

Abstract
While prostaglandins (PGs), short-range lipid signals, regulate single cell migration, their roles in collective migration remain unclear. To address this, we use Drosophila border cell migration, an invasive, collective migration that occurs during Stage 9 of oogenesis. Pxt is the Drosophila cyclooxygenase-like enzyme responsible for PG synthesis. Loss of Pxt results in both delayed border cell migration and elongated clusters, whereas somatic Pxt knockdown causes delayed migration and compacted clusters. These findings suggest PGs act in both the border cells and nurse cells, the substrate on which the border cells migrate. As PGs regulate the actin bundler Fascin, and Fascin is required for on-time migration, we assessed whether PGs regulate Fascin to promote border cell migration. Coreduction of Pxt and Fascin results in delayed migration and elongated clusters. The latter may be due to altered cell adhesion, as loss of Pxt or Fascin, or coreduction of both, decreases integrin levels on the border cell membranes. Conversely, integrin localization is unaffected by somatic knockdown of Pxt. Together these data lead to the model that PG signaling controls Fascin in the border cells to promote migration and in the nurse cells to maintain cluster cohesion.
INTRODUCTION
During invasive, collective cell migration multiple cells migrate as a group between other cells and/or through tissues. Such migration requires the coordination of numerous factors including cell polarity, cytoskeletal remodeling, and notably, adhesion dynamics. Migratory cells must properly adhere and release from their substrate to promote migration (Grashoff et al., 2010; De Pascalis and Etienne-Manneville, 2017). Collective migration also requires maintaining communication and adhesions between all the cells of the cluster (Mayor and Etienne-Manneville, 2016). While many factors regulating cell migration have been uncovered by studying single cell migration, in vivo, most cell migration occurs as collective migration, including during embryonic development, regeneration, and cancer metastasis (Friedl and Gilmour, 2009; Scarpa and Mayor, 2016). Thus, it is critical to define the factors regulating collective cell migration.
One regulator of cell migration is prostaglandins (PGs; Menter and Dubois, 2012). PGs are short-range lipid signals produced at their sites of action (Tootle, 2013). Cyclooxygenase (COX) enzymes convert arachidonic acid into the PG precursor, PGH2. This precursor is then acted upon by PG-type specific synthases to produce the different bioactive PGs. Each PG is secreted to bind and activate one or more G protein–coupled receptors to elicit different downstream signaling cascades to mediate various cellular and physiological outcomes. One PG, PGE2, is widely implicated in promoting cell migration during both development and cancer progression. Inhibition of COX activity or loss of PGE2 signaling alters cellular adhesion dynamics and blocks gastrulation in zebrafish (Cha et al., 2005, 2006; Speirs et al., 2010). Additionally, PGE2 regulates vascular maturation and angiogenesis (Ugwuagbo et al., 2019), homing of hematopoietic stem cells to their niche (North et al., 2007; Hoggatt et al., 2009), and macrophage migration (Digiacomo et al., 2015). Notably, the majority of these PG-dependent developmental migrations occur as collectives or groups of cells. PGs are also widely implicated in promoting cancer migration and metastasis (Menter and Dubois, 2012), both by functioning within the tumor cells and within the microenvironment (Li et al., 2012; Kobayashi et al., 2018). Cancer cells can migrate as both single cells and collectives (Friedl and Mayor, 2017; Pandya et al., 2017). Recent evidence suggests that collectively migrating cancer cells are more likely to establish metastatic tumors, are resistant to chemotherapies, and correlate with a poor prognosis (Giampieri et al., 2009; Alexander and Friedl, 2012; Khalil et al., 2017; Stuelten et al., 2018). As collective cell migration is important for normal development and contributes to cancer progression, and PGs regulate migration in both of these contexts, it is essential to establish a robust system for defining the mechanisms by which PGs regulate invasive, collective cell migration.
Drosophila oogenesis is an ideal in vivo model to uncover the roles of PGs (Tootle and Spradling, 2008; Spracklen and Tootle, 2015). Each female fly has two ovaries composed of chains of sequentially developing follicles or eggs (Spradling, 1993). Follicle development is divided into 14 morphological stages. Each follicle is made up of 16 germline-derived cells—one posterior oocyte and 15 nurse cells. These germ cells are surrounded by a layer of somatic epithelial cells termed follicle cells. The follicle cells can be divided into different subtypes, including outer follicle cells, stretch follicle cells, centripetal cells, and the border cell cluster, which includes both polar and border cells. Drosophila possess a single COX-like enzyme, Pxt (Tootle and Spradling, 2008). Loss of Pxt results in multiple defects during follicle development and results in female sterility (Tootle and Spradling, 2008; Tootle et al., 2011; Groen et al., 2012; Spracklen et al., 2014; Spracklen and Tootle, 2015).
During Drosophila oogenesis, the process of border cell migration has been widely used to uncover conserved mechanisms regulating invasive collective cell migration (Montell, 2003; Montell et al., 2012). During Stage 9 (S9), the anterior pair of polar cells specifies four to six follicle cells to differentiate into border cells; together these cells form a cluster, delaminate from the follicular epithelium, and collectively migrate from the anterior tip of the follicle, between the much larger nurse cells, to the nurse cell–oocyte boundary by S10A. During this migration, the follicle grows in size, the nurse cells become covered in squamous stretch follicle cells, and the outer follicle cells ultimately cover only the oocyte. The border cell cluster then migrates to the dorsal side of the oocyte and ultimately aids in the formation of the micropyle, the structure through which the sperm enters to fertilize the egg (Montell et al., 1992). Thus, border cell migration is required for fertility.
Here we utilize border cell migration to uncover the roles of PGs in invasive, collective cell migration. Examination of S10A follicles reveals PGs are required for regulating border cell cluster morphology, as loss of Pxt results in elongated clusters with cells being left along the migration path. To uncover the cause of these defects, border cell migration was examined during S9. Loss of Pxt results in delayed border cell migration during S9 and aberrant, elongated border cell clusters. Knockdown of Pxt in the somatic cells results in delayed border cell migration, but the clusters are more compact. These findings suggest that PGs act in both the germline and the somatic cells to regulate border cell migration. As PGs regulate Fascin (Drosophila singed) to control actin remodeling within the germline (Groen et al., 2012; Spracklen et al., 2019), and Fascin is required in both the border cells and nurse cells for on-time border cell migration during S9 (Lamb et al., 2020), we postulated that PGs regulate Fascin to control border cell migration. Indeed, dominant genetic interaction studies reveal that coreduction of Pxt and Fascin phenocopies loss of Pxt, resulting in delayed migration and elongated border cell clusters. One mechanism regulating cluster cohesion and morphology is cellular adhesion, including integrin-based adhesions (Dinkins et al., 2008; Llense and Martin-Blanco, 2008). We find loss of either Pxt or Fascin, or coreduction of both decreases the membrane enrichment of integrins on the border cells. Conversely, somatic RNAi knockdown of Pxt does not alter integrin localization. Together these data lead to the model that Pxt produces PGs that activate a signaling cascade to control Fascin in the border cells to promote on-time migration and in the nurse cells to promote integrin-based adhesions necessary for maintaining cluster cohesion.
RESULTS
Pxt regulates border cell cluster morphology
A common means of assessing border cell migration is to determine whether the cluster reaches the nurse cell–oocyte boundary by S10A. Prior work defining the roles of Pxt during Drosophila oogenesis reported that while the border cell cluster did reach the oocyte by S10A, the cluster had a long trail of cells remaining along the migration path (Tootle and Spradling, 2008). Extending from these studies, we sought to quantify the border cell defects when Pxt is lost.
For our analyses, we make use of two insertional pxt alleles: EY03052 (EY) and f01000 (f). Prior work characterizing these alleles has shown that pxtEY/EY exhibits a low level of pxt expression by both qRT-PCR and in situ hybridization (Tootle and Spradling, 2008). These same analyses revealed pxtf/f exhibited little to no pxt expression and immunoblotting revealed no protein product, suggesting pxtf is a null allele (Spracklen et al., 2014). Using these alleles, we assessed border cell migration at S10A by labeling the follicle cell nuclei, including the border cells, by immunofluorescence staining for Eyes absent (Eya). Eya marks the border cells or “rosette cells” within the cluster but does not mark the polar cells at the center of the cluster; thus, polar cells were excluded from our analyses. Although the border cell clusters in pxt mutant follicles reach the nurse cell–oocyte boundary, the clusters are abnormal. Border cells initiate migration but a few or long chains of the border cells detach from and trail behind the main cluster to ultimately remain between the nurse cells in pxt mutant S10 follicles (Figure 1, B–C′ compared with A–A′; yellow arrows and bracket). Only 8% of wild-type follicles exhibit multiple border cells trailing behind, while 76% of pxtf/f, 16% of pxtEY/EY, and 48% of pxtEY/f mutant follicles have trailing border cells (Figure 1, B–C′ compared with A–A′, and Supplemental Figure S1A). Specifically, we find that in pxtf/f follicles there is an average of 2.24 trailing border cells and in pxtEY/f follicles there is an average of 0.76, compared with 0.11 in wild type (Supplemental Figure S1A; p < 0.0001). In addition, the total number of border cells (in the cluster and trailing cells) is increased when Pxt is completely lost; wild-type follicles exhibit an average of 5.2 border cells while pxtf/f follicles exhibit an average of 9.1 (p < 0.0001) and pxtEY/EY follicles exhibit an average of 6.1 border cells (Supplemental Figure S1B; p = 0.0011). Notably, the number of polar cells within the border cell cluster is not changed (unpublished data). Together these data indicate Pxt contributes to border cell migration by regulating border cell number and cluster cohesion.
FIGURE 1:

Prostaglandins regulate border cell cluster integrity. (A–C′) Maximum projections of three confocal slices of S10 follicles of the indicated genotypes; anterior is to the left. (A–A′) wild type (yw). (B–B′) pxtEY03052/EY03052 (pxtEY/EY). (C–C′) pxtf01000/f01000 (pxtf/f). (A–C) Merged images: Eyes absent (Eya), white; phalloidin (F-actin), red; and DAPI (DNA), blue. (A′–C′) Eya, white; images were brightened by 50% in Photoshop for better visualization. The nuclei of the border, stretch follicle, and centripetal cells are marked by Eya staining; polar cells are not marked. By S10, the intact border cell cluster is normally located at the nurse cell/oocyte boundary (A–A′, cyan asterisk). In pxt mutants, despite the majority of the cluster reaching the boundary (cyan asterisk), cells are often left behind along the migration pathway (B–C′); the frequency of S10 follicles exhibiting trailing border cells is indicated in the top right of panels A′–C′. These cells can exist as single cells or pairs of cells being left behind (B–B′, yellow arrows), or long continuous chains of cells being left behind (C–C′, yellow bracket). Scale bars = 50 μm.
Pxt is required for on-time border cell migration and maintenance of cluster morphology
To determine how the border cell defects arise when Pxt is lost, we examined border cell migration during S9. While live imaging is an ideal approach to define defects during the invasive, collective migration of the border cells (Prasad and Montell, 2007), pxt mutant follicles proved difficult to keep alive during the live imaging process (unpublished data). Therefore, we assessed border cell migration from fixed immunofluorescence images. In wild-type follicles, the outer follicle cells (orange dashed line) are in line with the border cell cluster throughout S9 (Figure 2B), which indicates an on-time migration. Similarly, heterozygotes for mutations in pxt (pxt−/+) also exhibit on-time border cell migration (Figure 2C and unpublished data). Surprisingly, when either pxt allele is over the MKRS balancer chromosome, border cell migration is delayed (unpublished data and Supplemental Figure S2); we speculate this is due to a genetic interaction with a mutation on the balancer chromosome and pxt. Loss of Pxt by either homozygosity for either mutant allele (unpublished data) or transheterozygosity for both alleles (pxtEY/f) results in delayed border cell migration, as the border cells remain anterior to the outer follicle cells (Figure 2D and unpublished data).
FIGURE 2:

Prostaglandins are essential for on-time border cell migration during S9. (A) Diagram depicting the wild-type alignment of the position of the border cell cluster (green) with that of the outer follicle cells (teal) and the measurements used to quantify border cell migration. The position of the border cells is quantified by measuring the distance from the anterior tip of the follicle to the leading edge of the border cell cluster (A, cyan dashed line), and the position of the outer follicle cells is quantified by measuring from the anterior tip of the follicle to the anterior edge of the outer follicle cells (A, purple dashed line). The migration index is defined as the border cell distance (A, cyan) divided by the outer follicle cell distance (A, purple). Normal or on-time migration should result in a migration index of 1, while delayed migration will result in values <1 and accelerated migration will result in values >1. (B–D) Maximum projections of three confocal slices of S9 follicles of the indicated genotypes; anterior is to the left. (B) wild type (yw). (C) pxtEY/+. (D) pxtEY/f. Merged images: Fascin, green; phalloidin (F-actin), red; and DAPI (DNA), blue. Orange dashed lines indicate the position of the outer follicle cells. White arrow indicates the tail on the border cell cluster. (E, G) Graphs of the migration index quantification during S9 for the indicated genotypes. In G, follicles are binned into groups based on overall follicle length. Each circle represents a single border cell cluster; n = number of follicles. Each line indicates the average and the whiskers indicate the SD (SD). Dotted line at 1 indicates an on-time migration. ****, p < 0.0001; ***, p < 0.001 (two-tailed unpaired t test). (F) Graph of the follicle length vs outer follicle cell distance for wild-type (blue) and pxtEY/f (orange) follicles; each circle represents a single follicle, n = 99 for both genotypes, and the best-fit lines provided. Loss of Pxt results in significant migration delays (D, E). The follicle cell distance vs. follicle length is similar between wild-type and pxt mutant follicles, indicating that outer follicle cell morphogenesis is normal in pxt mutants and therefore, the migration index can be used to assess border cell migration defects during S9. Significant migration delays are observed throughout S9 in pxt mutant follicles (G). Scale bars = 50 μm.
To further characterize the border cell migration defects during S9 we developed a quantitative method of assessing migration from fixed immunofluorescence images (Figure 2A). Specifically, we measure the distance the border cells have migrated from the anterior end of the follicle and divide it by the distance the outer follicle cells are from the anterior of the follicle. We term this the migration index. A migration index value of ∼1 indicates on-time migration, while values less than 1 indicate delayed migration and values greater than 1 indicate accelerated migration. On average, wild-type clusters exhibit a migration index of 1.029 (Figure 2E). Loss of Pxt results in a significant delay in border cell migration with an average migration index of 0.7647 for pxtEY/EY and 0.7167 for pxtf/f follicles (Figure 2E; p < 0.0001). Additionally, transheterozygotes of the two alleles (pxtEY/f) exhibit a migration index of 0.78802 (Figure 2E; p < 0.0001). A migration index below 1 could result from either delayed border cell migration or increased outer follicle cell distance. To distinguish between these possibilities, we plotted the distance of the outer follicle cells versus follicle length for wild type (blue) and pxtEY/f (orange), and find that they exhibit a similar slope, indicating outer follicle cell behavior is normal in pxt mutants (Figure 2F). Together these findings indicate Pxt is essential for on-time border cell migration during S9.
To further characterize the role of Pxt in border cell migration, we assessed migration at different points during S9. Because the follicle increases in length throughout S9, we can use follicle length as an indicator of S9 progression. Using the follicle length, we binned S9 into three groups (from early to late S9) and compared the average migration indices of wild-type and pxt mutant follicles (Figure 2G). Loss of Pxt (all allelic combinations combined) results in delayed migration per our migration index quantification throughout the entirety of S9. This finding indicates Pxt is required throughout the whole process of border cell migration.
In addition to delayed migration, loss of Pxt also alters border cell cluster morphology during S9. Wild-type clusters are round and held tightly together (Figure 3A; Bianco et al., 2007; Prasad and Montell, 2007). We find that in pxt mutants the majority of border cell clusters are elongated at the trailing edge (61% compared with 23% in wild type; Figure 3B). To further quantify this defect, we measured the length of the main cluster (Figure 3, A and B; yellow lines) and found that compared with wild type, loss of Pxt results in significantly longer clusters (Figure 3C). Wild-type clusters averaged 33.32 µm in length while clusters in pxtf/f follicles averaged 40.20 µm (p = 0.0018), clusters in pxtEY/EY follicles averaged 46.13 µm (p = 0.0012), and clusters in pxtf/EY follicles averaged 44.25 µm (p = 0.0004). These data reveal that Pxt regulates the morphology of the border cell cluster and suggest that these shape defects may impair migration. Furthermore, this change in cluster morphology likely contributes to the increased number of unattached cells observed at S10A in pxt mutants (Supplemental Figure S1A).
FIGURE 3:
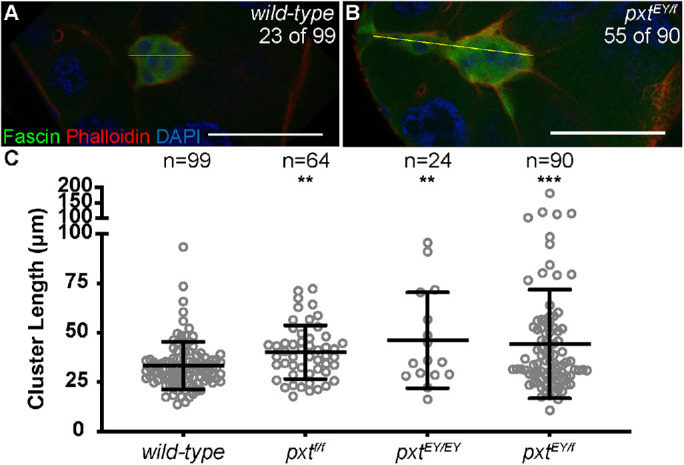
Prostaglandins regulate border cell cluster morphology. (A,B) Maximum projection of three confocal slices of S9 follicles of the indicated genotypes; anterior is to the left. (A) wild type (yw). (B) pxtEY/f. Merged images: Fascin, green; phalloidin (F-actin), red; and DAPI (DNA), blue. The frequency of S9 follicles exhibiting rearward elongated border cell clusters is indicated at the top right of the panels. The yellow lines in A and B each indicate a representative measurement of the assessment in C. (C) Graph of the quantification of primary cluster length for the indicated genotypes; note that cells left behind and fully detached from the cluster were not included in the measurements. Each circle represents a single border cell cluster; n = number of follicles. Each line indicates the average and the whiskers indicate the SD. ****, p < 0.0001; **, p < 0.01 (two-tailed unpaired t test). While wild-type follicles exhibit a round border cell cluster morphology (A, C), loss of Pxt results in significantly elongated clusters during S9 (B, C). Scale bars = 50 μm.
Prostaglandin signaling is necessary in the somatic cells for on-time border cell migration
Having found that Pxt is required for border cell migration during S9, we next sought to determine where Pxt activity is necessary. Pxt is expressed in all cells of the developing follicle (Tootle and Spradling, 2008). Thus, Pxt may function in the germline cells, the somatic cells, or both to promote proper border cell migration.
To assess where Pxt is required, we used the UAS/GAL4 system (Fischer et al., 1988) to knock down Pxt by RNAi in the somatic cells (c355 GAL4). A border cell–specific GAL4 (c306 GAL4) was not used because it failed to sufficiently knock down Pxt based on immunofluorescence analyses (unpublished data). As expected, the somatic GAL4-only control exhibits normal border cell migration (Figure 4A). RNAi knockdown of Pxt in the somatic cells results in delayed border cell migration (Figure 4B). Quantification of the migration index reveals that somatic knockdown of Pxt results in a significant delay with a migration index of 0.8428 compared with the control migration index of 1.100 (Figure 4C; p = 0.0035). This finding was verified using a second RNAi line (Supplemental Figure S3). We next assessed how somatic knockdown of Pxt affects cluster morphology. While qualitative analysis of the fixed images did not reveal striking cluster morphology defects when Pxt was knocked down (Figure 4B compared with A), quantitative analysis uncovered a surprising result. Somatic knockdown of Pxt results in a more condensed cluster, with an average length of 21.73 µm compared with 27.85 µm for the control clusters (Figure 4D; p = 0.0015). Interestingly, this phenotype does not seem to be due to either an increase in detached cells or a change in the number of cells in the cluster (Supplemental Figure S1, C and D). The second RNAi line exhibits cluster lengths similar to wild type (Supplemental Figure S3); the differences between the two RNAi lines are likely due to the knockdown efficiency. These findings reveal that somatic knockdown of Pxt is not sufficient to cause the elongated cluster morphology observed in the pxt mutant follicles. This difference may be due to insufficient loss of Pxt by RNAi knockdown and/or that Pxt in the germline modulates cluster morphology. Unfortunately, these RNAi constructs are under the control of the UASt promoter and cannot be easily expressed with germline GAL4 drivers. Additionally, available RNAi lines (TRiP) designed for germline expression fail to knock down Pxt as assessed by immunofluorescence (unpublished data). Further, our attempts to express the validated UASt RNAi lines using the method of combining a germline GAL4 driver with reduced Hsp70 (DeLuca and Spradling, 2018) have had limited success in knocking down Pxt. We find this “weak” germline knockdown of Pxt results in on-time migration, but may alter cluster morphology, as 42% of clusters are elongated (n = 45; unpublished data). Together these data suggest Pxt is required in the somatic cells to regulate on-time border cell migration, and may act in the nurse cells to control cluster cohesion.
FIGURE 4:
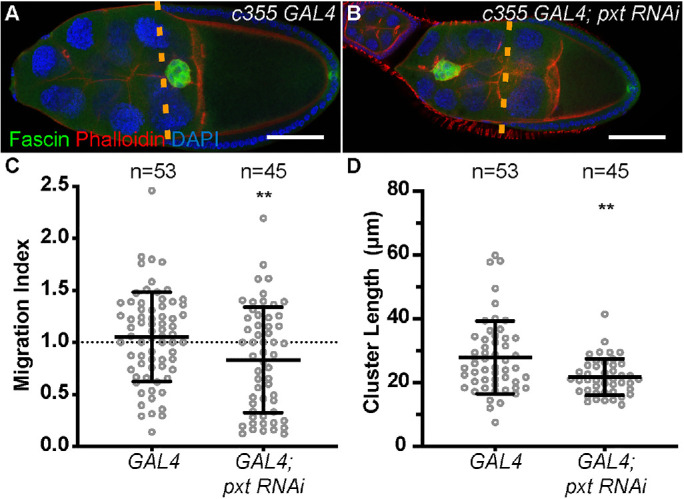
Pxt is required in the somatic cells for on-time border cell migration. (A, B) Maximum projection of three confocal slices of S9 follicles of the indicated genotypes; anterior is to the left. (A) Somatic GAL4 control (c355 GAL4/+). (B) Somatic knockdown of Pxt (c355 GAL4/+; pxt RNAi/+). Merged images: Fascin, green; phalloidin (F-actin), red; and DAPI (DNA), blue. Dashed orange lines indicate the position of the outer follicle cells. (C) Graph of the migration index quantification during S9 for the above indicated genotypes. Dotted line at 1 indicates an on-time migration. (D) Graph of the quantification of primary cluster length for the above indicated genotypes; measured as described in Figure 3. In C and D, each circle represents a single border cell cluster; n = number of follicles. Each line indicates the average and the whiskers indicate the SD. **, p < 0.01 (two-tailed unpaired t test). Somatic knockdown of Pxt (Vienna 14379) results in delayed border cell migration (B, C) and shorter cluster length (D), compared with somatic GAL4 controls (A, C, D). Scale bars = 50 μm.
Prostaglandins regulate Fascin to promote on-time border cell migration
We hypothesized that PGs may regulate the actin bundling protein Fascin to control border cell migration. This hypothesis is based on our prior finding that PGs regulate Fascin during S10B to promote actin remodeling (Groen et al., 2012), Fascin is highly expressed in the border cell cluster (Cant et al., 1994), and Fascin is required in both the border cells and the nurse cells for on-time border cell migration (Lamb et al., 2020). To test our hypothesis, we used dominant genetic interaction studies. Partial reduction of either Pxt (pxt−/+) or Fascin (fascin−/+) should not alter border cell migration. However, if PGs and fascin function together to promote border cell migration, then reduced levels of both (fascin−/+; pxt−/+) will display defects in migration. We performed immunofluorescence staining for Hu-li tai shao (Hts) and Fasciclin III (FasIII); this stain labels both the border cells and outer follicle cells and enables us to assess border cell migration in a similar manner to the Fascin stain used previously. As expected, heterozygous loss of Pxt or Fascin alone does not alter border cell migration using our migration index quantification (Figure 5C). However, partial loss of both Pxt and Fascin (fascin−/+; pxt−/+) results in significant border cell migration delays (Figure 5, A–C; average migration indices: 0.6977 for fascinsn28/+; pxtEY/+ compared with 0.9393 for pxtEY/+, p < 0.0001; and 0.3802 for fascinsn28/+; pxtf/+ compared with 1.023 for pxtf/+, p < 0.0001). Additionally, we observed altered border cell cluster morphology in follicles heterozygous for mutations in both pxt and fascin. Similar to the clusters in pxt mutant follicles, the clusters from double heterozygous follicles (fascin−/+; pxt−/+) display an elongated phenotype with posterior tails (Figure 5, D and E). Quantification reveals that the clusters from fascin−/+; pxt−/+ follicles have a significant increase in cluster length (Figure 5F; average cluster length: 39.69 µm for fascinsn28/+; pxtEY/+ compared with 28.23 µm for pxtEY/+, p = 0.0135; and 47.62 µm for fascinsn28/+; pxtf/+ compared with 27.71 µm for pxtf/+, p < 0.0001). These results indicate PGs and Fascin genetically interact to regulate both border cell migration and cluster morphology.
FIGURE 5:
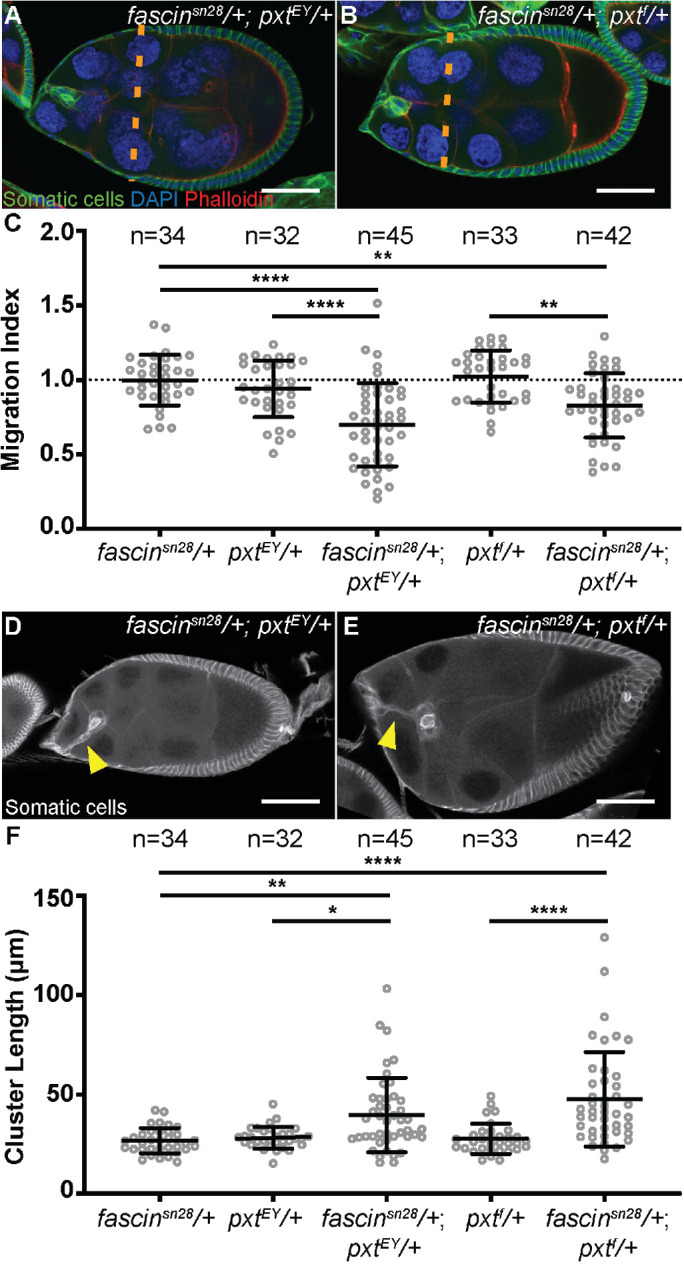
Prostaglandins regulate Fascin to promote border cell migration and control cluster morphology. (A, B) Maximum projections of two to four confocal slices of S9 follicles of the indicated genotypes; anterior is to the left. (A) fascinsn28/+; pxtEY/+. (B) fascinsn28/+; pxtf/+. Merged images: somatic cell stain (Hts and FasIII), green; DAPI (DNA), blue; and phalloidin (F-actin), red. Orange dashed lines indicate the position of the outer follicle cells. (C) Graph of the migration index quantification during S9 for the indicated genotypes. Dotted line at 1 indicates an on-time migration. Each circle represents a single border cell cluster; n = number of follicles. Each line indicates the average and the whiskers indicate the SD. ****, p < 0.0001; **, p < 0.01 (one-way ANOVA with Tukey’s multiple comparison test). (D, E) Maximum projections of two to four confocal slices of S9 follicles of the indicated genotypes stained with the somatic cell stain (Hts and FasIII, white). (D) fascinsn28/+; pxtEY/+. (E) fascinsn28/+; pxtf/+. Yellow arrowheads denote tails attached to the cluster. (F) Graph of the quantification of border cluster length from follicles of the indicated genotypes. Each circle represents a single border cell cluster; n = number of follicles. Each line indicates the average and the whiskers indicate the SD. ****, p < 0.0001; **, p < 0.01; and *, p < 0.05 (one-way ANOVA with Tukey’s multiple comparison test). While heterozygosity for mutations in pxt or fascin do not affect border cell migration or cluster morphology, double heterozygotes (fascin−/+; pxt−/+) exhibit delayed border cell migration (A–C) and elongated clusters (D–F). Scale bars = 50 μm.
Prostaglandins regulate Fascin to control integrin-based cellular adhesions
We next wanted to determine how Pxt and Fascin regulate border cell migration. One way by which border cell migration and cluster morphology are regulated is through cellular adhesions. One adhesion factor involved is E-cadherin (Niewiadomska et al., 1999; De Graeve et al., 2012; Cai et al., 2014). Both increased and decreased E-cadherin levels in either the border cells or the nurse cells inhibit border cell migration and loss of E-cadherin in the border cells results in elongated clusters (Niewiadomska et al., 1999; Cai et al., 2014). We find that E-cadherin localization and levels appear grossly normal in pxt mutants (Supplemental Figure S4), indicating altered E-cadherin adhesions are not causing the cluster morphology defects. Interestingly, we previously reported that fascin mutant follicles display increased E-cadherin at both the border cell and nurse cell membranes (Lamb et al., 2020). Thus, we hypothesize that Fascin’s function in regulating E-cadherin localization or expression is independent of PG signaling.
Another adhesion type that regulates border cell migration and morphology is integrin-based adhesions (Dinkins et al., 2008; Llense and Martin-Blanco, 2008; Assaker et al., 2010). Integrin receptors are composed of one alpha and one beta subtype. In the border cells, βPS-integrin (Drosophila myospheroid; Mys) and αPS3-integrin (Drosophila Scab; Scb) are enriched on the border cell membranes; RNAi knockdown of either results in delayed border cell migration during S9 and, in combination with reduced JNK signaling, elongated clusters (Dinkins et al., 2008; Llense and Martin-Blanco, 2008). While Pxt does not genetically interact with either integrin subunit (Supplemental Figure S5) and loss of Pxt does not alter JNK signaling (Supplemental Figure S6), we find that βPS-integrin localization is strikingly altered in the border cell clusters, but not in the outer follicle cells (unpublished data), of both pxt and fascin mutants. To account for potential staining variability, we stained wild-type and pxt or fascin mutant follicles for βPS-integrin in the same tube using Pxt or Fascin antibody staining, respectively, to differentiate the genotypes. In wild-type follicles, βPS-integrin exhibits strong and continuous stretches of membrane localization on the border cell cluster (Figure 6A). Conversely, loss of Pxt or Fascin results in reduced enrichment of βPS-integrin at the border cell membrane and increased cytoplasmic integrin (Figure 6, B and C). To quantify the differences in integrin localization, we measured integrin intensity along the border cell membranes and normalized it to the phalloidin intensity at the same location. We observed that pxt and fascin mutants display a significant decrease in integrin membrane intensity compared with the wild-type controls (Figure 6G). This data suggests that PGs and Fascin are needed for proper membrane enrichment of integrins on the border cell cluster.
FIGURE 6:
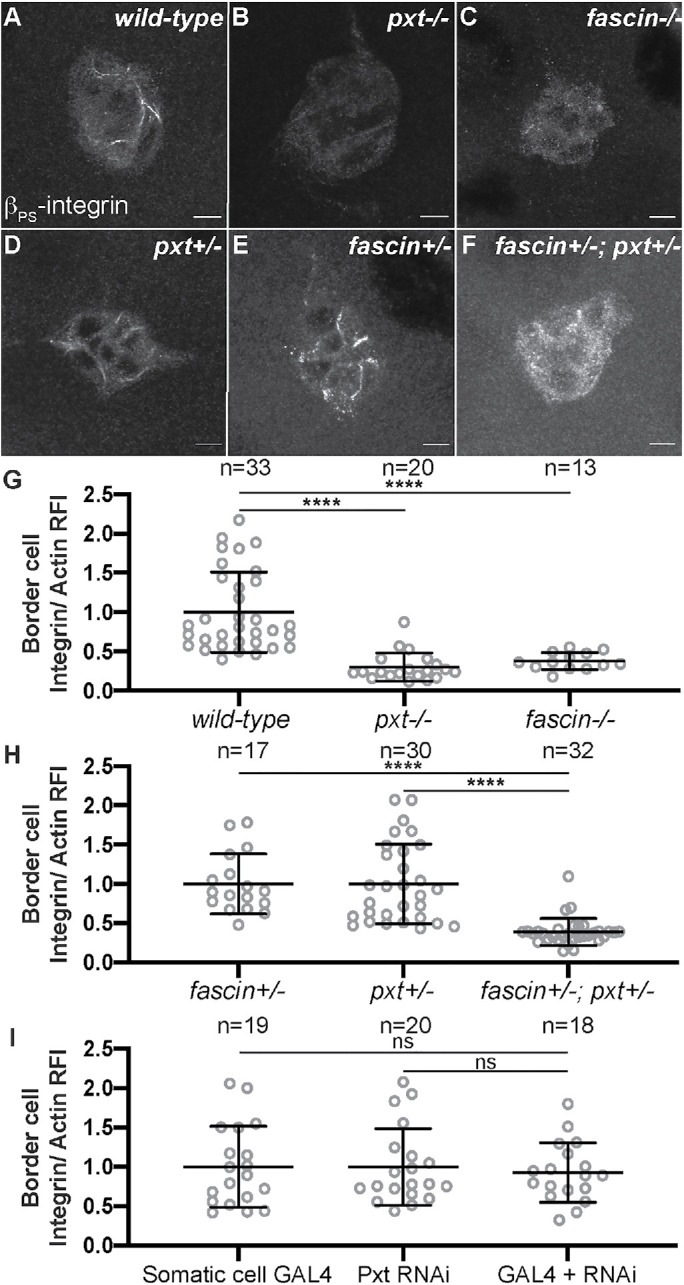
Prostaglandins and Fascin regulate integrin localization on the border cell cluster. (A-F) Maximum projection of three confocal slices of S9 border cell clusters of the indicated genotypes stained with βPS-integrin (white). (A) wild type (yw). (B) pxt−/− (pxtf/f). (C) fascin−/− (fascinsn28/sn28). (D) pxt+/− (pxtf/+). (E) fascin+/− (fascinsn28/+). (F) fascin+/−; pxt+/− (fascinsn28/+; pxtf/+). (G–I) Graphs of the quantification of βPS-integrin intensity along the border cell membranes of the indicated genotypes. Peak βPS-integrin intensity was quantified and normalized to phalloidin staining, three measurements were taken per cluster and averaged, and then normalized to the appropriate control. In G, pxt and fascin mutants were normalized to wild-type controls. In H, fascin+/−; pxt+/− mutants were normalized to the pxt+/− controls. In I, somatic Pxt RNAi knockdown (c355 GAL4/+; pxt RNAi/+) was normalized to the RNAi-only control (pxt RNAi/+). Each circle represents a single border cell cluster; n = number of follicles. Each line indicates the average and the whiskers indicate the SD. ****, p < 0.0001; ns indicates p > 0.05. (one-way ANOVA with Tukey’s multiple comparison test). pxt and fascin mutants show reduced βPS-integrin membrane localization compared with wild-type clusters (A–C, G). Pxt and Fascin genetically interact to regulate βPS-integrin membrane localization (D–F, H). Somatic Pxt knockdown does not alter βPS-integrin membrane localization (I). Scale bars = 10 μm.
We next asked whether PGs and Fascin genetically interact to regulate integrins on the border cell cluster. We assessed integrin localization, using the βPS-integrin antibody, in follicles heterozygous for mutations in pxt (pxt−/+) or fascin (fascin−/+) or heterozygous for both mutations (fascin−/+; pxt−/+). Partial loss of Fascin (fascin−/+) or Pxt (pxt−/+) exhibits normal enrichment of βPS-integrin along the border cell membranes (Figure 6, D and E, compared with A), whereas coreduction of Fascin and Pxt (fascin−/+; pxt−/+) results in decreased βPS-integrin localization to the border cell membranes (Figure 6F compared with B and C). Further, using the quantification method described above, coreduction of both Fascin and Pxt (fascin−/+; pxt−/+) results in a significant decrease in integrin membrane intensity compared with the single heterozygous controls (Figure 6H). These results indicate that PGs and Fascin act in the same pathway to control integrin-based adhesions on the border cell cluster.
To determine whether PGs produced within the somatic or germ cells regulate the integrin-based adhesions on the border cells, we assessed βPS-integrin localization when Pxt is knocked down in the somatic cells. We find somatic RNAi knockdown of Pxt does not affect the membrane enrichment of integrins on the border cells (Figure 6I). This finding indicates PG signaling in the border cell cluster does not regulate integrin-based adhesions, and supports the model that PG signaling in the germline regulates these adhesions to control cluster morphology.
DISCUSSION
Drosophila border cell migration has been widely used to identify factors regulating invasive, collective cell migration (Montell, 2003; Montell et al., 2012). Here we find that PG signaling is required for on-time border cell migration and normal cluster morphology. Specifically, loss of the COX-like enzyme Pxt results in both delayed border cell migration during S9 and aberrant, elongated clusters with cells occasionally being left behind along the migration path.
These border cell defects vary in severity across the two pxt alleles: pxtf and pxtEY. Both alleles result in delayed migration and increased cluster length during S9 (Figures 2 and 3). During S10, the stronger allele, pxtf, results in elongated clusters with too many cells (9.1 compared with 5.2; p < 0.0001), while the weaker allele, pxtEY, has a lower frequency of elongated clusters and only a slight increase in cell number (Figure 1 and Supplemental Figure S1; 6.1 border cells, p = 0.0011). Transalleles of pxtEY/f exhibit an intermediate phenotype. These data suggest that the phenotypic variation is primarily due to the level of Pxt loss.
The defects observed in pxt mutants are only partially recapitulated by RNAi knockdown of Pxt in the somatic cells. Somatic knockdown of Pxt causes delayed border cell migration during S9. However, the border cell clusters have the normal number of cells (Supplemental Figure S1) and are not elongated (Figure 4 and Supplemental Figure S3). Further, somatic knockdown with one Pxt RNAi line results in clusters with shorter lengths (Figure 4). The phenotypic differences between the pxt mutants and RNAi knockdown may be due to the RNAi failing to reduce Pxt sufficiently. However, if this were the case, the cluster length should be normal and not shorter. Additionally, weak germline knockdown results in on-time migration, but the clusters are elongated. It is unclear whether germline knockdown alters border cell number. However, as border cell number is regulated by JAK/STAT signaling within the border cells (Silver and Montell, 2001; Beccari et al., 2002; Silver et al., 2005), Pxt activity within the border cells, the nurse cells, or both may impinge upon this signaling cascade to control border cell number. Together these data lead us to hypothesize Pxt acts in both the somatic and germ cells to regulate border cell migration. Specifically, Pxt may function within the somatic cells, likely within the border cells themselves, to mediate on-time border cell migration, whereas Pxt may function within the germ cells, likely within the nurse cells on which the border cells migrate, to control cluster morphology.
Regulation of cell migration by PGs within both the migrating cells and their microenvironment is conserved across organisms. Cancer cells up-regulate COX enzyme expression and exhibit increased PG production (Cha and DuBois, 2007; Wang and Dubois, 2010; Menter and Dubois, 2012). Specifically, increased PG signaling is associated with increased in vitro cell migration and invasion that can be blocked by COX inhibitor treatment (Tsujii et al., 1997; Chen et al., 2001; Lyons et al., 2011). Increased PG production within the tumor cells is also associated with high levels of in vivo metastasis and poor patient outcomes (Rolland et al., 1980; Khuri et al., 2001; Gallo et al., 2002; Denkert et al., 2003). These data support a role for PGs within the migrating cells themselves. PGs also play a role in the tumor microenvironment, contributing to chronic inflammation and immune modulation (Wang and DuBois, 2018). Additionally, in a key study, Li et al. 2012 uncovered that PG signaling within the mesenchymal stroma cells plays critical roles in regulating the fate of the carcinoma cells and promoting the cells to undergo an epithelial to mesenchymal transition and invade the surrounding tissue (Li et al., 2012). Thus, it is critical to define how PGs act both within the migrating cells and their environment to control invasive migration.
One mechanism by which PGs may promote border cell migration is by regulating actin remodeling. Indeed, PG signaling promotes actin remodeling necessary for late-stage Drosophila follicle morphogenesis by controlling a number of actin-binding proteins, including the actin bundler Fascin (Groen et al., 2012; Spracklen et al., 2014; Spracklen et al., 2019). Fascin is highly up-regulated in the border cells (Cant et al., 1994) and Fascin is required in both the border cells and the nurse cells for on-time border cell migration during S9 (Lamb et al., 2020). These findings led us to hypothesize PG signaling in the border cells may regulate Fascin and thereby, the actin cytoskeleton. Supporting this hypothesis, dominant genetic interaction studies reveal that S9 follicles from double heterozygotes, fascin−/+; pxt−/+, phenocopy pxt mutants. Specifically, border cell migration is delayed during S9 and the clusters are elongated (Figure 5). These data indicate that PGs regulate Fascin to control both border cell migration and cluster morphology.
As somatic knockdown of Pxt results in delayed migration, we hypothesize PGs regulate the functions of Fascin within the border cells to promote migration. We recently found that Fascin controls protrusions during border cell migration (Lamb et al., 2020). Loss of Fascin results in protrusions extending from all sides of the cluster, and these protrusions are shorter in length and duration. Further, genetic evidence leads to the model that Fascin regulates protrusion dynamics by controlling the processivity of the actin elongation factor Enabled. As PGs regulate Enabled in the nurse cells (Spracklen et al., 2014, 2019), we speculate that PGs regulate the functions of Fascin to promote Enabled-dependent actin elongation required for generating the protrusions necessary for on-time migration.
PGs may also regulate Fascin to control border cell adhesions within the cluster, between the border cells and the nurse cells, or both. E-cadherin membrane localization and levels appear normal in pxt mutants (Supplemental Figure S4), and are increased in fascin mutants (Lamb et al., 2020). These data suggest that the PG pathway regulating Fascin to control migration and cluster morphology does not act through modulating E-cadherin–based adhesions. Instead, our data suggest that one downstream effector of PGs and Fascin is integrins. Loss of Pxt or Fascin alone results in decreased membrane enrichment of βPS-integrin on the border cells (Figure 6). Further, the same phenotype is observed in fascin−/+; pxt−/+ follicles (Figure 6). These data lead to the model that PGs regulate Fascin to control integrin-based adhesions on the border cells.
Integrins contribute to both border cell migration and cluster morphology. RNAi knockdown of βPS-integrin or αPS3-integrin results in delayed border cell migration during S9 (Dinkins et al., 2008). Additionally, in combination with altered JNK signaling, integrins are required to maintain tight cluster cohesion (Dinkins et al., 2008; Llense and Martin-Blanco, 2008). To determine which of these functions is downstream from the PG signaling pathway, we assessed integrin localization when Pxt was knocked down in the somatic cells. In this context, the membrane enrichment of βPS-integrin was normal (Figure 6). This finding, in conjunction with the data that somatic Pxt RNAi knockdown results in delayed migration and more compact border cell clusters (Figure 4), leads to the idea that PGs regulate Fascin in the germline to control integrin-based adhesions on the border cells, and this contributes to maintaining proper cluster cohesion.
PG signaling in the nurse cells could regulate integrin adhesions on the border cells by a number of mechanisms. For example, this pathway could signal to the border cells to affect integrin expression or trafficking to the cell surface. If either of these were the case, then the dominant genetic interactions between mutations in the integrin subunits and pxt would be expected to cause border cell migration defects. Instead, we find that migration is normal (Supplemental Figure S5). Additionally, our prior microarray analysis indicates PGs do not alter integrin expression during Drosophila oogenesis (Tootle et al., 2011). Another means of affecting integrin enrichment is decreased activation and thus, decreased clustering. Integrins are activated by binding to extracellular matrix (ECM) components (termed outside-in signaling) or by intracellular changes in the actin cytoskeleton connecting to the intracellular domains of the integrins (termed inside-out signaling; Harburger and Calderwood, 2009; Vicente-Manzanares et al., 2009). As there is little evidence of ECM surrounding the border cells or contributing to their migration (Medioni and Noselli, 2005), integrins on the border cells are likely activated by inside-out signaling. Both activation and clustering of integrins require interaction with the actin cytoskeleton and adaptor proteins, including Paxillin (Harburger and Calderwood, 2009; Vicente-Manzanares et al., 2009). Notably, microarray analysis of pxt mutant follicles revealed that paxillin is down-regulated (Tootle et al., 2011). Within the border cells, Paxillin expression is known to be regulated by JNK signaling (Llense and Martin-Blanco, 2008); however, our data indicates PGs do not act through the JNK pathway to regulate border cell migration and cluster morphology (Supplemental Figure S6). Further, in mammalian systems, both PGs (Mayoral et al., 2005; Bai et al., 2009, 2013; Liu et al., 2010) and Fascin (Anilkumar et al., 2003; Villari et al., 2015) mediate increased integrin adhesion stability. These data lead us to speculate that the decreased membrane enrichment of integrins in both the fascin and pxt mutants and double heterozygotes (fascin−/+; pxt−/+) is due to a loss of inside-out activation of the integrins.
Another means by which integrins may be activated is that PG signaling may alter the rigidity of the nurse cells. Integrin receptors sense and are activated by substrate stiffness (Sun et al., 2016; Kechagia et al., 2019). Further, the balance of force between the border cell cluster and the nurse cells must be maintained in order for normal cluster morphology, and misbalanced forces in either tissue lead to border cell migration delays and cluster elongation (Majumder et al., 2012; Aranjuez et al., 2016; Cai et al., 2016). Supporting a role for PGs in regulating nurse cell stiffness, during S10B, pxt mutants exhibit altered nurse cell levels and localization of active nonmuscle myosin II (Spracklen et al., 2019), a known regulator of nurse cell stiffness (Aranjuez et al., 2016). Myosin activity is also regulated by Fascin in other systems (Elkhatib et al., 2014). Additionally, PGs regulate Fascin to control nurse cell cortical actin integrity (Tootle and Spradling, 2008; Groen et al., 2012), which could contribute to the rigidity of the environment. Together these data lead us to speculate that within the nurse cells PGs regulate the functions of Fascin to maintain proper cortical actin and substrate stiffness, and in the absence of Pxt, the nurse cells are softer, resulting in reduced integrin-based adhesions on the border cells, which promotes the changes in cluster morphology.
In conclusion, this study leads to the model that PG signaling regulates Fascin in the somatic cells, including the border cells, to promote on-time collective migration, and in the nurse cells to control cluster morphology. Contributing to the latter is the regulation of integrin-based adhesions on the border cells. Thus, border cell migration provides a robust, in vivo system to delineate the downstream mechanisms by which PGs act within both the migratory cells and their substrate to regulate collective, invasive cell migration. These same mechanisms may contribute to cancer metastasis as both PGs (Rolland et al., 1980; Khuri et al., 2001; Gallo et al., 2002; Denkert et al., 2003) and Fascin (Hashimoto et al., 2004; Yoder et al., 2005; Okada et al., 2007; Li et al., 2008; Chan et al., 2010) are associated with highly aggressive cancers and poor patient outcomes.
MATERIALS AND METHODS
Fly stocks
Fly stocks were maintained on cornmeal/agar/yeast food at 21°C, except where noted. Before immunofluorescence, flies were fed wet yeast paste daily for 2–4 d. yw was used as the wild-type control. The following stocks were obtained from the Bloomington Drosophila Stock Center (Bloomington, IN): pxtEY03052 (BL15620), c355 GAL4 (BL3750), Df(3R)Hsp70A, Df(3R)Hsp70B (BL8841), mys10 (BL58806), and scb01288 (BL11035). The pxtf01000 stock was obtained from the Harvard Exelixis Collection. The UASt pxt RNAi (V14379; targets the transcript at 331–720 base pairs) and UASt pxt RNAi 2 (V104446; targets the transcript at 413–1064 base pairs) stocks were obtained from the Vienna Drosophila Resource Center. The fascinsn28 line was a generous gift form Jennifer Zanet (Université de Toulouse, Toulouse, France; Zanet et al., 2012), and the oskar GAL4 line (second chromosome) was a generous gift from Anne Ephrussi (European Molecular Biology Laboratory, Heidelber, Germany; Telley et al., 2012). Germline knockdown was achieved by crossing osk GAL4; Df(3R)Hsp70A, Df(3R)Hsp70B to the RNAi lines. Expression of the UAS pxt RNAi lines was achieved by crossing to the GAL4 line at room temperature and maintaining the adult progeny at 29°C for 3–5 d.
Immunofluorescence
Whole-mount Drosophila ovary samples were dissected into Grace’s insect medium (Lonza; Walkersville, MD or Thermo Fischer Scientific, Waltham, MA) and fixed for 10 min at room temperature in 4% paraformaldehyde in Grace’s insect medium. Briefly, samples were blocked by washing in antibody wash (1× phosphate-buffered saline [PBS], 0.1% Triton X-100, and 0.1% bovine serum albumin) six times for 10 min each at room temperature. Primary antibodies were incubated overnight at 4°C, except for βPS-integrin, which was incubated for ∼20–48 h at 4°C. The following primary antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the Department of Biology, University of Iowa (Iowa City, IA): mouse anti-Fascin 1:25 (sn7c; Cant et al., 1994); mouse anti–βPS-integrin 1:10 (CF.6G11; Brower et al., 1984); mouse anti-EYA 1:100 (eya10H6; Boyle et al., 1997); mouse anti–β-catenin (N2 7A1; Drosophila Armadillo) 1:100 (Riggleman et al., 1990); rat anti-DCAD2 1:10 (Oda et al., 1994); mouse anti-Hts 1:50 (1B1; Zaccai and Lipshitz, 1996); and mouse anti-FasIII 1:50 (7G10; Patel et al., 1987). Additionally, the following primary antibodies were used: mouse anti-pJun KM-1 (SC-822, Santa Cruz, Dallas, TX; Felix et al., 2015) and rabbit anti-Pxt 1:10000 (preabsorbed on pxtf/f ovaries at 1:20 and used at 1:500; Spracklen et al., 2014). After six washes in Triton antibody wash (10 min each), secondary antibodies were incubated overnight at 4°C or for ∼4 h at room temperature. The following secondary antibodies were used at 1:500–1:1000: AF488::goat anti-mouse, AF568::goat anti-mouse, AF647::goat anti-mouse, AF488::goat anti-rabbit, AF647::donkey anti-rabbit, AF488::donkey anti-rat, and AF633::goat anti-rabbit (Thermo Fischer Scientific). Alexa Fluor 647–, rhodamine- or Alexa Fluor 488–conjugated phalloidin (Thermo Fischer Scientific) was included with secondary antibodies at a concentration of 1:100–1:250. After six washes in antibody wash (10 min each), 4′,6-diamidino-2-phenylindole (DAPI; 5 mg/ml) staining was performed at a concentration of 1:5000 in 1× PBS for 10 min at room temperature. Ovaries were mounted in 1 mg/ml phenylenediamine in 50% glycerol, pH 9 (Platt and Michael, 1983). All experiments were performed a minimum of three independent times, except where noted in the figure legends.
Image acquisition and processing
Microscope images of fixed Drosophila follicles were obtained using LAS AF SPE Core software on a Leica TCS SPE mounted on a Leica DM2500 using an ACS APO 20×/0.60 IMM CORR -/D (Leica Microsystems, Buffalo Grove, IL), Zen software on a Zeiss 880 mounted on Zeiss Axio Observer.Z1 using Plan-Apochromat 20×/0.8 working distance (WD) = 0.55 M27, Plan-Apochromat 40×/1.3 oil Differential Interference Contrast (DIC) WD = 2.0 or Plan-Apochromat 63×/1.4 oil DIC f/ELYRA objectives, or Zen software on a Zeiss 700 LSM mounted on an Axio Observer.Z1 using a LD C-APO 40×/1.1 W/0 objective (Carl Zeiss Microscopy, Thornwood, NY). Maximum projections (two to five confocal slices), merged images, rotation, cropping, and distance measurements were performed using ImageJ software (Abramoff et al., 2004).
Image analyses
All image quantification was performed in a genotypically blinded manner, and where noted, in a double blinded manner, and was performed on a minimum of three independent experiments, except where noted in the figure legends.
Analysis of S10 clusters was performed on fixed confocal stacks of S10 follicles by counting the number of Eya stained nuclei visible within the main border cell cluster and those left between the nurse cells using ImageJ software (Abramoff et al., 2004). The data was compiled, graphs generated, and statistical analysis (two-tailed unpaired t test) was performed using GraphPad Prism version 7 or 8 (GraphPad Software, La Jolla, CA). In the graphs, the measurement for each follicle is represented as a circle, and the averages and standard deviations are indicated by lines and whiskers, respectively.
To assess border cell migration during S9 a number of measurements were performed on fixed confocal stacks using Image J software (Abramoff et al., 2004). Specifically, we measured the follicle length, the distance between the anterior tip of the follicle and the leading edge (posterior) of the border cell cluster (distance of the border cells), the distance between the anterior tip of the follicle and the anterior edge of the outer follicle cells (distance of the follicle cells), and the distance from the rear to the front of the border cell cluster (cluster length; detached cells were not included in the length measurement). The data was compiled, and the migration index was calculated in Microsoft Excel (Microsoft, Redmond, WA). The migration index = distance of the border cells/distance of the outer follicle cells. The migration index and cluster length data were compiled, graphs generated, and statistical analysis (two-tailed unpaired t test) was performed using GraphPad Prism version 7 or 8 (GraphPad Software). To assess migration at different points during S9, the migration index data was binned into three groups based on follicle length (200–250, 250–300, and 300–370 μm) for wild-type and all pxt−/− genetic backgrounds combined. In the graphs, the measurement for each follicle is represented as a circle, and the averages and standard deviations are indicated by lines and whiskers, respectively. To verify that any migration indices changes were due to altered border cell migration and not altered outer follicle cell position, the follicle length versus the distance of the outer follicle cells was plotted and analyzed in Microsoft Excel and GraphPad Prism 8.
Integrin membrane intensity was quantified using genotypically blinded confocal stacks of border cell clusters stained for both βPS-integrin and F-actin (phalloidin). Three curved line segments were drawn along the border cell cluster membranes with the highest integrin intensity. If a cluster did not have any significant integrin membrane localization, the lines were drawn on border cell membranes using the phalloidin channel. Each line was ∼5–8 μm long. The mean fluorescence intensity for both integrin and phalloidin were measured for each line. The integrin fluorescence intensity was normalized to phalloidin and then the values for the lines were averaged for each cluster. Averages were then normalized to the wild-type average for each experiment due to experimental variability. The data was compiled, graphs generated, and statistical analysis performed (one-way ANOVA with Tukey’s multiple comparison test) in GraphPad Prism version 7 or 8 (GraphPad Software).
Supplementary Material
Acknowledgments
We thank the Westside Fly Group and Dunnwald lab for helpful discussions, and the Tootle lab for helpful discussions and careful review of the manuscript. Stocks obtained from the Bloomington Drosophila Stock Center (National Institutes of Health [NIH] P40OD-018537) were used in this study. Resources provided by FlyBase (NIH U41HG-000739 and the British Medical Research Council) were used for this study. Information Technology Services–Research Services provides data storage support. This project is supported by NIH R01GM-116885. E.F.F. has been supported by the NIH Predoctoral Training Grant in Genetics T32GM008629 (PI Daniel Eberl), the University of Iowa Graduate College Post-Comps Fellowship, and Ballard and Seashore Fellowship. M.C.L. has been supported by the University of Iowa Summer Graduate Fellowship and the Anatomy and Cell Biology Department Graduate Fellowship. S.Q.M. is supported by the NIH Predoctoral Training Grant in Genetics T32GM008629 (PI Daniel Eberl).
Abbreviations used:
- COX
cyclooxygenase
- ECM
extracellular matrix
- Eya
Eyes absent
- F-actin
filamentous actin
- FasIII
Fasciclin III
- Hts
Hu-li tai shao
- PG
prostaglandin
- RFI
relative fluorescence intensity
- S9, 10, 10A
specific stages of oogenesis.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-10-0578) on May 20, 2020.
REFERENCES
- Abramoff MD, Magalhaes PJ, Ram SJ. (2004). Image processing with ImageJ. Biophotonics Int , 36–42. [Google Scholar]
- Alexander S, Friedl P. (2012). Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. Trends Mol Med , 13–26. [DOI] [PubMed] [Google Scholar]
- Anilkumar N, Parsons M, Monk R, Ng T, Adams JC. (2003). Interaction of fascin and protein kinase Cα: a novel intersection in cell adhesion and motility. EMBO J , 5390–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranjuez G, Burtscher A, Sawant K, Majumder P, McDonald JA. (2016). Dynamic myosin activation promotes collective morphology and migration by locally balancing oppositional forces from surrounding tissue. Mol Biol Cell , 1898–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaker G, Ramel D, Wculek SK, Gonzalez-Gaitan M, Emery G. (2010). Spatial restriction of receptor tyrosine kinase activity through a polarized endocytic cycle controls border cell migration. Proc Natl Acad Sci USA , 22558–22563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Wang J, Zhang L, Ma J, Zhang H, Xia S, Zhang M, Ma X, Guo Y, Rong R, et al (2013). Prostaglandin E(2) receptor EP1-mediated phosphorylation of focal adhesion kinase enhances cell adhesion and migration in hepatocellular carcinoma cells. Int J Oncol , 1833–1841. [DOI] [PubMed] [Google Scholar]
- Bai XM, Zhang W, Liu NB, Jiang H, Lou KX, Peng T, Ma J, Zhang L, Zhang H, Leng J. (2009). Focal adhesion kinase: important to prostaglandin E2-mediated adhesion, migration and invasion in hepatocellular carcinoma cells. Oncol Rep , 129–136. [PubMed] [Google Scholar]
- Beccari S, Teixeira L, Rorth P. (2002). The JAK/STAT pathway is required for border cell migration during Drosophila oogenesis. Mech Dev , 115–123. [DOI] [PubMed] [Google Scholar]
- Bianco A, Poukkula M, Cliffe A, Mathieu J, Luque CM, Fulga TA, Rorth P. (2007). Two distinct modes of guidance signalling during collective migration of border cells. Nature , 362–365. [DOI] [PubMed] [Google Scholar]
- Boyle M, Bonini N, DiNardo S. (1997). Expression and function of clift in the development of somatic gonadal precursors within the Drosophila mesoderm. Development , 971–982. [DOI] [PubMed] [Google Scholar]
- Brower DL, Wilcox M, Piovant M, Smith RJ, Reger LA. (1984). Related cell-surface antigens expressed with positional specificity in Drosophila imaginal discs. Proc Natl Acad Sci USA , 7485–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Chen SC, Prasad M, He L, Wang X, Choesmel-Cadamuro V, Sawyer JK, Danuser G, Montell DJ. (2014). Mechanical feedback through E-cadherin promotes direction sensing during collective cell migration. Cell , 1146–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Dai W, Prasad M, Luo J, Gov NS, Montell DJ. (2016). Modeling and analysis of collective cell migration in an in vivo three-dimensional environment. Proc Natl Acad Sci USA , E2134–E2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cant K, Knowles BA, Mooseker MS, Cooley L. (1994). Drosophila singed, a fascin homolog, is required for actin bundle formation during oogenesis and bristle extension. J Cell Biol , 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha YI, DuBois RN. (2007). NSAIDs and cancer prevention: targets downstream of COX-2. Annu Rev Med , 239–252. [DOI] [PubMed] [Google Scholar]
- Cha YI, Kim SH, Sepich D, Buchanan FG, Solnica-Krezel L, DuBois RN. (2006). Cyclooxygenase-1-derived PGE2 promotes cell motility via the G-protein-coupled EP4 receptor during vertebrate gastrulation. Genes Dev , 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha YI, Kim SH, Solnica-Krezel L, Dubois RN. (2005). Cyclooxygenase-1 signaling is required for vascular tube formation during development. Dev Biol , 274–283. [DOI] [PubMed] [Google Scholar]
- Chan C, Jankova L, Fung CL, Clarke C, Robertson G, Chapuis PH, Bokey L, Lin BP, Dent OF, Clarke S. (2010). Fascin expression predicts survival after potentially curative resection of node-positive colon cancer. Am J Surg Pathol , 656–666. [DOI] [PubMed] [Google Scholar]
- Chen WS, Wei SJ, Liu JM, Hsiao M, Kou-Lin J, Yang WK. (2001). Tumor invasiveness and liver metastasis of colon cancer cells correlated with cyclooxygenase-2 (COX-2) expression and inhibited by a COX-2-selective inhibitor, etodolac. Int J Cancer , 894–899. [DOI] [PubMed] [Google Scholar]
- De Graeve FM, Van de Bor V, Ghiglione C, Cerezo D, Jouandin P, Ueda R, Shashidhara LS, Noselli S. (2012). Drosophilaapc regulates delamination of invasive epithelial clusters. Dev Biol , 76–85. [DOI] [PubMed] [Google Scholar]
- DeLuca SZ, Spradling AC. (2018). Efficient expression of genes in the Drosophila germline using a UAS promoter free of interference by Hsp70 piRNAs. Genetics , 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denkert C, Winzer KJ, Muller BM, Weichert W, Pest S, Kobel M, Kristiansen G, Reles A, Siegert A, Guski H, Hauptmann S. (2003). Elevated expression of cyclooxygenase-2 is a negative prognostic factor for disease free survival and overall survival in patients with breast carcinoma. Cancer , 2978–2987. [DOI] [PubMed] [Google Scholar]
- De Pascalis C, Etienne-Manneville S. (2017). Single and collective cell migration: the mechanics of adhesions. Mol Biol Cell , 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digiacomo G, Ziche M, Dello Sbarba P, Donnini S, Rovida E. (2015). Prostaglandin E2 transactivates the colony-stimulating factor-1 receptor and synergizes with colony-stimulating factor-1 in the induction of macrophage migration via the mitogen-activated protein kinase ERK1/2. FASEB J , 2545–2554. [DOI] [PubMed] [Google Scholar]
- Dinkins MB, Fratto VM, Lemosy EK. (2008). Integrin alpha chains exhibit distinct temporal and spatial localization patterns in epithelial cells of the Drosophila ovary. Dev Dyn , 3927–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkhatib N, Neu MB, Zensen C, Schmoller KM, Louvard D, Bausch AR, Betz T, Vignjevic DM. (2014). Fascin plays a role in stress fiber organization and focal adhesion disassembly. Curr Biol , 1492–1499. [DOI] [PubMed] [Google Scholar]
- Felix M, Chayengia M, Ghosh R, Sharma A, Prasad M. (2015). Pak3 regulates apical-basal polarity in migrating border cells during Drosophila oogenesis. Development , 3692–3703. [DOI] [PubMed] [Google Scholar]
- Fischer JA, Giniger E, Maniatis T, Ptashne M. (1988). GAL4 activates transcription in Drosophila. Nature , 853–856. [DOI] [PubMed] [Google Scholar]
- Friedl P, Gilmour D. (2009). Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol , 445–457. [DOI] [PubMed] [Google Scholar]
- Friedl P, Mayor R. (2017). Tuning collective cell migration by cell-cell junction regulation. Cold Spring Harb Perspect Biol , a029199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo O, Masini E, Bianchi B, Bruschini L, Paglierani M, Franchi A. (2002). Prognostic significance of cyclooxygenase-2 pathway and angiogenesis in head and neck squamous cell carcinoma. Hum Pathol , 708–714. [DOI] [PubMed] [Google Scholar]
- Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. (2009). Localized and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol , 1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA. (2010). Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature , 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen CM, Spracklen AJ, Fagan TN, Tootle TL. (2012). Drosophila Fascin is a novel downstream target of prostaglandin signaling during actin remodeling. Mol Biol Cell , 4567–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harburger DS, Calderwood DA. (2009). Integrin signalling at a glance. J Cell Sci , 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Shimada Y, Kawamura J, Yamasaki S, Imamura M. (2004). The prognostic relevance of fascin expression in human gastric carcinoma. Oncology , 262–270. [DOI] [PubMed] [Google Scholar]
- Hoggatt J, Singh P, Sampath J, Pelus LM. (2009). Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood , 5444–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kechagia JZ, Ivaska J, Roca-Cusachs P. (2019). Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol , 457–473. [DOI] [PubMed] [Google Scholar]
- Khalil AA, Ilina O, Gritsenko PG, Bult P, Span PN, Friedl P. (2017). Collective invasion in ductal and lobular breast cancer associates with distant metastasis. Clin Exp Metastasis , 421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuri FR, Wu H, Lee JJ, Kemp BL, Lotan R, Lippman SM, Feng L, Hong WK, Xu XC. (2001). Cyclooxygenase-2 overexpression is a marker of poor prognosis in stage I non-small cell lung cancer. Clin Cancer Res , 861–867. [PubMed] [Google Scholar]
- Kobayashi K, Omori K, Murata T. (2018). Role of prostaglandins in tumor microenvironment. Cancer Metastasis Rev , 347–354. [DOI] [PubMed] [Google Scholar]
- Lamb MC, Anliker KK, Tootle TL. (2020). Fascin regulates protrusions and delamination to mediate invasive, collective cell migration in vivo . Dev Dyn, 10.1002/dvdy.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HJ, Reinhardt F, Herschman HR, Weinberg RA. (2012). Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov , 840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zheng H, Hara T, Takahashi H, Masuda S, Wang Z, Yang X, Guan Y, Takano Y. (2008). Aberrant expression of cortactin and fascin are effective markers for pathogenesis, invasion, metastasis and prognosis of gastric carcinomas. Int J Oncol , 69–79. [PubMed] [Google Scholar]
- Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. (2010). Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol , 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llense F, Martin-Blanco E. (2008). JNK signaling controls border cell cluster integrity and collective cell migration. Curr Biol , 538–544. [DOI] [PubMed] [Google Scholar]
- Lyons TR, O’Brien J, Borges VF, Conklin MW, Keely PJ, Eliceiri KW, Marusyk A, Tan AC, Schedin P. (2011). Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat Med , 1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder P, Aranjuez G, Amick J, McDonald JA. (2012). Par-1 controls myosin-II activity through myosin phosphatase to regulate border cell migration. Curr Biol , 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor R, Etienne-Manneville S. (2016). The front and rear of collective cell migration. Nat Rev Mol Cell Biol , 97–109. [DOI] [PubMed] [Google Scholar]
- Mayoral R, Fernandez-Martinez A, Bosca L, Martin-Sanz P. (2005). Prostaglandin E2 promotes migration adhesion in hepatocellular carcinoma cells. Carcinogenesis , 753–761. [DOI] [PubMed] [Google Scholar]
- Medioni C, Noselli S. (2005). Dynamics of the basement membrane in invasive epithelial clusters in Drosophila. Development , 3069–3077. [DOI] [PubMed] [Google Scholar]
- Menter DG, Dubois RN. (2012). Prostaglandins in cancer cell adhesion, migration, and invasion. Int J Cell Biol , 723419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell DJ. (2003). Border-cell migration: the race is on. Nat Rev Mol Cell Biol , 13–24. [DOI] [PubMed] [Google Scholar]
- Montell DJ, Rorth P, Spradling AC. (1992). slowborder cells, a locus required for a developmentally regulated cell migration during oogenesis, encodes Drosophila C/EBP. Cell , 51–62. [DOI] [PubMed] [Google Scholar]
- Montell DJ, Yoon WH, Starz-Gaiano M. (2012). Group choreography: mechanisms orchestrating the collective movement of border cells. Nat Rev Mol Cell Biol , 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomska P, Godt D, Tepass U. (1999). DE-Cadherin is required for intercellular motility during Drosophila oogenesis. J Cell Biol , 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, Weber GJ, Bowman TV, Jang IH, Grosser T, et al (2007). Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature , 1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Uemura T, Harada Y, Iwai Y, Takeichi M. (1994). A Drosophila homolog of cadherin associated with armadillo and essential for embryonic cell-cell adhesion. Dev Biol , 716–726. [DOI] [PubMed] [Google Scholar]
- Okada K, Shimura T, Asakawa K, Hashimoto S, Mochida Y, Suehiro T, Kuwano H. (2007). Fascin expression is correlated with tumor progression of extrahepatic bile duct cancer. Hepato-gastroenterology , 17–21. [PubMed] [Google Scholar]
- Pandya P, Orgaz JL, Sanz-Moreno V. (2017). Modes of invasion during tumour dissemination. Mol Oncol , 5–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NH, Snow PM, Goodman CS. (1987). Characterization and cloning of fasciclin III: a glycoprotein expressed on a subset of neurons and axon pathways in Drosophila. Cell , 975–988. [DOI] [PubMed] [Google Scholar]
- Platt J, Michael A. (1983). Retardation of fading and enhancement of intensity of immunofluorescence by p-phenylenediamine. J Histochem Cytochem , 840–842. [DOI] [PubMed] [Google Scholar]
- Prasad M, Montell DJ. (2007). Cellular and molecular mechanisms of border cell migration analyzed using time-lapse live-cell imaging. Dev Cell , 997–1005. [DOI] [PubMed] [Google Scholar]
- Riggleman B, Schedl P, Wieschaus E. (1990). Spatial expression of the Drosophila segment polarity gene armadillo is posttranscriptionally regulated by wingless. Cell , 549–560. [DOI] [PubMed] [Google Scholar]
- Rolland PH, Martin PM, Jacquemier J, Rolland AM, Toga M. (1980). Prostaglandin in human breast cancer: evidence suggesting that an elevated prostaglandin production is a marker of high metastatic potential for neoplastic cells. J Natl Cancer Inst , 1061–1070. [PubMed] [Google Scholar]
- Scarpa E, Mayor R. (2016). Collective cell migration in development. J Cell Biol , 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver DL, Geisbrecht ER, Montell DJ. (2005). Requirement for JAK/STAT signaling throughout border cell migration in Drosophila. Development , 3483–3492. [DOI] [PubMed] [Google Scholar]
- Silver DL, Montell DJ. (2001). Paracrine signaling through the JAK/STAT pathway activates invasive behavior of ovarian epithelial cells in Drosophila. Cell , 831–841. [DOI] [PubMed] [Google Scholar]
- Speirs CK, Jernigan KK, Kim SH, Cha YI, Lin F, Sepich DS, DuBois RN, Lee E, Solnica-Krezel L. (2010). Prostaglandin Gβγ signaling stimulates gastrulation movements by limiting cell adhesion through Snai1a stabilization. Development , 1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spracklen AJ, Kelpsch DJ, Chen X, Spracklen CN, Tootle TL. (2014). Prostaglandins temporally regulate cytoplasmic actin bundle formation during Drosophila oogenesis. Mol Biol Cell , 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spracklen AJ, Lamb MC, Groen CM, Tootle TL. (2019). Pharmaco-genetic screen to uncover actin regulators targeted by prostaglandins during Drosophila oogenesis. G3 , 3555–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spracklen AJ, Tootle TL. (2015). Drosophila—a model for studying prostaglandin signaling. In: Bioactive Lipid Mediators: Current Reviews and Protocols, ed. Yokomizo T., Murakami M., Springer Protocols, 181–197. [Google Scholar]
- Spradling A. (1993). Developmental genetics of oogenesis. In: The Development of Drosophila melanogaster, Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, 1–70. [Google Scholar]
- Stuelten CH, Parent CA, Montell DJ. (2018). Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat Rev Cancer , 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Guo SS, Fassler R. (2016). Integrin-mediated mechanotransduction. J Cell Biol , 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley IA, Gaspar I, Ephrussi A, Surrey T. (2012). Aster migration determines the length scale of nuclear separation in the Drosophila syncytial embryo. J Cell Biol , 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tootle TL. (2013). Genetic insights into the in vivo functions of prostaglandin signaling. Int J Biochem Cell Biol , 1629–1632. [DOI] [PubMed] [Google Scholar]
- Tootle TL, Spradling AC. (2008). Drosophila Pxt: a cyclooxygenase-like facilitator of follicle maturation. Development , 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tootle TL, Williams D, Hubb A, Frederick R, Spradling A. (2011). Drosophila eggshell production: identification of new genes and coordination by Pxt. PLoS One , e19943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujii M, Kawano S, DuBois RN. (1997). Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA , 3336–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugwuagbo KC, Maiti S, Omar A, Hunter S, Nault B, Northam C, Majumder M. (2019). Prostaglandin E2 promotes embryonic vascular development and maturation in zebrafish. Biol Open , bio039768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Choi CK, Horwitz AR. (2009). Integrins in cell migration—the actin connection. J Cell Sci , 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villari G, Jayo A, Zanet J, Fitch B, Serrels B, Frame M, Stramer BM, Goult BT, Parsons M. (2015). A direct interaction between fascin and microtubules contributes to adhesion dynamics and cell migration. J Cell Sci , 4601–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Dubois RN. (2010). Eicosanoids and cancer. Nat Rev Cancer , 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, DuBois RN. (2018). Role of prostanoids in gastrointestinal cancer. J Clin Invest , 2732–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder BJ, Tso E, Skacel M, Pettay J, Tarr S, Budd T, Tubbs RR, Adams JC, Hicks DG. (2005). The expression of fascin, an actin-bundling motility protein, correlates with hormone receptor-negative breast cancer and a more aggressive clinical course. Clin Cancer Res , 186–192. [PubMed] [Google Scholar]
- Zaccai M, Lipshitz HD. (1996). Differential distributions of two adducin-like protein isoforms in the Drosophila ovary and early embryo. Zygote , 159–166. [DOI] [PubMed] [Google Scholar]
- Zanet J, Jayo A, Plaza S, Millard T, Parsons M, Stramer B. (2012). Fascin promotes filopodia formation independent of its role in actin bundling. J Cell Biol , 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.