

Abstract
Antisense oligonucleotide therapies are important cancer treatments, which can suppress genes in cancer cells that are critical for cell survival. SF3B1 has recently emerged as a promising gene target that encodes a key splicing factor in the SF3B protein complex. Over 10% of cancers have lost one or more copies of the SF3B1 gene, rendering these cancers vulnerable after further suppression. SF3B1 is just one example of a CYCLOPS (Copy-number alterations Yielding Cancer Liabilities Owing to Partial losS) gene, but over 120 additional candidate CYCLOPS genes are known. Antisense oligonucleotide therapies for cancer offer the promise of effective suppression for CYCLOPS genes, but developing these treatments is difficult due to their limited permeability into cells and poor cytosolic stability. Here we develop an effective approach to suppress CYCLOPS genes by delivering antisense peptide nucleic acids (PNAs) into the cytosol of cancer cells. We achieve efficient cytosolic PNA delivery with the two main non-toxic components of the anthrax toxin: protective antigen (PA) and the 263-residue N-terminal domain of lethal factor (LFN). Sortase-mediated ligation readily enables the conjugation of PNAs to the C-terminus of the LFN protein. LFN and PA work together in concert to translocate PNAs into the cytosol of mammalian cells. Antisense SF3B1 PNAs delivered with the LFN/PA system suppress the SF3B1 gene and decrease cell viability, particularly of cancer cells with partial copy-number loss of SF3B1. Moreover, antisense SF3B1 PNAs delivered with a HER2-binding PA variant selectively target cancer cells that over-express the HER2 cell receptor, demonstrating receptor-specific targeting of cancer cells. Taken together, our efforts illustrate how PA-mediated delivery of PNAs provides an effective and general approach for delivering antisense PNA therapeutics and for targeting gene dependencies in cancer.
Graphical Abstract
INTRODUCTION
Suppressing gene transcription or translation with antisense therapeutics represents an important strategy for treating human disease, including cardiovascular disorders, amyloid diseases, and cancer.1 Although small molecules are typically unable to target genes, therapeutic oligonucleotides are a promising way to achieve gene suppression with high affinity and selectivity for complementary strands of DNA or RNA.
One set of gene targets that is increasingly important is called: Copy-number alterations Yielding Cancer Liabilities Owing to Partial LosS (CYCLOPS). Cancer cells with partial genomic copy-number loss of a CYCLOPS gene are sensitive after further suppression (Figure 1A–B), making CYCLOPS genes ideal targets for antisense therapeutics. These genes are also a rich source of potential targets since a typical cancer will exhibit partial copy loss of up to 47 CYCLOPS genes.2 CYCLOPS genes are characterized by their importance in cell survival and partial copy-number loss in cells. This partial copy loss occurs frequently in cancer cells3 and is usually clonal,4 in which all cells of the tumor exhibit lower expression of the gene relative to normal cells. After therapeutic gene suppression treatment, the normal cells survive (Figure 1A) but the cancer cells undergo cell death (Figure 1B).2
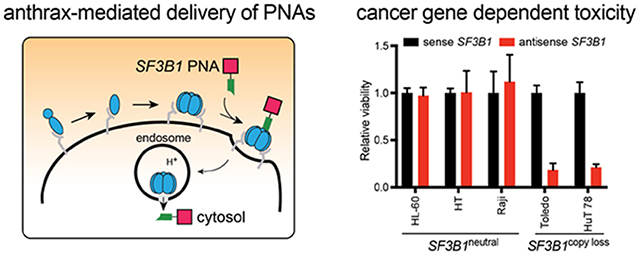
Figure 1. CYCLOPS gene dependency in copy-loss cells and further gene suppression enabled by anthrax-mediated delivery of antisense PNAs.
(A) Normal (diploid) cells express excess amounts of essential proteins, which enable the cells to survive after partial gene suppression. (B) Cancer (copy loss) cells express lower amounts of some essential proteins, which do not enable the cells to survive after gene suppression. (C) Anthrax-mediated translocation mechanism of PNAs delivered with LFN and PA, which enables therapeutic gene suppression. The blue ovals represent PA, the green polygon represents LFN, and the pink square represents the PNA.
The SF3B1 gene is a promising CYCLOPS target that encodes for a key splicing factor in the spliceosome that is critical for cell survival. This gene exhibits partial copy-number loss in 11% of all cancers5 and is also a well-established oncogene that is frequently mutated in leukemia,6 retinal melanoma,7 and breast cancer cells.8 Previously, we characterized mechanisms of cell vulnerability after SF3B1 gene suppression.3 Following suppression, RNA sequencing revealed splicing defects from increased intron retention, dysregulation of exon inclusion, and more alternative 3’ and 5’ splice site selection. We also found that actually reducing SF3B1 protein levels was essential to selectively promote cancer cell death, while inhibiting SF3B1 splicing activity did not reproduce the CYCLOPS phenotype. Similarly, we found that reducing PSMC2 protein levels, rather than inhibition of PSMC2 or the proteasome, was essential for promoting cancer cell death. In this paper, we leverage our tools for studying CYCLOPS genes toward developing a therapeutic approach to suppress their expression—anthrax-mediated delivery of peptide nucleic acids (PNAs).
PNAs are an attractive way to target CYCLOPS genes with high affinity and selectivity. The PNAs comprise N-(2-aminoethyl)glycine units, which gives a pseudopeptide backbone that replaces the sugar–phosphate groups of oligosaccharides. This backbone provides resistance to nuclease and protease degradation,9,10 and also promotes the formation of strong complexes with reversed-complementary strands of DNA or RNA.11,12
Although PNAs can suppress gene transcription and translation, their delivery into cells remains a major challenge.12,13 PNAs are neutrally-charged molecules under physiological conditions and exhibit limited permeability across biological barriers, including endosomal and plasma membranes. Increased cell uptake can be achieved through either electroporation14 or by incorporation of a membrane-permeable molecule, including the conjugation of a cell-penetrating peptide15 or the co-incubation with a lipid.16 These methods enhance PNA activity at micromolar concentrations, but have not yet provided a sufficiently robust and general approach that permits further development in the clinic.17 New, generalizable methods are needed for achieving efficient cytosolic delivery of PNAs to unlock their full therapeutic potential.
Nature has already evolved mechanisms for transporting large molecules across biological barriers. In particular, anthrax lethal toxin from Bacillus anthracis is an efficient two-component delivery system that comprises protective antigen (PA) and lethal factor (LF). Figure 1C illustrates the translocation mechanism of PA, which begins by the binding of PA83 to receptors on host cells, followed by proteolytic cleavage with a furin-family protease.18 The resulting PA63 fragment self-assembles to form a heptamer or octamer,19,20 and further assembles with LF. The complex then undergoes endocytosis, followed by an acid-promoted conformational rearrangement to form an ion-conductive transmembrane pore.21 LF then translocates through the PA pore into the cytosol. This anthrax machinery can be rendered non-toxic by removing the catalytic portion of LF on the C-terminus to give LFN.22,23 The LFN/PA delivery system has previously been shown to efficiently transport a wide range of cargo into mammalian cells, including proteins, peptides, and small molecules.24,25 One report even showed PA-mediated cell uptake, without LFN, of PNAs with Lys residues appended to the C-terminus.26 These constructs showed limited PNA activity in the micromolar concentration range, which precluded further development.
In this paper, we develop an efficient approach for using the LFN/PA machinery to deliver PNAs and perform gene suppression. We show that appending PNAs to LFN enhances the PNA activity to nanomolar concentrations. We also illustrate the therapeutic potential for using LFN/PA to enable antisense therapy to treat cancer. Since LFN and the PNAs cannot be generated recombinantly as fusion proteins, we used sortase-mediated ligation with SrtA* for PNA conjugation onto the C-terminus of LFN.27 The sortase ligation enabled rapid conjugation of each PNA onto LFN in less than 30 min. We then demonstrate that the LFN/PA machinery translocates the PNAs into the cytosol of mammalian cells. We demonstrate that the delivery of antisense PNAs promotes gene suppression and decreases cell viability of copy-number loss cells. In particular, we show that nanomolar concentrations of antisense SF3B1 PNAs delivered with LFN/PA can decrease cell viability of naturally occurring SF3B1 copy-number loss cancer cells. Moreover, we show that antisense SF3B1 PNAs delivered with a HER2-targeting PA can selectively target cancer cells that overexpress the HER2 receptor. These studies demonstrate how combining PNAs with the LFN/PA delivery system can enable antisense therapeutics for cancer and for other classes of human disease.
RESULTS
Protective antigen delivers PNAs into the cytosol of mammalian cells.
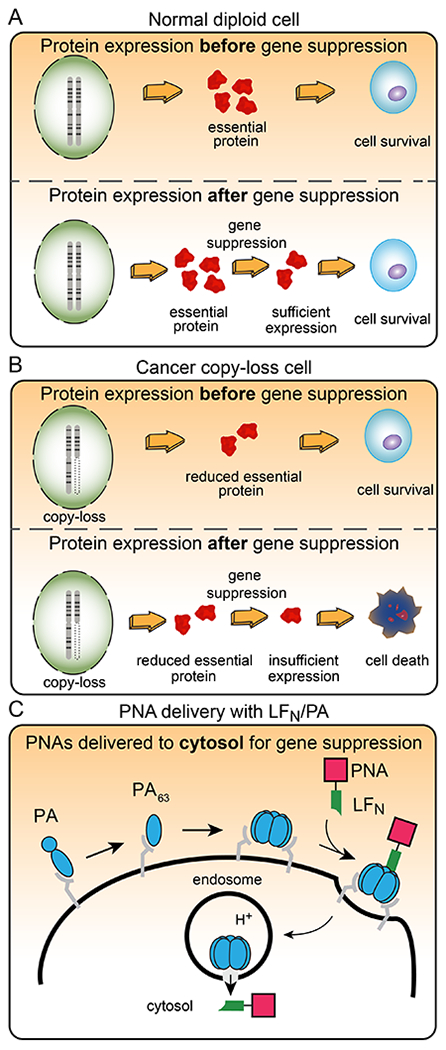
We began our study by investigating whether the LFN/PA delivery system can translocate PNAs into the cytosol of mammalian cells. To answer this question, we prepared nine PNA substrates using Fmoc-based solid-phase PNA synthesis with the four main nucleobase PNA units: adenine (A), cytosine (C), guanine (G), and thymine (T). We also incorporated five Gly residues on the N-terminus for sortase-mediated ligation, which gave G5-PNA 1, which is a negative control that lacks PNAs, and G5-PNAs 2–9 (Figure 2A; Figure S1). In addition, we prepared the fusion protein of LFN with the A chain of diphtheria toxin (DTA), which is enzyme that catalyzes the ADP-ribosylation of elongation factor 2 and inhibits protein synthesis. DTA serves as a reporter for evaluating translocation efficiency of the LFN/PA delivery system.
Figure 2. Design of LFN-PNA constructs for cytosolic delivery into mammalian cells.
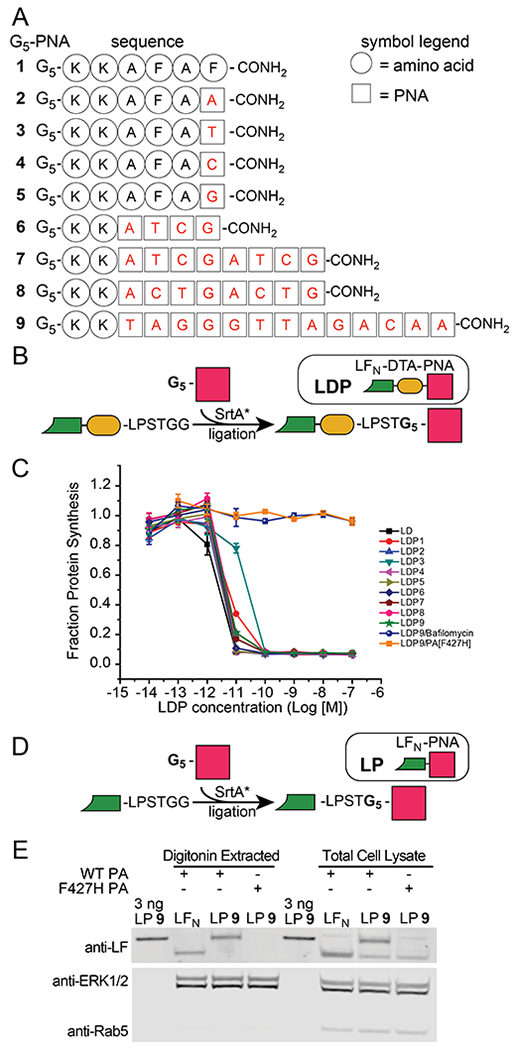
(A) Amino acid and nucleobase sequences of G5-PNAs 1–9, which contain the five N-terminal Gly residues for sortase-mediated ligation. (B) Preparation of LDPs 1–9 by sortase-mediated ligation with SrtA* of LFN-DTA-LPSTGG-H6 and G5-PNAs 1–9. (C) Protein synthesis inhibition assay of CHO-K1 cells incubated with varying concentrations of LDPs 1–9 in the presence of 20 nM PA, followed by the treatment with 3H-labelled leucine (3H-Leu) medium. The incorporation of 3H-Leu was measured with a scintillation counter to determine the level of protein synthesis inhibition, then was normalized to cells incubated with 20 nM PA (no LDP). F427H PA is a nonfunctional PA mutant; Bafilomycin A1 is a small molecule inhibitor of endosomal acidification. Data represent the mean of three replicate wells ± the standard deviation (± SD). (D) Preparation of LPs 9 and 10 by sortase-mediated ligation with SrtA* of LFN-LPSTGG-H6 and G5-PNAs 9 and 10. (E) Western blot analysis of HEK-293T cell lysate showing the translocation of LP 9. Cells were incubated with 250 nM LP 9 in the presence of 40 nM (wild-type) WT or F427H PA for 12 hours, followed by the treatment with a digitonin extraction or total lysis buffer (~1 million cells per lane). Purified LP 9 (3 ng) was also run on the gel for the purpose of quantification.
We then incorporated the G5-PNAs onto LFN-DTA to analyze PNA translocation based on DTA-mediated inhibition of protein synthesis. We used sortase-mediated ligation with SrtA* to append the G5-PNAs 1–9 onto the C-terminus of LFN-DTA, which gave nine LFN-DTA-PNA fusion proteins called LDPs 1–9 (Figure 2B, Figures S2 and S3).27,28
We then investigated the membrane translocation efficiency of LDPs 1–9 with a series of biological experiments.29,30 We began by performing an in cell protein synthesis inhibition assay, which measures translocation efficiency based on the amount of DTA delivered to the cytosol. CHO-K1 cells were incubated with serial dilutions of LDPs 1–9 in the presence of 20 nM PA for 2 hours, then were treated with 3H-labelled leucine (3H-Leu) medium. Measuring the scintillation from 3H-Leu incorporated into newly synthesized proteins reports on the total ribosomal protein synthesis, which is inversely proportional to the amount delivered of the LDP constructs. We found that LDPs 1–9 promoted reduction of protein synthesis at concentrations as low as 100 pM in the presence of 20 nM PA (Figure 2C). We next established whether LDP translocation occurs by a PA-dependent mechanism with two control experiments. CHO-K1 cells were incubated in the presence of LDP 9 with either: (1) 20 nM F427H PA, which is a nonfunctional PA mutant that is translocation deficient,31,32 or (2) 20 nM wild-type (WT) PA and Bafilomycin A1, which is an inhibitor of endosomal acidification. Neither experiment showed any inhibition of protein synthesis (Figure 2C), which demonstrated that a functional PA and an acidic endosome are both essential for translocation.
To further evaluate translocation efficiency, we performed a series of biological experiments with two LFN-PNAs (LPs). These LPs comprise G5-PNAs, without the fused DTA reporter, to permit studies of translocation efficiency that do not interfere with normal cellular function. For these studies, we prepared an LP 9 construct using sortase-mediated ligation with SrtA* (Figure 2D, Figures S2 and S3). We also prepared a variant of LP 9, called LP 10, that contains a scrambled PNA sequence (Figure S2). We then investigated the cytosolic delivery of LPs 9 and 10 by western blot analysis. HEK-293T cells were incubated with 250 nM LP 9 or 10 in the presence of 40 nM PA. After 12 h, the intracellular material was extracted using a digitonin or lysis buffer, as previously described.33 Immunostaining for the early endosome marker Rab5 and the cytosolic marker Erk1/2 indicated cytosolic delivery of both LP 9 (Figure 2E) and LP 10 (Figure S4).
We also quantified the amount of the LPs delivered into cells by establishing the relationship between protein loading and anti-LF staining intensity (Figure S5). Based on this relationship, we estimated that a total of 3 ng of LP 9 was delivered into 1 million cells (which corresponds with 51,000 molecules/cell or cytosolic concentration of 84 nM). In addition, SDS-PAGE analysis showed that the bands for LP 9 and purified LFN migrate at comparable molecular weights, which shows that the LP 9 remains largely intact after translocation (Figure 2E).
We then compared the amount of protein delivered to the cytosol vs. other cellular compartments (e.g., endosomes). In the western blot, we compared the intensity of the LP 9 band from the cytosolic (digitonin fraction) and total lysis fractions. The intensity was similar in both fractions, which shows that the majority of LP 9 was delivered into the cytosol (Figure 2E). We also compared the intensity of the LP 9 band when incubated in the presence of 40 nM of either WT or F427H mutant PA. The LP 9 band was absent from the F427H PA condition, which shows that cytosolic PNA delivery requires a functional PA (Figure 2E).
We also evaluated the cytosolic delivery of LDPs by western blot analysis, and further demonstrated that a functional PA and endosomal acidification is important for achieving cytosolic PNA delivery. HEK293-T or CHO-K1 cells were incubated with 100 nM LDPs 6–9 in the presence of 20 nM PA. Western blot analysis showed a prominent band for the LDPs, which decreased in the control experiments with the nonfunctional F427H PA and the endosome acidification inhibitor Bafilomycin A1 (Figure S6).
These results establish that the LFN/PA system efficiently translocated the PNAs into the cytosol of mammalian cells, regardless of length up to 15 base pairs or sequence for the PNAs studied. Encouraged by these results, we set out to leverage this platform for performing CYCLOPS gene suppression.
Anthrax-delivered PNAs effectively target cancer cell gene dependencies.
We evaluated whether LFN/PA-mediated delivery of PNAs can facilitate gene suppression and cancer cell death. We performed these studies with two isogenic cell lines derived from CAL-51 cells: CRISPRcopy loss cells that were edited using CRISPR to generate partial copy-number loss of SF3B1, and CRISPRneutral cells, which were edited using non-targeting guide RNAs.3
We then asked if the CRISPRcopy loss cells exhibit reduced cell viability due to lower amounts of the SF3B1 protein. We performed three main experiments to measure SF3B1 protein production and evaluate cell viability. We first evaluated cell viability after incubating the cells with spliceostatin A, which is a gene-splicing inhibitor that binds to the SF3B subcomplex.34 The CRISPRneutral and CRISPRcopy loss cells showed comparable sensitivity to splicing inhibition from Splicesostatin A at concentrations as low as 100 nM (Figure S7). Second, we measured the levels of SF3B1 protein in both the CRISPRneutral and CRISPRcopy loss cells by western blot analysis. The blot showed that the SF3B1 protein is produced by both cell lines, but is lower in the CRISPRcopy loss cells (Figure S8). Third, we characterized the sensitivity of CRISPRneutral and CRISPRcopy loss cells to partial SF3B1 gene suppression with siRNA. We transfected an SF3B1-targeting siRNA at concentrations ranging from 12.5 to 200 nM, then evaluated the levels of SF3B1 by quantitative PCR and cell viability analysis (Figure S9). The siRNA treatment promoted SF3B1 suppression and preferentially decreased cell viability of CRISPRcopy loss cells. These results show that the CRISPRneutral and CRISPRcopy loss cell lines are similar, but differ in sensitivity to SF3B1 gene suppression.
With these cell lines in hand, we investigated the ability of the LFN/PA delivery system to target the SF3B1 gene and promote suppression (see supporting information for the exact sequence of the reference transcript). When designing an antisense oligonucleotide, targeting a region that is 50 to 100 bases downstream of the start codon is generally more robust than targeting the start codon.35 Sequences closer to 5’ or 3’ regions are rich in regulatory protein binding sites that can have bound protein complexes, which would interfere with the complexation of a gene suppression oligonucleotide. We designed and prepared sense and antisense LP constructs (Figure 3A), based on a short hairpin RNA (shRNA) that we had previously found to effectively reduce SF3B1 expression.3 The antisense LP-SF3B1 construct is designed to hybridize at the 150 position on the SF3B1 gene transcript (Figure 3B; Figure S10); the sense LP-SF3B1 serves as a non-hybridizing control.36 We incorporated three additional Lys residues within the antisense LP-SF3B1 construct to enhance solubility. We prepared these constructs by synthesizing the corresponding PNAs, followed by performing sortase-mediated ligation with SrtA*.
Figure 3. Cytosolic delivery of antisense SF3B1 PNAs reduces SF3B1 protein production.
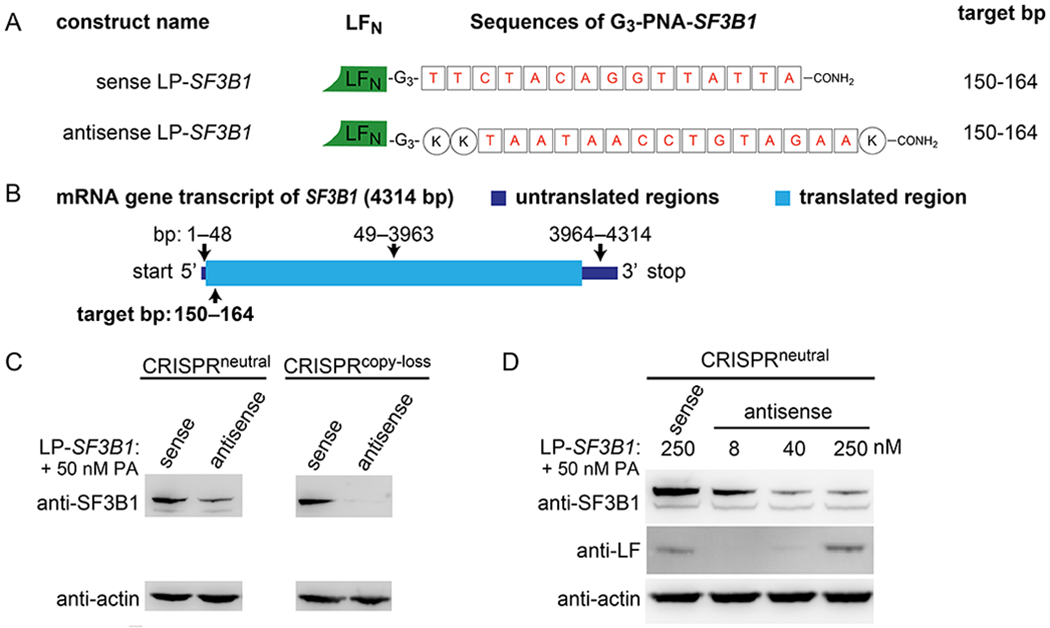
(A) Illustrations of the sense and antisense LP-SF3B1 sequences. (B) Cartoon illustration of the SF3B1 mRNA transcript with the translated and untranslated regions drawn according to scale for their respective lengths. (C) Immunoblot showing the suppression of the SF3B1 protein in CRISPRneutral and CRISPRcopy loss cell lysate after incubation with 250 nM sense or antisense LP-SF3B1 and 50 nM PA. (D) Immunoblot showing dose-dependent suppression of the SF3B1 protein in CRISPRneutral cell lysate after incubation with 8, 40, or 250 nM sense or antisense LP-SF3B1 and 50 nM PA. Cell lysates were collected after a four-day incubation time.
We then evaluated the delivery and activity of the sense and antisense LP-SF3B1 constructs. We quantified SF3B1 protein expression in both CRISPRneutral and CRISPRcopy loss cell lines after incubation with 250 nM sense or antisense LP-SF3B1 and 50 nM PA. Western blot analysis showed a decrease in SF3B1 protein with antisense LP-SF3B1, but not with sense LP-SF3B1 (Figure 3C). The sense LP-SF3B1-treated cells showed comparable levels of SF3B1 protein production as the untreated cells (not shown). We also quantified SF3B1 protein after varying the amount of antisense LP-SF3B1, which showed that this construct promotes a dose-dependent decrease in SF3B1 protein expression in both CRISPRneutral (Figure 3D) and CRISPRcopy loss cells (Figure S11). For these experiments, we also performed a corroboratory immunoblot detection of LFN in the lysates. These results showed bands associated with increasing amounts of LFN, which are faint at 40 nM, due to the limit of detection, but more prominent at 250 nM. The observed dose-dependant intensity of these bands suggests effective SF3B1 protein suppression by the LFN/PA system at concentrations below immunoblot detection limits. These results indicate that the antisense LP-SF3B1 can perform SF3B1 gene suppression that is mediated by an LFN/PA-dependent mechanism.
Antisense SF3B1 PNAs reduce gene splicing activity and cell viability.
We then performed a rescue experiment to evaluate the selectivity of LP-SF3B1 targeting for the SF3B1 gene. To perform this experiment, we asked if the death of CRISPRcopy loss cells could be prevented by amplifying SF3B1 protein expression. We transfected CRISPRcopy loss cells with an SF3B1 protein expression vector to promote exogenous expression of the SF3B1 protein. In addition, we separately transfected CRISPRcopy loss cells with a GFP protein expression vector to provide a negative control with unaltered SF3B1 expression. In both cell lines, we quantified the relative levels of SF3B1 expression and cell viability. After incubating the exogenous SF3B1 expression cells with 250 nM antisense LP-SF3B1 and 50 nM PA, western blot analysis showed increased levels of SF3B1 expression relative to the GFP control (Figure 4A). Cell viability analysis showed that the exogenous expression of SF3B1 helped to maintain cell viability (Figure 4B). These results indicate that LP-SF3B1 selectively targets the SF3B1 gene to perform gene suppression and promote cell death.
Figure 4. SF3B1 gene suppression decreases the viability of SF3B1 copy loss cells.
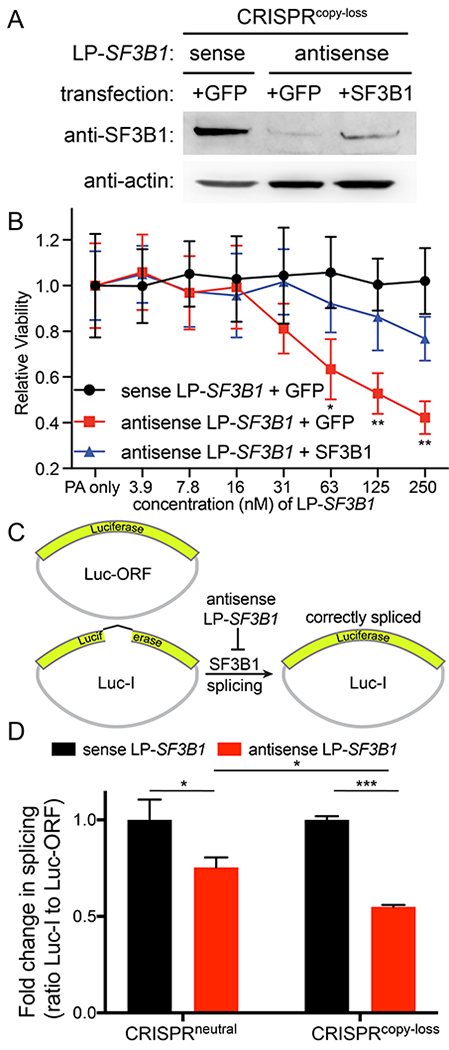
(A) Immunoblot of CRISPRcopy loss cell lysate with expression of either GFP or exogenous SF3B1 protein after incubation with 250 nM sense or antisense LP-SF3B1 in the presence of 50 nM PA. (B) Cell viability of CRISPRcopy loss cells expressing either GFP or exogenous SF3B1 protein upon incubation with varying concentrations of sense or antisense LP-SF3B1 in the presence of 50 nM PA. Cell viability was measured as the change in luminescence relative to 50 nM PA control (with no LFN-PNA) using a CellTiter-Glo viability assay. Data represent the mean of three replicate wells ± SD. (C) Cartoon illustration of luciferase reporters of pre-mRNA splicing: Luc-ORF contains a luciferase open-reading frame that does not require splicing for activity; Luc-I contains a luciferase open-reading frame with an intron sequence that requires splicing for activity. (D) Splicing activity after incubation with 250 nM sense (black) or antisense (red) LP-SF3B1 in the presence of 50 nM PA. Splicing activity was quantified as the ratio of Luc-I to Luc-ORF signal after normalizing to the control with 250 nM sense LP-SF3B1 (with no PA). Data represent the mean of three replicate wells ± SD. For all panels, *p<0.05, **p<0.01 and ***p<0.001.
We also evaluated whether antisense LP-SF3B1 can interfere with gene splicing. We quantified the splicing activity using dual luciferase reporters: one vector contained a luciferase ORF, interrupted by an intron (Luc-I), that required gene splicing to function; the other vector contained a control luciferase ORF (Luc-ORF) that did not require gene splicing to function (Figure 4C). Upon incubation with 250 nM antisense LP-SF3B1 and 50 nM PA, the CRISPRcopy loss cells showed a significant decrease in splicing activity when compared to CRISPRneutral cells (Figure 4D). CRISPRneutral cells incubated with antisense LP-SF3B1 also exhibited a decrease in splicing activity when compared to the sense LP-SF3B1 control. These results show that LFN/PA delivery of PNAs interferes with proper splicing activity by the SF3B1 protein complex (Figure 4D), which, in turn, reduces the viability of the SF3B1 gene-dependent cells (Figure 4B).
SF3B1 suppression selectively decreases viability of CYCLOPS cancer cells.
We then explored the therapeutic potential for delivering antisense LP-SF3B1 to kill SF3B1copy loss (SF3B1copy loss) cancer cells in vitro. We began these studies by incubating CRISPRneutral and CRISPRcopy loss cells with sense or antisense LP-SF3B1 in the presence of 50 nM PA (Figure 5A). In addition, we performed a side-by-side comparison of cell viability with 1 μM sense or antisense LP-SF3B1 in the presence of 50 nM PA (Figure 5B). The CRISPRcopy loss cells showed a significant decrease in cell viability after incubation with antisense LP-SF3B1, which was not observed with either the CRISPRneutral cells or the sense LP-SF3B1 control. These results indicate that the LFN/PA delivery platform can effectively target CYCLOPS vulnerabilities to promote cancer cell death.
Figure 5. Cytosolic delivery of antisense SF3B1 PNAs promotes the death of SF3B1copy loss cells.
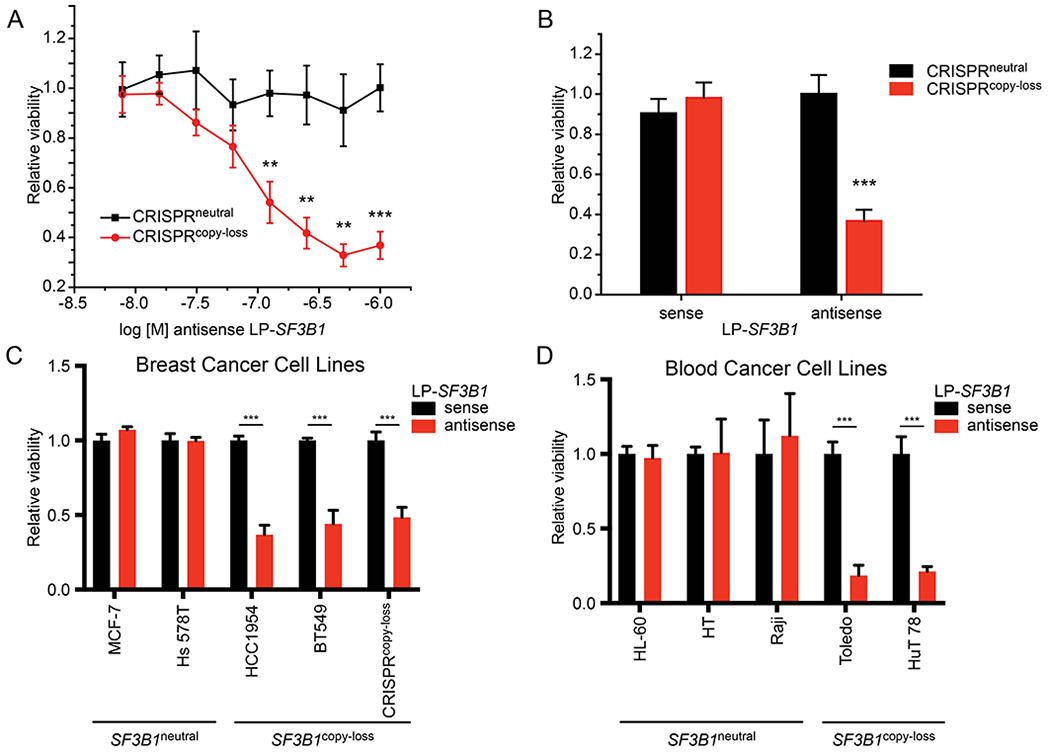
(A) Dose-response curve for relative cell viability of CRISPRneutral (black) and CRISPRcopy loss (red) cells incubated with varying concentrations of antisense LP-SF3B1 in the presence of 50 nM PA. Cell viability calculated as the change in luminescence relative to the treatment with LP-SF3B1 alone (not shown). Data represent the mean of two replicate wells ± SD. (B) Cell viability of cells incubated with 1 μM sense or antisense LP-SF3B1 in the presence of 50 nM WT PA. (C,D) Breast and blood cell lines with SF3B1neural or SF3B1copy loss incubated with 100 nM sense or antisense LP-SF3B1 in the presence of 50 nM WT PA. Cell viability was measured by CellTiter-Glo viability assay, which was normalized to untreated cells. Data represent the mean of three replicate wells ± SD; *p<0.05, **p<0.01, and ***p<0.001.
To demonstrate the robustness of cytosolic delivery provided by the LFN/PA system, we tested SF3B1 gene suppression across nine cancer cell lines from breast and blood lineages. These cell lines represent varied cellular contexts, including adherent or non-adherent growth conditions and with or without naturally occurring SF3B1copy loss. SF3B1 gene suppression with 100 nM antisense LP-SF3B1 promoted a 50% decrease in viability of SF3B1copy loss breast cancer cell lines (BT549 and HCC1954, Figure 5C) and also promoted an 80% decrease in viability of SF3B1copy loss blood cancer cell lines (Toledo and HUT 78, Figure 5D). These results demonstrate that the LFN/PA system provides effective PNA delivery across different cell types and promotes the death of SF3B1copy loss cells.
Targeted delivery of antisense SF3B1 PNAs into specific cancer cells.
The LFN/PA system can also be retargeted to act on specific cell types in a receptor-dependent fashion,37 which can prevent potential adverse effects from suppressing genes in normal cells.38 To demonstrate the cell-targeting feature of the LFN/PA deliver system, we evaluated the effect of suppressing the SF3B1 gene in cancer cells that display amplified levels of the HER2 receptor (HER2amp). We prepared a previously reported HER2-targeting analogue of PA, called mPA-ZHER2, in which PA is fused to an affibody that binds to the HER2 receptor.37 We incubated HER2neutral cells (BT549) with 50 nM mPA-ZHER2 alone or with 250 nM sense LP-SF3B1, which showed no decrease in cell viability; We also incubated HER2neutral cells with 50 nM mPA-ZHER2 and 250 nM antisense LP-SF3B1, which showed no decrease in cell viability (Figure 6A). In contrast, the HER2amp cells (HCC1954) cells gave a different result, which contain naturally-occurring partial copy loss of SF3B1 and increased expression levels of the HER2 receptor. These cells showed a 90% decrease in cell viability when incubated with 50 nM mPA-ZHER2 and antisense 250 nM LP-SF3B1 (Figure 6A). We also evaluated mPA-ZHER2 treatment of the CRISPRcopy loss cells, which are sensitive to SF3B1 gene suppression but express normal levels of the HER2 receptor. These cells did not show decreased viability after incubation with mPA-ZHER2 and antisense LP-SF3B1, but did show decreased viability after incubation with WT PA and antisense LP-SF3B1 (Figure 6B). These results show that the PA system can target specific cancer cells in a receptor-dependent fashion.
Figure 6. Retargeting antisense SF3B1 PNAs.

(A) Viability of HER2neutral or HER2amp cells after incubation with 250 nM sense or antisense LP-SF3B1 in the presence of 50 nM WT PA or mPA-ZHER2; (B) Viability of CRISPRcopy loss cells after incubation with 100 nM antisense LP-SF3B1 with 50 nM of either WT PA or mPA-ZHER2. (C, D) Viability of SF3B1copy loss cells after incubation with antisense PNA-SF3B1, TAT-PNA-SF3B1, or LP-SF3B1 (with 50 nM WT PA or mPA-ZHER2). The cell lines are indicated in the figure panels. Data for all panels represent the mean of three replicate wells ± SD; *p<0.05, **p<0.01 and ***p<0.001.
We also compared the LFN/PA system with a cell-penetrating peptide (CPP), because CPP-mediated PNA delivery has previously been shown.39 The antisense PNA-SF3B1 was conjugated to the TAT CPP with “click chemistry”, which gave the TAT-PNA-SF3B1 construct. SF3B1copy loss cells (CAL-51 CRISPRcopy loss and HCC1954) were then incubated with the antisense PNA-SF3B1 or TAT-PNA-SF3B1 constructs. Neither construct promoted a decrease in cell viability at concentrations as high as 5 μM. The cells were also incubated with LP-SF3B1 in the presence of 50 nM PA or mPA-ZHER2. Both cell lines exhibited a decrease in cell viability from the antisense LP-SF3B1 and WT PA, but only the HER2amp cell line (HCC1954) showed a decrease in viability after treatment with antisense LP-SF3B1 and mPA-ZHER2 (Figures 6C,D). These results show LFN/PA-mediated delivery of PNAs can be 100- to 1000-fold more efficient than a PNA alone or a CPP-PNA.
DISCUSSION
This work shows that the two non-toxic anthrax proteins, PA and LFN, enable robust, cytosolic delivery of antisense PNAs for therapeutic gene suppression. Other delivery systems require higher concentrations and exhibit a complex cell-uptake mechanism. Intracellular LFN-PNA delivery occurs by a straightforward, PA-dependent and acid-induced translocation mechanism. Upon entering the cell cytosol, PNA recognition of reversed-complementary strands suppresses translation of the gene target. In particular, PNA targeting of the SF3B1 CYCLOPS gene selectively induces cell vulnerability by reducing SF3B1 protein levels and, in turn, decreases viability of copy-loss cells over normal cells.
The SF3B1 gene is a promising gene target for CYCLOPS as well as in broader contexts. Mutations to the SF3B1 gene have been found in many cancer types, including chronic lymphocytic leukemia,6 myelodysplatic syndrome,40,41 breast cancer,42 uveal melanoma,43 and lung cancer.44 In addition, other cancer types have shown increased sensitivity to spliceosome modulators that are structurally related to spliceostatin A.45 These modulators are currently in clinical development.46 MYC-driven cancers can also display sensitivity to spliceosome modulators and to partial SF3B1 suppression.47 These recent developments show that PNA-mediated suppression of the SF3B1 gene may have important therapeutic properties for treating both SF3B1-mutant and MYC-driven cancers, and that additional cancer types may also be susceptible to SF3B1 suppression.
LFN/PA delivery of PNAs, which proved useful for suppressing SF3B1, provides a generally applicable strategy for suppressing other gene targets. Many cancers lack therapeutic targets that are readily “druggable”, but may still exhibit a CYCLOPS or non-CYCLOPS dependency on the expression of individual genes.2,3 We think the current work lays groundwork for developing therapeutic approaches that exploit these dependencies with PNAs delivered by LFN/PA.
To further illustrate the significance of this work, beyond targeting the SF3B1 gene, we set out to find new potential gene suppression targets in other cancer models. We analyzed previously-reported genomic data to explore the scope of the gene dependencies. We integrated 5711 genes from a previous shRNA screen of 216 cancer cell lines with characterized genome-wide genetic profiles.48 We designated regions as “gene dependencies” based on whether the sequences encoded were essential for cell survival. We then hypothesized that genes with somatic mutations may represent cancer-specific dependencies, because normal cells would not share these alterations. As cancer-specific dependencies, we considered whether these genes would be therapeutic targets in cancer (Figure 7A).
Figure 7.
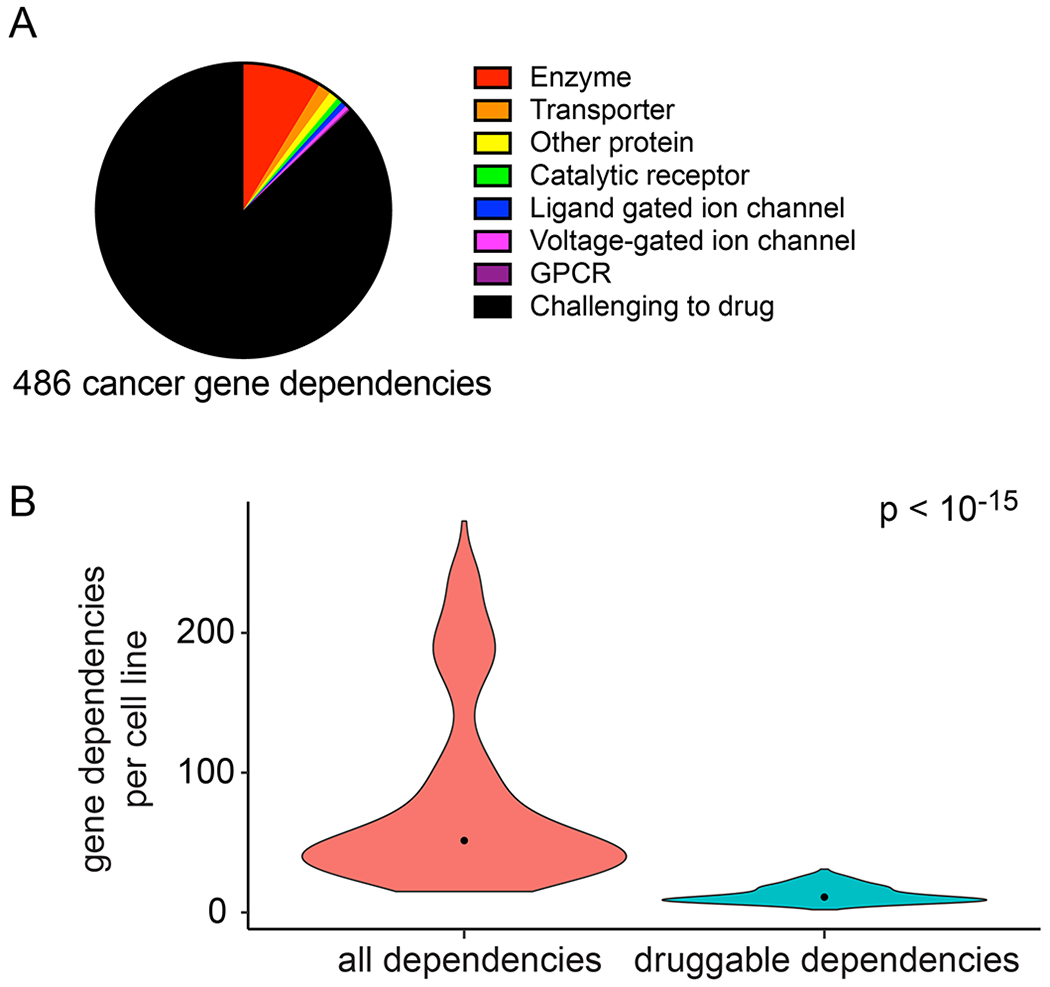
Cancer gene dependency targets. (A) Pie chart indicating the IUPHAR target classification for the 486 cancer gene dependencies identified. (B) Violin plot quantifying the number of potent gene dependencies per cell line. Candidate gene dependencies that belonged to an existing IUPHAR target class were considered “draggable”. Width of plots represents relative sample density, dots represent sample median. A t-test was used for comparing the gene dependencies in the two groups, which indicated that the number of druggable dependencies is significantly different (p<10−15).
We identified gene dependencies associated with mutations in six known driver genes (KRAS, PIK3CA, BRAF, NRAS, CTNNB1 and EGFR) or with any copy-number alteration genome wide. We found that mutations in five (KRAS, PIK3CA, BRAF, NRAS, and CTNNB1) of the six driver genes were associated with increased dependence on that driver; we also identified 342 additional gene dependencies associated with mutations in the driver genes. Integration of the mutation and copy number associated dependencies from our previous work yielded 486 unique context-specific gene dependencies, comprising 8% of all genes analyzed.
We found that the majority (87%) of these candidate cancer gene dependencies do not belong to a straightforward “druggable” protein target class. We classified each gene dependency by its protein target class using the IUPHAR drug targets and family database.49 Of the 486 context-specific gene dependencies, 423 were not classified as belonging to a protein family that could be targeted by a small molecule (Figure 7B). On average, each cell line had 12.7 dependencies that were in an existing IUPHAR druggable target class and 82.1 dependencies that were not (Figure 7B). This analysis illustrates the substantial opportunity to target gene dependencies for cancer, particularly with therapeutic antisense oligonucleotides.
CONCLUSION
In this paper, we showed that anthrax-mediated delivery of PNAs into the cytosol of cancer cells enables effective delivery of antisense oligonucleotides and gene suppression. Appending PNAs onto the C-terminus of LFN with sortase-mediated ligation enabled efficient preparation of the antisense therapeutic. LFN/PA facilitated the PNA delivery into the cytosol of cells, which also enabled targeting of the SF3B1 gene to promote suppression. Robust delivery of the PNAs with LFN/PA was further established across a panel of nine cancer cell lines from two tissue types. In addition, PNAs delivered with a HER2-targeting PA can selectively target cancer cells that overexpress the HER2 receptor. These results demonstrate that LFN/PA deliver PNAs into cells at nanomolar concentrations by a well-established translocation mechanism. Other delivery systems require higher concentrations, as supported by our experiment with the TAT cell-penetrating peptide that showed no PNA activity at micromolar concentrations.
Our results indicate that cell vulnerability is induced through PNA-mediated reduction of the SF3B1 protein, which may occur through one of several gene-suppression mechanisms: (1) Disruption of RNA transcription. We showed that the antisense SF3B1 PNA binds to the target DNA sequence, which suggests that PNAs may also bind to DNA in the cell.50 The PNA-DNA duplex could prevent transcription by obstructing RNA polymerase enzymes during extension of the mRNA transcript, and thus prevent complete transcription of SF3B1 mRNA and preclude subsequent translation.36 (2) Disruption of RNA translation. Rather than binding to DNA, the antisense SF3B1 PNA may instead bind to RNA. Formation of PNA–RNA duplexes could obstruct SF3B1 translation, and thus prevent ribosome processivity and inhibit production of the complete protein. (3) Disruption of mRNA splicing. Another potential suppression mechanism involves inhibition of proper splicing for newly transcribed mRNA.51 This mechanism is possible, because the antisense SF3B1 PNA targets a sequence located in exon 2 of the mRNA transcript. This exon also contains several key splicing motifs, including SRSF5, SRSF1, and SRSF2.52,53 PNA binding to this region could disrupt mRNA splicing, in which improper splicing occurs for exon 2 but not exon 1, and thus could prevent expression of normal protein due to the resultant frame shift. Each of these three mechanisms would prevent renewal of functional SF3B1 protein, leading to the reduction of SF3B1 protein levels over time. Elucidating which mechanism occurs is critical to understanding the role of PNAs in gene suppression, and is an ongoing part of research in our laboratories. Establishing the gene suppression mechanism is also critical for developing other therapeutic antisense PNAs, which is also part of ongoing research in our laboratories.
Neutralizing antibodies generated by the immune system are a critical challenge in the development of all macromolecular delivery systems, including for PA and LFN.54 Efforts to engineer proteins that bind to erythrocytes have enabled antigen-specific tolerance for the corresponding protein.55,56 Although these methods have not yet been developed for inducing LFN-specific tolerance, they may offer the key to preventing generation of neutralizing antibodies against PA and LFN and enabling therapeutic use.
The LFN/PA system provides an ideal PNA-delivery platform for performing CYCLOPS gene suppression, because it enables: (i) rapid conjugation of PNAs onto LFN using sortase-mediated ligation; (ii) efficient endosomal release of PNAs into the cytosol; (iii) delivery into a wide range of mammalian cell types; (iv) targeting of specific cell types in a receptor-dependent fashion, including HER2. Although in vivo studies are outside the scope of the current work, recent in vivo studies suggest that this delivery platform can be applied in therapeutic contexts.57,58,59
In addition, the two anthrax proteins LFN/PA provide a general oligonucleotide delivery platform that is valuable in broader contexts. Neither the length nor sequence of the PNAs studied appeared to affect translocation efficiency, which suggests that these proteins may effectively deliver other PNA sequences and additional classes of oligonucleotides. Also, the proof-of-principle studies described in this paper show promise for in vivo oligonucleotide delivery, and even for in vivo targeting to specific cell types. We anticipate that further development of this platform for oligonucleotide delivery, which is part of ongoing research in our laboratory, may have a significant impact for achieving gene suppression in both research and therapeutic settings.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by MIT start-up funds, MIT Reed Fund, Damon Runyon Cancer Research Foundation Innovation Award, and National Science Foundation (NSF) CAREER Award (CHE-1351807) to B.L.P, NIH grants R01 CA188228 (R.B.), F32 CA180653 (B.R.P.), F32 CA239362 (N.L.T.), the Sontag Foundation (R.B. & B.L.P.), the Melanoma Research Alliance (R.B.), The Gray Matters Brain Cancer Foundation (R.B.), the Pediatric Low-Grade Astrocytoma Fund, and the Friends of DFCI (B.R.P.). We also acknowledge support from the Bridge Project between the Koch Institute and Dana-Farber/Harvard Cancer Center (B.L.P.). We thank R. Collier (Harvard) for his contribution of laboratory equipment and the NERCE facility (grant: U54 AI057159) for expression of protein toxins. We also thank A. Loas, D. Lauffenburger, and J. Ling for their comments.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/
Experimental methods for the synthesis and purification of peptide nucleic acids, plasmid and siRNA construction, protein expression and purification, CellTiter-Glo luminescent cell viability assays, splicing luciferase reporter assays, and Western blot analysis (PDF)
B.L.P. is a founder of Amide Technologies and Resolute Bio.
REFERENCES
- (1).Gambari R (2014) Peptide nucleic acids: A review on recent patents and technology transfer. Expert Opin. Ther. Pat 24, 267–294. [DOI] [PubMed] [Google Scholar]
- (2).Nijhawan D, Zack TI, Ren Y, Strickland MR, Lamothe R, Schumacher SE, Tsherniak A, Besche HC, Rosenbluh J, Shehata S, Cowley GS, Weir BA, Goldberg AL, Mesirov JP, Root DE, Bhatia SN, Beroukhim R, and Hahn WC (2012) Cancer vulnerabilities unveiled by genomic loss. Cell 150, 842–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Paolella BR, Gibson WJ, Urbanski LM, Alberta JA, Zack TI, Bandopadhayay P, Nichols CA, Agarwalla PK, Brown MS, Lamothe R, Yu Y, Choi PS, Obeng EA, Heckl D, Wei G, Wang B, Tsherniak A, Vazquez F, Weir BA, Root DE, Cowley GS, Buhrlage SJ, Stiles CD, Ebert BL, Hahn WC, Reed R, and Beroukhim R (2017) Copy-number and gene dependency analysis reveals partial copy loss of wild-type sf3b1 as a novel cancer vulnerability. eLife 6, e23268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, Lazar AJ, Cancer Genome Atlas Research, N., Cherniack AD, Beroukhim R, and Meyerson M (2018) Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 33, 676–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).The Cancer Genome Atlas Research, N., Mclendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis GM, Olson JJ, Mikkelsen T, Lehman N, Aldape K, Alfred Yung WK, Bogler O, Vandenberg S, Berger M, Prados M, Muzny D, Morgan M, Scherer S, Sabo A, Nazareth L, Lewis L, Hall O, Zhu Y, Ren Y, Alvi O, Yao J, Hawes A, Jhangiani S, Fowler G, San Lucas A, Kovar C, Cree A, Dinh H, Santibanez J, Joshi V, Gonzalez-Garay ML, Miller CA, Milosavljevic A, Donehower L, Wheeler DA, Gibbs RA, Cibulskis K, Sougnez C, Fennell T, Mahan S, Wilkinson J, Ziaugra L, Onofrio R, Bloom T, Nicol R, Ardlie K, Baldwin J, Gabriel S, Lander ES, Ding L, Fulton RS, Mclellan MD, Wallis J, Larson DE, Shi X, Abbott R, Fulton L, Chen K, Koboldt DC, Wendl MC, Meyer R, Tang Y, Lin L, Osborne JR, Dunford-Shore BH, Miner TL, Delehaunty K, Markovic C, Swift G, Courtney W, Pohl C, Abbott S, Hawkins A, Leong S, Haipek C, Schmidt H, Wiechert M, Vickery T, Scott S, Dooling DJ, Chinwalla A, Weinstock GM, Mardis ER, Wilson RK, Getz G, Winckler W, Verhaak RGW, Lawrence MS, O’kelly M, Robinson J, Alexe G, Beroukhim R, Carter S, Chiang D, Gould J, Gupta S, Korn J, Mermel C, Mesirov J, Monti S, Nguyen H, Parkin M, Reich M, Stransky N, Weir BA, Garraway L, Golub T, Meyerson M, Chin L, Protopopov A, Zhang J, Perna I, Aronson S, Sathiamoorthy N, Ren G, Yao J, Wiedemeyer WR, Kim H, Won Kong S, Xiao Y, Kohane IS, Seidman J, Park PJ, Kucherlapati R, Laird PW, Cope L, Herman JG, Weisenberger DJ, Pan F, Van Den Berg D, Van Neste L, Mi Yi J, Schuebel KE, Baylin SB, Absher DM, Li JZ, Southwick A, Brady S, Aggarwal A, Chung T, Sherlock G, Brooks JD, Myers RM, Spellman PT, Purdom E, Jakkula LR, Lapuk AV, Marr H, Dorton S, Gi Choi Y, Han J, Ray A, Wang V, Durinck S, Robinson M, Wang NJ, Vranizan K, Peng V, Van Name E, Fontenay GV, Ngai J, Conboy JG, Parvin B, Feiler HS, Speed TP, Gray JW, Brennan C, Socci ND, Olshen A, Taylor BS, Lash A, Schultz N, Reva B, Antipin Y, Stukalov A, Gross B, Cerami E, Qing Wang W, Qin L-X, Seshan VE, Villafania L, Cavatore M, Borsu L, Viale A, Gerald W, Sander C, Ladanyi M, Perou CM, Neil Hayes D, Topal MD, Hoadley KA, Qi Y, Balu S, Shi Y, Wu J, Penny R, Bittner M, Shelton T, Lenkiewicz E, Morris S, Beasley D, Sanders S, Kahn A, Sfeir R, Chen J, Nassau D, Feng L, Hickey E, Zhang J, Weinstein JN, Barker A, Gerhard DS, Vockley J, Compton C, Vaught J, Fielding P, Ferguson ML, Schaefer C, Madhavan S, Buetow KH, Collins F, Good P, Guyer M, Ozenberger B, Peterson J, and Thomson E (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, Deluca DS, Zhang L, Zhang W, Vartanov AR, Fernandes SM, Goldstein NR, Folco EG, Cibulskis K, Tesar B, Sievers QL, Shefler E, Gabriel S, Hacohen N, Reed R, Meyerson M, Golub TR, Lander ES, Neuberg D, Brown JR, Getz G, and Wu CJ (2011) Sf3b1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med 365, 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Harbour JW, Roberson EDO, Anbunathan H, Onken MD, Worley LA, and Bowcock AM (2013) Recurrent mutations at codon 625 of the splicing factor sf3b1 in uveal melanoma. Nat. Genet 45, 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Maguire SL, Leonidou A, Wai P, Marchio C, Ng CK, Sapino A, Salomon AV, Reis-Filho JS, Weigelt B, and Natrajan RC (2015) Sf3b1 mutations constitute a novel therapeutic target in breast cancer. J. Pathol 235, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Demidov VV, Potaman VN, Frankkamenetskii MD, Egholm M, Buchard O, Sonnichsen SH, and Nielsen PE (1994) Stability of peptide nucleic-acids in human serum and cellular-extracts. Biochem. Pharmacol 48, 1310–1313. [DOI] [PubMed] [Google Scholar]
- (10).Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, and Nielsen PE (1993) Pna hybridizes to complementary oligonucleotides obeying the watson-crick hydrogen-bonding rules. Nature 365, 566–568. [DOI] [PubMed] [Google Scholar]
- (11).Nielsen PE, Egholm M, Berg RH, and Buchardt O (1991) Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 254, 1497–1500. [DOI] [PubMed] [Google Scholar]
- (12).Kaihatsu K, Janowski BA, and Corey DR (2004) Recognition of chromosomal DNA by pnas. Chem. Biol 11, 749–758. [DOI] [PubMed] [Google Scholar]
- (13).Corey DR (2007) Chemical modification: The key to clinical application of rna interference? J. Clin. Invest 117, 3615–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Joergensen M, Agerholm-Larsen B, Nielsen PE, and Gehl J (2011) Efficiency of cellular delivery of antisense peptide nucleic acid by electroporation depends on charge and electroporation geometry. Oligonucleotides 21, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shiraishi T, and Nielsen PE (2006) Enhanced delivery of cell-penetrating peptide-peptide nucleic acid conjugates by endosomal disruption. Nat. Prot 1, 633–636. [DOI] [PubMed] [Google Scholar]
- (16).Hamilton SE, Simmons CG, Kathiriya IS, and Corey DR (1999) Cellular delivery of peptide nucleic acids and inhibition of human telomerase. Chem. Biol 6, 343–351. [DOI] [PubMed] [Google Scholar]
- (17).Koppelhus U, and Nielsen PE (2003) Cellular delivery of peptide nucleic acid (pna). Adv Drug Deliver Rev 55, 267–280. [DOI] [PubMed] [Google Scholar]
- (18).Klimpel KR, Molloy SS, Thomas G, and Leppla SH (1992) Anthrax toxin protective antigen is activated by a cell-surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. U. S. A 89, 10277–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Milne JC, Furlong D, Hanna PC, Wall JS, and Collier RJ (1994) Anthrax protective antigen forms oligomers during intoxication of mammalian-cells. J. Biol. Chem 269, 20607–20612. [PubMed] [Google Scholar]
- (20).Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, Zhang TT, Williams ER, Berger JM, and Krantz BA (2009) The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol 392, 614–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Krantz BA, Trivedi AD, Cunningham K, Christensen KA, and Collier RJ (2004) Acid-induced unfolding of the amino-terminal domains of the lethal and edema factors of anthrax toxin. J. Mol. Biol 344, 739–756. [DOI] [PubMed] [Google Scholar]
- (22).Rabideau AE, Liao X, Akcay G, and Pentelute BL (2015) Translocation of non-canonical polypeptides into cells using protective antigen. Scientific reports 5, 11944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liao X, Rabideau AE, and Pentelute BL (2014) Delivery of antibody mimics into mammalian cells via anthrax toxin protective antigen. Chembiochem 15, 2458–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ballard JD, Collier RJ, and Starnbach MN (1996) Anthrax toxin-mediated delivery of a cytotoxic t-cell epitope in vivo. P Natl Acad Sci USA 93, 12531–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hobson JP, Liu SH, Rono B, Leppla SH, and Bugge TH (2006) Imaging specific cell-surface proteolytic activity in single living cells. Nat. Methods 3, 259–261. [DOI] [PubMed] [Google Scholar]
- (26).Wright DG, Zhang Y, and Murphy JR (2008) Effective delivery of antisense peptide nucleic acid oligomers into cells by anthrax protective antigen. Biochem. Biophys. Res. Commun 376, 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ling JJ, Policarpo RL, Rabideau AE, Liao X, and Pentelute BL (2012) Protein thioester synthesis enabled by sortase. J. Am. Chem. Soc 134, 10749–10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chen I, Dorr BM, and Liu DR (2011) A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. U. S. A 108, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Collier RJ, and Kandel J (1971) Structure and activity of diphtheria toxin .1. Thiol-dependent dissociation of fraction of toxin into enzymically active and inactive fragments. J. Biol. Chem 246, 1496–1503. [PubMed] [Google Scholar]
- (30).Wilson BA, and Collier RJ (1992) Diphtheria-toxin and pseudomonas-aeruginosa exotoxin-a - active-site structure and enzymatic mechanism. Curr Top Microbiol 175, 27–41. [DOI] [PubMed] [Google Scholar]
- (31).Krantz BA, Melnyk RA, Zhang S, Juris SJ, Lacy DB, Wu ZY, Finkelstein A, and Collier RJ (2005) A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 309, 777–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sun J, Lang AE, Aktories K, and Collier RJ (2008) Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. P Natl Acad Sci USA 105, 4346–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Adam SA, Marr RS, and Gerace L (1990) Nuclear-protein import in permeabilized mammalian-cells requires soluble cytoplasmic factors. J. Cell Biol 111, 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, Tani T, Horinouchi S, and Yoshida M (2007) Spliceostatin a targets sf3b and inhibits both splicing and nuclear retention of pre-mrna. Nat. Chem. Biol 3, 576–583. [DOI] [PubMed] [Google Scholar]
- (35).Elbashir SM, Harborth J, Weber K, and Tuschl T (2002) Analysis of gene function in somatic mammalian cells using small interfering rnas. Methods 26, 199–213. [DOI] [PubMed] [Google Scholar]
- (36).Doyle DF, Braasch DA, Simmons CG, Janowski BA, and Corey DR (2001) Inhibition of gene expression inside cells by peptide nucleic acids: Effect of mrna target sequence, mismatched bases, and pna length. Biochemistry 40, 53–64. [DOI] [PubMed] [Google Scholar]
- (37).Mccluskey AJ, Olive AJ, Starnbach MN, and Collier RJ (2013) Targeting her2-positive cancer cells with receptor-redirected anthrax protective antigen. Mol. Oncol 7, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kohn DB, Sadelain M, and Glorioso JC (2003) Occurrence of leukaemia following gene therapy of x-linked scid. Nat. Rev. Cancer 3, 477–488. [DOI] [PubMed] [Google Scholar]
- (39).Bendifallah N, Rasmussen FW, Zachar V, Ebbesen P, Nielsen PE, and Koppelhus U (2006) Evaluation of cell-penetrating peptides (cpps) as vehicles for intracellular delivery of antisense peptide nucleic acid (pna). Bioconjug. Chem 17, 750–758. [DOI] [PubMed] [Google Scholar]
- (40).Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, Mcgee C, Ellis P, Mudie LJ, Stephens PJ, Mclaren S, Massie CE, Tarpey PS, Varela I, Nik-Zainal S, Davies HR, Shlien A, Jones D, Raine K, Hinton J, Butler AP, Teague JW, Baxter EJ, Score J, Galli A, Della Porta MG, Travaglino E, Groves M, Tauro S, Munshi NC, Anderson KC, El-Naggar A, Fischer A, Mustonen V, Warren AJ, Cross NC, Green AR, Futreal PA, Stratton MR, and Campbell PJ (2011) Somatic sf3b1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med 365, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, and Ogawa S (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- (42).Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, Mcmichael JF, Miller CA, Lu C, Harris CC, Mclellan MD, Wendl MC, Deschryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, Mcdonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, and Mardis ER (2012) Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, and Bowcock AM (2013) Recurrent mutations at codon 625 of the splicing factor sf3b1 in uveal melanoma. Nat. Genet 45, 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, De Waal L, Sharifnia T, Brooks A, Greulich H, Banerji S, Zander T, Seidel D, Leenders F, Ansen S, Ludwig C, Engel-Riedel W, Stoelben E, Wolf J, Goparju C, Thompson K, Winckler W, Kwiatkowski D, Johnson BE, Janne PA, Miller VA, Pao W, Travis WD, Pass HI, Gabriel SB, Lander ES, Thomas RK, Garraway LA, Getz G, and Meyerson M (2012) Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, Mcconkey ME, Ali AM, Raza A, Yu L, Buonamici S, Smith PG, Mullally A, Wu CJ, Fleming MD, and Ebert BL (2016) Physiologic expression of sf3b1(k700e) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 30, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lee SC, and Abdel-Wahab O (2016) Therapeutic targeting of splicing in cancer. Nat. Med 22, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, Karlin KL, Dominguez-Vidana R, Hartman JD, Renwick A, Scorsone K, Bernardi RJ, Skinner SO, Jain A, Orellana M, Lagisetti C, Golding I, Jung SY, Neilson JR, Zhang XH, Cooper TA, Webb TR, Neel BG, Shaw CA, and Westbrook TF (2015) The spliceosome is a therapeutic vulnerability in myc-driven cancer. Nature 525, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, East-Seletsky A, Ali LD, Gerath WF, Pantel SE, Lizotte PH, Jiang G, Hsiao J, Tsherniak A, Dwinell E, Aoyama S, Okamoto M, Harrington W, Gelfand E, Green TM, Tomko MJ, Gopal S, Wong TC, Li H, Howell S, Stransky N, Liefeld T, Jang D, Bistline J, Hill Meyers B, Armstrong SA, Anderson KC, Stegmaier K, Reich M, Pellman D, Boehm JS, Mesirov JP, Golub TR, Root DE, and Hahn WC (2014) Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 1, 140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, Gray AJG, Bruce L, Alexander SPH, Anderton S, Bryant C, Davenport AP, Doerig C, Fabbro D, Levi-Schaffer F, Spedding M, Davies JA, and Nc I (2018) The iuphar/bps guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Res. 46, D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Good L, and Nielsen PE (1998) Inhibition of translation and bacterial growth by peptide nucleic acid targeted to ribosomal rna. Proc. Natl. Acad. Sci. U. S. A 95, 2073–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Perlman S, and Netland J (2009) Coronaviruses post-sars: Update on replication and pathogenesis. Nat. Rev. Microbiol 7, 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Woo PC, Lau SK, and Yuen KY (2006) Infectious diseases emerging from chinese wet-markets: Zoonotic origins of severe respiratory viral infections. Curr. Opin. Infect. Dis 19, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Chen M, and Manley JL (2009) Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol 10, 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Brady RA, Verma A, Meade BD, and Burns DL (2010) Analysis of antibody responses to protective antigen-based anthrax vaccines through use of competitive assays. Clin. Vaccine Immunol 17, 1390–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lorentz KM, Kontos S, Diaceri G, Henry H, and Hubbell JA (2015) Engineered binding to erythrocytes induces immunological tolerance to <em>e. Coli</em> asparaginase. Sci. Advances 1, e1500112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kontos S, Kourtis IC, Dane KY, and Hubbell JA (2013) Engineering antigens for in situ erythrocyte binding induces t-cell deletion. Proc. Natl. Acad. Sci. U. S. A 110, E60–E68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Liu S, Liu J, Ma Q, Cao L, Fattah RJ, Yu Z, Bugge TH, Finkel T, and Leppla SH (2016) Solid tumor therapy by selectively targeting stromal endothelial cells. Proc. Natl. Acad. Sci. U. S. A 113, E4079–E4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Liu S, Redeye V, Kuremsky JG, Kuhnen M, Molinolo A, Bugge TH, and Leppla SH (2005) Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat. Biotechnol 23, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Liu S, Ma Q, Fattah R, Bugge TH, and Leppla SH (2017) Anti-tumor activity of anthrax toxin variants that form a functional translocation pore by intermolecular complementation. Oncotarget 8, 65123–65131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.