

Graphical abstract
Keywords: Interactomics, Mitochondria, Psychotropic medication, Psychiatric disorders, Precision Psychiatry Medicine, Artificial intelligence
Abstract
Neuropsychiatric disorders (NPDs) such as bipolar disorder (BD), schizophrenia (SZ) and mood disorder (MD) are hard to manage due to overlapping symptoms and lack of biomarkers. Risk alleles of BD/SZ/MD are emerging, with evidence suggesting mitochondrial (mt) dysfunction as a critical factor for disease onset and progression. Mood stabilizing treatments for these disorders are scarce, revealing the need for biomarker discovery and artificial intelligence approaches to design synthetically accessible novel therapeutics. Here, we show mt involvement in NPDs by associating 245 mt proteins to BD/SZ/MD, with 7 common players in these disease categories. Analysis of over 650 publications suggests that 245 NPD-linked mt proteins are associated with 800 other mt proteins, with mt impairment likely to rewire these interactions. High dosage of mood stabilizers is known to alleviate manic episodes, but which compounds target mt pathways is another gap in the field that we address through mood stabilizer-gene interaction analysis of 37 prescriptions and over-the-counter psychotropic treatments, which we have refined to 15 mood-stabilizing agents. We show 26 of the 245 NPD-linked mt proteins are uniquely or commonly targeted by one or more of these mood stabilizers. Further, induced pluripotent stem cell-derived patient neurons and three-dimensional human brain organoids as reliable BD/SZ/MD models are outlined, along with multiomics methods and machine learning-based decision making tools for biomarker discovery, which remains a bottleneck for precision psychiatry medicine.
1. Introduction
Owing to the complex and mosaic nature of the human brain, numerous classes of mental illnesses exist, including neuropsychiatric disorders (NPDs) such as bipolar disorder (BD), schizophrenia (SZ) and mood disorder (MD) [1], [2]. These classes are categorized based on observable manifestations exemplified by uncontrolled anger, mood swings, brain seizures, or attention deficit, but in many cases are harder to diagnose due to overlapping symptoms [3], [4]. This dilemma often results in misdiagnosis due to the lack of clear disease biomarkers. Although the etiology of these disorders is still poorly understood, strong evidence points out to the contribution of an assortment of factors, including genetic susceptibility, epigenetic dysregulation, and environmental stimuli to mental disease onset and/or progression [5], [6]. Further, it remains unclear on the scope of mt pathways targeted by BD/SZ/MD psychotropic treatments, or how to strategize their administration. These issues necessitates the requirement of advanced multiomics and artificial intelligence (AI)-based research endeavours that can potentially accelerate biomarker discovery and therapeutic development for NPDs.
Altered brain function appears to be a consistent player in various NPDs, and epigenetic spatiotemporal dysregulation of gene expression was found to be a common player in NPDs, resulting in altered neuronal differentiation, brain development and circuitry formation, which commonly contribute to NPD pathophysiology [5]. The human brain consumes a staggering one-fifth of the body’s energy, which is supplied by mitochondria (mt) [7]. Thus, understanding how mt dysfunction is linked to NPDs becomes a focal point. Mt produce the vast majority of ATP in nerve cells, and therefore mt dysfunction may contribute to energy-depleted neuronal impairment associated with NPDs. For example, mutations in MRPL40, encoding mt ribosome component, leads to 25-fold increase in SZ risk, with ATP depletion considered the hallmark of mutated MRPL40 in patient forebrain-like excitatory neurons [8]. Beyond energy production, mt engage in fusion and fission, bidirectional trafficking along neuronal axons from soma to terminal synapses and vice versa, as well as clearance of damaged mt (mitophagy), to collectively sustain neuronal functions [7], [9], [10]. Thus, alterations in bioenergetics/mt dynamics are linked to NPDs. For example, DISC1 interaction with mt Rho (MIRO1, 2) or mitofusin (MFN1, 2) GTPases and trafficking adaptors (TRAK1, 2) links mt fusion and transport mechanisms in healthy neurons [11]. Single nucleotide polymorphisms (SNPs) and haplotypes in DISC1 also associate with BD and SZ, suggesting interrupted mt transport and fusion machineries in these disorders [12].
In addition, the glycogen synthase kinase-3β (GSK-3β), regulates healthy mt activities [13]. GSK-3β inhibition upregulates the transcriptional factor PGC-1α, which increases mt biogenesis [14]. This demonstrates that GSK-3β exhibits regulatory roles over transcription factors, with GSK-3β alteration reported to associate with NPDs [15]. While these examples shed light on the linkage between mt dysfunction and NPDs, major gaps remain, including the absence of extensive studies on mt interactome alterations in NPDs, and the scarcity in databases describing the pathological pathways restored by a specific treatment or combination therapies.
Despite these molecular points of convergence between distinct NPDs, differences still exist and segregate BD/SZ/MD from one another. For example, a genomic study conducted on both BD and SZ has elegantly dissected the differences between these disorders by identifying disease-specific genetic variants and the genetic footprint underlying various phenotypes within the same illness [16]. Another comparative transcriptomic study on BD and other major mental disorders revealed the downregulation of genes encoding receptors, channels or transporters, and the upregulation of genes involved in stress response in BD, suggesting a differential pathological background that discriminates between BD and other mood disorders [17]. This is in addition to gray matter deficits that appear to be more intense in SZ compared to BD [18].
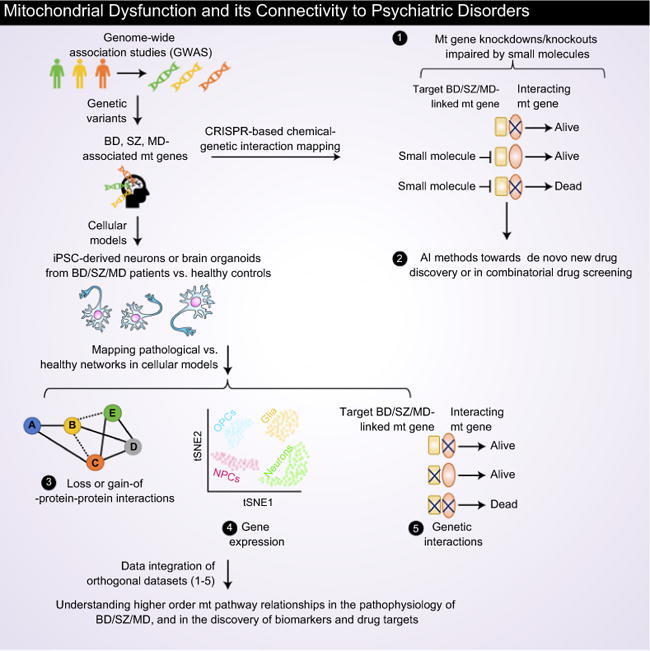
Although both convergent and divergent pathological mechanisms are involved in the onset of these mental illnesses, in this review, we discuss how mt proteins-linked to BD/SZ/MD that interact with other mt proteins are impacted by psychotropic drugs, revealing mt pathways that are commonly or uniquely affected by targeted medication. We also discuss human disease models (i.e. induced pluripotent stem cell (iPSC)-derived neurons and brain organoids) available for BD/SZ/MD, and how their integration with multiomics (chemical genetic interaction, gene expression, physical or genetic interactions) and AI (Fig. 1A) can facilitate the discovery of biomarkers and drug targets to improve precision psychiatry medicine.
Fig. 1.

Conceptual systems framework in NPDs (BD/SZ/MD), and lithium treatment amelioration of mitochondrial (mt) functions in distinct pathways. (A) Genetic variants from GWAS will allow the discovery of NPD-associated mt risk genes. A CRISPR-based chemical-genetic interaction screening can unveil NPD-relevant mt targets inhibited by a small molecule. Here, knockouts (or knockdowns) of the interacting gene of NPD-associated mt risk factors is lethal when cells are treated with small molecule. Artificial intelligence (AI) methods, such as deep learning can be employed towards de novo drug discovery and combinatorial drug screening. By utilizing iPSC-derived neurons or 3D brain organoids from NPD patients vs. healthy subjects, loss (dotted lines)-or gain (thick lines) of protein–protein interactions (PPIs), gene expression in distinct cell states (OPC, oligodendrocytes; NPCs, Neural progenitor cells), and genetic interaction networks (i.e. two functionally linked single gene knockouts show lethal phenotype) can be mapped to pinpoint specific or common pathological macromolecular complexes (identified from PPI networks) or pathways involved in the pathophysiology of BD/SZ/MD. Integration of PPIs with co-expression profiles from scRNA-seq should uncover functional modules and driver genes of NPDs. Notably, a gene knockout that show chemical-genetic interaction with a small molecule should display lethal with a mutation in the drug target gene. Thus, comparing genetic interactions with chemical-genetic interaction profiles will identify mt pathway targets modulated by small molecule. Collectively, integration of these orthogonal datasets in various combinations should generate a higher order pathway-level understanding of NPD-related mt genes and in the discovery of disease biomarkers and drug targets. (B) Lithium inhibits GSK-3 pathway, allowing the translocation of β-catenin to the nucleus and transcription of pro-survival factors. Lithium prevents mt translocation of GSK-3 and improves oxidative phosphorylation (OXPHOS). (C) Lithium impedes the phosphoinositol cycle, reducing myoinositol levels and, consequently, blocking the activation of PKC pathway. Inhibition of this pathway prevents destabilization of neuronal membranes, increased long-term potentiation, and induction of apoptosis. (D) BCL-2 and BDNF factors are improved via ERK activation. Increased expression of these two proteins ameliorates neurotransmission, avoids mt Ca2+ overload and prevents cytochrome c release.
2. Psychotropic treatments for NPD patients with MT damage
Frontline NPD treatments include anti-depressants, anti-psychotics, α2-adrenoceptor agonists, and other mood stabilizers. Due to increasing evidence for mt connectivity to NPDs [19], [20], [21], [22], we will discuss how various psychotropic treatments improve mt fitness, with emphasis given to lithium as a widely administered drug of choice.
Secondary messenger signal transduction abnormalities associate with depressive episodes due to 5-hydroxytryptamine receptor activation and monoamine receptor dysregulation, resulting in low mt capacity to synthesize ATP observed in depression-linked ailments such as BD and SZ [20], [21], [23], [24], [25]. Recently, a meta-analysis study conducted on 21 anti-depressants showed that select compounds (e.g. mirtazapine) exhibit higher response and lower dropout rates in adults affected by major depressive disorder (MDD), while other compounds showed lower efficacy and acceptability profiles, such as fluvoxamine, making them less promising therapeutic options [26]. However, efficacy varies considerably between sexes and age groups, with fluoxetine effectively reducing depressive symptoms in younger patients [26]. This is due to differential anti-depressant efficacy in improving mt fitness that varies over time. For example, single injection of fluoxetine improves citrate synthase activity in the striatum and mt complex I in the hippocampus, but after 28 days of treatment, this drug was no longer beneficial [27], [28].
Interestingly, sex and brain region-specific effects of fluoxetine were demonstrated by another study [29], with increased mt cytochrome oxidase 1 and 3 in the prefrontal cortex of female but not male rats, with conversely decreased expression in the hippocampus region of male and not female rats. Increased cytochrome oxidase and glutamate dehydrogenase levels were also observed in the hippocampus of rats treated with fluoxetine and desipramine [30]. Similarly, venlafaxine enhances anti-apoptotic and antioxidant mt gene expression, but also exhibited a detrimental impact on mt complex IV activity. Since mt fitness varies with ages, this might explain inconsistencies in potency and side effects of mt fitness-improving compounds when administered to different age groups [31].
Antipsychotics such as risperidone, quetiapine, olanzapine and others are routinely prescribed for BD and SZ to improve patient moods by minimizing psychosis [32]. However, they may deteriorate mt fitness through damaging mt membrane potential and altering secondary messenger signaling pathways, thus affecting protein kinase A and C (PKA, PKC) levels. Despite the controversy on whether antipsychotics improve or deteriorate mt functions [33], available data demonstrate a role for mt impairment in BD/SZ progression. Another important class of psychotropics is α2-adrenoceptor agonists, such as brimonidine, that regulate neurotransmitter release [34], exhibit neuroprotective effects by upregulating mitogen-activated protein kinase (MAPK) and AKT pathways, alleviate mt oxidative stress and inhibit pro-apoptotic mt signaling cascades [35]. Also, AKT activation dissociates BAD and BCL-2 at mt level and inhibits cytochrome c release, with ensuing activation of apoptotic pathways [35].
Lithium carbonate is the gold standard mood stabilizer for manic and depressive episodes of BD [36]. It also improves mood in SZ patients, animal or cellular models that recapitulate pathological SZ phenotypes [37]. Lithium has neurotrophic effect, which restores signal transduction cascades, hormonal or circadian regulation, and ion transport mechanisms [36]. Lithium also reverses pathological BD phenotypes (e.g. oxidative stress, inflammation, excitotoxicity, neuronal death). Yet, lithium targets in the cell remains poorly understood, including how it restores healthy mt protein–protein interactions (PPIs) in nerve cells [36], [38], [39], [40].
Lithium’s anti-apoptotic effect is driven by direct (i.e. modulation of transcription) or indirect (i.e. activation of protein kinase B/AKT) inhibition of GSK-3, which impacts the activity of cell survival-promoting factors (i.e. cyclic AMP response element binding protein (CREB) and β-catenin), improving neuronal viability [41], [42] (Fig. 1B). Intriguingly, GSK-3 can also translocate into mt and impact their permeability transition pore, thus regulating mt functions [15], [42]. Given the pro-oxidative and pro-inflammatory properties of GSK-3, its inhibition exerts protective effects against oxidative stress, mt dysfunction, and suppressing reactive oxygen species-induced apoptotic pathways. Lithium in addition increases mt complex I-II activities [42], [43], yet the molecular attributes or mtPPIs coordinating these observations remain understudied.
Studies propose that inositol monophosphatase, an enzyme of the phosphatidylinositol cycle, is inhibited by lithium, leading to reduced myoinositol levels and impeding PKC activation, which is involved in the regulation of neuronal processes affected in BD such as neuronal excitability, neurotransmitter release and neuroplasticity [44], [45], [46] (Fig. 1C). In this context, PKC inhibition by lithium reduces the phosphorylation of neurogranin, (a neuronal calmodulin-binding protein expressed in dendritic spines), which binds calmodulin to decrease long-term potentiation [47]. Among all PKC substrates, MARCKS is one of the most biologically significant (Fig. 1C), due to its implications in cytoskeletal remodeling. Lithium also reduces MARCKS expression, resulting in the stabilization of neuronal membranes [46], [47]. Phosphoinositol pathway inhibition ameliorates Ca2+ dynamics by reducing inositol triphosphate levels and impedes Ca2+ release from endoplasmic reticulum [48]. This lithium-mediated effect resolves the BD imbalanced Ca2+ homeostasis, leading to Ca2+ accumulation within mt beyond their buffering capacity, affecting proton export, ATP synthesis and the permeability transition pore, which results in releasing cytochrome c and apoptotic pathway activation [49]. Lithium moreover refines Ca2+ homeostasis, which is transported inside mt, possibly through Na+/Ca2+ exchangers, minimizing mt damage caused by elevated Ca2+ levels in NPD patients [50], [51].
Additionally, lithium activates extracellular-signal-regulated kinase (ERK) and MAPK pathways, which in turn upregulates the transcription of several factors involved in neurogenesis, cell survival, and synaptic transmission and plasticity, including BCL-2, through nuclear CREB stimulation [39], [52] (Fig. 1D). BCL-2 acts as pro-survival factor through limiting Ca2+ overload and impeding cytochrome c release from mt [53], [54] (Fig. 1D). Lithium also elevates the expression of mt-associated protein, BDNF (brain-derived neurotrophic factor) through ERK/MAPK pathway activation or β-catenin nuclear translocation, which downregulates pro-apoptotic mt signaling leading to neurotransmitter release such as glutamate, dopamine and serotonin [55] (Fig. 1B and D). Further, BDNF activation by lithium inhibits GSK-3β and increases CREB expression [55], which elevates BCL-2 and BDNF expression, while BCL-2 protects mt by inhibiting pro-apoptotic BAD and consequently elevates mt Ca2+ influx and cytochrome c release [55]. Also, lithium-mediated BDNF activation enables glutamatergic regulation and reduced excitatory neurotransmission [47], [56].
Dopamine neurotransmission is also regulated by lithium via the AKT/GSK-3β cascade, the phosphoinositol cycle and PKC signalling pathways downstream of dopamine receptors [46], [57], [58]. Loss-of-function of ANT1, a gene involved in the translocation of adenosine triphosphate and adenosine diphosphate across inner mt membrane, was recently identified as a risk factor for BD [59]. Mutations in ANT1 increase serotonin turnover and hyperactivity of serotonergic neurons, drawing a clear connection between mt dysfunction and altered neurotransmission in BD [59]. Notably, lithium increases serotonin biosynthesis, tryptophan uptake and neurotransmitter release [60], but further studies are crucial to verify if lithium or other mood stabilizers affect ANT1 activity. Lithium treatment also restores the pathological phenotype of POLG (gene encoding for the gamma polymerase) in mutated mice, a model used to recapitulate psychiatric behaviour and pathological pathways in BD, suggesting that lithium offers a beneficial effect in animal models of POLG [19], [60].
Valproic acid is another major BD mood stabilizer that reduces mt matrix pH and higher Ca2+ levels associated with BD due to mt 10398A > G polymorphism, which is resistant to lithium treatment [19]. Valproic acid is metabolized in the mt and improves mt oxygen consumption rate and membrane potential. Similar to lithium, valproic acid ameliorates mt Ca2+ homeostasis and inhibits apoptotic pathways by impeding cytochrome c release and increasing BCL-2 expression [19]. Valproic acid also acts as a histone deacetylase inhibitor by increasing BDNF expression, leading to histone 3 and 4 acetylation [19]. Together, these data strengthen a role for mood stabilizers in restoring altered mt pathways in NPDs.
3. NPD-linked MT proteins are targets of mood stabilizers
Although growing evidence links mt impairment to BD/SZ/MD, most biochemical studies are of small-scale nature. To gain understanding of NPDs from studies deposited in public databases, we analyzed mt-localized proteins from five sources: Mitocarta 2.0 [61], IMPI Q2 2018 [62], UniProt [63], Gene Ontology (GO) [64] and COMPARTMENTS resource [65]. Besides an overlap between the sources of mt annotations, we generated a list of 2,239 mt proteins (Fig. 2A; Supplemental Table S1), of which three-fourths (73%; 1627 of 2239) were in agreement in at least two data sources. We then deduced mt proteins associated with BD/SZ/MD by combining gene-disease annotations retrieved from the DisGeNET database [66], which were filtered at DisGeNET Score ≥ 0.005 and evidence index > 0.9, in more than one source. This led us to identify common and specific mt proteins in NPDs.
Fig. 2.

Mitochondrial (mt) proteins involved in BD/SZ/MD and their regional specificity. (A) Venn diagram showing the overlap of mt proteins compiled from five different sources (see Supplemental Table S1). (B) Mt proteins specific or common among BD/SZ/MD. (C, D) Heatmaps (C) and line graphs (D; dotted line represent median mRNA expression) of BD/SZ/MD-associated mt proteins showing regionalization of mRNA expression in human brain regions from RNA-sequencing (RNA-seq) datasets of Human Protein Atlas (integrated with GTex and Fantom) and single cell RNA-seq (scRNA-seq) data from Allen Brain Atlas. Bar graph (D, below) shows the number of BD/SZ/MD-related mt proteins that are above or below median mRNA expression across brain regions from the indicated repositories.
Among the 245 non-redundant mt proteins annotated for all three categories, 148 were specific to SZ, 34 to BD, and 9 for MD, while only 7 mt proteins were in common between BD, SZ and MD (Fig. 2B; Supplemental Table S2). Most of these mt proteins show regional enrichment (3.3 × 10−40 < p < 1.9 × 10−6) in different brain regions for cognate gene expression (Fig. 2C; Supplemental Table S3), with three-fourths (73%; 175 of 241) of mt genes from Human Protein Atlas and most (90%; 207 of 231) from Allen Brain Atlas showing higher abundance (i.e. above median mRNA expression; Fig. 2D), implicating BD/SZ/MD-linked mt protein roles in core brain activities. In addition, we observed three-fourths (189of245) of mt proteins involved in these NPDs encompass 838 PPIs from 657 publications (Fig. 3A and B; Supplemental Table S4), with many PPIs enriched (6.7 × 10−34 < p < 1.0 × 10−2; Supplemental Table S5) for mt related processes, providing additional support on mt involvement in NPDs. Despite the observable discrepancies between annotations in different databases (attributed to dissimilar prediction algorithms or data disparities), this approach highlights biomarkers specific to or common among pathologies, representing a solid basis to develop precision biomarker panels. For example, as shown in Supplemental Table S2, the mt orphan gene, C12orf65 is specifically impaired in SZ [67], and the protein coding gene HIP1R (Huntingtin-interacting protein 1-related protein) was found to be largely associated with BD [68].
Fig. 3.

BD/SZ/MD-associated mt proteins and mood stabilizers. (A, B) Mt proteins associated with BD/SZ/MD, and their interacting partners compiled from BioGRID database (A), and the number of publications (B) supporting those interactions. (C) Mood stabilizers targeting (either binding or inhibiting) BD/SZ/MD-associated mt proteins.
Next, we compiled 37 prescription and over-the-counter mood stabilizers from online pharmaceutical repository (www.drugs.com), which we shortlisted to 15 active ingredients upon eliminating redundant trade names using KEGG (Kyoto Encyclopedia of Genes and Genomes) drug index (Supplemental Table S6). The molecular basis and mt targets for many of these mood stabilizers in NPDs are unknown, hence we used the drug-gene interaction database [69] to examine if any drugs target the 245 BD/SZ/MD-related proteins. We observed 26 associations with 7 mood stabilizers (aripiprazole, carbamazepine, fluoxetine, lithium, olanzapine, risperidone, valproic acid) and 16 BD/SZ/MD-linked mt proteins (Fig. 3C), which had physical connectivity to 80 other mt proteins (Fig. 4A). Consistent with this, mt pathways such as signaling (e.g. neurotrophin, MAPK, mTOR, VEGF), apoptosis, axon guidance, and dopaminergic or glutamatergic synapses that were targeted by 2 or more mood stabilizers were enriched (7.5 × 10−6 < FDR adjusted p-value < 4.7 × 10−2, Fig. 4B). Since this observation suggests mood-stabilizing agents target mt pathways, testing compounds in combinations to target multiple mt pathways with synergistic effects would be a beneficial therapeutic strategy.
Fig. 4.

Mood stabilizers and mt protein or pathway targets, as well as the schematic of precision psychiatry medicine. (A) Physical connectivity of NPD-linked mt proteins that are targets of the mood stabilizers. (B) Enriched (see Supplemental Table S6 for Benjamini-Hochberg FDR adjusted p-value) mt pathways targeted alone or shared by the mood stabilizers. (C) NPD-associated mt proteins targeted by one or more mood stabilizing medications (panel i) are highlighted (panel ii-iv) with their interacting partners as sub-networks. (D) Precision psychiatry model is shown by integrating various data features, which is fed into AI (or machine learning) algorithms to stratify and identify best therapeutic treatment for each subject through patient matching to evaluate treatment response and disease prognosis (see main text).
Over half (9of16) of those NPD-associated mt proteins were found to be targeted by one mood stabilizer, while the rest were targeted by two or more mood stabilizing medications (Fig. 4C). For example, the myo-inositol monophosphatase (IMPase) enzyme, IMPA1, and its interacting partner, IMPA2, in manic-depressive BD, are targeted by lithium carbonate or citrate hydrate, consistent with IMPase inhibition by lithium in phosphatidylinositol signaling [70]. Besides IMPase, lithium acts on other candidate mt GSK-3α and -3ß isoforms [71], [72], [73] linked to BD/SZ/MD. As with the Hsp90 interactome database, GSK-3 isoforms interacting with HSP90 chaperone were targeted by lithium and/or fluoxetine (Fig. 4C). Lithium and fluoxetine co-treatment has been suggested to inhibit GSK-3 serine phosphorylation to promote adult hippocampal neurogenesis [74], revealing that dysregulated GSK-3 may impair neurogenesis by contributing to MDs, while neurogenesis is rescued by lithium and fluoxetine treatments [75]. Also, we found carbamazepine mood stabilizer (Fig. 4C) that targets NPD-related mt proteins (e.g. tumour necrosis factor, TNFα, and its co-complex member TNFRSF1A, TNF Receptor Superfamily Member 1A) to bind to the heat shock protein, HSPA1A, and its interacting partners HSPD1 and BCL-2 that mediate HSP70 [76]. Further, the BD-associated BCL-2, which is physically connected to histone deacetylase 2 (HDAC2), consistent with HDAC2 involvement in BCL-2 transcription [77], was targeted by valproic acid. Thus, mood stabilizers either directly bind or inhibit NPD-related mt proteins, or may disrupt their interaction with other mt proteins. However, network-based approaches should be analyzed with caution and require extensive evidence-based validations that uncover the scope of biological complexity and variability, whereas statistically inferred networks can be better suited to infer relationships between small molecules and/or drugs [78].
Despite systems biology insights, knowledge of NPDs is still limited due to the shortage of NPD disease models. Given the polygenic risk of these pathologies, animal models partially mimic neurophysiological, neuroanatomical and/or behavioral hallmarks of NPDs without providing ample basis for heterogeneous pathologies [79], [80]. This can be addressed through iPSC-derived neurons, which are becoming a promising tool to study NPD pathophysiology.
4. Disease models and systems or AI approaches to study NPDs
Since iPSCs mirror the polygenic risk factors of the patient, they can highlight developmental and differentiation neuronal alterations and evaluate treatment responses [79], [80]. For instance, electrophysiological characterization of iPSC-derived hippocampal dentate gyrus (DG) neurons were segregated into lithium-responsive and lithium-non-responsive BD patients, matching patient categories in 92% of cases [81]. Decreased excitability caused by lithium in responsive patients was determined by changes in the expression of several key genes pertaining to BD pathology, rescuing mt dysfunction [82]. In line with the neurodevelopmental hypothesis of BD [83], it has been demonstrated that miR-34a levels are increased in iPSC-derived neuronal progenitor cells of BD patients, silencing mRNA and protein levels of its target genes and compromising neuronal development, differentiation and morphology [84]. Moreover, dysregulation of Ca2+ signaling in neuronal development and synaptic plasticity, as well as elevated transcripts involved in the differentiation of ventral neuronal subtypes, collectively explain different specifications of BD neuronal stem cell fates when compared with dorsal telencephalic neuronal identity observed during differentiation in control subjects [85].
Although iPSC-derived neurons offer a promising platform, they cannot fully recapitulate higher-order brain functions. The complexity of neuronal cell types in particular brain regions cannot be reconstituted by iPSCs, which omits astrocytes, glial cells and oligodendrocytes, and limits synaptic connectivity in a monolayer culture [86]. Moreover, the application of epigenetic approaches on iPSC-derived neurons is still limited by the reprogramming process itself, since it might erase much of the epigenetics associated with these cell lines [87]. These limitations have catalyzed the generation of brain organoids, a refined model that offers close-to-natural multidimensional neuronal complexity. The structures essentially mirror human brain development [86], [88] so that it approximates regional connectivity by fusing different types of brain organoids into assembloids [89], and to epitomize brain complexity through the inclusion of astrocytes, oligodendrocytes, glial cells and neural progenitor cells [86], [88], [89]. As an example, brain organoids were derived from SZ patients to characterize pathogenic mechanisms during the first trimester of an in-utero brain development [90]. Abnormal proliferation of Ki67+ neural progenitor cells, depletion of neurons in the cortex, and reduced intracortical connectivity were the hallmarks of psychiatric patient-derived impaired brain organoids [90], suggesting that SZ has neurodevelopmental origins. The NPD field can also benefit from brain organoids to map disease progression, aiding the dissection of neuronal development over patient’s lifespan. Yet, the use of brain organoids in neuropsychiatric research has some intrinsic limitations. For instance, the three-dimensional (3D) tissues may still lack sufficient vascularization, circulation, and a necrotic core that inevitably appears inside organoids. In turn, this necrosis determines a depletion of proliferative neural progenitor cells, thus limiting neurodevelopmental studies [91]. Lastly, the recapitulation of the complex structure of brain layers and/or lobes still remains unattainable even with the current organoid technology [91].
5. Network-based multiomics to explore mt alterations in IPSCs or brain organoids of NPD patients for biomarker discovery
Most patients with mt disorders, in which genetic impairment of one or more mt metabolic pathways determines the onset of primary pathological defects, show frequent psychiatric symptoms linked to a specific illness, such as BD, MDD and panic or anxiety disorders [92]. Notably, mt encephalopathy, lactic acidosis and stroke-like episodes (MELAS) is a maternally inherited disease associated with several NPD symptoms and often exists as comorbidity with BD or SZ [92], [93]. The potential overlap between psychiatric illnesses and mt diseases suggests a point of convergence in disease pathogenesis. Yet, it is not known how many BD/SZ/MD-related mt genes genetically interact with mt pathways in a linear manner or if they function redundantly, but converge in a common pathological outcome that impacts patient brain fitness.
It is also unclear which mt protein complexes commonly or specifically drive BD/SZ/MD. These questions are of particular relevance in biomarker discovery, which remains an obstacle for therapeutic management. Thus, it becomes imperative to explore mtPPIs and altered genetic interactions to pinpoint specific or common pathological pathways involved in the onset and development of these diseases (Fig. 1A). As we have recently described [10], several network-based approaches can be used to map mt interactome perturbations in NPDs. For example, mt protein complexes can be studied under native conditions by biochemical fractionation and mass spectrometry (MS), which can capture strong, as well as weak, transient or low abundance interactors using cross-linkers in neuronal cells [94] and mammalian brain [95]. Notably, this method can map loss- or gain-of-function PPIs between NPD patients vs. healthy controls. Besides the advancement in proximity-dependent biotinylation assays (e.g. BioID, APEX, TurboID), affinity purification coupled with MS is another method that can pinpoint interactome alterations in psychiatric illnesses in near-physiological contexts. The latter technique was successfully used to reveal abnormalities in the SNAP25 (Synaptosome Associated Protein 25) interactome from SZ patients, where SNAP25 was found to interact with the mt ADP-ribosylation factor 1 (ARF1), contributing to SZ manifestations [96]. Despite the advantages yielded by these techniques, false positive and/or negative results are important issues of concern in interactomics that can be however diligently addressed via checkpoint computational or molecular strategies.
Recently, a new drug discovery platform, MaMTH-DS [97], was used to identify interactions between membrane proteins and how they change in response to a specific mutation or following the induction of specific stimulus, such as a hormone, agonist, or phosphorylation event [98], with potential usefulness in studying dynamic interactome changes in proteins involved in cell signaling, and how these pathways are affected in NPDs. Since more than one gene may contribute to NPD phenotypes, CRISPR-based genetic screens can be applied to reveal functional relationships between genes in coordinating a phenotype [99].
Although genome-wide association studies (GWAS) have provided insights into NPD biomarkers (e.g. transcription factors, scaffolding and receptor proteins), transcriptomic analysis becomes imperative from at least two perspectives: (1) validate GWAS data by sorting out their linkage to NPDs, and (2) reveal disease-enriched expression profiles. As an example, differential expression of Prominin 1/CD133, the ATP-binding cassette-sub-family G-member 2 (ABCG2) and long non-coding RNAs (lncRNAs) were enriched in BD patients, with implications in neuroplasticity [100]. Further, genes involved in GTPase binding were differentially expressed and enriched for BD-associated SNPs identified by GWAS, suggesting that differentially expressed patterns are not a consequence of BD or its treatment mechanism [100]. Thus, transcriptome analyses offer essential information by shedding light on the complexity of gene expression patterns and offer in-depth explanations of GWAS data.
Additional evidence suggests that ~71% of cortical tissue transcription level changes in BD and SZ patient samples are correlated [101], revealing overlapping drivers in NPDs, but also potential disease-specific divergent mechanisms. Such divergence may also reveal sex-specific variations within the same disease. For example, SZ is prevalent in men with worse prognosis than women, while poor prognoses in BD are more common for women than men, despite comparable incidence rates [101]. The divergence in SZ-biased brain transcriptome between females and males is becoming evident, with neuronal development and differentiation mechanisms, chemokine pathway and circadian rhythm being transcriptionally female-biased [101], as opposed to inflammatory response, organophosphates and neurotransmitter release as being male-biased SZ transcriptome profiles [101]. In contrast and in BD, macrophage activation and JAK/STAT (Janus kinase/signal transducers and activators of transcription) regulation mechanisms are transcriptionally female-biased, whereas neutrophil degranulation, NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) regulation and varied response to IL-6 (Interleukin 6) production appear to be male-biased in BD [101]. This reveals the contribution of transcriptomics as comparative analysis tool to uncover disease-specific biomarkers, including sex-specific drivers, for diagnosis and therapeutic intervention.
Evidence have also pointed to a role for epigenetics in the etiology of mental illnesses, for instance in the impact of epigenetic regulation of chromatin remodeling and DNA methylation on these pathologies [5], [6]. Further, MDD is caused by the lack of methylation at certain CpG sites in the exon 1 of BDNF gene [102], as well as reduced response to antidepressant drugs due to the absence of methylation at CpG site in exon 4 of the same gene [103]. Downregulation of miR-132 is instead associated with SZ development. Notably, miR-132 downregulation determines an increased expression of its target mRNAs, which are linked with synaptic long-term potentiation and depression, and neuronal CREB signaling, and DNA methylation [5], [104].
The DNA methylation profile differences were also observed in another epigenetic study conducted on monozygotic twins discordant for SZ and BD, highlighting differential epigenetic dysregulations among these mental illnesses [105]. However, the identification of epigenetic profiles caused by a specific environmental chemical and their correlation with a mental illness is still limited by the lack of large scale studies on this topic [6]. Nevertheless, small scale studies are gradually pointing to a role for early-life lead exposure in the developmental, behavioral, and neuromolecular changes that are among the SZ hallmarks [106]. For all these reasons, integrating epigenomics with other global and unbiased methods can shed more light on pathogenetic pathways involved in the onset of these debilitating psychiatric disorders. For example, redox proteomics and epigenomics could be used together to verify the impact of oxidative stress generated by mt dysfunction on DNA oxidation and methylation [107].
In addition to aforesaid omics approaches, proteomics can identify different biomarkers for various human diseases, including NPDs. For instance, 19 different proteomic studies (see references reviewed in [108]) in BD and MDD revealed 185 potential BD and MDD blood-based diagnostic protein biomarkers (i.e. comparison between patient vs. healthy control), as well as differential biomarkers (MDD vs. BD) pertaining to altered immune response, inflammation, metabolism, cell signaling, amongst others. As well, 103 unique proteins have been suggested as potential diagnostic biomarkers specific for BD (e.g. Apolipoprotein A-I, serotransferrin) and MDD (e.g. alpha-2-macroglobulin, Apolipoprotein B-100, Apolipoprotein D, BDNF). Yet, these proteomic approaches raise concerns: (1) MS-based data were extracted from blood samples, which may not reflect the differential proteins or PPI variations in relevant neuronal types of BD/SZ/MD patients; (2) hundreds of biomarkers from 19 proteomic studies may not offer meaningful clues, revealing the need for stringent computational-based filtration strategies; and (3) biomarker overlaps between diseases may not identify disease-specific diagnostics.
RNA-sequencing on a single-cell level (scRNA-seq) is gaining considerable momentum, permitting analysis of previously confounding brain tissue heterogeneity [109], which is harder to investigate via proteomic analyses alone. The use of scRNA-seq provides the opportunity to address brain tissue heterogeneity by analyzing the transcriptomic profile of each individual cell, starting from tens of thousands of cells. This technique can be integrated with interactomics by matching RNA expression profiles with the expression of interacting proteins derived from the PPI networks (Fig. 1A). Since the subunits of a protein complex tend to display similar or highly correlated co-expression profiles [110], this twofold approach can reveal functional modules, identify or prioritize proteins within a complex to be associated with disease, refine the identification of driver genes, and unveil the pathological mechanisms of specific disease phenotypes or cell types [111], [112], [113], [114]. In particular, application of these techniques in iPSC-derived neurons and brain organoids can be useful since these models can recapitulate neuronal differentiation and brain development mechanisms, respectively.
An attempt to connect SZ genomic findings with specific brain cell types as defined by scRNA-seq revealed 24 cell types associated to the etiology of SZ [115], including medium spiny neurons, pyramidal cells in hippocampal CA1 or somatosensory cortex and cortical interneurons. Similarly, scRNA-seq on mice cortex has recovered ~3 K transcripts per cell, categorizing transcript sequences into 8 different cell types: excitatory neurons, inhibitory neurons, oligodendrocytes and oligodendrocyte precursor cells, astrocytes, endothelial and smooth muscle cells, pericytes, microglia, and macrophages along with 30 other subtypes [116]. Such data can be overlaid with PPI networks to reveal connectivity between PPIs and their cognate expression profiles via scRNA-seq in specific neuronal cell types (Fig. 1A). Further, integrating gene expression profiles with human interactome data can facilitate the identification of disease modules, biomarkers, and druggable targets, as was the case in Type 2 diabetes [117].
6. New horizons of neuropsychiatry drug discovery beyond classical approaches
In contrast to classical approaches such as CRISPR-based chemical-genetic interaction mapping of BD/SZ/MD-related mt gene-drug interactions (Fig. 1A), AI and machine learning-based de novo drugs may offer new insights into drug discovery for NPDs [118], [119], [120], essentially generating computer-aided chemical designs for downstream validation. Yet, these computer-assisted structures are not supported by clear bench-synthesis methodologies, thus remain largely limited to virtual screening [121]. Similarly, libraries of small molecules in high-throughput screening are biased towards the manufacturer’s expertise, while overlooking compound categories may offer a more potent choice. To address these limitations, AI-augmented decision-making platforms coupled with machine learning processes such as reinforcement, semi-supervised, transfer, active or multi-task learning (for detailed review, see [122] can offer orders of magnitude increases in pool search. These platforms navigate synthetically accessible chemical space by subjecting commercially available molecules to chemical reactions at every step of the iterative virtual synthesis process, increasing likelihood of new drug discovery [123].
Advances in implementing AI learning algorithms (e.g. Bayesian model, Logistic regression, Support vector machines, Decision tree, Deep learning, Neural networks) are being achieved by assessing how small molecules can regulate gene expression with implication in drug discovery [122], [124]. This is essentially attained by training, for instance, neural networks, to predict differential gene expression in the presence of small molecules, combining molecular fingerprinting with gene descriptors from GO, which can be tailored for NPD treatments by modulating gene expression in select disease-linked candidates. The excitement behind AI and its potential usage in drug discovery mainly stems from its tendency to reduce costs associated with high-throughput screening, which consumes significant time and resources sometimes to no avail [122]. AI essentially attempts to minimize this hit-hunting process by offering an automated workflow to establish a virtual screening pipeline that examines possible target-effective compounds that are safe and sufficiently novel to rule out intellectual infringements. While it is hard to predict where AI is going to take us in the next decade, it is important to note that AI has not yet led to commercialized drugs, thus remaining a promising area that lacks proof-of-concept product outcomes [125].
7. Precision psychiatry medicine in the systems biology ERA
In an attempt to abolish the “one-size-fits-all” approach to clinical treatment, focus has been gradually shifting towards finding ways to administer the right drug to the right patient at the right time [126]. The comparison between some of the most commonly used medications to date, only showed 11 out of 17 met the criteria of clinical significance in tested pathologies [127]. Even further, there have also been instances where patients have responded negatively, and sometimes fatally, to therapies because of genetic variations that mediate adverse drug interactions [128]. Therefore, identifying clinically-actionable biomarkers, and tailoring specific drugs to known pathways becomes an attractive path.
Advancements in sequencing technology have made it possible to collect individual molecular profiles for disease diagnosis and therapeutic intervention as a feasible front [129], [130]. One of the most notable examples of this is the advent of imatinib, a tyrosine kinase inhibitor designed to treat cancer patients with chronic myeloid leukemia, which has increased the overall survival rates to 90% at 5 years and 86% at 8 years [131]. To reach a higher understanding of NPDs, systems biology enables characterization of entire biological systems associated with a specific disease [132] from integrative disease models by utilizing genomics, transcriptomics and proteomics [133]. Using these ‘omics’ technologies to analyze healthy and disease patients, multiple datasets can be generated for each disease and organized into networks that suggest distinct molecular function, biomarkers and drug targets [134], [135]. For NPDs in particular, systems approach allows studying these highly heterogeneous and multi-phenotypic diseases where clinically actionable biomarkers have been difficult to determine [136], [137].
Given the complexity of NPDs, future precision psychiatry needs to identify risk indicator biomarkers [138], [139]. Since several biomarkers can be linked to one specific illness and one biomarker can overlap between different diseases [140], [141], [142], data-driven approaches can fine-tune biomarker multivariate profiles determined by the heterogeneity of these disorders [139]. Omics data represent a global and an unbiased area that combines whole genome, transcriptome and proteome data, and integrates them with more targeted experimental approaches and other factors that dissect disease risk and prognosis (e.g. life style and environment) to uncover overlapping elements involved in the pathophysiology of psychiatric diseases. The combination of these data (Fig. 4D) with those deposited in literature and public repositories, along with biobanks and hospital records, is necessary to evaluate the weight of each criterion in the onset and development of psychiatric illnesses. AI and machine learning algorithms can be used to stratify and match patients by taking into consideration of individual characteristics. Eventually, patient stratification can reveal the best therapeutic treatment for each subject (Fig. 4D), paving the way for a precision psychiatry approach [143], [144], [145], [146].
8. Conclusions
Gene-disease information places mt dysfunction under the spotlight as a corner stone in the onset and progression of BD/SZ/MD. This calls for the continued development of reliable disease models that mimic patient disease states, and we have discussed possible advancements using both iPSC-derived neurons and 3D brain organoids for NPD research. Mental illnesses remain in dire need of potent treatments, and the molecular attributes of psychotropic medications, along with their restorative capacity of mt fitness in BD/SZ/MD patients, remain poorly studied. Systems biology is taking us a step closer to accelerating precision medicine by combining different layers of ‘omics’ analyses, along with other disease risk resource data, to compile lists of prioritized targets for novel therapeutic intervention.
CRediT authorship contribution statement
Mohan Babu: Conceptualization, funding acquisition, project administration and supervision, Investigation, writing - original draft, Writing - review & editing. Sadhna Phanse: Data curation and formal analysis. Mara Zilocchi: Investigation, writing - original draft, Writing - review & editing. Kirsten Broderick: Investigation, writing - original draft, Writing - review & editing. Khaled A. Aly: Investigation, writing - original draft, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements and Disclosures
We thank Matthew Jessulat from Babu’s lab for helpful discussions and comments. M.B. is a Canadian Institutes of Health Research (CIHR) New Investigator (MSH-130178). This work was supported by the CIHR (MOP-125952; FDN-154318) and National Institutes of Health (R01GM106019) research grants to M.B. The authors declare no competing financial interests.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.09.008.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Egervari G., Kozlenkov A., Dracheva S., Hurd Y.L. Molecular windows into the human brain for psychiatric disorders. Mol Psychiatry. 2019;24:653–673. doi: 10.1038/s41380-018-0125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fields R.D. White matter in learning, cognition and psychiatric disorders. Trends Neurosci. 2008;31:361–370. doi: 10.1016/j.tins.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doherty J.L., Owen M.J. Genomic insights into the overlap between psychiatric disorders: implications for research and clinical practice. Genome Med. 2014;6:29. doi: 10.1186/gm546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fried E.I., van Borkulo C.D., Cramer A.O., Boschloo L., Schoevers R.A., Borsboom D. Mental disorders as networks of problems: a review of recent insights. Soc Psychiatry Psychiatr Epidemiol. 2017;52:1–10. doi: 10.1007/s00127-016-1319-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuehner J.N., Bruggeman E.C., Wen Z., Yao B. Epigenetic regulations in neuropsychiatric disorders. Front Genet. 2019;10:268. doi: 10.3389/fgene.2019.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollander J.A., Cory-Slechta D.A., Jacka F.N., Szabo S.T., Guilarte T.R., Bilbo S.D. Beyond the looking glass: recent advances in understanding the impact of environmental exposures on neuropsychiatric disease. Neuropsychopharmacology. 2020;45:1086–1096. doi: 10.1038/s41386-020-0648-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Misgeld T., Schwarz T.L. Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron. 2017;96:651–666. doi: 10.1016/j.neuron.2017.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J., Ryan S.K., Deboer E., Cook K., Fitzgerald S., Lachman H.M. Mitochondrial deficits in human iPSC-derived neurons from patients with 22q11.2 deletion syndrome and schizophrenia. Transl Psychiatry. 2019;9:302. doi: 10.1038/s41398-019-0643-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheng Z.H., Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zilocchi M., Moutaoufik M.T., Jessulat M., Phanse S., Aly K.A., Babu M. Misconnecting the dots: altered mitochondrial protein-protein interactions and their role in neurodegenerative disorders. Expert Rev Proteomics. 2020;17:119–136. doi: 10.1080/14789450.2020.1723419. [DOI] [PubMed] [Google Scholar]
- 11.Norkett R., Modi S., Birsa N., Atkin T.A., Ivankovic D., Pathania M. DISC1-dependent regulation of mitochondrial dynamics controls the morphogenesis of complex neuronal dendrites. J Biol Chem. 2016;291:613–629. doi: 10.1074/jbc.M115.699447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hennah W., Thomson P., McQuillin A., Bass N., Loukola A., Anjorin A. DISC1 association, heterogeneity and interplay in schizophrenia and bipolar disorder. Mol Psychiatry. 2009;14:865–873. doi: 10.1038/mp.2008.22. [DOI] [PubMed] [Google Scholar]
- 13.Yang K., Chen Z., Gao J., Shi W., Li L., Jiang S. The key roles of GSK-3beta in regulating mitochondrial activity. Cell Physiol Biochem. 2017;44:1445–1459. doi: 10.1159/000485580. [DOI] [PubMed] [Google Scholar]
- 14.Undi R.B., Gutti U., Gutti R.K. LiCl regulates mitochondrial biogenesis during megakaryocyte development. J Trace Elem Med Biol. 2017;39:193–201. doi: 10.1016/j.jtemb.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Jope R.S., Roh M.S. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bipolar D, Schizophrenia Working Group of the Psychiatric Genomics Consortium. Electronic address drve, Bipolar D, Schizophrenia Working Group of the Psychiatric Genomics C (2018): Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell. 173:1705-1715. [DOI] [PMC free article] [PubMed]
- 17.Iwamoto K., Kakiuchi C., Bundo M., Ikeda K., Kato T. Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol Psychiatry. 2004;9:406–416. doi: 10.1038/sj.mp.4001437. [DOI] [PubMed] [Google Scholar]
- 18.Yu K., Cheung C., Leung M., Li Q., Chua S., McAlonan G. Are bipolar disorder and schizophrenia neuroanatomically distinct? an anatomical likelihood meta-analysis. Front Hum Neurosci. 2010;4:189. doi: 10.3389/fnhum.2010.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clay H.B., Sillivan S., Konradi C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci. 2011;29:311–324. doi: 10.1016/j.ijdevneu.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasahara T., Kato T. What can mitochondrial DNA analysis tell us about mood disorders? Biol Psychiatry. 2018;83:731–738. doi: 10.1016/j.biopsych.2017.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Allen J., Romay-Tallon R., Brymer K.J., Caruncho H.J., Kalynchuk L.E. Mitochondria and mood: mitochondrial dysfunction as a key player in the manifestation of depression. Front Neurosci. 2018;12:386. doi: 10.3389/fnins.2018.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossignol D.A., Frye R.E. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012;17:290–314. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimamoto A., Rappeneau V. Sex-dependent mental illnesses and mitochondria. Schizophr Res. 2017;187:38–46. doi: 10.1016/j.schres.2017.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petschner P., Gonda X., Baksa D., Eszlari N., Trivaks M., Juhasz G. Genes linking mitochondrial function, cognitive impairment and depression are associated with endophenotypes serving precision medicine. Neuroscience. 2018;370:207–217. doi: 10.1016/j.neuroscience.2017.09.049. [DOI] [PubMed] [Google Scholar]
- 25.Schulze T.G., Akula N., Breuer R., Steele J., Nalls M.A., Singleton A.B. Molecular genetic overlap in bipolar disorder, schizophrenia, and major depressive disorder. World J Biol Psychiatry. 2014;15:200–208. doi: 10.3109/15622975.2012.662282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cipriani A., Furukawa T.A., Salanti G., Chaimani A., Atkinson L.Z., Ogawa Y. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Focus (Am Psychiatr Publ) 2018;16:420–429. doi: 10.1176/appi.focus.16407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agostinho F.R., Reus G.Z., Stringari R.B., Ribeiro K.F., Ferraro A.K., Benedet J. Treatment with olanzapine, fluoxetine and olanzapine/fluoxetine alters citrate synthase activity in rat brain. Neurosci Lett. 2011;487:278–281. doi: 10.1016/j.neulet.2010.10.037. [DOI] [PubMed] [Google Scholar]
- 28.Agostinho F.R., Reus G.Z., Stringari R.B., Ribeiro K.F., Ferreira G.K., Jeremias I.C. Olanzapine plus fluoxetine treatment alters mitochondrial respiratory chain activity in the rat brain. Acta Neuropsychiatr. 2011;23:282–291. doi: 10.1111/j.1601-5215.2011.00569.x. [DOI] [PubMed] [Google Scholar]
- 29.Adzic M., Lukic I., Mitic M., Djordjevic J., Elakovic I., Djordjevic A. Brain region- and sex-specific modulation of mitochondrial glucocorticoid receptor phosphorylation in fluoxetine treated stressed rats: effects on energy metabolism. Psychoneuroendocrinology. 2013;38:2914–2924. doi: 10.1016/j.psyneuen.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 30.Villa R.F., Ferrari F., Bagini L., Gorini A., Brunello N., Tascedda F. Mitochondrial energy metabolism of rat hippocampus after treatment with the antidepressants desipramine and fluoxetine. Neuropharmacology. 2017;121:30–38. doi: 10.1016/j.neuropharm.2017.04.025. [DOI] [PubMed] [Google Scholar]
- 31.Tamasi V., Petschner P., Adori C., Kirilly E., Ando R.D., Tothfalusi L. Transcriptional evidence for the role of chronic venlafaxine treatment in neurotrophic signaling and neuroplasticity including also Glutamatergic [corrected] - and insulin-mediated neuronal processes. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0113662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapur S., Agid O., Mizrahi R., Li M. How antipsychotics work-from receptors to reality. NeuroRx. 2006;3:10–21. doi: 10.1016/j.nurx.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan S.T., McCarthy M.J., Vawter M.P. Psychiatric drugs impact mitochondrial function in brain and other tissues. Schizophr Res. 2020;217:136–147. doi: 10.1016/j.schres.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langer S.Z. alpha2-Adrenoceptors in the treatment of major neuropsychiatric disorders. Trends Pharmacol Sci. 2015;36:196–202. doi: 10.1016/j.tips.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 35.Wheeler L., WoldeMussie E., Lai R. Role of alpha-2 agonists in neuroprotection. Surv Ophthalmol. 2003;48(Suppl 1):S47–51. doi: 10.1016/s0039-6257(03)00004-3. [DOI] [PubMed] [Google Scholar]
- 36.Won E., Kim Y.K. An oldie but goodie: lithium in the treatment of bipolar disorder through neuroprotective and neurotrophic mechanisms. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18122679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo D.Z., Chang C.Y., Huang T.R., Studer V., Wang T.W., Lai W.S. Lithium for schizophrenia: supporting evidence from a 12-year, nationwide health insurance database and from Akt1-deficient mouse and cellular models. Sci Rep. 2020;10:647. doi: 10.1038/s41598-019-57340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vieta E., Berk M., Schulze T.G., Carvalho A.F., Suppes T., Calabrese J.R. Bipolar disorders. Nat Rev Dis Primers. 2018;4:18008. doi: 10.1038/nrdp.2018.8. [DOI] [PubMed] [Google Scholar]
- 39.Machado-Vieira R., Manji H.K., Zarate C.A., Jr. The role of lithium in the treatment of bipolar disorder: convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord. 2009;11(Suppl 2):92–109. doi: 10.1111/j.1399-5618.2009.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta A., Schulze T.G., Nagarajan V., Akula N., Corona W., Jiang X.Y. Interaction networks of lithium and valproate molecular targets reveal a striking enrichment of apoptosis functional clusters and neurotrophin signaling. Pharmacogenomics J. 2012;12:328–341. doi: 10.1038/tpj.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gould T.D., Manji H.K. Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology. 2005;30:1223–1237. doi: 10.1038/sj.npp.1300731. [DOI] [PubMed] [Google Scholar]
- 42.Luca A., Calandra C., Luca M. Gsk3 signalling and redox status in bipolar disorder: evidence from lithium efficacy. Oxid Med Cell Longev. 2016;2016:3030547. doi: 10.1155/2016/3030547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maurer I.C., Schippel P., Volz H.P. Lithium-induced enhancement of mitochondrial oxidative phosphorylation in human brain tissue. Bipolar Disord. 2009;11:515–522. doi: 10.1111/j.1399-5618.2009.00729.x. [DOI] [PubMed] [Google Scholar]
- 44.Jakobsson E., Arguello-Miranda O., Chiu S.W., Fazal Z., Kruczek J., Nunez-Corrales S. Towards a unified understanding of lithium action in basic biology and its significance for applied biology. J Membr Biol. 2017;250:587–604. doi: 10.1007/s00232-017-9998-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saiardi A., Mudge A.W. Lithium and fluoxetine regulate the rate of phosphoinositide synthesis in neurons: a new view of their mechanisms of action in bipolar disorder. Transl Psychiatry. 2018;8:175. doi: 10.1038/s41398-018-0235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saxena A., Scaini G., Bavaresco D.V., Leite C., Valvassori S.S., Carvalho A.F. Role of protein kinase C in bipolar disorder: A review of the current literature. Mol Neuropsychiatry. 2017;3:108–124. doi: 10.1159/000480349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szabo S.T., Machado-Vieira R., Yuan P., Wang Y., Wei Y., Falke C. Glutamate receptors as targets of protein kinase C in the pathophysiology and treatment of animal models of mania. Neuropharmacology. 2009;56:47–55. doi: 10.1016/j.neuropharm.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berridge M.J. Calcium signalling and psychiatric disease: bipolar disorder and schizophrenia. Cell Tissue Res. 2014;357:477–492. doi: 10.1007/s00441-014-1806-z. [DOI] [PubMed] [Google Scholar]
- 49.Giorgi C., Marchi S., Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018;19:713–730. doi: 10.1038/s41580-018-0052-8. [DOI] [PubMed] [Google Scholar]
- 50.Boyman L., Williams G.S., Khananshvili D., Sekler I., Lederer W.J. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol. 2013;59:205–213. doi: 10.1016/j.yjmcc.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiu C.T., Chuang D.M. Neuroprotective action of lithium in disorders of the central nervous system. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2011;36:461–476. doi: 10.3969/j.issn.1672-7347.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engel S.R., Creson T.K., Hao Y., Shen Y., Maeng S., Nekrasova T. The extracellular signal-regulated kinase pathway contributes to the control of behavioral excitement. Mol Psychiatry. 2009;14:448–461. doi: 10.1038/sj.mp.4002135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farah R., Khamisy-Farah R., Amit T., Youdim M.B., Arraf Z. Lithium's gene expression profile, relevance to neuroprotection A cDNA microarray study. Cell Mol Neurobiol. 2013;33:411–420. doi: 10.1007/s10571-013-9907-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen R.W., Chuang D.M. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J Biol Chem. 1999;274:6039–6042. doi: 10.1074/jbc.274.10.6039. [DOI] [PubMed] [Google Scholar]
- 55.Quiroz J.A., Machado-Vieira R., Zarate C.A., Jr., Manji H.K. Novel insights into lithium's mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010;62:50–60. doi: 10.1159/000314310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jope R.S. Anti-bipolar therapy: mechanism of action of lithium. Mol Psychiatry. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- 57.Beaulieu J.M., Sotnikova T.D., Yao W.D., Kockeritz L., Woodgett J.R., Gainetdinov R.R. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ashok A.H., Marques T.R., Jauhar S., Nour M.M., Goodwin G.M., Young A.H. The dopamine hypothesis of bipolar affective disorder: the state of the art and implications for treatment. Mol Psychiatry. 2017;22:666–679. doi: 10.1038/mp.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kato T.M., Kubota-Sakashita M., Fujimori-Tonou N., Saitow F., Fuke S., Masuda A. Ant1 mutant mice bridge the mitochondrial and serotonergic dysfunctions in bipolar disorder. Mol Psychiatry. 2018;23:2039–2049. doi: 10.1038/s41380-018-0074-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alda M. Lithium in the treatment of bipolar disorder: pharmacology and pharmacogenetics. Mol Psychiatry. 2015;20:661–670. doi: 10.1038/mp.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Calvo S.E., Clauser K.R., Mootha V.K. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith A.C., Robinson A.J. MitoMiner v3.1, an update on the mitochondrial proteomics database. Nucleic Acids Res. 2016;44:D1258–1261. doi: 10.1093/nar/gkv1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.The Gene Ontology C The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019;47:D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Binder JX, Pletscher-Frankild S, Tsafou K, Stolte C, O'Donoghue SI, Schneider R, et al. (2014): Compartments: unification and visualization of protein subcellular localization evidence. Database (Oxford). 2014:bau012. [DOI] [PMC free article] [PubMed]
- 66.Pinero J., Bravo A., Queralt-Rosinach N., Gutierrez-Sacristan A., Deu-Pons J., Centeno E. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017;45:D833–D839. doi: 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goncalves V.F., Cappi C., Hagen C.M., Sequeira A., Vawter M.P., Derkach A. A Comprehensive analysis of nuclear-encoded mitochondrial genes in schizophrenia. Biol Psychiatry. 2018;83:780–789. doi: 10.1016/j.biopsych.2018.02.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Provencal N., Shink E., Harvey M., Tremblay M., Barden N. Analysis of a variable number tandem repeat polymorphism in the huntingtin interacting protein-1 related gene for anticipation in bipolar affective disorder. Prog Neuro-Psychopharmacol Biol Psychiatry. 2004;28:1299–1303. doi: 10.1016/j.pnpbp.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 69.Cotto K.C., Wagner A.H., Feng Y.Y., Kiwala S., Coffman A.C., Spies G. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018;46:D1068–D1073. doi: 10.1093/nar/gkx1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sjoholt G., Ebstein R.P., Lie R.T., Berle J.O., Mallet J., Deleuze J.F. Examination of IMPA1 and IMPA2 genes in manic-depressive patients: association between IMPA2 promoter polymorphisms and bipolar disorder. Mol Psychiatry. 2004;9:621–629. doi: 10.1038/sj.mp.4001460. [DOI] [PubMed] [Google Scholar]
- 71.Klein P.S., Melton D.A. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tobe B.T.D., Crain A.M., Winquist A.M., Calabrese B., Makihara H., Zhao W.N. Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc Natl Acad Sci U S A. 2017;114:E4462–E4471. doi: 10.1073/pnas.1700111114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li X., Jope R.S. Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology. 2010;35:2143–2154. doi: 10.1038/npp.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eom T.Y., Jope R.S. Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol Psychiatry. 2009;66:494–502. doi: 10.1016/j.biopsych.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jope R.S. Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front Mol Neurosci. 2011;4:16. doi: 10.3389/fnmol.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang B., Liang P., Deng G., Tu Z., Liu M., Xiao X. Increased stability of Bcl-2 in HSP70-mediated protection against apoptosis induced by oxidative stress. Cell Stress Chaperones. 2011;16:143–152. doi: 10.1007/s12192-010-0226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duan H., Heckman C.A., Boxer L.M. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005;25:1608–1619. doi: 10.1128/MCB.25.5.1608-1619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oulas A., Minadakis G., Zachariou M., Sokratous K., Bourdakou M.M., Spyrou G.M. Systems bioinformatics: increasing precision of computational diagnostics and therapeutics through network-based approaches. Brief Bioinform. 2019;20:806–824. doi: 10.1093/bib/bbx151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Falk A., Heine V.M., Harwood A.J., Sullivan P.F., Peitz M., Brustle O. Modeling psychiatric disorders: from genomic findings to cellular phenotypes. Mol Psychiatry. 2016;21:1167–1179. doi: 10.1038/mp.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaiser T., Feng G. Modeling psychiatric disorders for developing effective treatments. Nat Med. 2015;21:979–988. doi: 10.1038/nm.3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stern S., Santos R., Marchetto M.C., Mendes A.P.D., Rouleau G.A., Biesmans S. Neurons derived from patients with bipolar disorder divide into intrinsically different sub-populations of neurons, predicting the patients' responsiveness to lithium. Mol Psychiatry. 2018;23:1453–1465. doi: 10.1038/mp.2016.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mertens J., Wang Q.W., Kim Y., Yu D.X., Pham S., Yang B. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 2015;527:95–99. doi: 10.1038/nature15526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O'Shea K.S., McInnis M.G. Neurodevelopmental origins of bipolar disorder: iPSC models. Mol Cell Neurosci. 2016;73:63–83. doi: 10.1016/j.mcn.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 84.Bavamian S., Mellios N., Lalonde J., Fass D.M., Wang J., Sheridan S.D. Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol Psychiatry. 2015;20:573–584. doi: 10.1038/mp.2014.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen H.M., DeLong C.J., Bame M., Rajapakse I., Herron T.J., McInnis M.G. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl Psychiatry. 2014;4 doi: 10.1038/tp.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Amin N.D., Pasca S.P. Building models of brain disorders with three-dimensional organoids. Neuron. 2018;100:389–405. doi: 10.1016/j.neuron.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 87.Kathuria A., Lopez-Lengowski K., Watmuff B., McPhie D., Cohen B.M., Karmacharya R. Synaptic deficits in iPSC-derived cortical interneurons in schizophrenia are mediated by NLGN2 and rescued by N-acetylcysteine. Transl Psychiatry. 2019;9:321. doi: 10.1038/s41398-019-0660-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pasca S.P. The rise of three-dimensional human brain cultures. Nature. 2018;553:437–445. doi: 10.1038/nature25032. [DOI] [PubMed] [Google Scholar]
- 89.Birey F., Andersen J., Makinson C.D., Islam S., Wei W., Huber N. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545:54–59. doi: 10.1038/nature22330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stachowiak E.K., Benson C.A., Narla S.T., Dimitri A., Chuye L.E.B., Dhiman S. Cerebral organoids reveal early cortical maldevelopment in schizophrenia-computational anatomy and genomics, role of FGFR1. Transl Psychiatry. 2017;7:6. doi: 10.1038/s41398-017-0054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qian X., Song H., Ming G.L. Brain organoids: advances, applications and challenges. Development. 2019;146 doi: 10.1242/dev.166074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manji H., Kato T., Di Prospero N.A., Ness S., Beal M.F., Krams M. Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci. 2012;13:293–307. doi: 10.1038/nrn3229. [DOI] [PubMed] [Google Scholar]
- 93.Gorman G.S., Chinnery P.F., DiMauro S., Hirano M., Koga Y., McFarland R. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080. doi: 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- 94.Moutaoufik M.T., Malty R., Amin S., Zhang Q., Phanse S., Gagarinova A. Rewiring of the human mitochondrial interactome during neuronal reprogramming reveals regulators of the respirasome and neurogenesis. iScience. 2019;19:1114–1132. doi: 10.1016/j.isci.2019.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pourhaghighi R., Ash P.E.A., Phanse S., Goebels F., Hu L.Z.M., Chen S. BraInMap elucidates the macromolecular connectivity landscape of mammalian brain. Cell Syst. 2020;10 doi: 10.1016/j.cels.2020.03.003. 333–350 e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ramos-Miguel A., Barakauskas V., Alamri J., Miyauchi M., Barr A.M., Beasley C.L. The SNAP25 interactome in ventromedial caudate in schizophrenia includes the mitochondrial protein ARF1. Neuroscience. 2019;420:97–111. doi: 10.1016/j.neuroscience.2018.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saraon P., Snider J., Kalaidzidis Y., Wybenga-Groot L.E., Weiss K., Rai A. A drug discovery platform to identify compounds that inhibit EGFR triple mutants. Nat Chem Biol. 2020 doi: 10.1038/s41589-020-0484-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Petschnigg J., Groisman B., Kotlyar M., Taipale M., Zheng Y., Kurat C.F. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat Methods. 2014;11:585–592. doi: 10.1038/nmeth.2895. [DOI] [PubMed] [Google Scholar]
- 99.Ramkumar P., Kampmann M. CRISPR-based genetic interaction maps inform therapeutic strategies in cancer. Transl Cancer Res. 2018;7:S61–S67. doi: 10.21037/tcr.2018.01.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Akula N., Barb J., Jiang X., Wendland J.R., Choi K.H., Sen S.K. RNA-sequencing of the brain transcriptome implicates dysregulation of neuroplasticity, circadian rhythms and GTPase binding in bipolar disorder. Mol Psychiatry. 2014;19:1179–1185. doi: 10.1038/mp.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lobentanzer S., Hanin G., Klein J., Soreq H. Integrative transcriptomics reveals sexually dimorphic control of the cholinergic/neurokine interface in schizophrenia and bipolar disorder. Cell Rep. 2019;29 doi: 10.1016/j.celrep.2019.09.017. 764–777 e765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fuchikami M., Morinobu S., Segawa M., Okamoto Y., Yamawaki S., Ozaki N. DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0023881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tadic A., Muller-Engling L., Schlicht K.F., Kotsiari A., Dreimuller N., Kleimann A. Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol Psychiatry. 2014;19:281–283. doi: 10.1038/mp.2013.58. [DOI] [PubMed] [Google Scholar]
- 104.Miller B.H., Zeier Z., Xi L., Lanz T.A., Deng S., Strathmann J. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc Natl Acad Sci U S A. 2012;109:3125–3130. doi: 10.1073/pnas.1113793109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dempster E.L., Pidsley R., Schalkwyk L.C., Owens S., Georgiades A., Kane F. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Human Mol Genet. 2011;20:4786–4796. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guilarte T.R., Opler M., Pletnikov M. Is lead exposure in early life an environmental risk factor for Schizophrenia? Neurobiological connections and testable hypotheses. Neurotoxicology. 2012;33:560–574. doi: 10.1016/j.neuro.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Andreazza A.C. Combining redox-proteomics and epigenomics to explain the involvement of oxidative stress in psychiatric disorders. Mol BioSyst. 2012;8:2503–2512. doi: 10.1039/c2mb25118c. [DOI] [PubMed] [Google Scholar]
- 108.Preece R.L., Han S.Y.S., Bahn S. Proteomic approaches to identify blood-based biomarkers for depression and bipolar disorders. Expert Rev Proteomics. 2018;15:325–340. doi: 10.1080/14789450.2018.1444483. [DOI] [PubMed] [Google Scholar]
- 109.Haque A., Engel J., Teichmann S.A., Lonnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9:75. doi: 10.1186/s13073-017-0467-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Simonis N., Gonze D., Orsi C., van Helden J., Wodak S.J. Modularity of the transcriptional response of protein complexes in yeast. J Mol Biol. 2006;363:589–610. doi: 10.1016/j.jmb.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 111.Mitra K., Carvunis A.R., Ramesh S.K., Ideker T. Integrative approaches for finding modular structure in biological networks. Nature Rev Genet. 2013;14:719–732. doi: 10.1038/nrg3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu C., Zhu J., Zhang X. Integrating gene expression and protein-protein interaction network to prioritize cancer-associated genes. BMC Bioinf. 2012;13:182. doi: 10.1186/1471-2105-13-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tyler S.R., Rotti P.G., Sun X., Yi Y., Xie W., Winter M.C. PyMINEr finds gene and autocrine-paracrine networks from human islet scRNA-Seq. Cell Rep. 2019;26 doi: 10.1016/j.celrep.2019.01.063. 1951–1964 e1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang T., Zhang D. Integrating omics data and protein interaction networks to prioritize driver genes in cancer. Oncotarget. 2017;8:58050–58060. doi: 10.18632/oncotarget.19481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Skene N.G., Bryois J., Bakken T.E., Breen G., Crowley J.J., Gaspar H.A. Genetic identification of brain cell types underlying schizophrenia. Nat Genet. 2018;50:825–833. doi: 10.1038/s41588-018-0129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hrvatin S., Hochbaum D.R., Nagy M.A., Cicconet M., Robertson K., Cheadle L. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat Neurosci. 2018;21:120–129. doi: 10.1038/s41593-017-0029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li J.W., Lee H.M., Wang Y., Tong A.H., Yip K.Y., Tsui S.K. Interactome-transcriptome analysis discovers signatures complementary to GWAS loci of type 2 diabetes. Sci Rep. 2016;6:35228. doi: 10.1038/srep35228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bzdok D., Meyer-Lindenberg A. Machine learning for precision psychiatry: opportunities and challenges. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3:223–230. doi: 10.1016/j.bpsc.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 119.Chandler C., Foltz P.W., Elvevag B. Using machine learning in psychiatry: the need to establish a framework that nurtures trustworthiness. Schizophr Bull. 2020;46:11–14. doi: 10.1093/schbul/sbz105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Graham S., Depp C., Lee E.E., Nebeker C., Tu X., Kim H.C. Artificial intelligence for mental health and mental illnesses: an overview. Curr Psychiatry Rep. 2019;21:116. doi: 10.1007/s11920-019-1094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vamathevan J., Clark D., Czodrowski P., Dunham I., Ferran E., Lee G. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18:463–477. doi: 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang X., Wang Y., Byrne R., Schneider G., Yang S. Concepts of artificial intelligence for computer-assisted drug discovery. Chem Rev. 2019;119:10520–10594. doi: 10.1021/acs.chemrev.8b00728. [DOI] [PubMed] [Google Scholar]
- 123.Toh T.S., Dondelinger F., Wang D. Looking beyond the hype: Applied AI and machine learning in translational medicine. EBioMedicine. 2019;47:607–615. doi: 10.1016/j.ebiom.2019.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Woo G., Fernandez M., Hsing M., Lack N.A., Cavga A.D., Cherkasov A. DeepCOP: deep learning-based approach to predict gene regulating effects of small molecules. Bioinformatics. 2020;36:813–818. doi: 10.1093/bioinformatics/btz645. [DOI] [PubMed] [Google Scholar]
- 125.Fleming N. How artificial intelligence is changing drug discovery. Nature. 2018;557:S55–S57. doi: 10.1038/d41586-018-05267-x. [DOI] [PubMed] [Google Scholar]
- 126.Abrahams E. Right drug-right patient-right time: personalized medicine coalition. Clin Transl Sci. 2008;1:11–12. doi: 10.1111/j.1752-8062.2008.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Leucht S., Helfer B., Gartlehner G., Davis J.M. How effective are common medications: a perspective based on meta-analyses of major drugs. BMC Med. 2015;13:253. doi: 10.1186/s12916-015-0494-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roden D.M., Wilke R.A., Kroemer H.K., Stein C.M. Pharmacogenomics: the genetics of variable drug responses. Circulation. 2011;123:1661–1670. doi: 10.1161/CIRCULATIONAHA.109.914820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lightbody G., Haberland V., Browne F., Taggart L., Zheng H., Parkes E. Review of applications of high-throughput sequencing in personalized medicine: barriers and facilitators of future progress in research and clinical application. Brief Bioinform. 2019;20:1795–1811. doi: 10.1093/bib/bby051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jorgensen J.T. Twenty years with personalized medicine: past, present, and future of individualized pharmacotherapy. Oncologist. 2019;24:e432–e440. doi: 10.1634/theoncologist.2019-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hehlmann R., Muller M.C., Lauseker M., Hanfstein B., Fabarius A., Schreiber A. Deep molecular response is reached by the majority of patients treated with imatinib, predicts survival, and is achieved more quickly by optimized high-dose imatinib: results from the randomized CML-study IV. J Clin Oncol. 2014;32:415–423. doi: 10.1200/JCO.2013.49.9020. [DOI] [PubMed] [Google Scholar]
- 132.Maldonado E.M., Taha F., Rahman J., Rahman S. Systems biology approaches toward understanding primary mitochondrial diseases. Front Genet. 2019;10:19. doi: 10.3389/fgene.2019.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Karahalil B. Overview of systems biology and omics technologies. Curr Med Chem. 2016;23:4221–4230. doi: 10.2174/0929867323666160926150617. [DOI] [PubMed] [Google Scholar]
- 134.Topol E.J. Individualized medicine from prewomb to tomb. Cell. 2014;157:241–253. doi: 10.1016/j.cell.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Suomalainen A., Elo J.M., Pietilainen K.H., Hakonen A.H., Sevastianova K., Korpela M. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 2011;10:806–818. doi: 10.1016/S1474-4422(11)70155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ahmed S.S., Ahameethunisa A.R., Santosh W., Chakravarthy S., Kumar S. Systems biological approach on neurological disorders: a novel molecular connectivity to aging and psychiatric diseases. BMC Syst Biol. 2011;5:6. doi: 10.1186/1752-0509-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Peedicayil J. Identification of biomarkers in neuropsychiatric disorders based on systems biology and epigenetics. Front Genet. 2019;10:985. doi: 10.3389/fgene.2019.00985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Biomarkers Definitions Working G Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 139.Fernandes B.S., Williams L.M., Steiner J., Leboyer M., Carvalho A.F., Berk M. The new field of 'precision psychiatry'. BMC Med. 2017;15:80. doi: 10.1186/s12916-017-0849-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fernandes B.S., Gama C.S., Kauer-Sant'Anna M., Lobato M.I., Belmonte-de-Abreu P., Kapczinski F. Serum brain-derived neurotrophic factor in bipolar and unipolar depression: a potential adjunctive tool for differential diagnosis. J Psychiatr Res. 2009;43:1200–1204. doi: 10.1016/j.jpsychires.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 141.Carvalho A.F., Kohler C.A., Brunoni A.R., Miskowiak K.W., Herrmann N., Lanctot K.L. Bias in peripheral depression biomarkers. Psychother Psychosom. 2016;85:81–90. doi: 10.1159/000441457. [DOI] [PubMed] [Google Scholar]
- 142.Carvalho A.F., Kohler C.A., Fernandes B.S., Quevedo J., Miskowiak K.W., Brunoni A.R. Bias in emerging biomarkers for bipolar disorder. Psychol Med. 2016;46:2287–2297. doi: 10.1017/S0033291716000957. [DOI] [PubMed] [Google Scholar]
- 143.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 144.Marzano L., Bardill A., Fields B., Herd K., Veale D., Grey N. The application of mHealth to mental health: opportunities and challenges. Lancet Psychiatry. 2015;2:942–948. doi: 10.1016/S2215-0366(15)00268-0. [DOI] [PubMed] [Google Scholar]
- 145.Johnson K.W., Shameer K., Glicksberg B.S., Readhead B., Sengupta P.P., Bjorkegren J.L.M. Enabling precision cardiology through multiscale biology and systems medicine. JACC Basic Transl Sci. 2017;2:311–327. doi: 10.1016/j.jacbts.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Buske O.J., Girdea M., Dumitriu S., Gallinger B., Hartley T., Trang H. PhenomeCentral: a portal for phenotypic and genotypic matchmaking of patients with rare genetic diseases. Hum Mutat. 2015;36:931–940. doi: 10.1002/humu.22851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.