p-PhTPCP)4 favors the primary benzylic site. b: representative examples of primary C–H functionalization at benzylic and allylic sites and adjacent to oxygen.
Site-Selective and Enantioselective C-H Functionalization of Unactivated C–H Bonds
With the advent of the new classes of catalysts, procedures have been developed to go beyond selective functionalization of electronically activated C–H bonds. Catalysts have been developed for site selective reactions to occur at either primary, secondary or tertiary C–H bonds that are unactivated.52,53,114 The optimum catalyst for tertiary C–H functionalization is Rh2(TCPTAD)4.53 The tertiary sites are the most electronically activated and Rh2(TCPTAD)4 is sufficiently small to be able to sterically access tertiary C–H bonds as long as they are in the periphery of the structure (Fig. 10). The reaction can be applied not only to relative simple substrates as illustrated in the formation of 71-75 (Fig 10a), but also, to complex structures, containing a large number of different C–H bonds (Fig. 10b). Examples to illustrate the level of possible site selectivity are the reactions with cholesteryl acetate (77) and vitamin E (79). These substrates have multiple tertiary C–H bonds and electronically activated sites such as allylic and benzylic. Yet the C–H functionalization occurs cleanly at the most accessible tertiary site to form 78 and 80, respectively. Detailed computational studies on the catalyst and the rhodium-carbene complex have been reported to explain the site-selectivity preference.53 The catalyst adopts a relatively shallow bowl shape as can be seen in the X-ray structure of 54 (Fig. 8b). The tertiary C–H bonds are electronically preferred, and when the carbene is bound to the catalyst, the most accessible C–H tertiary bond is able to approach the carbene but other tertiary and even electronically activated C–H bonds in the center of the substrate are unable to do so. The carbene bound complex causes distortion of the pseudo-C4 symmetric orientation of the ligands, due to π-stacking between the ligands and the carbene, and this is considered to be a key factor for the relatively high asymmetric induction generated in these reactions.
Figure 10. Rh2(TCPTAD)4 catalyzed functionalization of tertiary C–H bonds.
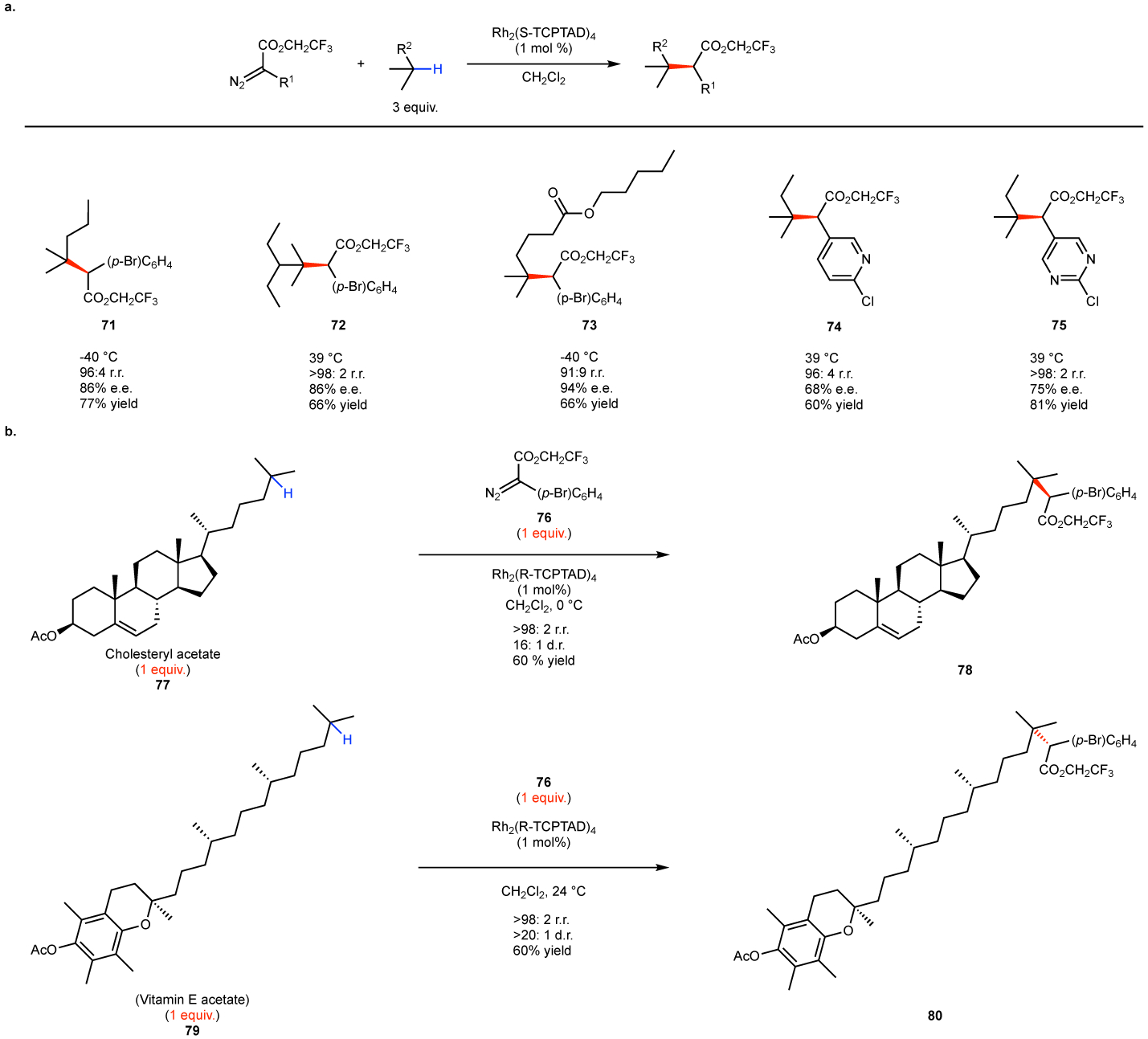
a: representative simple substrates. b: application to the C–H functionalization of complex substrates, containing multiple C–H bonds.
The 3,5-diaryl-TPCP catalyst 58 was found to be a very effective for selective functionalization of unactivated secondary C–H bonds.52 An impressive example of the selectivity possible with this catalyst is the reaction with pentane (Fig. 11). Even though pentane has two relatively similar methylene sites the reactions occurs cleanly at C-2 to form 81 with high levels of regioselectivity (the competing site is C1), diastereoselectivity and enantioselectivity. The reaction was applied to a variety of substrates, as illustrated for the formation of 82-88, and in all cases the reaction preferentially occurred at the most sterically accessible methylene site. Furthermore, these studies showed that the reaction at unactivated C–H bonds could be carried out in the presence of some functional groups such as esters, halides and silanes. The catalyst 58 adopts a D2 symmetric structure (Fig. 8d) and thus, the bound carbene is also in a bowl-shaped cavity. In this case, however, the bowl is steeper than in the case of Rh2(TCPTAD)4, and consequently, it is difficult for any tertiary C–H bond or even secondary C–H bonds unless one of the substituents is methyl to approach the carbene bound to the catalyst.
Figure 11.
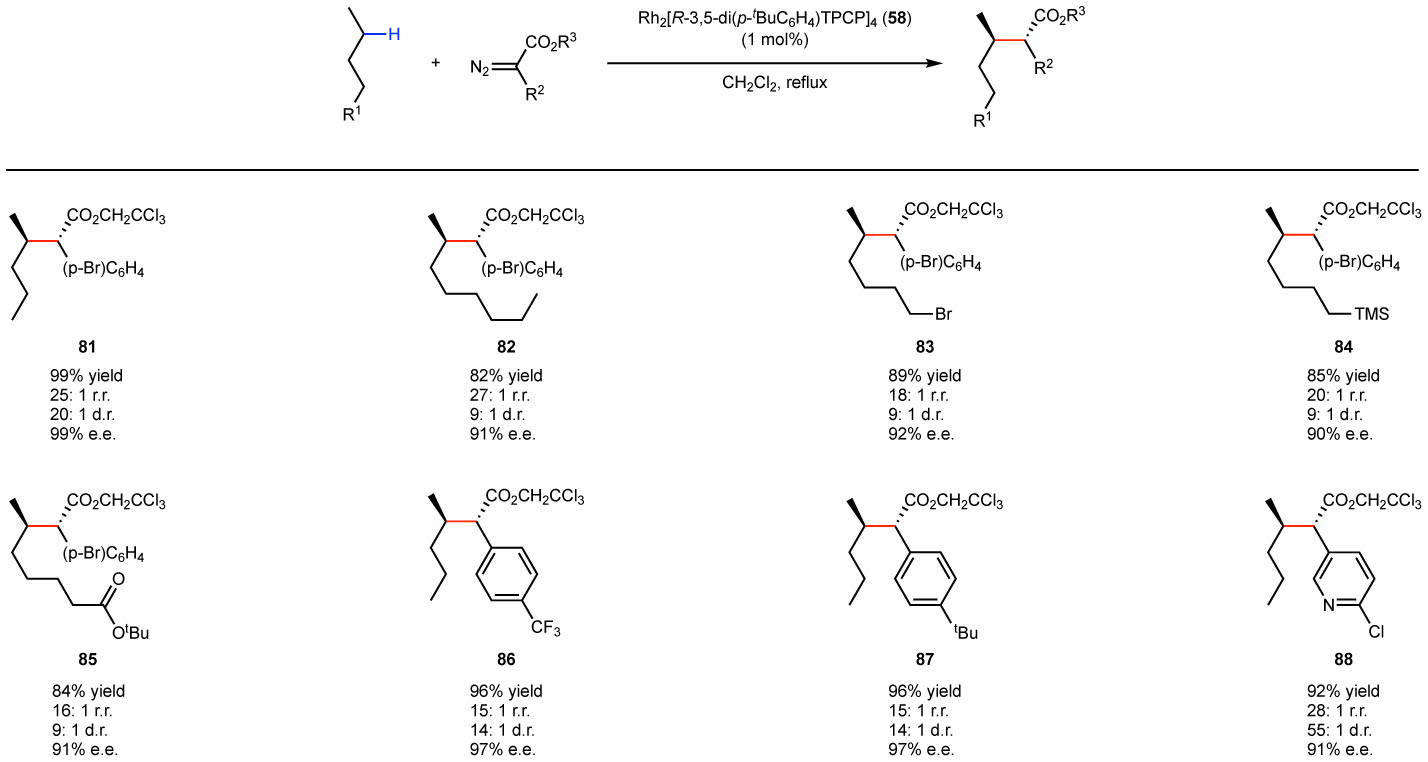
C–H functionalization of the most sterically accessible secondary C–H bond.
More recently, the ortho-chloro-TPCP catalysts, 59 and 60, have been shown to be even more selective than 3,5-diaryl-TPCP catalyst 58 for unactivated secondary C–H bonds.54 A good demonstration of the subtleties of steric control is the reaction with alkylbenzene derivatives (Fig. 12).65 The two preferred sites for reaction are the electronically activated benzylic site and the most sterically accessible unactivated C–H bonds. In the Rh2(TCPTAD)4-catalyzed reaction, a strong preference for the benzylic site is observed. However, in the case of the reaction catalyzed by the ortho-chloro-TPCP catalysts, the reaction preferentially occurs at the most accessible methylene site (Fig. 12). Of the ortho-chloro-TPCP catalysts, Rh2(2-Cl-5-BrTPCP)4 (60) is the best for high asymmetric induction, and its reactions can proceed with good regiocontrol, diastereocontrol and enantiocontrol. The reaction was applied to a range of alkylarenes and alkyl heteroarenes as illustrated in the representative examples to form 89-93 (Fig. 12a). In each case, the Rh2(2-Cl-5-BrTPCP)4 (61)-catalyzed reaction preferentially occurred at the C2 site. The synthetic possibilities of this transformation have been illustrated through the synthesis of the macrocyclic core of the cylindrocyclophanes class of natural products (Fig. 12b). The scheme consists of two carbene-induced C–H functionalization reactions, the first of which is an intermolecular to form 96 and the second is intramolecular to form the macrocycle 98, and between them they control the stereochemistry at four new stereogenic centers.
Figure 12. Selective functionalization of unactivated C–H bonds in the presence of benzylic C–H bonds.
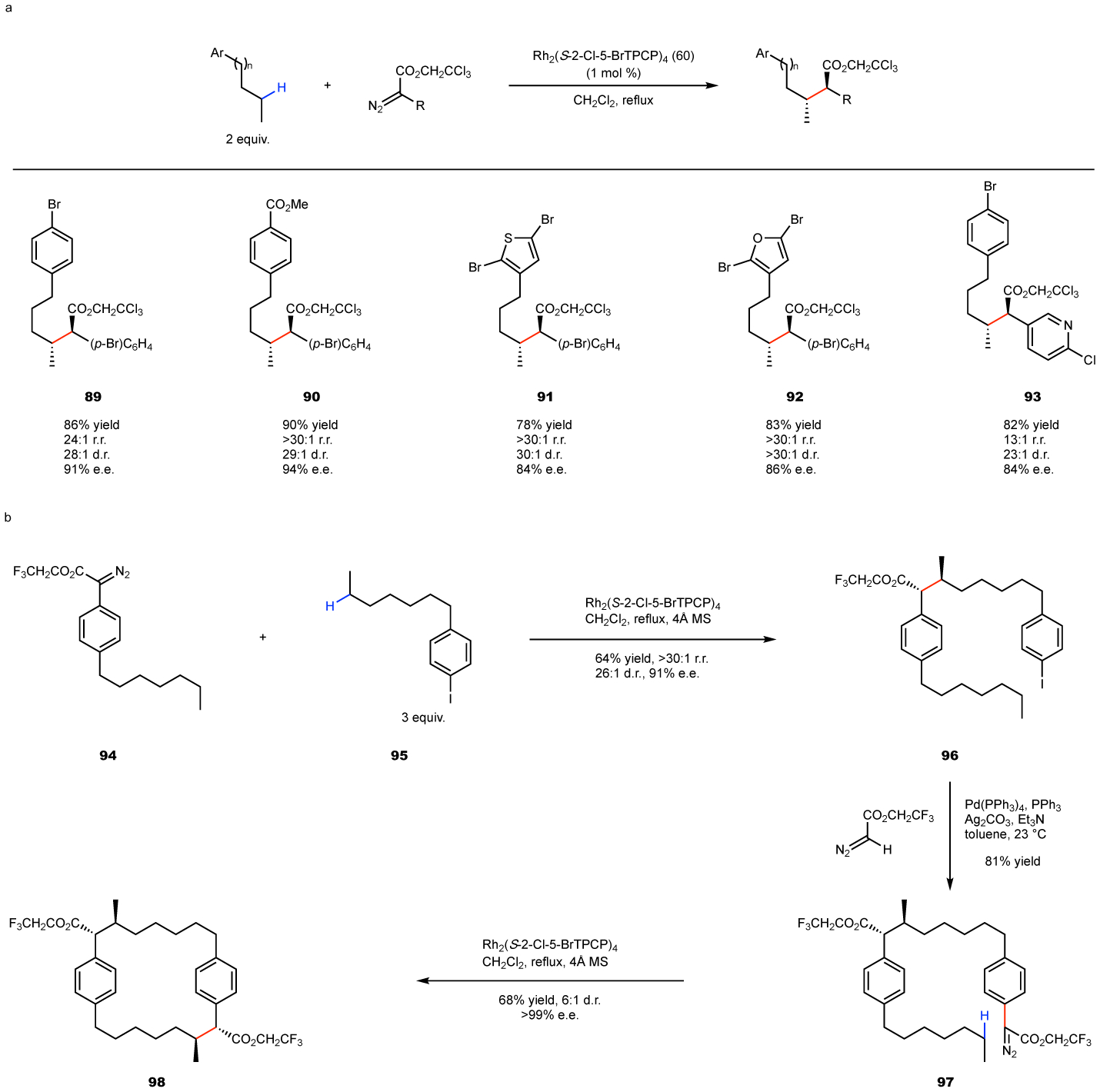
a: illustrative examples of functional group compatibility. b: demonstration of application to the synthesis of the macrocyclic core of the cylindrocyclophane natural products
The fully-p-substituted TPCP catalyst (61) is an even more bulky catalyst and is capable of blocking C–H functionalization at both tertiary and secondary sites. It leads to the preferential functionalization of primary C–H bonds, as illustrated in the simple substrates shown in Fig. 13a, generating the products 99-103 with exceptionally high asymmetric induction.114 Some illustrative examples of the potential of this chemistry are illustrated in the reaction with the chiral substrates 104 and 105 (Fig 13b). These substrates contain functional groups and even electronically activated C–H bonds, but the chemistry occurs cleanly at the most accessible primary C–H bonds. This catalyst gives very high asymmetric induction, and either diastereomeric series (106 and 107 or 108 and 109) can be generated, depending on which enantiomer of the catalyst 61 is used. The reaction can be extended to the steroid 110 and this substrate illustrates the subtleties of the catalyst control. The Rh-TCPTAD (54) reaction occurs at the most accessible tertiary C–H bond to form 111. In contrast, the fully-p-substituted TPCP (61)-catalyzed reaction occurs at the most accessible primary C–H bond to from 112.
Figure 13. Selective functionalization of unactivated primary C–H bonds.
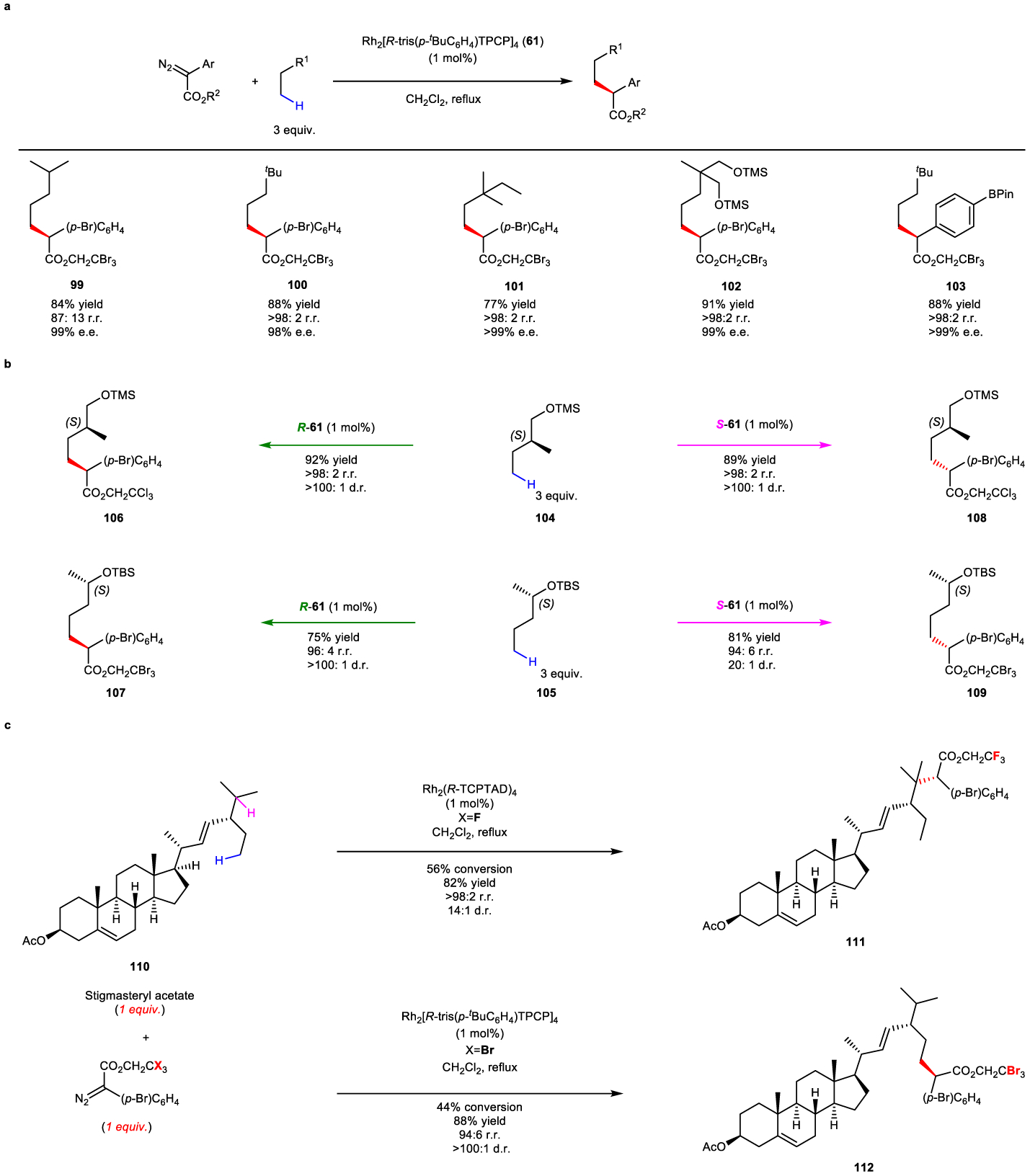
a: reactions of relatively simple substrates. b: reactions of chiral substrates to form either diastereomeric series of the products. b: illustrative examples of functional group compatibility. c: catalyst-controlled site selective functionalization of a steroid.
The expanded tool-box of catalysts enables even more challenging C–H functionalization reactions to be envisioned. A notoriously challenging site-selectivity problem has been the C–H functionalization of alkyl-substituted cyclohexanes.115–118 Even though it appears deceptively simple, the alkyl-substituted cyclohexane 113 actually has eleven different C–H bonds within the ring, ten of which would be secondary (Fig. 14a). Although, rich mixtures of products were formed with most of the dirhodium catalysts examined, Rh2(S-TPPTTL)4 (55) gave an exceptionally clean reaction.64 The reaction with tert-butyl cyclohexane resulted in an effective desymmetrization to form cleanly the C-3 equatorial substituted product 116 with high regioselectivity, diastereoselectivity and enantioselectivity (Fig 14b). The reaction was applied to a variety of cyclohexane derivatives and some representative example of the types of products formed (117-119) are shown in Fig. 14c. Rh2(S-TPPTTL)4 (55) is not an exceptionally bulky catalyst at the carbene site but the sixteen phenyl groups on the ligands generate a well-defined wall at the periphery of the catalyst, which has a dramatic effect in how substrates can approach. As can be seen in Fig. 8b, the chiral pocket for Rh2(S-TPPTTL)4 (55) has a relatively deep bowl shape, and if the substrate is relatively big it needs to be pointing out of the bowl. This can be seen in the transition state model for tert-butylcyclohexane reaction (Fig. 14d) where the large tert-butyl group is pointing out of the catalyst. If attack had occurred at C4 of the cyclohexane, the tert-butyl group would be pointing into the wall of the catalyst, generating a very unfavorable steric clash.
Figure 14. Desymmetrization of alkyl-substituted cycloalkanes.
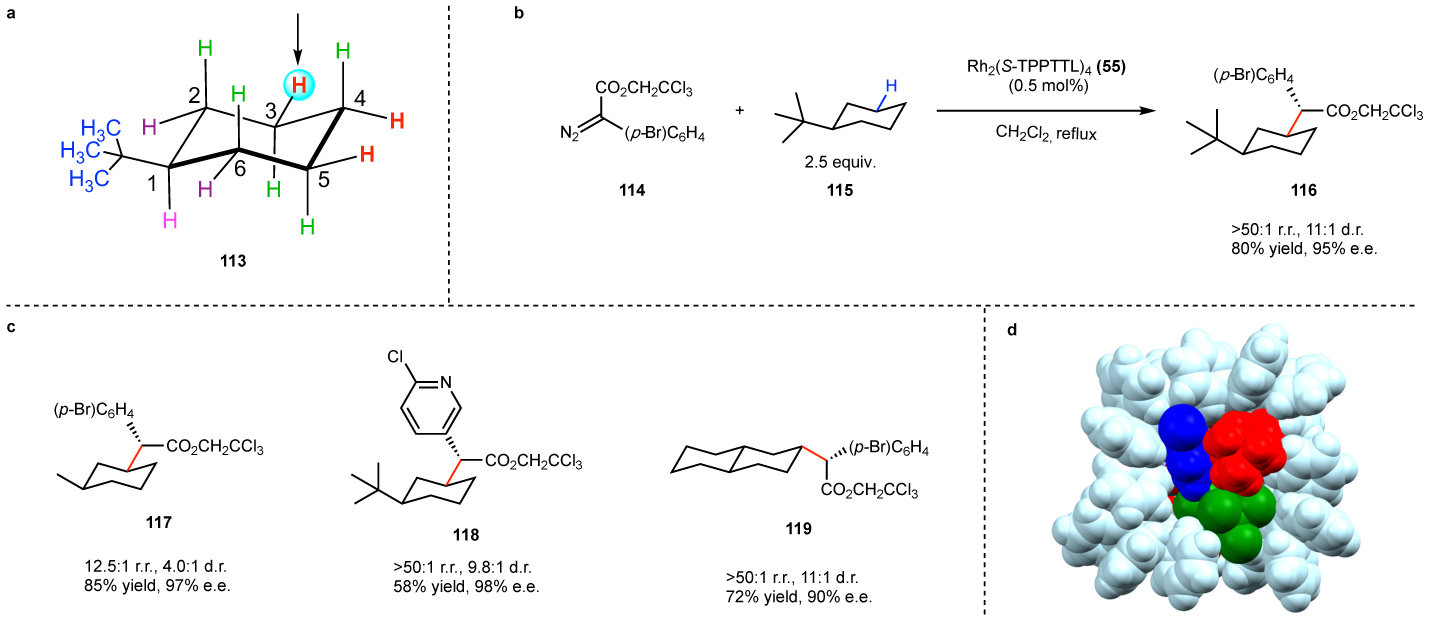
a: challenges associated with the reaction of alkylcycloalkanes. The system contains ten different secondary C–H bonds. b: C–H functionalization of tert-butylcyclohexane proceeds with exceptional site-selectivity and stereoselectivity. c: other representative examples, illustrating that heteroaryl derivatives and more elaborate cyclohexane derivatives can be used. d: computationally calculated transition state for the reaction of tert-butylcyclohexane at C3 with the rhodium bound carbene. The sixteen phenyl groups forming the wall of the catalyst are colored light blue. The trichloroethyl group of the carbene is green and the aryl group of the carbene is dark blue. The tert-butyl group of the cyclohexane is marked in red and is projecting out of the pocket. If attack had occurred at C4 of the cyclohexane, then, the tert-butyl group would have a steric clash with the wall of the catalyst.
The carbene C–H functionalization has been combined with other C–H functionalization strategies to rapidly generate synthetic complexity as illustrated in Fig. 15. A carbene C–H functionalization between 120 and 121 to form 122, followed by a palladium catalyzed C–H oxidation on 123 offers a flexible entry to the dihydrobenzofuran 124 (Fig. 15a),119 An intermolecular carbene C–H functionalization on the oxabicycle 125 generate 126 followed by an intramolecular C–H activation/Heck reaction generated the indoxamycin core 127 (Fig. 15b).120
Figure 15. Streamlined synthesis by sequential C–H functionalization reactions.
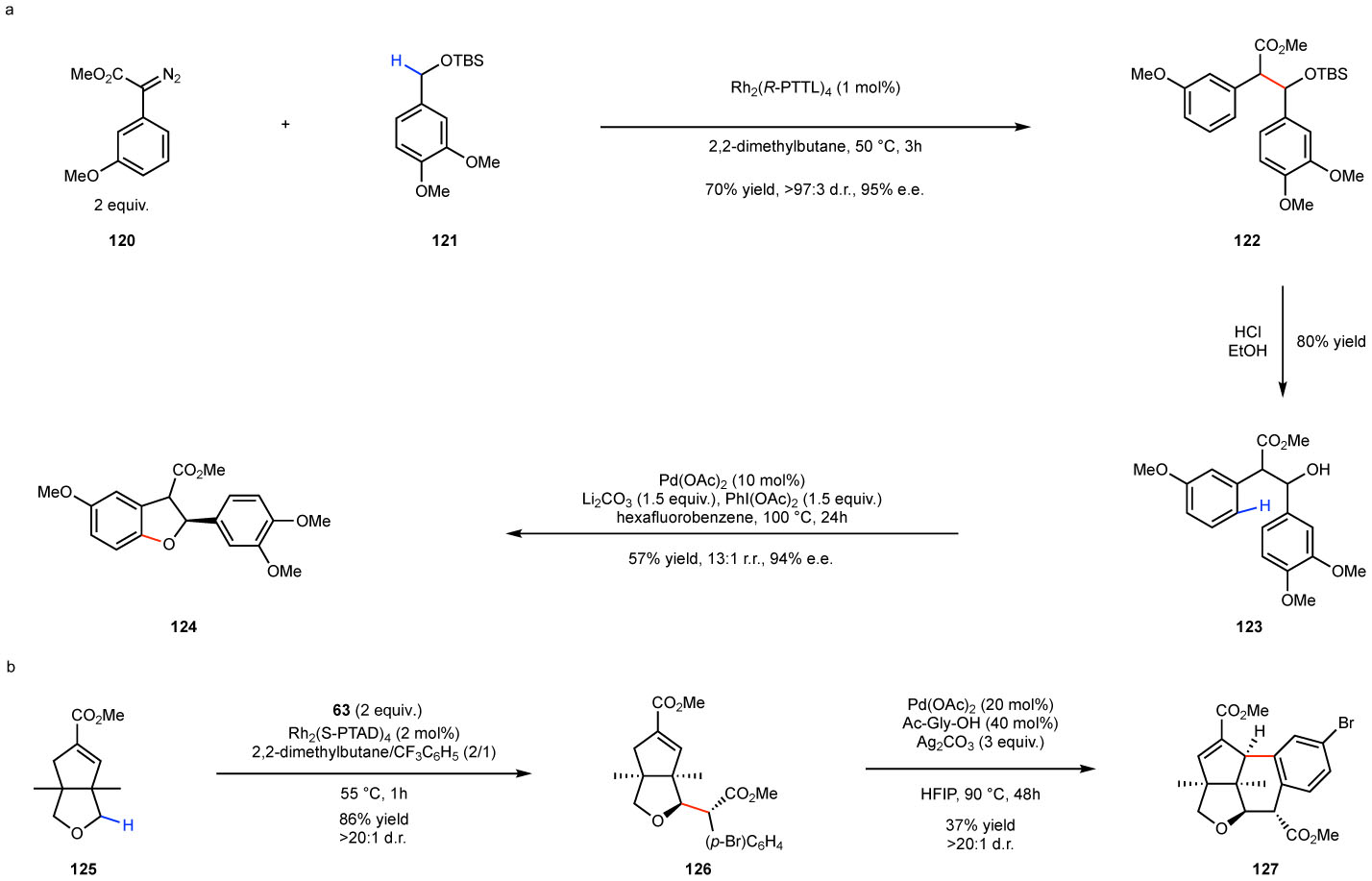
a: intermolecular carbene-induced intermolecular benzylic C–H functionalization followed by two palladium catalyzed C–H functionalizations. b: intermolecular carbene-induced intermolecular C–H functionalization followed by a palladium-catalyzed C–H activation/Heck reaction.
In conclusion, the rhodium-catalyzed reactions with donor/acceptor carbenes show that it is indeed possible to achieve site selective C–H functionalization at unactivated C–H bonds without resorting to the use of directing groups on the substrate. Indeed, a series of catalysts have been designed with different steric demands such that the site selectivity can be tailored by using the appropriate catalysts. The next challenge in this system is to incorporate electronic and coordinating influences into the catalyst design to achieve even more subtle control of site selectivity. Also, as the catalysts involve an expensive metal, conditions need to be developed so that the reactions can be conducted at extremely low catalyst loading, and ideally would be recyclable. The high levels of carbene-induced site-selectivities can currently only be achieved with donor/acceptor carbenes and dirhodium tetracarboxylate catalysts, a rather specialized reagent/catalyst combination. This work, however, should inspire further attempts to discover other reactive systems capable of similar levels of site selectivity.
Acknowledgements:
Financial support was provided by NSF under the CCI Center for Selective C-H Functionalization (CHE-1700982). Catalyst 55 was initially developed with financial support provided by NIH (GM-099142).
Footnotes
H.M.L.D. is a named inventor on a patent entitled, Dirhodium Catalyst Compositions and Synthetic Processes Related Thereto (US 8,974,428, issued March 10, 2015). K. L. has no competing financial interests.
References
- 1.Wencel-Delord J & Glorius F C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem 5, 369–375 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Godula K & Sames D C-H bond functionalization in complex organic synthesis. Science 312, 67–72 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Noisier AF & Brimble MA C-H functionalization in the synthesis of amino acids and peptides. Chem. Rev 114, 8775–8806 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Gutekunst WR & Baran PS C-H functionalization logic in total synthesis. Chem. Soc. Rev 40, 1976–1991 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi J, Yamaguchi AD & Itami K C-H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed 51, 8960–9009 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Davies HML & Morton D Recent Advances in C-H Functionalization. J. Org. Chem 81, 343–350 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Davies HML Du Bois J & Yu JQ C-H functionalization in organic synthesis. Chem. Soc. Rev 40, 1855–1856 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Ye J & Lautens M Palladium-catalysed norbornene-mediated C-H functionalization of arenes. Nat. Chem 7, 863–870 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Davies HML & Manning JR Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 451, 417–424 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karkas MD Electrochemical strategies for C-H functionalization and C-N bond formation. Chem. Soc. Rev 47, 5786–5865 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Revathi L, Ravindar L, Fang W-Y, Rakesh KP & Qin H-L Visible Light-Induced C-H Bond Functionalization: A Critical Review. Adv. Synth. Catal (2018). [Google Scholar]
- 12.Wei Y, Hu P, Zhang M & Su W Metal-Catalyzed Decarboxylative C-H Functionalization. Chem. Rev 117, 8864–8907 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Hofmann AW Ueber die Einwirkung des Broms in alkalischer Lösung auf die Amine. Ber. Deut. Chem. Ges 16, 558–560 (1883). [Google Scholar]
- 14.Löffler K Über eine neue BildungsweiseN-alkylierter Pyrrolidine. Ber. Deut. Chem. Ges 43, 2035–2048 (1910). [Google Scholar]
- 15.Corey EJ & Hertler WR The Synthesis of Dihydroconessine. A Method for Functionalizing Steroids at C18. J. Am. Chem. Soc 80, 2903–2904 (1958). [Google Scholar]
- 16.Buchschacher P, Kalvoda J, Arigoni D & Jeger O Direct Introduction of a Nitrogen Function at C-18 in a Steroid1. J. Am. Chem. Soc 80, 2905–2906 (1958). [Google Scholar]
- 17.Barton DHR, Beaton JM, Geller LE & Pechet MMA A new photochemical reaction. J. Am. Chem. Soc 82, 2640 (1960). [Google Scholar]
- 18.Wolff ME Cyclization of N-Halogenated Amines (The Hofmann-Löffler Reaction). Chem. Rev 63, 55–64 (1963). [Google Scholar]
- 19.Barton DHR, Hesse RH, Pechet MM & Smith LC The mechanism of the Barton reaction J. Chem. Soc., Perkin Trans 1 1159–1165 (1979). [Google Scholar]
- 20.Stateman LM, Nakafuku KM & Nagib DA Remote C-H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 50, 1569–1586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin J & MacMillan DWC Alcohols as alkylating agents in heteroarene C-H functionalization. Nature 525, 87–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davies HML Hansen T & Churchill MR Catalytic Asymmetric C-H Activation of Alkanes and Tetrahydrofuran. J. Am. Chem. Soc 122, 3063–3070 (2000). [Google Scholar]
- 23.Davies HML, Antoulinakis EG & Hansen T Catalytic Asymmetric Synthesis of Syn-Aldol Products from Intermolecular C-H Insertions between Allyl Silyl Ethers and Methyl Aryldiazoacetates. Org. Lett 1, 383–386 (1999). [Google Scholar]
- 24.Davies HML & Antoulinakis EG Asymmetric Catalytic C-H Activation Applied to the Synthesis ofSyn-Aldol Products. Org. Lett 2, 4153–4156 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Chan K-H, Guan X, Lo VK-Y & Che C-M Elevated catalytic activity of ruthenium(II)-porphyrin-catalyzed carbene/nitrene transfer and insertion reactions with N-heterocyclic carbene ligands. Angew. Chem. Int. Ed 53, 2982–2987 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Huang J-S, Zhou Z-Y, Che C-M & You X-Z Remarkably stable iron porphyrins bearing nonheteroatom-stabilized carbene or (alkoxycarbonyl)carbenes: isolation, X-ray crystal structures, and carbon atom transfer reactions with hydrocarbons. J. Am. Chem. Soc 124, 13185–13193 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Suematsu H & Katsuki T Iridium (III) catalyzed diastereo- and enantioselective C-H bond functionalization. J. Am. Chem. Soc 131, 14218–14219 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Axten JM, Ivy R, Krim L & Winkler JD Enantioselective Synthesis ofd-threo-Methylphenidate. J. Am. Chem. Soc 121, 6511–6512 (1999). [Google Scholar]
- 29.Davies HML, Hansen T, Hopper DW & Panaro SA Highly Regio-, Diastereo-, and Enantioselective C-H Insertions of Methyl Aryldiazoacetates into Cyclic N-Boc-Protected Amines. Asymmetric Synthesis of NovelC2-Symmetric Amines andthreo-Methylphenidate. J. Am. Chem. Soc 121, 6509–6510 (1999). [Google Scholar]
- 30.Cuthbertson JD & MacMillan DW The direct arylation of allylic sp(3) C-H bonds via organic and photoredox catalysis. Nature 519, 74–77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruckl T, Baxter RD, Ishihara Y & Baran PS Innate and guided C-H functionalization logic. Acc. Chem. Res 45, 826–839 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newhouse T & Baran PS If C-H bonds could talk: selective C-H bond oxidation. Angew. Chem. Int. Ed 50, 3362–3374 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diesel J, Finogenova AM & Nicolai Cramer N Nickel-Catalyzed Enantioselective Pyridone C-H Functionalizations Enabled by a Bulky N-Heterocyclic Carbene Ligand. J. Am. Chem. Soc 140, 4489–4493 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Lewis JC, Coelho PS & Arnold FH Enzymatic functionalization of carbon-hydrogen bonds. Chem. Soc. Rev 40, 2003–2021 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lyons TW & Sanford MS Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev 110, 1147–1169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hwang SJ, Cho SH & Chang S Synthesis of condensed pyrroloindoles via Pd-catalyzed intramolecular C-H bond functionalization of pyrroles. J. Am. Chem. Soc 130 (2008). [DOI] [PubMed] [Google Scholar]
- 37.McQuaid KM & Sames D C-H bond functionalization via hydride transfer: Lewis acid catalyzed alkylation reactions by direct intramolecular coupling of sp3 C-H bonds and reactive alkenyl oxocarbenium intermediates. J. Am. Chem. Soc 131, 402–403 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He G, Zhao Y, Zhang S, Lu C & Chen G Highly efficient syntheses of azetidines, pyrrolidines, and indolines via palladium catalyzed intramolecular amination of C(sp3)-H and C(sp2)-H bonds at gamma and delta positions. J. Am. Chem. Soc 134, 3–6 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Dick AR, Hull KL & Sanford MS A highly selective catalytic method for the oxidative functionalization of C-H bonds. J. Am. Chem. Soc 126, 2300–2301 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Desai LV, Malik HA & Sanford MS Oxone as an inexpensive, safe, and environmentally benign oxidant for C-H bond oxygenation. Org. Lett 8, 1141–1144 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Kalyani D & Sanford MS Regioselectivity in palladium-catalyzed C-H activation/oxygenation reactions. Org. Lett 7, 4149–4152 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Liu Y-J et al. Overcoming the limitations of directed C-H functionalizations of heterocycles. Nature 515, 389–393 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simmons EM & Hartwig JF Catalytic functionalization of unactivated primary C-H bonds directed by an alcohol. Nature 483, 70–73 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pesciaioli F, Dhawa U, Oliveira JCA, Yin R, John M & Ackermann L Enantioselective Cobalt(III)-Catalyzed C-H Activation Enabled by Chiral Carboxylic Acid Cooperation. Angew.Chem. Int.Ed 57,15425–15429 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Saint-Denis TG, Zhu RY, Chen G, Wu QF & Yu JQ Enantioselective C(sp(3))H bond activation by chiral transition metal catalysts. Science 359 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi H, Herron AN, Shao Y, Shao Q & Yu JQ Enantioselective remote meta-C-H arylation and alkylation via a chiral transient mediator. Nature 558, 581–585 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engle KM, Mei TS, Wasa M & Yu JQ Weak coordination as a powerful means for developing broadly useful C-H functionalization reactions. Acc. Chem. Res 45, 788–802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sigman MS & Werner EW Imparting catalyst control upon classical palladium-catalyzed alkenyl C-H bond functionalization reactions. Acc. Chem. Res 45, 874–884 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hickman AJ & Sanford MS Catalyst Control of Site Selectivity in the PdII/IV-Catalyzed Direct Arylation of Naphthalene. ACS Catal. 1, 170–174 (2011). [Google Scholar]
- 50.Hartwig JF Catalyst-Controlled Site-Selective Bond Activation. Acc. Chem. Res 50, 549–555 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neufeldt SR & Sanford MS Controlling site selectivity in palladium-catalyzed C-H bond functionalization. Acc. Chem. Res 45, 936–946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao K, Negretti S, Musaev DG, Bacsa J & Davies HML Site-selective and stereoselective functionalization of unactivated C-H bonds. Nature 533, 230–234 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Liao K et al. Site-selective and stereoselective functionalization of non-activated tertiary C-H bonds. Nature 551, 609–613 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Liao K et al. Site-Selective Carbene-Induced C-H Functionalization Catalyzed by Dirhodium Tetrakis(triarylcyclopropanecarboxylate) Complexes. ACS Catal. 8, 678–682 (2017). [Google Scholar]
- 55.Ravelli D, Fagnoni M, Fukuyama T, Nishikawa T & Ryu I Site-Selective C-H Functionalization by Decatungstate Anion Photocatalysis: Synergistic Control by Polar and Steric Effects Expands the Reaction Scope. ACS. Catal 8, 701–703 (2018). [Google Scholar]
- 56.Milan M, Salamone M, Costas M & Bietti M The Quest for Selectivity in Hydrogen Atom Transfer Based Aliphatic C-H Bond Oxygenation. Acc. Chem. Res 51, 1984–1995 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Ammann SE, Rice GT & White MC Terminal olefins to chromans, isochromans, and pyrans via allylic C-H oxidation. J. Am. Chem. Soc 136, 10834–10837 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cook AK, Schimler SD, Matzger AJ & Sanford MS Catalyst-controlled selectivity in the C-H borylation of methane and ethane. Science 351, 1421–1424 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Roizen JL, Harvey ME & Du Bois J Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C-H bonds. Acc. Chem. Res 45, 911–922 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qin C et al. D2-symmetric dirhodium catalyst derived from a 1,2,2-triarylcyclopropanecarboxylate ligand: design, synthesis and application. J. Am. Chem. Soc 133, 19198–19204 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Qin C & Davies HML Role of sterically demanding chiral dirhodium catalysts in site-selective C-H functionalization of activated primary C-H bonds. J. Am. Chem. Soc 136, 9792–9796 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Guptill DM & Davies HML 2,2,2-Trichloroethyl aryldiazoacetates as robust reagents for the enantioselective C-H functionalization of methyl ethers. J. Am. Chem. Soc 136, 17718–17721 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Fu L, Guptill DM & Davies HML Rhodium(II)-Catalyzed C-H Functionalization of Electron-Deficient Methyl Groups. J. Am. Chem. Soc 138, 5761–5764 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Fu J, Ren Z, Bacsa J, Musaev DG & Davies HML Desymmetrization of Cyclohexanes by Site- and Stereoselective C-H Functionalization. Nature just accepted. [DOI] [PubMed] [Google Scholar]
- 65.Liu W et al. Catalyst-Controlled Selective Functionalization of Unactivated C-H Bonds in the Presence of Electronically Activated C-H Bonds. J. Am. Chem. Soc 140, 12247–12255 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Davies HML & Morton D Guiding principles for site selective and stereoselective intermolecular C-H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev 40, 1857–1869 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Davies HML & Denton JR Application of donor/acceptor-carbenoids to the synthesis of natural products. Chem. Soc. Rev 38, 3061–3071 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davies HML & Beckwith RE Catalytic enantioselective C-H activation by means of metal-carbenoid-induced C-H insertion. Chem. Rev 103, 2861–2904 (2003). [DOI] [PubMed] [Google Scholar]
- 69.Davies HML Recent advances in catalytic enantioselective intermolecular C-H functionalization. Angew. Chem. Int. Ed 45, 6422–6425 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Doyle MP, Duffy R, Ratnikov M & Zhou L Catalytic carbene insertion into C-H bonds. Chem. Rev 110, 704–724 (2010). [DOI] [PubMed] [Google Scholar]
- 71.Scott LT & DeCicco GJ Intermolecular carbon-hydrogen insertion of copper carbenoids. J. Am. Chem. Soc 96, 322–323 (1974). [Google Scholar]
- 72.Demonceau A, Noels AF, Hubert AJ & Teyssie P Transition-metal-catalysed reactions of diazoesters. Insertion into C-H bonds of paraffins by carbenoids. J. Chem. Soc., Chem. Commun, 688–689 (1981). [Google Scholar]
- 73.Caballero A et al. Silver-catalyzed C-C bond formation between methane and ethyl diazoacetate in supercritical CO(2). Science 332, 835–838 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Callot HJ & Metz F Homologation of n-alkanes using diazoesters and rhodium(III)porphyrins. Enhanced attack on primary C H bonds. Tetrahedron Lett. 23, 4321–4324 (1982). [Google Scholar]
- 75.Callot HJ & Metz F Rhodium(II)2,4,6-triarylbenzoates: improved catalysts for the cyclopropanation of z-olefins. Tetrahedron 41, 4495–4501 (1985). [Google Scholar]
- 76.Callot HJ & Schaeffer E Rhodium(III)porphyrins and diazoalkanes - alkylrhodium(III)porphyrins and carbene transfer. New J. Chem 4, 311–314 (1980). [Google Scholar]
- 77.Caballero A et al. Highly regioselective functionalization of aliphatic carbon-hydrogen bonds with a perbromohomoscorpionate copper(I) catalyst. J. Am. Chem. Soc 125, 1446–1447 (2003). [DOI] [PubMed] [Google Scholar]
- 78.Taber DF & Ruckle RE Cyclopentane construction by dirhodium tetraacetate-mediated intramolecular C-H insertion: steric and electronic effects. J. Am. Chem. Soc 108, 7686–7693 (1986). [DOI] [PubMed] [Google Scholar]
- 79.Doyle MP, Kalinin AV & Ene DG Chiral Catalyst Controlled Diastereoselection and Regioselection in Intramolecular Carbon-Hydrogen Insertion Reactions of Diazoacetates. J. Am. Chem. Soc 118, 8837–8846 (1996). [Google Scholar]
- 80.Taber DF & Stiriba S-E Synthesis of Natural Products By Rhodium-Mediated Intramolecular C-H Insertion. Chem.-Eur. J 4, 990–992 (1998). [Google Scholar]
- 81.Breslow R & Gellman SH Intramolecular nitrene carbon-hydrogen insertions mediated by transition-metal complexes as nitrogen analogs of cytochrome P-450 reactions. J. Am. Chem. Soc 105, 6728–6729 (1983). [Google Scholar]
- 82.Espino CG, Wehn PM, Chow J & Du Bois J Synthesis of 1,3-Difunctionalized Amine Derivatives through Selective C-H Bond Oxidation. J. Am. Chem. Soc 123, 6935–6936 (2001). [Google Scholar]
- 83.Hashimoto S. i., Watanabe N, Sato T, Shiro M & Ikegami S Enhancement of enantioselectivity in intramolecular C-H insertion reactions of α-diazo β-keto esters catalyzed by chiral dirhodium(II) carboxylates. Tetrahedron Lett. 34, 5109–5112 (1993). [Google Scholar]
- 84.Doyle MP et al. Diastereocontrol for Highly Enantioselective Carbon-Hydrogen Insertion Reactions of Cycloalkyl Diazoacetates. J. Am. Chem. Soc 116, 4507–4508 (1994). [Google Scholar]
- 85.Bode JW, Doyle MP, Protopopova MN & Zhou Q-L Intramolecular Regioselective Insertion into Unactivated Prochiral Carbon-Hydrogen Bonds with Diazoacetates of Primary Alcohols Catalyzed by Chiral Dirhodium(II) Carboxamidates. Highly Enantioselective Total Synthesis of Natural Lignan Lactones. J. Org. Chem 61, 9146–9155 (1996). [Google Scholar]
- 86.Caballero A et al. Catalytic functionalization of low reactive C(sp(3))-H and C(sp(2))-H bonds of alkanes and arenes by carbene transfer from diazo compounds. Dalton Trans. 44, 20295–20307 (2015). [DOI] [PubMed] [Google Scholar]
- 87.Davies HML, Smith HD & Korkor O Tandem cyclopropanation/Cope rearrangement sequence. Stereospecific [3 + 4] cycloaddition reaction of vinylcarbenoids with cyclopentadiene. Tetrahedron Lett. 28, 1853–1856 (1987). [Google Scholar]
- 88.Davies HML & Doan BD Asymmetric synthesis of the tremulane skeleton by a tandem cyclopropanation/Cope rearrangement. Tetrahedron Lett. 37, 3967–3970 (1996). [Google Scholar]
- 89.Davies HML, Bruzinski PR & Fall MJ Effect of diazoalkane structure on the stereoselectivity of rhodium(II) (S)-N-(arylsulfonyl)prolinate catalyzed cyclopropanations. Tetrahedron Lett. 37, 4133–4136 (1996). [Google Scholar]
- 90.Lamb KN et al. Synthesis of Benzodihydrofurans by Asymmetric C-H Insertion Reactions of Donor/Donor Rhodium Carbenes. Chem.-Eur. J 23, 11843–11855 (2017). [DOI] [PubMed] [Google Scholar]
- 91.Soldi C et al. Enantioselective intramolecular C-H insertion reactions of donor-donor metal carbenoids. J. Am. Chem. Soc 136, 15142–15145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu D et al. Highly Chemo- and Stereoselective Catalyst-Controlled Allylic C-H Insertion and Cyclopropanation Using Donor/Donor Carbenes. Angew. Chem. Int. Ed 57, 12405–12409 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Zhu D et al. Enantioselective Intramolecular C-H Insertion of Donor and Donor/Donor Carbenes by a Nondiazo Approach. Angew. Chem. Int. Ed 55, 8452–8456 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Hansen J, Autschbach J & Davies HML Computational study on the selectivity of donor/acceptor-substituted rhodium carbenoids. J. Org. Chem 74, 6555–6563 (2009). [DOI] [PubMed] [Google Scholar]
- 95.Davies HML, Venkataramani C, Hansen T & Hopper DW New strategic reactions for organic synthesis: catalytic asymmetric C-H activation alpha to nitrogen as a surrogate for the mannich reaction. J. Am. Chem. Soc 125, 6462–6468 (2003). [DOI] [PubMed] [Google Scholar]
- 96.Davies HML, Ren P & Jin Q Catalytic Asymmetric Allylic C-H Activation as a Surrogate of the Asymmetric Claisen Rearrangement. Org. Lett 3, 3587–3590 (2001). [DOI] [PubMed] [Google Scholar]
- 97.Davies HML & Ren P Catalytic Asymmetric C-H Activation of Silyl Enol Ethers as an Equivalent of an Asymmetric Michael Reaction. J. Am. Chem. Soc 123, 2070–2071 (2001). [DOI] [PubMed] [Google Scholar]
- 98.Davies HML, Yang J & Nikolai J Asymmetric C-H insertion of Rh(II) stabilized carbenoids into acetals: A C-H activation protocol as a Claisen condensation equivalent. J. Organomet. Chem 690, 6111–6124 (2005). [Google Scholar]
- 99.Hansen JH et al. On the mechanism and selectivity of the combined C-H activation/Cope rearrangement. J. Am. Chem. Soc 133, 5076–5085 (2011). [DOI] [PubMed] [Google Scholar]
- 100.Davies HML & Lian Y The combined C-H functionalization/Cope rearrangement: discovery and applications in organic synthesis. Acc. Chem. Res 45, 923–935 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davies HML & Jin Q Highly diastereoselective and enantioselective C-H functionalization of 1,2-dihydronaphthalenes: a combined C-H activation/Cope rearrangement followed by a retro-Cope rearrangement. J. Am. Chem. Soc 126, 10862–10863 (2004). [DOI] [PubMed] [Google Scholar]
- 102.Lian Y & Davies HML Combined C-H functionalization/Cope rearrangement with vinyl ethers as a surrogate for the vinylogous Mukaiyama aldol reaction. J. Am. Chem. Soc 133, 11940–11943 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davies HML, Dai X & Long MS Combined C-H activation/cope rearrangement as a strategic reaction in organic synthesis: total synthesis of (−)-colombiasin a and (−)-elisapterosin B. J. Am. Chem. Soc 128, 2485–2490 (2006). [DOI] [PubMed] [Google Scholar]
- 104.Davies HML D. X, Long MS Combined C-H activation/cope rearrangement as a strategic reaction in organic synthesis: total synthesis of (−)-colombiasin a and (−)-elisapterosin B. J. Am. Chem. Soc 128, 2485–2490 (2006). [DOI] [PubMed] [Google Scholar]
- 105.Thu H-Y et al. Highly selective metal catalysts for intermolecular carbenoid insertion into primary C-H bonds and enantioselective C-C bond formation. Angew. Chem. Int. Ed 47, 9747–9751 (2008). [DOI] [PubMed] [Google Scholar]
- 106.Hansen J & Davies HML High Symmetry Dirhodium(II) Paddlewheel Complexes as Chiral Catalysts. Coord Chem. Rev 252, 545–555 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Watanabe N, Ogawa T, Ohtake Y, Ikegami S & Hashimoto S Dirhodium(II) Tetrakis[N-phthaloyl-(S)-tert-leucinate]: A Notable Catalyst for Enantiotopically Selective Aromatic Substitution Reactions of α-Diazocarbonyl Compounds. Synlett 1996, 85–86 (1996). [Google Scholar]
- 108.Yamawaki M, Tsutsui H, Kitagaki S, Anada M & Hashimoto S Dirhodium(II) tetrakis[N-tetrachlorophthaloyl-(S)-tert-leucinate]: a new chiral Rh(II) catalyst for enantioselective amidation of C-H bonds. Tetrahedron Lett. 43, 9561–9564 (2002). [Google Scholar]
- 109.Reddy RP & Davies HML Dirhodium tetracarboxylates derived from adamantylglycine as chiral catalysts for enantioselective C-h aminations. Org. Lett 8, 5013–5016 (2006). [DOI] [PubMed] [Google Scholar]
- 110.Lindsay VN, Lin W & Charette AB Experimental evidence for the all-up reactive conformation of chiral rhodium(II) carboxylate catalysts: enantioselective synthesis of cis-cyclopropane alpha-amino acids. J. Am. Chem. Soc 131, 16383–16385 (2009). [DOI] [PubMed] [Google Scholar]
- 111.Lindsay VNG & Charette AB Design and Synthesis of Chiral Heteroleptic Rhodium(II) Carboxylate Catalysts: Experimental Investigation of Halogen Bond Rigidification Effects in Asymmetric Cyclopropanation. ACS Catal. 2, 1221–1225 (2012). [Google Scholar]
- 112.DeAngelis A, Dmitrenko O, Yap GP & Fox JM Chiral crown conformation of Rh(2)(S-PTTL)(4): enantioselective cyclopropanation with alpha-alkyl-alpha-diazoesters. J. Am. Chem. Soc 131, 7230–7231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.DeAngelis A et al. The chiral crown conformation in paddlewheel complexes. Chem. Commun 46, 4541–4543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liao K et al. Design of Catalysts for Site-Selective and Enantioselective Functionalization of Non-Activated Primary C-H Bonds. Nat. Chem 10, 1048–1055 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Banerjee A, Sarkar S & Patel BK C-H functionalisation of cycloalkanes. Org. Biomol. Chem 15, 505–530 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Salamone M, Ortega VB & Bietti M Enhanced Reactivity in Hydrogen Atom Transfer from Tertiary Sites of Cyclohexanes and Decalins via Strain Release: Equatorial C-H Activation vs Axial C-H Deactivation. J. Org. Chem 80, 4710–4715 (2015). [DOI] [PubMed] [Google Scholar]
- 117.Milan M, Bietti M & Costas M Highly Enantioselective Oxidation of Nonactivated Aliphatic C-H Bonds with Hydrogen Peroxide Catalyzed by Manganese Complexes. ACS Cent. Sci 3, 196–204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dantignana V et al. Chemoselective Aliphatic C-H Bond Oxidation Enabled by Polarity Reversal. ACS Cent. Sci 3, 1350–1358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang H, Li G, Engle KM, Yu JQ & Davies HML Sequential C-H functionalization reactions for the enantioselective synthesis of highly functionalized 2,3-dihydrobenzofurans. J. Am. Chem. Soc 135, 6774–6777 (2013). [DOI] [PubMed] [Google Scholar]
- 120.Bedell TA et al. Rapid Construction of a Benzo-Fused Indoxamycin Core Enabled by Site-Selective C-H Functionalizations. Angew. Chem. Int. Ed 55, 8270–8274 (2016). [DOI] [PubMed] [Google Scholar]