

Summary
Impairment of circadian rhythms impacts carcinogenesis. SMAD4, a clock-controlled gene and central component of the TGFβ canonical pathway, is frequently mutated in pancreatic ductal adenocarcinoma (PDA), leading to decreased survival. Here, we used an in vitro PDA model of SMAD4-positive and SMAD4-negative cells to investigate the interplay between circadian rhythms, the TGFβ canonical signaling pathway, and its impact on tumor malignancy. Our data show that TGFβ1, SMAD3, SMAD4, and SMAD7 oscillate in a circadian fashion in SMAD4-positive PDA cells, whereas altering the clock impairs the mRNA dynamics of these genes. Furthermore, the expression of the clock genes DEC1, DEC2, and CRY1 varied depending on SMAD4 status. TGFβ pathway activation resulted in an altered clock, cell-cycle arrest, accelerated apoptosis rate, enhanced invasiveness, and chemosensitivity. Our data suggest that the impact of TGFβ on the clock is SMAD4-dependent, and SMAD3, SMAD4, DEC1, and CRY1 involved in this cross-talk affect PDA patient survival.
Subject Areas: Cell Biology, Chronobiology, Cancer
Graphical Abstract
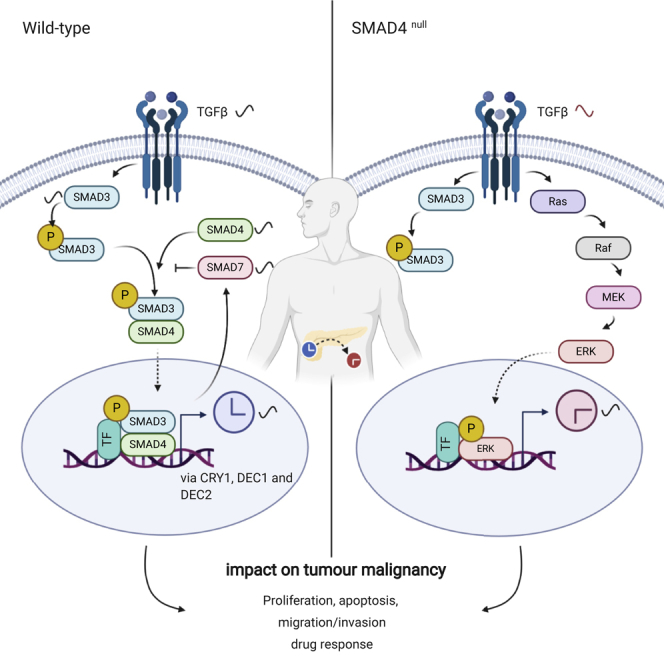
Highlights
-
•
Transcripts of the canonical TGFβ pathway are rhythmically expressed in SMAD4+/+ PDA
-
•
A reciprocal interplay exists between the TGFβ pathway and the circadian clock in SMAD4+/+ PDA
-
•
SMAD4-WT and SMAD4-Null cells exibit differential circadian regulation of cell-fate decisions
-
•
Drug administration in PDA cells shows time- and SMAD4-dependent effects
Cell Biology; Chronobiology; Cancer
Introduction
In mammals, an internal biological timing system, the circadian clock coordinates the time of behavioral activities and physiological processes. At the molecular level, the circadian clock is composed of a set of genes and their protein products, interconnected by transcriptional/translational feedback loops (Fuhr et al., 2015). This core-clock network (CCN) generates endogenous 24-h oscillations in the expression of genes and proteins and is involved in the circadian regulation of various cellular processes, including the cell cycle (El-Athman et al., 2017), apoptosis (Gery et al., 2005; Wang et al., 2016), DNA repair (Di Micco et al., 2011), the epithelial-to-mesenchymal transition (EMT) (Mao et al., 2012), metabolism (Fuhr et al., 2018; Reinke and Asher, 2019), and immunity (Abreu et al., 2018). Its aberrant function impacts cell functioning and can lead to the development and progression of several diseases including cancer (Davis et al., 2019; Sulli et al., 2019; Yalcin et al., 2020). Genes involved in cell cycle regulation (e.g., MYC, WEE1, and INK4A), immune function (e.g., TNF), and metabolism (e.g., SIRT3, CPT1, and PDH) show a rhythmic pattern of expression and are known clock-controlled genes (CCGs).
The transforming growth factor β (TGFβ) transduction pathway is among the clock-controlled pathways involved in oncogenic transformation. The TGFβ pathway is implicated in the maintenance of tissue homeostasis, regulation of fetal development, immune system control, wound repair, and EMT (Massague, 2008). The basic elements of the canonical TGFβ pathway include TGFβ cytokines, receptors, and Smads (Smad4, receptor-Smads, and inhibitory-Smads). TGFβ activates downstream pathways by binding to two pairs of receptors (type I and type II receptors). Once the receptors are activated, reporter-Smads (R-Smads) undergo phosphorylation, which results in signal propagation. The activated R-Smads (Smad2 and Smad3) translocate to the nucleus and form a complex with Smad4. Activated Smad complexes regulate gene expression by DNA-binding activity, and inhibitory-Smads (Smad7) prevent this complex formation (Massague, 2008; Prunier et al., 2019).
In normal and premalignant cells, TGFβ functions as tumor suppressor via the regulation of cytostasis, differentiation, cell cycle, apoptosis, and suppression of inflammation, and malfunctions in the TGFβ signaling pathway can result in tumorigenesis (Cheng et al., 2001; Massague, 2008; Principe et al., 2017; Scandura et al., 2004; Siegel and Massague, 2003).
Pancreatic ductal adenocarcinoma (PDA) is a malignant disease with poor prognosis and low overall survival rate, and it is the fourth leading cause of cancer death in the European Union and the United States (Coveler et al., 2016). More than half of the patients with PDA harbor mutations in SMAD4. The inactivation of SMAD4 in tumors is a common event in advanced-stage PDA and is linked to poorer prognosis (Singh et al., 2012).
Interestingly, several components of the TGFβ canonical pathway are circadian regulated in different organisms (Akagi et al., 2017; Chen et al., 2015; Sato et al., 2019). Previous studies reported circadian expression of TGFβ1 and Smad3 (transcripts or proteins) in mouse brown adipocyte (Nam et al., 2015), mouse kidney (Sato et al., 2019), and mouse heart (Sato et al., 2017). In addition, the oscillating pattern of TGFβ1 was altered after the disruption of Clock (Chen et al., 2015). However, it remains unclear whether components of the TGFβ canonical signaling (including TGFβ1, SMAD4, SMAD3, and SMAD7) are clock regulated in human cancer cells and whether circadian disruption affects these elements in cancer cells with consequences on tumor malignancy. Work from our group indicates that Smad4, the core mediator of the TGFβ canonical pathway, is regulated indirectly by the circadian clock (Lehmann et al., 2015). Wu et al. reported the binding of Smad3 or Smad4 to the Smad-binding elements in the DEC1 promoter in pancreatic cancer cells, further contributing to their circadian regulation (Wu et al., 2012). These studies pointed to a link bridging the circadian and TGFβ pathway.
Despite current findings regarding rhythmicity in elements of the TGFβ pathway and the functionality of this pathway, the reciprocal interplay between the TGFβ/SMAD4 pathway, the circadian clock, and its impact on tumor progression remains unclear in PDA.
Here, we investigated the influence of a dysregulated biological clock on PDA progression using an in vitro cellular model system. For this, we used SMAD4 wild-type and mutant PDA cell lines, derived respectively, from the primary tumor and the metastatic lesions of patients with PDA. We further explored the impact of clock dysregulation on the TGFβ/SMAD4 canonical pathway. Our results show that elements of the TGFβ canonical pathway (including SMAD3, SMAD4, SMAD7, and TGFβ1) are expressed in a circadian manner in SMAD4 wild-type PDA cells. In addition, we found that the rhythmic expression of the above-mentioned transcripts was altered upon the dysregulation of the circadian clock, suggesting that these TGFβ canonical elements are under circadian modulation. Our data point to a modulation of the TGFβ canonical pathway via the clock through interactions with the transcription factors DEC1, DEC2, and the core-clock gene CRY1. Also, we explored the influence of the canonical TGFβ pathway on the clock phenotype in these cells. Our results indicated that the activation of the TGFβ canonical pathway through SMAD4 overexpression and TGFβ induction results in a faster clock in PDA cells. Also, genetic modifications of SMAD4 (knockdown or overexpression) altered the expression of the core-clock genes BMAL1, PER2, NR1D1, and CRY1, and of the clock- and TGFβ-controlled genes, DEC1 and DEC2.
Furthermore, we investigated proliferation, apoptosis, migration, and invasion in clock-disrupted (shBMAL1, shPER2, and shNR1D1 knockdown) and SMAD4 up- or downregulated PDA cells, as well as the effects of a dysregulated clock on drug sensitivity in both SMAD4-proficient and SMAD4-deficient PDA cell lines. Our data provide evidence for SMAD4- and clock-dependent drug response in PDA cells, and highlight the role of the bidirectional interaction between the biological clock and the TGFβ/SMAD4 canonical pathway in cell cycle, apoptosis, and cancer metastasis in pancreatic cancer cells, which may further affect patient survival.
Results
Bidirectional Interplay between the Circadian Clock and TGFβ Canonical Pathway
To investigate the impact of a dysregulated clock in cancer metastasis and cell fate decisions in a pancreatic cancer cellular model, we used an established in vitro model of SMAD4-proficient (Panc1) and SMAD4-deficient (AsPC1) pancreatic adenocarcinoma cells (PDA), derived from different anatomical patient lesions (primary tumor and metastasis ascites, respectively) representing PDA tumors at different stages. Panc1 (ATCC: CRL-1469) is derived from the primary tumor of a male patient. The cell line AsPC1 (ATCC: CRL-1682) was established from ascites of a female patient with PDA. The doubling time of both cell lines is very similar and close to 52 h (Lieber et al., 1975; Watanabe et al., 2012). In addition, we analyzed cell growth of wild-type Panc1 and AsPC1 in our work using cell nucleus fluorescence labeling, which shows similar growth curves within 72 h for both cell lines (n ± SEM, n = 8, Figure S1D). Hence, both cell lines show similar cell cycle dynamics, making them suitable for our study. Of note, both cell lines carry mutated forms of KRAS, p16, and TP53 (Berrozpe et al., 1994; Kita et al., 1999; Sun et al., 2001). Furthermore, our preliminary work for this study (via computational network analysis) showed that, among the highly mutated genes in PDA (above 5% mutation rate), SMAD4 is tightly correlated with the CCN and has an impact on patient outcome (Cancer Genome Atlas Research Network, 2017; Lehmann et al., 2015).
Both cell lines showed oscillations, but with smaller amplitudes and shorter period for the Panc1 cells (BMAL1: 26.1 ± 0.3 h, n = 3) when compared with the AsPC1 cells (BMAL1: 27.4 ± 0.6 h, n = 3, Figure 1A). The oscillation parameters of PER2:Luc are provided in Table 1. We performed a 48 h time course RT-qPCR analysis, which confirmed the phenotypes observed in our luminescence data for BMAL1 and PER2 with an anti-phasic oscillation in both PDA cells (Figures 1A and 1B, Tables 1 and 2). Thus, according to our data, the endogenous time machinery operates differentially in these PDA cell lines.
Figure 1.

Panc1 and AsPC1 Cell Lines Harbor Different Clock Phenotypes
(A) Panc1 and AsPC1 cells were lentivirally transduced with a BMAL1 promoter (green) or PER2 promoter (orange)-driven luciferase construct. Bioluminescence was measured for 5 consecutive days. Depicted is one representative replicate for each condition.
(B) The 48 h time course RT-qPCR measurements of selected core-clock genes (BMAL1, PER2, and NR1D1) in Panc1 and AsPC1 WT cells.
(C) The relative mRNA levels of SMAD4 compared with AsPC1.
(D) RT-qPCR measurements after knockdown of core-clock genes (shBMAL1, shPER2, and shNR1D1) in Panc1 and AsPC1 cells.
Data are expressed as mean ± SEM, n = 3, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figures S1 and S2.
Table 1.
Circadian Parameters of PER2 Promoter Activity in PDA Cells (Chronostar Analysis)
| Panc1 | AsPC1 | |
|---|---|---|
| Period (h) | 26.6 ± 2.7 | 26.4 ± 0.4 |
| Phase (h) | 22.1 ± 2.7 | 5.2 ± 0.5 |
| Amplitude | 0.2 ± 0.0 | 0.2 ± 0.1 |
| CC | 0.9 ± 0.1 | 0.9 ± 0.1 |
| Phase shift relative to BMAL1 (h) | 15.0 | 6.8 |
Table 2.
Circadian Parameters Retrieved with Cosinor Analysis for a 48 h Time Course RT-qPCR Data (BMAL1, PER2, and NR1D1).
| Cosinor Analysis | |||
|---|---|---|---|
| Panc1 |
|||
| BMAL1 | PER2 | NR1D1 | |
| Phase (h) | 17.0 ± 0.5 | 4.1 ± 0.7 | 14.4 ± 0.5 |
| Period (h) | 19.0 ± 1.0 | 18.3 ± 1.5 | 18.6 ± 0 |
| Amplitude | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.1 ± 0.1 |
| p Value | <0.0001 | <0.0001 | 0.045 |
| AsPC1 |
|||
|---|---|---|---|
| BMAL1 | PER2 | NR1D1 | |
| Phase (h) | 14.4 ± 0.3 | 0 ± 3.6 | 20 ± 0.0 |
| Period (h) | 22.3. ± 0.9 | 24.8 ± 0.0 | 20.0 ± 1.3 |
| Amplitude | 0.3 ± 0.0 | 0.2 ± 0.1 | 0.2 ± 0.1 |
| p Value | <0.0001 | <0.0001 | 0.0003 |
Next, to explore the impact of clock disruption in the TGFβ pathway, we produced knockdown PDA cell lines for the core-clock genes BMAL1, PER2, and NR1D1 and evaluated the output in terms of gene expression (Figures 1D, S1B, and S1C). SMAD4 expression in Panc1 cells was significantly higher when compared with that of AsPC1 (Figure 1C, ∗∗∗p < 0.001). We confirmed that AsPC1 is a SMAD4-deficient cell line at the protein level (Figure S1A), as previously reported (Schutte et al., 1996; Sun et al., 2001). Furthermore, SMAD4 expression was downregulated in Panc1-shBMAL1 and Panc1-shNR1D1 cells at 3 h after cell synchronization (Figure 1D, Panc1, ∗∗p < 0.01, n = 3). Interestingly, the downregulation of BMAL1 and NR1D1 impacts the rhythmic expression of SMAD4 in the SMAD4-proficient PDA cells (Figure 2A).
Figure 2.

Panc1 and AsPC1 Cells as a Model for Investigating the Cross-Talk between the Circadian Clock and the TGFβ Canonical Pathway
(A) The 24 h time course RT-qPCR was carried out in Panc1 cells (A) containing shCtrl empty vector, shBMAL1, and shNR1D1 constructs. Relative expression value of TGFβ signaling-associated genes (SMAD4, TGFβ1, SMAD3, and SMAD7) is shown compared with shCtrl cells at each time point.
(B, C, E, F, H, and I) SMAD4-KD or SMAD4-OE cells were lentivirally transduced with a BMAL1- or a PER2-promoter-driven luciferase construct. Bioluminescence was measured for 5 consecutive days with TGFβ1 or corresponding empty vehicle (0.1% BSA). Depicted are the periods for Panc1 (B, C, E, and F) and for AsPC1 (H and I) of BMAL1 or PER2 promoter activities, as indicated. One representative replicate for each condition is provided in Figure S4.
(D, G, and J) Effect of SMAD4-KD or SMAD4-OE on the expression of clock genes in Panc1 (D and G) and AsPC1 (J) cells.
Data are expressed as mean ± SEM, n = 3, one-way ANOVA (B, C, E, F, H, and I) or multiple t test (D, G, and J); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figures S3, S4, and S5A–S5B.
We further evaluated the consequences of these perturbations on the clock phenotype (Figure S2). The downregulation of BMAL1 and PER2 in Panc1 cells resulted in an almost complete loss of oscillations, whereas NR1D1 KD (knockdown) showed no significant effect on the rhythmicity of BMAL1. The downregulation of BMAL1 in AsPC1 cells led to a complete loss of oscillations, and AsPC1-shNR1D1 cells depicted a significant shorter period.
Our results point to a connection between core-clock elements and SMAD4. To further investigate this interplay, we quantified the expression of SMAD3, SMAD4, SMAD7, and TGFβ1 in our in vitro model (Figure S3). According to previous studies in other model systems (Chen et al., 2015; Nam et al., 2015), Clock-Bmal1 heterodimers or Bmal1 activate the expression of Tgfβ, Smad3 by binding to their promoter regions. Particularly, the robust circadian rhythmicity of mTgfβ1 is abolished in mClock-KO mice (Chen et al., 2015). Such findings suggest the existence of a circadian regulation of these TGFβ signaling components at the transcriptome level. Thereby, we hypothesized that this phenomenon might also exist in human PDA cancer cells. We found rhythmic expression of TGFβ1 in both the SMAD4-proficient and SMAD4-deficient PDA cells (Figures S3A and S3E). Surprisingly, we also observed circadian oscillations in the expression of SMAD3, SMAD4, and SMAD7 in SMAD4 WT cells (Panc1, Figures S3B–S3D and Table 3). Interestingly, SMAD3 and TGFβ1 transcripts oscillated with slightly different phases and similar periods as BMAL1 in SMAD4-proficient cells (Panc1, Figure S3, Tables 2 and 3). However, the oscillation of TGFβ1 exhibited longer periods (∼27 h) when compared with BMAL1 (22.3 ± 0.9 h) in SMAD4-deficient cells (AsPC1, Figure S3E), implying that the correlation between the rhythmic expression of TGFβ1 and BMAL1 is likely SMAD4 dependent. Furthermore, we compared the expression level of these components for the PDA cell lines in a 39 h time course (Figures S3F–S3H). The mRNA levels of SMAD3 and SMAD7 (Figures S3G and S3H) were much reduced in AsPC1 when compared with Panc1, whereas TGFβ1 was highly expressed in AsPC1 when compared with Panc1 (Figure S3F). These data point to the inactivation of the TGFβ canonical pathway in SMAD4-deficient PDA cells, which may result in altered interaction with the clock machinery and ultimately support further progression towards malignant phenotypes.
Table 3.
Circadian Parameters Retrieved with Cosinor Analysis for a 33 h Time Course RT-qPCR Data (TGFβ1, SMAD3, SMAD4, and SMAD7).
| Cosinor Analysis | ||||
|---|---|---|---|---|
| Panc1 |
||||
| TGFβ1 | SMAD3 | SMAD7 | SMAD4 | |
| Phase (h) | 1.7 ± 1.5 | 19.1 ± 0.8 | 11.85 ± 1.3 | 12.6 ± 1.1 |
| Period (h) | 20.0 ± 3.8 | 21.8 ± 2.6 | 22.8 ± 4.7 | 21.7 ± 3.4 |
| Amplitude | 0.1 ± 0.1 | 0.3 ± 0.1 | 0.2 ± 0.1 | 0.3 ± 0.2 |
| p Value | 0.0015 | 0.0004 | 0.0001 | 0.0256 |
| AsPC1 |
|
|---|---|
| TGFβ1 | |
| Phase (h) | 4.6 ± 0.3 |
| Period (h) | 27.0 |
| Amplitude | 0.2 ± 0.0 |
| p Value | <0.0001 |
We further explored the reciprocal interplay between the core-clock and the TGFβ canonical pathway and analyzed the effect of perturbing the core-clock elements on the expression of several components of the TGFβ canonical pathway (SMAD3, SMAD7, and TGFβ1).
A former study suggested Bmal1 and Nr1d1 to be key clock elements bridging the clock and the TGFβ canonical pathway in mouse adipocytes (Nam et al., 2015). Hence, we performed a 24 h time course RT-qPCR for BMAL1 and NR1D1 KDs and shCtrl in Panc1 cells. The expression of TGFβ1, SMAD3, SMAD4, and SMAD7 exhibited a large variation after the BMAL1 and NR1D1 KDs when compared with shCtrl (Figure 2A). In particular, in Panc1-shBMAL1 and Panc1-shNR1D1 cells, the expression of SMAD4 reached its peak at 18 h after synchronization. As previously reported, Bmal1 binding to the promoter region of Smad3 (resulting in activation of Smad3) occurred at a specific time point (CT8 in mice) (Nam et al., 2015). Our results show that clock dysregulation (BMAL1 KD) repressed SMAD3 and SMAD7 transcripts at 21 h after synchronization, suggesting that the inactivation of the TGFβ canonical pathway (particularly SMAD7 as an indicator of the TGFβ pathway activation) occurs at a specific time point after the clock disruption (Figure 2A). The TGFβ1 transcript was overexpressed upon BMAL1 KD over the time of the measurements (Figure 2A). These data indicated that the disruption of BMAL1 and NR1D1 modified the time-dependent variations of elements of the TGFβ pathway (Figures 2A and S3). We observed a similar expression pattern of SMAD3 and SMAD7 upon BMAL1 and NR1D1 KDs, indicating that these elements may assist to set up a rapid reciprocal cross-talk between the TGFβ canonical pathway and the circadian clock (Figure 2A).
We then measured BMAL1- and PER2-promoter activity in SMAD4-KD and overexpression (SMAD4-OE) cells when compared with the corresponding control conditions (shCtrl or oeCtrl) and with additional TGFβ1 stimulation, to explore the impact of the TGFβ pathway on the clock phenotype. Overexpression efficiency was analyzed at the transcript and protein levels (Figure S5A and S5B). The activation of the TGFβ canonical pathway by SMAD4-OE and additional TGFβ1 stimulation significantly shortened the period of the oscillations (Figures 2E, 2F, 2H, 2I, and S4A–S4C, ∗p < 0.05, n = 3). In addition, SMAD4-KD in Panc1 led to a shorter period when compared with shCtrl (Figures 2B and 2C, ∗∗p < 0.01), pointing to a putative role of SMAD4 in the regulation of the circadian clock via BMAL1.
In addition, we evaluated the impact of SMAD4 in the regulation of core-clock elements BMAL1, PER2, NR1D1, and CRY1 and further analyzed the expression of the clock genes DEC1 and DEC2 (Figures 2D, 2G, and 2J), reported as mediators in the above cross-talk (Kon et al., 2008; Sato et al., 2016, 2019). Interestingly, most of the core-clock genes showed significant alterations for Panc1 SMAD4-OE and SMAD4-KD (Figures 2D and 2G). Overall, CRY1 was upregulated, whereas DEC1 and DEC2 were downregulated in SMAD4-OE PDA cells (Figures 2D and 2J, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3). In agreement, we observed the opposite results in Panc1-shSMAD4 (Figure 2G, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3). These results further reinforce the existence of a bidirectional connection between the core-clock and the SMAD4-dependent pathways via the regulation of CRY1, DEC1, and DEC2.
Dysregulation of the Core-Clock Genes BMAL1, PER2, and NR1D1, and Perturbations in SMAD4 Impact Cell Proliferation and Apoptosis in PDA Cells
We further evaluated the effects of disrupting core-clock genes and SMAD4 in cell fate decisions by analyzing cell proliferation and apoptosis. Interestingly, SMAD4-proficient (Panc1) and SMAD4-deficient (AsPC1) cells showed different effects on proliferation upon the different knockdowns (Figure 3A). Proliferation increased in Panc1 cells after BMAL1 and NR1D1 KDs, whereas this was not observed in AsPC1 knockdown cells (Figure 3A). Panc1-shSMAD4 cells showed increased proliferation when compared with the shCtrl. However, SMAD4-OE in both PDA cells showed no significant difference in the proliferative ability compared with oeCtrl. These results indicate that the non-canonical pathway (which includes RAS, MAPK, JNK) might be activated after SMAD4 KD, leading to an increase in proliferation. To verify our assumption, we measured RAS and phosphorylated-ERK (p-ERK) proteins in Panc1-shCtrl and Panc1-shSMAD4 cells with and without a 24-h TGFβ1 stimulation. Our results show that the TGFβ1 stimulation activates the phosphorylation of ERK for both Panc1-shCrtl and Panc1-shSMAD4 (Figures S6A and S6C). In addition, SMAD4-KD (particularly stimulated with TGFβ1) indeed upregulated the expression of RAS and p-ERK, when compared with control cells (Figures S6A–S6C). These data suggest the activation of the Ras pathway after the downregulation of SMAD4. In addition, the apoptotic profiles of PDA cells were altered upon core-clock dysregulation. The downregulation of BMAL1, PER2, and NR1D1 increased apoptosis when compared with shCtrl (Figures 3B, 3C, and 3E). SMAD4 OE promoted apoptosis in both PDA cells (Figures 3B, 3D, and 3F). These results implied that the upregulation of SMAD4 and the disruption of core-clock genes induces apoptosis in our vitro model.
Figure 3.

Dysregulation of the Core-Clock Genes (BMAL1, PER2, and NR1D1) and SMAD4 Leads to Differential Cell Proliferation and Apoptosis in Panc1 and AsPC1 Cells
(A) Proliferation analyses of PDA cell lines for KD conditions (shBMAL1, shPER2, shNR1D1, and shSMAD4), SMAD4-OEs, and corresponding empty vectors (shCtrl and oeCtrl, respectively).
(B) Apoptosis analysis of PDA cell lines after KDs or OEs. Data are expressed as mean ± SEM, n ≥ 6 (A and B).
(C–F) Representative images of apoptosis assays for Panc1 shCtrl and knockdown cells shBMAL1, shPER2, shNR1D1, shSMAD4 (C) or Panc1 oeCtrl and SMAD4-OE cells (D) and AsPC1 shCtrl and knockdown cells shBMAL1, shPER2, shNR1D1 (E) or AsPC1 oeCtrl and SMAD4-OE cells (F), with fluorescently labeled caspase3/7 with (objective 20×) at time points 0, 72, and 120 h, respectively. Data are expressed as mean ± SEM, n ≥ 6 (A and B).
Clock Dysregulation and the Activation of the TGFβ/SMAD4 Pathway Impacts Apoptosis and Cell Cycle in PDA Cells
To further characterize the putative effects of dysregulating the TGFβ/SMAD4 signal transduction in our model, we stimulated the pathway with TGFβ1 in addition to SMAD4-KD or SMAD4-OE and analyzed cell proliferation and apoptosis. The activation of the pathway (SMAD4-OE with TGFβ1 stimulation) did not alter proliferation in Panc1 cells, and its inactivation (SMAD4-KD with or without TGFβ1) promoted proliferation (Figures 4A and 4B). Interestingly, we observed increased proliferation in SMAD4-deficient cells (AsPC1) with TGFβ1 stimulations compared with SMAD4-OE cells (Figure 4C). Thus, we hypothesized that TGFβ1 triggers the downstream elements of the TGFβ non-canonical signaling pathway (e.g., RAS, PI3K) leading to increased proliferation in SMAD4-deficient cells.
Figure 4.

Clock Disruption and the Activation of the TGFβ/SMAD4 Pathway Affects Apoptosis and Cell Cycle in PDA Cells
(A–C) Proliferation analysis of PDA cells containing SMAD4-OE (A, C) or SMAD4-KD (B) constructs and their corresponding empty vectors (shCtrl or oeCtrl) stimulated with additional TGFβ1 (10ng/ml) or its solvent.(n = 8; mean ± SEM).
(E–G) Apoptosis analysis of PDA cell lines after SMAD4-OE (E, G) or SMAD4-KD (F) and their corresponding empty vectors (shCtrl or oeCtrl) with 24-h stimulation with TGFβ1 or its solvent (n = 3, mean ± SEM).
(D and H) Cell cycle measurements after clock gene KDs (shBMAL1, shPER2, shNR1D1) and its empty control (shCtrl) in Panc1 (D) and AsPC1 (H) cells.
(I–K) Cell cycle measurements of SMAD4-KD and SMAD4-OE and corresponding control conditions (shCtrl and oeCtrl) in Panc1 (I, J) and AsPC1 (K) cells after a 24-h stimulation with TGFβ1 (10 ng/mL) or its solvent. (D and H–K) Phase distributions were compared with their respective control conditions (n =3, mean ± SEM, two-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
See also Figures S5C, S5D, and S6.
The stimulation of TGFβ1 induced apoptosis particularly in SMAD4-positive PDA cells (including SMAD4-OE and wild-type, Figures 4E–4G).
We analyzed the cell cycle of the PDA cells in greater detail in all the aforementioned conditions. Our analysis showed significant cell-type-specific alterations upon KD of clock genes when compared with control cells. For the SMAD4-proficient cell line (Panc1), the percentage of cells in S phase decreased significantly and G2/M phase increased after PER2 KD (Figure 4I, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3), whereas the percentage of cells in S phase increased after NR1D1, in agreement with the proliferation results. In AsPC1 cells, we observed a significant increase of S phase and decrease of G1 phase in PER2 KD cells (Figure 4K, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3), and no significant alterations upon the KD of BMAL1 and NR1D1, in agreement with our proliferation results (Figure 3A). The differential phenotypes observed might result from the different genomic background of both cell lines (in particular the SMAD4 alteration) that subsequently impacts the circadian pathway, leading to an overall differential proliferative phenotype.
Next, we investigated the impact of TGFβ on the cell cycle. Panc1 cells depicted increased G1/S arrest upon TGFβ1 stimulation (Figures 4D and S5C). Interestingly, unlike the impact of TGFβ1 that has been reported in previous studies for other model systems (Alexandrow and Moses, 1995; Mukherjee et al., 2010), and also found in SMAD4-proficient cells, a 24-h TGFβ1 stimulation increased the percentage of cells in G2/M phase in shNR1D1 cells (Figure S5C), pointing towards differential regulation of the cell cycle by TGFβ upon NR1D1 downregulation. Additional TGFβ1 did not cause G1/S arrest in SMAD4-deficient cells (AsPC1, Figures 4H and S5D); this is distinct from the observations in SMAD4-proficient cells (Panc1) Instead, we observed an increase in G2/M or S phase in all KD conditions (shBMAL1, shPER2, and shNR1D1) and shCtrl cells upon TGFβ stimulation when compared with non-stimulated cells (Figures 4H and S5D). These results point to a SMAD4 dependency regarding the effects of TGFβ on cell cycle distribution. We further analyzed the cell cycle in SMAD4-KD and SMAD4-OE conditions with or without additional TGFβ1 stimulation (Figures 4I–4K). For Panc1, the additional TGFβ1 and upregulation of SMAD4 led to increased number of cells in G1/S phase (Figures 4I and 4J). However, the impairment of the TGFβ canonical pathway (SMAD4 KD) increased the number of cells in S phase particularly when compared with the activation of TGFβ canonical pathway (shCtrl with TGFβ1): G1 decreased and S phase increased (Figure 4J). In SMAD4-deficient cells (AsPC1), SMAD4 OE with additional TGFβ1 stimulation resulted in an increased G1 and decreased S phase when compared with oeCtrl cells (Figure 4K). However, in AsPC1 oeCtrl cells, TGFβ1 stimulation led to an increase in the percentage of cells in S phase (Figure 4K) and consistently a higher proliferative potential (Figure 4C) when compared to non-stimulation oeCtrl. This finding further supports the activation of the non-canonical TGFβ pathway in the absence of SMAD4, which triggers the activation of the oncogene RAS (Figure S6) in the downstream pathway and subsequently affects cell cycle and proliferation.
These results demonstrate that TGFβ canonical and non-canonical pathways regulate the cell cycle, proliferation, and apoptosis in a SMAD4-dependent manner.
The Dysregulation of the Core Clock Impacts Cell Migration in a SMAD4-Dependent Manner
Next, we aimed to examine if the activation of TGFβ/SMAD4 pathway may contribute to the formation of tumor metastasis. We used a real-time cell recording approach to measure the invasive and migration properties of these PDA cells, analyzed known EMT markers, and investigated morphological cellular alterations (Figure 5). TGFβ1 stimulation significantly increased the migration speed in both PDA cells, particularly in SMAD4-OE cells (Figures 5B, 5C, 5E, and 5F, ∗p < 0.05, ∗∗∗p < 0.001, n = 8). For AsPC1 oeCtrl cells, TGFβ1 stimulation did not significantly promote migration likely due to its intrinsic SMAD4 deficiency. In contrast, Panc1 cells showed a significantly slower migration speed after SMAD4 KD (Figure 5D, ∗∗p < 0.01, n = 8) and TGFβ stimulation was not able to induce the migrative potential in SMAD4 KD cells (Figures 5A and 5D), in agreement with previous data by Takano and colleagues who reported a role for SMAD4 in the expression of EMT marker genes in pancreatic cancer (Takano et al., 2007). In addition, the resembled phenomenon was found in invasion assays as well; TGFβ1 stimulation or SMAD4 overexpression promoted the invasiveness of PDA cells (Figures 5H and 5I), whereas SMAD4-KD without additional TGFβ1 impaired invasive ability (Figure 5G). Altogether, these data suggest that the presence of intrinsic SMAD4 and TGFβ in the tumor microenvironment is crucial for tumor invasiveness and cell motility. Cells that undergo EMT have a greater tendency to invade and metastasize (Yang et al., 2020), thus we verified our results by quantifying EMT biological makers and examining alterations in cell morphology. The cadherins are major adhesion molecules anchoring in the cytomembrane. Epithelial cells express E-cadherin, whereas cells that show mesenchymal features express N-cadherin and R-cadherin (Wheelock et al., 2008). Cancer cells can enhance or reduce their metastatic potential through cadherin switching, which is one characteristic of the EMT. Also Vimentin, a cytoskeletal molecule responsible for maintaining cell integrity and resistance against stress (Satelli and Li, 2011), has been described as an EMT biomarker. Additionally, we also analyzed the transcription factors SNAIL and SLUG that are sensitive to microenvironmental stimuli and function as EMT switchers (Lamouille et al., 2014).
Figure 5.

Migration and Invasion Analysis of SMAD4-KD and SMAD4-OE PDA Cells With or Without TGFβ1 Stimulations and Corresponding Expression Levels of EMT Markers
(A–C) Migration assays were performed with the IncuCyte S3 Live Cell System Analysis using a scratch wound assay. Partial representation of the scratch wound assay for SMAD4-KD (A) or SMAD4-OE (B, C) and their control (shCtrl and oeCtrl) conditions in PDA cells with additional TGFβ1 or its solvent stimulation at 0, 24, 48 h. Images were obtained with the IncuCyte S3 Software. Yellow mask indicates the wound boundaries. Blue mask indicates the initial scratch wound area.
(D–F) Average cell migration speed distribution in Panc1 (D, E) and AsPC1 (F) cells containing SMAD4-KD or SMAD4-OE constructs and their empty vectors (shCtrl and oeCtrl) with additional TGFβ1 or its solvent stimulation, (n ≥ 8, mean ± SEM; two-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001)..
(G–I) Invasion assays were carried out using a chemotaxis cell invasion assay for the IncuCyte S3 Live Cell System Analysis. Trans-well invasion assays for SMAD4-KD (G) or SMAD4-OE (H, I) and their control (shCtrl and oeCtrl) conditions in PDA cells were measured every 2h. Quantification was performed by measuring the total phase area of the bottom layer of the inner chamber within 72 h. Data were normalized for the initial value (n ≥ 6, mean ± SEM).
(J–L) Expression levels of EMT and cancer stemness-related genes (E-cadherin, N-cadherin, Vimentin, SNAIL, SLUG,and CD133) in SMAD4-KD (J) or SMAD4-OE ( K, L) conditions compared with their control conditions, shCtrl and oe-Ctrl respectively (n = 3, mean ± SEM, t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
See also Figure S7.
CD133 has been commonly used as an important biomarker to identify cancer stem cells (Glumac and LeBeau, 2018). We found a significant overexpression of CD133 and E-cadherin and a downregulation of Vimentin and SLUG in Panc1 SMAD4-KD cells (Figure 5J, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3). In contrast, E-cadherin expression decreased, whereas N-cadherin, Vimentin, SNAIL, and SLUG were significantly increased in both PDA cells after SMAD4-OE (Figures 5K and 5L, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3), pointing to the initiation of the EMT process and gain of motility in SMAD4-OE cells, increasing the invasiveness potential of these cells. Furthermore, the decrease in the expression level of CD133 after SMAD4-OE indicated a reduction of CSCs (cancer stemness cells) along with the increased levels of SMAD4. Interestingly, we observed morphological changes in the cells (from cuboidal shaped to spindle shaped), after the activation of the TGFβ canonical pathway (Figure S7). The morphological cellular change may facilitate cells to traverse the cellular matrix into intercellular spaces. Consistently, the upregulation of Vimentin pointed to an alteration of the cytoskeleton in SMAD4-OE cells (Figures 5K and 5L). However, this morphological change was not observed in the SMAD4-deficient or SMAD4-KD cells.
Altogether, these results reinforce the important role of SMAD4 and TGFβ in the EMT process and PDA cancer metastasis; on the other hand, the absence of SMAD4 enhanced tumor stemness in our PDA model system.
We then further analyzed the possible role of the interplay between the TGFβ pathway and the biological clock in the formation of cancer metastasis. Interestingly, cell migration decreased upon PER2 KD, but increased after NR1D1 KD in SMAD4-proficient cells (Panc1, Figures 6A and 6C, ∗∗∗p < 0.001, n = 3), in contrast to the results in SMAD4-deficient cells (AsPC1, Figures 6B and 6D, ∗∗∗p < 0.001, n = 3). The TGFβ stimulation promoted the migration properties in clock genes KDs Panc1cells (Figure 6E, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3). TGFβ stimulation did not alter the migration ability of AsPC1 cells containing empty vectors (Figures 5F and 6F). On the other hand, TGFβ stimulation repressed migration in shBMAL1 and shPER2 and induced it in shNR1D1 cells (Figure 6F, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3), indicating that the interactions of the clock and the TGFβ non-canonical pathway may modify migration properties in SMAD4-deficient cells (AsPC1).
Figure 6.

Dysregulation of Core-Clock Genes Affects Migration of Panc1 and AsPC1
(A, B, E, and F) Migration properties of shCtrl and knockdown cells (shBMAL1, shPER2, and shNR1D1) of Panc1 and AsPC1 with stimulation of TGFβ1 (10 ng/mL) or its solvent. Migration assays were performed using a scratch wound assay for the IncuCyte S3 Live Cell System Analysis. Average cell migration speed distribution from Panc1 and AsPC1 cells (n ≥ 8, mean ± SEM; (A and B): one-way ANOVA; (E and F): t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(C and D) Partial representation of the scratch wound assay for Panc1 (C) and AsPC1 (D) knockdown (shBMAL1, shPER2 and shNR1D1) and control (shCtrl) cells at 0, 24, and 48 h. Images were obtained with the IncuCyte S3 Software. Yellow mask indicates the wound boundaries. Blue mask indicates the initial scratch wound area.
(G, H, K, and L) Invasion assays were carried out using a chemotaxis cell invasion assay for the IncuCyte S3 Live Cell System Analysis. Invasion assay for shCtrl and knockdown (shBMAL1, shPER2 and shNR1D1) conditions in Panc1 (G, K) and AsPC1 (H, L) cells with or without TGFβ1 stimulation (10 ng/mL). Quantification was performed by measuring the total phase area of the bottom layer of the inner chamber within 72 h. Data were normalized to the initial value and presented as mean ± SEM, n ≥ 6.
(I and J) mRNA levels of CD133, Vimentin, E-cadherin, and N-cadherin in PDA cells were analyzed after knockdown of core-clock genes (shBMAL1, shPER2, shNR1D1). Data shown as comparison to the shCtrl (mean ± SEM, n = 3, t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
See also Figure S8.
Therefore, we examined the expression level of clock genes after a 24-h TGFβ stimulation compared to Ctrl. We found that TGFβ stimulation upregulated NR1D1 expression in AsPC1 (SMAD4-deficient) NR1D1 KD cells (Figure S8D, ∗∗∗p < 0.001, n = 3). Considering the decreased migration potential observed upon NR1D1 KD (Figure 6B), the overexpression of NR1D1 resulting from TGFβ stimulation (Figure S8D) might be the cause of the increase in migration in NR1D1 KD cells (Figure 6F).
We further carried out trans-well invasion assays, which are more similar to the in vivo scenario. The results supported the conclusion from the migration assays (Figures 6G, 6H, 6K, and 6L).
We quantified the expression of N-cadherin, E-cadherin, Vimentin, SNAIL, and SLUG. Although the expression of E-cadherin was not significantly altered in PER2 KD Panc1 cells, the expression of key MET (mesenchymal-epithelial transition, the reverse process of EMT) markers (N-cadherin, Vimentin, and SLUG) were significantly reduced, which hints toward initiation of MET in these cells, compared to the control cell line (Figures 6I and S8A, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Although N-cadherin expression was not significantly altered (Figure 6I), the significant overexpression of SNAIL and SLUG in Panc1 cells upon NR1D1 KD (Figure S8A) suggested that NR1D1 KD initiated the EMT process in Panc1 cells, which is further supported by our results on migration and invasion. In AsPC1 cells, N-cadherin expression increased significantly upon PER2 KD, whereas NR1D1 KD led to the downregulation of SLUG (Figures 6J and S8B, ∗p < 0.05, ∗∗∗p < 0.001, n = 3). Thus, PER2 KD triggered the EMT process in AsPC1 (SMAD4 deficient) cells, as supported by our results from the migration and invasion assays. Interestingly, N-cadherin was significantly increased, but SLUG was suppressed in AsPC1 shBMAL1 cells (Figures 6J and S8, ∗∗∗p < 0.001). However, there was no significant increase of the migration ability of shBMAL1 when compared with shCtrl (Figure 6B). Furthermore, CD133 was overexpressed in PER2 KD for both PDA cell lines (Figures 6I and 6J, ∗p < 0.05), pointing to a role of PER2 as a potential suppressor of tumor stemness in these cells.
In addition, TGFβ impacts the expression of clock genes in a SMAD4-dependent manner (Figures S8C and S8D). BMAL1 and NR1D1 were upregulated in SMAD4-proficient cells (Panc1), and PER2 was upregulated in SMAD4-deficient cells (AsPC1). These alterations on clock gene expression observed in shCtrl cells were not present upon BMAL1 and NR1D1 KDs in Panc1 and PER2 KD in AsPC1. The expression of clock genes in AsPC1 shBMAL1 revealed no significant difference with or without additional TGFβ. These results indicated that TGFβ upregulated the expression of core-clock genes differentially via the canonical and non-canonical pathway in our PDA cell model system.
The Knockdown of Core-Clock Genes and Modifications of the TGFβ Canonical Pathway Impact Treatment Response and Patient Survival
The main problem in current PDA standard therapy with gemcitabine is the rapid development of chemoresistance. Gemcitabine, an analog of deoxycytidine, leads to DNA fragmentation and cell death by inhibiting DNA chain elongation (Noble and Goa, 1997). We further investigated whether SMAD4 or clock dysfunction impacts the response of gemcitabine in our vitro model system. Our results showed that SMAD4-OE PDA cells were remarkably more sensitive to gemcitabine compared with oeCtrl, whereas shSMAD4 cells were resistant to gemcitabine (Figures 7A–7C). Next, we explored the differential drug response of these two cell lines after core-clock gene KD. Although the gemcitabine IC50 of AsPC1(23.9μM) is much higher than that of Panc1 (9.5μM), AsPC1 shCtrl cells revealed higher viability after 72-h treatment when compared with Panc1 shCtrl. This further confirms an increased resistance to gemcitabine in the absence of SMAD4, in PDA cells. Unlike AsPC1, Panc1 clock gene KD cells showed significant resistance to gemcitabine treatment compared with shCtrl cells (Figures 7D and 7E, ∗∗∗p < 0.001). These results emphasize the importance of circadian dysfunction in gemcitabine resistance in SMAD4-proficient PDA cells. The dysregulation of the circadian clock in Panc1 cells increased the resistant to gemcitabine, when compared with SMAD4-deficient cells (AsPC1), suggesting that SMAD4 impacts gene expression programs associated with the circadian clock and drug response in Panc1, which are distinct from AsPC1. These data highlight a novel role of the circadian clock in fine-tuning drug effectiveness in PDA with an emphasis on SMAD4. Moreover, to test whether drug administration at different times had different effects, we selected the time points 17, 20, and 23 h based on the time course RT-qPCR and bioluminescence data for the two cell lines, which show different expression of the core-clock genes measured (Figure S9A). Our data show that the timing of treatment administration has different effects in cell survival depending also on the additional perturbations generated to the cells. At particular times, the SMAD4-proficient PDA cells are more resistant to treatment, whereas for the SMAD4-deficient AsPC1 cells this effect was not observed (Figures S9B and S9C). After downregulation of SMAD4 in Panc1 (Figure S9B), the differential drug response to different treatment time points is significantly impaired. These results point to a role of SMAD4 in the time-dependent effect of treatment administration.
Figure 7.

The Downregulation of Core-Clock Genes and the Alteration of SMAD4 Expression Affects Drug Response in Panc1 and AsPC1 Cells
Cytotoxicity assays were performed using either an IncuCyte Red Cytotoxicity Reagent or NucLight Rapid Red Reagent for the IncuCyte S3 Live Cell System Analysis
(A–C) Cytotoxicity analysis for Panc1 (A, B) and AsPC1 (C) cells containing SMAD4-KD or SMAD4-OE constructs, in comparison with the corresponding empty controls (shCtrl and oeCtrl). 0 h after cell synchronization, gemcitabine was dissolved in IncuCyte Red Cytotoxicity Reagent and added to PDA cells (IC50: Panc1, 9.5 μM, and AsPC1, 23.9 μM). A cytotoxic index was calculated on IncuCyte S3 Live-Cell Analysis System in red phases. Data are expressed as mean ± SEM, n ≥ 6.
(D and E) Cytotoxicity analysis for Panc1 (D) and AsPC1 (E) cells containing knockdown constructs (shBMAL1, shPER2 and shNR1D1) or its empty vector (shCtrl). Cell nuclei were labeled with NucLight Rapid Red Reagent. At 0 h after cell synchronization, gemcitabine (IC50: Panc1, 9.5 μM, and AsPC1, 23.9 μM) was added into the cells. 72 h after treatment, the number of viable cells per well was quantified with IncuCyte S3 Live-Cell Analysis System. Compared with the shCtrl (mean ± SEM, n = 6, one-way ANOVA, ∗∗∗p < 0.001). See also Figure S9.
(F) Impact of alterations in our 10 candidate genes on progression-free survival (PFS) in a TCGA cohort of patients with pancreatic adenocarcinoma.
(G) Impact of SMAD4 mutation on PFS in a TCGA cohort of patients with pancreatic adenocarcinoma when compared with patients who do not have SMAD4 mutation (log rank p < 0.05).
(H) Impact of variation in candidate gene expression on overall survival in a TCGA cohort of patients with pancreatic adenocarcinoma. Survival curves were plotted using a Cox model that includes coxph (Surv (times, died) ~ gene + grade1 + grade2 + grade3 + age). Statistically significant results (p < 0.05) are shown in the figure. The project ID used for Kaplan Meier survival analysis (F and H) is retrieved from TCGA PAAD, dbGaP Study Accesion No: phs000178 (https://portal.gdc.cancer.gov/projects/TCGA-PAAD).
We then analyzed the impact of alterations in the clock genes BMAL1, PER2, NR1D1, CRY1, DEC1, and DEC2, as well as elements of the TGFβ pathway, TGFβ1, SMAD3, SMAD4, and SMAD7 in patient survival. For that we quantified the mutational frequencies of these 10 genes from a cohort of patients with pancreatic adenocarcinoma (184 samples from the PanCancer study, Study Accession No: phs000178, https://portal.gdc.cancer.gov/projects/TCGA-PAAD) (Cancer Genome Atlas Research Network, 2017) curated from The Cancer Genome Atlas (TCGA) database. Among these candidate genes, SMAD4 showed the highest mutation rate in the population (34%).
Patients with SMAD4 mutation (mostly deletion) had a significant worse progression-free survival (PFS, p < 0.05) (Figure 7G). This suggested that SMAD4 mutation in the advanced stage of the disease facilitates cancer progression. Interestingly if we compare the survival between the group of patients having no mutations for any of the candidate genes with the patients having mutations in at least one of the candidate genes, our results show a statistically significant influence on PFS (p < 0.05, Figure 7F). These results suggest that SMAD4 mutation alone had a significant impact on the PFS, which is likely to be increased with the influence of additional mutations in our other candidate genes (Figures 7F and 7G).
To study the influence of alterations in gene expression for the set of genes described earlier in survival of patients, we performed a cox-regression-based survival analysis based on clinical parameters that include age, tumor grade, and gender in addition to the gene expression levels (Figure 7H). To evaluate high- and low-risk groups based on gene expression changes, we applied OncoLnc to 177 available mRNA datasets (RNA-Seq v.2) from the TCGA, Pancreatic Adenocarcinoma, patient cohort, which we divided into two equal cohorts (N = 87 low, N = 87 high) representing high- and low-expression groups, respectively (Figure 7H). The clock genes CRY1 and DEC1, and TGFβ signaling family members SMAD3 and SMAD4, showed significant impact on survival (Figure 7H, log rank test p < 0.05) for PDA patients suggesting a potential clinical value for targeting these genes in a therapeutic context. Together with the results from our in vitro model, the expression of the clock genes DEC1 and CRY1 also revealed a SMAD4 dependency. These data highlight an important role for the cross-talk between the endogenous molecular clock and the TGFβ canonical pathway and its likely contribution to the progression and prognosis of PDA.
Discussion
A dysregulated clock in patients with cancer was shown to be associated with poorer prognosis and a shorter lifespan. Pancreatic cancer is one of the major causes of cancer mortality worldwide. Several studies have identified disruptions in clock genes, which are correlated with cancer progression in pancreatic cancer (Jiang et al., 2016; Relles et al., 2013; Tavano et al., 2015; Wu et al., 2012). Among these genes are members of the core-clock machinery (BMAL1 and PER2), as well as clock-regulated genes (SIRT1 and DEC1), which either show abnormal expression or altered rhythmic pattern resulting in cancer progression and EMT (Jiang et al., 2016; Relles et al., 2013; Tavano et al., 2015; Wu et al., 2012). However, the detailed mechanism of circadian clock regulation in pancreatic cancer is still unclear. TGFβ is involved in oncogenic transformation and cancer progression, and it plays a vital role in cancer homeostasis. As tumors grow and progress, genetic alterations often occur on TGFβ canonical signaling components (including SMAD4 mutations), leading to the activation of pro-oncogenic pathways such as RAS, PI3K, and MAPK (Principe et al., 2017). These genetic modifications eventually override the growth inhibitory effects of the TGFβ canonical pathway, further contributing to tumor development. Although it is known that elements of TGFβ canonical pathway are involved in circadian regulation (Akagi et al., 2017; Chen et al., 2015; Sato et al., 2019), the molecular mechanisms underlying this cross-talk in human cancers remain largely uncharacterized.
In the present study, we first characterized the circadian phenotype of pancreatic cancer cells, with similar proliferation profiles, originated from a primary tumor (Panc1) and metastasis ascites (AsPC1). The cells derived from the primary tumor show a differential clock phenotype when compared with the cells derived from the metastasis ascites, pointing to a differential regulation of the clock machinery in different types, genomic background, and stages of cancer, in agreement with previous reports for other cancer types (Fuhr et al., 2018; Relogio et al., 2014).
To explore the possible mechanisms connecting the circadian clock and tumor progression in pancreatic cancer, we focused on the CCG SMAD4. As the core mediator of the TGFβ canonical pathway, SMAD4 is known to assist the modulation of cancer progression (reviewed in Xu et al., (2009)). We hypothesized that SMAD4 could affect the circadian phenotype and subsequently malignant phenotypes of PDA cells. Hence, we established a pancreatic cancer in vitro model consisting of SMAD4-positive (Panc1) and SMAD4-negative (AsPC1) cell lines and investigated this interplay through gene knockdown and overexpression of core-clock elements, TGFβ and SMAD4. Our data show that TGFβ1, SMAD3, SMAD4, and SMAD7 exhibit circadian oscillation at the transcript level in Panc1 cells, pointing to the circadian regulation of the TGFβ pathway in SMAD4-proficient PDA cells. Chen et al. reported that the transcript of TGFβ1 oscillates in a circadian fashion in mouse embryonic fibroblasts, whereas the oscillation is perturbed in Clock-deficient mice (Chen et al., 2015). Furthermore, the authors showed that binding of the Bmal1-Clock heterodimer to mTGFβ1 exhibits a circadian rhythmicity in mouse renal cells (Chen et al., 2015). Another study showed enriched Bmal1 binding on promoter regions of TGFβ1 and Smad3, which occurred at the similar time as Bmal1 E-box binding to Nr1d1 (at CT8) in mouse brown adipocytes (Nam et al., 2015). These results indicate that Bmal1 activates the expression of components of the TGFβ canonical pathway (via TGFβ1 and Smad3) in parallel with the activation of Nr1d1. However, whether mRNA dynamics of TGFβ signaling elements are under the circadian modulation in human cancers is still largely uncharacterized. Interestingly, our results show that the circadian oscillation (period of circa 24 h) of elements of the TGFβ pathway is only detectable in SMAD4-positive cells and not in SMAD4-deficient cells. In the absence of extrinsic TGFβ, transcription of SMAD7 can be induced by the intrinsic TGFβ following a circadian pattern. TGFβ1 transcript also shows a similar period with a different phase than SMAD7. This further suggests that autocrine TGFβ may activate the transcription of SMAD7 in a circadian fashion. We also detected circadian rhythms in BMAL1 mRNA of SMAD4-positive cells, resembling our observation in transcripts of TGFβ canonical elements (SMAD3 and TGFB1). This supported our hypothesis that SMAD4 may serve as a mediator element bridging the clock and the TGFβ signaling pathway.
It has been previously shown that the core-clock genes Bmal1 and Nr1d1 are involved in the TGFβ canonical pathway in mouse brown adipose (Nam et al., 2015). In agreement, we show that clock perturbation via small hairpin RNA knockdown of core-clock elements (BMAL1 and NR1D1) resulted in abolished oscillations of TGFβ1, SMAD3, SMAD4, and SMAD7, confirming the existence of bidirectional connection between core-clock elements and the TGFβ canonical pathway in human PDA cells. In addition, the transcription factors, DEC1 and DEC2, inhibit PER via protein-protein interactions with BMAL1 and/or competition for E-box elements (Honma et al., 2002). Of note, DEC1/2 is a downstream element of TGFβ canonical pathway as well (Adorno et al., 2009; Kon et al., 2008; Prunier et al., 2019; Zawel et al., 2002). To identify the specific bridging elements between the core-clock and the TGFβ signaling, we further analyzed the expression of the clock genes DEC1 and DEC2 (Figures 2D, 2G, and 2J). We observed a negative correlation between SMAD4 and DEC1/2 expression through SMAD4 downregulation and overexpression assays, which reinforced the mediating role of DEC1/2 via TGFβ pathway in regulating the circadian clock.
We further investigated whether TGFβ activation and SMAD4 expression could affect the oscillatory phenotype of BMAL1 and PER2 promoter activity in both SMAD4-positive and SMAD4-deficient pancreatic cancer cell lines using live bioluminescence recordings. Surprisingly, TGFβ stimulation alone did not cause major changes in the period of oscillations for both cell lines. However, the period was significantly shorter when we combined TGFβ stimulation with SMAD4 overexpression in both cell lines, indicating that the TGFβ-clock interplay might be SMAD4 dependent.
Downregulation of core-clock genes resulted in an overall higher proliferative and apoptotic activity in SMAD4-positive cells. Of note, SMAD4 downregulation only affected cell proliferation and not apoptosis in these cells. We speculate that reduced expression of SMAD4 might activate the non-SMAD TGFβ signaling pathway, including the MAPK/ERK, resulting in higher cell proliferation and growth (reviewed in Zhang, 2017). We confirmed the activation of the Ras-mediated TGFβ non-canonical pathway by measuring the expression of RAS and p-ERK, and particularly after stimulation with TGFβ, further pointing to the activation of the RAS/ERK pathway.
In SMAD4-deficient cells, the apoptotic rate was also increased upon knockdown of clock genes. However, cell proliferation was only notably increased upon PER2 downregulation in AsPC1. Our cell cycle analysis also confirmed this finding showing higher percentage of shPER2 cells in the S phase. The overall differential proliferative properties and cell cycle distributions upon the KD of clock genes in both PDA cells suggest that cell fate determination via the clock is altered in SMAD4 mutant (AsPC1) cells. SMAD4 overexpression, on the other hand, resulted in remarkably higher cellular apoptosis in both cell lines. This confirms the apoptotic role of the canonical TGFβ signaling pathway mediated by SMAD4, as described in previous studies, and hints to the central role of SMAD4 in regulating the canonical TGFβ signaling pathway (Du et al., 2018; Pang et al., 2011). This effect is more evident in our SMAD4-deficient cells, with a notable increase in apoptosis upon SMAD4 overexpression with additional TGFβ1 stimulation.
Previous reports indicate a prominent role for the TGFβ signaling pathway in regulating cell cycle progression (Buenemann et al., 2001; Mukherjee et al., 2010; Voss et al., 1999). Indeed, we showed in our PDA model that cell-cycle arrest was triggered when cells were stimulated with TGFβ and SMAD4 was overexpressed. We observed an opposite effect after SMAD4 downregulation resulting in more S-phase cells and proliferation. Furthermore, TGFβ stimulation alone inhibited G1/S cell cycle propagation only in SMAD4-positive cells. These results highlight the determinant role of SMAD4 in regulating cell cycle proliferation and apoptosis via downstream genes of TGFβ canonical and non-canonical signaling pathway (e.g., P21, c-MYC, and RAS) (Alexandrow and Moses, 1995; Mukherjee et al., 2010; Principe et al., 2017; Voss et al., 1999), further pointing to a differential regulation of TGFβ pathway in different stages of pancreatic cancer, as reported in the present or former studies in a SMAD4-dependent manner.
Regulating EMT and cell invasion is one of the key functions of TGFβ signaling pathway in cancer cells (Lee et al., 2013; Xu et al., 2009). Cells undergoing EMT lose expression of E-cadherin and other components needed for epithelial cell junction stability and are able to produce a mesenchymal cell cytoskeleton acquiring motility and invasive properties (Massague, 2008). The reverse process of EMT, MET, involves the activation of E-cadherin and suppression of N-cadherin (Yao et al., 2011). Hence, we analyzed migration/invasion properties upon SMAD4 expression and TGFβ stimulation in both cell models. TGFβ stimulation alone was sufficient to enhance cell migration in SMAD4-positive cells, whereas in SMAD4-deficient cells, both SMAD4 overexpression and TGFβ stimulation resulted in the highest migratory and invasion activities, suggesting that SMAD4 expression is necessary for enhancing cancer metastasis. We confirmed this finding through SMAD4 downregulation and showed that cell migration was not affected upon TGFβ stimulation. After measuring the expression of EMT marker genes (e.g., E-Cad, N-Cad, SNAIL, SLUG) in pancreatic cancer (Takano et al., 2007), we found that SMAD4 overexpression reduced the expression of epithelial marker genes (E-Cad) and enhanced the expression of genes involved in mesenchymal transition (N-Cad, Vim, SNAIL, and SLUG) in both cell lines, indicating that SMAD4 mediates invasion by regulating the expression of key marker genes involved in EMT. We observed the opposite effect in EMT gene expression after SMAD4 knockdown, which reinforces the role of SMAD4 in cancer invasion. In addition, we sought to explore another cancer hallmark, tumor stemness via the analysis of CD133 (Tabu et al., 2010). In our model system, we observed a downregulation of CD133 in PDA cells with SMAD4 overexpression. More interestingly, increased CD133 expression after SMAD4 KD implied the acquisition of cancer stem-like hallmark in our study, in agreement with previous studies (Chen et al., 2014).
Cancer metastasis is the cumulative result of multiple changes in tumor cells and their microenvironment. Cell migration was differentially regulated in SMAD4-positive and SMAD4-deficient cells upon PER2 and NR1D1 downregulation (Figure 6). In SMAD4-positive cells, cell migration was decreased upon PER2 knockdown and increased after NR1D1 downregulation, whereas we observed an opposite effect in the cell line deficient for SMAD4. Interestingly, TGFβ activation also implied an overall opposite effect in the core-clock knockdown cells from SMAD4-positive and SMAD4-negative PDA: promoting migration in Panc1 cells and inhibiting it in AsPC1 cells. These results show that cancer cell properties seem to be SMAD4-dependent upon clock dysregulation, highlighting the reciprocal role of the circadian clock and SMAD4 in pancreatic cancer. Notably, the downregulation of PER2 results in the increased expression of CD133 in both PDA cells, hinting at the crucial role of PER2 in cancer stemness independent of SMAD4 (Katamune et al., 2019). Hence, based on our results on proliferation, apoptosis, cell cycle, and cellular metastatic proprieties, we speculated that TGFβ might alter the circadian clock differently via the canonical (in Panc1) and non-canonical (in AsPC1) pathways.
Finally, we tested whether treatment response in pancreatic cancer is clock and/or SMAD4 dependent. Results from previous clinical trials concluded that the loss of SMAD4 is a negative prognostic indicator, which is strongly associated with poor chemosensitivity (Shin et al., 2017; Singh et al., 2012; Yamada et al., 2015). Extrinsic DNA damage due to gemcitabine administration imposes threats to cellular homeostasis involving the regulation of both the TGFβ signaling pathway (Li et al., 2019; Liu et al., 2019) and clock components (Jiang et al., 2018; Ka et al., 2017; Mazzoccoli et al., 2016; Sulli et al., 2018). To elucidate the molecular mechanisms that drive chemoresistance in this PDA model, we hypothesized that during the exposure to gemcitabine, clock genes and TGFβ canonical components participate in the regulation of DNA repair, as well as in the maintenance of genomic stability.
A previous study showed that patients with PDA with SMAD4 loss have poorer PFS in the gemcitabine-based treatment subgroup (Ormanns et al., 2017). Consistently, we observed that SMAD4-deficient cells are resistant to the chemotherapy with gemcitabine. Although the intrinsic mechanism has not been fully understood yet, based on our apoptosis and cell cycle data, we assume that it relates to the effect in the cell cycle due to the anti-apoptotic role of SMAD4.
Clock perturbation via the knockdown of core-clock genes aggravates chemoresistance, notably only in SMAD4-proficient pancreatic cancer cells. Associated with the fact that genetic alterations (mostly inactivation) of SMAD4 often occur in the advanced stage of pancreatic cancers, we concluded that the molecular clock functions as a repressor of chemoresistance of gemcitabine while SMAD4 is not mutated (e.g., cancers in the early stage). Hence, loss of Smad4 function along with TGFβ stimulation (as observed for AsPC1, SMAD4-deficient cells) activates the non-canonical pathway (RAS) and increases CSCs (CD133 upregulation), further contributing to the acquisition of malignant properties and drug resistance. These findings highlight the important role of SMAD4 in affecting gene expression programs associated with the circadian clock and drug response in SMAD4-proficient cells, which are distinct from those in SMAD4-deficient cells. Furthermore, our results emphasize the importance of circadian clock dysfunction in gemcitabine resistance in SMAD4-proficient PDA cells. Our data indicate that drug response and cell fate determination in pancreatic cancer cells with WT or mutated SMAD4, upon the dysregulation of the circadian clock, resulted in a significant differential change in cell survival rate and invasiveness, highlighting the importance of the associations between the TGFβ canonical pathway, biological clock, and drug timing affecting chemosensitivity in a cancer context.
The information that we obtained from our in vitro model system aids to the understanding of the impact of the clock in the SMAD4 network and consequently in cell fate decisions. In addition, and to better show the potential of our results beyond the in vitro experiments presented, we performed a computational analysis with publicly available datasets for patients with pancreatic cancer. In our PDA model system, the overexpression of SMAD4 resulted in upregulation of CRY1 and downregulation of DEC1; the downregulation of SMAD4 led to suppression of CRY1 and overexpression of DEC1. These results agree with our survival analysis for a cohort of patients with PDA that indicated a better prognosis (PFS, p < 0.05) for the groups of patients with high expression of CRY1, low expression of DEC1, and high expression of SMAD4. In addition, the genomic alteration of SMAD4 (mostly deletion) alone led to a worse PFS (p = 0.0265), which is aggravated when combined with alterations of other genes bridging the clock and the TGFβ canonical pathway (p = 0.0147). This points to a relevant role of the molecular clock, together with the TGFβ pathway, in contributing to the progression and prognosis of PDA. Altogether, our data show that the circadian clock regulates the TGFβ/SMAD4 pathway in PDA cells with likely consequences on cancer progression (invasion and EMT) and patient survival and suggest a profound role for SMAD4 as an intermediate component linking the clock machinery to the TGFβ pathway. This information may be relevant for patients with pancreatic cancer undergoing chemotherapy to increase the effectiveness of the therapy based on SMAD4 mutation and the circadian clock.
Limitations of the Study
We consider the lack of in vivo data as a limitation of our study, with the caveat that mice (as nocturnal animals) have a different clock compared with humans (as diurnal animals) and hence results from an in vivo mouse model regarding alteration of the circadian clock may not be directly relevant or properly translated to human disease models. This is particularly relevant when comparing drug timing, drug toxicity, and drug efficacy between nocturnal and diurnal models (reviewed in Dallmann et al., 2016). In addition, we have previously shown that in circadian studies, the host-cancer interaction in in vivo models (using xenografts) has an impact on the circadian phenotype of the xenograft, which adds complexity to the interpretation of the results with regards to the circadian machinery (Basti et al., 2020).
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Angela Relógio (angela.relogio@charite.de).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate datasets/code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Rosa Schmuck from the Chirurgischen Klinik, Charité – Universitätsmedizin Berlin, for providing the cell lines, and Dr. Sam Thiagalingam from the department of medicine, Boston University, for providing pBabe-puro-Smad4-Flag. The work in A.R.'s group was funded by the German Federal Ministry of Education and Research (BMBF)—eBio-CIRSPLICE - FKZ031A316, and by the Dr. Rolf M. Schwiete Stiftung. Y.L. was additionally funded by Jinan Huaiyin Hospital of Shandong Province. Y.L., A.B., and M.Y. were additionally funded by the Berlin School of Integrative Oncology (BSIO) of the Charité – Universitätsmedizin Berlin. We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité – Universitätsmedizin Berlin.
Author Contributions
Conceptualization, A.R.; Methodology, A.R. and Y.L.; Investigation, Y.L., A.B., and M.Y.; Validation, Y.L., A.B., and A.R.; Writing – Original Draft, Y.L., A.B., and A.R.; Writing – Review & Editing, Y.L., A.B., M.Y., and A.R.; Funding Acquisition, A.R.; Resources, A.R.; Supervision, A.R.
Declaration of Interests
The authors declare that they have no conflict of interest.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101551.
Supplemental Information
References
- Abreu M., Basti A., Genov N., Mazzoccoli G., Relogio A. The reciprocal interplay between TNFalpha and the circadian clock impacts on cell proliferation and migration in Hodgkin lymphoma cells. Sci. Rep. 2018;8:11474. doi: 10.1038/s41598-018-29847-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adorno M., Cordenonsi M., Montagner M., Dupont S., Wong C., Hann B., Solari A., Bobisse S., Rondina M.B., Guzzardo V. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- Akagi R., Akatsu Y., Fisch K.M., Alvarez-Garcia O., Teramura T., Muramatsu Y., Saito M., Sasho T., Su A.I., Lotz M.K. Dysregulated circadian rhythm pathway in human osteoarthritis: NR1D1 and BMAL1 suppression alters TGF-beta signaling in chondrocytes. Osteoarthr. Cartil. 2017;25:943–951. doi: 10.1016/j.joca.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrow M.G., Moses H.L. Transforming growth factor beta and cell cycle regulation. Cancer Res. 1995;55:1452–1457. [PubMed] [Google Scholar]
- Basti A., Fior R., Yalin M., Povoa V., Astaburuaga R., Li Y., Naderi J., Godinho Ferreira M., Relogio A. The core-clock gene NR1D1 impacts cell motility in vitro and invasiveness in A zebrafish xenograft colon cancer model. Cancers (Basel) 2020;12:853. doi: 10.3390/cancers12040853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrozpe G., Schaeffer J., Peinado M.A., Real F.X., Perucho M. Comparative analysis of mutations in the p53 and K-ras genes in pancreatic cancer. Int. J. Cancer. 1994;58:185–191. doi: 10.1002/ijc.2910580207. [DOI] [PubMed] [Google Scholar]
- Buenemann C.L., Willy C., Buchmann A., Schmiechen A., Schwarz M. Transforming growth factor-beta1-induced Smad signaling, cell-cycle arrest and apoptosis in hepatoma cells. Carcinogenesis. 2001;22:447–452. doi: 10.1093/carcin/22.3.447. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185–203 e113. doi: 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.D., Yeh J.K., Peng M.T., Shie S.S., Lin S.L., Yang C.H., Chen T.H., Hung K.C., Wang C.C., Hsieh I.C. Circadian CLOCK mediates activation of transforming growth factor-beta signaling and renal fibrosis through cyclooxygenase 2. Am. J. Pathol. 2015;185:3152–3163. doi: 10.1016/j.ajpath.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Chen Y.W., Hsiao P.J., Weng C.C., Kuo K.K., Kuo T.L., Wu D.C., Hung W.C., Cheng K.H. SMAD4 loss triggers the phenotypic changes of pancreatic ductal adenocarcinoma cells. BMC Cancer. 2014;14:181. doi: 10.1186/1471-2407-14-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T., Shen H., Rodrigues N., Stier S., Scadden D.T. Transforming growth factor beta 1 mediates cell-cycle arrest of primitive hematopoietic cells independent of p21(Cip1/Waf1) or p27(Kip1) Blood. 2001;98:3643–3649. doi: 10.1182/blood.v98.13.3643. [DOI] [PubMed] [Google Scholar]
- Coveler A.L., Rossi G.R., Vahanian N.N., Link C., Chiorean E.G. Algenpantucel-L immunotherapy in pancreatic adenocarcinoma. Immunotherapy. 2016;8:117–125. doi: 10.2217/imt.15.113. [DOI] [PubMed] [Google Scholar]
- Dallmann R., Okyar A., Levi F. Dosing-time makes the poison: circadian regulation and pharmacotherapy. Trends Mol. Med. 2016;22:430–445. doi: 10.1016/j.molmed.2016.03.004. [DOI] [PubMed] [Google Scholar]
- Davis K., Roden L.C., Leaner V.D., van der Watt P.J. The tumour suppressing role of the circadian clock. IUBMB Life. 2019;71:771–780. doi: 10.1002/iub.2005. [DOI] [PubMed] [Google Scholar]
- Di Micco R., Sulli G., Dobreva M., Liontos M., Botrugno O.A., Gargiulo G., dal Zuffo R., Matti V., d'Ario G., Montani E. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011;13:292–302. doi: 10.1038/ncb2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X., Pan Z., Li Q., Liu H., Li Q. SMAD4 feedback regulates the canonical TGF-beta signaling pathway to control granulosa cell apoptosis. Cell Death Dis. 2018;9:151. doi: 10.1038/s41419-017-0205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Athman R., Genov N.N., Mazuch J., Zhang K., Yu Y., Fuhr L., Abreu M., Li Y., Wallach T., Kramer A. The Ink4a/Arf locus operates as a regulator of the circadian clock modulating RAS activity. PLoS Biol. 2017;15:e2002940. doi: 10.1371/journal.pbio.2002940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhr L., Abreu M., Pett P., Relogio A. Circadian systems biology: when time matters. Comput. Struct. Biotechnol. J. 2015;13:417–426. doi: 10.1016/j.csbj.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhr L., El-Athman R., Scrima R., Cela O., Carbone A., Knoop H., Li Y., Hoffmann K., Laukkanen M.O., Corcione F. The circadian clock regulates metabolic phenotype rewiring via HKDC1 and modulates tumor progression and drug response in colorectal cancer. EBioMedicine. 2018;33:105–121. doi: 10.1016/j.ebiom.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gery S., Gombart A.F., Yi W.S., Koeffler C., Hofmann W.K., Koeffler H.P. Transcription profiling of C/EBP targets identifies Per2 as a gene implicated in myeloid leukemia. Blood. 2005;106:2827–2836. doi: 10.1182/blood-2005-01-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glumac P.M., LeBeau A.M. The role of CD133 in cancer: a concise review. Clin. Transl. Med. 2018;7:18. doi: 10.1186/s40169-018-0198-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma S., Kawamoto T., Takagi Y., Fujimoto K., Sato F., Noshiro M., Kato Y., Honma K. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. 2002;419:841–844. doi: 10.1038/nature01123. [DOI] [PubMed] [Google Scholar]
- Jiang W., Zhao S., Jiang X., Zhang E., Hu G., Hu B., Zheng P., Xiao J., Lu Z., Lu Y. The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the p53 tumor suppressor pathway. Cancer Lett. 2016;371:314–325. doi: 10.1016/j.canlet.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Jiang W., Zhao S., Shen J., Guo L., Sun Y., Zhu Y., Ma Z., Zhang X., Hu Y., Xiao W. The MiR-135b-BMAL1-YY1 loop disturbs pancreatic clockwork to promote tumourigenesis and chemoresistance. Cell Death Dis. 2018;9:149. doi: 10.1038/s41419-017-0233-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka N.L., Na T.Y., Na H., Lee M.H., Park H.S., Hwang S., Kim I.Y., Seong J.K., Lee M.O. NR1D1 recruitment to sites of DNA damage inhibits repair and is associated with chemosensitivity of breast cancer. Cancer Res. 2017;77:2453–2463. doi: 10.1158/0008-5472.CAN-16-2099. [DOI] [PubMed] [Google Scholar]
- Katamune C., Koyanagi S., Hashikawa K.I., Kusunose N., Akamine T., Matsunaga N., Ohdo S. Mutation of the gene encoding the circadian clock component PERIOD2 in oncogenic cells confers chemoresistance by up-regulating the Aldh3a1 gene. J. Biol. Chem. 2019;294:547–558. doi: 10.1074/jbc.RA118.004942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita K., Saito S., Morioka C.Y., Watanabe A. Growth inhibition of human pancreatic cancer cell lines by anti-sense oligonucleotides specific to mutated K-ras genes. Int. J. Cancer. 1999;80:553–558. doi: 10.1002/(sici)1097-0215(19990209)80:4<553::aid-ijc12>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Kon N., Hirota T., Kawamoto T., Kato Y., Tsubota T., Fukada Y. Activation of TGF-beta/activin signalling resets the circadian clock through rapid induction of Dec1 transcripts. Nat. Cell Biol. 2008;10:1463–1469. doi: 10.1038/ncb1806. [DOI] [PubMed] [Google Scholar]
- Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Choi J.H., Joo C.K. TGF-beta1 regulates cell fate during epithelial-mesenchymal transition by upregulating survivin. Cell Death Dis. 2013;4:e714. doi: 10.1038/cddis.2013.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann R., Childs L., Thomas P., Abreu M., Fuhr L., Herzel H., Leser U., Relogio A. Assembly of a comprehensive regulatory network for the mammalian circadian clock: a bioinformatics approach. PLoS One. 2015;10:e0126283. doi: 10.1371/journal.pone.0126283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Liu Y., Chiang Y.J., Huang F., Li Y., Li X., Ning Y., Zhang W., Deng H., Chen Y.G. DNA damage activates TGF-beta signaling via ATM-c-cbl-mediated stabilization of the type II receptor TbetaRII. Cell Rep. 2019;28:735–745.e734. doi: 10.1016/j.celrep.2019.06.045. [DOI] [PubMed] [Google Scholar]
- Lieber M., Mazzetta J., Nelson-Rees W., Kaplan M., Todaro G. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer. 1975;15:741–747. doi: 10.1002/ijc.2910150505. [DOI] [PubMed] [Google Scholar]
- Liu Q., Lopez K., Murnane J., Humphrey T., Barcellos-Hoff M.H. Misrepair in context: TGFbeta regulation of DNA repair. Front. Oncol. 2019;9:799. doi: 10.3389/fonc.2019.00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L., Dauchy R.T., Blask D.E., Slakey L.M., Xiang S., Yuan L., Dauchy E.M., Shan B., Brainard G.C., Hanifin J.P. Circadian gating of epithelial-to-mesenchymal transition in breast cancer cells via melatonin-regulation of GSK3beta. Mol. Endocrinol. 2012;26:1808–1820. doi: 10.1210/me.2012-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoccoli G., Colangelo T., Panza A., Rubino R., De Cata A., Tiberio C., Valvano M.R., Pazienza V., Merla G., Augello B. Deregulated expression of cryptochrome genes in human colorectal cancer. Mol. Cancer. 2016;15:6. doi: 10.1186/s12943-016-0492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P., Winter S.L., Alexandrow M.G. Cell cycle arrest by transforming growth factor beta1 near G1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction. Mol. Cell Biol. 2010;30:845–856. doi: 10.1128/MCB.01152-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam D., Guo B., Chatterjee S., Chen M.H., Nelson D., Yechoor V.K., Ma K. The adipocyte clock controls brown adipogenesis through the TGF-beta and BMP signaling pathways. J. Cell Sci. 2015;128:1835–1847. doi: 10.1242/jcs.167643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble S., Goa K.L. Gemcitabine. A review of its pharmacology and clinical potential in non-small cell lung cancer and pancreatic cancer. Drugs. 1997;54:447–472. doi: 10.2165/00003495-199754030-00009. [DOI] [PubMed] [Google Scholar]
- Ormanns S., Haas M., Remold A., Kruger S., Holdenrieder S., Kirchner T., Heinemann V., Boeck S. The impact of SMAD4 loss on outcome in patients with advanced pancreatic cancer treated with systemic chemotherapy. Int. J. Mol. Sci. 2017;18:1094. doi: 10.3390/ijms18051094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang L., Qiu T., Cao X., Wan M. Apoptotic role of TGF-beta mediated by Smad4 mitochondria translocation and cytochrome c oxidase subunit II interaction. Exp. Cell Res. 2011;317:1608–1620. doi: 10.1016/j.yexcr.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Principe D.R., Diaz A.M., Torres C., Mangan R.J., DeCant B., McKinney R., Tsao M.S., Lowy A., Munshi H.G., Jung B. TGFbeta engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene. 2017;36:4336–4348. doi: 10.1038/onc.2016.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunier C., Baker D., Ten Dijke P., Ritsma L. TGF-beta family signaling pathways in cellular dormancy. Trends Cancer. 2019;5:66–78. doi: 10.1016/j.trecan.2018.10.010. [DOI] [PubMed] [Google Scholar]
- Reinke H., Asher G. Crosstalk between metabolism and circadian clocks. Nat. Rev. Mol. Cell Biol. 2019;20:227–241. doi: 10.1038/s41580-018-0096-9. [DOI] [PubMed] [Google Scholar]
- Relles D., Sendecki J., Chipitsyna G., Hyslop T., Yeo C.J., Arafat H.A. Circadian gene expression and clinicopathologic correlates in pancreatic cancer. J. Gastrointest. Surg. 2013;17:443–450. doi: 10.1007/s11605-012-2112-2. [DOI] [PubMed] [Google Scholar]
- Relogio A., Thomas P., Medina-Perez P., Reischl S., Bervoets S., Gloc E., Riemer P., Mang-Fatehi S., Maier B., Schafer R. Ras-mediated deregulation of the circadian clock in cancer. PLoS Genet. 2014;10:e1004338. doi: 10.1371/journal.pgen.1004338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satelli A., Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato F., Bhawal U.K., Yoshimura T., Muragaki Y. DEC1 and DEC2 crosstalk between circadian rhythm and tumor progression. J. Cancer. 2016;7:153–159. doi: 10.7150/jca.13748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato F., Kohsaka A., Takahashi K., Otao S., Kitada Y., Iwasaki Y., Muragaki Y. Smad3 and Bmal1 regulate p21 and S100A4 expression in myocardial stromal fibroblasts via TNF-alpha. Histochem. Cell Biol. 2017;148:617–624. doi: 10.1007/s00418-017-1597-x. [DOI] [PubMed] [Google Scholar]
- Sato F., Otsuka T., Kohsaka A., Le H.T., Bhawal U.K., Muragaki Y. Smad3 suppresses epithelial cell migration and proliferation via the clock gene Dec1, which negatively regulates the expression of clock genes Dec2 and Per1. Am. J. Pathol. 2019;189:773–783. doi: 10.1016/j.ajpath.2019.01.006. [DOI] [PubMed] [Google Scholar]
- Scandura J.M., Boccuni P., Massague J., Nimer S.D. Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc. Natl. Acad. Sci. U S A. 2004;101:15231–15236. doi: 10.1073/pnas.0406771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte M., Hruban R.H., Hedrick L., Cho K.R., Nadasdy G.M., Weinstein C.L., Bova G.S., Isaacs W.B., Cairns P., Nawroz H. DPC4 gene in various tumor types. Cancer Res. 1996;56:2527–2530. [PubMed] [Google Scholar]
- Shin S.H., Kim H.J., Hwang D.W., Lee J.H., Song K.B., Jun E., Shim I.K., Hong S.M., Kim H.J., Park K.M. The DPC4/SMAD4 genetic status determines recurrence patterns and treatment outcomes in resected pancreatic ductal adenocarcinoma: a prospective cohort study. Oncotarget. 2017;8:17945–17959. doi: 10.18632/oncotarget.14901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel P.M., Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Singh P., Srinivasan R., Wig J.D. SMAD4 genetic alterations predict a worse prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas. 2012;41:541–546. doi: 10.1097/MPA.0b013e318247d6af. [DOI] [PubMed] [Google Scholar]
- Sulli G., Lam M.T.Y., Panda S. Interplay between circadian clock and cancer: new frontiers for cancer treatment. Trends Cancer. 2019;5:475–494. doi: 10.1016/j.trecan.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulli G., Rommel A., Wang X., Kolar M.J., Puca F., Saghatelian A., Plikus M.V., Verma I.M., Panda S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature. 2018;553:351–355. doi: 10.1038/nature25170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C., Yamato T., Furukawa T., Ohnishi Y., Kijima H., Horii A. Characterization of the mutations of the K-ras, p53, p16, and SMAD4 genes in 15 human pancreatic cancer cell lines. Oncol. Rep. 2001;8:89–92. doi: 10.3892/or.8.1.89. [DOI] [PubMed] [Google Scholar]
- Tabu K., Kimura T., Sasai K., Wang L., Bizen N., Nishihara H., Taga T., Tanaka S. Analysis of an alternative human CD133 promoter reveals the implication of Ras/ERK pathway in tumor stem-like hallmarks. Mol. Cancer. 2010;9:39. doi: 10.1186/1476-4598-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano S., Kanai F., Jazag A., Ijichi H., Yao J., Ogawa H., Enomoto N., Omata M., Nakao A. Smad4 is essential for down-regulation of E-cadherin induced by TGF-beta in pancreatic cancer cell line PANC-1. J. Biochem. 2007;141:345–351. doi: 10.1093/jb/mvm039. [DOI] [PubMed] [Google Scholar]
- Tavano F., Pazienza V., Fontana A., Burbaci F.P., Panebianco C., Saracino C., Lombardi L., De Bonis A., di Mola F.F., di Sebastiano P. SIRT1 and circadian gene expression in pancreatic ductal adenocarcinoma: effect of starvation. Chronobiol. Int. 2015;32:497–512. doi: 10.3109/07420528.2014.1003351. [DOI] [PubMed] [Google Scholar]
- Voss M., Wolff B., Savitskaia N., Ungefroren H., Deppert W., Schmiegel W., Kalthoff H., Naumann M. TGFbeta-induced growth inhibition involves cell cycle inhibitor p21 and pRb independent from p15 expression. Int. J. Oncol. 1999;14:93–101. doi: 10.3892/ijo.14.1.93. [DOI] [PubMed] [Google Scholar]
- Wang F., Li C., Yongluo, Chen L. The circadian gene clock plays an important role in cell apoptosis and the DNA damage response in vitro. Technol. Cancer Res. Treat. 2016;15:480–486. doi: 10.1177/1533034615585433. [DOI] [PubMed] [Google Scholar]
- Watanabe M., Sheriff S., Lewis K.B., Cho J., Tinch S.L., Balasubramaniam A., Kennedy M.A. Metabolic profiling comparison of human pancreatic ductal epithelial cells and three pancreatic cancer cell lines using NMR based metabonomics. J. Mol. Biomark. Diagn. 2012;3 doi: 10.4172/2155-9929.S3-002. S3-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock M.J., Shintani Y., Maeda M., Fukumoto Y., Johnson K.R. Cadherin switching. J. Cell Sci. 2008;121:727–735. doi: 10.1242/jcs.000455. [DOI] [PubMed] [Google Scholar]
- Wu Y., Sato F., Yamada T., Bhawal U.K., Kawamoto T., Fujimoto K., Noshiro M., Seino H., Morohashi S., Hakamada K. The BHLH transcription factor DEC1 plays an important role in the epithelial-mesenchymal transition of pancreatic cancer. Int. J. Oncol. 2012;41:1337–1346. doi: 10.3892/ijo.2012.1559. [DOI] [PubMed] [Google Scholar]
- Xu J., Lamouille S., Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin M., El-Athman R., Ouk K., Priller J., Relogio A. Analysis of the circadian regulation of cancer hallmarks by a cross-platform study of colorectal cancer time-series data reveals an association with genes involved in huntington's disease. Cancers (Basel) 2020;12:963. doi: 10.3390/cancers12040963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S., Fujii T., Shimoyama Y., Kanda M., Nakayama G., Sugimoto H., Koike M., Nomoto S., Fujiwara M., Nakao A. SMAD4 expression predicts local spread and treatment failure in resected pancreatic cancer. Pancreas. 2015;44:660–664. doi: 10.1097/MPA.0000000000000315. [DOI] [PubMed] [Google Scholar]
- Yang J., Antin P., Berx G., Blanpain C., Brabletz T., Bronner M., Campbell K., Cano A., Casanova J., Christofori G. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020;21:341–352. doi: 10.1038/s41580-020-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D., Dai C., Peng S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011;9:1608–1620. doi: 10.1158/1541-7786.MCR-10-0568. [DOI] [PubMed] [Google Scholar]
- Zawel L., Yu J., Torrance C.J., Markowitz S., Kinzler K.W., Vogelstein B., Zhou S. DEC1 is a downstream target of TGF-beta with sequence-specific transcriptional repressor activities. Proc. Natl. Acad. Sci. U S A. 2002;99:2848–2853. doi: 10.1073/pnas.261714999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.E. Non-smad signaling pathways of the TGF-beta family. Cold Spring Harb. Perspect. Biol. 2017;9:a022129. doi: 10.1101/cshperspect.a022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets/code.