

Abstract
We report a family in which two brothers had an undiagnosed genetic disorder comprised of dysmorphic features, microcephaly, severe intellectual disability (non-verbal), mild anemia, and cryptorchidism. Both developed osteosarcoma. Trio exome sequencing (using blood samples from the younger brother and both parents) was performed and a nonsense NM_000489.4: c.7156C>T (p.Arg2386*) mutation in the ATRX gene was identified in the proband (hemizygous) and in the mother’s peripheral blood DNA (heterozygous). The mother is healthy, does not exhibit any clinical manifestations of ATR-X syndrome and there was no family history of cancer. The same hemizygous pathogenic variant was confirmed in the affected older brother’s skin tissue by subsequent Sanger sequencing. Chromosomal microarray studies of both brothers’ osteosarcomas revealed complex copy number alterations consistent with the clinical diagnosis of osteosarcoma. Recently, somatic mutations in the ATRX gene have been observed as recurrent alterations in both osteosarcoma and brain tumors. However, it is unclear if there is any association between osteosarcoma and germline ATRX mutations, specifically in patients with constitutional ATR-X syndrome. This is the first report of osteosarcoma diagnosed in two males with ATR-X syndrome, suggesting a potential increased risk for cancer in patients with this disorder.
Keywords: ATR-X syndrome, cancer predisposition syndrome, chromosomal microarray, intellectual disability, osteosarcoma, tumor, whole exome sequencing
1 |. INTRODUCTION
The X-linked alpha-thalassemia/intellectual disability syndrome (ATR-X syndrome, MIM: 301040 and 309580) is characterized by severe intellectual disability, mild Hemoglobin H disease, dysmorphic facies, genital anomalies, and skeletal abnormalities. The ATR-X syndrome is caused by germline mutations in the ATRX gene (Gibbons, Picketts, Villard, & Higgs, 1995; Picketts et al., 1996; Villard et al., 1996). Somatic variants in the ATRX gene have recently been observed as recurrent alterations in both osteosarcoma and brain tumors (Chen et al., 2014; Wiestler et al., 2013). To date, there have been no reported cases of osteosarcoma in patients with constitutional ATR-X syndrome so the association between the two conditions is unclear. We describe a family in which two brothers inherited a putative inactivating mutation in the ATRX gene from their unaffected mother. Both children showed phenotypic features of ATR-X syndrome and both developed osteosarcoma.
2 |. CLINICAL REPORT
The family pedigree is shown in Figure 1A. The proband (younger brother) in this family had microcephaly (−4 SD), and dysmorphic features including mild hypertelorism, downturned mouth, coarse facial features, in addition to cryptorchidism, and micropenis. His brother was also dysmorphic with coarse facial features, microcephaly, ambiguous genitalia, and coarctation of the aorta in conjunction with a bicuspid aortic valve. Both brothers had severe intellectual disability (non-verbal) and mild microcytic anemia. The mother is phenotypically normal with no clinical features of the ATR-X syndrome. A three-generation family history was queried and there was no history of cancer in the family. Both parents are of Armenian descent and consanguinity was denied.
FIGURE 1.
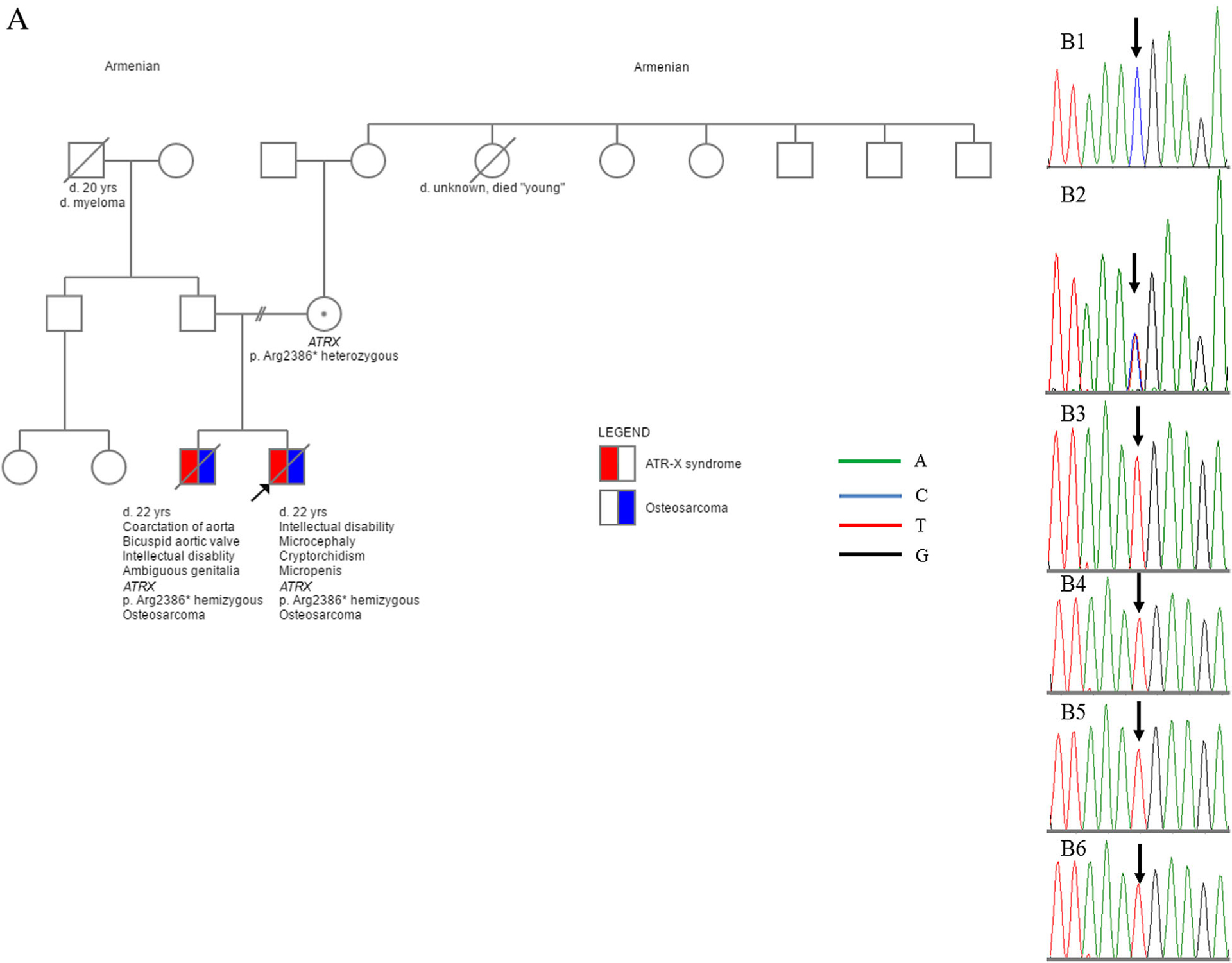
Family pedigree (A) and Sanger sequencing results on both brothers’ germline cells and osteosarcoma tissues (B). The c.7156C > T (p.Arg2386*) variant was identified/confirmed in the blood sample from the mother (B2, heterozygous) and the younger brother (B3, hemizygous), osteosarcoma tissue from the younger brother (B4, hemizygous), normal skin tissue from the older brother (B5, hemizygous), and the second osteosarcoma tissue from the older brother (B6, hemizygous). B1: Normal control sequence.
Prior genetic testing was performed over a period of 15 years, and included karyotype, constitutional chromosomal microarray and an intellectual disability gene panel without any conclusive findings.
At 17 years of age, the proband’s older brother was seen by an orthopedic surgeon when both parents noted that he was refusing to bear weight on his right leg. An X-ray revealed a destructive lesion in the right proximal tibia and a subsequent biopsy demonstrated whorls of interlacing plump spindle and stellate cells with large, hyperchromatic, pleomorphic nuclei, and abundant, often atypical mitoses. Only a few foci (<5%) contained lacy intercellular osteoid. The final diagnosis was fibroblastic osteosarcoma. Given the patient’s profound intellectual disability, the family and caregivers opted not to treat the patient with chemotherapy. Instead, he underwent an above-the-knee amputation of the right leg for palliative care.
Three-and-a-half years after his first amputation, a second lesion, arising in the left proximal tibia revealed histologic features similar to the first tumor but also contained focal myoblastic-appearing cells. Desmin stains were diffusely positive on the biopsy, but MyoD1 stains revealed only non-specific cytoplasmic positivity. No definite osteoid was found on the biopsy of the second tumor, which was also SATB2-negative on subsequent staining. An above the—knee amputation was performed on his left leg. The post treatment excision exhibited definite osteoid formation without obvious myoblastic differentiation, consistent with a final diagnosis of osteosarcoma. Since the histologic and immunophenotypic findings between the first and second tumors were different, they were considered to be two primary tumors. Subsequently, the patient developed pulmonary metastases that progressed slowly. He died 5 years following his diagnosis at 22 years of age.
Due to the significant cognitive impairment noted in the older sibling, the parents closely tracked the proband’s developmental milestones and significant development delay was noted before 12 months of age. At age 21 years, his mother noted a mass in the proband’s right thigh. He was subsequently hospitalized for a fractured right femur and radiographic imaging was suggestive of osteosarcoma. Pathology review revealed an epithelioid osteosarcoma, primarily composed of cells with oval eccentric nuclei and voluminous lightly eosinophilic cytoplasm. Scattered intercellular foci with lacy osteoid were observed. Immunohistochemistry revealed diffuse, membranous epithelial membrane antigen positivity, diffuse SATB2 and BAF47 (SMARCB1) positivity, and negative cytokeratin 20 expression. A right hip disarticulation was performed for palliative care and histologic evaluation was notable for a peculiar rosette-like pattern. He was not treated with chemotherapy and developed pulmonary metastases that progressed rapidly. He survived one year after diagnosis.
3 |. METHODS
Trio-exome sequencing was performed using peripheral blood DNA from the younger brother and both parents after pre-test genetic counseling and informed consent was obtained from the family. The exome sequencing library was generated using the Agilent SureSelect Human All Exon V6 capture kit. Captured DNA fragments were then sequenced using the Illumina NextSeq 500, with 2 × 100 basepair (bp) paired-end reads. Single nucleotide variants (SNVs) and small insertions and deletions (<10 bp) were detected using the FreeBayes variant caller (Garrison & Marth, 2012) after mapping the reads to the human reference genome (GRCh37/hg19) using bwa-mem (Li, 2013). The mean coverage was 151× across the RefSeq protein-coding exons and splice junctions. Approximately 98% of exonic regions were reliably sequenced with at least 10× coverage. To identify the potential disease-causing variants, a total of 663 genes were prioritized for analysis by searching the HPO phenotype-disease association databases with physician-selected Human Phenotype Ontology (HPO) terms: ambiguous genitalia, cryptorchidism, severe intellectual disability, microcephaly, and osteosarcoma. Variants within exonic regions and splice-site junctions (5 bp into introns) and minor allele frequency <1%, mainly based on the ExAC databases (Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org) [July, 2016]), were further annotated and analyzed using a commercial tool (Cartagenia 4.2, Cartagenia, Cambridge). Sequencing variants were analyzed and classified following the ACMG guidelines (Richards et al., 2015).
Multiplex ligation-dependent probe amplification (MLPA) analysis of the TP53 gene (MRC-Holland) was performed according to the manufacturer’s protocols using peripheral blood from the proband.
Chromosomal microarray analysis (CMA) was performed on the frozen tissue from the primary osteosarcoma of the younger brother and the second tumor from the older brother using the Affymetrix CytoScan HD platform following validated standard protocols (Affymetrix, Santa Clara). Additionally, CMA was performed on the FFPE tissue from the first tumor of the older brother using the Affymetrix Oncoscan platform (Affymetrix, Santa Clara).
4 |. RESULTS
Trio exome sequencing of the proband (younger brother) and both parents revealed a nonsense NM_000489.4:c.7156C>T (p.Arg2386*) variant in the ATRX gene in the proband (hemizygous) and in the mother’s peripheral blood (heterozygous). No other pathogenic or likely pathogenic variants associated with the patient’s primary clinical concerns were identified. Several variants of unknown clinical significance were reviewed and noted (Supplemental Material & Supplemental Table SI). Since a small subset of osteosarcomas occur in individuals with cancer predisposition syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, Rothmund-Thomson syndrome type 2, RAPADILINO syndrome, Werner syndrome, Bloom syndrome, Paget disease of bone, and Diamond Blackfan anemia, the corresponding genes (TP53, RB1, RECQL4, RECQL2, RECQL3, SQSTM1) as well as genes associated with Diamond Blackfan (Calvert et al., 2012) were specifically analyzed. The results were all negative (Supplemental Table SII). There was no evidence for a deletion of the coding exons in the TP53 locus, however single exon deletions in the genes listed above could not be formally ruled out.
The nonsense variant in the ATRX gene was subsequently confirmed using Sanger sequencing (Figure 1B). This variant has not been observed in the general population but has been reported as disease-causing in at least two individuals with ATR-X syndrome (Brett et al., 2014; Gibbons et al., 1995). This variant is not present in the ExAC database, and is predicted to cause loss of normal protein function either through protein truncation or nonsense-mediated mRNA decay. Therefore, it is classified as a pathogenic variant in this family.
Sanger sequencing on the older brother’s normal skin FFPE tissue showed the same hemizygous nonsense variant. Additionally, this c.7156C>T (p.Arg2386*) variant was identified in the osteosarcoma from the proband and the second osteosarcoma from the older brother, as shown in Figure 1B.
Chromosomal microarray analysis on all three tumors showed complex copy number alterations with a high level of genomic instability (Figure 2), consistent with the clinical diagnosis of osteosarcoma. The copy number alterations in the first and second tumors from the older brother were distinct, and thus consistent with two primary tumors (Figure 2B and C). The first tumor from the older brother (Figure 2B) showed a deletion of 13q and gain of 17p, while the tumor from the proband (Figure 2A) and the 2nd tumor from the older brother (Figure 2C) both demonstrated loss of heterozygosity (LOH) of 13q including RB1 and LOH of 17p, including TP53, although the copy numbers were different. We did not perform tumor sequencing to rule out somatic mutations in RB1 and TP53; however the exome sequencing analysis was negative for a germline mutation.
FIGURE 2.
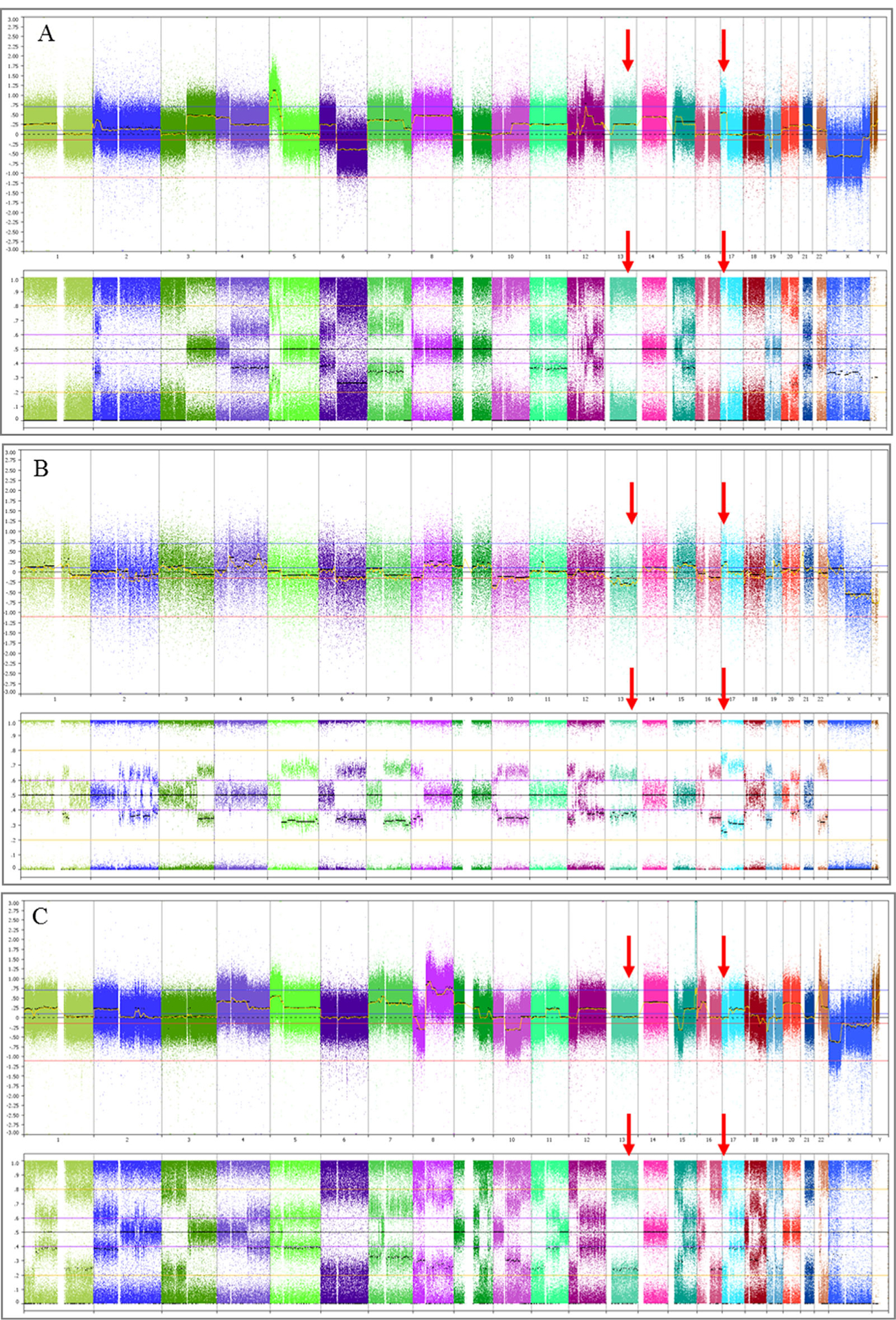
Chromosomal microarray whole genome view of the osteosarcoma from the younger brother. (A, frozen tissue), the first (B, FFPE tissue) and the second (C, frozen tissue) osteosarcoma from the older brotherTop view in all three graphics: Total copy number view showing gains and losses (from left to right: chromosomes 1–22 followed by X and Y); Bottom view: Allele difference (A and C) or B-allele frequency plots (B) confirming the gains and losses observed in the top copy number view and demonstrating LOH. All three tumors showed complex copy number alterations with a high level of genomic instability consistent with osteosarcoma. Red arrows indicate the alterations in 13q and 17p in the three tumors. The tumor from the proband (A) demonstrated gain and LOH of 13q and 17p, including RB1 and TP53, respectively. The first tumor from the older brother (B) had a deletion of chromosome13 and gain of 17p. The second tumor from the older brother (C) showed copy number copy number neutral LOH for 13q and 17p.
5 |. DISCUSSION
The clinical findings in both siblings were consistent with ATR-X syndrome (Gibbons & Higgs, 2000; Wada, Kubota, Fukushima, & Saitoh, 2000), and appear to be related to the identified germline pathogenic nonsense variant in the ATRX gene inherited from their mother.
The protein encoded by ATRX demonstrates a SWI/SNF-like chromatin remodeling function that has been implicated in multiple cellular roles including gene expression, DNA replication, and histone variant deposition (Clynes & Gibbons, 2013; Clynes, Higgs, & Gibbons, 2013; Ratnakumar & Bernstein, 2013). Somatic variations including point mutations, focal deletions, and structural variants that affect the coding region of the ATRX gene have been reported as recurrent alterations in osteosarcomas (Chen et al., 2014). Loss of ATRX has been correlated with alternative lengthening of telomeres (ALT), which is associated with telomere maintenance and impaired cell growth in ALT cancer cells (Episkopou et al., 2014; Heaphy et al., 2011; Lovejoy et al., 2012; Ramamoorthy & Smith, 2015). However, it is currently unknown whether germline pathogenic variants in the ATRX gene or the ATR-X syndrome are associated with cancer predisposition to osteosarcoma or to other tumors. In this family, the fact that the proband had osteosarcoma and the older brother had two primary osteosarcomas, and that all the known osteosarcoma predisposition genes were negative by whole exome sequencing suggests an association between the germline ATRX mutation and osteosarcoma. The observation that the mother has not been affected with cancer may be explained by the presence of a normal copy of ATRX on the other X chromosome and/or skewed X inactivation. The formal possibility exists that there is another locus that predisposed both brothers to osteosarcoma in addition to the ATR-X syndrome, however due to the limited study material, investigation of the intronic or regulatory regions of the other cancer predisposition genes was not performed, and single exon deletions or duplications would not have been detected at either the chromosomal microarray level or by exome sequencing.
Despite the previously described mounting evidence linking ATRX to ALT, the crucial demonstration of a causal role for ATRX in the activation of ALT has not been reported. It may be of interest to perform telomere length testing for patients with germline ATRX mutations. Although knockdown of ATRX in cell lines was not sufficient to convert them to the ALT cell state (Lovejoy et al., 2012; O’Sullivan & Almouzni, 2014; O’Sullivan et al., 2014), it is possible that loss of ATRX may be part of an early event for ALT activation. Recent studies have shown that ATRX is frequently lost in cancer cells that use ALT to maintain telomeres (Ramamoorthy & Smith, 2015). Telomere dysfunction and ATRX deficiency may result in extensive genomic instability, which is a hallmark of ALT cancer cells (Lovejoy et al., 2012; Ramamoorthy & Smith, 2015). The complex copy number alterations and LOH seen in both brothers’ tumors were characteristic of conventional osteosarcoma (Biegel, Womer, & Emanuel, 1989; Perry et al., 2014) although the underlying mechanism for the chromosomal instability has not yet been clearly defined. This is the first report of osteosarcoma diagnosed in two individuals with ATR-X syndrome, suggesting a possible increased risk for cancer in patients with this syndrome. The identification of other individuals with ATR-X germline mutations and cancer is needed to confirm this association.
Supplementary Material
ACKNOWLEDGMENTS
We thank the family for their invaluable collaboration and for their permission to share the data. We also thank Cindy Fong and Dolores Estrine for their technical assistance in Sanger sequencing and chromosomal microarray analysis, respectively.
Footnotes
CONFLICTS OF INTEREST
None.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- Biegel JA, Womer RB, & Emanuel BS (1989). Complex karyotypes in a series of pediatric osteosarcomas. Cancer Genetics and Cytogenetics, 38(1), 89–100. [DOI] [PubMed] [Google Scholar]
- Brett M, McPherson J, Zang ZJ, Lai A, Tan ES, Ng I, … Tan EC (2014). Massively parallel sequencing of patients with intellectual disability, congenital anomalies and/or autism spectrum disorders with a targeted gene panel. Public Library of Science, 9(4), e93409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert GT, Randall RL, Jones KB, Cannon-Albright L, Lessnick S, & Schiffman JD (2012). At-risk populations for osteosarcoma: The syndromes and beyond. Sarcoma, 2012, 152382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, … Dyer MA (2014). Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Reports, 7(1), 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes D, & Gibbons RJ (2013). ATRX and the replication of structured DNA. Current Opinion in Genetics and Development, 23(3), 289–294. [DOI] [PubMed] [Google Scholar]
- Clynes D, Higgs DR, & Gibbons RJ (2013). The chromatin remodeller ATRX: A repeat offender in human disease. Trends in Biochemical Sciences, 38(9), 461–466. [DOI] [PubMed] [Google Scholar]
- Episkopou H, Draskovic I, Van Beneden A, Tilman G, Mattiussi M, Gobin M, … Decottignies A (2014). Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Research, 42(7), 4391–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G 2012. Haplotype-based variant detection from short-read sequencing arXiv preprint arXiv . 1207.3907. [Google Scholar]
- Gibbons RJ, & Higgs DR (2000). Molecular-clinical spectrum of the ATR-X syndrome. American Journal of Medical Genetics, 97(3), 204–212. [DOI] [PubMed] [Google Scholar]
- Gibbons RJ, Picketts DJ, Villard L, & Higgs DR (1995). Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell, 80(6), 837–845. [DOI] [PubMed] [Google Scholar]
- Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, … Meeker AK (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science, 333(6041), 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM arXiv preprint arXiv:1303.3997. [Google Scholar]
- Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T … Consortium ALTSC (2012). Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genetics, 8(7), e1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan RJ, & Almouzni G (2014). Assembly of telomeric chromatin to create ALTernative endings. Trends in Cell Biology, 24(11), 675–685. [DOI] [PubMed] [Google Scholar]
- O’Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, & Karlseder J (2014). Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nature Structural and Molecular Biology, 21(2), 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, … Janeway KA (2014). Complementary genomic approaches highlight the PI3 K/mTOR pathway as a common vulnerability in osteosarcoma. Proceedings of the National Academy of Sciences of the United States of America, 111(51), E5564–E5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picketts DJ, Higgs DR, Bachoo S, Blake DJ, Quarrell OW, & Gibbons RJ (1996). ATRX encodes a novel member of the SNF2 family of proteins: Mutations point to a common mechanism underlying the ATR-X syndrome. Human Molecular Genetics, 5(12), 1899–1907. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy M, & Smith S (2015). Loss of ATRX suppresses resolution of telomere cohesion to control recombination in ALT cancer cells. Cancer Cell, 28(3), 357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnakumar K, & Bernstein E (2013). ATRX: The case of a peculiar chromatin remodeler. Epigenetics, 8(1), 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Committee ALQA (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villard L, Toutain A, Lossi AM, Gecz J, Houdayer C, Moraine C, & Fontes M (1996). Splicing mutation in the ATR-X gene can lead to a dysmorphic mental retardation phenotype without alpha-thalassemia. American Journal of Human Genetics, 58(3), 499–505. [PMC free article] [PubMed] [Google Scholar]
- Wada T, Kubota T, Fukushima Y, & Saitoh S (2000). Molecular genetic study of Japanese patients with X-linked alpha-thalassemia/mental retardation syndrome (ATR-X). American Journal of Medical Genetics, 94(3), 242–248. [PubMed] [Google Scholar]
- Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister SM, Platten M, … Wick W (2013). ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathologica, 126(3), 443–451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.