

Abstract
Experimental and prospective epidemiologic evidence suggest that arsenic exposure has diabetogenic effects. However, little is known about how family exposure to arsenic may affect risk for type 2 diabetes (T2D)-related outcomes in adulthood. We evaluated the association of both maternal and offspring arsenic exposure with fasting glucose and incident T2D in 466 participants of the Strong Heart Family Study. Total arsenic (ΣAs) exposure was calculated as the sum of inorganic arsenic (iAs) and methylated (MMA, DMA) arsenic species in maternal and offspring baseline urine. Median maternal ΣAs at baseline (1989–91) was 7.6 μg/g creatinine, while median offspring ΣAs at baseline (2001–03) was 4.5 μg/g creatinine. Median offspring glucose in 2006–2009 was 94 mg/dL, and 79 participants developed T2D. The fully adjusted mean difference (95% CI) for offspring glucose was 4.40 (−3.46, 12.26) mg/dL per IQR increase in maternal ΣAs vs. 2.72 (−4.91- 10.34) mg/dL per IQR increase in offspring ΣAs. The fully adjusted odds ratio (95%CI) of incident T2D was 1.35 (1.07, 1.69) for an IQR increase in maternal ΣAs and 1.15 (0.92, 1.43) for offspring ΣAs. The association of maternal ΣAs with T2D outcomes were attenuated with adjustment for offspring adiposity markers. Familial exposure to arsenic, as measured in mothers 15-20 years before offspring follow-up, is associated with increased odds of offspring T2D. More research is needed to confirm findings and better understand the importance of family exposure to arsenic in adult-onset diabetes.
Keywords: American Indians, arsenic, indigenous populations, fasting glucose, insulin resistance, prospective cohort studies, prenatal exposures
1. Introduction
Exposure to inorganic arsenic (iAs) is a major public health concern in the US and globally. iAs is a recognized toxicant and carcinogen common in groundwater and some foods (1,2). Studies in Taiwan, Bangladesh and Mexico have found evidence to support an association between moderate to high water arsenic levels (≥50 μg/L) and type 2 diabetes (T2D) (3,4). More recently, exposure to low-moderate and low water arsenic levels (<50 μg/L) has been associated with increased risk for, and poor control of, T2D in the United States (5), Denmark (6), and Mexico (7).
The concept of the Developmental Origins of Health and Disease (DOHaD) purports that early-life influences may induce long-term metabolic changes and increase T2D risk in adulthood (8). Studies have shown that arsenic exposure likely induces epigenetic modifications, typically in the form of DNA methylation that can affect gene expression. This differential gene expression can be passed down from mother to child, thus potentially influencing offspring health later in life (9). The hypothesis that epigenetic modifications influence disease risk later in life provides part of the foundation for the DOHaD concept (8).
Epidemiologic and experimental studies have demonstrated that arsenic readily passes through the placenta and can cause developmental toxicity (10,11). Prenatal arsenic exposure has been associated with adverse birth outcomes, including low birthweight (12-19), a risk factor for T2D (20). Prenatal and early-life arsenic exposure have also been associated with various adverse health outcomes later in life, including increased risk for lung disease, cancer, and cardiovascular disease (21). No epidemiologic research has been done, however, on the effects of family exposure to arsenic on adult T2D-related outcomes.
In this study, we evaluated the association of maternal arsenic exposure with adult offspring fasting plasma glucose and insulin resistance among American Indian communities in the Strong Heart Study (SHS) and Strong Heart Family Study (SHFS). The SHS recruited adult men and women from tribal communities in Arizona, Oklahoma and North and South Dakota who were 45 to 74 years of age. The SHFS recruited relatives of SHS members who were 14 years and older. Historical data support that urine arsenic concentrations determined in samples from 1989-1991, the baseline visit of the SHS, were constant over a 10-year period (22). Based on water arsenic data, we also anticipate that urine arsenic measured at baseline for mothers can reflect decades of prior exposure, including the pregnancy period and early years of life of the offspring (8). The family connections between the SHS and the SHFS allow us to link maternal exposures, as determined in the SHS, with offspring outcomes determined in the SHFS, providing a unique opportunity to evaluate the role of maternal arsenic exposure in the current burden of T2D affecting many tribal and rural communities in the United States and globally. We hypothesized that higher urine arsenic levels in mothers would be associated with increased fasting plasma glucose levels and increased insulin resistance in their adult children.
2. Research Design and Methods
2.1. Study Population
The SHS is a population-based prospective cohort study of tribal members from American Indian communities in Arizona, Oklahoma and North and South Dakota. The cohort consists of 4,549 participants ages 45-74 at baseline from 13 tribes who were recruited and examined in 1989-1991. The SHFS is an extension of the SHS, in which family members of SHS participants who had at least 5 living siblings including 3 original SHS participants were invited to participate. This cohort consists of 3,838 participants from 96 families who were recruited and examined in 1998-1999 and 2001-2003. Participants recruited in 1998-1999 had follow-up visits in 2001-2003 and 2006-2009, while participants recruited in 2001-2003 had one follow-up visit in 2009-2009. Maternal arsenic values were based on SHS visit 1 (1989-1991), and offspring arsenic values were based on the baseline SHFS visit (1998-1999 or 2001-2003). Fasting glucose and homeostasis model assessment for insulin resistance (HOMA2-IR) values were obtained from the follow-up SHFS visit (2006-2009). Due to the constant arsenic exposure over decades in these populations, the maternal arsenic values may reflect in utero exposure. Details about the methods and design of the two studies have been previously published (22,23,24). Protocols were approved by the Indian Health Service, institutional review boards, and participating communities. All participants provided informed consent.
Pedigree information was used to link SHFS participants with at least one parent in the SHS. Due to tribal request, data from one tribe was not used (n = 1033 in SHS, n = 919 in SHFS). Offspring ages 14-93 (median 36) who had mothers in the SHS were selected for this study (n = 1886). We excluded those with missing offspring ΣAs (n = 476) and maternal ΣAs (n = 896) measurements, as well as offspring missing visit 5 (2006-2009) fasting glucose (n = 12) and HOMA2-IR (n = 1). We further excluded those missing other covariates (n = 35), including offspring and maternal eGFR, BMI, and waist circumference, and maternal fasting glucose. Finally, we excluded those with incident diabetes at visit 5 (n = 79) from analyses with HOMA2-IR. After all exclusions, a total of 466 participants were used in fasting glucose analyses, and 387 participants were used in HOMA2-IR analyses presented here (Supplemental Figure 1). Participants who were excluded did not differ appreciably from those who were included in the analysis (Supplemental Table 2).
2.2. Urine Arsenic Measurements
Spot urine samples were collected in polypropylene tubes, frozen within 1–2 hours of collection, shipped in dry ice, and stored at −70°C in the Penn Medical Laboratory, MedStar Research Institute, Washington, DC, USA (23). The freezers were operating under a strict quality control system to guarantee secure sample storage. For arsenic analyses, urine samples were thawed, and up to 1.0 mL was transferred to a small vial, transported on dry ice to the Trace Element Laboratory, Graz University, Austria, and stored at < −70°C until analysis. Quality control and quality assurance methods have been described previously (24).
We measured urine arsenic species concentrations of arsenite, arsenate, methylarsonate (MMA), and dimethylarsinate (DMA) using high performance liquid chromatography/inductively coupled plasma-mass spectrometry (HPLC/ICPMS). Urine arsenobetaine was measured using HPLC/ICPMS together with other more rare arsenic cations. The concentrations measured for mother and offspring arsenobetaine were low (median (IQR): 0.75 (0.50 – 1.50) μg/L for mothers and 0.55 (0.34 – 1.25) μg/L for offspring), confirming that seafood consumption in this population is infrequent. We used the sum of inorganic and methylated (MMA and DMA) arsenic species as the biomarker of inorganic arsenic exposure.
The inter-assay coefficients of variation, evaluated by including the same reference urine sample in each batch of samples, were better than 5% for all species (24). The limits of detection (LOD) for arsenite, arsenate, MMA, DMA, and urine arsenobetaine were 0.1 μg/L. Of the 466 participants used for the analyses, 19 (4.1%), 14 (3.0%), and 1 (0.21%) had offspring arsenobetaine, iAs, and MMA values below the LOD, respectively. Six (1.3%) and 19 (4.1%) participants had maternal arsenobetaine and iAs values below the LOD, respectively. No participants had offspring DMA, or maternal MMA or DMA below the LOD. For participants with concentrations below the LOD, we imputed the corresponding limit of detection divided by the square root of two.
2.3. Fasting Glucose and Insulin Resistance
Fasting plasma samples were collected from all participants at each examination after a 12-hour fast and stored at <−70°C (25). Glucose was determined by enzymatic methods using reagent kits from Boehringer Mannheim Diagnostic (Indianapolis, IN) on a chemistry analyzer (23). Insulin was measured using overnight radioimmunoassay. HOMA2-IR values were calculated with the computed solved model for HOMA2-IR using fasting glucose and insulin values (26). HOMA2-IR measurements at follow-up were excluded for participants with T2D because HOMA2-IR correlates well with insulin sensitivity in populations without T2D but not among those with T2D (27). Fasting glucose was measured in all participants. Both variables were assayed at MedStar Research Institute, Washington, DC (28).
T2D status was also assessed at visit 5 of the SHFS, defined as fasting plasma glucose ≥ 126 mg/dL, self-reported physician diagnosis or self-reported use of insulin or oral diabetes treatment (5). Of those included in this study, 79 had been diagnosed with T2D at visit 5, 103 had impaired fasting glucose levels (100 ≤ fasting plasma glucose < 126 mg/dL), and 284 had normal fasting glucose levels. Due to the small number of participants with T2D, we lacked power to directly assess diabetes as the outcome. Instead, we chose to use continuous fasting glucose and HOMA2-IR as outcomes for this study.
2.4. Other Variables
Baseline information on sociodemographic data (age, sex, study region), smoking history (never, former and current smoking), and body mass index (kg/m2) were obtained during the SHS and SHFS questionnaires and physical exams collected and performed by trained and certified personnel using standardized protocols (23,25). Urine creatinine was measured at the laboratory of the National Institute of Diabetes and Digestive and Kidney Diseases Epidemiology and Clinical Research Branch (Phoenix, AZ, USA) using an automated alkaline picrate methodology run on a rapid flow analyzer (23).
2.5. Statistical Methods
We estimated inorganic arsenic exposure (ΣAs) as the sum of urine iAs, MMA, and DMA. We divided ΣAs by urine creatinine to account for urine dilution. Because the distribution of ΣAs was right-skewed, we log-transformed the variable for analysis of both maternal and offspring ΣAs.
Maternal arsenic values were based on SHS visit 1 (1989-1991). Offspring arsenic values were based on the baseline SHFS visit (1998-1999 [n = 147] or 2001-2003 [n = 319]). Fasting glucose and HOMA2-IR values were obtained from the follow-up SHFS visit (2006-2009).
We used Spearman correlation coefficients to describe the unadjusted association between maternal and offspring arsenic exposure, as well as between maternal and offspring arsenic exposure and offspring fasting glucose and HOMA2-IR.
We used generalized estimation equations (GEE) with an independent correlation structure to assess the associations of both maternal ΣAs and offspring ΣAs with offspring fasting glucose levels. A sensitivity analysis using an exchangeable correlation structure showed similar results. We estimated the mean difference in offspring glucose levels per interquartile range (IQR) increase in ΣAs. We also used GEE to assess the associations of both maternal ΣAs and offspring ΣAs with offspring log-transformed HOMA2-IR. Here we estimated the geometric mean ratio (GMR) of HOMA2-IR per IQR increase in ΣAs by multiplying the beta coefficients by the IQR in log-transformed ΣAs and then exponentiating the coefficients.
Models were run with progressive adjustments. First, we adjusted for offspring sex and age at baseline as well as for maternal eGFR for maternal models or for offspring eGFR for offspring models. Adjustment for eGFR was included because kidney function impairs the excretion of ΣAs levels in the urine (29). Second, we further adjusted offspring and maternal ΣAs models for maternal BMI and maternal fasting glucose at baseline. This was to ensure that the associations observed were due to maternal urine arsenic, not to maternal BMI, and because mothers with increased fasting glucose levels may have different urinary As excretion compared to those with normal fasting glucose levels. Third, to assess the independent association of maternal arsenic exposure from adult offspring arsenic exposure, the models for maternal urine arsenic were adjusted for offspring baseline urine arsenic levels (2001-2003 for most SHFS participants), and models for offspring urine arsenic were adjusted for maternal urine arsenic (1989-1991). Additionally, a sensitivity analysis controlling for year of offspring ΣAs measurement was performed to ensure that a possible decrease in arsenic exposure during this time did not affect the associations.
Offspring waist circumference and offspring BMI were added separately to the fully adjusted model (model 3), to assess whether the association of maternal arsenic exposure with offspring glucose and HOMA2-IR could be mediated by offspring adiposity. Models with possible mediator variables were compared to fully adjusted model to determine whether addition of mediator variables attenuated the association. Another sensitivity analysis restricted offspring to BMI < 25 kg/m2 to further investigate whether the association is consistent among participants with lower BMI. A third sensitivity analysis was performed to investigate whether offspring diabetes medication usage might confound the relationships of offspring ΣAs and maternal ΣAs with offspring fasting glucose. A four-level variable indicating whether participants were taking oral diabetes medication, insulin, both, or neither was added to the fully adjusted model to adjust for offspring diabetes medication usage. Finally, one further sensitivity analysis stratified the sample by study center to ensure that the directions of the associations are consistent across every study site.
Exploratory interaction analyses were additionally performed. Effect modification by sex and study center was assessed by including interaction terms to fully adjusted models for both maternal and offspring exposure with HOMA2-IR and fasting glucose outcomes. We additionally assessed the possible interaction between maternal and offspring ΣAs exposure using two types of models. First, we recoded a 4-level categorical variable of combined exposure: category 1 was defined as offspring and maternal ΣAs below the median, category 2 was offspring ΣAs below the median but maternal ΣAs above the median, category 3 was offspring ΣAs above the median but maternal ΣAs below the median, and category 4 was offspring and maternal ΣAs above the median. Second, we also ran models for maternal and offspring continuous ΣAs variables including an interaction term for both of them in the model.
Finally, we conducted exploratory analyses for offspring incident T2D and incident T2D plus IFG compared with NFG at visit 5. Here we estimated odds ratio (OR) of T2D or of T2D+IFG per IQR increase in ΣAs by multiplying the beta coefficients by the IQR and then exponentiating the coefficients Though the number of SHFS participants with T2D (79) and with T2D+IFG (182) is relatively small, this analysis directly explores the relationship between maternal arsenic exposure and adult T2D status later in life.
All statistical analyses were performed using the R software version 3.5.2. GEE analysis was performed using the ‘geepack’ package.
3. Results
3.1. Participant Characteristics
Offspring age ranged from 15 to 69 (mean: 41) years old, while mothers’ age ranged from 45 to 73 (mean: 56). Mean (standard deviation [SD]) offspring and maternal BMI were 31.6 (7.1) kg/m2 and 31.9 (5.7) kg/m2, respectively. Sixty percent of offspring were female (Table 1).
Table 1:
Median (IQR) values for maternal and offspring age, BMI, ΣAs, estimated glomerular filtration rate, waist circumference, and fasting glucose
| Maternal Median (IQR) |
Offspring Median (IQR) |
|
|---|---|---|
| Age (years) | 55.60 (49.62, 62.60) | 40.61 (35.81, 46.60) |
| BMI (kg/m2) | 31.64 (27.77, 35.55) | 30.48 (26.78, 35.13) |
| ΣAs (μg/g creatinine) | 7.57 (5.13, 12.91) | 4.53 (2.98, 7.54) |
| eGFR (mL/min) | 79.30 (69.37, 91.97) | 88.96 (77.32, 97.34) |
| Waist Circ. (cm) | 106 (98, 115) | 99.00 (91, 112.75) |
| Fasting Glc (mg/dL) | 116 (103, 177) | 94.00 (87, 108) |
The median (interquartile range [IQR]) baseline concentrations of maternal and offspring ΣAs were 7.57 (5.13, 12.91) and 4.53 (2.98, 7.54) μg/g creatinine, respectively. (Table 1). Total urinary arsenic concentrations were highest among mothers from Arizona (median 21.8 μg/g creatinine), followed by mothers from North and South Dakota (9.2 μg/g creatinine) and Oklahoma (6.2 μg/g creatinine) (Figure 1). This is expected, as groundwater in Arizona has higher arsenic concentrations than the other states in the SHS (22). The analyses stratified by center showed that the associations remained consistent across centers, with somewhat stronger associations in North/South Dakota, the region with higher within region variability in arsenic exposure. (Supplemental Table 3).
Figure 1:
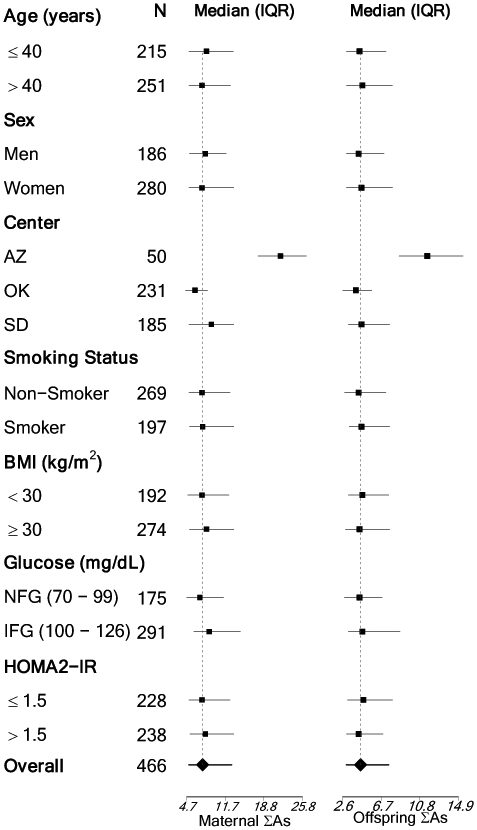
Median and interquartile range (IQR) for maternal and offspring total arsenic by offspring characteristics
Age, smoking status, and BMI were measured at baseline, while fasting glucose and HOMA2-IR are from SHFS visit 5
Maternal ΣAs and offspring ΣAs were significantly correlated (r = 0.48, p-value < 0.001) (Figure 2). At study visit 5, the median (IQR) offspring glucose and HOMA2-IR were 94 (87 – 108) μg/dL and 1.38 (0.78 – 2.22), respectively.
Figure 2:

Spearman correlation matrix for maternal total arsenic, offspring total arsenic, offspring glucose, and offspring HOMA2-IR
3.2. Association of Maternal and Offspring ΣAs with Offspring Fasting Glucose
Maternal ΣAs and offspring ΣAs were significantly correlated with offspring fasting glucose (r = 0.19, p-value < 0.001, and r = 0.12 respectively, p-value = 0.008) (Figure 2). After adjustment for offspring sex and age, and for maternal eGFR, the mean difference (95% CI) in offspring glucose was 7.83 (0.01, 15.65) mg/dL for an interquartile range (IQR) increase in maternal ΣAs (Table 2). While adjustment for maternal BMI and maternal fasting glucose had a minor impact (mean difference (95% CI) 6.79 (−0.70, 14.28) mg/dL), further adjustment for offspring ΣAs attenuated the effect estimate to 4.40 (−3.46, 12.26) mg/dL. Mean differences for the relationship between adult offspring ΣAs and fasting glucose followed a similar trend, with a minimally and fully adjusted mean differences (95% CI) of 5.40 (−1.75, 12.55) mg/dL and 2.72 (−4.91, 10.34) mg/dL respectively (Table 2).
Table 2:
Mean difference (95% CI) in offspring glucose for IQR increase in total ΣAs, and geometric mean ratio (95% CI) of offspring HOMA-IR by ΣAs.
| Glucose (n = 466) | HOMA2-IR (n = 387) | |||
|---|---|---|---|---|
| Maternal ΣAs | Offspring ΣAs | Maternal ΣAs | Offspring ΣAs | |
| Model 1 | 7.83 (0.01, 15.65) | 5.40 (−1.75, 12.55) | 1.04 (0.92, 1.17) | 0.93 (0.84, 1.03) |
| Model 2 | 6.79 (−0.70, 14.28) | 4.73 (−2.30, 11.75) | 1.00 (0.90, 1.11) | 0.90 (0.81, 1.00) |
| Model 3 | 4.40 (−3.46, 12.26) | 2.72 (−4.91, 10.34) | 1.04 (0.93, 1.16) | 0.89 (0.80, 0.99) |
| Model 4 | 2.12 (−4.36, 8.59) | 4.88 (−2.07, 11.84) | 1.02 (0.93, 1.13) | 0.96 (0.87, 1.07) |
| Model 5 | 1.68 (−4.82, 8.18) | 4.25 (−2.68, 11.18) | 1.01 (0.93, 1.11) | 0.95 (0.86, 1.04) |
Model 1 is adjusted for offspring sex and age at baseline visit, and maternal eGFR at baseline for maternal models or offspring eGFR for offspring models
Model 2 = Model 1 + maternal BMI (kg/m2) and maternal fasting glucose (mg/dL) at baseline
Model 3 = Model 2 + offspring ΣAs in maternal models or maternal ΣAs in offspring models (fully adjusted model)
Model 4 = Model 3 + offspring BMI at baseline
Model 5 = Model 3 + offspring waist circumference at baseline
Geometric mean ratios (95% CI) are reported per an increase equal to the IQR in ΣAs distribution.
Generalized estimation equations (GEE) used to account for family clustering.
In the maternal ΣAs and offspring glucose models, the addition of offspring BMI and waist circumference attenuated the mean difference (95% CI) to 2.12 (−4.36, 8.59) mg/dL and 1.68 (−4.82, 8.18) mg/dL for an IQR increase in maternal ΣAs respectively. In the offspring ΣAs and glucose models, the addition of BMI and waist circumference strengthened the association to a mean difference (95% CI) of 4.88 (−2.07, 11.84) mg/dL and 4.25 (−2.68, 11.18) mg/dL (Table 2).
When restricting to individuals with BMI < 25 k/m2, the mean difference in fasting glucose levels was larger in offspring models than in maternal models, though none were statistically significant (Supplemental Table 3). The addition of offspring diabetes medication usage attenuated the mean difference (95% CI) in fasting glucose levels to 3.89 (−2.43, 10.20) mg/dL in maternal models, and 1.79 (−4.48, 8.06) mg/dL in offspring models (Supplemental Table 3).
3.3. Association of Maternal and Offspring ΣAs with Offspring HOMA2-IR
Neither maternal ΣAs nor offspring ΣAs were correlated with offspring HOMA2-IR (Figure 2). In linear regression models, after adjusting for offspring sex and age and maternal eGFR, the GMR (95% CI) of offspring HOMA2-IR was 1.04 (0.92, 1.17) for an IQR increase in maternal ΣAs, and it was 0.93 (0.84, 1.03) for an IQR increase in offspring ΣAs (Table 2). The fully adjusted model was largely unchanged for maternal ΣAs (GMR (95% CI): 1.04 (0.93, 1.16)), and was slightly strengthened for offspring ΣAs (GMR (95% CI): 0.89 (0.80, 0.99)) (Table 2).
For maternal ΣAs, the addition of offspring BMI and waist circumference attenuated the GMR (95% CI) to 1.02 (0.93, 1.13) and 1.01 (0.93, 1.11) for an IQR increase in maternal ΣAs respectively. Similarly, for offspring ΣAs, the addition of BMI and waist circumference attenuated the association to a GMR (95% CI) of 0.96 (0.87, 1.07) and 0.95 (0.86, 1.04) (Table 2).
When restricting to individuals with BMI < 25 kg/m2, the GMR for HOMA2-IR was larger in offspring models than in maternal models, with maternal ΣAs exposure showing an inverse association (GMR (95% CI): 0.75 (0.59, 0.95) (Supplemental Table 3).
We observed consistent results in the sensitivity analysis adjusting for year of offspring ΣAs measurement (data not shown).
3.4. Effect Modification Analysis
Compared to participants with low maternal and low offspring ΣAs levels, the mean difference in fasting plasma glucose was 8.16 (95% CI: −4.12, 20.44) mg/dL for those with both high maternal and offspring ΣAs, 2.16 (95% CI: −9.04, 13.36) mg/dL for those with high maternal but low offspring ΣAs, and −1.23 (95% CI: −11.88, 9.43) mg/dL for those with low maternal but high offspring ΣAs (Supplemental Table 1).
Compared to participants with low maternal and low offspring ΣAs levels, the GMR for HOMA2-IR was 0.83 (95% CI: 0.55, 1.25) for those with both high maternal and offspring ΣAs, 1.04 (95% CI: 0.56, 1.92) for those with high maternal but low offspring ΣAs, and 0.81 (95% CI: 0.54, 1.21) for those with low maternal but high offspring ΣAs (Supplemental Table 1).
The coefficient (95% CI) for the interaction between continuous maternal and offspring ΣAs on fasting glucose was 0.05 (−0.01, 0.12) (p-value = 0.12), and on HOMA2-IR was −0.22 (−0.63, 0.19) (p-value = 0.53). Interaction by sex and study center was assessed in an exploratory manner (and limited due to the small sample size) with no significant interaction detected, although power for interactions might be limited due to small sample sizes, especially in Arizona.
3.5. Incident Diabetes
In fully adjusted logistic regression models, the OR (95% CI) of incident offspring T2D was 1.35 (1.07, 1.69) for an IQR increase in maternal ΣAs, and it was 1.15 (0.92, 1.43) for an IQR increase in offspring ΣAs. The fully adjusted OR (95% CI) of incident offspring T2D + IFG was 1.42 (1.18, 1.72) for an IQR increase in maternal ΣAs, and it was 1.03 (0.85, 1.24) for an IQR increase in offspring ΣAs (Table 3)
Table 3:
Odds ratios (95% CI) of incident diabetes or diabetes + IFG at visit 5 by urinary ΣAs at baseline
| Diabetes (79/387) | Diabetes and IFG (182/284) | |||
|---|---|---|---|---|
| (n = 466) | Maternal ΣAs | Offspring ΣAs | Maternal ΣAs | Offspring ΣAs |
| Model 1 | 1.43 (1.16, 1.78) | 1.27 (1.03, 1.58) | 1.45 (1.15, 1.83) | 1.18 (0.99, 1.40) |
| Model 2 | 1.43 (1.15, 1.78) | 1.26 (1.01, 1.56) | 1.42 (1.18 1.71) | 1.15 (0.96, 1.38) |
| Model 3 | 1.35 (1.07, 1.69) | 1.15 (0.92, 1.43) | 1.42 (1.18 1.72) | 1.03 (0.85, 1.24) |
| Model 4 | 1.20 (0.95, 1.53) | 1.26 (1.00, 1.58) | 1.32 (1.09 1.59) | 1.12 (0.91, 1.37) |
| Model 5 | 1.18 (0.94, 1.49) | 1.23 (0.98, 1.54) | 1.30 (1.08 1.56) | 1.09 (0.88, 1.35) |
Model 1 is adjusted for offspring sex and age at baseline visit, and maternal eGFR at baseline for maternal models or offspring eGFR for offspring models
Model 2 = Model 1 + maternal BMI (kg/m2) and maternal fasting glucose (mg/dL) at baseline
Model 3 = Model 2 + offspring ΣAs in maternal models or maternal ΣAs in offspring models (fully adjusted model)
Model 4 = Model 3 + offspring BMI at baseline
Model 5 = Model 3 + offspring waist circumference at baseline
Odds ratios (95% CI) are reported per an increase equal to the IQR in ΣAs distribution.
Numbers in brackets are cases and non-cases
GEE used to account for family clustering.
The addition of offspring BMI and offspring waist circumference supported partial mediation by both variables in maternal ΣAs models, which is consistent with results from glucose analyses. In offspring models, the association increased with the addition of offspring BMI and waist circumference, which was also consistent with other analyses (Table 3).
4. Discussion
In this population of American Indians from Oklahoma, Arizona, and North and South Dakota, maternal ΣAs was prospectively associated with offspring fasting glucose and incident diabetes. Results were consistent after adjustment for offspring age and sex, and maternal eGFR, as well as, although no longer significant for fasting glucose, after adjustment for maternal BMI and maternal glucose. Higher offspring ΣAs was non-significantly associated with higher offspring glucose and incident T2D. Maternal ΣAs was positively but non-significantly associated with HOMA-IR, while offspring ΣAs was inversely and significantly associated with HOMA-IR. Both for plasma glucose and incident T2D, the magnitude of the association was markedly attenuated with adjustment for offspring BMI –and especially for waist circumference–suggesting possible mediation by central adiposity. Although limited by a small sample size and limited statistical power, these findings support the DOHaD hypothesis that in-utero exposures can lead to adverse health outcomes later in life.
Urinary arsenic levels were lower in the offspring (median ΣAs 4.53 μg/g creatinine) compared to the maternal (7.57 μg/g creatinine). These reductions in exposure are positive for the population, however, results from this study support that maternal arsenic exposure may affect risk for adult T2D outcomes. The mechanism behind these associations are unknown, but it is hypothesized that arsenic-induced epigenetic DNA modifications passed down from mother to offspring could influence offspring T2D-related outcomes (30, 31).
Recent research has shown that various mechanisms could explain the relationship between As exposure and genetic imprinting. Arsenic exposure can both increase and decrease promotor region methylation in various genes, resulting in either downregulation or upregulation of those genes, and thus various health effects. These epigenetic changes have the potential to be passed down across generations (9). Epigenetic changes in several genes have been associated with both As exposure and increased T2D risk through various pathways. Differential methylation of genes involved in regulation of insulin production (PDX1, INS) and secretion (VAMP2) has been associated with As exposure (32). Future research should investigate these associations further, as well as explore other potential imprinted genes related to As exposure and T2D development.
One potential mechanism for the relationship between maternal ΣAs exposure and adult offspring T2D-related outcomes is through low birth weight. Numerous studies have shown that maternal arsenic exposure is related to low birth weight (12, 15), some suggesting that a molecular mechanism for this involves both increase and decreased gene expression. One study in particular showed that in utero As exposure was associated with increased expression of the gene AQP9, which increases cellular As uptake. Increased expression of AQP9 is followed by decreased expression of the gene ENPP2, which is associated with decreased infant birth weight (19). Other potential mechanisms exist, and confirmation of these genetic biomarkers will require further research.
Both low and high birth weight have been widely associated with development of T2D later in life, though the mechanisms of this association are debated (33). Potential mechanisms involve impaired programming of neuroendocrine circuits in infants. One widely supported hypothesis is that low birthweight babies are subjected to overfeeding, resulting in both rapid weight gain, which is associated with overweight and obesity later in life, and impaired programming of circuits regulating appetite control, body weight, and metabolism (34). High birthweight babies are also likely subject to this impaired programming, but due to exposure to maternal hyperglycemia in utero (35). In our study, unfortunately data on birth weight was not available. In analyses restricted to participants with BMI < 25 kg/m2, the association of maternal As exposure with fasting glucose was weaker compared to the overall association, supporting that the association between maternal arsenic exposure and offspring glucose could be mediated by an impact of maternal arsenic on offspring BMI.
Additional research should assess the possible mediation of low birth weight and offspring weight gain on the relationship between maternal arsenic exposure and adult T2D and related outcomes.
In this study, we observed opposite effects for maternal As and offspring As on insulin resistance. The results for maternal As, but not for offspring As, are consistent with a higher risk for insulin resistance, a major underlying mechanism for T2D. The inverse association between adult arsenic exposure and HOMA2-IR was also found in studies in Mexico (4), and could potentially be related to As affecting pancreatic function.
To our knowledge, this study is the first to prospectively evaluate the association between maternal arsenic exposure and adult T2D-related outcomes. Previous studies have shown significant effects of maternal arsenic exposure during pregnancy on decreased birth weight (36), increased risk of infection in infants, higher infant mortality (37), and increased occurrence of lung disease, cardiovascular disease, and cancer in childhood and later in life (38, 39, 40). Our findings provide evidence that maternal arsenic exposure may also play a role in risk for adult-onset T2D. Further, our results suggest that offspring BMI and waist circumference may partially mediate the associations of maternal ΣAs with offspring fasting glucose and insulin resistance, as well as offspring ΣAs with insulin resistance.
Strengths of this study include the standardized protocol, high quality of laboratory methods, and length of follow-up time to allow for analysis of outcomes in adult offspring. However, there are also several limitations. First, there is a slight possibility of collinearity between maternal ΣAs and offspring ΣAs, as both exposures are significantly correlated. Their correlation, however, is moderate (r= 0.48). Additionally, maternal exposure was estimated from a single maternal urine sample. Although studies have shown urine ΣAs to be relatively stable over time (22), we were unable to measure maternal arsenic during pregnancy as the participants in the SHS original cohort were recruited when they were 45 years and older. For this reason, we were unable to investigate the role of arsenic exposure occurring during pregnancy. In a sensitivity analysis adjusting for T2D medication, the associations remained, although attenuated, after adjustment. Finally, other potential confounders include diet and genetics. However, dietary sources of arsenic in the population have shown to explain less than 4% of urinary As levels, supporting water as the main source of arsenic in SHS communities (41).
In conclusion, maternal arsenic exposure was non-significantly associated with adult offspring fasting glucose levels among a small sample of men and women from American Indian communities in Arizona, Oklahoma, and North and South Dakota. The association was independent of offspring adult arsenic exposure and somewhat stronger for maternal exposure, although the associations were not significant. These non-significant associations were also attenuated after adjustment for offspring BMI and waist circumferences, suggesting potential mediation by central adiposity. Further research is necessary to confirm these findings and to better understand the biological mechanisms behind the observed associations, including the possible role of long-term epigenetic effects of early life arsenic exposure.
Supplementary Material
Highlights.
Maternal arsenic exposure may play a role in risk for adult-onset type 2 diabetes
Maternal arsenic exposure has a greater effect on diabetes than adult arsenic
Adult offspring BMI may mediate the association
Long-term epigenetic effects of maternal arsenic exposure are likely involved
Acknowledgements
This study was supported by the National Institute of Environmental Health Sciences (P42ES010349, P30ES009089, R01ES028758, R01ES025216).
Footnotes
No potential conflicts of interest relevant to this article were reported.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.NTP (National Toxicology Program). Report on carcinogens, Fourteenth edition. Research Triangle Park, NC: US Department of Health and Human Services, Public Health Service, 2016. [Google Scholar]
- 2.IARC (International Agency for Research on Cancer). Some drinking water disinfectants and contaminants, including arsenic IARC Monographs on the evaluation of carcinogenic risks to humans. Lyon: IARC, 2002. [PMC free article] [PubMed] [Google Scholar]
- 3.Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, et al. Evaluation of the association between arsenic and diabetes: a National Toxicology Program workshop review. Environ Health Perspect 2012; 120:1658–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Del Razo LM, Garcia-Vargas GG, Valenzuela OL, Hernandez Castellanos E, et al. Exposure to arsenic in drinking water is associated with increased prevalence of diabetes: a cross-sectional study in the Zimapán and Lagunera regions in Mexico. Environ Health 2011; 10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grau-Perez M, Kuo CC, Gribble MO, et al. Association of low-moderate arsenic exposure and arsenic metabolism with incident diabetes and insulin resistance in the Strong Heart Family Study. Env Health Perspect 2017; 125:127004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brauner EV, Nordsborg RB, Andersen ZJ, Tjonneland A, Loft S, Raaschou-Nielsen O. Long term exposure to low-level arsenic in drinking water and diabetes incidence: A prospective study of the Diet, Cancer, and Health Cohort. Env Health Perspect 2014;122:1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coronado-González JA, Del Razo LM, García-Vargas G, Sanmiguel-Salazar F, Escobedo-de la Peña J. Inorganic arsenic exposure and type 2 diabetes mellitus in Mexico. Env Research 2007; 104: 383–389. [DOI] [PubMed] [Google Scholar]
- 8.Wadhwa PD, Buss C, Entringer S, Swanson J. Developmental Origins of Health and Disease: Brief History of the Approach and Current Focus on Epigenetic Mechanisms. Semon Reprod Med 2009; 27:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smeester L, Yosim AE, Nye MD, Hoyo C, Murphy SK, Fry RC. Imprinted Genes and the Environment: Links to the Toxic Metals Arsenic, Cadmium and Lead. Genes 2014; 5:477–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hall M, Gamble M, Slavkovich V, Liu X, Levy D, Cheng Z, van Geen A, Yunus M, Rahman M, Pilsner JR, and Graziano J, Determinants of arsenic metabolism: blood arsenic metabolites, plasma folate, cobalamin, and homocysteine concentrations in maternal-newborn pairs. Environ Health Perspect, 2007. 115(10): p. 1503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Concha G, Vogler G, Lezcano D, Nermell B, and Vahter M, Exposure to inorganic arsenic metabolites during early human development. Toxicol Sci, 1998. 44(2): p. 185–90. [DOI] [PubMed] [Google Scholar]
- 12.Hopenhayn C, Ferreccio C, Browning SR, Huang B, Peralta C, Gibb H, and Hertz-Picciotto I, Arsenic exposure from drinking water and birth weight. Epidemiology, 2003. 14(5): p. 593–602. [DOI] [PubMed] [Google Scholar]
- 13.Yang CY, Chang CC, Tsai SS, Chuang HY, Ho CK, and Wu TN, Arsenic in drinking water and adverse pregnancy outcome in an arseniasis-endemic area in northeastern Taiwan. Environ Res, 2003. 91(1): p. 29–34. [DOI] [PubMed] [Google Scholar]
- 14.Rahman A, Vahter M, Smith AH, Nermell B, Yunus M, El Arifeen S, Persson LA, and Ekstrom EC, Arsenic exposure during pregnancy and size at birth: a prospective cohort study in Bangladesh. Am J Epidemiol, 2009. 169(3): p. 304–12. [DOI] [PubMed] [Google Scholar]
- 15.Huyck KL, Kile ML, Mahiuddin G, Quamruzzaman Q, Rahman M, Breton CV, Dobson CB, Frelich J, Hoffman E, Yousuf J, Afroz S, Islam S, and Christiani DC, Maternal arsenic exposure associated with low birth weight in Bangladesh. J Occup Environ Med, 2007. 49(10): p. 1097–104. [DOI] [PubMed] [Google Scholar]
- 16.Claus Henn B, Ettinger AS, Hopkins MR, Jim R, Amarasiriwardena C, Christiani DC, Coull BA, Bellinger DC, and Wright RO, Prenatal Arsenic Exposure and Birth Outcomes among a Population Residing near a Mining-Related Superfund Site. Environ Health Perspect, 2016. 124(8): p. 1308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan H, Piao F, Zhang X, Li X, Li Q, Xu L, Kitamura F, and Yokoyama K, Prenatal exposure to arsenic and its effects on fetal development in the general population of Dalian. Biol Trace Elem Res, 2012. 149(1): p. 10–5. [DOI] [PubMed] [Google Scholar]
- 18.Laine JE, Bailey KA, Rubio-Andrade M, Olshan AF, Smeester L, Drobna Z, Herring AH, Styblo M, Garcia-Vargas GG, and Fry RC, Maternal arsenic exposure, arsenic methylation efficiency, and birth outcomes in the Biomarkers of Exposure to ARsenic (BEAR) pregnancy cohort in Mexico. Environ Health Perspect, 2015. 123(2): p. 186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fei DL, Koestler DC, Li Z, Giambelli C, Sanchez-Mejias A, Gosse JA, Marsit CJ, Karagas MR, and Robbins DJ, Association between In Utero arsenic exposure, placental gene expression, and infant birth weight: a US birth cohort study. Environ Health, 2013. 12: p. 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi Donghua, Fang Hongjuan, Zhao Yaqun, Zhong Liyong. Birth weight and type 2 diabetes: A meta-analysis. Exp Ther Med. 2017. December; 14(6): 5313–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naujokas M, Anderson B, Ahsan H, et al. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem. Env Health Perspect 2013; 121:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navas-Acien A, Umans JG, Howard BV, Goessler W, et al. Urine arsenic concentrations and species excretion patterns in American Indian communities over a 10-year period: the Strong Heart Study. Env Health Perspect 2009; 117:1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee ET, Welty TK, Fabsitz R, Cowan LD, Le NA, et al. The Strong Heart Study. A study of cardiovascular disease in American Indians: design and methods. Am J Epidemiol 1990; 132:141–1155. [DOI] [PubMed] [Google Scholar]
- 24.Scheer J, Findenig S, Goessler W, Francesconi K, Howard B, Umans JG, Pollak J, Tellez-Plaza M, Silbergeld EK, Guallar E, Navas-Acien A. Arsenic species and selected metals in human urine: validation of HPLC/ICPMS and ICPMS procedures for a long-term population-based epidemiological study. Anal Methods 2012; 4:406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howard BV, Welty TK, Fabsitz RR, Cowan LD, Oopik AJ, Le N, Yeh J, Savage PJ, Lee ET. Risk Factors for Coronary Heart Disease in Diabetic and Nondiabetic Native Americans. Diabetes 1992; 41:4–11. [DOI] [PubMed] [Google Scholar]
- 26.Levy JC, Matthews DR, Hermans MP. Correct homeostasis model assessment (HOMA). Diabetes Care 1998; 21:2192–2192. [DOI] [PubMed] [Google Scholar]
- 27.Resnick HE, Bergman RN, Henderson JA, Nez-Henderson P, Howard BV. Utility of a surrogate measure of insulin resistance in American Indians: the Strong Heart Study. Ethn Dis 2002;12(4):523–529. [PubMed] [Google Scholar]
- 28.North KE, Howard BV, Welty TK, Best LG, et al. Genetic and Environmental Contributions to Cardiovascular Disease Risk in American Indians: The Strong Heart Family Study. Am J Epidemiol 2003; 157:303–314. [DOI] [PubMed] [Google Scholar]
- 29.Zheng LY, Umans JG, Yeh F, Francesconi KA, et al. The Association of Urine Arsenic with Prevalent and Incident Chronic Kidney Disease: Evidence from the Strong Heart Study. Epidemiology 2015; 26:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kile ML, Houseman EA, Baccarelli AA, Quamruzzaman Q, et al. Effect of prenatal arsenic exposure on DNA methylation and leukocyte subpopulations in cord blood. Epigenetics 2014; 9:774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kushal A, Zhang H, Karmaus WJJ, Everson TM, et al. Genome-wide DNA methylation at birth in relation to in utero arsenic exposure and the associated health in later life. Env Health 2017; 16:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin EM, Styblo M, Fry RC. Genetic and epigenetic mechanisms underlying arsenic-associated diabetes mellitus: a perspective of the current evidence. Epigenomics 2017; 9:701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harder T, Rodekamp E, Schellong K, Dudenhausen J, Plagemann A. Birth Weight and Subsequent Risk of Type 2 Diabetes: A Meta-Analysis. Am J Epidemiol 2007; 165:849–857. [DOI] [PubMed] [Google Scholar]
- 34.Plagemann A, Harder T, Rake A, et al. Perinatal increase of hypothalamic insulin, acquired malformation of hypothalamic galaninergic neurons, and syndrome X-like alterations in adulthood of neonatally overfed rats. Brain Res 1999; 836:146–55. [DOI] [PubMed] [Google Scholar]
- 35.Silverman BL, Rizzo T, Green OC, et al. Long-term prospective evaluation of offspring of diabetic mothers. Diabetes 1991; 40(suppl 2):121–5. [DOI] [PubMed] [Google Scholar]
- 36.Rahman A, Vahter M, Smith AH, Nermell B, Yunus M, El Arifeen S, Persson L, Ekström E. Arsenic Exposure During Pregnancy and Size at Birth: A Prospective Cohort Study in Bangladesh. Am J Epidemiol 2009; 169:304–312. [DOI] [PubMed] [Google Scholar]
- 37.Farzan SF, Korrick S, Li Z, Enelow R, Gandolfi AJ, Madan J, Nadeau K, Karagas M. In utero arsenic exposure and infant infection in a United States cohort: A prospective study. Environ Res 2013; 126:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dauphiné DC, Ferreccio C, Guntur S, Yuan Y, Hammond SK, Balmes J., et al. Lung function in adults following in utero and childhood exposure to arsenic in drinking water: preliminary findings. Int Arch Occup Environ Health 2011; 84:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan Y, Marshall G, Ferreccio C, Steinmaus C, Selvin S, Liaw J, Bates MN, Smith AH. Acute Myocardial Infarction Mortality in Comparison with Lung and Bladder Cancer Mortality in Arsenic-exposed Region II of Chile from 1950 to 2000. Am J Epidemiol 2007; 166:1381–1391. [DOI] [PubMed] [Google Scholar]
- 40.Liaw J, Marshall G, Yuan Y, Ferreccio C, Steinmaus C, Smith AH. Increased childhood liver cancer mortality and arsenic in drinking water in Northern Chile. Cancer Epidemiol Biomarkers Prev 2009; 17:1982–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nigra AE, Olmedo P, Grau-Perez M, O’Leary R, O’Leary M, Fretts AM, et al. Dietary determinants of inorganic arsenic exposure in the Strong Heart Family Study. Environ Res 2019; 177: 108616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.